 Claire V. Mulholland1,2
Claire V. Mulholland1,2 Abigail C. Shockey3
Abigail C. Shockey3 Htin L. Aung2,4Ray T. Cursons1
Htin L. Aung2,4Ray T. Cursons1 Ronan F. O’Toole5,6Sanjay S. Gautam5Daniela Brites7,8
Ronan F. O’Toole5,6Sanjay S. Gautam5Daniela Brites7,8 Sebastien Gagneux7,8
Sebastien Gagneux7,8 Sally A. Roberts9Noel Karalus10Gregory M. Cook2,4
Sally A. Roberts9Noel Karalus10Gregory M. Cook2,4 Caitlin S. Pepperell3,11*
Caitlin S. Pepperell3,11* Vickery L. Arcus1,2*
Vickery L. Arcus1,2*- 1School of Science, University of Waikato, Hamilton, New Zealand
- 2Maurice Wilkins Centre for Molecular Biodiscovery, The University of Auckland, Auckland, New Zealand
- 3Department of Medical Microbiology and Immunology, School of Medicine and Public Health, University of Wisconsin–Madison, Madison, WI, United States
- 4Department of Microbiology and Immunology, School of Biomedical Sciences, University of Otago, Dunedin, New Zealand
- 5School of Medicine, University of Tasmania, Hobart, TAS, Australia
- 6School of Molecular Sciences, La Trobe University, Melbourne, VIC, Australia
- 7Swiss Tropical and Public Health Institute, Basel, Switzerland
- 8University of Basel, Basel, Switzerland
- 9LabPLUS, Auckland City Hospital, Auckland, New Zealand
- 10Waikato Hospital, Hamilton, New Zealand
- 11Department of Medicine, Division of Infectious Diseases, School of Medicine and Public Health, University of Wisconsin–Madison, Madison, WI, United States
Mycobacterium tuberculosis (Mtb) is a globally distributed bacterial pathogen whose population structure has largely been shaped by the activities of its obligate human host. Oceania was the last major global region to be reached by Europeans and is the last region for which the dispersal and evolution of Mtb remains largely unexplored. Here, we investigated the evolutionary history of the Euro-American L4.4 sublineage and its dispersal to the South Pacific. Using a phylodynamics approach and a dataset of 236 global Mtb L4.4 genomes we have traced the origins and dispersal of L4.4 strains to New Zealand. These strains are predominantly found in indigenous Māori and Pacific people and we identify a clade of European, likely French, origin that is prevalent in indigenous populations in both New Zealand and Canada. Molecular dating suggests the expansion of European trade networks in the early 19th century drove the dispersal of this clade to the South Pacific. We also identify historical and social factors within the region that have contributed to the local spread and expansion of these strains, including recent Pacific migrations to New Zealand and the rapid urbanization of Māori in the 20th century. Our results offer new insight into the expansion and dispersal of Mtb in the South Pacific and provide a striking example of the role of historical European migrations in the global dispersal of Mtb.
Introduction
Tuberculosis (TB) is caused by the bacterial pathogen Mycobacterium tuberculosis (Mtb) and other members of the Mtb complex (MTBC). The MTBC comprises seven human adapted lineages, which show strong phylogeographic structure and vary in the extent of their global distribution (Hirsh et al., 2004; Hershberg et al., 2008; Gagneux, 2018). The most widely globally dispersed MTBC lineage is lineage 4 (L4), also known as the “Euro-American” lineage (Gagneux et al., 2006; O’Neill et al., 2019). Spatial and temporal patterns of L4 dispersal suggest that it was spread through European colonial migrations to Africa and the Americas (Hirsh et al., 2004; Gagneux et al., 2006; Hershberg et al., 2008; Pepperell et al., 2011; Brynildsrud et al., 2018; O’Neill et al., 2019). Oceania was the last major region to be reached by Europeans and includes the > 1000 islands of Polynesia scattered across the central and southern Pacific Ocean. Little is known about the origins and dispersal of Mtb in this region. It is commonly assumed that Mtb was introduced to Polynesia with the arrival of European sailors and settlers, however, TB-like lesions in skeletons predating European arrival challenge this view (Buckley et al., 2010).
New Zealand is the largest country in Polynesia and is home to the local indigenous Māori people and the largest diaspora of communities of Pacific peoples globally. This provides a unique setting for the investigation of Mtb dispersal and transmission in this region. Mtb genotypes in New Zealand Europeans, Māori and Pacific people in New Zealand are dominated by L4 strains (Yen et al., 2013), consistent with introduction of present-day strains by Europeans. Māori and Pacific People in New Zealand are disproportionately affected by TB and collectively account for ∼70% of New Zealand born TB cases (ESR, 2018). Around three-quarters of Mtb isolates from Māori and Pacific People have shared molecular typing patterns indicative of recent transmission. The largest Mtb cluster in New Zealand identified by related 24-loci MIRU-VNTR typing patterns is known as the “Rangipo” cluster. This strain predominantly occurs in Māori, accounting for around one-quarter of Māori TB cases (J Sherwood, ESR, personal communication). Two other large clusters known as the “Southern Cross” and “Otara” clusters predominately occur in Pacific people. Globally, indigenous people are generally found to have higher rates of TB than non-indigenous people (Tollefson et al., 2013). Understanding how the pathogen was dispersed and is maintained among indigenous populations is important for designing improved strategies for TB control in these often disproportionally affected populations.
The most common L4 sublineage in New Zealand is L4.4, which accounts for 43% of New Zealand born L4 cases (Stucki et al., 2016). Here, we show that the Rangipo and Otara clusters both belong to the L4.4 sublineage. We sought to examine the global evolutionary history of L4.4 and trace the dispersal of these strains to New Zealand. For this purpose, we analyzed a genomic dataset of 236 Mtb L4.4 isolates from 19 different countries including 23 recent and newly sequenced genomes from New Zealand clinical isolates belonging to the Rangipo and Otara clusters. We find the New Zealand strains belong to a L4.4.1.1 “S-type” sublineage clade that includes the DS6Quebec lineage, which was dispersed to Western Canadian First Nations by French-Canadian fur traders in the 18th–19th centuries (Pepperell et al., 2011). Our results suggest migration of this clade to the South Pacific was driven by the expansion of European trade networks in the 19th century, with the whaling trade serving as a likely dispersal route to indigenous Pacific populations. Our findings show the dispersal and expansion of these strains in the Pacific is related to historical and social drivers of TB transmission and provide new insights into the dispersal of this globally successful human pathogen.
Results
Phylogeny of the New Zealand Mtb Strains
We first performed single nucleotide polymorphism (SNP) based lineage assignment on 34 New Zealand Mtb Rangipo, Otara and Southern Cross strain genomes, these included sequences from previous work (Gautam et al., 2017; Mulholland et al., 2017) and new sequencing data. This assigned Rangipo and Otara isolates to the L4.4.1.1 (S-type) sublineage and Southern Cross to L4.3.3 (LAM). A total of 23 New Zealand L4.4 genomes (16 Rangipo, 7 Otara) spanning a 22-year period (1991–2013) met mapping quality thresholds and were included in downstream analyses (Supplementary Table S1 and Supplementary Figure S1). A whole-genome SNP phylogeny of these strains shows the Rangipo and Otara strains form two well-differentiated monophyletic clades with differing phylogenetic structures (Supplementary Figure S2). The Rangipo cluster is characterized by short terminal branches and low genetic diversity (pairwise SNPs 0–12, median 4) suggesting recent clonal expansion and temporally short transmission chains, consistent with its association with local outbreaks. Conversely, Otara isolates have long terminal branches and higher genetic diversity (pairwise SNPs 1–102, median 91). This is consistent with predominant reactivation disease due to a locally endemic strain rather than a recent transmission cluster as previously thought based on MIRU-VNTR typing.
Global Phylogeny of the L4.4 Sublineage
To investigate the origins and dispersal of the New Zealand L4.4 strains, we compiled a dataset comprising our 23 New Zealand Rangipo and Otara strain genomes and 213 L4.4 genomes from 18 different countries representing all five major global regions (Supplementary Figure S3 and Supplementary Table S2). WGS reads were mapped to the H37Rv reference genome and repetitive genomic regions were removed prior to alignment. High quality variant sites were extracted producing a 9024 bp SNP alignment used to infer a global L4.4 maximum likelihood phylogeny (Figure 1A and Supplementary Figure S4). This shows L4.4 comprises three well-differentiated sublineages, L4.4.1.1, L4.4.1.2 and L4.4.2 (pairwise FST values between lineages 0.50–0.57), consistent with the Coll classification system (Coll et al., 2014).
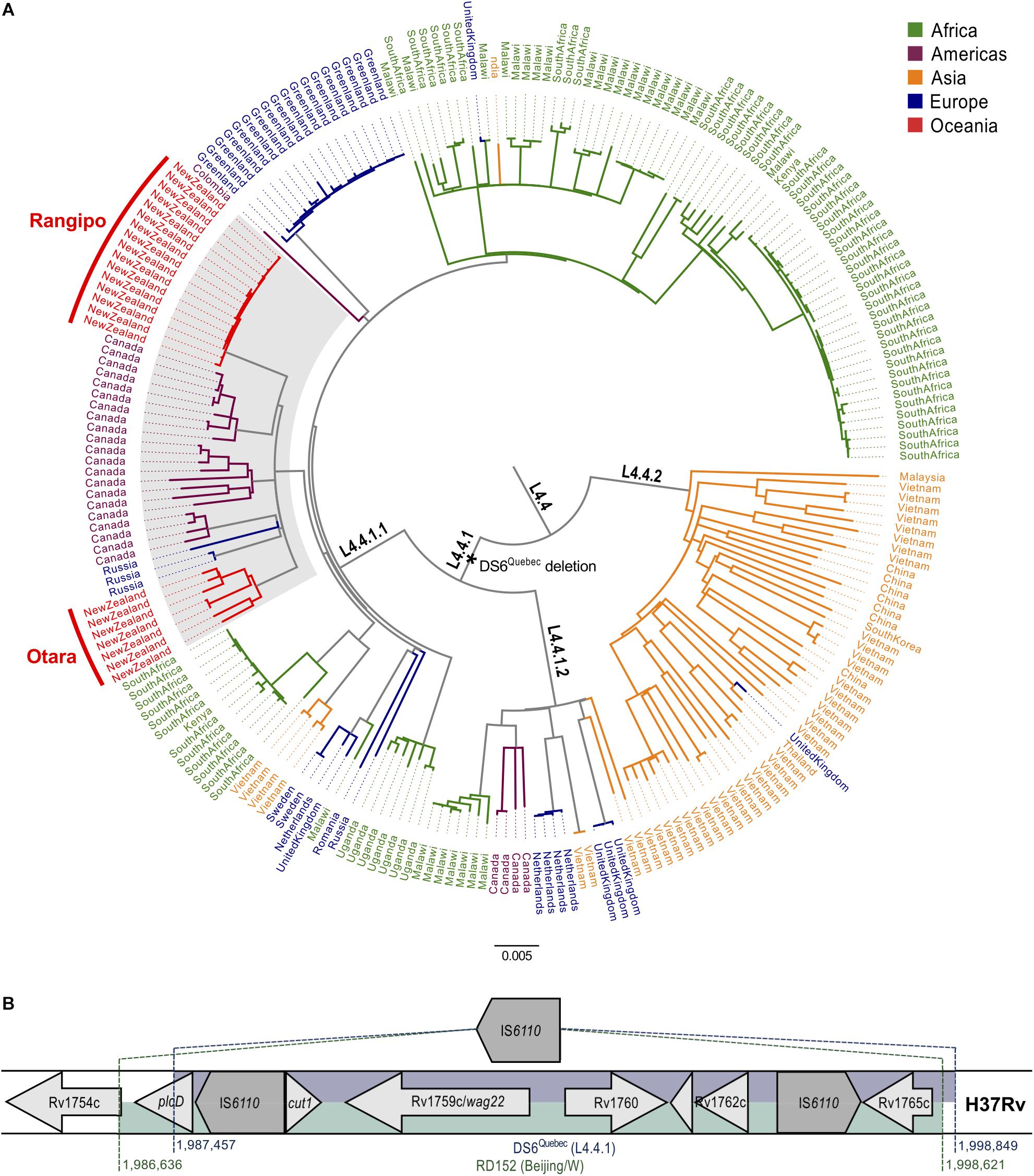
Figure 1. Global phylogeny of the Mycobacterium tuberculosis complex L4.4 sublineage and genomic structure and phylogenetic placement of the DS6Quebec deletion. (A) Whole genome SNP maximum likelihood phylogeny of L4.4 comprised of 236 isolates from 19 different countries, including 23 isolates from the New Zealand Rangipo and Otara clusters. Scale bar indicates substitutions per polymorphic site. A black asterisk indicates the DS6Quebec deletion and the CS clade is highlighted in gray. Branches and tip labels are colored by geographic region and lineages labeled according to the nomenclature of Coll et al. (2014). A black circle indicates the node position of the most recent common ancestor of L4.4 (rooted to H37Rv, not shown). (B) Schematic of the DS6Quebec deletion. Genomic regions in H37Rv that are deleted by the DS6Quebec deletion and the RD152 deletion in the Beijing/W lineage are shown.
L4.4 has previously been observed at high proportions in parts of Asia and Africa (Stucki et al., 2016). Consistent with this observation, a large proportion of isolates in our dataset come from these regions (Supplementary Figure S3). Our results reveal differing global distributions and population structures of the L4.4 sublineages. We find L4.4.2 is essentially restricted to Eastern Asia consistent with in situ growth and diversification of this sublineage. Conversely, both L4.4.1.1 and L4.4.1.2 are relatively well distributed globally, indicative of high rates of migration and efficient dispersal.
Within L4.4.1.1, we identified a clade comprised predominantly of isolates from New Zealand and Canada. The Canadian isolates belong to the DS6Quebec lineage, which is endemic in French Canadians in Quebec and Western Aboriginal Canadian populations, and is characterized by the presence of the DS6Quebec deletion (Nguyen et al., 2004; Pepperell et al., 2011). We named the clade encompassing the New Zealand strains and Canadian DS6Quebec lineage isolates the “CS” clade (colonial S-type) for its association with European colonial activities in Canada (Pepperell et al., 2011) and the Pacific (shown in this work). Examination of mapped sequencing reads found that both of the New Zealand clusters carry the DS6Quebec deletion and the presence of the deletion in the Rangipo and Otara strains was further confirmed by PCR and Sanger sequencing. The Rangipo and Otara clusters are not monophyletic within the CS clade, consistent with at least two separate introductions into New Zealand.
Phylogenetic Placement of the DS6Quebec Deletion
The DS6Quebec deletion removes an approximately 11.4 kb region truncating or removing the genes from Rv1755c/plcD to Rv1765c (between positions 1987457 to 1998849 in H37Rv) (Nguyen et al., 2004; Figure 1B). Examination of mapped reads found that all L4.4.1.1 and L4.4.1.2, but not L4.4.2 genomes, harbored the DS6Quebec deletion, showing that this is a characteristic deletion of L4.4.1. One L4.4.1.1 genome had an earlier start to the deletion (position 1987142) indicating a subsequent small deletion event. This same region is also removed by the similar but evolutionarily independent ∼12 kb RD152 deletion (positions 1986636 to 1998621) in L2/Beijing strains (Figure 1B; Tsolaki et al., 2005). The genomic region affected by RD152 and DS6Quebec is highly variable and is associated with frequent insertion of IS6110 elements (Ho et al., 2000), suggesting homologous recombination between adjacent IS6110 elements as the likely mechanism responsible for these similar but evolutionarily distinct deletion events.
Temporal Evolution of the L4.4.1.1 Sublineage and the CS Clade
The polytomy at the root of the CS clade and the polyphyletic nature of the New Zealand and Canadian isolates implies dispersal of several closely related strains from a common origin. It is likely that the DS6Quebec lineage was introduced to Canada from France (Pepperell et al., 2011), suggesting a similar European origin for the New Zealand strains. French whalers had a notable presence in New Zealand and Polynesia during the South Pacific whaling era (1790–1860) (Haines, 2010) and the arrival of whalers and other traders in the region is associated with the introduction of new diseases, including TB (Lange, 1984; Chappell, 1997). We hypothesized the CS clade may have been introduced to the Pacific via this route. To further explore this hypothesis, the temporal evolution and dispersal of the L4.4.1.1 sublineage was investigated by Bayesian evolutionary analysis using BEAST2 (Bouckaert et al., 2014) with an alignment of 3161 variable nucleotide positions from 117 L4.4.1.1 genomes, which included all New Zealand isolates and global L4.4.1.1 isolates with known year of isolation at the time of analysis (Supplementary Table S3). Both root-to-tip regression (R2 = 0.229) and date randomization tests detected sufficient temporal signal in the data set for calibration of the molecular clock by tip-dating (Supplementary Figure S5).
The L4.4.1.1 phylogeny, mutation rate and node ages were inferred using strict and relaxed molecular clocks with different coalescent demographic models. Nucleotide substitution rate was modeled using the general time reversible (GTR) model. All models produced similar rate and date estimates (median 6.15 × 10–8–6.64 × 10–8 substitutions per site per year (sub/site/yr); widest 95% highest posterior density (HPD) intervals over all models, 4.23 × 10–8–9.08 × 10–8) (Table 1). Model comparison using path sampling determined that the strict clock with the Bayesian skyline demographic model provided the best fit to the data (Supplementary Table S5). Under this model we estimated a substitution rate of 6.28 × 10–8 sub/site/yr (95% HPD, 4.54 × 10–8–8.10 × 10–8), resulting in a time to most recent common ancestor (TMRCA) estimate of 1492 for L4.4.1.1 (95% HPD, 1325–1629). Our substitution rate estimate is similar to the results from other studies using contemporary L4 and mixed lineage MTBC genomes, all of which produced median rate estimates of ∼7 × 10–8–1 × 10–7 sub/site/yr (Ford et al., 2013; Pepperell et al., 2013; Roetzer et al., 2013; Walker et al., 2013; Eldholm et al., 2015).
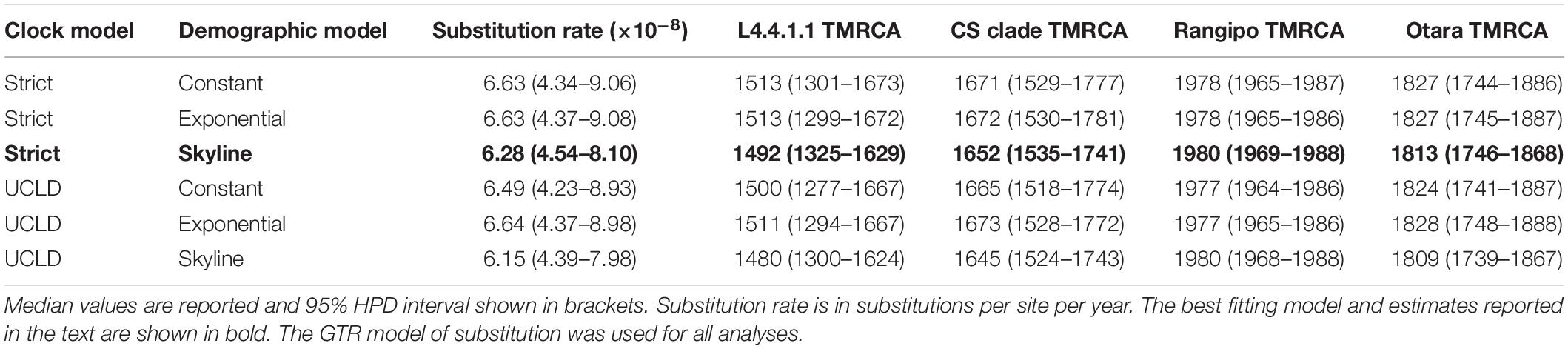
Table 1. Mycobacterium tuberculosis complex L4.4.1.1 sublineage substitution rate and time to most recent common ancestor (TMRCA) estimates.
The Bayesian skyline plot suggests the L4.4.1.1 sublineage underwent a rapid population expansion following its emergence, and corresponding migration analyses show a spike in migration at this time (Figures 2A,B). This was followed by a period where the population size remained consistent until another period of population growth in the 19th century, during which time migration tapers off. Our phylogeographic reconstruction is indicative of connectivity between Africa and Europe and shows a pattern of dispersal from Europe to Canada and Oceania (Figures 2C,D). These patterns of connectivity and the dispersal of L4.4.1.1 through Africa and Europe are consistent with previous reconstructions of the migratory history of L4 (O’Neill et al., 2019).
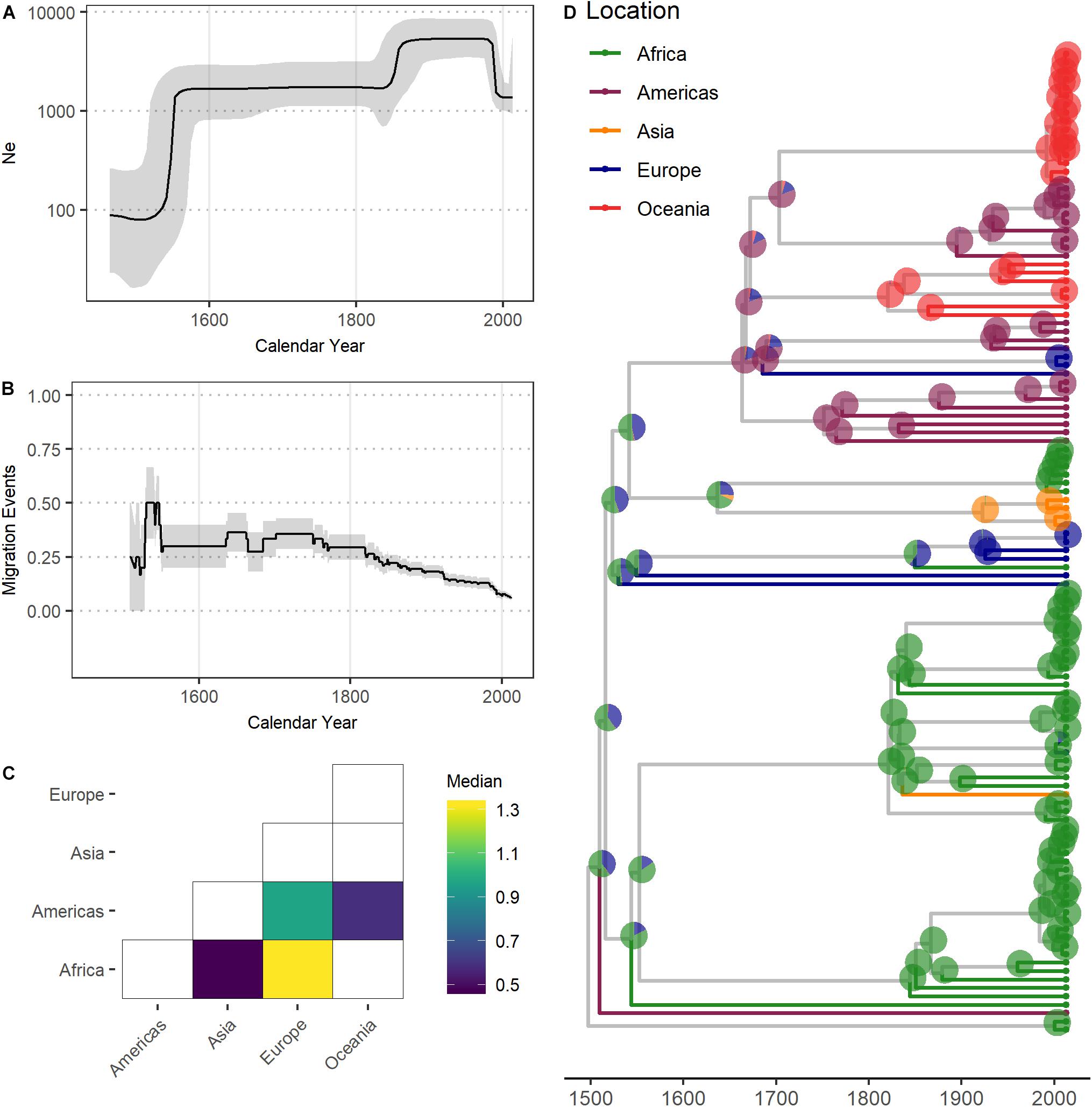
Figure 2. Demographic analysis of the Mycobacterium tuberculosis complex L4.4.1.1 sublineage. (A) Effective population size (Ne) through time of L4.4.1.1. Median Ne and 95% highest posterior density pictured as black line and gray shading, respectively. X-axis in calendar years. (B) Migration events through time of L4.4.1.1. Black line depicts the rate of migration through time, calculated as the sum of migration events occurring across every year of the phylogeny divided by the total number of branches during each year of the phylogeny. Gray shading depicts the rates inferred after the addition or subtraction of a single migration event. X-axis in calendar years. (C) Migration matrices of L4.4.1.1. Heatmap of pairwise relative migration rates between UN regions. Only relative rates with Bayes factor > 5 shown. (D) MCC phylogeny of L4.4.1.1. Tips and terminal branches colored according to UN region of isolation. Pie charts on nodes colored according to geographic state probabilities. X-axis in calendar years.
Our estimated TMRCA of the CS clade is 1652 (95% HPD, 1535–1741) and the TMRCA of Rangipo and the closest Canadian clade was 1691 (95% HPD, 1588–1776) (Table 1 and Supplementary Figure S6). This is coincident with the French migration to Quebec between 1608–1760 (Charbonneau et al., 1993), and is thus consistent with a European, likely French, origin of the CS clade (Figure 3). The TMRCA estimate for the Otara strain is 1813 (95% HPD, 1746–1868), which coincides with arrival of European whalers and other traders, including sealers, bêche-de-mer and sandalwood traders, to the Pacific region the early 19th century (Campbell, 2011; Fischer, 2013). Our TMRCA estimate of the Rangipo strain is 1980 (95% HPD, 1969–1988), indicating this strain is either a relatively recent introduction or clonal expansion from a previously introduced, unsampled, CS strain.
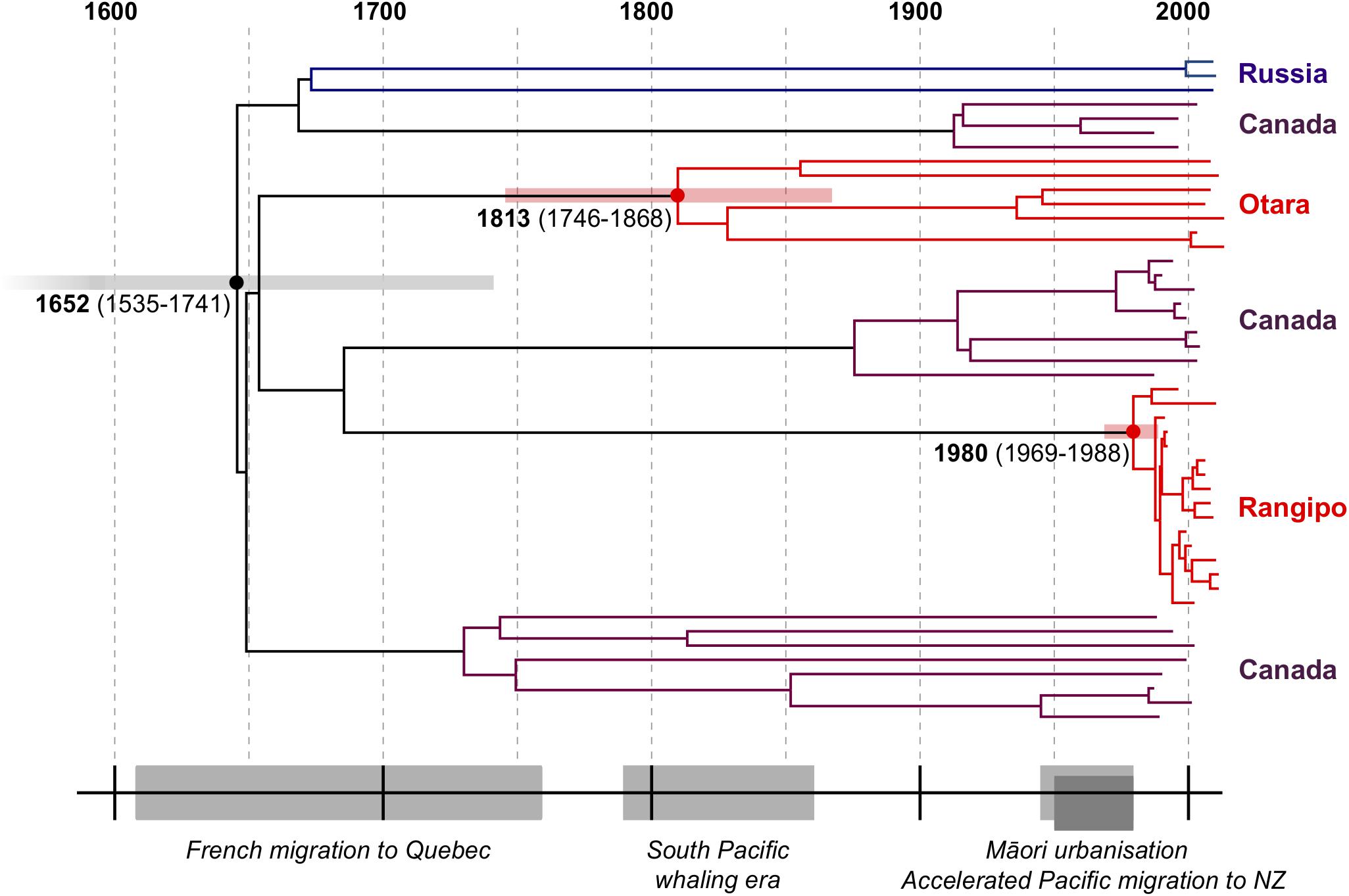
Figure 3. Dated Bayesian phylogeny of the CS clade and historical timeline. Median TMRCA estimates and 95% HPD intervals are shown for the CS, Rangipo, and Otara clades. Events shown include the French migration to Quebec (1608–1760), the South Pacific whaling era (1790–1860), the rapid urbanization of Māori in the 20th century (1945–1980), and the surge in Pacific migration into New Zealand in the 1950s–1970s.
Discussion
Using a phylodynamics inference from WGS data, we have performed a global characterization of the L4.4 sublineage and identify a L4.4.1.1 sublineage clade that is common in indigenous populations in Canada and Polynesia. Molecular dating estimated the MRCA for this clade, which we term the “CS” clade, to have existed in the mid-17th century, which is coincident with the French migration to Quebec and thus consistent with a French origin as previously reported for the DS6Quebec lineage (Pepperell et al., 2011; Figure 3). Our results indicate multiple migrations of closely related CS strains out of Europe to Canada and to the South Pacific, providing a striking example of the role of European colonial and trade migrations in driving the global spread of L4.
The early 19th century TMRCA estimate for the Otara strain fits with an introduction to the Pacific by French/European whalers or other traders (Figure 3). To date, the Otara strain has predominantly been identified in Pacific people living in New Zealand. Considering the recent history of migration to New Zealand from the Pacific Islands, our results suggest this strain was initially dispersed to the Pacific Islands from Europe and subsequently migrated to New Zealand. Although limited data are available on early migration from the Pacific, only very small numbers of Pacific people began to settle in New Zealand in the 1800s and in 1916 there were only 151 Pacific Island people in New Zealand (United Nations and Economic Social Commission for Asia and the Pacific, 1985). The Pacific population in New Zealand began to slowly increase in the first half of the 20th century until an upsurge in migration in the 1950s–1970s (Dunsford et al., 2011), and by 1976 the Pacific population in New Zealand had increased to 61,354 (United Nations and Economic Social Commission for Asia and the Pacific, 1985). The TMRCA of the Otara strain is therefore consistent with initial dispersal to the Pacific Islands by Europeans in the early 1800s. Long internal branches in the phylogeny stretching back to the 19th century suggest multiple subsequent introductions to New Zealand that may have accompanied more recent Pacific migrations.
Unlike New Zealand, which began to receive large influxes of European, predominantly British and Irish, migrants following British annexation in 1840 (United Nations and Economic Social Commission for Asia and the Pacific, 1985), other Polynesian islands did not experience the same en masse arrival of European emigrants, lending further support to this being a trade-associated introduction. The whaling trade was also the only French economic activity of any scale in the South Pacific during the early 19th century (Maclellan, 1998), the most significant years of which were 1832–1846 (Foucrier, 2005). As with the Canadian fur trade, commercial success of the whaling industry depended on establishing productive social and economic relationships with the local people. During the whaling era, large numbers of Pacific peoples relocated from villages to harbor settlements for trade and employment opportunities, and Pacific men were often recruited as crew on whaling ships accounting for up to one-fifth of European whaling crews (Chappell, 1997; Fischer, 2013). Intermarriage also played a central role in industry establishment and success in both the Canadian fur trade and New Zealand whaling (Stevens and Wanhalla, 2017). Such interactions would have established strong social ties conducive for the dispersal of Mtb. Accordingly, contact with European trade vessels and ports have been implicated in the introduction of TB and other infectious diseases into the South Pacific (Lange, 1984; Chappell, 1997).
The L4.4.1.1 sublineage defined by SNP genotyping corresponds to the S lineage, also known as the “S-type,” classified by spoligotyping (Brudey et al., 2006; Coll et al., 2014). Molecular typing has shown that the S lineage also has a notable presence in French Polynesia, accounting for over one-third of Mtb isolates in Tahiti (10/27, 37%) (Osman et al., 2017). Tahiti was made a French protectorate in 1842 and a colony in 1880, and was an important commerce hub provisioning European whaling and trade vessels in the early 19th century. Although no WGS data were available for inclusion in phylogenetic analyses, we speculate that this lineage may have been introduced to Tahiti via the same historical migrations that introduced the New Zealand CS strains to Polynesia. In addition to CS strains in Canada and New Zealand, the CS clade also contains isolates from Russia. Unlike New Zealand and Canada where CS strains occur at relatively high frequencies in indigenous populations, L4.4 is rare in Russia (Casali et al., 2014; Stucki et al., 2016). Historically, Western Europe and Russia have been culturally and politically more connected and trade between them dates back to ancient times (Öhberg, 1955). Russia was also engaged the colonial fur trade (Rich, 1955), providing possible avenues for dispersal of CS strains.
The Rangipo cluster has been responsible for numerous TB outbreaks for over the last 30 years (De Zoysa et al., 2001; McElnay et al., 2004; Colangeli et al., 2014) and is an important source of TB in Māori. Unlike the older endemic Otara strain, our results indicate that the Rangipo cluster arose from a relatively recent clonal expansion, due to either a more recent introduction or emergence from a previously introduced CS strain. Between 1840–1843 the majority of French whaling voyages included New Zealand (70/81, 86.4%) (Foucrier, 2005) and French whaling provides a conceivable route for historical introduction of CS strains into New Zealand from France. Alternatively, Rangipo may be a more recent introduction. The TMRCA follows a period of mass migration to New Zealand from the Pacific Islands in the 1950s–1970s offering another plausible route. Although it is evident that the Rangipo cluster has ultimately emerged from a strain of European origin, more in-depth sampling of L4.4.1.1 isolates from both New Zealand and the Pacific may provide a clearer picture of the route this strain took from Europe to New Zealand and will shed additional light on the dispersal of this sublineage in the South Pacific region.
The Rangipo strain was named for its association with a large TB outbreak in the late-1990s involving cases who had spent time in the Rangipo prison (De Zoysa et al., 2001). Prior to this, health professionals were aware of clusters of infection caused by this strain first appearing in the early 1990s (N. Karalus, personal communication). Our TMRCA estimate for the Rangipo cluster predates this outbreak, although its introduction into the prison environment has presumably helped contribute to its further spread. The TMRCA of the Rangipo strain coincides with major demographic changes in the Māori population that occurred in the mid-20th century. Māori TB mortality rates declined sharply in the mid-1900s (MacLean, 1964), which presumably would have imposed a bottleneck on the Mtb population. Along with falling TB rates, between 1945–1980 Māori also experienced one of the fastest rates of urbanization of any population in the world (Pool, 1991). This was accompanied by significant social and environmental changes including overcrowded housing and increased prison incarceration rates, both of which are TB risk factors (Clark et al., 2002; Baussano et al., 2010). These host social and environmental changes are intricately tied to the colonial history of New Zealand. Globally, indigenous people generally have a higher prevalence of “proximate determinants” of TB such as smoking and food insecurity (Cormier et al., 2019). These may be substantial contributors to the high burden of TB in these communities and their prevalence is often linked to upstream social factors (Cormier et al., 2019). The temporal association between the emergence of the Rangipo cluster and the urbanization of Māori suggests that human social phenomena are important contributors to the expansion and dispersal of Mtb in indigenous populations. A similar pattern is also observed in Canada, whereby Mtb population expansion in indigenous Canadian populations occurred concomitant with major environmental and social changes affecting host populations (Pepperell et al., 2011).
Both Rangipo and Otara are Māori place names and Otara is a city that is home to large populations of Pacific people, associating these names with Māori and Pacific people more generally. Our results show that these strains are a product of European contact and colonization and highlight the pejorative naming of these strains with Māori names. Naming diseases by place of origin stigmatizes the associated population and the name “Rangipo” also further perpetuates the stigma attached to the disease by associating it with prison and criminality. Stigma increases the emotional suffering of TB patients and has implications for TB control efforts, for example by affecting health-seeking behaviors and adherence to treatment (Craig et al., 2017). The findings of this work point to the appropriateness of renaming these clusters to refrain from further stigmatizing communities where TB is present and perpetuating stigma associated with the disease, and further work will seek to formally rename these clusters in consultation with Māori.
Recently, Brynildsrud et al. (2018) reconstructed the migratory history of the L4 sublineage, including isolates from Europe, Africa, the Americas and Southeast Asia. Global dispersal of L4 was found to be dominated by historical migrations out of Europe and dispersal of L4 to Africa and the Americas occurred concomitant with European colonial migrations (Brynildsrud et al., 2018). We observe the same scenario with the introduction of L4.4 to the South Pacific and the CS clade provides a striking example of the role of European expansion in the global dispersal of Mtb. Our analyses reveal the migration of several closely related CS strains out of Europe in the 17th–19th centuries to remote and unconnected populations driven by European colonial migrations and expanding trade networks. In a separate study by O’Neill et al. (2019), the evolutionary history of L4 was found to be characterized by rapid diffusion and high rates of migration, with range expansion contributing to the growth of this lineage. Consistent with this, our results suggest efficient dispersal of L4.4 and a more extensive demographic analysis of the L4.4.1.1 sublineage revealed increased population growth concurrent with a spike in migration in the 16th century following emergence of this lineage. This timing is coincident with the European age of exploration, providing a plausible factor that may have contributed to the growth and dispersal of this sublineage. A similar pattern of increased population growth and migration during this era has also been detected for L4 as a whole (O’Neill et al., 2019). We detect L4.4.1.1 population growth in the 19th century that could be attributable to various colonial activities around this time involving countries represented in our sample; the French-Canadian fur trade (1710–1870) (Innis, 1999), the South Pacific whaling trade (1790–1860) (Haines, 2010), and the rapid occupation and colonization of much of the African continent during the New Imperialism period (1876–1912) (Pakenham, 2015). The later population decline in the late 20th century coincides with the dramatic decline in TB incidence in the developed world over the last century.
The phylogeography of bacterial pathogens can provide valuable insights into the migratory history of their human hosts. Most notably, Helicobacter pylori has been identified as reliable marker to deduce human population movements, providing valuable insights into ancient human migrations in the Pacific and globally (Falush et al., 2003; Moodley et al., 2009). In this study, we identify multiple migrations of several closely related Mtb strains to geographically distant and unconnected indigenous populations driven by European colonial and trade expansion in the 17–19th centuries. The presence of the CS clade in indigenous Pacific populations provides a potential marker of these historical migrations and reasserts the role of European migrations in the global dispersal of L4 Mtb. Our results highlight the power of phylodynamic methods and the utilization of public WGS data repositories to trace recent migrations of Mtb in high resolution at both the global and local scale, uncovering human movements and social changes that have contributed to the dispersal and success of Mtb in indigenous Pacific populations.
Materials and Methods
Genomic Data
New Zealand L4.4 Genomes
We have recently sequenced 18, seven and five Mtb isolates from the New Zealand Rangipo, Otara and Southern Cross clusters, respectively, on the Illumina MiSeq platform (Mac Aogáin et al., 2016; Gautam et al., 2017; Mulholland et al., 2017). An additional four Rangipo genomes were sequenced for this study using paired-end 250-bp reads on the Illumina MiSeq platform using the NexteraTM XT DNA Library Preparation Kit (Illumina Inc., CA, United States) as previously described (Aung et al., 2016). These four strains have been previously sequenced on the SOLiD platform (Colangeli et al., 2014) and were re-sequenced here on Illumina for inclusion in this study. Sequencing data were submitted to the National Centre for Biotechnology Information (NCBI) Nucleotide Archive (PRJNA356104).
MTBC lineage was determined with KvarQ (v0.12.3a1) (Steiner et al., 2014) using the coll14 testsuite (Coll et al., 2014). This classified Rangipo and Otara isolates as belonging to the L4.4.1.1 sublineage. Southern Cross isolates belonged to L4.3.3 and were excluded from further analysis. Nineteen previously sequenced Rangipo and Otara genomes and all four new Rangipo genomes met mapping quality thresholds for inclusion in this study (depth of coverage > 25X, > 75% reads mapped to the reference genome, < 10% missing calls in alignments), resulting in a dataset of 23 New Zealand L4.4 strains included in further analyses (Supplementary Table S1). The Rangipo strain isolates (n = 16) span a 20-year collection period (1991–2011) and include isolates from different geographical locations. MIRU-VNTR data was available for seven of these isolates, all of which share the 24-loci MIRU-VNTR profile that distinguishes the Rangipo cluster (233325153324-341444223362). The remaining isolates had been identified as Rangipo strain based on IS6110-RFLP typing and/or contact tracing information. Otara isolates (n = 7) spanned an 10-year collection period (2003–2013) and represent four variant MIRU-VNTR typing profiles distinguishable by a single locus.
Global L4.4 Genomes
Global L4.4 genomes included 23 newly sequenced genomes from Canada and 190 publicly available genomes from published studies (Bryant et al., 2013a, b; Clark et al., 2013; Walker et al., 2013; Zhang et al., 2013; Casali et al., 2014; Guerra-Assunção et al., 2015a, b; Bjorn-Mortensen et al., 2016; Brynildsrud et al., 2018; Holt et al., 2018) and Broad Institute sequencing initiatives (broadinstitute.org) (Supplementary Table S2). Publicly available genomes were assembled from a list of 13,067 L4 genomes (Stucki et al., 2016) from which those belonging to L4.4 were selected after being identified with KvarQ (n = 401) (Supplementary Figure S1). Country and year of isolation were obtained from the NCBI BioSample database. Genomes identified as low or mixed coverage by KvarQ, missing country data, known cross-sectional and longitudinal isolates from the same patient were excluded, and if more than one genome sequence was available for a sample only the first listed was used (n = 139 genomes retained). Literature and GMTV database (Chernyaeva et al., 2014) searches, coupled with screening using KvarQ, were also performed to identify additional L4.4 genomes (n = 78). BLAST searches identified eight previously sequenced Canadian SUMu strains (Pepperell et al., 2013) belonging to the L4.4 sublineage. Fastq data was unavailable for these genomes therefore new Canadian Mtb sequence data was included [23 new and two recently published genomes (Brynildsrud et al., 2018)]. These isolates were selected to encompass a broad range of collection dates and IS6110-RFLP molecular typing patterns. Canadian Mtb genomes were sequenced using paired-end 250-bp reads on an Illumina HiSeq 2500 using the NexteraTM XT DNA Library Preparation Kit (Illumina Inc., CA, United States) as previously described (Doroshenko et al., 2018). New sequencing data were submitted to the NCBI Nucleotide Archive (PRJNA573497). All 23 new Canadian genomes and 190 publicly available L4.4 genomes passed mapping quality thresholds (depth of coverage > 25X, > 75% reads mapped to the reference genome, and < 10% missing calls in alignments) and were included in downstream analyses.
Reference Genome Alignment and Variant Calling
Reference genome alignment and variant calling was performed using the Reference Guided Assembly Pepperell Lab Pipeline1. Publicly available genomes were downloaded from NCBI using Fastq-dump (v2.5.2). Raw reads were trimmed with TrimGalore! (v0.4.0)2 using quality threshold of 15 and reads less than 20 bp long were discarded. Trimmed reads were mapped to the H37Rv reference genome (NC_000962.3) (Cole et al., 1998) using BWA-MEM (v0.7.12) (Li, 2013). Duplicates were removed using Picard tools (v1.138)3 and local realignment was performed using GATK (v3.4.46) (DePristo et al., 2011). Mapping quality was assessed using Qualimap (v2.2.1) (Garcia-Alcalde et al., 2012). Genomes were excluded if the depth of coverage was < 25X or if < 75% of trimmed reads mapped to the reference genome. Variants were called using Pilon (v1.16) (Walker et al., 2014) using a minimum depth threshold of 10, base quality threshold of 20 and mapping quality threshold of 40. VCF files generated by Pilon were converted to FASTA format using in house scripts that treat ambiguous calls and deletions as missing data (pilonVCFtoFasta.py).
Bases in repetitive regions of the Mtb genome (Supplementary Table S4) were masked using Bedtools (v2.18) (Quinlan and Hall, 2010) and removed from FASTA sequences prior to alignment. Variant sites were extracted from concatenated whole genome alignments using SNP-sites (v2.3.2) (Page et al., 2016). Genomes with missing data at > 10% of sites were excluded from further analyses and only sites where at least 90% of isolates had high quality base calls were included in phylogenetic and molecular dating analyses. VCF and bam files were manually examined for the presence of the DS6Quebec deletion using SAMtools tview (v1.2) (Li et al., 2009) and Artemis (v16.0.0) (Rutherford et al., 2000) (positions 1987457 to 1998849) (Nguyen et al., 2004).
Maximum Likelihood Phylogenetic Inference
Maximum likelihood trees were inferred using PhyML 3.1 (v3.1) (Guindon and Gascuel, 2003) with x1000 bootstrap replicates using the GTR substitution model as this was the best fitting model based on the Bayesian information criterion in jmodeltest2 (v2) (Darriba et al., 2012). A phylogeny of 23 New Zealand L4.4.1.1 sublineage genomes was inferred from a 345 bp whole-genome SNP alignment. Pairwise SNP distances were calculated from these variant sites using the poppr:bitwise.dist (v2.8.1) package in R (Kamvar et al., 2014) using the “missing_match = T” option to count sites with missing data as matching. A 9024 bp SNP alignment was used to infer a global L4.4 phylogeny, this included all high-quality New Zealand and global L4.4 genomes (n = 236, Supplementary Table S2) and H37Rv. The R-package PopGenome (v2.6.1) (Pfeifer et al., 2014) was used to calculate pairwise fixation indices (FST) from variant sites to estimate population separation between lineages, specifying groups by lineage as determined by KvarQ.
Bayesian Phylogenetic Analyses
Bayesian evolutionary analysis of the L4.4.1.1 sublineage was performed in BEAST2 (Bouckaert et al., 2014) using 3161 variant sites extracted from a 3949977 bp alignment of 117 L4.4.1.1 genomes with known year of isolation (Supplementary Table S3). XML-input files were manually modified to specify the number of invariant sites as calculated by scaling the number of non-SNP sites in the full alignment by the frequency of each base.
Assessment of Temporal Signal for Tip-Based Calibration
The molecular clock was calibrated using tip dates covering a 26-year period (1987–2013). To determine if the temporal signal was sufficient for accurate molecular dating, the dataset was assessed using root-to-tip regression and date randomization (Supplementary Figure S5). A maximum likelihood tree was first constructed in PhyML and then Tempest (v1.5) (Rambaut et al., 2016) was used to root the phylogeny and determine root-to-tip distance for regression analysis against tip date, revealing a modest temporal signal in the data (R2 = 0.229). The CS clade sample subset (n = 47) showed weaker temporal signal than the full L4.4.1.1 dataset (R2 = 0.139). To further validate the temporal signal, sampling dates were randomized 20 times and analyzed with BEAST2 (v2.4) using a strict clock and constant demographic model with the same parameters for the random and real dates. Estimates of the substitution rate and TMRCA showed no overlap in the 95% HPD between the real and randomized dates, indicating that the data contains sufficient temporal signal for tip-based calibration.
Molecular Dating
Mutation rates and divergence times were estimated using MCMC sampling in BEAST2 (v2.4) with the BEAGLE library (Ayres et al., 2012). Analyses were performed using the GTR substitution model, strict and relaxed molecular clocks [uncorrelated relaxed clock with a log-normal distribution (UCLD)] (Drummond et al., 2006), and coalescent constant, exponential and Bayesian skyline (Drummond et al., 2005) demographic models. To correctly place the root as determined with high confidence bootstrap support in the maximum likelihood phylogeny, two monophyletic taxon sets were created to ensure the most deeply rooted L4.4.1.1 clade was placed as an outgroup. Uniform prior distributions were defined for the substitution rate (1 × 10–10–1 × 10–6 sub/site/yr) and effective population size (upper bound of 1 × 1010). For the Bayesian skyline model, the Jeffrey’s (1/X) prior was deselected for the population size parameter as this an improper prior and therefore unsuitable for model evaluation using path sampling. Default priors were used for the remaining parameters. To estimate posterior distributions, three independent chains were run for 100–350 million states sampling every 10000 states. The first 10% of states were discarded as burn-in and chains were assessed for convergence and sufficient mixing (effective sample size > 200 for all parameters) (Supplementary Figure S8). Samples from the three independent chains were combined and parameter estimation based on the combined chain. Median estimates are reported unless otherwise specified. The maximum clade credibility (MCC) tree was estimated from combined tree samples in TreeAnnotator (Supplementary Figures S6, S7).
The performance of various clock and demographic models was evaluated by path sampling analysis in BEAST2 (v2.5) (Lartillot and Philippe, 2006). For each model, 100 path steps were specified using the proportions of a β(0.3, 1.0) distribution and two separate runs were performed per model to check for consistency. The MCMC was also run in the absence of data to sample prior distributions for each model. Comparison of marginal posterior and prior distributions showed a strong signal from the data indicating our results are just not an artifact reflecting the prior (Supplementary Figure S9). The effect of the prior on parameter estimation was also examined by using different upper bounds and the default 1/X prior for the effective population size. Congruent rate and date estimates were obtained when the varying prior parameters on population size demonstrating the robustness of our estimates to this prior specification (Supplementary Figure S10).
Phylogeographic Inference
Ancestral reconstruction was performed using BEAST2 (v2.4), with UN region for each isolate modeled as a discrete trait. Analyses were performed using the GTR model of nucleotide substitution, a strict molecular clock with the estimated substitution rate of 6.28 × 10–8 sub/site/yr and BSP demographic models. Migration rates over time were inferred from an MCC tree. As described in O’Neill et al. (2019), migration events were defined as a change in the most probable reconstructed state from parent to child node. Only nodes with a posterior probability > 80% were considered. Median heights of the parent and child nodes were treated as the range of time in which a migration event could occur. Migration rates through time were inferred by summing the number of migration events during each year of the phylogeny, divided by the total number of branches in existence during each year of the phylogeny. The Bayesian stochastic search variable selection method (BSSVS) (Lemey et al., 2009) implemented in BEAST2 was used to identify well-supported migration rates between UN regions in the phylogeographic analyses. SpreaD3 (v0.9.7rc) (Vrancken et al., 2016) was used to calculate Bayes factor for each pairwise rate.
Data Availability Statement
New genome sequencing data generated for this study were deposited in the NCBI BioProject database (IDs: PRJNA356104 and PRJNA573497). Individual accession numbers for genomes analyzed in this study are given in Supplementary Tables S1–S3. The reference guided assembly Pepperell Lab Pipeline is available at GitHub (https://github.com/pepperell-lab/RGAPepPipe).
Author Contributions
VA, CP, GC, and RC conceived and designed the study. CM performed the phylogenetic and molecular dating analyses and wrote the manuscript. AS performed the phylogeographic inference. DB, SG, and CM compiled the global dataset. NK and SR provided bacterial isolates. HA sequenced isolates. RO’T and SSG provided sequencing data. CM and AS prepared the figures. VA, CP, DB, and SG provided critical comments about the manuscript. All authors reviewed the manuscript.
Funding
CM was supported by the Maurice Wilkins Centre Ph.D. Scholarship. CP was funded by the National Institutes of Health/National Institute of Allergy and Infectious Diseases (Grant Number 1R01AI113287-01A1). HA was supported by the New Zealand Health Research Council (Grant Numbers 15/648 and 18/024), and the New Zealand Lottery Health Research Grant (Grant Number 353021). RO’T and SSG were funded by the Royal Hobart Hospital Research Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Dr. Jill Sherwood, ESR (The Institute of Environmental Science and Research, New Zealand), for providing public health data.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02778/full#supplementary-material
Footnotes
- ^ https://github.com/pepperell-lab/RGAPepPipe
- ^ https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
- ^ https://broadinstitute.github.io/picard/
References
Aung, H. L., Tun, T., Moradigaravand, D., Koser, C. U., Nyunt, W. W., Aung, S. T., et al. (2016). Whole-genome sequencing of multidrug-resistant Mycobacterium tuberculosis isolates from Myanmar. J. Glob. Antimicrob. Resist. 6, 113–117. doi: 10.1016/j.jgar.2016.04.008
Ayres, D. L., Darling, A., Zwickl, D. J., Beerli, P., Holder, M. T., Lewis, P. O., et al. (2012). BEAGLE: an application programming interface and high-performance computing library for statistical phylogenetics. Syst. Biol. 61, 170–173. doi: 10.1093/sysbio/syr100
Baussano, I., Williams, B. G., Nunn, P., Beggiato, M., Fedeli, U., and Scano, F. (2010). Tuberculosis incidence in prisons: a systematic review. PLoS Med. 7:e1000381. doi: 10.1371/journal.pmed.1000381
Bjorn-Mortensen, K., Soborg, B., Koch, A., Ladefoged, K., Merker, M., Lillebaek, T., et al. (2016). Tracing Mycobacterium tuberculosis transmission by whole genome sequencing in a high incidence setting: a retrospective population-based study in East Greenland. Sci. Rep. 6:33180. doi: 10.1038/srep33180
Bouckaert, R., Heled, J., Kuhnert, D., Vaughan, T., Wu, C. H., Xie, D., et al. (2014). BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 10:e1003537. doi: 10.1371/journal.pcbi.1003537
Brudey, K., Driscoll, J. R., Rigouts, L., Prodinger, W. M., Gori, A., Al-Hajoj, S. A., et al. (2006). Mycobacterium tuberculosis complex genetic diversity: mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol. 6:23. doi: 10.1186/1471-2180-6-23
Bryant, J. M., Harris, S. R., Parkhill, J., Dawson, R., Diacon, A. H., van Helden, P., et al. (2013a). Whole-genome sequencing to establish relapse or re-infection with Mycobacterium tuberculosis: a retrospective observational study. Lancet Respir. Med. 1, 786–792. doi: 10.1016/S2213-2600(13)70231-5
Bryant, J. M., Schurch, A. C., van Deutekom, H., Harris, S. R., de Beer, J. L., de Jager, V., et al. (2013b). Inferring patient to patient transmission of Mycobacterium tuberculosis from whole genome sequencing data. BMC Infect. Dis. 13:110. doi: 10.1186/1471-2334-13-110
Brynildsrud, O. B., Pepperell, C. S., Suffys, P., Grandjean, L., Monteserin, J., Debech, N., et al. (2018). Global expansion of Mycobacterium tuberculosis lineage 4 shaped by colonial migration and local adaptation. Sci. Adv. 4:eaat5869. doi: 10.1126/sciadv.aat5869
Buckley, H. R., Tayles, N., Halcrow, S. E., Robb, K., and Fyfe, R. (2010). The people of Wairau Bar: a re-examination. J. Pacif. Archaeol. 1, 1–20.
Campbell, I. C. (2011). Worlds Apart: A History of the Pacific Islands. Christchurch: Canterbury University Press.
Casali, N., Nikolayevskyy, V., Balabanova, Y., Harris, S. R., Ignatyeva, O., Kontsevaya, I., et al. (2014). Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat. Genet. 46, 279–286. doi: 10.1038/ng.2878
Chappell, D. A. (1997). Double Ghosts: Oceanian Voyagers on Euroamerican Ships. Armonk, NY: M.E. Sharpe, Inc.
Charbonneau, H., Boleda, M., and Bates, R. L. (1993). The First French Canadians: Pioneers in the St. Lawrence Valley. Newark, NJ: University of Delaware Press.
Chernyaeva, E. N., Shulgina, M. V., Rotkevich, M. S., Dobrynin, P. V., Simonov, S. A., Shitikov, E. A., et al. (2014). Genome-wide Mycobacterium tuberculosis variation (GMTV) database: a new tool for integrating sequence variations and epidemiology. BMC Genomics 15:308. doi: 10.1186/1471-2164-15-308
Clark, M., Riben, P., and Nowgesic, E. (2002). The association of housing density, isolation and tuberculosis in Canadian First Nations communities. Int. J. Epidemiol. 31, 940–945. doi: 10.1093/ije/31.5.940
Clark, T. G., Mallard, K., Coll, F., Preston, M., Assefa, S., Harris, D., et al. (2013). Elucidating emergence and transmission of multidrug-resistant tuberculosis in treatment experienced patients by whole genome sequencing. PLoS One 8:e83012. doi: 10.1371/journal.pone.0083012
Colangeli, R., Arcus, V. L., Cursons, R. T., Ruthe, A., Karalus, N., Coley, K., et al. (2014). Whole genome sequencing of Mycobacterium tuberculosis reveals slow growth and low mutation rates during latent infections in humans. PLoS One 9:e91024. doi: 10.1371/journal.pone.0091024
Cole, S. T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., et al. (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544. doi: 10.1038/31159
Coll, F., McNerney, R., Guerra-Assuncao, J. A., Glynn, J. R., Perdigao, J., Viveiros, M., et al. (2014). A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat. Commun. 5:4812. doi: 10.1038/ncomms5812
Cormier, M., Schwartzman, K., N’Diaye, D. S., Boone, C. E., dos Santos, A. M., Gaspar, J., et al. (2019). Proximate determinants of tuberculosis in indigenous peoples worldwide: a systematic review. Lancet Glob. Health 7, e68–e80. doi: 10.1016/S2214-109X(18)30435-2
Craig, G. M., Daftary, A., Engel, N., O’Driscoll, S., and Ioannaki, A. (2017). Tuberculosis stigma as a social determinant of health: a systematic mapping review of research in low incidence countries. Int. J. Infect. Dis. 56, 90–100. doi: 10.1016/j.ijid.2016.10.011
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9:772. doi: 10.1038/nmeth.2109
De Zoysa, R., Shoemack, P., Vaughan, R., and Vaughan, A. (2001). A prolonged outbreak of tuberculosis in the North Island. N. Z. Publ. Health Rep. 8, 1–3.
DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. doi: 10.1038/ng.806
Doroshenko, A., Pepperell, C. S., Heffernan, C., Egedahl, M. L., Mortimer, T. D., Smith, T. M., et al. (2018). Epidemiological and genomic determinants of tuberculosis outbreaks in First Nations communities in Canada. BMC Med. 16:128. doi: 10.1186/s12916-018-1112-9
Drummond, A. J., Ho, S. Y., Phillips, M. J., and Rambaut, A. (2006). Relaxed phylogenetics and dating with confidence. PLoS Biol. 4:e88. doi: 10.1371/journal.pbio.0040088
Drummond, A. J., Rambaut, A., Shapiro, B., and Pybus, O. G. (2005). Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22, 1185–1192. doi: 10.1093/molbev/msi103
Dunsford, D., Park, J., Littleton, J., Friesen, W., Herda, P., Neuwelt, P., et al. (2011). Better Lives: The Struggle for Health of Transnational Pacific Peoples in New Zealand, 1950-2000. Auckland: University of Auckland.
Eldholm, V., Monteserin, J., Rieux, A., Lopez, B., Sobkowiak, B., Ritacco, V., et al. (2015). Four decades of transmission of a multidrug-resistant Mycobacterium tuberculosis outbreak strain. Nat. Commun. 6:7119. doi: 10.1038/ncomms8119
ESR, (2018). Tuberculosis in New Zealand: Annual Report 2015. Porirua: Institute of Environmental Science and Research Ltd (ESR).
Falush, D., Wirth, T., Linz, B., Pritchard, J. K., Stephens, M., Kidd, M., et al. (2003). Traces of human migrations in Helicobacter pylori populations. Science 299, 1582–1585. doi: 10.1126/science.1080857
Ford, C. B., Shah, R. R., Maeda, M. K., Gagneux, S., Murray, M. B., Cohen, T., et al. (2013). Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat. Genet. 45, 784–790. doi: 10.1038/ng.2656
Foucrier, A. (2005). The French and the Pacific world, 17th-19th Centuries: Explorations, Migrations and Cultural Exchanges. Aldershot: Ashgate Variorum.
Gagneux, S. (2018). Ecology and evolution of Mycobacterium tuberculosis. Nat. Rev. Microbiol. 16:202. doi: 10.1038/nrmicro.2018.8
Gagneux, S., DeRiemer, K., Van, T., Kato-Maeda, M., de Jong, B. C., Narayanan, S., et al. (2006). Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 103, 2869–2873. doi: 10.1073/pnas.0511240103
Garcia-Alcalde, F., Okonechnikov, K., Carbonell, J., Cruz, L. M., Gotz, S., Tarazona, S., et al. (2012). Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics 28, 2678–2679. doi: 10.1093/bioinformatics/bts503
Gautam, S. S., Mac Aogain, M., Bower, J. E., Basu, I., and O’Toole, R. F. (2017). Differential carriage of virulence-associated loci in the New Zealand Rangipo outbreak strain of Mycobacterium tuberculosis. Infect. Dis. 49, 680–688. doi: 10.1080/23744235.2017.1330553
Guerra-Assunção, J. A., Crampin, A., Houben, R., Mzembe, T., Mallard, K., Coll, F., et al. (2015a). Large-scale whole genome sequencing of M. tuberculosis provides insights into transmission in a high prevalence area. eLife 4:e05166. doi: 10.7554/eLife.05166
Guerra-Assunção, J. A., Houben, R. M. G. J., Crampin, A. C., Mzembe, T., Mallard, K., Coll, F., et al. (2015b). Recurrence due to relapse or reinfection with Mycobacterium tuberculosis: a whole-genome sequencing approach in a large, population-based cohort with a high HIV infection prevalence and active follow-up. J. Infect. Dis. 211, 1154–1163. doi: 10.1093/infdis/jiu574
Guindon, S., and Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. doi: 10.1080/10635150390235520
Haines, D. (2010). “Lighting up the World? Empires and Islanders in the Pacific Whaling Industry, 1790-1860,” in Maritime History as Global History, eds M. Fusaro and A. Polónia (Oxford: Liverpool University Press), 159–176. doi: 10.2307/j.ctt21pxjhv.11
Hershberg, R., Lipatov, M., Small, P. M., Sheffer, H., Niemann, S., Homolka, S., et al. (2008). High functional diversity in Mycobacterium tuberculosis driven by genetic drift and human demography. PLoS Biol. 6:e311. doi: 10.1371/journal.pbio.0060311
Hirsh, A. E., Tsolaki, A. G., DeRiemer, K., Feldman, M. W., and Small, P. M. (2004). Stable association between strains of Mycobacterium tuberculosis and their human host populations. Proc. Natl. Acad. Sci. U.S.A. 101, 4871–4876. doi: 10.1073/pnas.0305627101
Ho, T. B., Robertson, B. D., Taylor, G. M., Shaw, R. J., and Young, D. B. (2000). Comparison of Mycobacterium tuberculosis genomes reveals frequent deletions in a 20 kb variable region in clinical isolates. Yeast 17, 272–282. doi: 10.1002/1097-0061(200012)17:4<272::AID-YEA48>3.0.CO;2-2
Holt, K. E., McAdam, P., Thai, P. V. K., Thuong, N. T. T., Ha, D. T. M., Lan, N. N., et al. (2018). Frequent transmission of the Mycobacterium tuberculosis Beijing lineage and positive selection for the EsxW Beijing variant in Vietnam. Nat. Genet. 50, 849–856. doi: 10.1038/s41588-018-0117-9
Innis, H. A. (1999). The Fur Trade in Canada: An Introduction to Canadian Economic History. Toronto: University of Toronto Press.
Kamvar, Z. N., Tabima, J. F., and Grunwald, N. J. (2014). Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281. doi: 10.7717/peerj.281
Lange, R. (1984). Plagues and pestilence in polynesia - the 19th-century cook Islands experience. Bull. Hist. Med. 58, 325–346.
Lartillot, N., and Philippe, H. (2006). Computing Bayes factors using thermodynamic integration. Syst. Biol. 55, 195–207. doi: 10.1080/10635150500433722
Lemey, P., Rambaut, A., Drummond, A. J., and Suchard, M. A. (2009). Bayesian phylogeography finds its roots. PLoS Comput. Biol. 5:e1000520. doi: 10.1371/journal.pcbi.1000520
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv [Preprint].
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Mac Aogáin, M., Gautam, S. S., Bower, J. E., Basu, I., and O’Toole, R. F. (2016). Draft genome sequence of a New Zealand Rangipo strain of Mycobacterium tuberculosis. Genome Announc. 4:e00657-16. doi: 10.1128/genomeA.00657-16
MacLean, F. S. (1964). Challenge for Health: A History of Public Health in New Zealand. Wellington: Government Printer.
McElnay, C., Thornley, C., and Armstrong, R. (2004). A community and workplace outbreak of tuberculosis in Hawke’s Bay in 2002. N. Z. Med. J. 117, U1019.
Moodley, Y., Linz, B., Yamaoka, Y., Windsor, H. M., Breurec, S., Wu, J. Y., et al. (2009). The peopling of the Pacific from a bacterial perspective. Science 323, 527–530. doi: 10.1126/science.1166083
Mulholland, C. V., Ruthe, A., Cursons, R. T., Durrant, R., Karalus, N., Coley, K., et al. (2017). Rapid molecular diagnosis of the Mycobacterium tuberculosis Rangipo strain responsible for the largest recurring TB cluster in New Zealand. Diagn. Microbiol. Infect. Dis. 88, 138–140. doi: 10.1016/j.diagmicrobio.2017.03.012
Nguyen, D., Brassard, P., Menzies, D., Thibert, L., Warren, R., Mostowy, S., et al. (2004). Genomic characterization of an endemic Mycobacterium tuberculosis strain: evolutionary and epidemiologic implications. J. Clin. Microbiol. 42, 2573–2580. doi: 10.1128/JCM.42.6.2573-2580.2004
Öhberg, A. (1955). Russia and the World Market in the Seventeenth Century. Scand. Econ. Hist. Rev. 3, 123–162. doi: 10.1080/03585522.1955.10411474
O’Neill, M. B., Shockey, A. C., Zarley, A., Aylward, W., Eldholm, V., Kitchen, A., et al. (2019). Lineage specific histories of Mycobacterium tuberculosis dispersal in Africa and Eurasia. Mol. Ecol. 28, 3241–3256. doi: 10.1101/210161
Osman, D. A., Phelippeau, M., Drancourt, M., and Musso, D. (2017). Diversity of Mycobacterium tuberculosis lineages in French Polynesia. J. Microbiol. Immunol. Infect. 50, 199–206. doi: 10.1016/j.jmii.2015.05.018
Page, A. J., Taylor, B., Delaney, A. J., Soares, J., Seemann, T., Keane, J. A., et al. (2016). SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. Genomics 2:e000056. doi: 10.1099/mgen.0.000056
Pepperell, C. S., Casto, A. M., Kitchen, A., Granka, J. M., Cornejo, O. E., Holmes, E. C., et al. (2013). The role of selection in shaping diversity of natural M. tuberculosis populations. PLoS Pathog. 9:e1003543. doi: 10.1371/journal.ppat.1003543
Pepperell, C. S., Granka, J. M., Alexander, D. C., Behr, M. A., Chui, L., Gordon, J., et al. (2011). Dispersal of Mycobacterium tuberculosis via the Canadian fur trade. Proc. Natl. Acad. Sci. U.S.A. 108, 6526–6531. doi: 10.1073/pnas.1016708108
Pfeifer, B., Wittelsburger, U., Ramos-Onsins, S. E., and Lercher, M. J. (2014). PopGenome: an efficient Swiss army knife for population genomic analyses in R. Mol. Biol. Evol. 31, 1929–1936. doi: 10.1093/molbev/msu136
Pool, I. (1991). Te Iwi Maori: A New Zealand Population, Past, Present and Projected. Auckland: Auckland University Press.
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Rambaut, A., Lam, T. T., Max Carvalho, L., and Pybus, O. G. (2016). Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2:vew007. doi: 10.1093/ve/vew007
Rich, E. E. (1955). Russia and the colonial fur trade. Econ. Hist. Rev. 7, 307–328. doi: 10.1111/j.1468-0289.1955.tb01533.x
Roetzer, A., Diel, R., Kohl, T. A., Ruckert, C., Nubel, U., Blom, J., et al. (2013). Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: a longitudinal molecular epidemiological study. PLoS Med. 10:e1001387. doi: 10.1371/journal.pmed.1001387
Rutherford, K., Parkhill, J., Crook, J., Horsnell, T., Rice, P., Rajandream, M.-A., et al. (2000). Artemis: sequence visualization and annotation. Bioinformatics 16, 944–945. doi: 10.1093/bioinformatics/16.10.944
Steiner, A., Stucki, D., Coscolla, M., Borrell, S., and Gagneux, S. (2014). KvarQ: targeted and direct variant calling from fastq reads of bacterial genomes. BMC Genomics 15:881. doi: 10.1186/1471-2164-15-881
Stevens, K., and Wanhalla, A. (2017). Intimate relations: kinship and the economics of shore whaling in Southern New Zealand, 1820-1860. J. Pacif. Hist. 52, 135–155. doi: 10.1080/00223344.2017.1366820
Stucki, D., Brites, D., Jeljeli, L., Coscolla, M., Liu, Q., Trauner, A., et al. (2016). Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat. Genet. 48, 1535–1543. doi: 10.1038/ng.3704
Tollefson, D., Bloss, E., Fanning, A., Redd, J. T., Barker, K., and McCray, E. (2013). Burden of tuberculosis in indigenous peoples globally: a systematic review. Int. J. Tuberc. Lung Dis. 17, 1139–1150. doi: 10.5588/ijtld.12.0385
Tsolaki, A. G., Gagneux, S., Pym, A. S., Goguet de la Salmoniere, Y. O., Kreiswirth, B. N., Van Soolingen, D., et al. (2005). Genomic deletions classify the Beijing/W strains as a distinct genetic lineage of Mycobacterium tuberculosis. J. Clin. Microbiol. 43, 3185–3191. doi: 10.1128/JCM.43.7.3185-3191.2005
United Nations and Economic Social Commission for Asia and the Pacific, (1985). The Population of New Zealand: Country Monograph Series, No. 12. New York, NY: United Nations.
Vrancken, B., Baele, G., Lemey, P., Bielejec, F., Suchard, M. A., and Rambaut, A. (2016). SpreaD3: interactive visualization of spatiotemporal history and trait evolutionary processes. Mol. Biol. Evol. 33, 2167–2169. doi: 10.1093/molbev/msw082
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Walker, T. M., Ip, C. L., Harrell, R. H., Evans, J. T., Kapatai, G., Dedicoat, M. J., et al. (2013). Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect. Dis. 13, 137–146. doi: 10.1016/S1473-3099(12)70277-3
Yen, S., Bower, J. E., Freeman, J. T., Basu, I., and O’Toole, R. F. (2013). Phylogenetic lineages of tuberculosis isolates in New Zealand and their association with patient demographics. Int. J. Tuberc. Lung Dis. 17, 892–897. doi: 10.5588/ijtld.12.0795
Keywords: tuberculosis, pathogen, phylodynamics, phylogeography, indigenous people, Pacific, New Zealand
Citation: Mulholland CV, Shockey AC, Aung HL, Cursons RT, O’Toole RF, Gautam SS, Brites D, Gagneux S, Roberts SA, Karalus N, Cook GM, Pepperell CS and Arcus VL (2019) Dispersal of Mycobacterium tuberculosis Driven by Historical European Trade in the South Pacific. Front. Microbiol. 10:2778. doi: 10.3389/fmicb.2019.02778
Received: 02 September 2019; Accepted: 14 November 2019;
Published: 04 December 2019.
Edited by:
Daniel Yero, Autonomous University of Barcelona, SpainReviewed by:
Joseph Crispell, University College Dublin, IrelandTakayuki Wada, Nagasaki University, Japan
Copyright © 2019 Mulholland, Shockey, Aung, Cursons, O’Toole, Gautam, Brites, Gagneux, Roberts, Karalus, Cook, Pepperell and Arcus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caitlin S. Pepperell, Y3NwZXBwZXJAbWVkaWNpbmUud2lzYy5lZHU=; Vickery L. Arcus, dmljLmFyY3VzQHdhaWthdG8uYWMubno=