 Guillermo Nogueira-López1
Guillermo Nogueira-López1 Fabiola Padilla-Arizmendi1
Fabiola Padilla-Arizmendi1 Sarah Inwood1,2
Sarah Inwood1,2 Sarah Lyne1Johanna M. Steyaert1,3Maria Fernanda Nieto-Jacobo1,4Alison Stewart1,5
Sarah Lyne1Johanna M. Steyaert1,3Maria Fernanda Nieto-Jacobo1,4Alison Stewart1,5 Artemio Mendoza-Mendoza1*
Artemio Mendoza-Mendoza1*- 1Bio-Protection Research Centre, Lincoln University, Lincoln, New Zealand
- 2Department of Biochemistry, University of Otago, Dunedin, New Zealand
- 3Lincoln Agritech Ltd, Lincoln, New Zealand
- 4Plant & Food Research Gerald St, Lincoln, New Zealand
- 5Foundation For Arable Research, Templeton, New Zealand
Species of the genus Trichoderma are ubiquitous in the environment and are widely used in agriculture, as biopesticides, and in the industry for the production of plant cell wall-degrading enzymes. Trichoderma represents an important genus of endophytes, and several Trichoderma species have become excellent models for the study of fungal biology and plant–microbe interactions; moreover, are exceptional biotechnological factories for the production of bioactive molecules useful in agriculture and medicine. Next-generation sequencing technology coupled with systematic construction of recombinant DNA molecules provides powerful tools that contribute to the functional analysis of Trichoderma genetics, thus allowing for a better understanding of the underlying factors determining its biology. Here, we present the creation of diverse vectors containing (i) promoter-specific vectors for Trichoderma, (ii) gene deletions (using hygromycin phosphotransferase as selection marker), (iii) protein localization (mCherry and eGFP, which were codon-optimized for Trichoderma), (iv) gene complementation (neomycin phosphotransferase) and (v) overexpression of encoding gene proteins fused to fluorescent markers, by using the Golden Gate cloning technology. Furthermore, we present the design and implementation of a binary vector for Agrobacterium-mediated transformation in Trichoderma to increase the homologous recombination rate and the generation of a novel selection marker based on carboxin resistance.
Introduction
Eukaryotic models, including ascomycetes fungi; for example, Saccharomyces cerevisiae, Aspergillus nidulans, and Neurospora crassa, have provided a comprehensive understanding of several biological processes that have been important for scientific research and applications (Nayak et al., 2006; Karathia et al., 2011; Zámborszky et al., 2014). Within the ascomycetes fungi, the genus Trichoderma has been widely used in agriculture and industry for its ability to display outstanding properties as biocontrol and biofertilizer agents (Druzhinina et al., 2011; Schmoll et al., 2016). Trichoderma spp. are distributed worldwide and more frequently found in the soil and/or rhizosphere, acting as free-living organisms. They can also colonize plant roots (Brotman et al., 2013), thus creating an endophyte-plant beneficial interaction. In general, the colonization of plant roots by Trichoderma spp. is beneficial to the host plant by enhancing plant growth and conferring resistance to biotic and abiotic stresses (Hermosa et al., 2012). In addition to the plant growth promotion abilities of Trichoderma, it also acts as a biocontrol agent of several plant pathogens (Rubio et al., 2014; Guigon-Lopez et al., 2015; Malmierca et al., 2016), diminishing signs and symptoms of the disease.
The availability of 25 genome sequences of Trichoderma spp.1 and2 has greatly assisted the genetic study of the Trichoderma genus (Mukherjee et al., 2013; Schmoll et al., 2016; Kubicek et al., 2019). However, the main challenge in genomics is to assign a function to predicted genes to reveal new insights into fungal biology. Functional gene characterization involves, in addition to in the generation of gene knockouts, studies of protein localization, identification of interaction partners, gene overexpression and complementation studies of the gene in question. Gene disruption by the substitution of gene sequences via homologous recombination is one of the most popular strategies to start the characterization of genes (Kück and Hoff, 2010).
To study gene function, the scientific community generally relies on the construction of recombinant DNA molecules using conventional cloning methodology that is based on restriction-digestion and ligation procedures. Although this strategy has been utilized to explore gene function with the generation of vectors to create deletion and/or over-expression mutants of the target genes, this technique has several disadvantages (e.g., time-consuming and retention of restriction endonuclease sites) when multi-targeted DNA fragments are ligated and inserted step-by-step into the vector.
The efficiency of homologous recombination during transformation in filamentous fungi is very low; usually less than 5% (Kück and Hoff, 2010) and the existing resistance markers are limited, and not all screening markers are useful for filamentous fungi. To overcome these limitations in Trichoderma, a streamlined knockout-construction system was created that included the construction of vectors using yeast mediated electroporation and the transformation of a T. reesei strain deficient in non-homologous end joining (Δtku70), using three different selection markers (the hygromycin phosphotransferase hph gene, acetamidase-encoding amdS gene, and the pyr4 gene encoding orotidine-5′-monophosphate decarboxylase) (Schuster et al., 2012).
The increase of functional genomics studies in the last decade has led to the development of more efficient and accurate cloning techniques that overcome the main issues of conventional cloning procedures including the Gateway and the Golden Gate cloning systems (Hartley et al., 2000; Walhout et al., 2000; Engler et al., 2008). The Gateway cloning system has been utilized for the analysis of functional genes in planta (Curtis and Grossniklaus, 2003) and the identification of functional genes during plant–microbe interactions. For example, in the fungus Magnaporthe oryzae, the creation of pFPL vectors using the Gateway system has facilitated the localization of effector proteins during plant infection (Gong et al., 2015). Nevertheless, one of the limitations of the Gateway system compared to the Golden Gate strategy is the retention of specific recombination site sequences in the final construct (Reece-Hoyes and Walhout, 2018). The Golden Gate cloning theory reported by Engler et al. (2008) allows for the creation of vectors containing more than two molecules in a single restriction-ligation reaction with the use of type IIs restriction enzymes; for example, BsaI which cleaves outside of its recognition sequence 5′-GGTCTC-3′, resulting in 5′ or 3′ DNA overhangs that are four bases in length. This one-pot, one-step method creates recombinant vectors in less than one day compared with conventional cloning procedures which take several days (Engler et al., 2008) and permits the assembly of multiple DNA fragments with high efficiency (Engler et al., 2009; Weber et al., 2011). The Golden Gate cloning approach has been successfully implemented in plants (Emami et al., 2013; Lampropoulos et al., 2013), fungi (Terfrüchte et al., 2014; Sarkari et al., 2017; Hernanz-Koers et al., 2018; Egermeier et al., 2019) and bacteria (Wang et al., 2018).
Recently it was reported that codon usage strongly correlates with both protein and mRNA levels genome-wide in Neurospora crassa. Moreover, gene codon optimization in this fungus, resulted in strong up-regulation of protein and RNA levels, suggesting that codon usage is an essential determinant of gene expression (Zhou et al., 2016).
For the transformation of Trichoderma spp., several markers have been developed including auxotrophic markers (e.g., L-arginine, L-lysine and uridine biosynthesis pathways) that require an auxotrophic mutant as a parental strain (Baek and Kenerley, 1998; Jørgensen et al., 2014; Derntl et al., 2015). However, drug resistance markers have an advantage compared to auxotrophic markers by removing this restriction to a specific parental strain. In Trichoderma, selectable drug resistance markers have been developed for its transformation such as hph (hygromycin phosphotransferase) (Mach et al., 1994), nptII (neomycin phosphotransferase II, geneticin G418 resistance) (Gruber et al., 2012) and β-tubulin-2 (benomyl resistance) (Peterbauer et al., 1992); however, resistance to these antimicrobial agents can differ between strains and species.
Succinate dehydrogenase (SDH) catalyses electron transfer from succinate to quinone during aerobic respiration (Singer et al., 1971). Carboxin is a specific inhibitor of this enzyme from several different organisms, including fungi (Broomfield and Hargreaves, 1992; Keon et al., 1994; Topp et al., 2002; Kilaru et al., 2009; Shima et al., 2009, 2011). Carboxin binds to the enzyme SDH, preventing electron transfer, and effectively killing the organism, as it cannot generate energy. Direct mutagenesis of the SDH gene, confers resistance to carboxin as described in other fungal models, including Ustilago maydis (Broomfield and Hargreaves, 1992; Keon et al., 1994; Topp et al., 2002), Aspergillus oryzae (Shima et al., 2009; Shima et al., 2011), and Coprinopsis cinerea (Kilaru et al., 2009), making it suitable as a drug resistance marker in fungi.
In this study, we aimed to improve a vector system for cloning and expression of any desired gene to study Trichoderma’s biology by using the Golden Gate strategy. The creation of entry vectors that contain specific molecules such as promoters, markers, terminators, tags and resistance cassettes were used for the generation of vectors to achieve gene deletion, protein localization and gene complementation. This was coupled with Agrobacterium-mediated transformation (AMT) to increase homologous recombination using Trichoderma virens as a model. Additionally, a new selection marker (carboxin resistance) was developed for the genetic transformation of T. virens.
Materials and Methods
Microorganisms, Media and Culture Conditions
T. virens Gv 29.8 (Kindly provided by Charles Kenerley, Texas A&M University, United States), T. sp. “atroviride B” LU132 (Braithwaite et al., 2017), and mutants were grown on potato dextrose agar (PDA) (Difco, Fisher Scientific, United States) at 25°C under a cycle of 12 h light and 12 h dark for 7 days to induce conidiation. Conidia were collected using sterile nanopure water and filtered through a double layer of sterile Miracloth (Millipore Merck, United States). The conidial concentration was determined using a Neubauer chamber. Escherichia coli cells transformants were grown on Luria Bertani (LB) medium supplemented with either 100 μg/mL ampicillin (PanReac AppliChem, Germany) or 50 μg/mL kanamycin (Sigma Aldrich, United States) at 37°C depending on the vector. Agrobacterium tumefaciens EHA105 cells transformants were grown on LB medium containing 25 μg/mL kanamycin and 25 μg/mL rifampicin (Sigma Aldrich, United States) at 28°C.
Improvements in Codon Usage in Trichoderma
Codon usage and GC content of T. virens and T. atroviride were taken from the Codon Usage Database3 and provided by GenScript® (Piscataway, NJ, United States) to codon-optimized the following the fluorescent markers eGFP, mCherry, and 3X eGFP, and epitope tags 3X c-Myc, 3X FLAG and 3X HA by using the OptimumGeneTM algorithm. These sequences were cloned into in the pUC57 cloning vector via EcoRV. Any restriction sites present in the multiple cloning site (MCS) from the pBluescript II KS (±) in the optimized sequences were removed (Supplementary Material).
Creation of Entry Vectors
To create the TrichoGate entry vectors, synthetic genes produced by GenScript (eGFP, mCherry, 3X-eGFP, 3X-HA, 3X-FLAG, and 3X-c-Myc), promoters [translation elongation factor EF-1 α (tef1α) promoter and glycerol-3-phosphate dehydrogenase (gpdh) promoter], terminators [nopaline synthase terminator (T-nos) and tryptophan synthase transcription terminator (TtrpC)] and resistant cassettes were amplified by PCR using specific primers (Supplementary Table 1). These specific primers contained BsaI recognition sequences and specific four bp overhang flanking sequences (5′-GGTCTC-n-NNNN-3′) which were introduced in front of the specific forward primer sequences (Supplementary Table 1). The sequence, GGTCTC is the BsaI recognition site, while NNNN are the specific overhangs (indicated in color in the Supplementary Tables). The nucleotide (n) represents the extra-base between BsaI and the specific overhang. For the reverse primers, the same sequence (5′-GGTCTC-n-NNNN-3′) was added in front of the reverse complement of the sequence of interest (Supplementary Tables 1, 2).
PCR products were amplified using high-fidelity PrimeSTAR HS DNA Polymerase (Takara, Japan) with the following cycling conditions: 98°C for 1 min; followed by 35 cycles of 98°C for 10 s; 60°C for 15 s; 72°C for 2 min, and a final extension at 72°C for 10 min. PCR products were purified using Wizard SV Gel and PCR Clean-Up System (Promega, United States). The purified molecules were quantified using a NanoDrop (NanoDrop Technologies Inc., United States), and cloned into the pICH41021 vector, a derivative of pUC19 with a mutated BsaI recognition sequence in the ampicillin resistance cassette (kindly provided by Sylvestre Marillonnet), or pCR-Blunt vector (Thermo Fisher Scientific, United States). The ligation reaction was carried out in a total volume of 20 μL containing: 1 μL (5U) of T4 DNA ligase (Thermo Fisher Scientific, United States), 2 μL of 10X T4 DNA ligase buffer, 100 ng of linear vector pICH41021 (previously linearized with SmaI) or pCR-Blunt vector as manufacturer’s instructions, and 200 ng of the purified molecule (1:2 ratio of vector:insert). The recombinant vectors were transformed into E. coli TOPO F’ using chemically competent cells. Selection of positive colonies was performed on LB plates supplemented with 100 μg/mL ampicillin, IPTG (Zymo Research, United States) and X-Gal (PanReac AppliChem, Germany). The vectors were isolated, quantified and digested with BsaI to verify the insertion and vector fragment sizes before sequencing.
Creation of Destination pTrichoGate-1 Vector
pTrichoGate-1 was created by the substitution of the hygromycin resistance cassette from pYT6, a derivative plasmid from pCAMBIA1380 (Tanaka et al., 2007), with the lacZ ß-galactosidase gene from pAGM1311 (Addgene, United States). First, we amplified two PCR fragments using pYT6 as a template and Phusion Taq polymerase (NEB, United Kingdom). The PCR products were flanked by Acc651 and BsaI restriction sites (with different four overhangs and compatible with the BsaI sites flanking lacZ in pAGM1311). Primers oAM-LU-700 and oAM-LU-701 bind near to the BsaI site in pYT6 and contained two point mutations that eliminate the BsaI recognition site (GGTCTC) and created a new Acc651 recognition site (GGTACC) (Supplementary Table 2). Primers oAM-LU-696 or oAM-LU-695 were, however, designed to include the BsaI restriction site with compatible 4 nucleotide overhangs to directionally subclone the lacZ-BsaI fragment obtained from pAGM1311 (Supplementary Table 2). Thus the primer combination oAM-LU-700 and oAM-LU-696 produced a 3,578 bp PCR product while the combination oAM-LU-701/oAM-LU-695 generated a 2,852 bp PCR product. The PCR were obtained by using the following cycling conditions: 98°C for 1 min; followed by 35 cycles of 98°C for 30 s; 60°C for 15 s; 72°C for 2 min, and a final extension at 72°C for 10 min. These PCR products were cut with BsaI and Acc65I, then purified with the Wizard SV Gel and PCR Clean-Up System (Promega, United States) and ligated together with the 596 bp lacZ-BsaI fragment obtained from pAGM1311 by T4 DNA ligase (Thermo Fisher Scientific, United States). The resulting 6,990 bp ligation vector (called pTrichoGate-1 vector) was transformed into E. coli and verified with restriction enzymes and sequencing.
Creation of Final Vectors
Destination vectors pAGM1311 (Addgene, United States) or pTrichoGate-1 were used to insert the desired construct to create the final vectors. Both vectors contain the lacZ ß-galactosidase gene bordered by BsaI restriction sites and the kanamycin resistance gene for selection in E. coli (Supplementary Figure 1). One-tube restriction-ligation reactions were set up independently for each construct as follows: in a total volume of 10 μL containing 10 U of BsaI-HF (NEB, United Kingdom), 10 U T4 DNA ligase (Thermo Fisher Scientific, United States), 1 μL of 10X T4 DNA ligase buffer, (∼80 ng) PCR products [in the case of Sm1, TvTF1 (ORFs) and terpene synthases flanks] and entry vectors (∼75 ng) containing DNA fragments and destination vector (∼50 ng) were cut and ligated in the same reaction. The reaction was incubated for 2 min at 37°C and 5 min at 16°C; both steps were repeated 40 times, followed by final digestion for 5 min at 50°C and then heat inactivation for 5 min at 80°C. The recombinant vectors were transformed into E. coli chemically competent cells using 10 μL of restriction-ligation product. Selection of positive colonies was performed on LBA plates supplemented with 50 μg/mL kanamycin (Sigma Aldrich, United States), IPTG and X-gal. The vectors from positive E. coli colonies were isolated and verified by digestion with EcoRV or BsaI enzymes. DNA fragments were separated by 0.8% agarose gel electrophoresis. The vectors were sequenced before being used for Trichoderma protoplast transformation (i.e., using linearized vectors).
For pTrichoGate-1 derivative vectors (e.g., knockout construct vectors), electrocompetent cells from A. tumefaciens EHA105 were used. Electroporation was carried out, using the “Agr” setting (cuvette 0.1 mm, 2.2 kV and single pulse) as per the manufacturer’s instructions on a MicroPulser Electroporator (Bio-Rad, United States). The resulting transformants were selected on LB medium containing 25 μg/mL kanamycin and 25 μg/mL rifampicin. Before the fungal transformation, the pTrichoGate-1 derivative plasmids cloned in Agrobacterium were re-isolated and re-confirmed as previously described.
Knockout Vectors
For the gene knockout strategy, putative terpene synthase (TS) encoding genes (called here TS1 and TS2) from T. virens were chosen as an example. PCR primers were designed to amplify 1–1.3-kb of the flanking regions of the gene of interest (Supplementary Table 1). The PCR reaction was carried out using Phusion Taq Polymerase (Thermo Fisher Scientific, United States) with the following cycling conditions: 98°C for 1 min; followed by 35 cycles of 98°C for 5 s, 62°C for 15 s and 72°C for 1 min; and a final extension at 72°C for 10 min. Each primer combination (Supplementary Table 3) was designed to produce PCR products having flanking BsaI restriction sites to generate appropriate four base overhangs for the correct orientation for the ligation of the hygromycin B phosphotransferase gene (hph) from pTrichoGate-21 and the destination vector pTrichoGate-1 (Table 1 and Supplementary Figure 2). The PCR products from both borders corresponding to the desired gene to be knocked out were purified, quantified and then combined with pTrichoGate-1 and pTrichoGate-21; the one-tube restriction-ligation reaction performed as described above (Supplementary Figure 2).
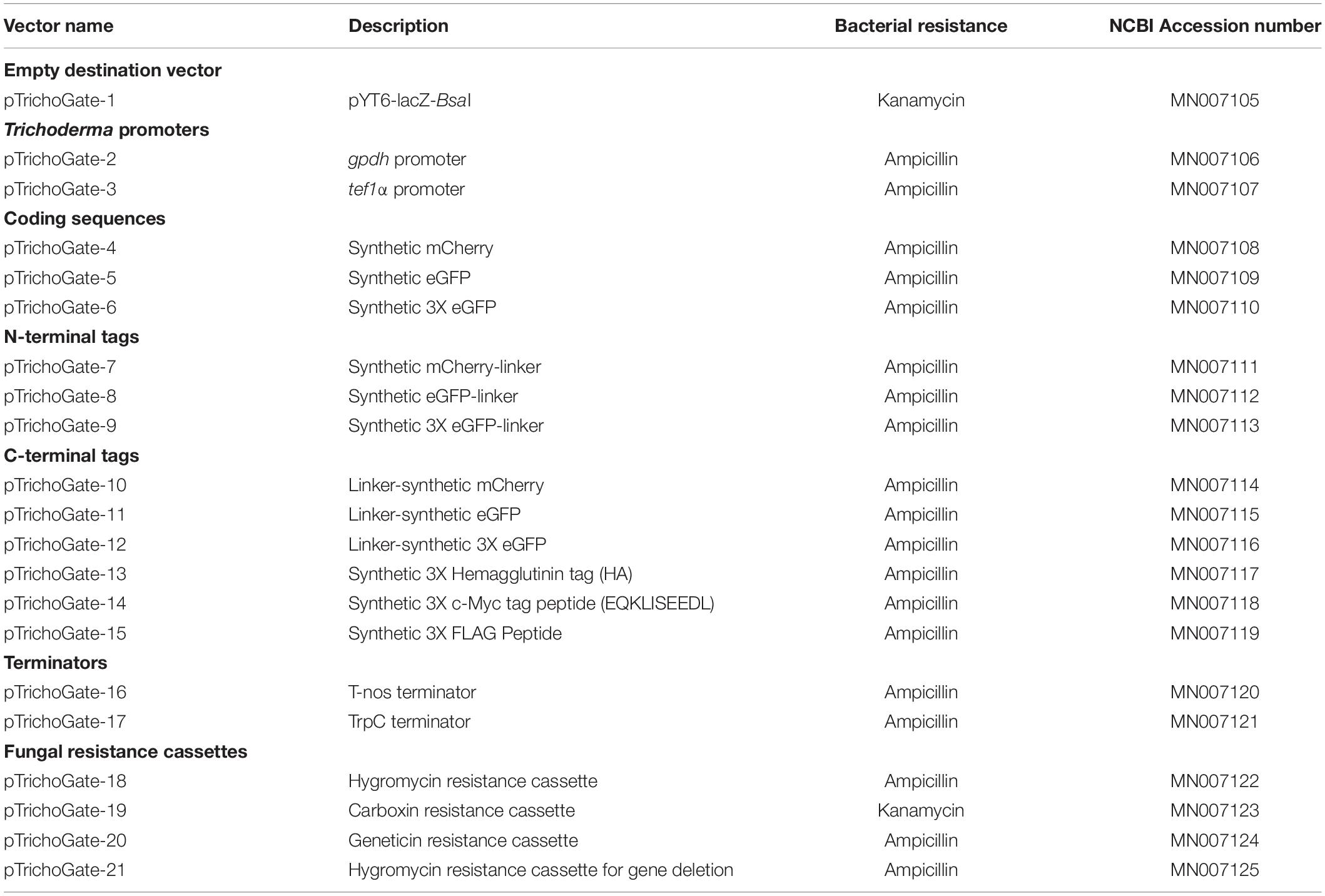
Table 1. Entry and destination vectors optimized for functional genetics in Trichoderma.
Fluorescent Marker Vectors
For the creation of the fluorescent marker vector, the vectors pTrichoGate-2 (gpdh promoter), pTrichoGate-4 (synthetic mCherry) and pTrichoGate-16 (nopaline synthase terminator, T-nos) and pTrichoGate-18 (hygromycin resistance cassette) were combined in a single tube with pAGM1311 and one-tube restriction-ligation reaction performed as described above, using 50 ng for each vector. The reaction was transformed into E. coli using chemically competent cells and the resulting plasmids isolated and analyzed by restriction enzymes before being used for T. virens protoplast transformation using the method described below.
Localization Vector
The open reading frame from the proteinaceous elicitor secreted protein Sm1 (ID 110852) that encodes a polypeptide of 138 amino acids with predicted mass of 14.4 kDa, an isoelectric point of 5.76 and contains a signal peptide of 18 amino acids (Djonovic et al., 2006); and the fungal transcription factor 1 (TvTF1) (ID 158742), which contains the classical nuclear localization signal, from T. virens were amplified by PCR using primer combination (Sm1 Loc_F and Sm1 Loc_R) and (TF Loc_F and TF Loc_R), respectively (Supplementary Table 1). The PCR reaction was carried out with Phusion Taq Polymerase (Thermo Fisher Scientific, United States) using T. virens DNA as template with the following cycling conditions: 98°C for 1 min; followed by 35 cycles of 98°C for 5 s, 62°C for 15 s and 72°C for 1 min; and a final extension at 72°C for 10 min. Reverse primers Sm1 Loc_R and TF Loc_R were designed to hybridize with the last 21 nucleotides of the Sm1 or TvTF1 gene without the last three nucleotides which form the stop codon. For the Sm1 localization, the following genetic material was combined in a single tube and the one-tube restriction-ligation reaction performed: the PCR product from SM1, the vectors pTrichoGate-2 (gpdh promoter), pTrichoGate-10 (Linker-synthetic mCherry), pTrichoGate-16 (T-nos terminator), pTrichoGate-18 (hygromycin resistance cassette) and pAGM1311. For the TvTF1 localization, the following molecules were used for the one-tube restriction-ligation reaction performed: the tvtf1 PCR product, pTrichoGate-3 (tef1α promoter), pTrichoGate-5 (Linker-synthetic eGFP), pTrichoGate-16 (T-nos terminator), pTrichoGate-18 (hygromycin resistance cassette) and pAGM1311 (Supplementary Figure 2). The fluorescent markers (mCherry and eGFP) from the vectors pTrichoGate-10 and pTrichoGate-11 were designed to contain a glycine and serine-rich flexible linker (GSAGSAAGSGEF) located at the N-terminus of the synthetic mCherry or synthetic eGFP (Waldo et al., 1999; Chen et al., 2013). The one-tube restriction-ligation reactions were transformed into E. coli using chemically competent cells and the plasmid isolated and analyzed by restriction enzymes before T. virens protoplast transformation.
Complementation Vector
To complement the 2-oxoglutarate (2OG)-Fe dioxygenase 1 (Tv2OG1) in the T. virens knockout mutant genome (Nogueira-Lopez, et al., in preparation), primers (Comp2OG_F and Comp2OG_R) were designed using the published T. virens genome database to amplify approximately 1.5 kb native promoter and 0.5 kb terminator sequences from the target gene region with the addition of a BsaI restriction site located on the 5′ and 3′ end flanks (Supplementary Table 1). Genomic DNA from T. virens was used as template. The complementing region was ligated to pAGM1311 and pTrichoGate-20, which contains the neomycin phosphotransferase encoding gene (nptII) (conferring geneticin G-418 resistance and termed GtR) under the control of the tryptophan gene promoter (PtrpC) and terminator (TtrpC) (Supplementary Figure 2). The reaction was transformed into E. coli using chemically competent cells and the plasmid isolated and analyzed by restriction enzymes before being used for T. virens protoplast transformation.
Carboxin Resistance Cassette
Direct mutagenesis of T. virens succinate dehydrogenase-encoding sdhTv gene (Protein ID88418, JGI T. virens) to confer resistance to carboxin was performed as described by Broomfield and Hargreaves (1992) in U. maydis. For the creation and implementation of the carboxin resistance cassette with Golden Gate methodology, five-point mutations were generated to eliminate four internal BsaI restriction sites in the carboxin resistance cassette sequence and to create the point mutation in the sdhTv iron-sulfur subunit protein (Ip) subunit (SdhB) that substitutes the His (CAC) codon for Leu (CTC) in codon 246, conferring resistance to carboxin (Broomfield and Hargreaves, 1992) (Supplementary Figure 3). Double-joint PCR methodology described by Yu et al. (2004) with modifications was used to eliminate BsaI sites and generate codon substitution by adding nucleotide changes in the primers. First-round PCR: amplification of 5′- and 3′ regions; six independent PCR products with 23–31 bases of homologous sequence overlapping with the ends of the next PCR product (for the primer combination, see Supplementary Figure 3A). Second-round PCR: fusion of six fragments into three using flanking primer combinations (CbxR1-CbxR4, CbxR5-CbxR8) for each independent PCR product. Third round PCR: amplification of the fused fragments into one by using LF and RF flank primers (CbxR1-CbxR12) (Supplementary Figure 3). PCR products were amplified using Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific, United States) with the following cycling conditions: 98°C for 1 min: followed by 35 cycles of 98°C for 10 s; 60°C for 30 s; 72°C for 30 s, and a final extension at 72°C for 10 min. The carboxin resistance cassette was subcloned into pCR-Blunt vector, linearized with blunt ends (Thermo Fisher Scientific, United States) according to the manufacturer’s instructions.
Nucleotide Sequence Accession Numbers
The DNA sequences of pTrichoGate-1 to pTrichogate-21 have been submitted to the GenBank database under accession numbers MN007105–MN007125 and are displayed in Table 1. These plasmids are available in Addgene4.
Sensitivity of T. virens Conidia to Carboxin
The antibiotic sensitivity of T. virens wild-type (WT) to carboxin (Sigma Aldrich, United States) was evaluated using four different media: PDA medium, AY medium (144 mM acetate–acetic acid buffer at pH 6.5, 5 g l–1 yeast extract and 15 g l–1 bacteriological agar), EY medium (10 ml l–1 ethanol, 5 g l–1 yeast extract and 15 g l–1 bacteriological agar) and GY medium (10 g l–1 glucose, 5 g l–1 yeast extract and 15 g l–1 bacteriological agar) (Shima et al., 2009). Carboxin resistance was tested on the different media mentioned above supplemented with different concentrations of carboxin (0, 50, 150, and 250 μg carboxin, diluted in DMSO per mL of medium). T. virens WT conidia (1 × 106) were homogeneously spread onto plates contacting the media mentioned above and incubated at 25°C for 7 days under a light/dark cycle. The samples were observed using a stereomicroscope (SZX12 Olympus, Japan) to confirm growth and germination inhibition. For each analysis, three replicates were used with different antibiotic concentrations.
Protoplast Transformation
The strategy used for Trichoderma protoplast transformation is described in the Supplementary Material. For the transformation of Trichoderma protoplasts, 10 μg of DNA containing the desired linearized vector was used. The digested vectors were visualized to confirm single digestion and then purified using Wizard SV Gel and PCR Clean-Up System (Promega, United States). For T. virens, positive transformants were recovered on recovering medium PDA (PDB supplemented with 0.7% agar and 0.7M Sucrose) amended with 100 μg/mL hygromycin, Vogel’s medium (supplemented with 0.7% agar) with 750 μg/mL geneticin G418 or solid AY-medium (supplemented with 0.7% agar and 0.7 M Sorbitol) with 350 μg/mL carboxin. For T. atroviride, the transformants were recovered on PDA amended with 100 μg/mL hygromycin. Trichoderma mutants were single spore purified as follows: conidia from Trichoderma positive mutants were grown on PDA supplemented with antibiotics for 5 days at 25°C under a 12 h day/night cycle to induce conidiation. To obtain mononucleate mutants, aliquots of conidial suspensions (1 × 103 cells, 100 μL) were plated on PDA amended with antibiotics and plates were incubated in darkness. After 18 h, colonies were observed using a stereomicroscope. A single colony was selected and inoculated to PDA with antibiotics. Single spore purification was repeated ten times to ensure the isolation of mononucleate strains. To confirm the stability of the mutation in Trichoderma mutants, all confirmed mutants were sub-cultured (10 times) on PDA without antibiotics and then transferred to PDA with antibiotics. The DNA of stable mutants was isolated using standard methodologies, and then confirmed by PCR and/or Southern blot. This methodology was used for the genetic transformation of T. virens with a fluorescent marker, localization, complementation and novel resistance marker vectors, but also for T. atroviride LU132 with mCherry.
Agrobacterium-Mediated Transformation
Agrobacterium-mediated transformation with modifications was performed for the generation of the T. virens knockout mutants and according to the method reported by de Groot et al. (1998), with improvements as described in Zeilinger (2004). After vector verification, 0.1 mL of pre-cultured A. tumefaciens cells containing desired vectors were inoculated into the minimal medium (MM) [10 mM K2HPO4, 10 mM KH2PO4, 2 mM MgSO4, 2.5 mM NaCl, 0.7 mM CaCl2, 10 mM D-glucose, 9 μM FeSO4, 4 mM(NH4)2SO4] supplemented with 25 μg/mL kanamycin and 25 μg/mL rifampicin and incubated overnight at 28°C with shaking at 250 rpm. The culture was centrifuged at 3,000 × g for 10 min at room temperature. A. tumefaciens cells were re-suspended in 50 mL of induction media with acetosyringone (IMAS broth) [MM, MES buffer (40 mM), 50% glycerol, acetosyringone] to an optical density of OD660 = 0.15, and the culture was incubated for up to 4 h at 28°C with shaking at 200 rpm until OD660 of 0.3 was reached. Trichoderma protoplasts were created according to the method described in the Supplementary Material. Equal volumes containing 100 μL of each A. tumefaciens culture and T. virens protoplast suspension (1 × 106 protoplasts) were plated onto sterile cellophane placed on IMAS agar and incubated at 23°C for 48 h in complete darkness. The circular cellophane was cut into eight triangular size pieces and transferred to PDA plates supplemented with 100 μg/mL hygromycin B and 300 μg/mL Timentin (GlaxoSmithKline, United Kingdom), and then incubated at 25°C for 2 days in complete darkness. T. virens colonies grown after incubation were transferred to PDA amended with antibiotics and subsequently purified to mitotic stability by ten rounds of single-spore isolation.
Culture Conditions for Protein Extraction
T. virens (WT and Sm1-Linker-mCherry) conidia, were inoculated onto PDA plates and incubated at 25°C with a 12 h day/night cycles for 7 days. Conidia were harvested as described above. The conidial suspension (1 × 106 conidia) was inoculated into 100 mL flasks containing 50 mL of PDB and incubated for 72 h at 25°C with shaking (160 rpm). The mycelia from the culture media were harvested by filtration and washed three times using sterile nanopure water, and then frozen in liquid nitrogen and stored at −80°C until protein extraction. The supernatant (50 mL) from the culture media was filtered through a double layer of sterile Miracloth and then filtered again through a 0.45 μm nylon filter (ReliaPrep, Ahlstrom-Mujnksjö, Finland) to remove any mycelia. The collected supernatant was stored at −80°C until protein extraction.
Protein Extraction
Mycelia Protein Extraction
For total protein extraction from T. virens mycelia, the methodology described by Bhadauria and Peng (2010) was implemented with modifications. Frozen mycelia (0.5 g) were ground into a fine powder using liquid nitrogen in precooled conditions. The powder was homogenized in 5 mL of ice-cold Tris/EDTA extraction buffer, containing 10 mM Tris-HCL pH 8, 1 mM EDTA, 2% w/v polyvinylpolypyrrolidone (PVPP) and with 0.3% (v/v) Pefabloc (Sigma Aldrich, United States), and then samples were centrifuged at 5,000 × g for 30 min at 4°C. The supernatant was collected, and proteins precipitated with 4 volumes of cold (−20°C) 10% w/v trichloroacetic acid (TCA)/acetone with 0.007% w/v DTT. Samples were centrifuged at 3,000 × g for 10 min; the pellet was washed three times with ice-cold acetone containing 0.007% w/v DTT. Protein pellets were dried to evaporate any remaining acetone and resuspended in rehydration buffer containing 7 M Urea, 2 M thiourea, 20 mM DTT and 2% w/v Triton X-100.
Supernatant Protein Extraction
For protein extraction of the T. virens secreted fraction, the methodology described by Medina and Francisco (2008) was implemented with modifications. For protein precipitation, 20 mL of broth supernatant was mixed with an equal volume of cold 20% (w/v) TCA solution. The mixture was incubated for 2 h at −20°C to allow protein precipitation and then centrifuged for 10 min at 14,000 × g. The supernatant was decanted, and the pellet was washed with 1 mL of cold 70% ethanol. The samples were vortexed and centrifuged for 3 min; this step was repeated three times. The final pellet was dried with the addition of 1 mL of acetone; the samples were vortexed and centrifuged for 1 min. The acetone was decanted, and the pellet air-dried for 30 min and then resuspended in the rehydration buffer as described above.
Western Blot
For Western blot analysis, 20 μg of protein was loaded into each well of a precast SDS-PAGE gel (Bio-Rad, United States), along with a molecular weight marker (PageRuler Plus Prestained Protein Ladder, Thermo Fisher Scientific, United States), and run at 100 V for 1 hr. Transfer of proteins to a PVDF blotting membrane (Amersham Hybond P; GE Healthcare Life Sciences, United Kingdom) was achieved as follows: (1) the gel was placed in 1 X transfer buffer pH 8.3 (25 mM Tris, 190 mM glycine and 20% methanol); (2) the transfer and the development was performed as described in a general protocol for western blotting (Bio-Rad, United States). For antibody incubation, the membrane was blocked in 5% skim milk in Tris-buffered saline Tween 20 buffer (TBST) (20 mM Tris pH 7.5, 150 mM NaCl and 0.1% Tween 20). The membrane was then incubated with the addition of 1:10,000 (v/v) anti-mCherry or anti-eGFP antibodies (BioVision, United States) dilution in TBST. The blot was rinsed in TBST and then incubated with 1:10,000 (v/v) HRP-linked secondary antibody (Cell Signaling Technology, United States) solution, using TBST for the dilution. A chemiluminescent substrate (500 μL luminol enhancer + 500 μL peroxide solution) was applied to the blot according to the manufacturer’s recommendations (Amersham ECL Prime Western Blotting Detection Reagent; GE Healthcare Life Sciences, United Kingdom).
Microscopic Visualization of Trichoderma virens Transformants
T. virens mutants (Sm1-Loc and TvTF1-Loc) were examined using fluorescent microscopy (Olympus BX51, Olympus, Japan). Colonies (3 days old) were incubated as described above. Fresh mycelia were analyzed under fluorescent microscopy to localize fluorescent protein mCherry and eGFP fusions within cells. Fluorescent proteins detection was observed by exciting at 488 and 561 nm. The detection wavelengths were in the range of 500–535 and 580–630 nm for eGFP and mCherry, respectively. Samples were analyzed using cellSens software (Olympus, Japan).
Results
TrichoGate Cloning Strategy
The TrichoGate system was developed to create a simple, efficient and reliable toolbox that enables assembly of synthetic DNA molecules into cloning vectors for the transformation of Trichoderma by using the Golden Gate cloning strategy. In this study, entry vectors (pICH41021 or pCR-Blunt) were used as recipients of synthetic DNA molecules (promoters, markers, tags, terminators and resistance cassettes) that were designed to be flanked by type II recognition sites by PCR. Each synthetic DNA molecule was flanked by a specific four nucleotide overhang region at the 5′- and 3′-end that was complementary to the next overhang in the next molecule to define the orientation and position in the final construct (Figure 1). To ensure a sufficient expression level of markers (mCherry, eGFP, 3X eGFP) and tags (3X HA, 3X FLAG and 3X c-Myc), we codon-optimized using the Codon Usage Database5 (see Tables in Supplementary Material). Chemically competent E. coli cells were transformed with the entry vectors containing target DNA molecules. All molecules used in this study, including promoters (tef1α and gpdh), markers (mCherry, eGFP, 3X eGFP, N-linker-mCherry, mCherry-linker-C, N-linker-eGFP, eGFP-linker-C, N-linker-3X eGFP and 3X eGFP-linker-C), tags (3X HA, 3X FLAG and 3X c-Myc), terminators (T-nos and TtrpC) and resistance cassettes (HygR, GtR and CbxR) were successfully subcloned into entry vectors (pICH41021 or pCR-Blunt) and ≥80% of the clones analyzed showed the correct construct (data not shown). To corroborate the efficiency and viability of the TrichoGate system in Trichoderma, these modules were used for the generation of different cloning approaches to study functionality of the selected proteins in Trichoderma: (i) gene deletion, (ii) gene overexpression, (iii) gene complementation, (iv) protein localization using fluorescent markers and (v) protein tagging with non-fluorescent markers. Each restriction-ligation was performed with their independent modules previously described above. Transformation of E. coli cells with the restriction-ligation products of the six independent constructs showed that (≥60%) of positive E. coli colonies contained the desired construct by using either pAGM1311 or pTrichoGate-1 as a receptor vector (Figure 2 and Supplementary Figure 2).
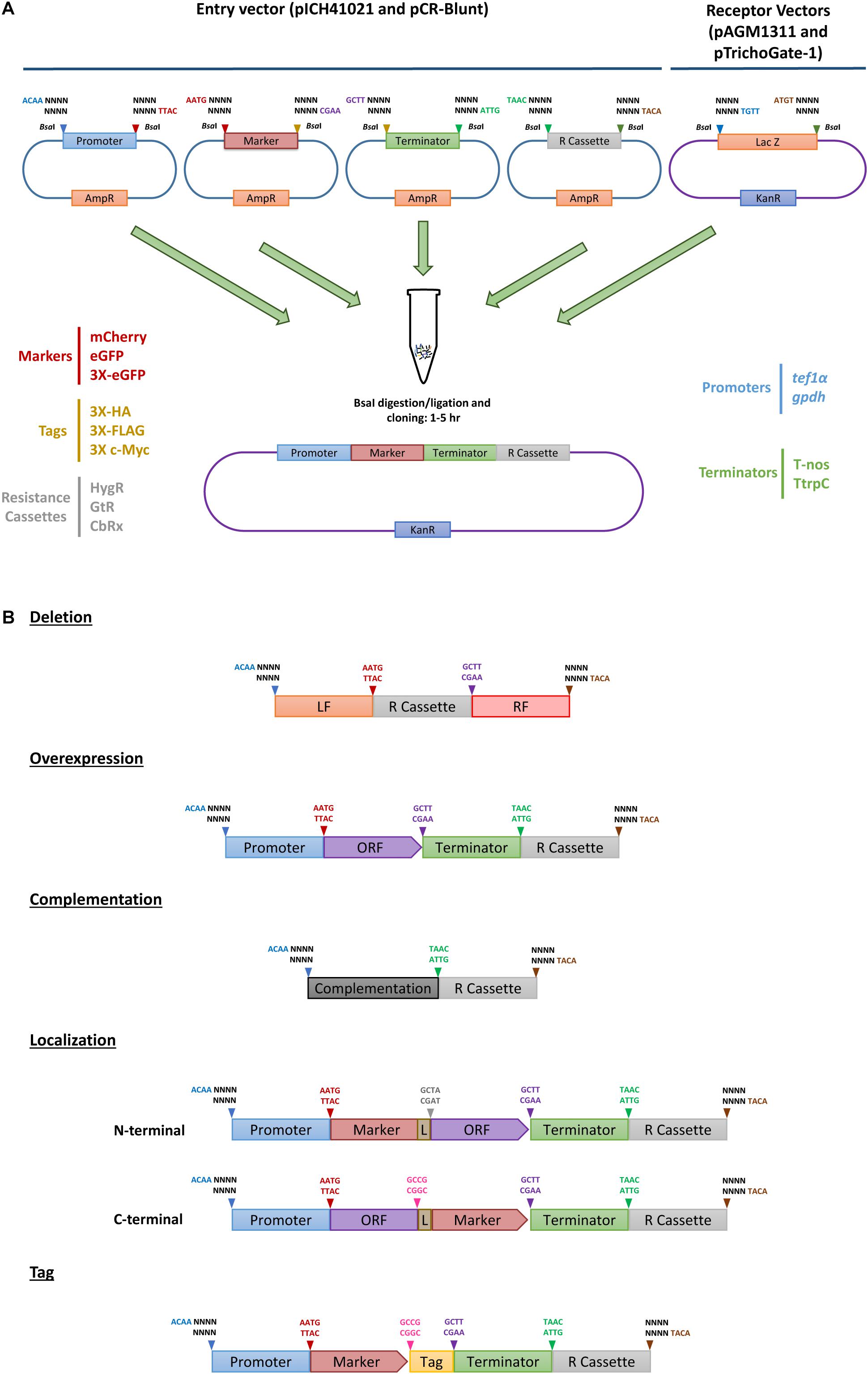
Figure 1. TrichoGate cloning system. Schematic representation of the TrichoGate cloning strategy to accomplish genomic modification in T. virens. (A) Implementation of TrichoGate cloning system using type II restriction enzyme (BsaI), target DNA sequences containing BsaI sites in their flanking regions with unique four bases overhangs must be subcloned in linearized entry vectors (pICH41021 or pCR-Blunt) carrying the ampicillin (AmpR) or kanamycin resistance (KanR) gene for selection in E. coli, respectively. TrichoGate vectors were constructed using up to five entry vectors (pICH41021 or pCR-Blunt) and/or PCR products containing synthetic DNA molecules of interest (e.g., promoters, markers, ORFs, terminators, tags and resistance cassettes) and one of the receptor vectors (pAGM1311 or pTrichoGate-1) that carry the KanR gene for selection in E. coli. BsaI restriction-ligation was carried out in one single microcentrifuge tube containing target DNA molecules for up to 5 h. (B) Examples of hypothetically generated vectors using BsaI restriction/ligation reaction for functional gene analysis in T. virens (e.g., gene deletion, gene complementation, overexpression, localization and immunoprecipitation).
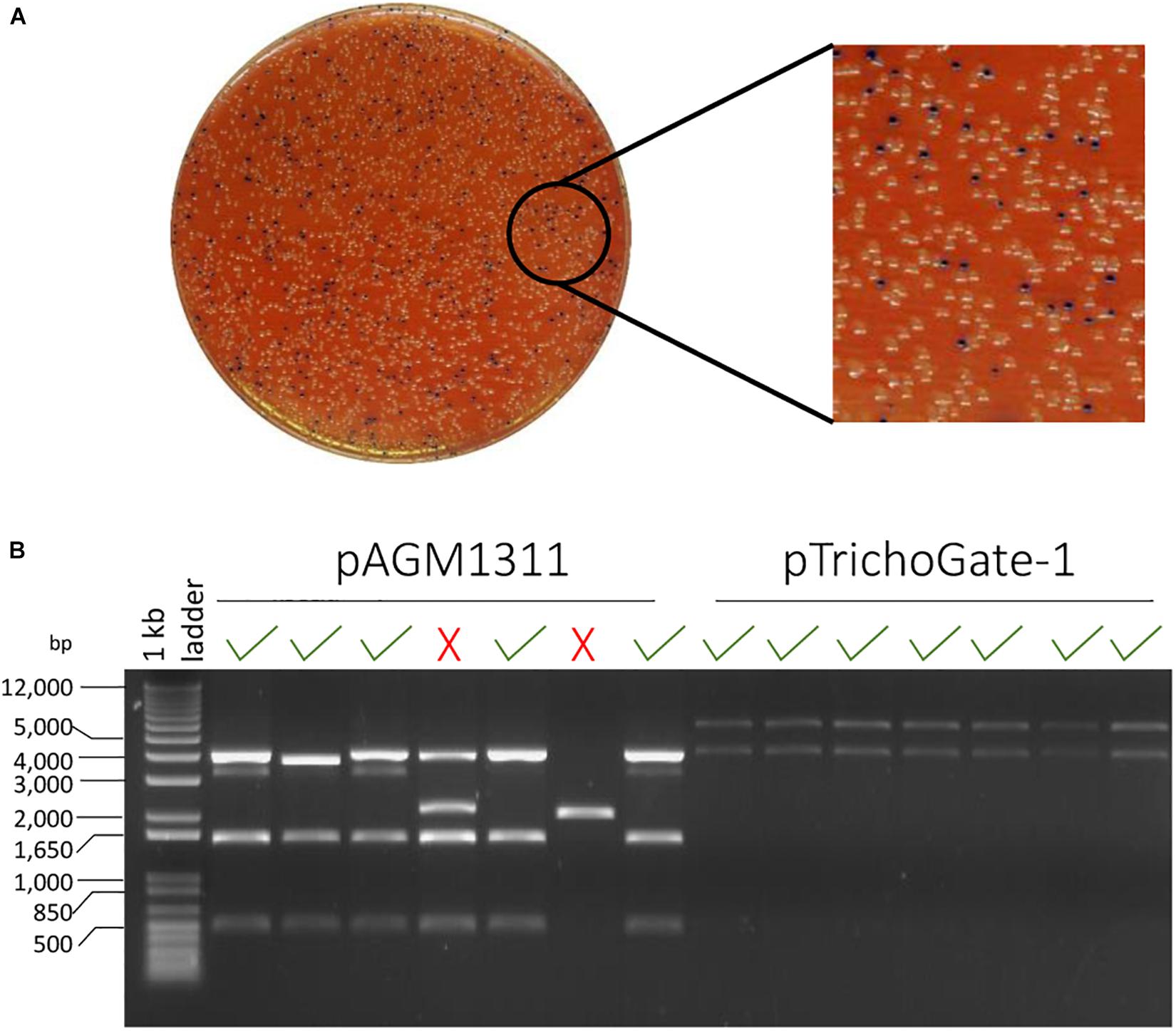
Figure 2. The efficiency of TrichoGate cloning system. (A) Luria-Bertani (LB) plate supplemented with 50 μg/mL kanamycin for selection of E. coli transformants containing constructs that were created using TrichoGate cloning system. Blue/white screening, E. coli cells transformed with vectors containing correct recombinant DNA should be white. (B) Restriction of plasmid DNA of minipreps from seven white colonies digested by EcoRV and run on a 1% agarose gel to verify insertion of the correct (√) or incorrect (X) construct by using two different receptor vectors: pAGM1311 and pTrichoGate-1. DNA from 70% (pAGM1311) and 100% (pTrichoGate-1) from white colonies analyzed have the desire restriction pattern.
Deletion and Complementation Studies in T. virens
Deletion of T. virens Terpene Synthases
The deletion of two terpene synthases (ts1 and ts2) encoding genes of T. virens was performed by homologous recombination replacing the open reading frame of each targeted gene with the PtrpC/hph fragment using Agrobacterium-mediated transformation. A total of 25 stable transformants of each deletion, were screened for their capability of continuously growing in PDA plates supplemented with 100 μg/mL hygromycin B and stable transformants from each deletion were screened for the insertion of the hph by PCR (Supplementary Figure 4 and data not shown). Nine and ten stable transformants of potential TS1 and TS2 deletion mutants, respectively, were randomly selected to confirm homologous recombination of the HygR cassette by Southern Blot. The hybridization results of the labeled hph probe showed a single band (7.3 kb) in five TS1 mutants when gDNA was digested with BamHI, and a single band (4.5 kb) was shown in seven TS2 mutants when gDNA was digested with BsaI and HindIII; thus, validating the replacement of the native genes with the HygR cassette. No fragments were shown for their parental strain (Figure 3). Overall, these results demonstrate that by using the Golden Gate cloning strategy coupled with Agrobacterium-mediate transformation, we were able to obtain a percentage of homologous recombination above 50%.
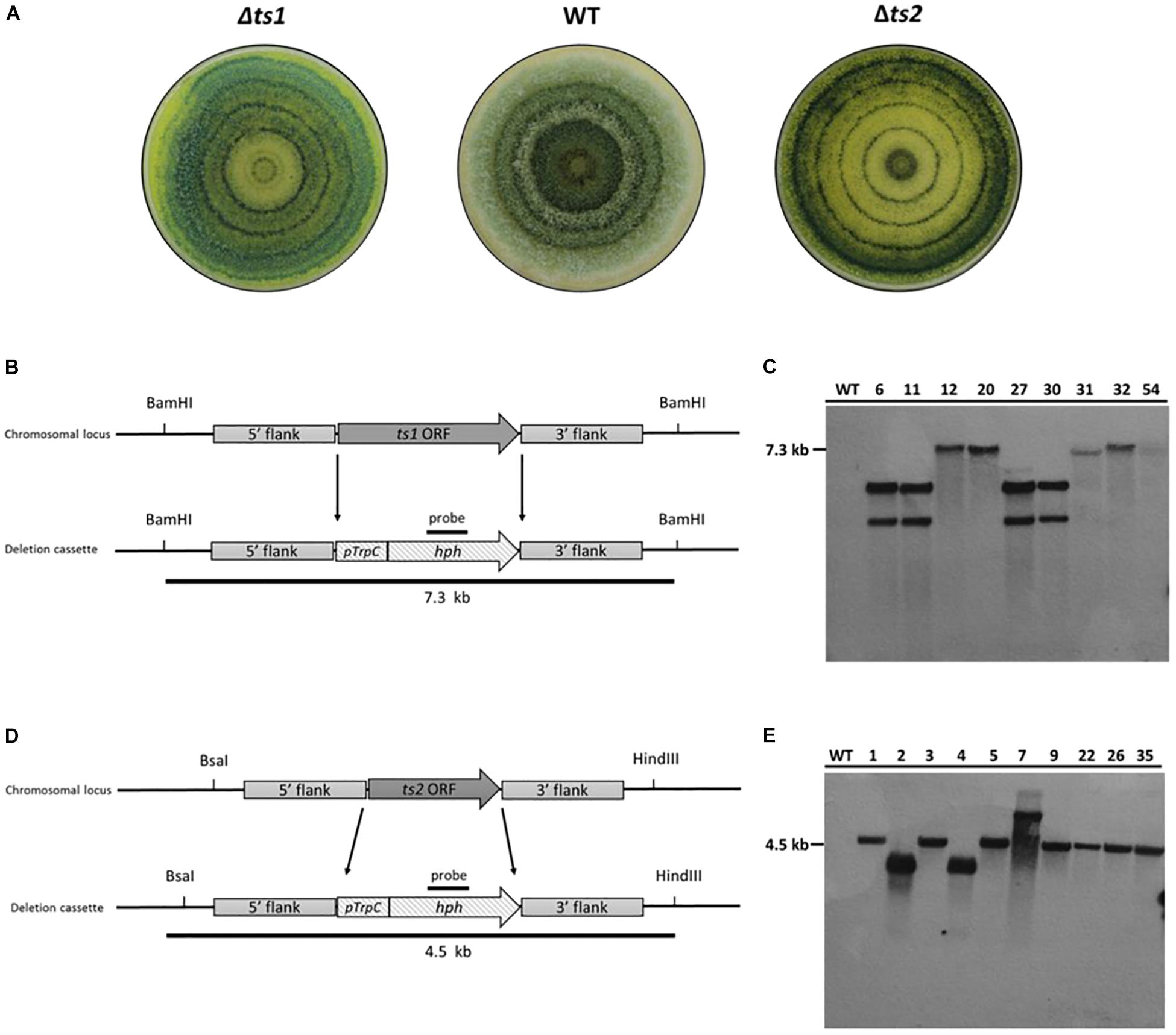
Figure 3. Confirmation of homologous recombination event of the HygR cassette in deletion mutants by Southern blot. (A) Colony appearance growing on axenic media (PDA) under light/dark cycle. (B–D) Schematic representation of gene deletion strategy. The ORF of each targeted gene is replaced by the homologous integration of the PtrpC/hph fragment (1.4 kb) that confers resistance to hygromycin B. (B) A 7.3 kb fragment would result for the ts1 deletion mutants when digested with BamHI. (C) Southern blot analysis of T. virens WT strain and ts1 deletion mutants (6, 11, 12, 20, 27, 30, 31, 32, and 54) (D) A 4.5 kb fragment would result for the ts2 deletion mutants when digested with BsaI and HindIII. (E) Southern blot of T. virens WT strain and ts2 deletion mutants (1, 2, 3, 4, 5, 7, 9, 22, 26, and 35). Expected fragments sizes for each deletion event along the left side of the films.
To prove homologous integration on both the 5′ and 3′ ends of the deletion constructs, a couple of additional PCR reactions was make. For the verification of the double recombination in the 5′ region, the primer combination TS1 Ext-LF/oAM-LU217 or TS2 Ext-LF/oAM-LU217 (Supplementary Table 1 and Supplementary Material) generated a PCR product in the positive recombinants. For the verification of the 3′ double recombination the primer combination oAM-LU218/TS1 Ext RF or combination oAM-LU218/TS2 Ext RF (Supplementary Table 1 and Supplementary Material) were used instead (Supplementary Figure 4).
Complementation of T. virens 2OG-Fe Dioxygenase
The complementation of the 2OG-Fe dioxygenase (tv2og1) encoding gene of T. virens was achieved by inserting the WT allele into the T. virens Δtv2og1 strain (Nogueira-Lopez et al., in preparation). The complementing construct contained the neomycin phosphotransferase encoding gene (nptII) that conferred resistance to geneticin G-418 and was used as a selectable marker. Twenty-five transformants were screened for the ability to grow continuously and sporulate in Vogel’s medium supplemented with 750 μg/mL geneticin G-418. Genetic analysis (Southern blot) of ten randomly selected transformants was performed to corroborate the insertion of the tv2og1 ORF and GtR cassette (Figure 4). DNA from the WT and Δtv2og1 strains was used as a positive and negative control, respectively. The hybridization results of DNA previously digested with PstI showed that all screened transformants were identified as positive recombinants, confirming the presence of the tv2og1 gene and GtR (2.4 kb band). However, all contained more than one ectopic integration of the tv2og1 complementing construct (Figure 4). The WT and Δtv2og1 showed the expected fragment sizes of 7.3 and 12 kb, respectively. These results confirmed the integration of the complementation construct into the T. virens genome by combining TrichoGate vectors and protoplast transformation.
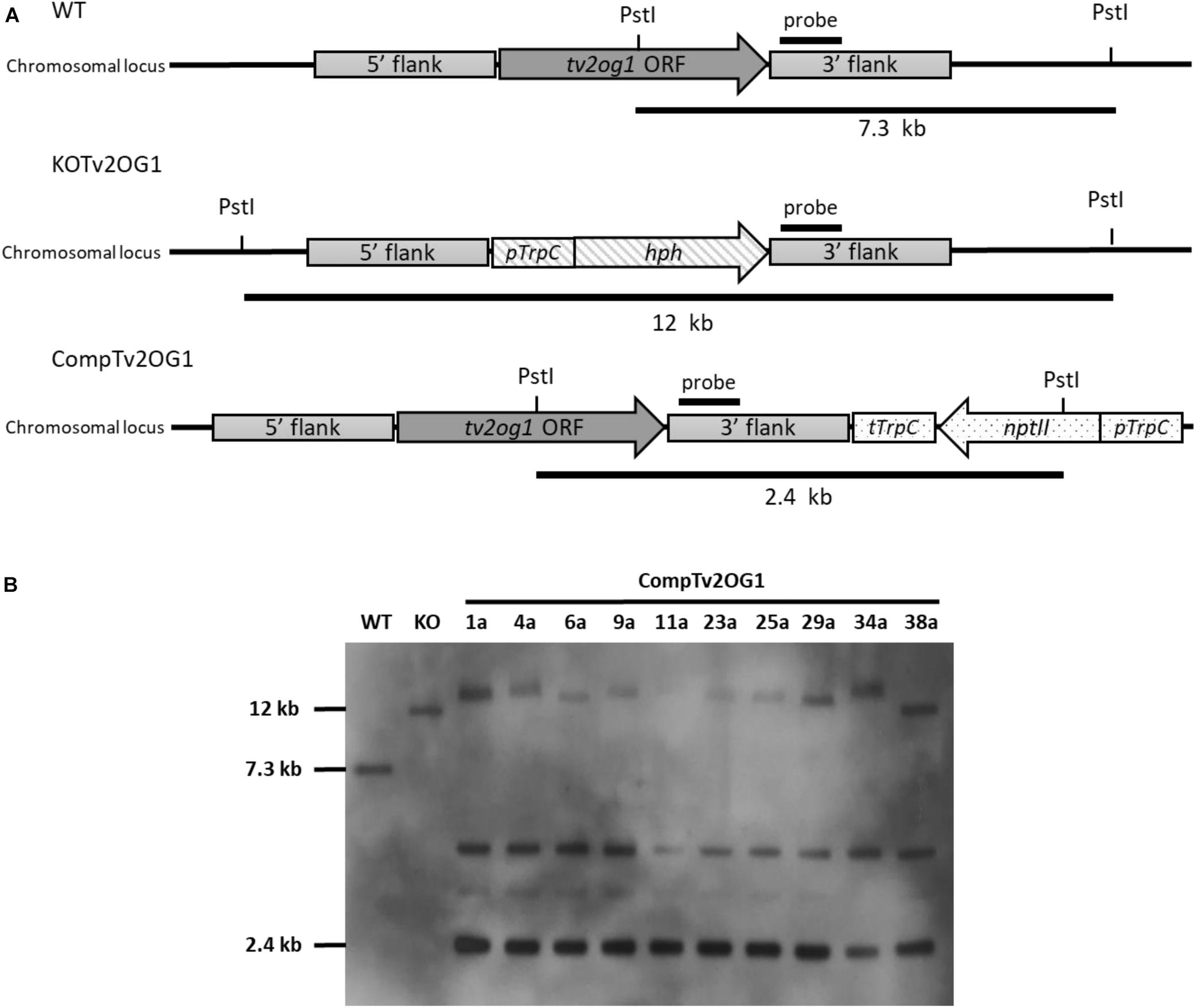
Figure 4. Southern blot confirmation of complementing mutants of the tv2og1 gene in T. virens. (A) Schematic representation of the gene deletion and complementation strategy. For gene deletion, tv2og1 was replaced by the homologous integration of the pTrpC/hph fragment (1.4 kb) that confers resistance to hygromycin B. For gene complementation, the Δtv2og1 null mutant was transformed with the complementing construct created using the TrichoGate system. The complementing region containing a 1.5 kb promoter and 0.5 kb terminator fragments, along with the tv2og1 ORF, was fused to the neomycin phosphotransferase encoding gene (nptII) (conferring resistance to geneticin G-418) under control of the tryptophan gene promoter (pTrpC) and tryptophan gene terminator (tTrpC). (B) Southern blot of the T. virens WT and the tv2og1 deletion and complementing mutants was carried out using a fragment of tv2og1 terminator as a probe. The blot shows the 2.4 kb fragment size expected in T. virens WT strain, the 12 kb fragment size expected for the T. virens tv2og1 null mutant strain and the 2.5 kb fragment expected in T. virens Ctv2og1 strains when gDNA was digested with PstI. An extra band was present in all ten complementing mutants suggesting ectopic integrations of the GtR cassette in the T. virens genome.
Expression of Codon-Optimized mCherry in Trichoderma
The validation of the TrichoGate technology was first analyzed by testing genetic transformation of T. virens protoplasts with a linearized TrichoGate vector containing the gpdh promoter, codon-optimized mCherry, T-nos terminator and HygR cassette. Stable T. virens hygromycin resistant colonies that were able to grow in the presence of 100 μg/mL hygromycin were screened, and the presence of the hph gene was verified by PCR (data not shown). Five-day old cultures of T. virens-mCherry strains were used to observe mCherry expression in both conidia and mycelia under a fluorescence microscope. The expression of mCherry as the reporter gene under the control of the constitutive gpdh promoter was visualized in T. virens (Figures 5A,B). Remarkably, the mCherry-tagged colonies displayed intense cytosolic fluorescence homogeneously distributed in both hypha and conidia cells (Figures 5A,B). To assess the functionality of the new vectors in other Trichoderma species, we designed a plasmid carrying the mCherry encoding gene (pTrichoGate-4) under the control of the gpdh1 (pTrichoGate-2) promoter and T-nos terminator (pTrichoGate-16). This vector was linearized and transformed into T. sp. “atroviride B” LU132 protoplasts. The expression of mCherry was analyzed by fluorescence microscopy (Supplementary Figure 5), indicating that these vectors are suitable for use in other Trichoderma species.
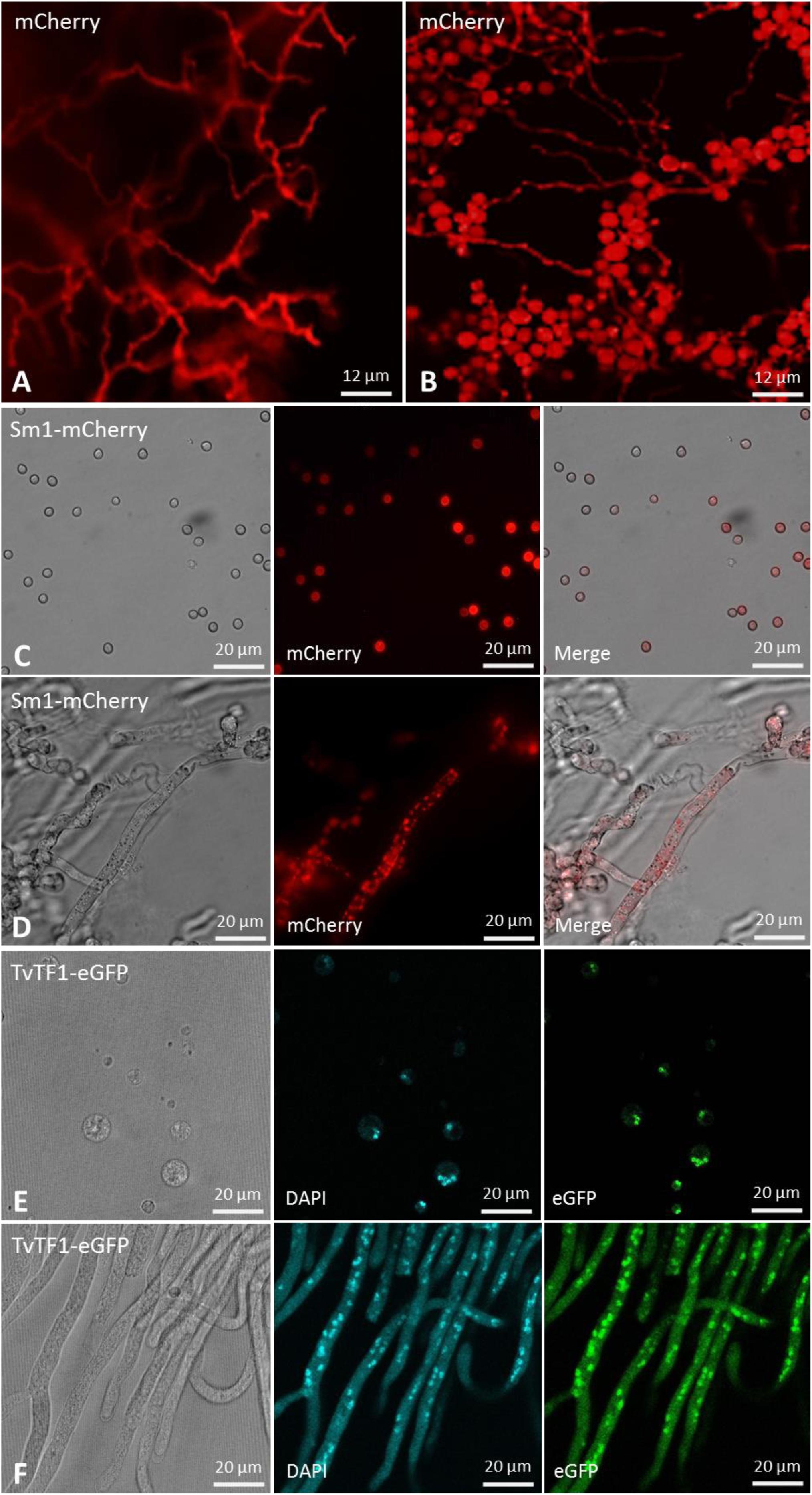
Figure 5. Microscopic observation of the T. virens mutants transformed with a fluorescent marker or protein localization vector generated through the TrichoGate system. Fluorescent microscopy of T. virens mycelia and conidia to verify the expression of the mCherry fluorescent marker under the control of the glyceraldehyde 3-phosphate dehydrogenase (gpdh) promoter from T. virens. Red fluorescence corresponding to the mCherry marker observed in T. virens (A) mycelia and (B) conidia. Microscopic observation of the Sm1-linker-mCherry expressed in T. virens (C) conidia and (D) mycelia; left bright-field, middle fluorescence and right merged images. Microscopic observation of the TvTF1-linker-eGFP expressed in T. virens (E) protoplasts and (F) mycelia; left bright-field and right fluorescence images. To visualize mCherry, 587 nm of excitation and 610 nm emission were used; while for the eGFP 484 nm excitation and 507 nm emission were used. No autofluorescence was observed in T. virens mycelia and conidia using either red or green channels. were visualized using an Olympus BX51 compound microscope. Images were captured using an Olympus DP70 digital camera system and processed with the CellSens software (Olympus).
Using Codon-Optimized Fluorescent Markers for Protein Localization Studies in T. virens
To test the effectiveness of the TrichoGate system to localize proteins in Trichoderma, we designed chimeric proteins between proteins from T. virens and fluorescent markers encoding in the TrichoGate plasmids. We choose Sm1, a well small-secreted protein that contains a signal peptide for secretion (Djonovic et al., 2006). The second protein chosen was the transcription factor TRIVIDRAFT_158742 (called here TvTF1), which contains a classical Nuclear Localization Signal in the N-terminal region (RTSKRRCIH)6. Localization vectors were created successfully using the TrichoGate strategy (Supplementary Figure 2). Sm1 was fuse to mCherry while TvTF1 to eGFP using a linker between the Trichoderma proteins and the fluorescent markers (Figure 5). Genetic transformation of T. virens protoplasts was performed using linearized localization vectors (Sm1 and TvTF1). Amplification of mCherry (730 bp) and eGFP (729 bp) ORF were used to confirm integration of the vector in the T. virens genome of the positive mutants (data not shown). Protein extractions of mycelia (Sm1-Loc and TvTF1-Loc mutants) and the supernatant fraction (Sm1-Loc mutants) were performed to verify protein localization and secretion. Western blot results showed that both fusions tagged proteins were present in the total protein extracts [Sm1-linker-mCherry fusion (∼41 kDa)] and [TvTF1-linker-eGFP (∼79 kDa)] from T. virens mycelia, and the chimeric protein Sm1-linker-mCherry was identified in the secreted fraction (Supplementary Figure 6). To visualize protein localization during fungal development, Sm1-linker-mCherry and TvTF1-linker-eGFP fusion proteins were expressed in T. virens independently, under the constitutive gpdh and tef1α promoter, respectively (Supplementary Figure 6). Fluorescence observations revealed that SM1 is expressed and localized in the cytosol or is attached to the fungal membrane (Figures 5C,D), and TvTF1 is localized in the nucleus (Figures 5E,F).
Generation of a Novel Carboxin Resistant Marker in T. virens
Carboxin Sensitivity in T. virens
The sensitivity of T. virens to carboxin was determined before the transformation. Four different media were evaluated to establish which medium was the most effective in synergy with carboxin to inhibit T. virens germination and growth. Carboxin sensitivity tested after 7 d.p.i on germination and growth of T. virens WT growing on PDA, EY and GY plates containing 0 to 250 μg/mL carboxin concentration, showed that T. virens germination and growth was not affected. In addition, the AY plates with the highest concentration of carboxin (250 μg/mL) resulted in T. virens germination inhibition (no geminated conidia or hyphae were detected) (Figure 6A). However, after 14 d.p.i small colonies were observed on AY medium at 250 μg/mL. Therefore, AY medium amended with 0–500 μg/mL carboxin was tested; no T. virens spore germination was observed from 350 to 500 μg/mL after 14 d.p.i (Figure 6B). Based on these results, AY medium amended with 350 μg/mL carboxin was used on for the isolation of carboxin-resistant mutants.
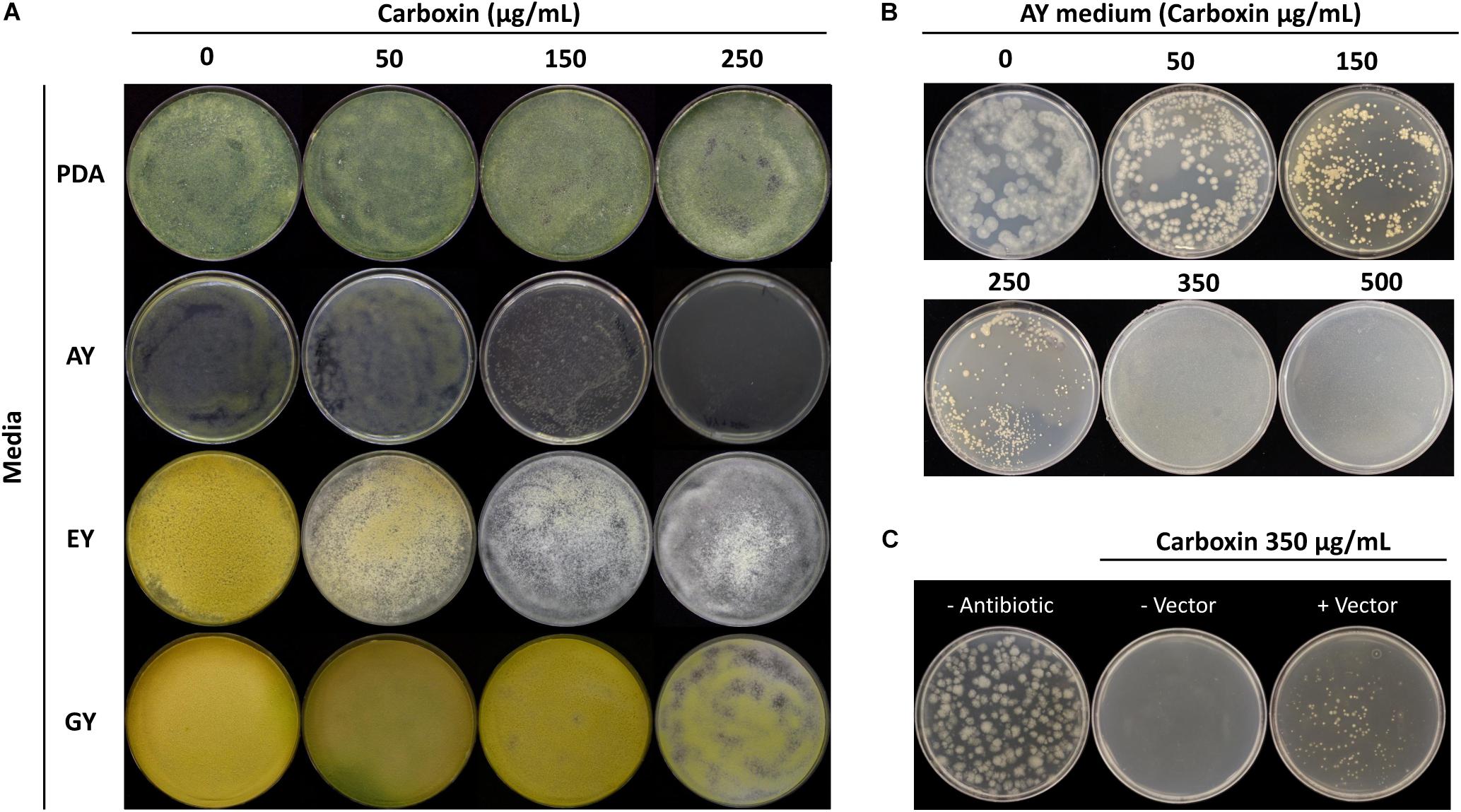
Figure 6. Trichoderma virens carboxin resistance. (A) Carboxin effect for mycelial growth of T. virens on different media [potato dextrose agar (PDA), acetate-acetic/yeast extract medium (AY), ethanol/yeast extract medium (EY) and glucose/yeast extract medium (GY)] containing various concentrations of carboxin. The wild-type (WT) strain was cultured for 7 days. (B) Carboxin sensitivity in T. virens WT on AY medium after 14 d.p.i. (C) Selection of carboxin resistance transformants of T. virens. Protoplasts were transformed with linearized pTrichoGate-19 vector, and then cultured on AY medium containing carboxin 350 μg/mL after 7 d.p.i.
Design of Carboxin Resistance Marker for T. virens
The generation of the plasmid containing the carboxin resistance marker for T. virens was achieved using double-joint PCR to create a point mutation in the SdhBTv subunit (substituting His for Leu in codon 246, Supplementary Figure 3); additional point mutations were necessary to remove the endogenous BsaI recognition sites without affecting the coding sequence. Three rounds of PCR were performed to generate the point mutations desired (Supplementary Figure 3A). By using standard molecular procedures, the entire CbxR cassette was amplified and subcloned into a pCR-Blunt backbone. The final product was sequenced and the point mutations verified (Supplementary Figure 3B) and called pTrichoGate-19. This construct was used for transform T. virens protoplasts as described above with 10 μg of the linearized pTrichoGate-19 vector (carboxin resistance cassette). Mutants emerged on selective medium 7 d.p.i and were transferred onto new AY selection plates amended with 350 μg carboxin/mL (Figure 6C). This demonstrated that pTrichoGate-19 was efficient in the generation of T. virens resistant mutants to carboxin.
Discussion
The aim of this study was the design of a Golden Gate strategy for the analysis of functional genes in T. virens. As a result, the TrichoGate cloning system has provided a reliable, fast and effective method for the construction of different vectors (e.g., deletion, complementation, overexpression and localization) to study the genetics and biology of Trichoderma.
Linear DNA construction has been a time-limiting step that has been overcome throughout the implementation of the Golden Gate cloning system. Overall, Golden Gate is a simple, highly efficient and low-cost cloning strategy that speeds up the genetic manipulation of plants, fungi and bacteria by using only one type IIs restriction endonuclease for the assembly of multi genetic constructs, thus reducing the experimental steps in cloning processes (Lampropoulos et al., 2013; Andreou and Nakayama, 2018; Hernanz-Koers et al., 2018). For the design of the TrichoGate cloning system in T. virens, the commercially available endonuclease BsaI (isoschizomer: Eco31I) was selected based on: (i) the generation of sticky ends after hydrolysis (with six bp separated by a single random nucleotide from the site of hydrolysis and generates a four nucleotide overhang) that permits efficient ligation reactions, and (ii) the restriction and the recognition sites which are not separated by more than ten bases, allowing its incorporation into primers of suitable length. Moreover, the design of specific inverse oriented BsaI sites (on primers) allows the directional fusion of the flanking regions to receptor vectors (Terfrüchte et al., 2014). However, the use of other type IIs restriction enzymes is also suitable for the implementation of Golden Gate methodology (e.g., BbsI or BsmBI) (Sarrion-Perdigones et al., 2011; Housden and Perrimon, 2016).
In order to implement an efficient cloning system, all synthetic DNA molecules including promoters, markers, tags, terminators and resistance cassettes used through this technique were subcloned into the entry vectors for their use in the construction of multipurpose vectors for the study of T. virens. The design of modules containing genetic elements of interest has been previously implemented by Weber et al. (2011), demonstrating the reliability of the system to assemble complex constructs in a single digestion-ligation reaction, thus reducing the time of the construct generation (Engler et al., 2008). Similarly, the development of the GreenGate system highlights the use of modules including promoters, N- and C-terminal tags, coding sequence, terminator and resistance cassettes for the generation of plant transformation vectors (Lampropoulos et al., 2013).
Generation of Time-Effective Knockout and Complementation Mutants in T. virens
The generation of deletion mutants in filamentous fungi is particularly challenging due to the low percentages of homologous recombination events. In Trichoderma spp. specifically, this percentage is approximately 2% (Mach and Zeilinger, 1998). However, in this work it was demonstrated that the generation of deletion constructs in T. virens using the TrichoGate cloning strategy coupled with Agrobacterium-mediated transformation that should be applicable for any Trichoderma species, resulted in an increase of homologous recombination events compared to conventional protoplast transformation methods, which generates a large number of transformants with ectopic integrations (Mach and Zeilinger, 1998). The results were consistent with previous reports in T. atroviride, where using Agrobacterium-mediated transformation increased the rate of homologous recombination (>60%) (Zeilinger, 2004).
The use of the Golden Gate cloning system to generate deletion constructs in fungi and bacteria as a strategy to overcome usual limitations of conventional methods has been implemented before in U. maydis, A. nidulans (Terfrüchte et al., 2014), Penicillium digitatum (Hernanz-Koers et al., 2018) and Bacillus anthracis (Wang et al., 2018). Moreover, the importance of flanking regions length when the Golden Gate cloning system is applied in filamentous fungi has been proved to be 1 kb for U. maydis, reaching 59% of homologous recombination events. In contrast, the use of shorter flanks led to increased rates of ectopic integrations (Terfrüchte et al., 2014). The results of the TrichoGate cloning system demonstrate that the homologous recombination event was higher than 50% when flanking regions from 1.0 to 1.3 kb were used for the generation of deletion mutants in T. virens. Furthermore, the construction of a complementation vector containing the geneticin resistance cassette was successfully implemented in T. virens, which should assist with functional analysis of targeted genes in Trichoderma that require efficient complementation vectors. The use of the geneticin resistance cassette as a selection marker for the genetic manipulation in Trichoderma spp. has been reported in the past for the complementation of a VELVET protein gene (vel1) (Mukherjee and Kenerley, 2010) and the generation of mutants of a constitutively activated version of the Gα subunit-encoding tga3 gene (Gruber et al., 2012).
The Golden Gate cloning system has used to develop a multiplex CRISPR vectors for various downstream applications. This method was called ASAP-cloning (Adaptable System for Assembly of multiplexed Plasmid) (Zuckermann et al., 2018). The TrichoGate system can be modified for CRISPR applications by the integration of a self-replication fragment in the recipient plasmid; additionally, this plasmid can contain the U6 promoter, the gRNA scaffold and the terminator, although these components can be part of different entry vectors. By combining the TrichoGate and the system of subcloning reported by Zuckermann et al. (2018), is possible to integrate one or multiple gRNA expression cassettes using a PCR-on-ligation reaction (Zuckermann et al., 2018).
Fluorescent Marker and Localization of Trichoderma Proteins
Trichoderma spp. closely interact with plants and other microorganisms (Harman et al., 2004). Microscopic observations of plant-fungi interactions have allowed the detection of specialized fungal structures that facilitate root colonization as well as fungal growth in both inter- and intracellular spaces (Nogueira-Lopez et al., 2018). In addition, mycoparasitic Trichoderma spp. utilize infective structures for the penetration of the cell wall of the host fungus (Guzman-Guzman et al., 2019). Therefore, the creation of fluorescent marker vectors (e.g., mCherry, eGFP and 3X eGFP) by using the TrichoGate cloning system may facilitate the monitoring of Trichoderma during the colonization of plant tissues and mycoparasitism. For example, the application of green fluorescent protein reporter systems during the study of three-way (Trichoderma-pathogen-plant) interactions (Lu et al., 2004) showed the effectiveness of the GFP marker for the in situ evaluation of T. atroviride-plant and/or T. atroviride-microbe interactions, including the results of codon optimization on mCherry fluorescent marker created in this study, showed an optimal visualization of T. virens and T. atroviride (mycelia and conidia).
Plant–microbe interactions are regulated by the secretion of several molecules that facilitate and maintain the interactions; among them, a variety of proteins that regulate the crosstalk between plants and microbes (Doehlemann and Hemetsberger, 2013). Therefore, the construction of protein-localization vectors in Trichoderma will assist in developing a deeper understanding of its interaction with plants. Localization vectors designed using the TrichoGate cloning system and the subsequent T. virens transformation enabled the localization of the Sm1-linker-mCherry and TvTF1-linker-eGFP recombinant proteins to corroborate the functionality of the fusion-protein linker. In addition, it was confirmed that SM1 was found in the secreted fraction under axenic conditions, which has been previously reported by Djonovic et al. (2006). Furthermore, Sm1 has been identified as part of the apoplastic secretome during the T. virens-maize interaction (Nogueira-Lopez et al., 2018). Moreover, TvTF1 [Zn2-Cys6 fungal transcription factor involved in secondary metabolism in T. virens (Nieto-Jacobo et al., in preparation)] was visualized in the nucleus (Figures 4E,F). The findings derived from this research show the potential of the use of the TrichoGate cloning system to successfully localize fungal proteins of interest to characterize Trichoderma-plant interactions.
Carboxin Resistance in T. virens
Hygromycin resistance is one of the most common and reliable antibiotic markers used in Trichoderma spp. for the generation of mutants. However, the development of novel resistance markers is indispensable for the genetic study of filamentous fungi in order to enable serial gene deletions and re-transformations. Therefore, a novel resistance-based vector such as carboxin could be a viable alternative for genetic transformation in Trichoderma. The carboxin resistance marker has been widely used in other fungal species such as U. maydis (Broomfield and Hargreaves, 1992), A. oryzae (Shima et al., 2009), Mycosphaerella graminicola (Skinner et al., 1998) and M. oryzae (Guo et al., 2016). Carboxin inhibits the respiratory chain by altering the enzyme activity of SDH. The results showed that T. virens sensitivity to carboxin increased on acetate-containing media in comparison with the other media tested; acetate is the carbon source used as direct substrate in Krebs cycle, but this pathway is blocked via inhibition of the SDH activity by carboxin that may have lethal effect on fungi as previously reported in A. oryzae (Shima et al., 2009). Therefore, for the development of the transformation system, the AY medium was ideal for recovering T. virens carboxin-resistant mutants. Furthermore, the results showed that the point mutation (His to Leu) of the sdhTv gene that encodes the SDH iron-sulfur subunit of T. virens mutants conferred resistance to carboxin, suggesting a similar role in fungicide resistance as studied in other fungal models (Broomfield and Hargreaves, 1992; Skinner et al., 1998; Shima et al., 2009; Guo et al., 2016). Demonstrating that the carboxin resistance cassette may be suitable as a selection marker for the generation of mutants in T. virens.
Conclusion
In conclusion, the present study demonstrates the functionality of a high-efficiency cloning system using the Golden Gate methodology applied to Trichoderma called TrichoGate. This system facilitates the generation of multipurpose vectors including localization of protein studies, tagging proteins for immune precipitation studies, gene deletion, overexpression of particular genes and gene complementations. Our system, together with gene editing using CRISPR/Cas9 technology in Trichoderma will be essential tools to conduct comprehensive studies of its biology.
Data Availability Statement
The datasets generated for this study can be found in the GenBank MN007105–MN007125.
Author Contributions
GN-L, AM-M, FP-A, SI, MN-J, and SL performed the experimental work. GN-L and AM-M designed the experiments. GN-L, AM-M, JS, FP-A, SI, SL, and MN-J discussed and interpreted the results. GN-L and AM-M designed the research. AM-M, JS, and AS contributed to the chemicals and scientific advice. GN-L, FP-A, and AM-M wrote the manuscript. All authors reviewed the final version of the manuscript.
Funding
This work was supported by the Lincoln University Research Fund, the Bio-protection Research Centre through the Tertiary Education Commission to AM-M and Meadow Mushrooms Scholarship Lincoln University to GN-L. GN-L was supported by Conacyt-Mexico for his Ph.D. studies at Lincoln University. GN-L acknowledge the Bio-protection Research Centre for the writing scholarship received. FP-A was supported for her Ph.D. studies by the Bio-protection Research Centre through the Tertiary Education Commission. JS was supported by MBIE LVLX1702.
Conflict of Interest
JS was employed by Lincoln Agritech Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We want to acknowledge Kee Hoon Sohn (Pohang University of Science and Technology, South Korea) for the introduction of this strategy; Charles Kenerley (Texas A&M University, United States) for having provided us with Trichoderma virens Gv28.9 strain and Sylvestre Marillonnet (Leibniz Institute of Plant Biochemistry, Halle, Germany) for pICH41021 vector. Alfredo Herrera-Estrella (Langebio, Mexico) for providing pUEO8 and Barry Scott (Massey University, New Zealand) for the pII99 containing the geneticin resistance cassette.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02794/full#supplementary-material
Footnotes
- ^ https://genome.jgi.doe.gov/Trichoderma/Trichoderma.info.html
- ^ https://www.uniprot.org/proteomes/
- ^ http://www.kazusa.or.jp/codon/
- ^ https://www.addgene.org/
- ^ http://www.kazusa.or.jp/codon/
- ^ http://nls-mapper.iab.keio.ac.jp/cgi-bin/NLS_Mapper_y.cgi
References
Andreou, A. I., and Nakayama, N. (2018). Mobius assembly: a versatile Golden-Gate framework towards universal DNA assembly. PLoS One 13:e0189892. doi: 10.1371/journal.pone.0189892
Baek, J. M., and Kenerley, C. M. (1998). The arg2 gene of Trichoderma virens: cloning and development of a homologous transformation system. Fungal Genet. Biol. 23, 34–44. doi: 10.1006/fgbi.1997.1025
Bhadauria, V., and Peng, Y. L. (2010). Optimization of a protein extraction technique for fungal proteomics. Indian J. Microbiol. 50(Suppl. 1), 127–131. doi: 10.1007/s12088-010-0072-3
Braithwaite, M., Johnston, P. R., Ball, S. L., Nourozi, F., Hay, A. J., Shoukouhi, P., et al. (2017). Trichoderma down under: species diversity and occurrence of Trichoderma in New Zealand. Aus. Plant Pathol. 46, 11–30. doi: 10.1007/s13313-016-0457-9
Broomfield, P. L., and Hargreaves, J. A. (1992). A single amino-acid change in the iron-sulphur protein subunit of succinate dehydrogenase confers resistance to carboxin in Ustilago maydis. Curr. Genet. 22, 117–121. doi: 10.1007/BF00351470
Brotman, Y., Landau, U., Cuadros-Inostroza, Á, Takayuki, T., Fernie, A. R., Chet, I., et al. (2013). Trichoderma-plant root colonization: escaping early plant defense responses and activation of the antioxidant machinery for saline stress tolerance. PLoS Pathog. 9:e1003221. doi: 10.1371/journal.ppat.1003221
Chen, X., Zaro, J. L., and Shen, W. C. (2013). Fusion protein linkers: property, design and functionality. Adv. Drug Deliv. Rev. 65, 1357–1369. doi: 10.1016/j.addr.2012.09.039
Curtis, M. D., and Grossniklaus, U. (2003). A gateway cloning vector set for high-throughput functional analysis of genes in planta. Plant Physiol. 133, 462–469. doi: 10.1104/pp.103.027979
de Groot, M. J., Bundock, P., Hooykaas, P. J., and Beijersbergen, A. G. (1998). Agrobacterium tumefaciens-mediated transformation of filamentous fungi. Nat. Biotechnol. 16, 839–842. doi: 10.1038/nbt0998-839
Derntl, C., Kiesenhofer, D. P., Mach, R. L., and Mach-Aigner, A. R. (2015). Novel strategies for genomic manipulation of Trichoderma reesei with the purpose of strain engineering. Appl. Environ. Microbiol. 81, 6314–6323. doi: 10.1128/AEM.01545-15
Djonovic, S., Pozo, M. J., Dangott, L. J., Howell, C. R., and Kenerley, C. M. (2006). Sm1, a proteinaceous elicitor secreted by the biocontrol fungus Trichoderma virens induces plant defense responses and systemic resistance. Mol. Plant Microbe Interact. 19, 838–853. doi: 10.1094/mpmi-19-0838
Doehlemann, G., and Hemetsberger, C. (2013). Apoplastic immunity and its suppression by filamentous plant pathogens. New Phytol. 198, 1001–1016. doi: 10.1111/nph.12277
Druzhinina, I. S., Seidl-Seiboth, V., Herrera-Estrella, A., Horwitz, B. A., Kenerley, C. M., Monte, E., et al. (2011). Trichoderma: the genomics of opportunistic success. Nat. Rev. Microbiol. 9, 749–759. doi: 10.1038/nrmicro2637
Egermeier, M., Sauer, M., and Marx, H. (2019). Golden Gate-based metabolic engineering strategy for wild-type strains of Yarrowia lipolytica. FEMS Microbiol. Lett. 366, fnz022. doi: 10.1093/femsle/fnz022
Emami, S., Yee, M.-C., and Dinneny, J. R. (2013). A robust family of Golden Gate Agrobacterium vectors for plant synthetic biology. Front. Plant Sci. 4:339. doi: 10.3389/fpls.2013.00339
Engler, C., Gruetzner, R., Kandzia, R., and Marillonnet, S. (2009). Golden Gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS One 4:e5553. doi: 10.1371/journal.pone.0005553
Engler, C., Kandzia, R., and Marillonnet, S. (2008). A one pot, one step, precision cloning method with high throughput capability. PLoS One 3:e3647. doi: 10.1371/journal.pone.0003647
Gong, X., Hurtado, O., Wang, B., Wu, C., Yi, M., Giraldo, M., et al. (2015). pFPL vectors for high-throughput protein localization in fungi: detecting cytoplasmic accumulation of putative effector proteins. Mol. Plant Microbe Interact. 28, 107–121. doi: 10.1094/MPMI-05-14-0144-TA
Gruber, S., Omann, M., Rodrìguez, C. E., Radebner, T., and Zeilinger, S. (2012). Generation of Trichoderma atroviride mutants with constitutively activated G protein signaling by using geneticin resistance as selection marker. BMC Res. Notes 5:641. doi: 10.1186/1756-0500-5-641
Guigon-Lopez, C., Vargas-Albores, F., Guerrero-Prieto, V., Ruocco, M., and Lorito, M. (2015). Changes in Trichoderma asperellum enzyme expression during parasitism of the cotton root rot pathogen Phymatotrichopsis omnivora. Fungal Biol. 119, 264–273. doi: 10.1016/j.funbio.2014.12.013
Guo, M., Zhu, X., Li, H., Tan, L., and Pan, Y. (2016). Development of a novel strategy for fungal transformation based on a mutant locus conferring carboxin-resistance in Magnaporthe oryzae. AMB Expr. 6:57. doi: 10.1186/s13568-016-0232-x
Guzman-Guzman, P., Porras-Troncoso, M. D., Olmedo-Monfil, V., and Herrera-Estrella, A. (2019). Trichoderma species: versatile plant symbionts. Phytopathology 109, 6–16. doi: 10.1094/phyto-07-18-0218-rvw
Harman, G. E., Howell, C. R., Viterbo, A., Chet, I., and Lorito, M. (2004). Trichoderma species-opportunistic, avirulent plant symbionts. Nat. Rev. Microbiol. 2, 43–56. doi: 10.1038/nrmicro797
Hartley, J. L., Temple, G. F., and Brasch, M. A. (2000). DNA cloning using in vitro site-specific recombination. Genome Res. 10, 1788–1795. doi: 10.1101/gr.143000
Hermosa, R., Viterbo, A., Chet, I., and Monte, E. (2012). Plant-beneficial effects of Trichoderma and of its genes. Microbiology 158(Pt 1), 17–25. doi: 10.1099/mic.0.052274-0
Hernanz-Koers, M., Gandia, M., Garrigues, S., Manzanares, P., Yenush, L., Orzaez, D., et al. (2018). FungalBraid: a GoldenBraid-based modular cloning platform for the assembly and exchange of DNA elements tailored to fungal synthetic biology. Fungal Genet. Biol. 116, 51–61. doi: 10.1016/j.fgb.2018.04.010
Housden, B. E., and Perrimon, N. (2016). Design and generation of donor constructs for genome engineering in Drosophila. Cold Spring Harb. Protoc 2016:pdb.prot090787. doi: 10.1101/pdb.prot090787
Jørgensen, M. S., Skovlund, D. A., Johannesen, P. F., and Mortensen, U. H. (2014). A novel platform for heterologous gene expression in Trichoderma reesei (Teleomorph Hypocrea jecorina). Microbial. Cell Factor. 13:33. doi: 10.1186/1475-2859-13-33
Karathia, H., Vilaprinyo, E., Sorribas, A., and Alves, R. (2011). Saccharomyces cerevisiae as a model organism: a comparative study. PLoS One 6:e16015. doi: 10.1371/journal.pone.0016015
Keon, J. P., Broomfield, P. L., White, G. A., and Hargreaves, J. A. (1994). A mutant form of the succinate dehydrogenase iron-sulphur protein subunit confers resistance to carboxin in the maize smut pathogen Ustilago maydis. Biochem. Soc. Trans. 22, 234–237. doi: 10.1042/bst0220234
Kilaru, S., Collins, C. M., Hartley, A. J., Burns, C., Foster, G. D., and Bailey, A. M. (2009). Investigating dominant selection markers for Coprinopsis cinerea: a carboxin resistance system and re-evaluation of hygromycin and phleomycin resistance vectors. Curr. Genet. 55, 543–550. doi: 10.1007/s00294-009-0266-6
Kubicek, C. P., Steindorff, A. S., Chenthamara, K., Manganiello, G., Henrissat, B., Zhang, J., et al. (2019). Evolution and comparative genomics of the most common Trichoderma species. BMC Genomics 20:485. doi: 10.1186/s12864-019-5680-7
Kück, U., and Hoff, B. (2010). New tools for the genetic manipulation of filamentous fungi. Appl. Microbiol. Biotechnol. 86, 51–62. doi: 10.1007/s00253-009-2416-7
Lampropoulos, A., Sutikovic, Z., Wenzl, C., Maegele, I., Lohmann, J. U., and Forner, J. (2013). GreenGate-a novel, versatile, and efficient cloning system for plant transgenesis. PLoS One 8:e83043. doi: 10.1371/journal.pone.0083043
Lu, Z., Tombolini, R., Woo, S., Zeilinger, S., Lorito, M., and Jansson, J. K. (2004). In vivo study of Trichoderma-pathogen-plant interactions, using constitutive and inducible green fluorescent protein reporter systems. Appl. Environ. Microbiol. 70, 3073–3081. doi: 10.1128/AEM.70.5.3073-3081.2004
Mach, R. L., Schindler, M., and Kubicek, C. P. (1994). Transformation of Trichoderma reesei based on hygromycin B resistance using homologous expression signals. Curr. Genet. 25, 567–570. doi: 10.1007/BF00351679
Mach, R. L., and Zeilinger, S. (1998). “Genetic transformation of Trichoderma and Gliocladium,” in Trichoderma and Gliocladium: Basic Biology, Taxonomy and Genetics, eds C. P. Kubicek and G. E. Harman, (London: Taylor & Francis), 226–241.
Malmierca, M. G., Izquierdo-Bueno, I., McCormick, S. P., Cardoza, R. E., Alexander, N. J., Barua, J., et al. (2016). Trichothecenes and aspinolides produced by Trichoderma arundinaceum regulate expression of Botrytis cinerea genes involved in virulence and growth. Environ. Microbiol. 18, 3991–4004. doi: 10.1111/1462-2920.13410
Medina, M. L., and Francisco, W. A. (2008). “Isolation and enrichment of secreted proteins from filamentous fungi,” in 2D PAGE: Sample Preparation and Fractionation, ed. A. Posch, (Totowa, NJ: Humana Press), 275–285. doi: 10.1007/978-1-60327-210-0_22
Mukherjee, P. K., Horwitz, B. A., Herrera-Estrella, A., Schmoll, M., and Kenerley, C. M. (2013). Trichoderma research in the genome Era. Annu. Rev. Phytopathol. 51, 105–129. doi: 10.1146/annurev-phyto-082712-102353
Mukherjee, P. K., and Kenerley, C. M. (2010). Regulation of morphogenesis and biocontrol properties in Trichoderma virens by a VELVET protein. Vel1. Appl. Environ. Microbiol. 76, 2345. doi: 10.1128/AEM.02391-09
Nayak, T., Szewczyk, E., Oakley, C. E., Osmani, A., Ukil, L., Murray, S. L., et al. (2006). A versatile and efficient gene-targeting system for Aspergillus nidulans. Genetics 172:1557. doi: 10.1534/genetics.105.052563
Nogueira-Lopez, G., Greenwood, D. R., Middleditch, M., Winefield, C., Eaton, C., Steyaert, J. M., et al. (2018). The Apoplastic secretome of Trichoderma virens during interaction with maize roots shows an inhibition of plant defence and scavenging oxidative stress secreted proteins. Front. Plant Sci. 9:409. doi: 10.3389/fpls.2018.00409
Peterbauer, C. K., Heidenreich, E., Baker, R. T., and Kubicek, C. P. (1992). Effect of benomyl and benomyl resistance on cellulase formation by Trichoderma reesei and Trichoderma harzianum. Can. J. Microbiol. 38, 1292–1297. doi: 10.1139/m92-213
Reece-Hoyes, J. S., and Walhout, A. J. M. (2018). Gateway recombinational cloning. Cold Spring Harb. Protoc. 2018:pdb.top094912. doi: 10.1101/pdb.top094912
Rubio, M. B., Quijada, N. M., Perez, E., Dominguez, S., Monte, E., and Hermosa, R. (2014). Identifying beneficial qualities of Trichoderma parareesei for plants. Appl. Environ. Microbiol. 80, 1864–1873. doi: 10.1128/aem.03375-13
Sarkari, P., Marx, H., Blumhoff, M. L., Mattanovich, D., Sauer, M., and Steiger, M. G. (2017). An efficient tool for metabolic pathway construction and gene integration for Aspergillus niger. Bioresour. Technol. 245(Pt B), 1327–1333. doi: 10.1016/j.biortech.2017.05.004
Sarrion-Perdigones, A., Falconi, E. E., Zandalinas, S. I., Juárez, P., Fernández-del-Carmen, A., Granell, A., et al. (2011). GoldenBraid: an iterative cloning system for standardized assembly of reusable genetic modules. PLoS One 6:e21622. doi: 10.1371/journal.pone.0021622
Schmoll, M., Dattenböck, C., Carreras-Villaseñor, N., Mendoza-Mendoza, A., Tisch, D., Alemán, M. I., et al. (2016). The genomes of three uneven siblings: footprints of the lifestyles of three Trichoderma species. Microbiol. Mol. Biol. Rev. 80, 205–327. doi: 10.1128/MMBR.00040-15
Schuster, A., Bruno, K. S., Collett, J. R., Baker, S. E., Seiboth, B., Kubicek, C. P., et al. (2012). A versatile toolkit for high throughput functional genomics with Trichoderma reesei. Biotechnol. Biof. 5:1. doi: 10.1186/1754-6834-5-1
Shima, Y., Ito, Y., Hatabayashi, H., Koma, A., and Yabe, K. (2011). Five carboxin-resistant mutants exhibited various responses to carboxin and related fungicides. Biosci. Biotechnol. Biochem. 75, 181–184. doi: 10.1271/bbb.100687
Shima, Y., Ito, Y., Kaneko, S., Hatabayashi, H., Watanabe, Y., Adachi, Y., et al. (2009). Identification of three mutant loci conferring carboxin-resistance and development of a novel transformation system in Aspergillus oryzae. Fungal Genet. Biol. 46, 67–76. doi: 10.1016/j.fgb.2008.10.005
Singer, T. P., Gutman, M., and Kearney, E. B. (1971). Succinate dehydrogenase. XVIII. Regulation of succinate dehydrogenase activity by reduced coenzyme Q10. Biochemistry 10, 2726–2733. doi: 10.1021/bi00790a011
Skinner, W., Bailey, A., Renwick, A., Keon, J., Gurr, S., and Hargreaves, J. (1998). A single amino-acid substitution in the iron-sulphur protein subunit of succinate dehydrogenase determines resistance to carboxin in Mycosphaerella graminicola. Curr. Genet. 34, 393–398. doi: 10.1007/s002940050
Tanaka, A., Wrenn, R. E., Takemoto, D., and Scott, B. (2007). “Agrobacterium tumefaciens T-DNA mediated transformation of Epichloë festucae,” in Proceedings of the 6th International Symposium on Fungal Endophytes of Grasses Grassland Research and Practice Series, No. 13, eds A. J. Popay and E. R. Thon (Christchurch: New Zealand Grassland Association), 469–472.
Terfrüchte, M., Joehnk, B., Fajardo-Somera, R., Braus, G. H., Riquelme, M., Schipper, K., et al. (2014). Establishing a versatile Golden Gate cloning system for genetic engineering in fungi. Fungal Genet. Biol. 62, 1–10. doi: 10.1016/j.fgb.2013.10.012
Topp, C. N., Ruiz-Herrera, J., Martinez-Espinoza, A. D., and Gold, S. E. (2002). Integration of the gene for carboxin resistance does not impact the Ustilago maydis-maize interaction. Curr. Microbiol. 44, 67–70. doi: 10.1007/s00284-001-0076-7
Waldo, G. S., Standish, B. M., Berendzen, J., and Terwilliger, T. C. (1999). Rapid protein-folding assay using green fluorescent protein. Nat. Biotechnol. 17, 691–695. doi: 10.1038/10904
Walhout, A. J. M., Temple, G. F., Brasch, M. A., Hartley, J. L., Lorson, M. A., van den Heuvel, S., et al. (2000). “GATEWAY recombinational cloning: application to the cloning of large numbers of open reading frames or ORFeomes,” in Methods in Enzymology, eds J. Thorner, S. D. Emr, and J. N. Abelson, (Cambridge, MA: Academic Press), 575–IN577.
Wang, T., Wang, D., Lyu, Y., Feng, E., Zhu, L., Liu, C., et al. (2018). Construction of a high-efficiency cloning system using the Golden Gate method and I-SceI endonuclease for targeted gene replacement in Bacillus anthracis. J. Biotechnol. 271, 8–16. doi: 10.1016/j.jbiotec.2018.02.006
Weber, E., Engler, C., Gruetzner, R., Werner, S., and Marillonnet, S. (2011). A modular cloning system for standardized assembly of multigene constructs. PLoS One 6:e16765. doi: 10.1371/journal.pone.0016765
Yu, J.-H., Hamari, Z., Han, K.-H., Seo, J.-A., Reyes-Domínguez, Y., and Scazzocchio, C. (2004). Double-joint PCR: a PCR-based molecular tool for gene manipulations in filamentous fungi. Fungal Genet. Biol. 41, 973–981. doi: 10.1016/j.fgb.2004.08.001
Zámborszky, J., Csikász-Nagy, A., and Hong, C. I. (2014). Neurospora crassa as a model organism to explore the interconnected network of the cell cycle and the circadian clock. Fungal Genet. Biol. 71, 52–57. doi: 10.1016/j.fgb.2014.08.014
Zeilinger, S. (2004). Gene disruption in Trichoderma atroviride via Agrobacterium-mediated transformation. Curr. Genet. 45, 54–60. doi: 10.1007/s00294-003-0454-8
Zhou, Z., Dang, Y., Zhou, M., Li, L., Yu, C. H., Fu, J., et al. (2016). Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc. Natl. Acad. Sci. U.S.A. 113, E6117–E6125. doi: 10.1073/pnas.1606724113
Keywords: Golden Gate cloning technology, Trichoderma, Agrobacterium-mediated transformation, carboxin resistance, TrichoGate, functional genomics, synthetic biology
Citation: Nogueira-López G, Padilla-Arizmendi F, Inwood S, Lyne S, Steyaert JM, Nieto-Jacobo MF, Stewart A and Mendoza-Mendoza A (2019) TrichoGate: An Improved Vector System for a Large Scale of Functional Analysis of Trichoderma Genes. Front. Microbiol. 10:2794. doi: 10.3389/fmicb.2019.02794
Received: 20 June 2019; Accepted: 18 November 2019;
Published: 10 December 2019.
Edited by:
Jessy L. Labbé, Oak Ridge National Laboratory (DOE), United StatesReviewed by:
Yaohua Zhong, Shandong University, ChinaPaul Daly, Westerdijk Fungal Biodiversity Institute, Netherlands
Copyright © 2019 Nogueira-López, Padilla-Arizmendi, Inwood, Lyne, Steyaert, Nieto-Jacobo, Stewart and Mendoza-Mendoza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Artemio Mendoza-Mendoza, YXJ0ZW1pby5tZW5kb3phQGxpbmNvbG4uYWMubno=