
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 18 February 2020
Sec. Aquatic Microbiology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.00157
 Delei Song1
Delei Song1 Yunhui Zhang1Ji Liu1Haohui Zhong1Yanfen Zheng1Shun Zhou1Min Yu1,2
Yunhui Zhang1Ji Liu1Haohui Zhong1Yanfen Zheng1Shun Zhou1Min Yu1,2 Jonathan D. Todd3*
Jonathan D. Todd3* Xiao-Hua Zhang1,2,4*
Xiao-Hua Zhang1,2,4*The microbial cycling of dimethylsulfoniopropionate (DMSP) and its gaseous catabolites dimethylsulfide (DMS) and methanethiol (MeSH) are important processes in the global sulfur cycle, marine microbial food webs, signaling pathways, atmospheric chemistry, and potentially climate regulation. Many functional genes have been identified and used to study the genetic potential of microbes to produce and catabolize these organosulfur compounds in different marine environments. Here, we sampled seawater, marine sediment and hydrothermal sediment, and polymetallic sulfide in the eastern Chinese marginal seas and analyzed their microbial communities for the genetic potential to cycle DMSP, DMS, and MeSH using metagenomics. DMSP was abundant in all sediment samples, but was fivefold less prominent in those from hydrothermal samples. Indeed, Yellow Sea (YS) sediment samples had DMSP concentrations two orders of magnitude higher than in surface water samples. Bacterial genetic potential to synthesize DMSP (mainly in Rhodobacteraceae bacteria) was far higher than for phytoplankton in all samples, but particularly in the sediment where no algal DMSP synthesis genes were detected. Thus, we propose bacteria as important DMSP producers in these marine sediments. DMSP catabolic pathways mediated by the DMSP lyase DddP (prominent in Pseudomonas and Mesorhizobium bacteria) and DMSP demethylase DmdA enzymes (prominent in Rhodobacteraceae bacteria) and MddA-mediated MeSH S-methylation were very abundant in Bohai Sea and Yellow Sea sediments (BYSS) samples. In contrast, the genetic potential for DMSP degradation was very low in the hydrothermal sediment samples—dddP was the only catabolic gene detected and in only one sample. However, the potential for DMS production from MeSH (mddA) and DMS oxidation (dmoA and ddhA) was relatively abundant. This metagenomics study does not provide conclusive evidence for DMSP cycling; however, it does highlight the potential importance of bacteria in the synthesis and catabolism of DMSP and related compounds in diverse sediment environments.
Approximately eight billion tons of the organosulfur compound dimethylsulfoniopropionate (DMSP) is produced in Earth’s surface oceans (Galí et al., 2015). Many marine phytoplankton, bacteria, corals, and some plants produce DMSP as an anti-stress compound (Zhang et al., 2019). Once released into the environment, DMSP is a key marine nutrient (Curson et al., 2011a; Zhang et al., 2019), a chemoattractant for marine organisms (Seymour et al., 2010), and it is a major precursor for the climate active gases dimethylsulfide (DMS) and methanethiol (MeSH). DMS is the primary marine source of sulfur delivered to the atmosphere (Lovelock et al., 1972), where DMS oxidative products act as cloud condensation nuclei and may affect the climate (Stefels et al., 2007; Vallina and Simó, 2007). DMSP and the trace gases DMS and MeSH are important components of the global sulfur cycle, and metabolic pathways (Yoch, 2002).
DMSP is synthesized by three known pathways (Figure 1): the methionine (Met) methylation pathway in plants and bacteria (Otte, 2004; Lyon et al., 2011; Williams et al., 2019), a transamination pathway in marine bacteria and algae (Curson et al., 2017, 2018; Zhang et al., 2019), and a decarboxylation pathway in one dinoflagellate (Uchida et al., 2012). The key methylthiohydroxybutyrate S-methyltransferase enzyme of the Met transamination pathway is catalyzed by three distinct S-adenosyl-Met (SAM)-dependent S-methyltransferase enzymes in different organisms. These are DsyB in some alpha-proteobacteria (Curson et al., 2017), DSYB in algae and corals (Curson et al., 2018), and TpMMT in the diatom Thalassiosira pseudonana (Kageyama et al., 2018). Recently, the Met methyltransferase, MmtN of the Met methylation pathway was identified in some Gram-positive bacteria, alpha-proteobacteria, and gamma-proteobacteria (Liao and Seebeck, 2019; Williams et al., 2019). Currently, no enzymes of the Met decarboxylation pathway are known. The dsyB, DSYB, and mmtN genes are robust reporters for the ability of an organism to produce DMSP, but the function of TpMMT encoded by the gene TmMT2 was only confirmed in T. pseudonana. Recent analysis of metagenomic data predicts that 1 and 0.1% of surface saltmarsh sediment bacteria contain dsyB gene and mmtN, respectively (Williams et al., 2019). Furthermore, Williams et al. predicted that ∼104 and 108 bacteria per mL/g of surface seawater or sediment have the genetic potential to produce DMSP. They propose bacteria as significant DMSP producers in both these environments but that their role is much more important in the sediment than the seawater. Curson et al. (2018) found that there were ∼two-fold more algal DSYB transcripts than those for the bacterial dsyB gene in North Pacific Ocean coastal seawater samples. This supports algae as the major DMSP producers in photic seawater.
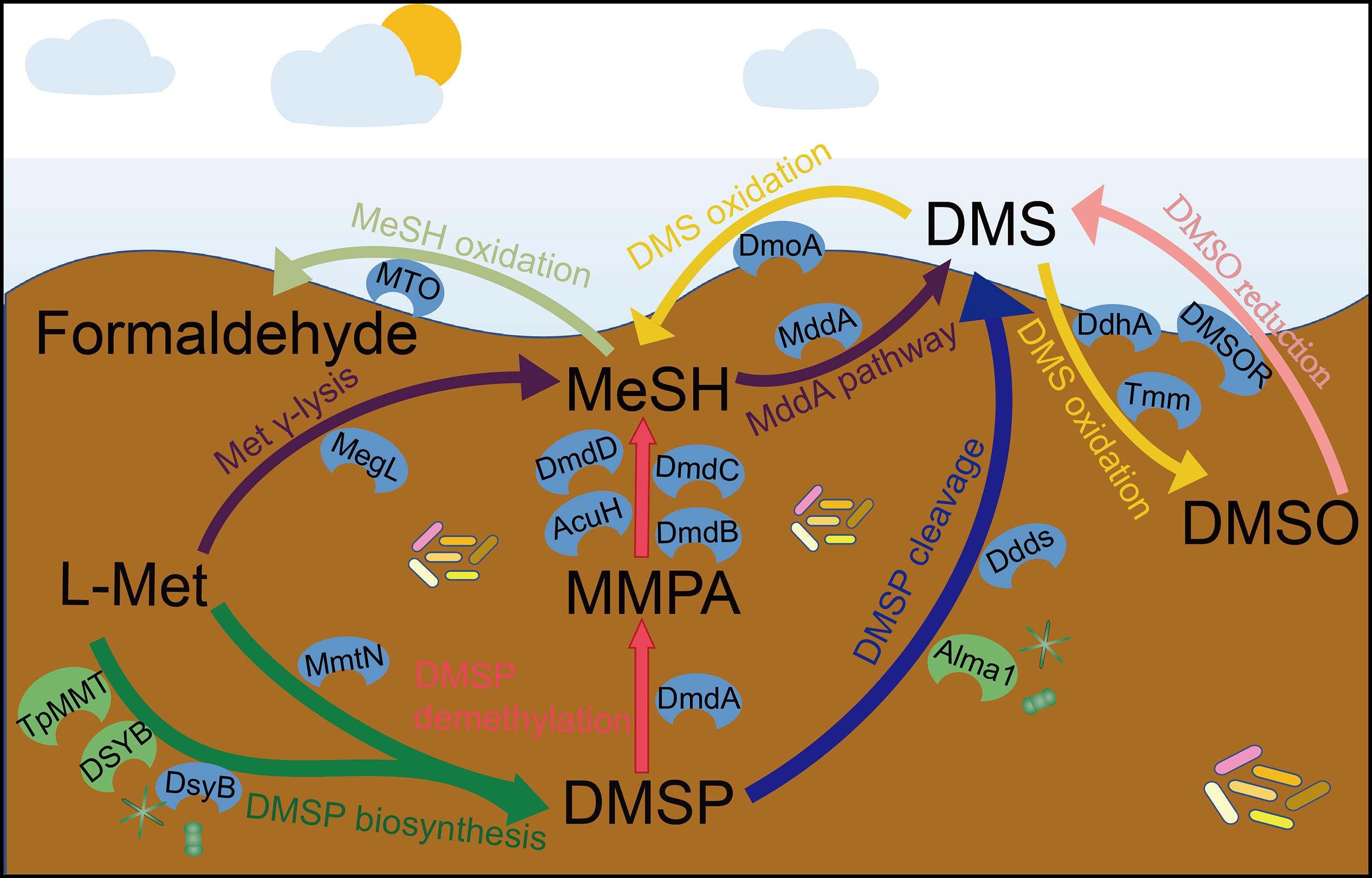
Figure 1. The known steps and key enzymes of each pathway in the production and cycling of DMSP and related compounds. Colored arrows represent individual pathways. The known key enzymes of these pathways are indicated; blue color are largely bacterial whereas those in green are eukaryotic. For DMSP synthesis, the two major pathways are represented by a split arrow. DMS, dimethylsulfide; DMSP, dimethylsulfoniopropionate; DMSO, dimethyl sulfoxide; MMPA, methylmercaptopropionate; MeSH, methanethiol; Ddds, various DMSP lyases; L-Met, L-methionine; Alma1, DMSP lyases; DsyB, DSYB; TpMMT, methylthiohydroxybutryrate SAM-dependent methyltransferase; MmtN, Met-methylating enzymes; MddA, MeSH S-methyltransferase; DmoA, dimethylsulfide monooxgenase; MTO, MeSH oxidase; DdhA, dimethylsulfide dehydrogenase; Tmm, trimethylamine monooxygenase; DMSOR, dimethyl sulfoxide reductase; MegL, methionine γ-lyase; DmdA, DMSP demethylase; DmdB, MMPA-CoA ligase; DmdC, MMPA-CoA dehydrogenase; DmdD, methylthioacryloyl-CoA hydratase; AcuH, acryloyl-CoA hydratase.
DMSP is degraded via the cleavage and demethylation pathways in heterotrophic bacteria and some algae for the cleavage pathway (Figure 1; Johnston, 2015; Zhang et al., 2019). In the DMSP cleavage pathway, eight distinct DMSP lyase enzymes generate DMS and either 3-hydroxypropionate (3-HP) in the case of the DddD enzyme (Todd et al., 2007), or acrylate with DddL (Curson et al., 2008), DddP (Todd et al., 2009), DddQ (Todd et al., 2011), DddY (Curson et al., 2011a, b), DddW (Todd et al., 2012), DddK (Sun et al., 2016; Schnicker et al., 2017), and the algal DMSP lyase Alma1 (Alcolombri et al., 2015). The ddd genes are found in a wide range of proteobacteria [mainly alpha-proteobacteria with Roseobacters and SAR11 (Candidatus Pelagibacter) being important representatives] and some fungi in the case of dddP (Todd et al., 2009). The DMSP demethylation pathway that can result in the generation of MeSH (Reisch et al., 2011a) is initiated by the DmdA enzyme that was identified in Ruegeria pomeroyi (Howard et al., 2006) and is common in many marine alpha-proteobacteria, including SAR11 bacteria (Reisch et al., 2008, 2011b). Many diverse bacteria that do not always have the capacity to demethylate DMSP (lacking dmdA in their genomes) contain dmdBCD/acuH, which encode enzymes that degrade the product of DMSP demethylation, methylmercaptopropionate (MMPA), to generate MeSH (Shao et al., 2019). Thus, the presence of dmdBCD is considered an indicator of MMPA degradation rather than of DMSP. Most previous metagenomic studies of DMSP cycling focus on seawater, particularly surface seawater, to assess the distribution and diversity of DMSP degradation genes (Howard et al., 2006; Moran et al., 2012; Cui et al., 2015; Nowinski et al., 2019). These studies generally report that dmdA dominates the seawater DMSP catabolic gene pool, with ∼33% of marine bacteria predicted to contain dmdA from SAR11 clade and Roseobacter group bacteria (Howard et al., 2006, 2008; Nowinski et al., 2019). To date, no one single DMSP lyase gene is as abundant as dmdA in any marine metagenome but in total ∼20% of bacteria are predicted have one of the 7 known bacterial DMSP lyase genes (Curson et al., 2018). DddP is the most abundant environmental DMSP lyase and is often found in the marine Roseobacter clade (Cui et al., 2015; Kudo et al., 2018).
Methanethiol can be modified through two different pathways catalyzed by the MeSH oxidase MTO or the MeSH S-methylase MddA enzymes (Figure 1). The MeSH degrading enzyme MTO, found in Thiobacillus, Rhodococcus, and Hyphomicrobium strains, oxidizes MeSH to yield formaldehyde (Suylen et al., 1987; Gould and Kanagawa, 1992; Kim et al., 2000; Lee et al., 2002; Eyice et al., 2018). Metagenomics analysis suggested that the mtoX gene was widely distributed in seawater (0.4–45.6% of bacteria), freshwater (5.3% of bacteria), soil environments (∼6.3% of bacteria; Eyice et al., 2018), and saltmarsh sediment (4.0%; Carrión et al., 2019). In contrast, MddA was identified in Pseudomonas deceptionensis (Carrión et al., 2015, 2017), but many diverse aerobic and anaerobic bacteria and cyanobacteria also contain MddA and likely methylate MeSH to generate DMS (Drotar et al., 1987; Kiene and Hines, 1995; Stets et al., 2004; Carrión et al., 2015). The mddA gene is present in varied environmental metagenomes, but thus far has been found to be much more abundant in terrestrial than in marine environments (Carrión et al., 2015, 2017, 2019). For example, 5–76% of bacteria in soil metagenomes were predicted to contain mddA, compared to 9.6% of bacteria from surface saltmarsh sediment samples and ≤ 0.5% of bacteria in seawater metagenomes. Another DMSP-independent pathway for the production of DMS is the reduction of DMSO by the DMSO reductase (DMSOR) enzyme present in some marine heterotrophic bacteria and bacteria associated to anaerobic environments (Griebler, 1997; Kappler and Schäfer, 2014).
Some bacteria consume DMS and three DMS-degrading enzymes have been discovered (Figure 1). DMS can be oxidized to generate DMSO by DMS dehydrogenase (DdhA) in, e.g., Rhodovulum sulfidophilum (McDevitt et al., 2002), or by trimethylamine monooxygenase (Tmm) in many Roseobacters and notably SAR11 bacteria (Lidbury et al., 2016). Some alpha- and beta-proteobacteria and Actinobacteria (Visscher and Taylor, 1993; Borodina et al., 2000; Boden et al., 2011) can oxidize DMS to generate MeSH via the DMS monooxygenase enzyme DmoA (Boden et al., 2011). DmoA was predicted to occur in 0.5% of surface saltmarsh bacteria, whereas the DdhA was far more abundant in these samples (13.3% of bacteria), as was Tmm (2.1% of bacteria) (Carrión et al., 2019).
It is clear from Carrión et al. (2019); Wilkening et al. (2019), and Williams et al. (2019) that coastal marine sediments may be important sites for organosulfur cycling. However, very few studies have examined the genetic potential of marine sediments for the cycling of DMSP and related molecules. In this study, we examine marine surface sediment samples from the eastern Chinese marginal sea, i.e., the Bohai Sea and Yellow Sea sediments (BYSS) and the Okinawa Trough hydrothermal field sediment and polymetallic sulfide (OTSP) for their standing stock DMSP concentration, microbial diversity, and genetic potential to cycle organosulfur compounds. Furthermore, we contrast these data to those generated from the surface seawater from the same sites. Very little is known about organosulfur cycling in deep-sea hydrothermal environments. The closest reported study examines the surface water metagenome of Kueishantao shallow-sea hydrothermal field and found that the DMSP demethylase affiliated to Roseobacter and SAR11 clade bacteria was present (Tang et al., 2013). Our study shows that: DMSP concentrations were far higher in marine sediment than those in surface waters per unit volume; DMSP-producing bacteria with DsyB were present in most seawater and sediment samples; bacteria with the genetic potential to cleave DMSP generating DMS were far more abundant than those with dmdA in all tested sediment, but that dmdA was more prominent in surface waters; bacteria with mddA were very abundant in marine sediment but not in seawater; DMSP catabolic potential via known pathways was rare in OTSP samples, yet genes for DMS, DMSO, and MeSH cycling pathways were more abundant.
The eastern Chinese marginal seas are a semi-enclosed marginal region of the western Pacific Ocean, and includes the Bohai Sea (BS), the Yellow Sea (YS), and the East China Sea (Figure 2). The mean depths of the BS and the YS are < 20 and ∼50 m, respectively. Both are fed by 30–40 rivers, notably the Yellow River, which empties into the BS. Both seas are surrounded by areas of high population and economic development in China and the Korean Peninsula (Figure 2), with significant effects on their ecosystems (Liu et al., 2011; Xu, 2011).
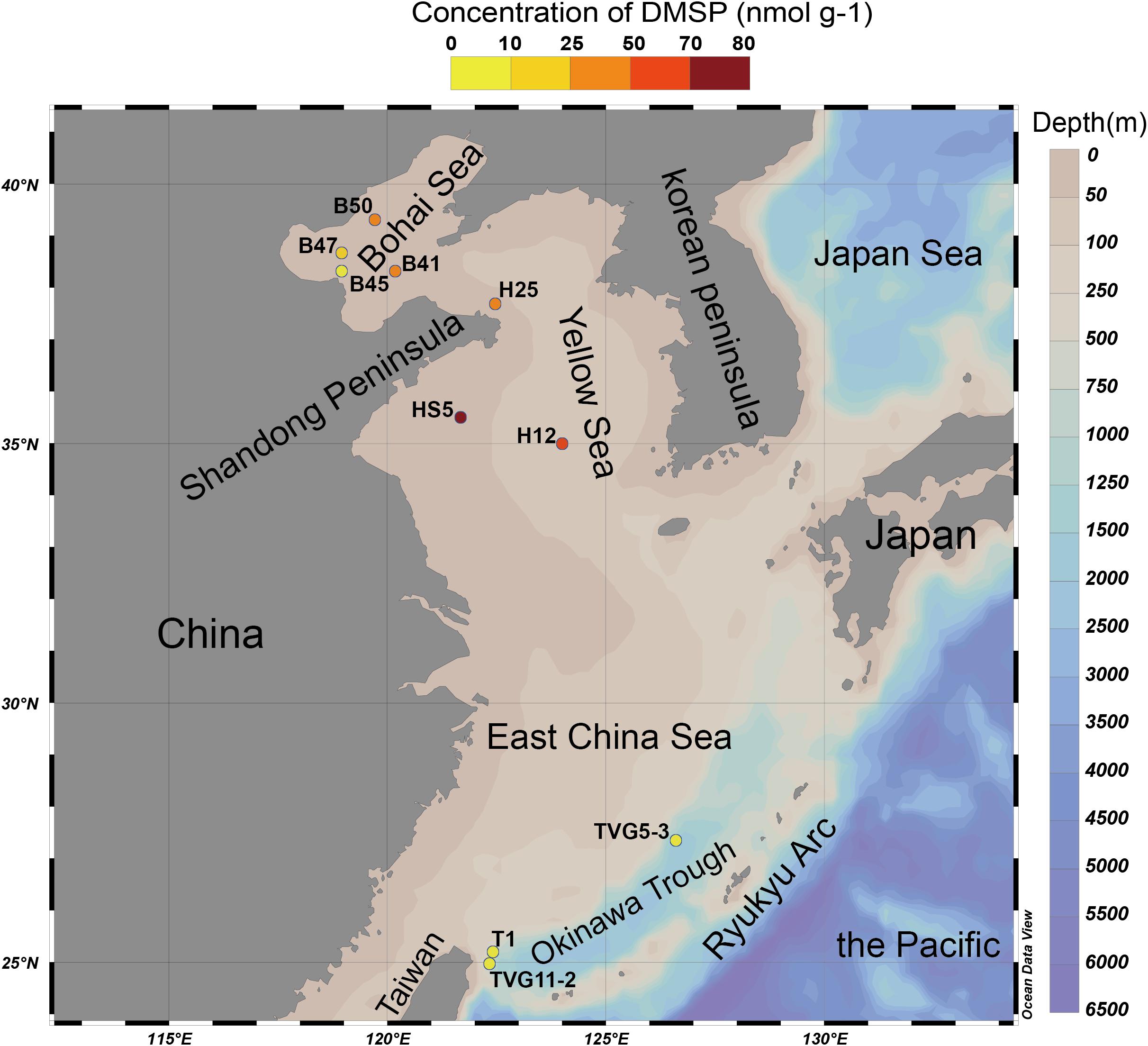
Figure 2. The Eastern Chinese marginal seas sampling stations. “B” refers to Bohai Sea, “H” to Yellow Sea, and “T” to Okinawa Trough. The sites B41, B45, B47, B50, and T1 were sampled only for sediment; sites HS5 and H25 were sampled for water samples (surface and bottom) and sediment samples; and sites TVG11-2 and TVG5-3 were sampled for sediment and polymetallic sulfide. Stations plotted in Ocean Data View (Schlitzer, 2002). The total DMSP concentration in the surface sediment of each site is indicated by the colored circle according to the DMSP concentration scale.
The Okinawa Trough is located between the East China Sea and the Ryukyu Arc (Figure 2). To date, more than 20 hydrothermal vents have been discovered in this region (Miyazaki et al., 2017b), many of these are in the Iheya ridge hydrothermal field (Stations of T1 and TVG11-2; Figure 2) (Kawagucci et al., 2011). This deep-sea hydrothermal vent environment has mean depths of around 1300 m, and is characterized by a thick blanket of muddy sediment of volcanic sands containing minor sulfide components (Sakai et al., 1990; Gamo et al., 1991; Ishibashi et al., 1995). Other deep-sea (mean depth of 1206 m) hydrothermal vents were found in the Tangyin hydrothermal field (the station of TVG5-3), which is located at the southwest of the Okinawa Trough, close to Taiwan island (Figure 2; Zeng, 2015). This site is characterized by lead-enriched polymetallic sulfides and abundant sulfate minerals (Ishibashi et al., 2015).
The sediment samples (top 5 cm surface sediment, predicted to span zones with the highest DMSP concentration; Wilkening et al., 2019) used in this study were collected in the YS (at sites termed H12, HS5, H25) and the BS (B41, B45, B47, B50) by grab sampler (Cuong and Obbard, 2006) and in the Okinawa Trough hydrothermal field at sites TVG11-2, TVG5-3, and T1 from sediment and polymetallic sulfide (see Figure 2 and Table 1 for locations and details of the sampling stations). The reported maximal photic zone depth of YS is ∼30 m (Jin et al., 2013), and the BS is ∼14 m (Shang et al., 2011). Thus, all sediment samples in this study were taken from below the photic zone, the sampling depths are shown in Table 1. The surface water samples (0 m) and bottom water samples (Table 1) were collected in the YS (H12, HS5) using a Sea logger CTD (Conductivity–Temperature–Depth, Sea-Bird SBE911) rosette water sampler; 20 L Seawater was filtered serially through 3 μm (TSTP, 142 mm, Millipore) and 0.2 μm (GTTP, 142 mm, Millipore) polycarbonate membranes (Zhang et al., 2008). The communities collected on the 3 and 0.2 μm filters were designated as particle-associated and free-living fractions, respectively. All filters were stored in liquid nitrogen onboard and at −80°C in the laboratory. Here, we integrated data from both the > 3μm and 0.2–3 μm seawater fractionations because the difference between these fractions was not the focus of this study.
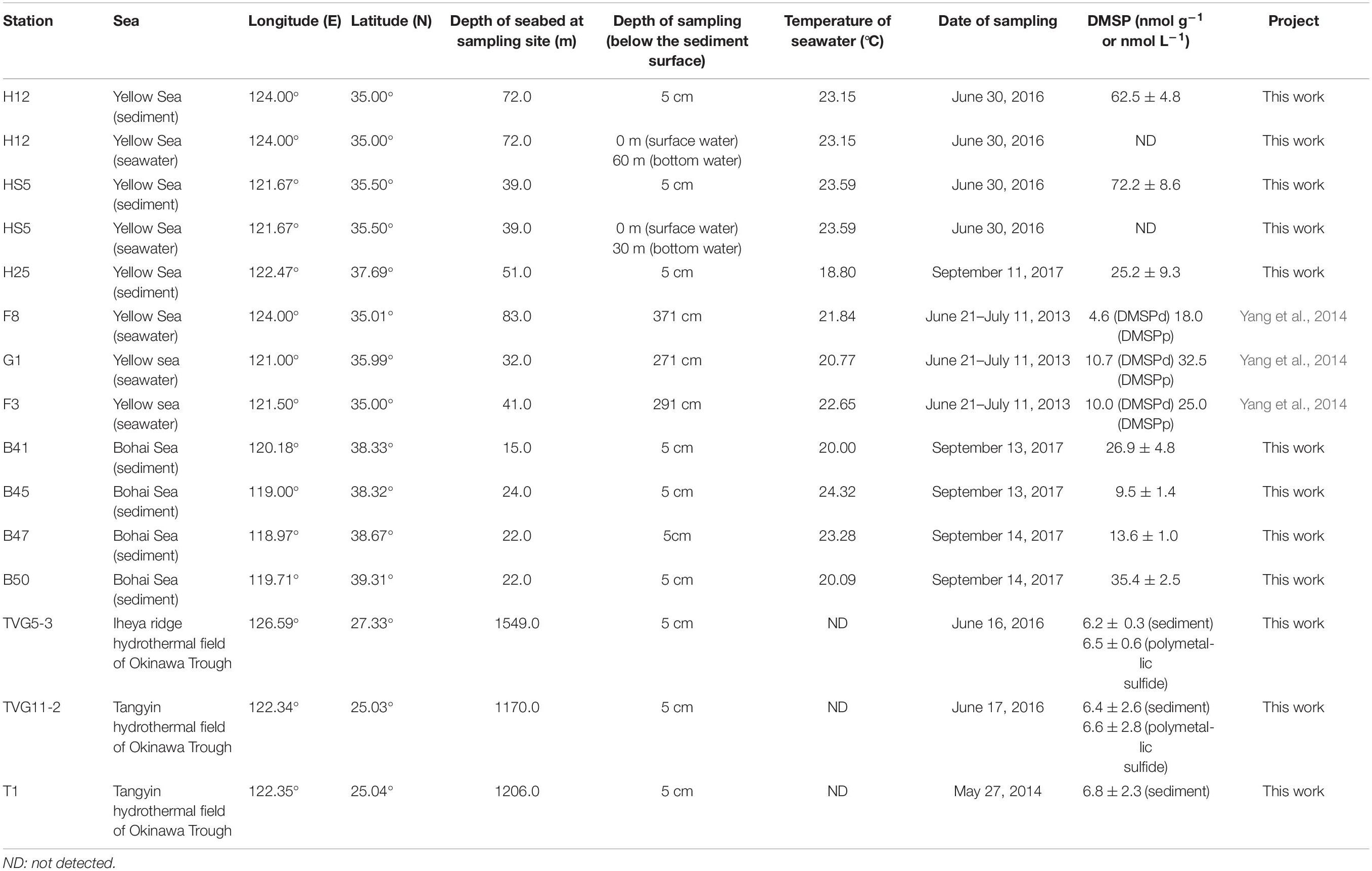
Table 1. Data on sediment sampling sites in three different seas.
The total DMSP concentrations in sediment and polymetallic sulfide samples were assessed by measurement of headspace DMS derived from DMSP by alkaline lysis, in 2.0 mL capacity vials (Agilent) containing 0.2 mL liquid samples, by using a flame photometric detector (Agilent 7890A GC fitted with a 7693 autosampler) and a HP-INNOWax 30 m × 0.320 mm capillary column (Agilent Technologies J&W Scientific). Briefly, the sediment or polymetallic sulfide samples (0.1–0.15 g) were mixed with 100 μL ultrapure water; 100 μL of 10 mol L–1 NaOH solution was added to the sample-water mixture and was incubated overnight with oscillation at room temperature to allow complete lysis DMSP to DMS. An eight-point calibration curve of DMS standards was used (Liu et al., 2018), and the detection limit for headspace DMS was 0.015 nmol. No steps were taken to account for endogenous DMS in the samples, thus, these assays represent total DMSP plus endogenous DMS. However, according to the previous studies (Nedwell et al., 1994; Kiene and Linn, 2000). DMSP always predominates over DMS in marine samples, thus, it is most likely the majority of DMS detected in this study is derived from DMSP. The DMSP concentrations in surface seawaters were not tested but were reported for the YS previously by Yang et al. (2014).
To compare the number of bacteria in seawater and sediment, quantitative polymerase chain reaction (qPCR) of the 16S rRNA gene was performed on StepOne ABI (Applied Biosystems, Foster City, CA, United States). The abundance of 16S rRNA gene was quantified using the primer set Eub338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and Eub518R (5′-ACTCCTACGGGAGGCAGCAG-3′) (Yin et al., 2013). qPCR reactions were performed in triplicates in a 20 μL system using 10 μL 2 × SYBR Premix Ex Taq II (Takara Bio Inc.), 0.4 μL 50 × ROX reference dye, 0.2–0.4 μM of each primer, 2 μL 1/10 diluted template DNA. The PCR reaction conditions were as described in Liu et al. (2018). Amplification efficiency for the 16S rRNA gene is 0.93, with an R2 value of 0.99.
Total DNA of seawater and sediment samples were extracted using the method described by Zhou et al. (1996). Briefly, to extract DNA from water (20 L), biomass was washed from half of each filter using 15 mL extraction buffer [100 mM Tris-HCl (pH 8.0), 100 mM sodium EDTA (pH 8.0), 100 mM sodium phosphate (pH 8.0), 1.5 M NaCl, 1% CTAB] before centrifuging at 5000 × g for 20 min at room temperature. The concentrated biomass was ground in liquid nitrogen, proteinase K and SDS were added in sequence, and the samples were incubated at 37 and 65°C, respectively, followed by phenol-chloroform extraction. DNA was precipitated with 0.6 volume of isopropanol, washed with 70% ethanol, air dried, and dissolved in TE buffer. The concentration and integrity of genomic DNA were analyzed by a Nanodrop 2000 spectrophotometer (Thermo Scientific, Denmark) and agarose gel electrophoresis (AGE), respectively.
To extract DNA from sediment and polymetallic sulfide, the samples (60 g) were ground in liquid nitrogen for temporary freeze and then collected into a filter containing 15 mL extraction buffer. Proteinase K and SDS were added in sequence with incubation at 37 and 65°C, respectively, followed by centrifugation at 6000 × g for 20 min at room temperature. The rest of the protocol was the same as for DNA extraction from water samples.
A total of 16 DNA samples (4 from seawater, 10 from sediments, and 2 from polymetallic sulfide) were sent to BGI (BGI, Shenzhen, China) for metagenomic sequencing. Metagenomic shotgun sequencing was performed on the Illumina HiSeq X-Ten platform, with 2 × 150 bp paired-end reads. First, DNA purity and integrity (no obvious disorganized bands and the main band ≥ 20 kb) were analyzed using AGE and quantification of DNA concentration (≥ 15 ng/μL) and quantity (≥ 1.5 μg) was performed using Qubit. The qualified DNA samples are randomly broken into fragments of approximately 350 bp in length using a Covaris focused ultrasound system. DNA fragments were end-repaired by addition of End Repair Mix and purified by QIA quick PCR Purification Kit (Qiagen). A-Tailing Mix was used to add an adenine at the 3′ end, and then the sequencing linker was ligated to both ends of the DNA fragment. Fragment selection was performed using 2% agarose gel and QIA Quick Gel Extraction kit (QIAGEN), and PCR amplification of several cycle was performed. The PCR product was purified again on a 2% agarose gel and the target fragment was recovered using QIA Quick Gel Extract on kit (QIAGEN). Finally, Agilent 2100 Bioanalyzer and ABI StepOnePlus Real-Time PCR System were used for quality control and quantification of sample libraries. Qualified libraries were sequenced using the Illumina Miseq PE300 platform (MiSeq Reagent Kit v3) at Major bio BiFo-Pharm Technology Co., Ltd., Shanghai, China. Sequences were processed with the pipeline of UPARSE (Edgar, 2013). The clean data basic information of all samples has been shown in Supplementary Table S1.
All the raw reads containing > 10% of undefined bases, > 40% of low-quality bases, and that had > 15 bases matching the adapters were removed. The quality-filtered reads were assembled using Megahit (Li et al., 2015) with a kmer size of 49. Contigs > 500 bp were retained. Gene prediction was performed using MetaGeneMark (Zhu et al., 2010) with default parameters1. Sequences were clustered at 95% identity and 90% coverage using CD-Hit (Li and Godzik, 2006). The longest sequence of each cluster was selected as a representative sequence of the cluster in order to create a non-redundant robust gene database. Then metagenomics reads of each sample were mapped to the non-redundant gene database using SoapAligner (Qin et al., 2012; Graham et al., 2017). Those sequences with less than two reads mapped were discarded and the resultant quality-filtered sequences were used for downstream analysis. The abundance of each gene was calculated using the calculation method (Supplementary Figure S1) and normalized by the number of mapped reads of the target gene to the gene length.
The initial paired-end reads from these samples were co-assembled using Megahit (Li et al., 2015), the principle of co-assembly is that all 3 and 0.2 μm samples from surface or bottom seawater at the same location in the YS were combined, as were all YS sediment samples, all BS sediment samples, and all Okinawa Trough samples. Subsequently, these reads were mapped to contigs using BWA (Short sequence alignment software; Li and Durbin, 2009), and then, grouped into metagenome assembled genomes (MAGs) by using the very sensitive mode of MetaBAT (Kang et al., 2015). Completeness and contamination of MAGs were assessed using CheckM, and MAGs with a completeness ≥ 80% and contamination ≤ 10% (high completion MAGs) were considered for further analysis. NCBI-nr (released August 2016) protein database and DIAMOND software (Buchfink et al., 2014) were used to determine the taxonomy of MAGs, followed by the MEGAN software (Huson et al., 2007; Liu et al., 2019) (version 4.6) system classification of the lowest common ancestor (LCA) algorithm to ensure the taxonomy. The data on MAGs are shown in Supplementary Table S2.
The filtered genes were compared against the NCBI-nr (released August 2016) protein database using DIAMOND software (Buchfink et al., 2014) BLASTp method, and matches with the lowest e-value minimum alignment results were selected for subsequent analysis. Since each sequence may have multiple alignment results through the above method, the MEGAN software (Huson et al., 2007; Liu et al., 2019) (version 4.6) system classification of the LCA algorithm was used to ensure its biological significance. According to the LCA annotation results and the gene abundance data, the abundance information of each sample at each classification level was obtained (Supplementary Figure S2). The abundance of a species in a sample was calculated by summing the abundances of genes annotated to the species in the sample. The calculation of the relative abundance of taxa possessing a certain DMSP cycling gene was calculated based on the assumption that the abundance of a species equals the total abundances of genes annotated to this species in the same sample.
To explore the genetic potential of DMSP cycling, hidden Markov Model (HMM)-based searches for homologs in metagenome datasets and MAGs were performed using HMMER tools (version 3.12; Kumaresan et al., 2018). Ratified DddD, DddK, DddL, DddP, DddQ, DddY, DddW, Alma1 (Symbiodinium and Emiliania), DsyB, DSYB, MmtN, DmdA, DmdB, DmdC, DmdD, AcuH, MddA, DmoA, DdhA, Tmm, MTO, and DMSOR sequences were used as training sequences to create the HMM profiles. All the training protein sequences are shown in Supplementary Table S3. Profile HMM-based searches eliminate the bias associated with single sequence BLAST queries (Eddy, 2011). Separate cut-off E-values were confirmed by blasting between functionally verified protein sequences (training sequences; Supplementary Table S4). All retrieved homolog sequences were aligned to the training sequences using the HMM alignment and this was used to construct an approximately maximum likelihood phylogenetic tree inferred using MEGAN v7.0.26 to further ensure the accuracy of the Hmm-based method. Candidate genes were removed if they did not align most closely to the ratified enzymes compared to proteins with different functions. Furthermore, the two databases KEGG and COG were also used to further ensure the reliability of the HMM method for gene function prediction. To estimate the proportion of bacteria and eukaryotes represented in each metagenome, selections of RecA proteins with a cut-off of E ≤ e–50 (Curson et al., 2017) and β-Actin proteins with a cut-off of E ≤ e–60 were used as probes to BLAST the same metagenome databases. The numbers of genes of interest detected in the metagenome data from each sample can be found in Supplementary Table S5. Additionally, the retrieved DsyB, DSYB, DmdA, and Ddd homolog sequences were aligned to the training sequences using the counterparts HMM alignment and the sequences that could not be sufficiently aligned were discarded, this was used to construct an approximately maximum likelihood phylogenetic tree inferred using FastTree v2.1 (Price et al., 2010). The taxa possessing each candidate gene was found using the taxonomic annotation and the abundance of taxa in each sample was calculated.
The metagenome data used in this study have been deposited at NCBI/BioProject, the Bio-Project accession numbers of these metagenome data are as follows: YS (PRJNA428417), BS (PRJNA514927), and Okinawa Trough hydrothermal field (PRJNA514953). The accession numbers of MAGs see the Supplementary Table S2. All the training sequences used to create the HMM profiles are available within the Supplementary Materials in the Supplementary Table S3.
Marine surface sediment samples from the YS (25.2–72.2 nmol g–1) and BS (9.5–35.4 nmol g–1) contained relatively high DMSP standing stock concentrations (Table 1). Unfortunately, we did not measure the DMSP levels in the surface waters for these sites on this cruise. However, these sediment values (Table 1) are about two orders of magnitude higher per mass unit than those reported for the surface waters (DMSPd, 4.6–10.7 nmol L–1; DMSPp, 18.0–32.5 nmol L–1; Table 1) in the YS on an earlier cruise (Yang et al., 2014). The sediment levels are ∼three-fold lower than the DMSP levels reported for surface saltmarsh sediments in Williams et al. (2019). In comparison, the DMSP concentration in OTSP samples (6.2–6.8 nmol g–1; Table 1) was ∼10-fold lower than the BYSS samples (Table 1 and Figure 2) and was similar to the levels previously reported for 4.5 km deep surface sediment from the Challenger Deep of the Mariana Trench (Williams et al., 2019). There was no noticeable difference in the DMSP levels of the hydrothermal sediment and polymetallic sulfide samples. Even the OTSP samples had far higher DMSP standing stock concentrations than surface seawater samples (Table 1), suggesting that marine sediments are highly active sites for DMSP cycling. As DMSP is rapidly degraded by abundant DMSP catabolic enzymes in the seawater (Kiene, 1996; Ledyard and Dacey, 1996), it is likely that much of the DMSP in the sediment may be derived from biosynthesis rather than sinking particles. However, further studies measuring the rates of DMSP synthesis and catabolism are required to test this hypothesis.
Metagenomics was carried out on the surface (SW) and bottom seawater (BW) from sites H12 and HS5 in the YS, and all surface sediment samples taken in this study (Table 1). The metagenomes were examined for the genetic potential to cycle DMSP and related compounds. In both the SW and BW samples dsyB, and thus the potential to produce DMSP, was predicted to be present in <1.0% of bacteria (Figure 3B). The dominant seawater dsyB genes in metagenome assembled genomes (MAGs) were from alpha-proteobacterial Roseospirillum and Thalassobaculum bacteria (Supplementary Figures S2,S3). Similar percentages of bacteria with dsyB were found in the sediment and polymetallic sulfide samples (average 1.3 and 0.9% in BYSS and OTSP, respectively; Figure 4). 16S rRNA qPCR analysis suggested that there were lower bacterial numbers in SW (1.53 × 108–1.91 × 108 copies L–1) than in the BW (6.39 × 108–7.92 × 108 copies L–1), but there were far greater bacterial numbers per equivalent mass in the sediment samples (4.62 × 107–1.39 × 108 copies g–1). Thus, there are likely far more DMSP-producing bacteria per unit area, playing a more significant role in DMSP synthesis, in the sediment than the seawater environments. Different bacterial dsyB genes, clustering with those from Pseudooceanicola, Roseovarius, and Roseospirillum, dominated in the BYSS sediment compared to those in the seawater (Supplementary Figure S3). The potential DMSP-producing bacteria dominating in the hydrothermal sediments were also distinct with their dsyB genes predicted to be in Caenispirillum, Albimonas, and Oceanicola bacteria (Figure 5).
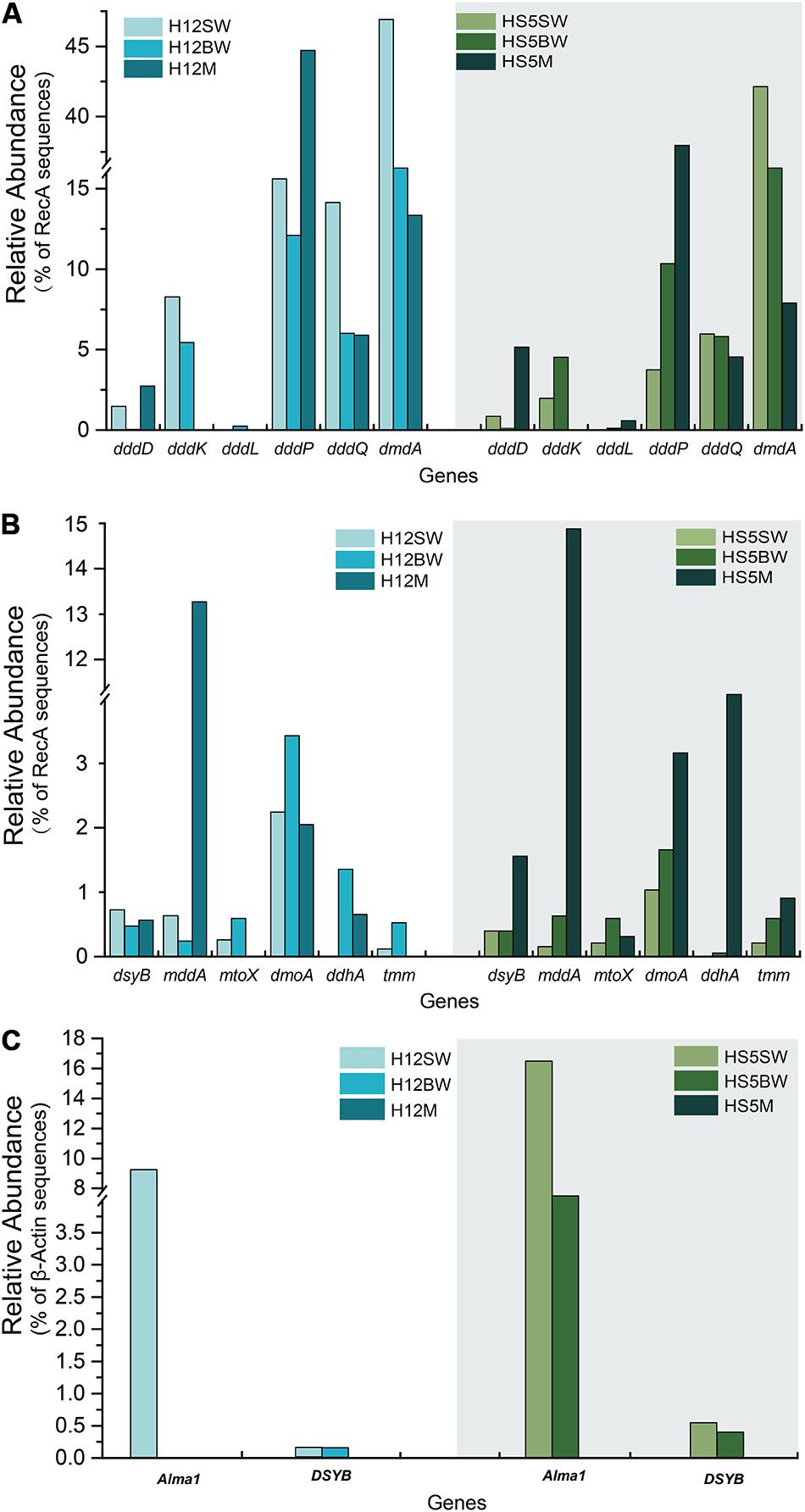
Figure 3. Comparison of relative gene frequencies in sediment and seawater samples from the Yellow Sea. (A) The relative abundance of bacterial DMSP catabolic genes (ddd and dmdA genes) in sediment and water samples from the Yellow Sea. (B) The relative abundance of bacterial organosulfur cycling genes in sediment and water in the Yellow Sea. (C) The relative abundance of algal genes in sediment and water in the Yellow Sea. SW, surface water; BW, bottom water; M, sediment.
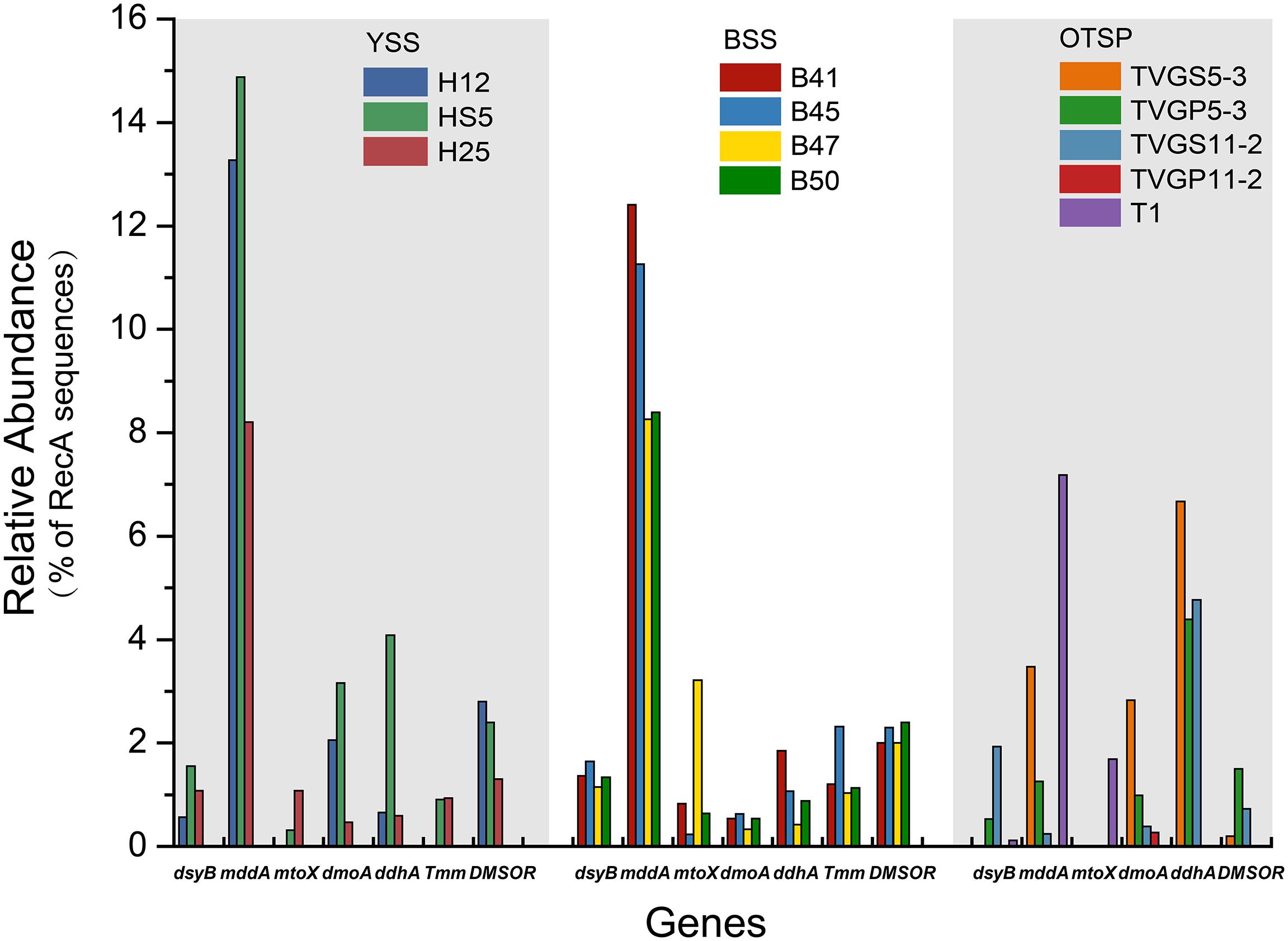
Figure 4. Relative abundances of known genes for the production and cycling of DMSP and related compounds in YSS, BSS, and OTSP sampling stations. YSS, sediments of the Yellow Sea; BSS, sediments of the Bohai Sea; OTSP, sediments and polymetallic sulfides of Okinawa Trough hydrothermal vents; TVGS, sediment samples in Okinawa Trough hydrothermal vents; TVGP, polymetallic sulfide samples in Okinawa Trough hydrothermal vents.
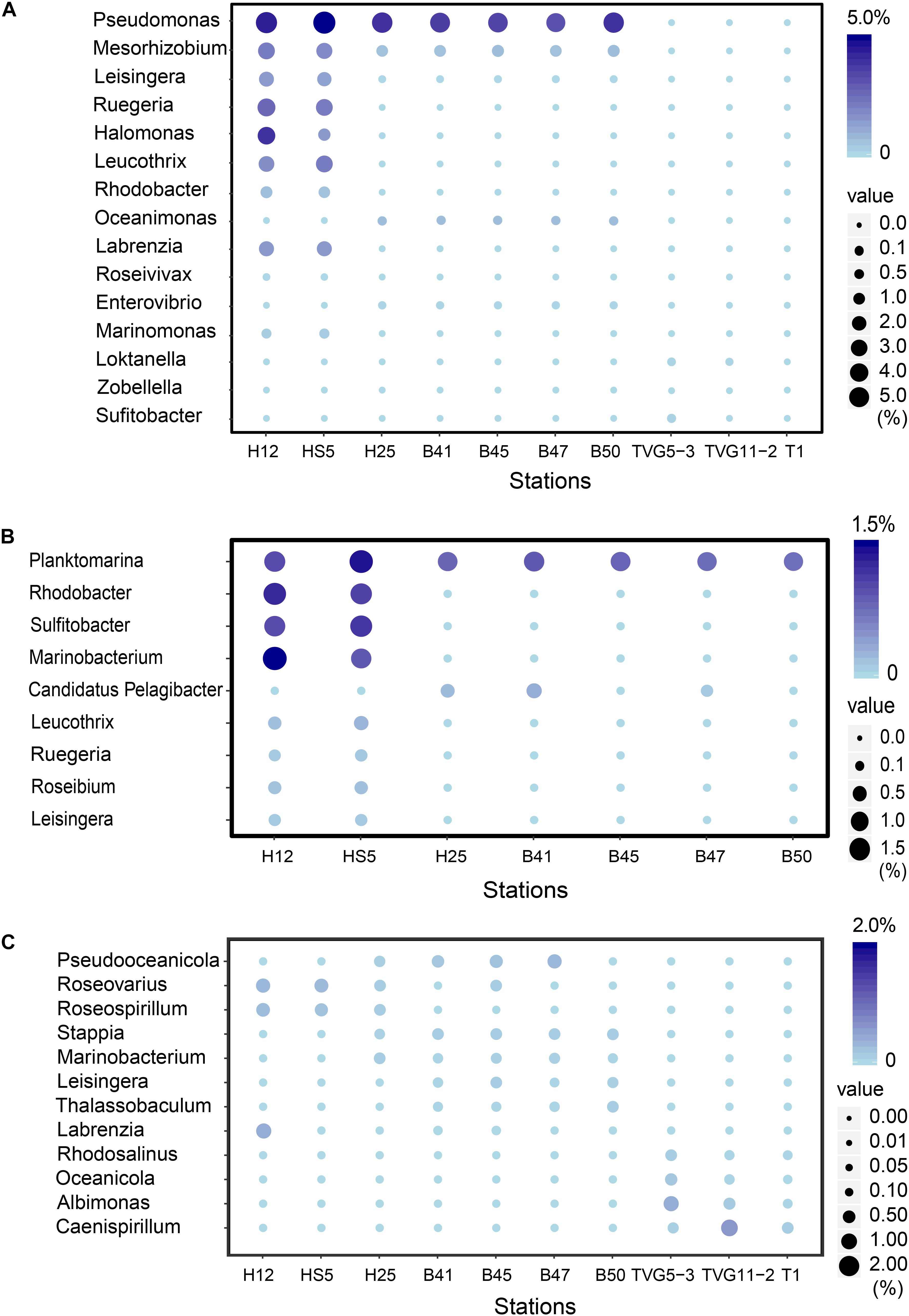
Figure 5. The most abundant bacterial genera predicted to be involved in the production and catabolism of DMSP in all sediment and polymetallic sulfide samples. (A) The most abundant bacterial genera possessing “ddd” genes. (B) The most abundant bacterial genera possessing dmdA. (C) The most abundant bacterial genera possessing dsyB.
The bacterial mmtN gene was absent in all of the metagenome sequences, suggesting that DMSP synthesis via MmtN is not a major pathway in the tested environments. The algal DSYB gene was present at similar levels (0.2–0.6% of eukaryotes, Figure 3C) in the SW and BW samples but was not found in the BYSS or OTSP metagenomes. The detected DSYB genes were most homologous to those in Fragilariopsis and Alexandrium (Supplementary Figures S3,S4). This is consistent with Diatom and Dinophyceae phytoplankton being important producers in the tested water samples, but less so in the surface sediments in this study.
As expected from previous research (Howard et al., 2006, 2008; Moran et al., 2012; Cui et al., 2015) the dmdA gene, conferring the genetic potential to demethylate DMSP, is far more abundant than any other DMSP lyase gene in both the SW (average 44.5%) and BW (average 35.5%) samples (Figure 3A). Interestingly, the percentage of bacteria with dmdA declines with seawater depth and is always lowest in the sediment samples (average ∼9.2% in BYSS; Figure 6). Furthermore, dmdA is undetectable in the OTSP samples indicating that demethylation is, at best, only a minor component of DMSP catabolism in the Okinawa Trough hydrothermal field (Figure 6). Alternatively, there may be DmdA isozymes in these environments, but there is no evidence that such enzymes exist and further work is needed to test this hypothesis. The genes involved in the catabolism of MMPA and generation of MeSH, dmdB, dmdC, and AcuH had high relative abundances in the investigated samples. In contrast to dmdA, the dmdB (38.3%), dmdC (38.5%), and AcuH (8.5%) genes were relatively less abundant in the seawater compared to the YBSS samples, with average abundances of 50.2, 39.7, and 18.1%, respectively (Figure 7A). Furthermore, in the OTSP samples where dmdA was undectable, dmdB (average 8.9%), dmdC (average 11.2%), and AcuH (average 7.8%) genes were relatively abundant. These data suggest that MMPA may be a significant source of MeSH in these marine environments and in some cases perhaps is independent of DMSP catabolism, e.g., in the OTSP samples. The dmdD (present in up to 2.1% of bacteria) is far less abundant than acuH and did not show any significant differences in relative abundance between YS sediment and seawater samples (Figure 7B; P > 0.05). These data suggest that AcuH is the major methylthioacryloyl-CoA hydratase responsible for generating MeSH in these environments. The high abundances of dmdB, dmdC, and acuH may be linked to the high plasticity and flexibility of the corresponding enzymes (Reisch et al., 2011a; Bullock et al., 2014; Shao et al., 2019).
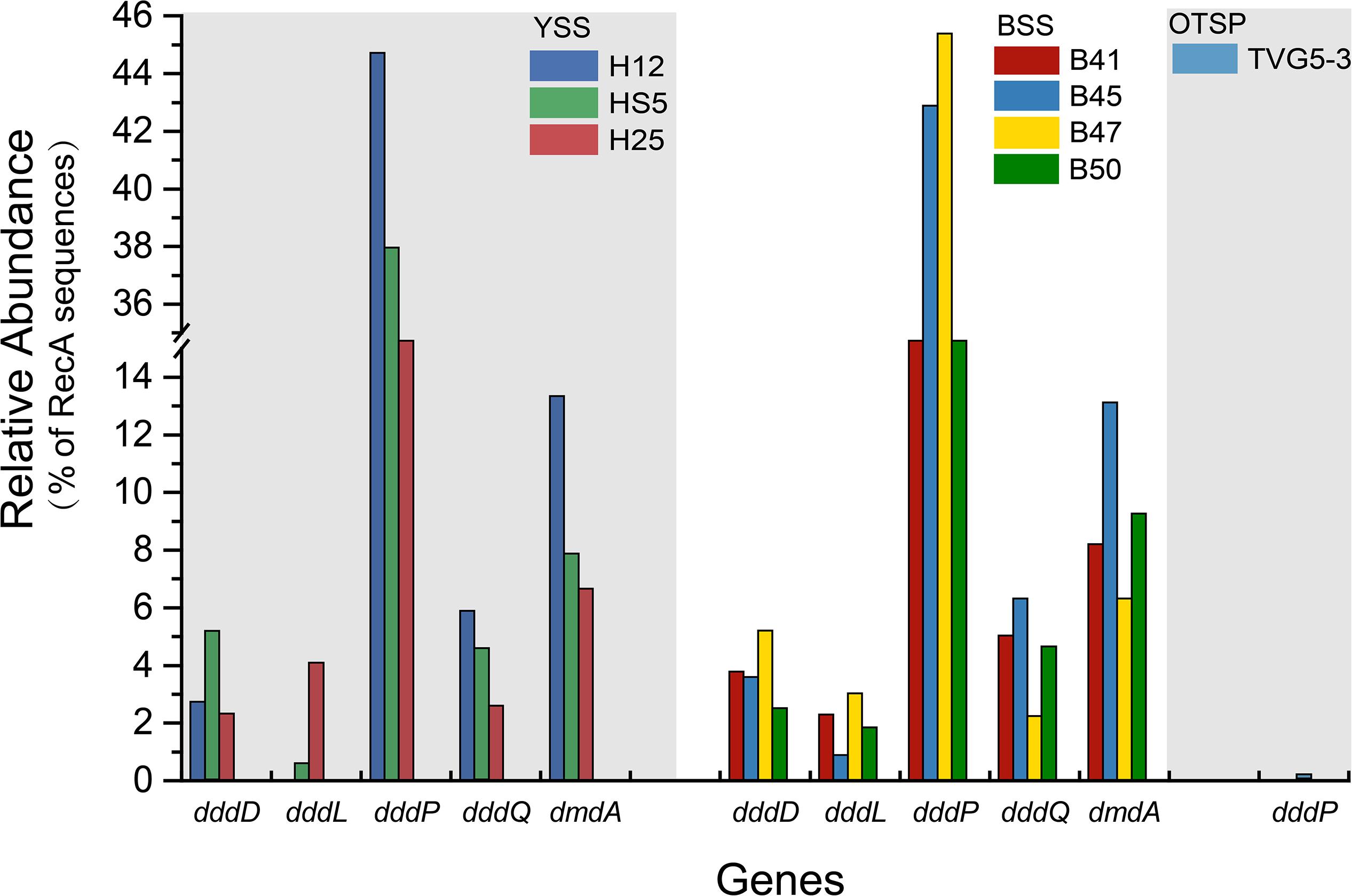
Figure 6. Relative abundances of DMSP catabolic genes in YSS, BSS, and OTSP sampling stations. YSS, sediment of Yellow Sea; BSS, sediment of Bohai Sea; OTSP, sediment and polymetallic sulfide of Okinawa Trough hydrothermal vents.
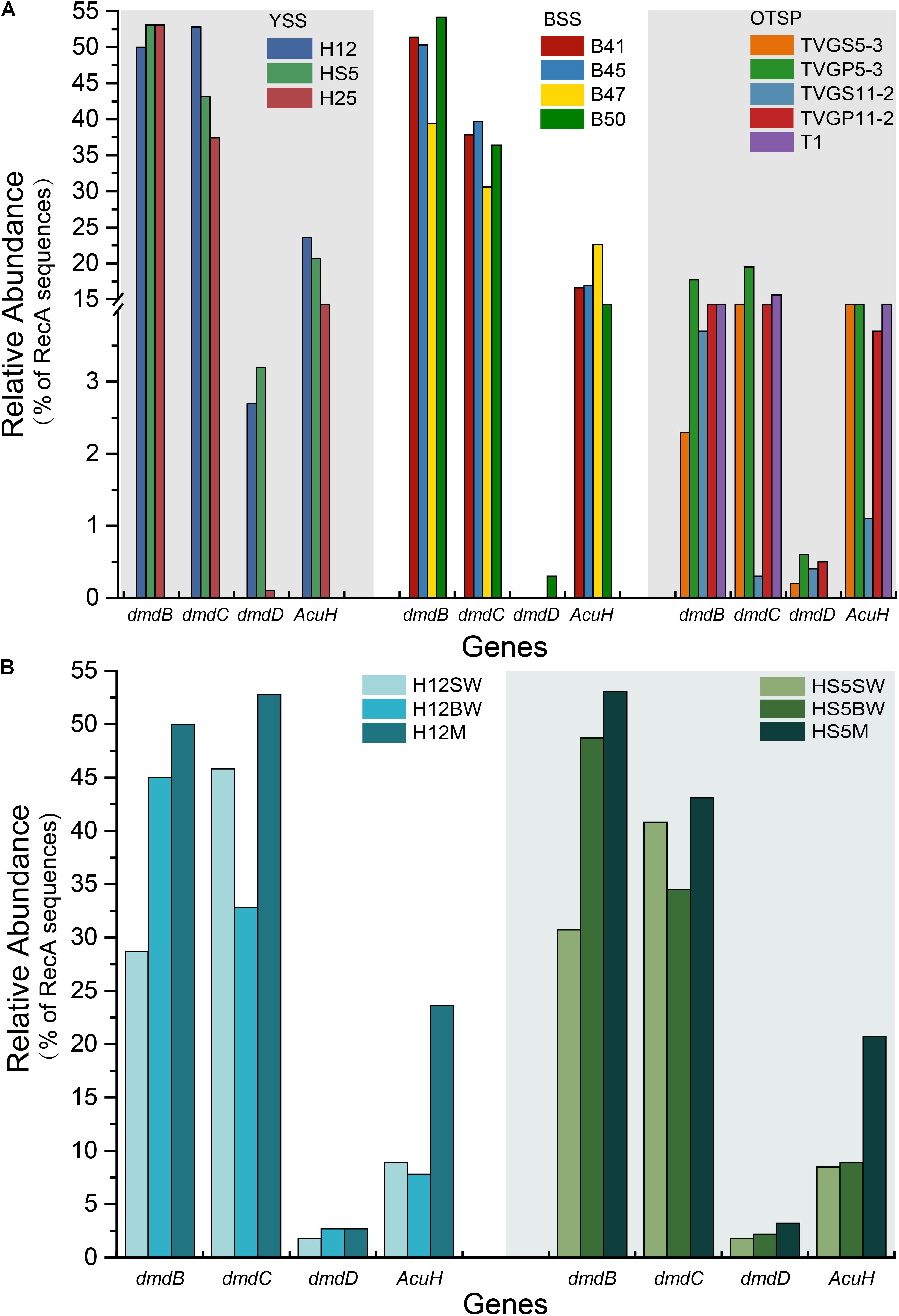
Figure 7. Relative abundances of DMSP demethylation genes in YSS, BSS, and OTSP sampling stations. (A) The relative abundance of the bacterial the dmdBCD and acuH genes in YSS, sediment of Yellow Sea; BSS, sediment of Bohai Sea; OTSP, sediment and polymetallic sulfide of Okinawa Trough hydrothermal vents. (B) The relative abundance of the bacterial dmdBCD and acuH genes in sediment and water in the Yellow Sea.
In both SW and BW samples, dddP was the most abundant DMSP lyase gene, predicted to be present in 4.0–15.6% of bacteria (Figure 3A). In contrast to dmdA, the abundance of the dddP gene increased in the sediment compared to the seawater samples. In fact, in all sediment samples dddP not dmdA was the dominant DMSP catabolic gene. Other DMSP lyase genes were also relatively abundant including dddQ (6.0–14.1% of bacteria), dddK (2.0–8.3% of bacteria), and dddD (up to 1.5%) (Figure 3A) in the SW and BW samples. In contrast, dddL was only predicted to be in < 0.3% of SW and BW bacteria and no homologs of dddY and dddW were found in the SW, BW samples, or sediment environments (Figure 3A). The eukaryotic DMSP lyase gene Alma1, most similar to Emiliania huxleyi Alma1, was detected in the SW and some BW samples, but not in any YS sediment samples, and when detected Alma1 was predicted to be in ∼4–16% of eukaryotes in these waters (Figure 3C).
There were no obvious differences between the census of DMSP lyase genes in the Yellow Sea sediment (YSS) versus the Bohai Sea sediment (BSS) samples (P > 0.05). The dddP gene (predicted to be on average in 37.8% of bacteria; Figure 6) was the most abundant bacterial cleavage gene, followed by dddQ (average 4.4%), dddD (average 3.9%), and dddL (average 2.2%) (Figure 6). No dddY, dddK, and dddW homologs were observed in BYSS samples. In contrast to the YSS samples where Alma1 is absent, very low levels of Alma1 were detected in 50% of the BSS samples, and this equated to ∼0.8% of eukaryotes in the samples containing this gene (Supplementary Table S5). These data suggest that bacteria and algae are likely important producers of DMS from DMSP in the YS photic waters, but bacteria mainly drive this process in these tested sediments. Alpha-proteobacteria were clearly important in seawater and sediment DMSP catabolic processes. The dmdA gene was predominantly in SAR11, SAR116 (Candidatus Puniceispirillum), and Rhodobacterales bacteria (Supplementary Figure S5) in both seawater and sediment. The ddd genes were predominantly in SAR11 and Rhodobacteraceae in seawater (Supplementary Figure S3), whereas they were mainly in alpha-proteobacterial Rhizobiales and Rhodobacterales bacteria as well as gamma-proteobacterial Pseudomonadales bacteria in the sediment (Supplementary Figures S6–S10).
Despite DMSP being detected in hydrothermal field samples and at much higher levels than in the YS seawater, most DMSP catabolic genes, like dmdA, were not detected in the majority of hydrothermal OTSP samples. Only one dddP sequence was detected in an Iheya ridge sample (Figure 6). This solitary dddP gene sequence clustered with the Sulfitobacter dddP (Figure 5A). These data suggest that either DMSP lysis, like DMSP demethylation, is not an important process in these hydrothermal sediments or that other unknown DMSP lyases exist in these environments. Further work studying the process rates of DMSP synthesis and degradation is needed to address these questions.
To further predict DMSP metabolic pathways in the investigated samples, we constructed 78 high-quality genomic bins, hereafter referred to as MAGs, from the combined contigs of the samples with a contaminant threshold of 10% and a completeness threshold of 80% (Supplementary Table S2). The dmdB and dmdC genes are found in most MAGs (Supplementary Table S2), which is likely linked to the high plasticity and flexibility of the enzymes encoded by these two genes. Reisch et al. (2011a) reported that the bacteria possessing dmdB and dmdC can utilize MMPA to produce MeSH, further supporting MMPA as a likely important source of MeSH in the marine environment. Furthermore, only two of these MAGs (B_bin.4 and B_bin.42; Supplementary Table S2) from sediment samples also contain dmdA suggesting that such bacteria contain an isoform DMSP demethylase enzyme or that MMPA may be abundant in these sediments. DMSP lyase genes dddP (HS5M_bin.12, B_bin.98, B_bin.4 and B_bin.42), dddD (B_bin.33 and B_bin.42), dddQ (B_bin.4 and B_bin.42), and dddL (B_bin.86) were found in MAGs of the YBSS. These data suggested that DMSP lysis is likely an important process in these sediment samples. However, no DMSP catabolic gene has been found in MAGs of OTSP. These data suggested that DMSP demethylation and cleavage are only a minor component of DMSP cycle in the Okinawa Trough hydrothermal field.
Methanethiol is biologically modified via two different pathways, i.e., the MeSH-dependent DMS production (MddA) and MeSH oxidation (MTO) pathways. In both the SW and BW, the mddA gene was predicted to be present in 0.2∼0.6% of bacteria (Figure 3B). These levels are similar to those predicted for surface ocean bacteria by Carrión et al. (2015). A similar abundance was detected for the mtoX gene (0.2∼0.6%) in the same environments. As expected (Carrión et al., 2015, 2019), the mddA gene was detected at much higher frequencies in the BYSS samples (average ∼11.0% of bacteria, Figure 4, no significant difference between BS and YS, P > 0.05) than in seawater, but mddA was always less abundant than the DMSP lyase dddP (average 37.8%; Figure 6). In contrast, abundance of the mtoX gene was not obviously different in the BYSS (average ∼0.8% of bacteria; Figure 4) compared to the seawater samples. These data are in accordance with Carrión et al. (2019), and suggest that the main biological fate of MeSH in tested BYSS and seawater samples is likely tied to the production of DMS along with the incorporation of MeSH into proteins (Kiene et al., 1999).
In contrast to known DMSP catabolic genes, the mddA and mtoX genes were detected in all hydrothermal vent sediment samples, with average relative abundances of 2.4 and 1.7% (Figure 4), respectively (no significant difference between sediment and polymetallic sulfide, P > 0.05). It would be interesting to know where the MeSH substrate for these processes comes from in these samples where no dmdA genes were identified. According to the analysis of MAGs, two MAGs (B_bin.33 and B_bin.40; Supplementary Table S2) from sediment samples contain mddA gene, and two contain the MeSH oxidation gene mtoX (B_bin.47 and B_bin.12) suggesting that MeSH removal may be an important process in marine sediment.
The three key genes for DMS oxidation dmoA (1.0∼3.4%), ddhA (0.1∼1.4%), and tmm (0.1%∼0.6%) were detected in all SW and BW samples (Figure 3B). There was a trend in HS5 samples where the abundance of these three genes increased with water depth and was maximal in the surface sediment, but this was not the case in H12 site samples (Figure 3B). Nevertheless, these DMS oxidation genes were always most abundant in the sediment samples and there were no significant differences between their abundance in the BS and YS sediments (P > 0.05). Bacteria with dmoA, ddhA, and tmm were predicted to represent on average ∼1.2, 1.4, and 1.2% of the communities in the BYSS sediment samples (Figure 4). Within seawater samples, the DMS monooxygenase gene dmoA was mainly predicted to be in Belnapia, Candidatus Entotheonella, and Microbacterium bacteria (Supplementary Figure S3), but, Alcaligenes and Pseudomonas within BYSS samples (Figure 8). The profile of bacteria predicted to contain the DMS dehydrogenase gene ddhA was also different between the seawater and sediment samples being in Rhodocyclales in SW and BW samples (Supplementary Figure S3), whilst in Mesorhizobium and Sterolibacterium in BYSS samples (Figure 8). Most tmm sequences were mapped to Sedimentitalea and Pseudomonas, and there was no obvious difference between seawater and the BYSS samples (Figure 8). The three genes mddA (H12swf_bin.13, H12BWF_bin.49 and OT_bin.42), ddhA (B_bin.47 and B_bin.12), and tmm (H12bwf_bin.29, H12BWP_bin.26 and B_bin.4) were also found in the MAGs of BS-YS samples. The dmoA gene was present at similar abundances in the hydrothermal OTSP samples (average 0.9%) compared to the BYSS samples, but the abundance of ddhA slightly increased to 3.1% (average; no significant difference between hydrothermal sediment and polymetallic sulfide, P > 0.05). There were no tmm genes detected in the OTSP samples. The dmoA gene in hydrothermal samples was mainly predicted to be from Alcaligenes and Stenotrophomonas (Figure 8). The two MAGs with dmoA (OT_bin.42 and 66, Supplementary Table S2) are phylogenetically similar to Stenotrophomonas, and a homolog of dmoA was found in the OT_bin.42 (completeness: 82.91%, contamination: 0). Which is most similar to Stenotrophomonas are present in hydrothermal sediments and likely oxidize DMS. Sorangiineae, Pseudomonas, and Sulfurovum were the dominant bacteria predicted to possess ddhA in the OTSP. Additionally, homologs of DMSOR (Satoh and Kurihara, 1987; Mcewan et al., 1991; Sambasivarao and Weiner, 1991) were predicted to be in BYSS (1.3∼2.8%) and OTSP bacteria (0.2∼1.5%; Figure 5), but this gene was absent in the seawater metagenome samples. There was no significant difference in the abundance of DMSOR and DMS oxidation genes in the BYSS samples. However, in the hydrothermal samples, DMS oxidation genes were far more abundant (P > 0.05). These data suggest that DMS oxidation processes occur throughout the water column and together with DMSO reduction in sediment samples but further experiments involving process measurement and RNA/protein work are required to prove this.
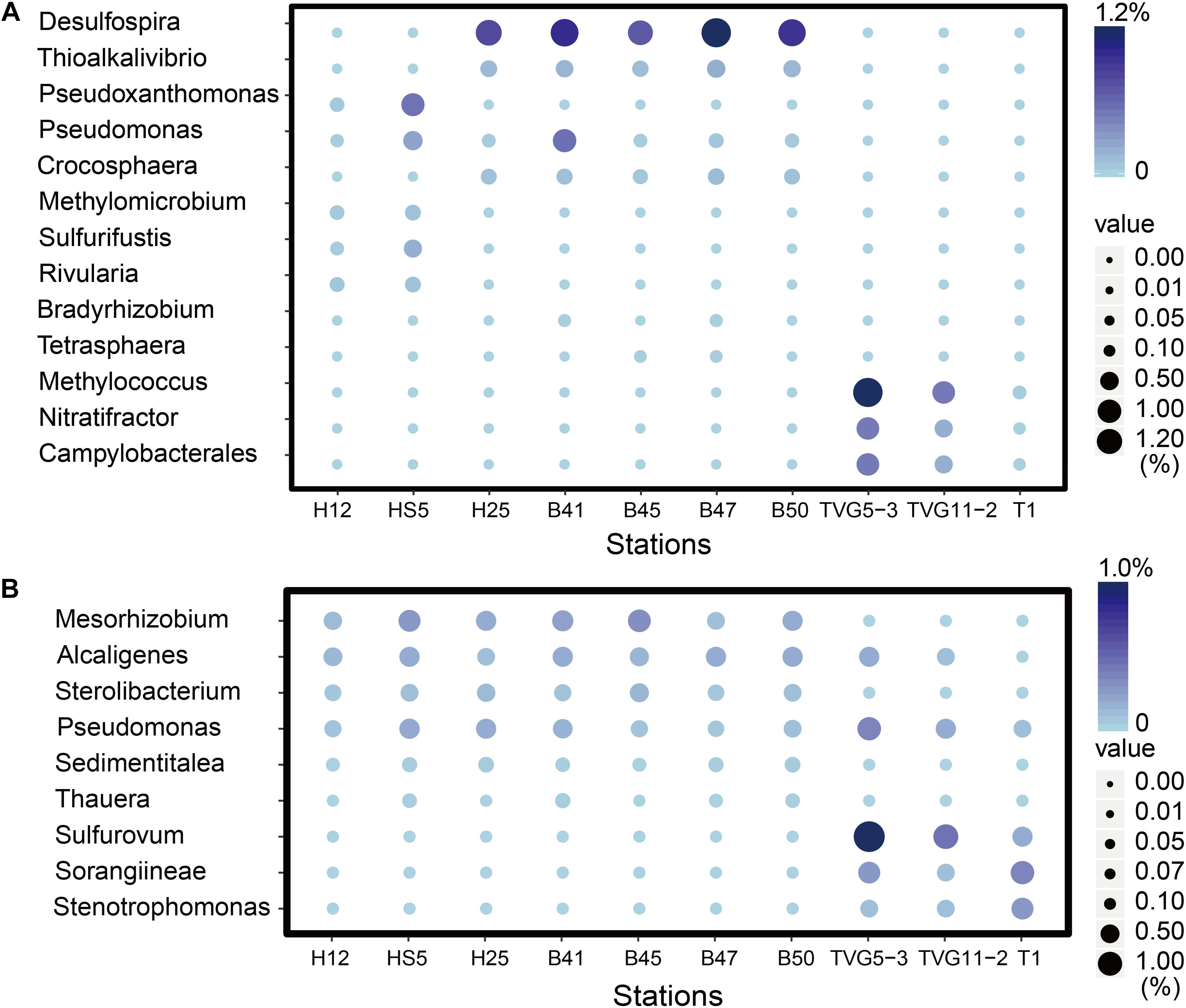
Figure 8. The most abundant genera predicted to oxidize DMS and methylate MeSH in all sediment and polymetallic sulfide samples. (A) The most abundant bacterial genera potentially degrading methanethiol (MeSH); MddA, Desulfospira, Thioalkalivibrio, Pseudoxanthomonas, Pseudomonas, Crocosphaera Sulfurifustis, Nitratifractor, Campylobacterales; MTO, Methylomicrobium, Rivularia, Bradyrhizobium, Tetrasphaera, Methylococcus. (B) The most abundant genera potentially oxidizing DMS; DmoA, Alcaligenes, Pseudomonas, Stenotrophomonas; DdhA, Mesorhizobium, Sterolibacterium, Sorangiineae, Pseudomonas, Sulfurovum; Tmm, Sedimentitalea, Pseudomonas, Thauera.
To date, most of the metagenomic studies on DMSP cycling have focused on seawater (Howard et al., 2006, 2008; Todd et al., 2009; Kudo et al., 2018), while only very few study sediments (Visscher et al., 1995) and these are heavily focused on saltmarsh sediments (Carrión et al., 2019; Williams et al., 2019). Such studies are focused on bacteria since the vast majority of known genes involved in the cycling of DMSP and related compounds were discovered in bacteria and are predominantly bacterial, with a few exceptions notably Alma1, dddP and DSYB, and TpMT2 (Curson et al., 2011a, 2017, 2008; Johnston, 2015; Zhang et al., 2019), which occur in eukaryotes. Here we examined the metabolic potential to cycle DMSP and related compounds in marine and hydrothermal surface sediment versus surface waters to provide insights in the organisms with the genetic potential to drive these processes.
The data in this study show that DMSP is an abundant molecule in BYSS, far more so than in any seawater sample from these YS study sites (Yang et al., 2014). BYSS samples had similar proportions of bacteria with the genetic potential (containing dsyB) to synthesize DMSP (1.3%) to those in the tested seawater (P > 0.05; Figure 3B). However, considering the far higher numbers of bacteria in sediment (4.62 × 107–1.39 × 108 16S rRNA gene copies g–1) than in seawater (1.53 × 108–7.92 × 108 16S rRNA gene copies L–1) reflected in 16S rRNA qPCR analysis, there is likely a far higher bacterial biomass potentially synthesizing DMSP in the sediment than in any of the water column samples per equivalent sample mass. Thus, the BYSS bacteria are likely making a far larger contribution to the total DMSP levels in this environment than those in the photic seawater samples. Rhodobacteraceae bacteria containing dsyB were the most likely bacterial producers of DMSP in both the seawater and sediment samples. The mmtN gene was not detected in any samples suggesting that DMSP synthesis via MmtN was not a major pathway in the tested environments. Phytoplankton, whose DSYB genes were detected in the water column at low levels, are likely the major contributors to DMSP production in this environment. In contrast, the BYSS sediments are likely aphotic and their metagenomes contained no algal DSYB sequences suggesting that these eukaryotes play a less significant role in sediment DMSP production. Further work studying the transcription and/or protein abundance of the different DMSP synthesis genes products is required to test these hypotheses. It is possible that DMSP in the sediment is largely derived from sinking particles and not from synthesis. Considering that DMSP is rapidly degraded in seawater (Kiene, 1996; Ledyard and Dacey, 1996) and the abundance of DMSP catabolic genes in the seawater samples, it is likely that much of the DMSP in the sediment is a consequence of synthesis. Another important point to be mindful of is that when determining DMSP in seawater and sediment by alkaline hydrolysis, we actually measure the DMS derived from DMSP plus the endogenous DMS already in the sample. Unfortunately, we did not measure the endogenous DMS in our environmental samples, thus have no way of knowing the proportion of each these two influential compounds. In seawater samples, ∼9% of DMSP plus DMS was apportioned to volitiles (Kiene and Linn, 2000) and in sediments endogenous DMS can be undetectable (Carrión et al., 2017, 2019) or up to ∼four-fold lower than DMSP (Nedwell et al., 1994). Given DMSP always predominates over DMS in marine samples, we believe that most of the DMS detected in DMSP plus DMS samples are likely to derive from DMSP. However, it is possible that the levels of endogenous DMS were quite different between samples, for instance, in the hydrothermal field samples that are rich in hydrogen sulfide (H2S), H2S methylation to produce MeSH, and further methylation to DMS by, e.g., MddA, may be more prominent. It is important in future studies that precautions are taken to account for endogenous DMS, together with synthesis and catabolic rate assays to better predict the organosulfur cycling in these environments.
The potential to catabolize DMSP was abundant in all seawater and BYSS samples. DMSP demethylation appeared to be the dominant pathway in the seawater samples with dmdA predicted to be present in ∼40.0% of bacteria, which is consistent with the studies of Howard et al. (2006, 2008), Cui et al. (2015), and Steiner et al. (2019). Seawater bacteria with the potential to cleave DMSP were also abundant with dddP being the most abundant DMSP lyase gene (4.0–15.6%) but dddQ (6.0–14.1%) and dddK (8.3%) were also well represented. Interestingly, the relative abundance of dmdA decreased with water depth and was fourfold less abundant in BYSS than in seawater samples. Given the likely higher numbers of bacteria in the sediment, the decrease in dmdA relative abundance may not result in such a dramatic decrease in bacteria with the potential to demethylate DMSP. However, in the BYSS samples, dmdA abundance was much lower than that of dddP (P < 0.01; Figure 6). Therefore, it is likely that there is a sediment shift where DMSP cleavage may be the major sediment DMSP catabolic pathway as appose to DMSP demethylation in the seawater (Kiene and Linn, 2000; Kiene et al., 2000). Remarkably, the relative abundance of dddP in sediment was almost threefold more than in seawater (P < 0.01; Figure 3A). The reason for this shift is unknown, but it may be a consequence of the reduction in the abundance of SAR11 in the sediment and an increase in gamma-proteobacteria such as Pseudomonas containing dddP, or perhaps to the increased DMSP levels experienced in these samples. It was noteworthy that BYSS bacteria predicted to contain dddP, Pseudomonas, Mesorhizobium, and Leisingera, are distinct from previous studies that showed the Roseobacter clade to be the major bacteria possessing dddP (Todd et al., 2009; Varaljay, 2012; Zeng et al., 2016).
Given the abundance of DMSP catabolic genes in seawater and sediment, there are likely to be catabolites, such as DMS and MeSH available in these environments. None of these DMSP metabolites were measured in this study. It is clear that the genetic potential to methylate MeSH through mddA is ∼30-fold higher in BYSS than in seawater samples. Thus, the MeSH-dependent DMS production pathway is likely an important process in BYSS samples, but it is not so significant in seawater samples. Perhaps this is a consequence of DMSP and/or H2S (both MeSH precursors), being available at lower levels in the seawater compared to the sediment and thus the likely lower levels of MeSH being generated in seawater that is mainly incorporated into proteins. The relative abundance of mddA seen in seawater and sediment samples here are similar to those reported in Carrión et al. (2015, 2019). The mddA gene is found in a wide range of bacterial taxa which vary dependent on the environment of study. For example, in soil samples, Mycobacterium, Bradyrhizobium, Cyanothece, and Pseudomonas bacteria with mddA are abundant (Carrión et al., 2015, 2017); whereas Rhodopseudomonas and Thioalkalivibrio were the main taxa containing mddA in saltmarsh sediment (Carrión et al., 2019). In BYSS samples, the main bacteria containing mddA were predicted to be Desulfospira, Thioalkalivibrio, Crocosphaera, and Pseudomonas (Figure 5). Carrión et al. (2017, 2019) found that seawater and marine and saltmarsh sediments only generated DMS when samples were incubated with MeSH. This indicates that ordinarily much of the DMS generated from these processes is removed by other microbial processes. Indeed, dmoA, tmm, and ddhA genes were also relatively abundant in the marine samples and encode for enzymes that modify and thus remove DMS.
In general, this metagenomics study has shown that there is a very significant potential to catabolize DMSP and its catabolites in both BYSS and seawater samples, but there is a switch-over in terms of the dominant genes and potentially their catabolic pathways between these environments—DMSP demethylation dominating in photic waters to DMSP lysis and MeSH-dependent DMS production in BYSS. Given the vastly higher DMSP concentration and numbers of bacteria in sediment, it is likely that the BYSS environments are centers of high activity for organosulfur cycling. All of which warrant further future investigation.
This study represents the first to analyze organosulfur cycling potential in deep sea hydrothermal sediment samples by metagenomics. DMSP levels were 10-fold lower in the OTSP hydrothermal sediment than they were in the BYSS samples, but these were still far higher than those in the seawater samples. OTSP DMSP levels were similar to 4.5 km deep ocean sediment samples assayed in Williams et al. (2019), perhaps indicating that depth may be a factor in the reduced DMSP sediment concentration. Further studies of deep ocean sites are required to test this hypothesis. The profile of DMSP synthesis and catabolic potential was very different in the BYSS and OTSP samples. In studies to date on seawater and saltmarsh sediment samples, DMSP synthesis genes were always far less abundant than those for catabolism (Curson et al., 2017, 2018; Williams et al., 2019). In OSTP samples, the reverse was found to be the case, with dsyB being more abundant than that of the only detected DMSP catabolic gene (dddP; Figure 5). The abundance of dsyB did not change between the BYSS and OTSP samples. However, dddP was ∼360-fold reduced in its abundance (to 0.1%) compared to BYSS samples. To our knowledge, this is the first report of a marine metagenomics study, where the samples had appreciable DMSP levels, but lack a dmdA homolog. Previous studies have shown that DMSP demethylation is the major bacterial DMSP catabolic pathway in seawater environments (Kiene et al., 1999; Niki et al., 2000; Zubkov et al., 2001), accounting for ∼70% DMSP catabolism (Kiene and Linn, 2000). The data presented here suggest either that (i) DMSP catabolism is not an important process in these hydrothermal samples or (ii) novel DMSP lyases and/or DMSP demethylase enzymes exist in hydrothermal sediment samples. We know that there are likely other DMSP lyases to be identified in bacteria, since Liu et al. (2016) identified bacteria from East China Sea seawater with DMSP lyase activity, but which lack known ddd and Alma1 genes (Liu et al., 2018). In contrast, González et al. (2019) considered it would be highly unlikely to find an environmentally significant alternative enzyme to DmdA that would carry out the DMSP demethylation reaction. Further work involving process measurements from hydrothermal samples is required to determine if DMSP cleavage and/or demethylation is an important process in this environment, but these data indicate they are not. It is also possible that DMSP plays a more important role in environmental stress tolerance in the microbial communities of OTSP samples which experience considerably higher temperature and hydrostatic pressure than those of the shallower BYSS samples.
The mddA gene is far more abundant than dddP in OTSP samples (Figures 4, 5), suggesting that in these samples, MeSH may be more important for the generation of DMS than DMSP in such deep-sea hydrothermal sediment and polymetallic sulfide environments. If this is the case, a key question is where is the MeSH coming from in these hydrothermal sediments considering the DMSP demethylation is absent? MeSH can be produced from the methylation of H2S (Drotar et al., 1987) or degradation of Met and S-methylcysteine (Bremner and Steele, 1978; Ferchichi et al., 1985; Taylor and Kiene, 1989). Okinawa Trough hydrothermal vents have been reported to be rich in H2S (Miyazaki et al., 2017a), additionally, Kawagucci et al. (2013) found that H2S content reached 4.5 mM kg–1 in hydrothermal sediments in the northern Iheya. Therefore, we propose that much of the MeSH in the Okinawa hydrothermal field is likely to be produced from H2S methylation. Alternatively, given that the dmdBC and acuH genes are relatively abundant in the OTSP samples, it is possible that MMPA is also an important source of MeSH. Future studies should assay the availability of MMPA in diverse marine environments to determine its potential importance. These hydrothermal sediments present a case similar to those in terrestrial soil environments where MddA-dependent DMS pathways are more abundant than those for DMSP cleavage. A major difference though is that the hydrothermal samples studied here are marine and contain DMSP at appreciable levels. Further studies are required to determine if the MeSH methylation pathway is important in the OTSP environment.
The DMS oxidation genes ddhA and dmoA, but not tmm are also relatively abundant in OTSP samples further supporting the hypothesis that DMS is an important metabolite in these samples. The DMS dehydrogenase gene ddhA is more abundant than the DMS monooxygenase gene dmoA in these samples and this is in agreement with the findings of Carrión et al. (2019) working on saltmarsh sediment. Together these data support the existence of a unique organosulfur cycling profile in the deep ocean hydrothermal sediments to those in BYSS samples.
The sediment sampling regime in this study combined 0–5 cm deep sediment, which likely spanned large gradients in DMSP and DMS (Wilkening et al., 2019; Williams et al., 2019), oxygen, and diverse microbes. For this study, we have to consider these together, thus, the DMSP plus DMS content and microbial community data will be an average not reflecting that in the different sediment zones. For example, the DMSP and DMS concentration is likely to be far higher in the top ∼1 cm than in the deeper anoxic zone (Wilkening et al., 2019; Williams et al., 2019), which will likely have a distinct microbial profile. Nevertheless, the data presented here are convincing that the tested sediments are centers of high microbial organosulfur cycling activity. It would be interesting to study more precisely how microbial organosulfur cycling changes within the different sediment zones.
Another major limitation of this metagenomics study is that it is all only predictive and lacks process and/or data on the transcription and translation of the key organosulfur cycling genes discussed. Additionally, the metagenomic data presented here have no analytical replication and thus only provide a snapshot of metabolic potential. However, it does provide useful insights and hypotheses on organosulfur cycling in marine and hydrothermal sediments that warrant further investigation in the future.
The datasets generated for this study can be found in the Bio-Project of these metagenome data as follows: Yellow Sea (PRJNA428417), Bohai Sea (PRJNA514927), and Okinawa Trough hydrothermal field (PRJNA514953).
X-HZ and JT designed the experiments, analyzed the data, and wrote the manuscript. DS measured the DMSP concentration, did the quantification of 16S rRNA genes, performed the metagenomic analysis, analyzed the data, and wrote the manuscript. JL, YHZ, SZ, and YFZ analyzed the data and wrote the manuscript. HZ performed the metagenomic sequence assembly and binning. MY extracted DNA and performed metagenomic sequencing for hydrothermal field samples. All authors edited and approved the final manuscript.
X-HZ was funded by the National Key Research and Development Program of China (2016YFA0601303 and 2018YFC0310701), the National Natural Science Foundation of China (91751202 and 41730530), and the Marine S&T Fund of Shandong Province for Pilot National Laboratory for Marine Science and Technology (Qingdao) (2018SDKJ0406-4). JT was funded by Natural Environmental Research Council grants (NE/N002385, NE/P012671, and NE/S001352).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank all of the scientists and crews on the R/V DongFangHong 2 and Kexue Yihao for their assistance with sampling during the three cruises. Sincerely thanks to Long Wang and Ying Su (Ocean University of China) for their help on sample collection from the Okinawa Trough hydrothermal field and the Yellow Sea. We also thank Prof. Andrew W. B. Johnston (University of East Anglia) for his critical comments on this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00157/full#supplementary-material
Alcolombri, U., Ben-Dor, S., Feldmesser, E., Levin, Y., Tawfik, D. S., and Vardi, A. (2015). Identification of the algal dimethyl sulfide–releasing enzyme: a missing link in the marine sulfur cycle. Science 348, 1466–1469. doi: 10.1126/science.aab1586
Boden, R., Borodina, E., Wood, A. P., Kelly, D. P., Murrell, J. C., and Schäfer, H. (2011). Purification and characterization of dimethylsulfide monooxygenase from Hyphomicrobium sulfonivorans. J. Bacteriol. 193, 1250–1258. doi: 10.1128/JB.00977-10
Borodina, E., Kelly, D. P., Rainey, F. A., Ward-Rainey, N. L., and Wood, A. P. (2000). Dimethylsulfone as a growth substrate for novel methylotrophic species of Hyphomicrobium and Arthrobacter. Arch. Microbiol. 173, 425–437. doi: 10.1007/s002030000165
Bremner, J. M., and Steele, C. G. (1978). Role of microorganisms in the atmospheric sulfur cycle. Adv. Microb. Ecol. 4, 155–201. doi: 10.1007/978-1-4615-8222-9_4
Buchfink, B., Xie, C., and Huson, D. H. (2014). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Bullock, H. A., Reisch, C. R., Burns, A. S., Moran, M. A., and Whitman, W. B. (2014). Regulatory and functional diversity of methylmercaptopropionate coenzyme A ligases from the dimethylsulfoniopropionate demethylation pathway in Ruegeria pomeroyi DSS-3 and other proteobacteria. J. Bacteriol. 196, 1275–1285. doi: 10.1128/JB.00026-14
Carrión, O., Curson, A. R., Kumaresan, D., Fu, Y., Lang, A. S., Mercadé, E., et al. (2015). A novel pathway producing dimethylsulphide in bacteria is widespread in soil environments. Nat. Commun. 6:6579. doi: 10.1038/ncomms7579
Carrión, O., Pratscher, J., Curson, A. R., Williams, B. T., Rostant, W. G., Murrell, J. C., et al. (2017). Methanethiol-dependent dimethylsulfide production in soil environments. ISME J. 11, 2379–2390. doi: 10.1038/ismej.2017.105
Carrión, O., Pratscher, J., Richa, K., Rostant, W. G., Farhan Ul Haque, M., Murrell, J. C., et al. (2019). Methanethiol and dimethylsulfide cycling in stiffkey saltmarsh. Front. Microbiol. 10:1040. doi: 10.3389/fmicb.2019.01040
Cui, Y., Suzuki, S., Omori, Y., Wong, S. K., Ijichi, M., Kaneko, R., et al. (2015). Abundance and distribution of dimethylsulfoniopropionate-degrading genes and the corresponding bacterial community structure at dimethyl sulfide hotspots in the tropical and subtropical Pacific Ocean. Appl. Environ. Microbiol. 81, 4184–4194. doi: 10.1128/AEM.03873-14
Cuong, D. T., and Obbard, J. P. (2006). Metal speciation in coastal marine sediments from singapore using a modified BCR-sequential extraction procedure. Appl. Geochem. 21, 1335–1346. doi: 10.1016/j.apgeochem.2006.05.001
Curson, A. R., Liu, J., Bermejo Martinez, A., Green, R. T., Chan, Y., Carrion, O., et al. (2017). Dimethylsulfoniopropionate biosynthesis in marine bacteria and identification of the key gene in this process. Nat. Microbiol. 2:17009. doi: 10.1038/nmicrobiol.2017.9
Curson, A. R., Rogers, R., Todd, J. D., Brearley, C. A., and Johnston, A. W. (2008). Molecular genetic analysis of a dimethylsulfoniopropionate lyase that liberates the climate-changing gas dimethylsulfide in several marine α–proteobacteria and Rhodobacter sphaeroides. Environ. Microbiol. 10, 757–767. doi: 10.1111/j.1462-2920.2007.01499.x
Curson, A. R., Sullivan, M. J., Todd, J. D., and Johnston, A. W. (2011b). DddY, a periplasmic dimethylsulfoniopropionate lyase found in taxonomically diverse species of Proteobacteria. ISME J. 5, 1191–1200. doi: 10.1038/ismej.2010.203
Curson, A. R., Todd, J. D., Sullivan, M. J., and Johnston, A. W. (2011a). Catabolism of dimethylsulphoniopropionate: microorganisms, enzymes and genes. Nat. Rev. Microbiol. 9, 849–859. doi: 10.1038/nrmicro2653
Curson, A. R., Williams, B. T., Pinchbeck, B. J., Sims, L. P., Martínez, A. B., and Rivera, P. P. L. (2018). DSYB catalyses the key step of dimethylsulfoniopropionate biosynthesis in many phytoplankton. Nat. Microbiol 3, 430–439. doi: 10.1038/s41564-018-0119-5
Drotar, A., Burton, G. A., Tavernier, J. E., and Fall, R. (1987). Widespread occurrence of bacterial thiol methyltransferases and the biogenic emission of methylated sulfur gases. Appl. Environ. Microbiol. 53, 1626–1631. doi: 10.1128/aem.53.7.1626-1631.1987
Eddy, S. R. (2011). Accelerated profile HMM searches. PLoS Comput. Biol. 7:e1002195. doi: 10.1371/journal.pcbi.1002195
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–1000. doi: 10.1038/nmeth.2604
Eyice, O., Myronova, N., Pol, A., Carrión, O., Todd, J. D., Smith, T. J., et al. (2018). Bacterial SBP56 identified as a Cu-dependent methanethiol oxidase widely distributed in the biosphere. ISME J. 12, 145–160. doi: 10.1038/ismej.2017.148
Ferchichi, M., Hemme, D., Nardi, M., and Pamboukdjian, N. (1985). Production of methanethiol from methionine by Brevibacterium linens CNRZ 918. J. Gen. Microbiol. 131, 715–723. doi: 10.1099/00221287-131-4-715
Galí, M., Devred, E., Levasseur, M., Royer, S.-J., and Babin, M. (2015). A remote sensing algorithm for planktonic dimethylsulfoniopropionate (DMSP) and an analysis of global patterns. Remote Sens. Environ. 171, 171–184. doi: 10.1016/j.rse.2015.10.012
Gamo, T., Sakai, H., Kim, E.-S., Shitashima, K., and Ishibashi, J. (1991). High alkalinity due to sulfate reduction in the CLAM hydrothermal field, Okinawa trough. Earth Planet. Sci. Lett. 107, 328–338. doi: 10.1016/0012-821x(91)90080-2
González, J. M., Hernández, L., Manzano, I., and Pedrós-Alió, C. (2019). Functional annotation of orthologs in metagenomes: a case study of genes for the transformation of oceanic dimethylsulfoniopropionate. ISME J. 13, 1183–1197. doi: 10.1038/s41396-019-0347-6
Gould, W. D., and Kanagawa, T. (1992). Purification and properties of methyl mercaptan oxidase from Thiobacillus thioparus TK-m. J. Gen. Microbiol. 138, 217–221. doi: 10.1099/00221287-138-1-217
Graham, E. D., Heidelberg, J. F., and Tully, B. J. (2017). BinSanity: unsupervised clustering of environmental microbial assemblies using coverage and affinity propagation. Peer J. 5:e3035. doi: 10.7717/peerj.3035
Griebler, C. (1997). Dimethylsulfoxide (DMSO) reduction: a new approach to determine microbial activity in freshwater sediments. J. Microbiol. Methods 29, 31–40. doi: 10.1016/s0167-7012(97)00990-1
Howard, E. C., Henriksen, J. R., Buchan, A., Reisch, C. R., Bürgmann, H., Welsh, R., et al. (2006). Bacterial taxa that limit sulfur flux from the ocean. Science 314, 649–652. doi: 10.1126/science.1130657
Howard, E. C., Sun, S., Biers, E. J., and Moran, M. A. (2008). Abundant and diverse bacteria involved in DMSP degradation in marine surface waters. Environ. Microbiol. 10, 2397–2410. doi: 10.1111/j.1462-2920.2008.01665.x
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386. doi: 10.1101/gr.5969107
Ishibashi, J., Sano, Y., Wakita, H., Gamo, T., Tsutsumi, M., and Sakai, H. (1995). Helium and carbon geochemistry of hydrothermal fluids from the Mid-Okinawa trough back arc basin, southwest of Japan. Chem. Geol. 123, 1–15. doi: 10.1016/0009-2541(95)00051-m
Ishibashi, J. I., Ikegami, F., Tsuji, T., and Urabe, T. (2015). “Hydrothermal activity in the Okinawa trough back-arc basin: geological background and hydrothermal mineralization,” in Subseafloor Biosphere Linked to Hydrothermal Systems, (Tokyo: Springer), 337–359. doi: 10.1007/978-4-431-54865-2_27
Jin, J., Liu, S. M., Ren, J. L., Liu, C. G., Zhang, J., and Zhang, G. L. (2013). Nutrient dynamics and coupling with phytoplankton species composition during the spring blooms in the Yellow Sea. Deep Sea Res. Part II Top. Stud. Oceanogr. 97, 16–32. doi: 10.1016/j.dsr2.2013.05.002
Kageyama, H., Tanaka, Y., Shibata, A., Waditee-Sirisattha, R., and Takabe, T. (2018). Dimethylsulfoniopropionate biosynthesis in a diatom Thalassiosira pseudonana: Identification of a gene encoding MTHB-methyltransferase. Arch. Biochem. Biophys. 645, 100–106. doi: 10.1016/j.abb.2018.03.019
Kang, D. D., Froula, J., Egan, R., and Wang, Z. (2015). MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. Peer J. 3:e1165. doi: 10.7717/peerj.1165
Kappler, U., and Schäfer, H. (2014). “Transformations of dimethylsulfide,” in The Metal-Driven Biogeochemistry of Gaseous Compounds in the Environment, eds P. Kroneck and M. E. Sosa Torres, (Dordrecht: Springer), 279–313. doi: 10.1007/978-94-017-9269-1_11
Kawagucci, S., Chiba, H., Ishibashi, J. I., Yamanaka, T., Toki, T., Muramatsu, Y., et al. (2011). Hydrothermal fluid geochemistry at the Iheya North field in the mid-Okinawa trough: implication for origin of methane in subseafloor fluid circulation systems. Geochem. J. 45, 109–124. doi: 10.2343/geochemj.1.0105
Kawagucci, S., Ueno, Y., Takai, K., Toki, T., Ito, M., and Inoue, K. (2013). Geochemical origin of hydrothermal fluid methane in sediment-associated fields and its relevance to the geographical distribution of whole hydrothermal circulation. Chem. Geol. 339, 213–225. doi: 10.1016/j.chemgeo.2012.05.003
Kiene, R. P. (1996). “Turnover of dissolved DMSP in estuarine and shelf waters of the northern Gulf of Mexico.” in Biological and Environmental Chemistry of DMSP and Related Sulfonium Compounds, eds P. K. Ronald, T. V. Pieter, D. K. Maureen, and O. K. Gunter, (Boston, MA: Springer Press), 337–349. doi: 10.1007/978-1-4613-0377-0_29
Kiene, R. P., and Hines, M. E. (1995). Microbial formation of dimethyl sulfide in anoxic sphagnum peat. Appl. Environ. Microbiol. 61, 2720–2726. doi: 10.1128/aem.61.7.2720-2726.1995
Kiene, R. P., and Linn, L. J. (2000). The fate of dissolved dimethylsulfoniopropionate (DMSP) in seawater: tracer studies using 35S-DMSP. Geochim. Cosmochim. Acta 64, 2797–2810. doi: 10.1016/s0016-7037(00)00399-9
Kiene, R. P., Linn, L. J., and Bruton, J. A. (2000). New and important roles for DMSP in marine microbial communities. J. Sea Res. 43, 209–224. doi: 10.1016/s1385-1101(00)00023-x
Kiene, R. P., Linn, L. J., González, J., Moran, M. A., and Bruton, J. A. (1999). Dimethylsulfoniopropionate and methanethiol are important precursors of methionine and protein-sulfur in marine bacterioplankton. Appl. Environ. Microbiol. 65, 4549–4558. doi: 10.1128/aem.65.10.4549-4558.1999
Kim, S. J., Shin, H. J., Kim, Y. C., Lee, D. S., and Yang, J. W. (2000). Isolation and purification of methyl mercaptan oxidase from Rhodococcus rhodochrous for mercaptan detection. Biotechnol. Bioprocess. Eng. 5, 465–468. doi: 10.1007/bf02931949
Kudo, T., Kobiyama, A., Rashid, J., Reza, M. S., Yamada, Y., Ikeda, Y., et al. (2018). Seasonal changes in the abundance of bacterial genes related to dimethylsulfoniopropionate catabolism in seawater from Ofunato Bay revealed by metagenomic analysis. Gene 665, 174–184. doi: 10.1016/j.gene.2018.04.072
Kumaresan, D., Stephenson, J., Doxey, A. C., Bandukwala, H., Brooks, E., and Hillebrand, V. A. (2018). Aerobic proteobacterial methylotrophs in Movile Cave: genomic and metagenomic analyses. Microbiome 6:1. doi: 10.1186/s40168-017-0383-2
Ledyard, K. M., and Dacey, J. W. (1996). Microbial cycling of DMSP and DMS in coastal and oligotrophic seawater. Limnol. Oceanogr. 41, 33–40. doi: 10.4319/lo.1996.41.1.0033
Lee, H.-H., Kim, S.-J., Shin, H.-J., Park, J.-Y., and Yang, J.-W. (2002). Purification and characterisation of methyl mercaptan oxidase from Thiobacillus thioparus for mercaptan detection. Biotechnol. Bioprocess. Eng. 7, 375–379. doi: 10.1007/bf02933525
Li, D., Liu, C.-M., Luo, R., Sadakane, K., and Lam, T. W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Liao, C., and Seebeck, F. P. (2019). In vitro reconstitution of bacterial DMSP biosynthesis. Angew. Chem. Int. Ed Engl. 131, 3591–3594. doi: 10.1002/ange.201814662
Lidbury, I., Kröber, E., Zhang, Z., Zhu, Y., Murrell, J. C., Chen, Y., et al. (2016). A mechanism for bacterial transformations of DMS to DMSO: a missing link in the marine organic sulfur cycle. Environ. Microbiol. 18, 2754–2765. doi: 10.1111/1462-2920.13354
Liu, J., Wang, Y., Liu, Y., and Zhang, X. H. (2016). Ahrensia marina sp. nov., a dimethylsulfoniopropionate-cleaving bacterium isolated from seawater, and emended descriptions of the genus Ahrensia and Ahrensia kielensis. Int. J. Syst. Evol. Microbiol. 66, 874–880. doi: 10.1099/ijsem.0.000805
Liu, J., Zheng, Y., Lin, H., Wang, X., Li, M., Liu, Y., et al. (2019). Proliferation of hydrocarbon-degrading microbes at the bottom of the Mariana Trench. Microbiome 7:47. doi: 10.1186/s40168-019-0652-3
Liu, J. L., Liu, J., Zhang, S. H., Liang, J., Lin, H., Song, D. L., et al. (2018). Novel insights into bacterial dimethylsulfoniopropionate catabolism in the East China Sea. Front. Microbiol. 9:3206. doi: 10.3389/fmicb.2018.03206
Liu, S., Lou, S., Kuang, C. P., Huang, W. R., Chen, W. J., Zhang, J., et al. (2011). Water quality assessment by pollution-index method in the coastal waters of Hebei Province in western Bohai Sea, China. Mar. Pollut. Bull. 62, 2220–2229. doi: 10.1016/j.marpolbul.2011.06.021
Lovelock, J. E., Maggs, R. J., and Rasmussen, R. A. (1972). Atmospheric dimethyl sulphide and the natural sulphur cycle. Nature 237, 452–453. doi: 10.1038/237452a0
Lyon, B. R., Lee, P. A., Bennett, J. M., DiTullio, G. R., and Janech, M. G. (2011). Proteomic analysis of a sea-ice diatom: salinity acclimation provides new insight into the dimethylsulfoniopropionate production pathway. Plant Physiol. 157, 1926–1941. doi: 10.1104/pp.111.185025
McDevitt, C. A., Hanson, G. R., Noble, C. J., Cheesman, M. R., and McEwan, A. G. (2002). Characterization of the redox centers in dimethyl sulfide dehydrogenase from Rhodovulum sulfidophilum. Biochemistry 41, 15234–15244. doi: 10.1021/bi026221u
Mcewan, A. G., Ferguson, S. J., and Jackson, J. B. (1991). Purification and properties of dimethyl sulphoxide reductase from Rhodobacter capsulatus. A periplasmic molybdoenzyme. Biochem. J. 274(Pt 1), 305–307. doi: 10.1042/bj2740305
Miyazaki, J., Kawagucci, S., Makabe, A., Takahashi, A., Kitada, K., and Torimoto, J. (2017a). Deepest and hottest hydrothermal activity in the Okinawa trough: the Yokosuka site at Yaeyama Knoll. R. Soc. Open Sci 4:171570. doi: 10.1098/rsos.171570
Miyazaki, J., Makabe, A., Matsui, Y., Ebina, N., Tsutsumi, S., Ishibashi, J. I., et al. (2017b). WHATS-3: an improved flow-through multi-bottle fluid sampler for deep-sea geofluid research. Front. Earth. Sci. 5:45. doi: 10.3389/feart.2017.00045
Moran, M. A., Reisch, C. R., Kiene, R. P., and Whitman, W. B. (2012). Genomic insights into bacterial DMSP transformations. Annu. Rev. Mar. Sci. 4, 523–542. doi: 10.1146/annurev-marine-120710-100827
Nedwell, D. B., Shabbeer, M. T., and Harrison, R. M. (1994). Dimethyl Sulphide in North Sea Waters and Sediments. Estuar. Coast Shelf. Sci. 39, 209–217. doi: 10.1006/ecss.1994.1059
Niki, T., Kunugi, M., and Otsuki, A. (2000). DMSP-lyase activity in five marine phytoplankton species: its potential importance in DMS production. Mar. Biol. 136, 759–764. doi: 10.1007/s002279900235
Nowinski, B., Motard-Côté, J., Landa, M., Preston, C. M., Scholin, C. A., Birch, J. M., et al. (2019). Microdiversity and temporal dynamics of marine bacterial dimethylsulfoniopropionate genes. Environ. Microbiol. 21, 1687–1701. doi: 10.1111/1462-2920.14560
Otte, M. L. (2004). Dimethylsulphoniopropionate (DMSP) and related compounds in higher plants. J. Exp. Bot. 55, 1919–1925. doi: 10.1093/jxb/erh178
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). Fast tree 2 – approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Qin, J., Li, Y., Cai, Z., Li, S., Zhu, J., Zhang, F., et al. (2012). A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60. doi: 10.1038/nature11450
Reisch, C. R., Moran, M. A., and Whitman, W. B. (2008). Dimethylsulfoniopropionate-dependent demethylase (DmdA) from Pelagibacter ubique and Silicibacter pomeroyi. J. Bacteriol. 190, 8018–8024. doi: 10.1128/jb.00770-08
Reisch, C. R., Moran, M. A., and Whitman, W. B. (2011a). Bacterial catabolism of dimethylsulfoniopropionate (DMSP). Front. Microbiol. 2:172. doi: 10.3389/fmicb.2011.00172
Reisch, C. R., Stoudemayer, M. J., Varaljay, V. A., Amster, I. J., Moran, M. A., and Whitman, W. B. (2011b). Novel pathway for assimilation of dimethylsulphoniopropionate widespread in marine bacteria. Nature 473, 208–211. doi: 10.1038/nature10078
Sakai, H., Gamo, T., Kim, E. S., Shitashima, K., Yanagisawa, F., Tsutsumi, M., et al. (1990). Unique chemistry of the hydrothermal solution in the mid-Okinawa trough Backarc Basin. Geop. Res. Lett. 17, 2133–2136. doi: 10.1029/gl017i012p02133
Sambasivarao, D., and Weiner, J. H. (1991). Dimethyl sulfoxide reductase of Escherichia coli: an investigation of function and assembly by use of in vivo complementation. J. Bacteriol. 173, 5935–5943. doi: 10.1128/jb.173.19.5935-5943.1991
Satoh, T., and Kurihara, F. N. (1987). Purification and properties of dimethylsulfoxide reductase containing a molybdenum cofactor from a photodenitrifier, Rhodopseudomonas sphaeroides f.s. denitrificans. J. Biochem. 102, 191–197. doi: 10.1093/oxfordjournals.jbchem.a122
Schlitzer, R. (2002). Interactive analysis and visualization of geoscience data with Ocean data view. Comput. Geosci. 28, 1211–1218. doi: 10.1016/S0098-3004(02)00040-7
Schnicker, N. J., De Silva, S. M., Todd, J. D., and Dey, M. (2017). Structural and biochemical insights into dimethylsulfoniopropionate cleavage by cofactor-bound DddK from the prolific marine bacterium Pelagibacter. Biochemistry 56, 2873–2885. doi: 10.1021/acs.biochem.7b00099
Seymour, J. R., Simo, R., Ahmed, T., and Stocker, R. (2010). Chemoattraction to dimethylsulfoniopropionate throughout the marine microbial food web. Science 329, 342–345. doi: 10.1126/science.1188418
Shang, S., Lee, Z., and Wei, G. (2011). Characterization of MODIS-derived euphotic zone depth: results for the China Sea. Remote Sens. Environ. 115, 180–186. doi: 10.1016/j.rse.2010.08.016
Shao, X., Cao, H.-Y., Zhao, F., Ming, P., Wang, P., and Li, C. Y. (2019). Mechanistic insight into 3-methylmercaptopropionate metabolism and kinetical regulation of demethylation pathway in marine dimethylsulfoniopropionate-catabolizing bacteria. Mol. Microbiol. 111, 1057–1073. doi: 10.1111/mmi.14211
Stefels, J., Steinke, M., Turner, S., Malin, G., and Belviso, S. (2007). Environmental constraints on the production and removal of the climatically active gas dimethyl sulphide (DMS) and implications for ecosystem modelling. Biogeochemistry 83, 245–275. doi: 10.1007/s10533-007-9091-5
Steiner, P. A., Sintes, E., Simó, R., De Corte, D., Pfannkuchen, D. M., Ivanèiæ, I., et al. (2019). Seasonal dynamics of marine snow-associated and free-living demethylating bacterial communities in the coastal northern Adriatic Sea. Environ. Microbiol. Rep. 11, 699–707. doi: 10.1111/1758-2229.12783
Stets, E. G., Hines, M. E., and Kiene, R. P. (2004). Thiol methylation potential in anoxic, low-pH wetland sediments and its relationship with dimethylsulfide production and organic carbon cycling. FEMS Microbiol. Ecol. 47, 1–11. doi: 10.1016/S0168-6496(03)00219-8
Sun, J., Todd, J. D., Thrash, J. C., Qian, Y., Qian, M. C., and Temperton, B. (2016). The abundant marine bacterium Pelagibacter simultaneously catabolizes dimethylsulfoniopropionate to the gases dimethyl sulfide and methanethiol. Nat. Microbiol 1:16065. doi: 10.1038/nmicrobiol.2016.65
Suylen, G. M. H., Large, P. J., Vandijken, J. P., and Kuenen, J. G. (1987). Methyl mercaptan oxidase, a key enzyme in the metabolism of methylated sulfur-compounds by Hyphomicrobium EG. J. Gen. Microbiol. 133, 2989–2997. doi: 10.1099/00221287-133-11-2989
Tang, K., Liu, K., Jiao, N., Zhang, Y., and Chen, C. T. (2013). Functional metagenomic investigations of microbial communities in a shallow-sea hydrothermal system. PLoS One. 8:e72958. doi: 10.1371/journal.pone.0072958
Taylor, B. F., and Kiene, R. P. (1989). “Microbial metabolism of dimethyl sulfide,” in Biogenic Sulfur in the Environment, Vol. 13, ed. E. Saltzman et al. (Washington, DC: American Chemical Society), 202–221. doi: 10.1021/bk-1989-0393.ch013
Todd, J. D., Curson, A. R., Dupont, C. L., Nicholson, P., and Johnston, A. W. (2009). The dddP gene, encoding a novel enzyme that converts dimethylsulfoniopropionate into dimethyl sulfide, is widespread in ocean metagenomes and marine bacteria and also occurs in some ascomycete fungi. Environ. Microbiol. 11, 1376–1385. doi: 10.1111/j.1462-2920.2009.01864.x
Todd, J. D., Curson, A. R., Kirkwood, M., Sullivan, M. J., Green, R. T., and Johnston, A. W. (2011). DddQ, a novel, cupin-containing, dimethylsulfoniopropionate lyase in marine roseobacters and in uncultured marine bacteria. Environ. Microbiol. 13, 427–438. doi: 10.1111/j.1462-2920.2010.02348.x
Todd, J. D., Kirkwood, M., Newton-Payne, S., and Johnston, A. W. (2012). DddW, a third DMSP lyase in a model Roseobacter marine bacterium. Ruegeria pomeroyi DSS-3. ISME J. 6, 223–226. doi: 10.1038/ismej.2011.79
Todd, J. D., Rogers, R., Li, Y. G., Wexler, M., Bond, P. L., Sun, L., et al. (2007). Structural and regulatory genes required to make the gas dimethyl sulfide in bacteria. Science 315, 666–669. doi: 10.1126/science.1135370
Uchida, A., Ooguri, T., Ishida, T., Kitaguchi, H., and Ishida, Y. (2012). Biological and Environmental Chemistry of DMSP and Related Sulfonium Compounds, Vol. 9. Berlin: Springer Science and Business Media, 97–107.
Vallina, S. M., and Simó, R. (2007). Strong relationship between DMS and the solar radiation dose over the global surface ocean. Science 315, 506–508. doi: 10.1126/science.1133680
Varaljay, V. A. (2012). Quantitative Analysis of Bacterial DMSP-Degrading Gene Diversity, Abundance, and Expression in Marine Surface Water Environments. Doctoral thesis, University of Georgia, Atlanta, GA
Visscher, P., Barrie, F. T., and Ronald, P. K. (1995). Microbial consumption of dimethyl sulfide and methanethiol in coastal marine sediments. FEMS Microbiol. Ecol. 18, 145–153. doi: 10.1016/0168-6496(95)00051-b
Visscher, P. T., and Taylor, B. F. (1993). A new mechanism for the aerobic catabolism of dimethyl sulfide. Appl. Environ. Microbiol. 59, 3784–3789. doi: 10.1128/aem.59.11.3784-3789.1993
Wilkening, J. V., Turchyn, A. V., Redeker, K. R., Mills, J. V., Antler, G., and Carrión, O. (2019). The Production and fate of volatile organosulfur compounds in sulfidic and ferruginous sediment. J. Geophys. Res. Biogeosci 124, 3390–3402. doi: 10.1029/2019JG005248
Williams, B. T., Cowles, K., Bermejo Martínez, A., Curson, A. R., Zheng, Y., and Liu, J. (2019). Bacteria are important dimethylsulfoniopropionate producers in coastal sediments. Nat. Microbiol. 4, 1815–1825. doi: 10.1038/s41564-019-0527-1
Xu, S. S. (2011). Decline Mechanisms of Fishery Resources in the Bohai Sea Under Anthropogenic Activities. Doctoral thesis, Graduate University of Chinese Academy of Sciences, Beijing.
Yang, G. P., Song, Y. Z., Zhang, H. H., Li, C. X., and Wu, G. W. (2014). Seasonal variation and biogeochemical cycling of dimethylsulfide (DMS) and dimethylsulfoniopropionate (DMSP) in the Yellow Sea and Bohai Sea. J. Geophys. Res. Oceans 119, 8897–8915. doi: 10.1002/2014jc010373
Yin, Q., Fu, B., Li, B., Shi, X., Inagaki, F., and Zhang, X. H. (2013). Spatial variations in microbial community composition in surface seawater from the ultra-oligotrophic center to rim of the South Pacific Gyre. PLoS One 8:e55148. doi: 10.1371/journal.pone.0055148
Yoch, D. C. (2002). Dimethylsulfoniopropionate: its sources, role in the marine food web, and biological degradation to dimethylsulfide. Appl. Environ. Microbiol 68, 5804–5815. doi: 10.1128/aem.68.12.5804-5815.2002
Zeng, Y. X., Qiao, Z. Y., Yu, Y., Li, H. R., and Luo, W. (2016). Diversity of bacterial dimethylsulfoniopropionate degradation genes in surface seawater of Arctic Kongsfjorden. Sci. Rep. 6:33031. doi: 10.1038/srep33031
Zeng, Z. G. (2015). “New hydrothermal field in the Okinawa Trough,” in Proceedings of the 25th Goldschmidt Conference, Abstracts, Prague, 3573.
Zhang, H. H., Yang, G. P., and Zhu, T. (2008). Distribution and cycling of dimethylsulfide (DMS) and dimethylsulfoniopropionate (DMSP) in the sea-surface microlayer of the Yellow Sea. China Spring. Cont. Shelf. Res. 28, 2417–2427. doi: 10.1016/j.csr.2008.06.003
Zhang, X. H., Liu, J., Liu, J., Yang, G., Xue, C. X., and Curson, A. R. (2019). Biogenic production of DMSP and its degradation to DMS—their roles in the global sulfur cycle. Sci. China Life Sci. 62, 1296–1319. doi: 10.1007/s11427-018-9524-y
Zhou, J., Bruns, M. A., and Tiedje, J. M. (1996). DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 62, 316–322. doi: 10.1128/aem.62.2.316-322.1996
Zhu, W., Lomsadze, A., and Borodovsky, M. (2010). Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 38, 132–132. doi: 10.1093/nar/gkq275
Zubkov, M. V., Fuchs, B. M., Archer, S. D., Kiene, R. P., Amann, R., and Burkill, P. H. (2001). Linking the composition of bacterioplankton to rapid turnover of dissolved dimethylsulphoniopropionate in an algal bloom in the North Sea. Environ. Microbiol. 3, 304–311. doi: 10.1046/j.1462-2920.2001.00196.x
Keywords: DMSP, DMS, sulfur cycle, metagenome, marine sediment, marginal seas
Citation: Song D, Zhang Y, Liu J, Zhong H, Zheng Y, Zhou S, Yu M, Todd JD and Zhang X-H (2020) Metagenomic Insights Into the Cycling of Dimethylsulfoniopropionate and Related Molecules in the Eastern China Marginal Seas. Front. Microbiol. 11:157. doi: 10.3389/fmicb.2020.00157
Received: 15 October 2019; Accepted: 22 January 2020;
Published: 18 February 2020.
Edited by:
Levente Bodrossy, CSIRO Oceans and Atmosphere, AustraliaReviewed by:
Michaela A. Mausz, University of Warwick, United KingdomCopyright © 2020 Song, Zhang, Liu, Zhong, Zheng, Zhou, Yu, Todd and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan D. Todd, Sm9uYXRoYW4uVG9kZEB1ZWEuYWMudWs=; Xiao-Hua Zhang, eGh6aGFuZ0BvdWMuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.