
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 07 November 2019
Sec. Virology
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.02461
 Mahmuda Akter1*
Mahmuda Akter1* Nathan Brown2
Nathan Brown2 Martha Clokie2
Martha Clokie2 Mahmuda Yeasmin1Tokee M. Tareq1
Mahmuda Yeasmin1Tokee M. Tareq1 Ramani Baddam1
Ramani Baddam1 Muhammad A. K. Azad1,3Amar N. Ghosh4
Muhammad A. K. Azad1,3Amar N. Ghosh4 Niyaz Ahmed1Kaisar A. Talukder1,5
Niyaz Ahmed1Kaisar A. Talukder1,5Shigellosis, caused by Shigella boydii type 1, is understudied and underreported. For 3 years, GEMS study identified 5.4% of all Shigella as S. boydii. We showed the prevalent serotypes of S. boydii in Bangladesh and phage-based diagnosis of S. boydii type 1, a rapid and low-cost approach. Previously typed 793 clinical S. boydii strains were used for serotype distribution. Twenty-eight environmental water samples were collected for isolation of Shigella phages. Forty-eight serotypes of Shigella and other enteric bacteria were used for testing the susceptibility to phage MK-13. Electron microscopy, restriction enzyme analysis, whole genome sequencing (WGS), and annotation were performed for extensive characterization. S. boydii type 1 is the second most prevalent serotype among 20 serotypes of S. boydii in Bangladesh. We isolated a novel phage, MK-13, which specifically lyses S. boydii type 1, but doesn’t lyse other 47 serotypes of Shigella or other enteric bacteria tested. The phage belongs to the Myoviridae family and distinct from other phages indicated by electron microscopy and restriction enzyme analysis, respectively. MK-13 genome consists of 158 kbp of circularly permuted double-stranded DNA with G + C content of 49.45%, and encodes 211 open reading frames including four tRNA-coding regions. The genome has 98% identity with previously reported phage, ΦSboM-AG3, reported to have a broader host range infecting most of the S. boydii and other species of Shigella tested. To our knowledge, MK-13 is the first phage reported to be used as a diagnostic marker to detect S. boydii type 1, especially in remote settings with limited laboratory infrastructure.
Shigellosis is an important cause of morbidity and mortality among preschool-aged and older children, and adults (Mani et al., 2016). Two recent studies, MAL-ED and GEMS, conducted in Bangladesh and other countries, identified Shigella as one of the four leading pathogens (Kotloff, 2017; Kotloff et al., 2017). GEMS identified Shigella and pathogenic Escherichia coli as the cause of moderate-to-severe diarrhea in children <5 years in Bangladesh among other countries (Kotloff et al., 2013). Worldwide, the annual burden of Shigella is estimated to be 164.7 million cases, with 163.2 million from developing countries, resulting in 1.1 million deaths, and 69% of which are in children <5 years of age (Kotloff et al., 1999). The exact demography is unclear, however, as a previous study conducted in six Asian countries showed that the burden of shigellosis was 2.1 per 1000 population per year for all ages, and for children under 5 years, it was 13.2 per 1000 population every year (von Seidlein et al., 2006).
Shigella is highly infectious because 10 Shigella colonies are enough to cause disease (DuPont et al., 1989). There are four species of Shigella, based on biochemical and serological properties: S. dysenteriae, S. flexneri, S. boydii, and S. sonnei (Livio et al., 2014). These species are further classified into 15, 23 (including subtypes), 20, and 1 serotype, respectively (Talukder and Azmi, 2012; Shahnaij et al., 2018). S. boydii has been reported less frequently worldwide compared to other Shigella species (Bratoeva et al., 1992; Ranjbar et al., 2008) and there are very few published studies available on few serotypes of S. boydii (Kania et al., 2016). GEMS showed that, over 3 years, 5.4% (61/1130) of all Shigella were identified as S. boydii (Livio et al., 2014). Although this is a small contribution compared to the other three Shigella species, S. boydii still makes up a significant component of the overall Shigella burden (Baker et al., 2015).
Shigella spp. are currently identified by biochemical tests and suspected colonies are confirmed by serotyping (Grimont et al., 2007) using commercially available antisera. Most Shigella O-antigens serologically cross-react with O-antigens of some E. coli strains (Liu et al., 2008), making identification difficult. Both demonstrate similar biochemical properties and can cause dysentery using the same mechanism (Ud-Din and Wahid, 2014). The 16S rRNA sequence similarities of S. flexneri, S. boydii, and S. sonnei with E. coli were reported to be 99.8, 99.7, and 99.9%, respectively (Fukushima et al., 2002). S. boydii and S. dysenteriae were considered physiologically similar, but differed biochemically by the mannitol test (Muthuirulandi Sethuvel et al., 2017). Although various molecular methods have been proposed in the past years, discriminating between species is still difficult (Pettengill et al., 2015; Muthuirulandi Sethuvel et al., 2017). Recently developed whole-genome sequence (WGS) based methods showed better discrimination between closely related species and provided clinically relevant information (Hasman et al., 2014). The k-mer-based identification approach derived from WGS data effectively differentiated Shigella from E. coli and accurately provided information on phylogenetic relationship (Chattaway et al., 2017). However, these are no cost-effective methods and those that do exist require specialized training and equipment.
The WHO designated Shigella as a priority area for research and development of new drugs (World Health Organization, 2017). To better understand different serotypes of Shigella, it is important to type or distinguish them accurately as immunity to Shigella is serotype specific (Mani et al., 2016). Shigella serotype-specific lytic phages are useful for typing Shigella at serotype level and thus constitute a powerful diagnostic tool. Phage typing is a rapid, consistent, and reproducible technique requiring minimal specialized equipment (Anderson and Williams, 1956; Adams, 1959; Ahmed et al., 1995).
The purpose of this work was to show the prevalence of 20 different serotypes of S. boydii in Bangladesh and diagnosis of S. boydii serotypes without using antisera. In this study, we have isolated phage MK-13 and evaluated its use for diagnosis of S. boydii type 1, one of the prevalent serotypes of S. boydii in Bangladesh. This is the first report of the isolation and whole genome sequence analysis of a phage that lyse only strains of S. boydii type 1 but not other 19 serotypes of S. boydii or other species of Shigella. So far, no serotype-specific phage has been reported for diagnosis of S. boydii type 1.
A total of 6475 strains of different serotypes of Shigella were isolated and identified at the Enteric and Food Microbiology Laboratory from patients of all ages with diarrhea attending the Dhaka treatment center of the International Centre for Diarrhoeal Disease Research, Bangladesh (icddr,b) hospital and diagnostic unit between 1999 and 2015 following the standard microbiological, biochemical, and serological methods (World Health Organization, 1987; Talukder et al., 2002). Of 6475 strains, 877 were found as S. boydii. From 877, 793 strains were serotyped and further analyzed in this study (Supplementary Table S1). It should be mentioned here that all the samples couldn’t be collected in 2014 and 2015, and therefore, the isolation rate is low in these 2 years. All the strains were stored in Tryptic Soy Broth (TSB) with 0.3% yeast extract (TSBY) including 15% glycerol at −80°C in icddr,b bio repository facility.
A total of 342 strains of clinical Shigella, E. coli, Salmonella, Vibrio, Klebsiella, Proteus, and Yersinia of which 294 strains belonged to 48 different serotypes of Shigella randomly selected from different years and 48 other enteric bacteria were used for testing phage susceptibility (Table 1 and Supplementary Table S2). These strains were collected from the Enteric and Food Microbiology laboratory of icddr,b, Dhaka, Bangladesh. S. boydii type 1, K-473 was used as host strain for detection of phage MK-13.
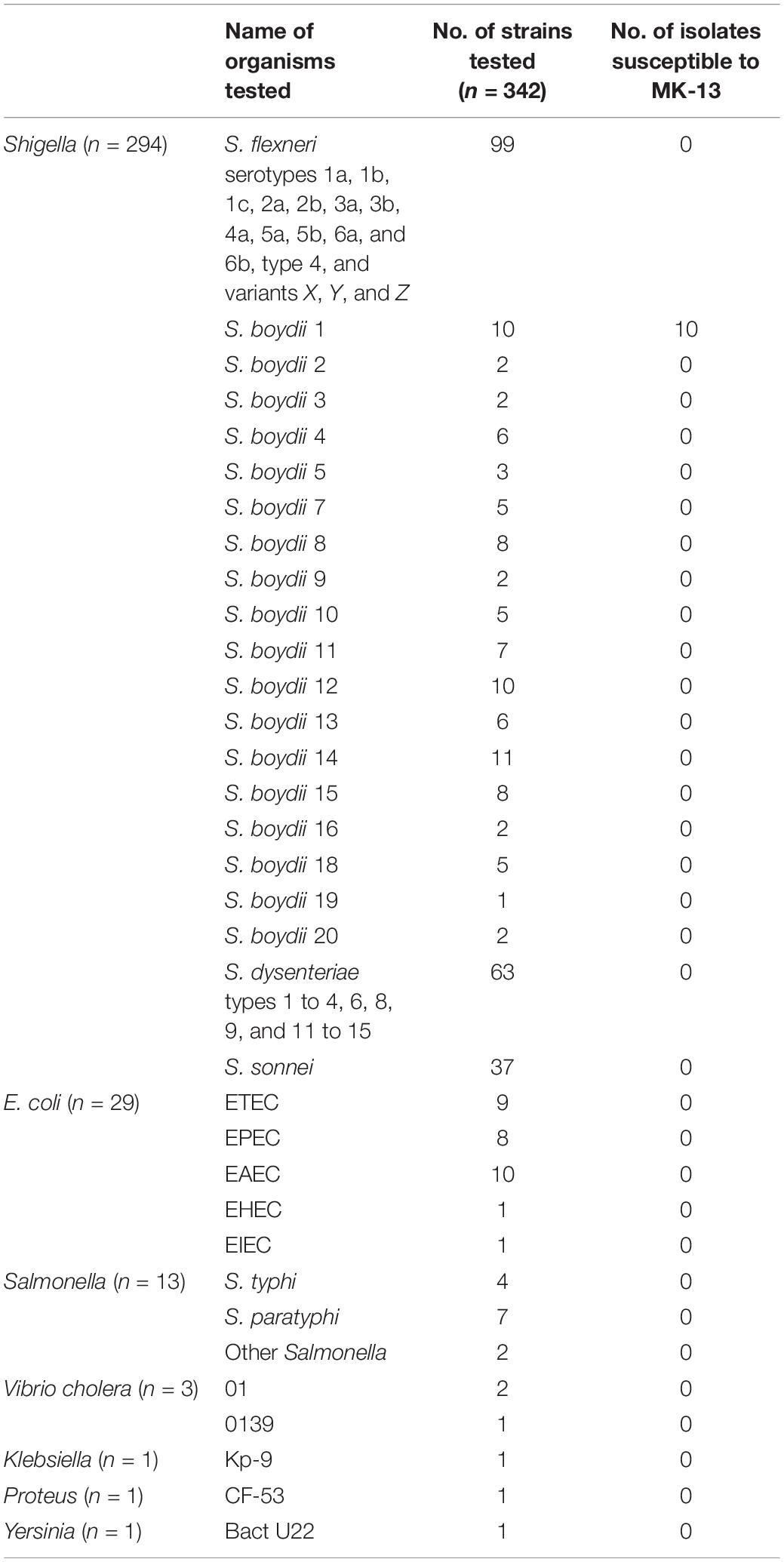
Table 1. Strains used for testing the susceptibility of MK-13.
All enteric bacteria were identified and isolated following the standard microbiological and biochemical methods (World Health Organization, 1987). Shortly, stool samples were first sub-cultured on different agar media such as MacConkey agar, Shigella-Salmonella (SS) agar, xylose lysine deoxycholate (XLD) agar, thiosulfate citrate bile salts sucrose (TCBS), taurocholate tellurite gelatin agar (TTGA), and incubated at 37°C overnight for all plates. The suspected colonies were then confirmed by short biochemical tests, oxidase reaction, and serology. Different E. coli pathotypes were identified according to the protocol standardized by the MAL-ED study investigators (Houpt et al., 2014). All the isolated strains were stored in TSBY including 15% glycerol at −80°C in icddr,b bio repository facility.
Shigella isolates were serotyped using two kits: a commercially available antisera-kit (Denka Seiken, Tokyo, Japan) and type- and group-specific monoclonal antibody reagents (Reagensia AB, Stockholm, Sweden). Strains were sub-cultured onto MacConkey agar (Difco) plates, and after 16–18 h of incubation, slide agglutination tests were performed following Shigella flexneri serotyping scheme as described previously (Talukder and Azmi, 2012). For the other three species, the manufacturer’s guidelines were followed.
Twenty-eight water samples were collected in sterile containers from different rivers, canals, and lakes in and around Dhaka City, Bangladesh, between February 2006 and February 2007 for isolation of Shigella-specific phages. Within 3 h of collection, water samples were mixed with equal volume of 2 × Luria-Bertani (LB) broth and overnight culture of Shigella (one strain of S. flexneri per serotype 2a, 2b, 3a, 1c; S. boydii 1, 12; S. dysenteriae 1, and S. sonnei) and incubated overnight at 37°C. The suspensions were then centrifuged at low speed, carefully transferred to another tube, and filtered through 0.22-μm pore-sized filters (Millipore). Filtered supernatants were then screened for presence of phages. Briefly, 500 μl of logarithmic-phase cells of the host strain in LB broth was mixed with 3.5-ml aliquots of soft agar (LB broth containing 0.8% Bactoagar, Difco), and the mixture was overlaid on previously prepared LB agar plates. Filtered supernatants were then inoculated on the plates and incubated for 16–17 h at 37°C. A sample was counted as positive for phages when plaque was observed on the bacterial lawn (Sambrook et al., 1989).
The samples positive for phages were then serially diluted, mixed with soft agar, and overlaid on hard bottom agar and incubated for 16–17 h at 37°C as described above. Next day, lytic plaque zones were collected by cutting the soft layer from the plate using sterile cut tips and placing them separately in 500 μl of logarithmic-phase cells, vortexed, and incubated at 37°C for 20 min. Then 3 ml of LB broth was added and incubated overnight at 37°C. Then, the culture was centrifuged at 10,000 × g for 20 min, and the supernatant was filtered through 0.22-μm pore-sized filters (Millipore) to exclude bacteria. The number of phage particles in the filtered supernatant was determined by testing serial dilutions of the supernatant with the host strain. This method was repeated for three successive times to obtain purified phages.
After purification, the phages were tested using 48 different serotypes of Shigella and then the selected phages were tested on other enteric bacteria (Table 1). Briefly, an overnight culture of the host strain was diluted 1:100 in fresh LB broth and grown at 37°C for 4 h. From this, 500 μl was added to 3 ml of LB broth and mixed with 3.5-ml aliquots of soft agar, and the mixture was overlaid on previously prepared LB agar plates. Each overlay was allowed to solidify for 15 min. Ten microliters of each purified phage at a titer of 103 PFU/ml was spotted onto the bacterial overlay, dried, and incubated overnight at 37°C. The tests were repeated three times to confirm the results.
A high-titer phage preparation (∼1011 PFU/ml) was obtained using the plate lysis procedure as described previously (Albert et al., 1996). Carbon-coated copper grids were used for morphological studies of bacteriophage. Grids were subjected to glow discharge before negative staining with 2% uranyl acetate. Samples were examined in a FEI Tecnai12BioTwin transmission electron microscope operating at an acceleration voltage of 100 kV and fitted with a SIS Megaview III CCD camera. All measurements were done using analySIS software (Ghosh et al., 2005).
Phage nucleic acid was extracted following a previously described method (Faruque et al., 2003). Briefly, the filtrates were mixed with one-fourth volume of a solution containing 20% polyethylene glycol (PEG-6000) and 10% NaCl. Then, the mixture was centrifuged at 13,000 × g and the pellet was dissolved in a solution containing 20 mM Tris–HCl (pH 7.5), 60 mM KCl, 10 mM MgCl2, and 10 mM NaCl, and digested with DNase I and RNase A at 37°C for 2 h. The solution was then extracted with phenol-chloroform. Ethanol was used to precipitate total nucleic acids, which were suspended in deionized water and purified using the SV Minipreps DNA purification system (Promega, Madison, United States). To initially check for diversity, the isolated phage nucleic acids were digested with EcoRI, XbaI, HindIII, MluI, SalI, and HaeIII (Invitrogen Corporation, Carlsbad, CA) according to the manufacturer’s recommendations (Sambrook et al., 1989) and analyzed by agarose gel electrophoresis following standard procedures.
The phage genome was sequenced at the University of Leicester, United Kingdom, using the Illumina MiSeq platform. The genomic library was prepared using the Illumina Truseq Nano DNA library Preparation Kit as per the manufacturer’s instructions and sequenced with the MiSeq Reagent Kit v3 (600 cycles) to yield 300-bp paired-end short DNA reads.
The quality of the raw sequencing reads was checked using FastQC (Andrews, 2010). The sequencing reads were filtered based on a threshold mean Phred quality score of 20 using skewer (Jiang et al., 2014). Reads were assembled with SPAdes-3.10.1 (Bankevich et al., 2012). Although reads were assembled using multiple k-mer lengths, the longest contig was assembled using k-mers of 127 bp in length and thus used for further analysis. We only considered the longest contig as being the phage genome as the coverage of the other contigs was 5–10 times lower. The phage genome was circularly permuted, resulting in direct repeats at each end. The repeats were identified and trimmed with the apc.pl script written by Joe Fass1. Open reading frames were annotated on the phage genome sequence with PROKKA (Seemann, 2014) and then fine-tuned using the RAST annotation server (Aziz et al., 2008). The COG functional classification was performed by submitting the protein sequences to NCBI Batch CD-search Marchler-Bauer et al. (2015). The visualization of genomic organization is made using DNA plotter (Carver et al., 2008).
Genomic organization of MK-13 phage was visualized using DNA plotter (Guy et al., 2010; R Core Team, 2013). Also, accessory genome-based phylogeny was built as there was no single gene of MK-13 that is shared among all currently available 35 Shigella phage genomes within NCBI (Supplementary Table S3). All the phage genome annotation was done using PROKKA, then using the gff files, core genome analysis was performed using tool Roary (Page et al., 2015) using default parameters. As we are unable to find any single core gene among all the genomes, we adopted the phylogeny based on gene presence–absence matrix. Therefore, we used the phylogeny tree generated by Roary (newick flat file) and visualized in R package “ggtree.” FastTree was used for the phylogeny construction, which infers approximate-maximum-likelihood phylogenetic trees. These accessory genes may or may not be shared between two phages and whether the particular sets of phages have high or low number of shared genes should be a reasonable indicator of their relatedness. The Genbank file of Shigella phage ΦSboM-AG3 was downloaded from NCBI (accession number FJ373894.1). The comparison of this phage against MK-13 was performed using EasyFig (Sullivan et al., 2011).
The MK-13 phage genome is deposited under accession number MK509462 in NCBI Genbank database.
Among 6475 Shigella, 13.54% (877/6475) were identified as S. boydii. Among 793 strains of S. boydii, S. boydii type 12 is the prevalent serotype (27.6%) followed by S. boydii type 1 (11.7%), type 4 (9.2%), type 14 (8.6%), type 18 (7.6%), type 5 (7.3%), type 11 (6.3%), type 8 (4.8%), type 2 (4.2%), type 13 (2.8%), types 15 and 20 (2.4% each), and others are below 2% (Figure 1).
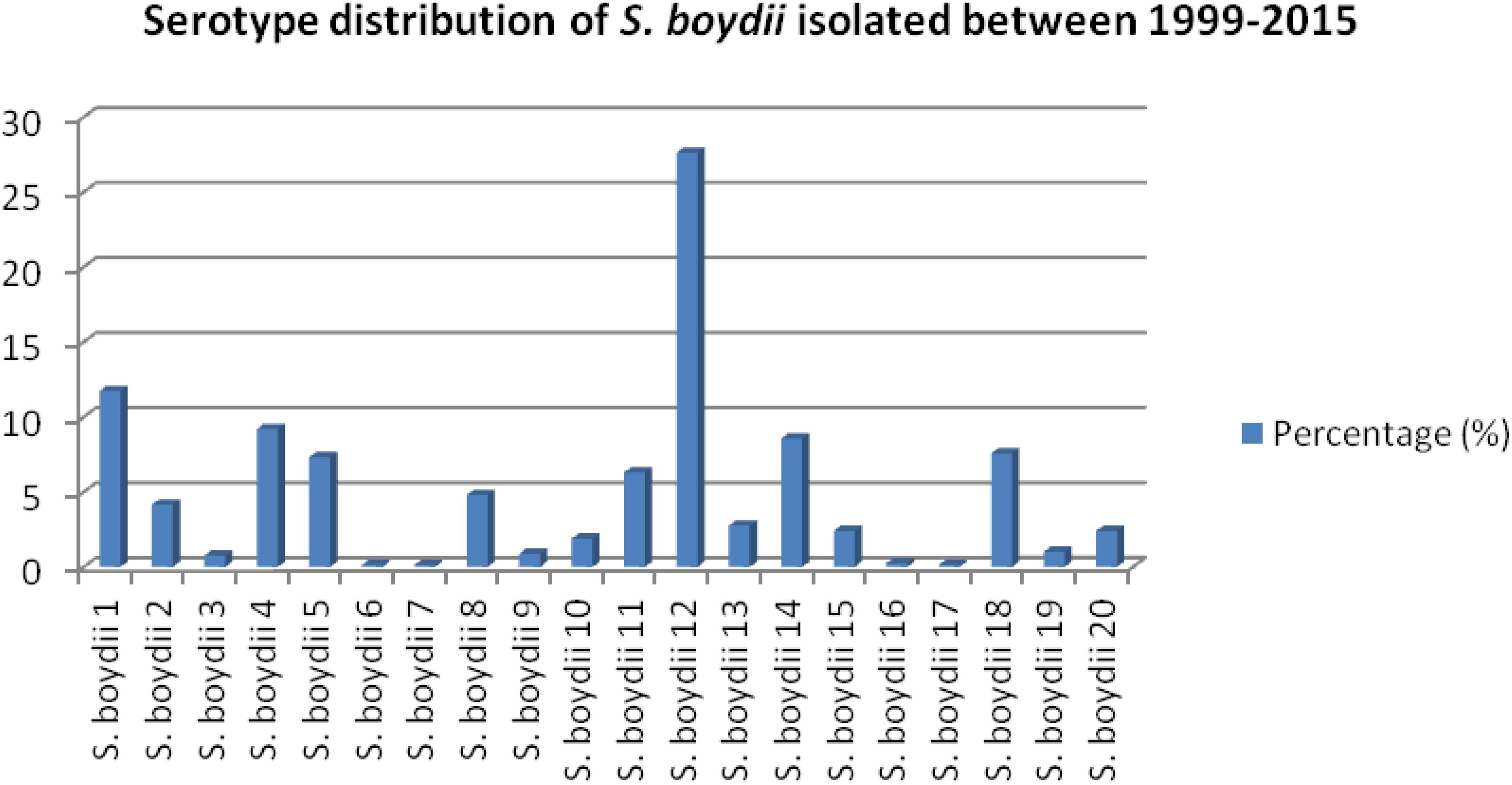
Figure 1. Prevalence of different serotypes of S. boydii in Bangladesh.
From 28 water samples, 84 Shigella-specific phages were isolated. Of these, 15 different types of phages were selected for purification. Among these, only one phage designated as MK-13 was found to be specific for S. boydii type 1, which was selected for further host range characterization.
The purified MK-13 phage lysed all strains of S. boydii type 1 only, but not the other 47 serotypes of Shigella or other enteric bacteria tested (Table 1). Phage MK-13 produced completely clear plaques with outer turbid zone and about 3 mm diameter on a lawn of S. boydii type 1. When grown in LB broth with control host, the phage produced a titer of 109 PFU/ml. Plaques of MK-13 are slightly bigger than most of the other phages tested (Figure 2).

Figure 2. Plaque morphology of MK-13 phage.
Electron microscopic examination of the phage showed that the phage has an icosahedral head, and a long contractile tail (Figure 3). The head is approximately 85 nm in diameter and the contractile tail is approximately 110 nm long and 15 nm in diameter. Therefore, the phage belongs to Myoviridae family.

Figure 3. Electron micrograph showing the morphology of Shigella boydii type 1 specific phage.
MK-13 phage DNA was sensitive to HaeIII, MluI, and HindIII restriction enzymes, but resistant to EcoRI, XbaI, and SalI. This pattern of sensitivity was different from that of DNA from other phages tested. Restriction profile of MK-13 phage DNA is shown in Figure 4.
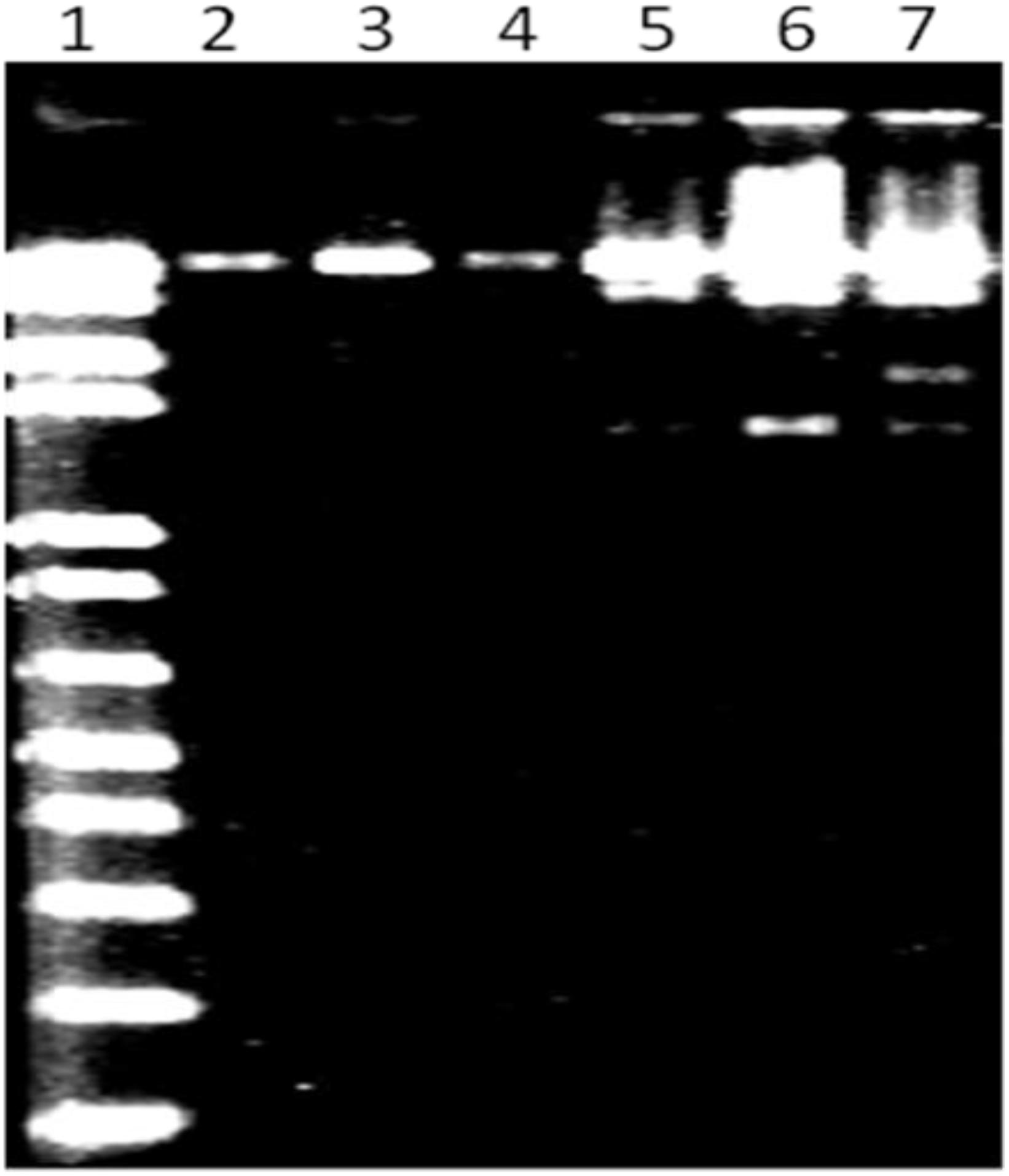
Figure 4. MluI restriction profile of MK-13 phage DNA (lane 7). Lane 1 shows 1-kb Plus DNA ladder (Invitrogen) and lanes 2 to 6 show some of the other phages isolated in this study.
The MK-13 phage (GenBank accession number: MK509462) contains a double-stranded linear DNA genome composed of 158,755 bp with a G + C content of 49.45%. A total of 211 open reading frames (ORFs) were identified, of which 4 included tRNA coding regions with specificity for asparagine (GTT), isoleucine (GAT), serine (TGA), and tyrosine (GTA). The remaining 207 ORFs encoded putative proteins. For the initial characterization of the phages, the contigs generated were automatically annotated with RAST. The annotated proteins were classified into different groups mainly as DNA replication, recombination, nucleotide metabolism, transcription, translation, phage structure and packaging, lysis, and phage–host interactions. Genes involved in the lysogenic process such as an integrase, repressor, or holin were not recognized in this phage. A list of phage structural proteins and some replication and regulatory proteins are shown in Supplementary Table S4 and a DNA plotter is presented in Figure 5. Annotation of MK-13 phage identified most ORFs as “hypothetical proteins” without putative functions, which is common among highly novel phages.
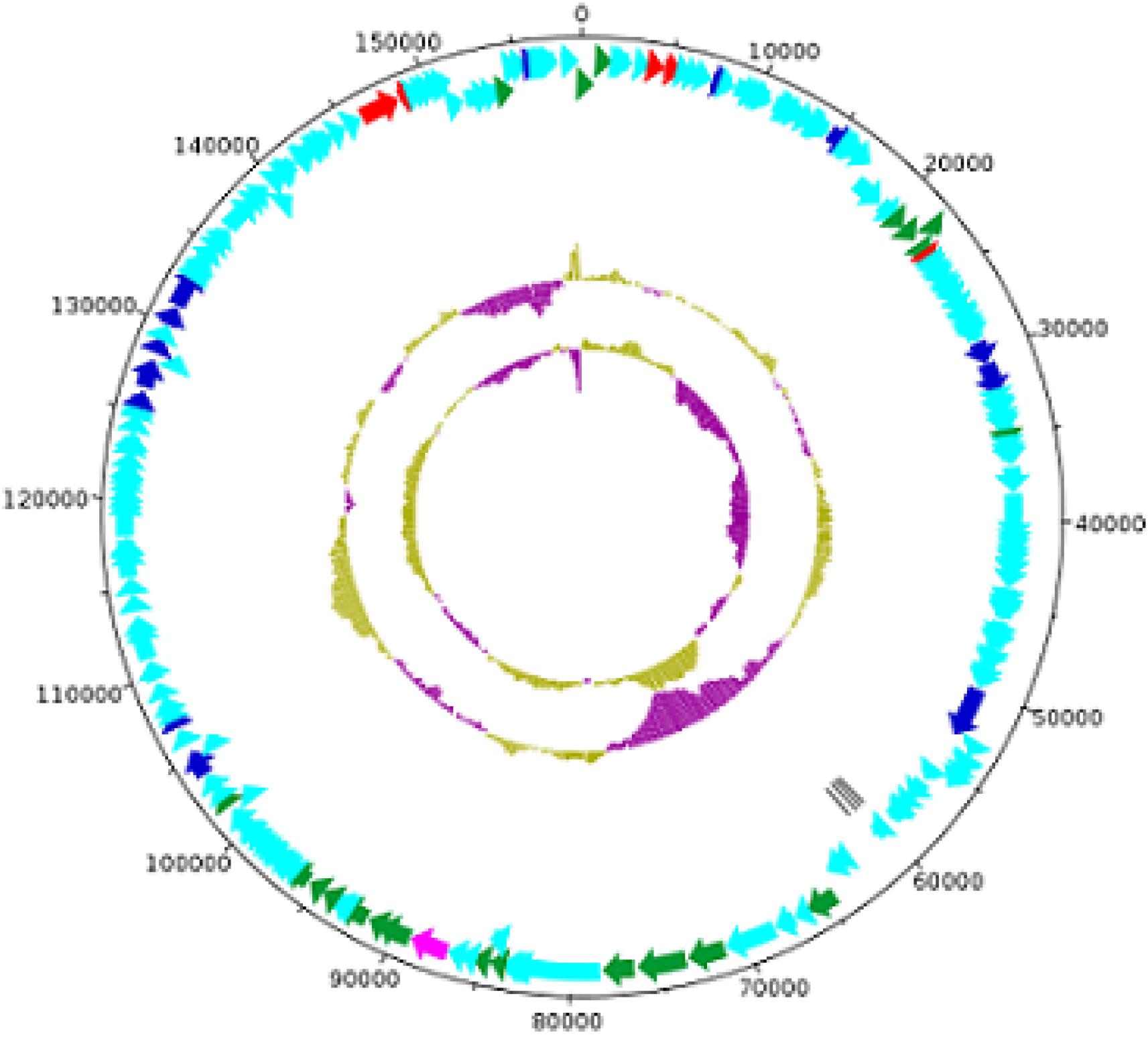
Figure 5. Circles from inside to outside indicate GC skew, GC content, tRNA genes on reverse strand, CDS on reverse strand, and CDS on forward strand. The COG functional categories are represented–blue (L: replication, recombination, and repair), red (F: nucleotide transport and metabolism), and pink (X: prophages and transposons). Structural genes of MK-13 phage are shown in green color.
BLAST analysis revealed that the newly sequenced phage MK-13 was distinct from the publicly available phages in databases except for ΦSboM-Ag3. At the time of writing this manuscript, 35 complete genomes of Shigella phages were found in public databases. Only two of these phages, PSb–1 (Jun et al., 2014) and ΦSboM-Ag3 (Anany et al., 2011), were found to infect S. boydii. Phage PSb–1 infects ATCC 8700 (S. boydii type 2) and ATCC 35966 (S. boydii type 18) but not the other species of Shigella or other bacteria tested. Whereas phage ΦSboM-AG3 was reported to have a broader host range including most of the S. boydii, S. flexneri, S. dysenteriae, and S. sonnei tested. Phylogenetic tree constructed with MK-13 and other Shigella-specific phages revealed that MK-13 phage is closely related to phage ΦSboM-AG3. RAST annotation showed that MK-13 had 89% query coverage with 98% sequence identity with ΦSboM-AG3. The phage family, genome length, GC content, tRNAs, CDS, and most of the other properties of phage MK-13 are similar to phage ΦSboM-AG3 but distant from Shigella phage PSb–1 (Table 2).
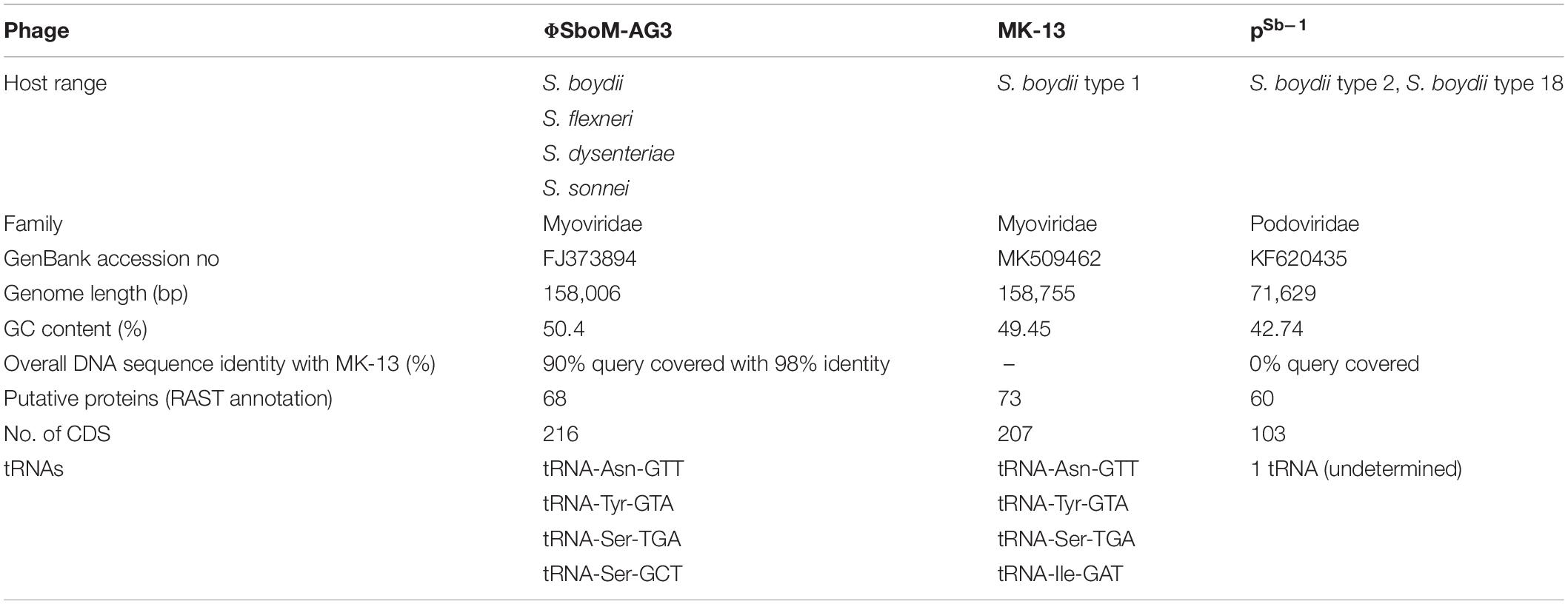
Table 2. Phenotypic and genomic properties of MK-13 compared with bacteriophages ΦSboM-AG3 and PSb–1.
The MK-13 genome displayed two inversions compared to ΦSboM-AG3 genome. The first inversion includes the genomic region of around 41 kb nucleotides of MK-13, whereas it comprised more than 42 kb nucleotides in ΦSboM-AG3. Interestingly, the DNA ligase protein in the first inversion had the most variability among all the core proteins of ΦSboM-AG3 and MK-13. In case of the second and largest inversion, the MK-13 genome was longer (116 kb) than the ΦSboM-AG3 (115 kb). The breakpoints of the genomes were adjacent to the phage rII lysis inhibitor genes. The phylogenetic tree and gene synteny are shown in Figures 6, 7, respectively.
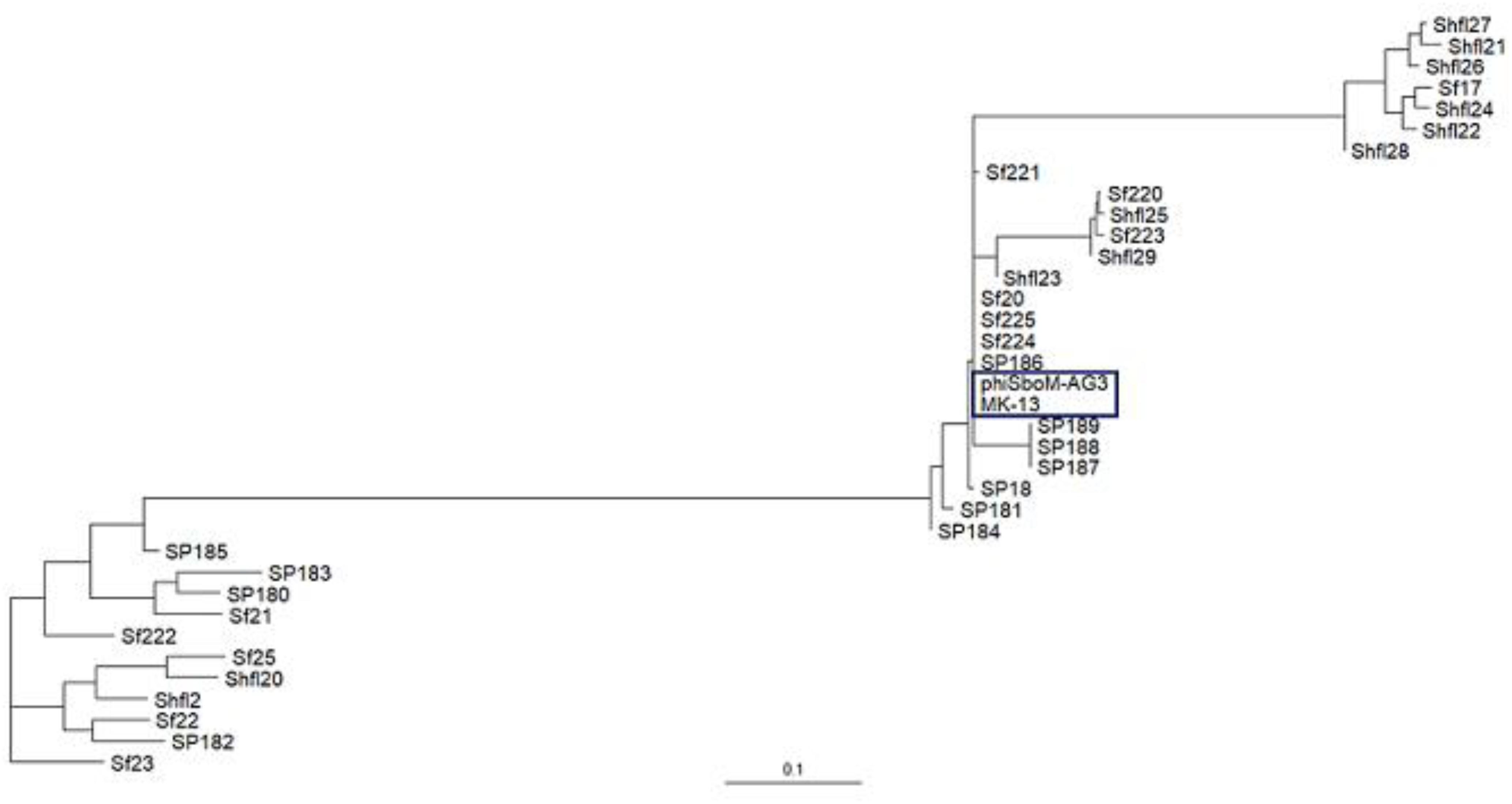
Figure 6. Phylogenetic interrelationship among the selected Shigella phages. The dendrogram represents the maximum-likelihood phylogenetic tree, which is based on the gene presence–absence data. The blue color box is used to indicate the selected phages of Shigella boydii. The scale bar represents accessory gene content similarity across the branch length.
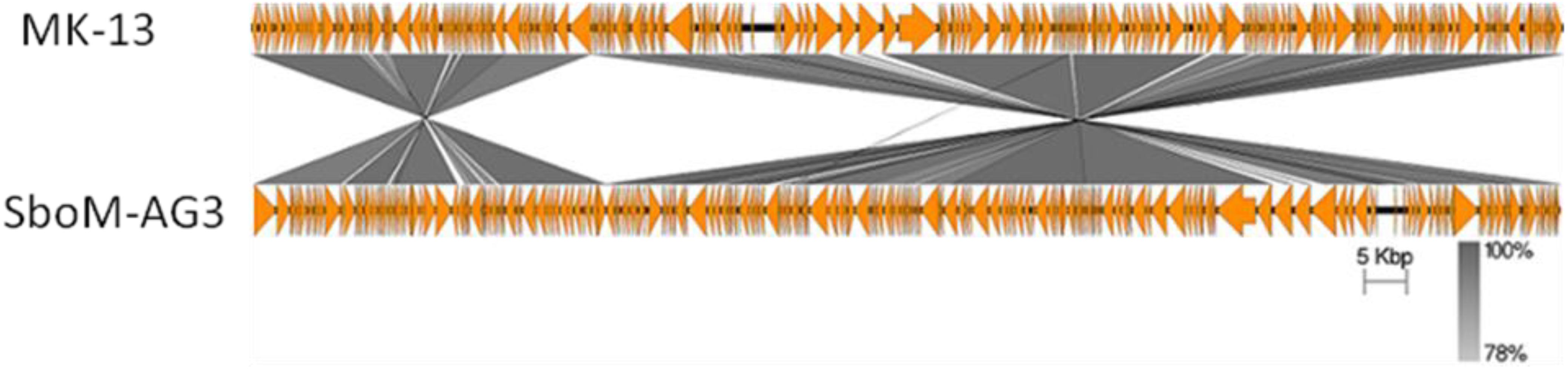
Figure 7. Genomic comparison of phage MK-13 with S. boydii phage ΦSboM-AG3. The gray color intensity is proportional to the sequence similarity as indicated in the scale below.
Phage MK-13 genome encodes four tRNA genes, of which three match with ΦSboM-Ag3 phage but did not match with any of the tRNA of PSb–1 (Tables 2, 3). Function-based comparison through RAST showed that only two proteins are common among the two phages specific for S. boydii (Table 4).
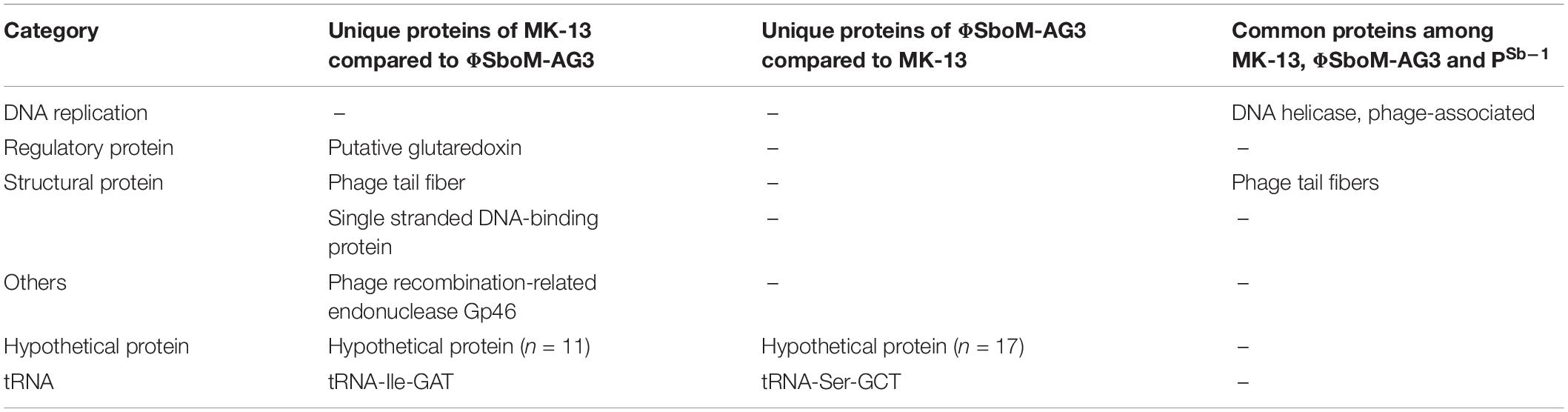
Table 3. Function based comparison among the 3 phages specific for S. boydii.
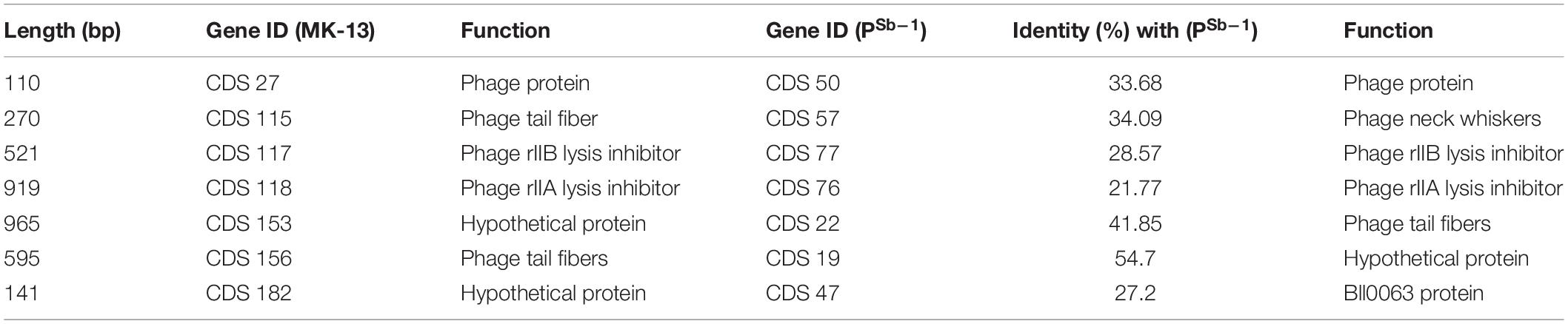
Table 4. Common proteins among MK-13 and PSb–1 with their functions.
Sequence-based comparison through RAST among MK-13 and ΦSboM-AG3 phages showed that the main differences among the CDSs are in hypothetical proteins. A total of 11 hypothetical proteins and one phage protein are found unique for MK-13, which did not match with any CDS of ΦSboM-AG3. However, 17 hypothetical proteins of ΦSboM-AG3 are unique compared to MK-13. BLAST alignment of the unique hypothetical proteins of MK-13 is shown in Supplementary Table S5.
Another CDS of MK-13 has been detected, which showed 97% identity with 78% query cover with ΦSboM-AG3 where their functions were different. Function of this sequence in MK-13 is ribose-phosphate pyrophosphokinase (EC 2.7.6.1), which showed closest match to more than 30 phages. However, the function in ΦSboM-AG3 is nicotinamide phosphoribosyltransferase (EC 2.4.2.12), which didn’t show any close match to other phage sequences. Sequence-based comparison among the MK-13 and PSb–1 phages showed seven CDSs from MK-13 matched with PSb–1 with 21–55% identity, of which functions of three CDSs are same (Table 4). Six CDSs of ΦSboM-AG3 matched with CDSs of PSb–1 with 21–53% identity having similar functions. From these six CDSs of ΦSboM-AG3, five also matched with CDSs of MK-13 with similar functions (Table 5). Common proteins among MK_13 and ΦSboM-AG3 are listed in Supplementary Table S6.

Table 5. Common proteins among ΦSboM-AG3 and PSb–1 having similar functions.
Of all the enteric bacteria including 20 serotypes of S. boydii tested in this study, only S. boydii type 1 was susceptible to phage MK-13. Hence, this phage appears to be a useful diagnostic tool (Olsen et al., 1993). Most of the other phages tested infected a broader spectrum of Shigella strains belonging to different species or serotypes.
Electron microscopic examination and WGS of the phage revealed that it belonged to the Myoviridae family (Hendrix, 2005). Its virion morphology is very similar to phage ΦSboM-AG3 (Anany et al., 2011). The MK-13 and ΦSboM-AG3 genomes shared nucleotide identity and gene synteny, providing evidence that they are closely related. However, only one structural protein, phage tail fiber, is the unique protein that is not found in phage ΦSboM-AG3. This protein may have a role in a narrow host range specificity of MK-13. ΦSboM-AG3 was reported to have a broader host range, where MK-13 showed specificity to its single host. Phage PSb–1 is highly specific to S. boydii, but its genome is only distantly related to the MK-13 genome. Breakpoints in synteny between the MK-13 and the ΦSboM-AG3 genomes were adjacent to phage rII lysis inhibitor genes. Mutation of these genes is known to alter the speed of lysis (Dressman and Drake, 1999; Chen and Young, 2016). Mosaicism (breaks in synteny) in phage genomes is a common reported phenomenon and may contribute to phage evolution and adaptation (Hatfull, 2008; Belcaid et al., 2010).
To detect S. boydii type 1 in an unknown sample using the current standard antisera approach, a set of polyvalent and another set of monovalent antisera are necessary for a series of agglutination tests to confirm a strain as S. boydii type 1 whereas 2 μl at 106 titer of MK-13 phage can alternatively be used for detection of S. boydii type 1, which is faster and cheaper. The production, purchasing, and shipment of antisera from the producer company are costly and time consuming. In contrast, phage production is easier, cheaper, and efficient. Developing a Shigella phage typing scheme will improve the speed, efficiency, and accuracy instead of the current standard Shigella serotyping scheme. Phage typing could provide better results than serotyping and have greater discriminative power compared to other commonly used methods. Phage MK-13 will be useful to confirm the diagnosis of infection caused by S. boydii type 1 and for differentiating it from other Shigella serotypes, and Shigella like-organisms, E. coli, and other non-lactose fermenting colonies.
Several typing techniques are used to identify different bacteria but the cost compared to phage typing is prohibitive. Though serotyping is the most prevalent effective diagnostic in resource-poor settings, phage typing has been proven as a rapid and low-cost approach for efficient diagnosis, epidemiological surveillance, and outbreak investigation but needs trained and experienced technicians. The technique has been adopted successfully within certain contexts in microbiology, such as Staphylococcus outbreaks in hospitals and nurseries in the 1950s (Hillier, 2006). In another study, for detection of Vibrio parahaemolyticus, the authors combined the technique with a coupled enzyme system in vitro because of its rapid detection, high specificity, and simplicity in operation (Peng et al., 2014).
The limitations of our study were the host specificity of MK-13 was not tested for S. dysenteriae types 5, 7, and 10; S. flexneri 4b; and S. boydii type 6 and 17, which were not available in the lab. These serotypes are not prevalent in Bangladesh. The precise bacterial epitope to which phage MK-13 binds is unknown, and it is not clear whether this epitope is encoded in a non-S. boydii type 1 strain that was not tested in this study.
Previously described Shigella phages infected a broader spectrum of Shigella belonging to different species or serotypes (Goodridge, 2013; Jun et al., 2016; Hamdi et al., 2017; Soffer et al., 2017). This study is the first one to address Shigella phage for typing of Shigella for diagnosis of shigellosis at the serotype level. The phage, MK-13, isolated in this study can be used as a cheap and reliable diagnostic marker for detection of shigellosis caused by S. boydii type 1 as an easier, cheaper, and efficient method in less developed countries, especially in rural areas. Although not the focus of this study, the phage may also have potential to remove bacterial contamination in water and foods that are not cooked before eaten, and in epidemiological application and treating. Further work will focus on testing more strains of different S. boydii serotypes and to identify the precise bacterial epitope to which phage MK-13 binds.
The datasets generated for this study can be found in the MK509462.
Experimental protocol was approved by the Research Review Committee (RRC) and the Ethics Review Committee (ERC) of the icddr,b (Protocol Number PR-14040). All methods were conducted in accordance with the guidelines of the RRC and ERC.
MA: conception, study design, laboratory experiments, genomic data analysis, writing of the original manuscript, editing, finalizing, and took overall responsibilities of the project as principal investigator. NB: library preparation, whole genome sequencing, genomic data assembly, review of annotation, and editing of the manuscript. MC: whole genome sequencing and review, and editing of the manuscript. MY: laboratory experiments. TT: genomic data analysis. RB: review of genomic data analysis and editing of the manuscript. MAA: laboratory experiments. AG: all preparation and operation for the TEM. NA: planning, supervision, facility for genomic data analysis, and review and editing of the manuscript. KT: conception, study design, manuscript writing and editing, and provided the laboratory facilities to carry out this study. All authors read and approved the final manuscript.
This work was supported by the Swedish International Development Cooperation Agency (Sida), University of Leicester, and other core donors of icddr,b. Current donors providing unrestricted support include the Government of the People’s Republic of Bangladesh, Global Affairs Canada (GAC), Swedish International Development Cooperation Agency (Sida), and the Department for International Development (UK Aid).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We gratefully acknowledge the core donors for their support and commitment to icddr,b for its operations and research.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02461/full#supplementary-material
Ahmed, R., Sankar-Mistry, P., Jackson, S., Ackermann, H., and Kasatiya, S. (1995). Bacillus cereus phage typing as an epidemiological tool in outbreaks of food poisoning. J. Clin. Microbiol. 33, 636–640.
Albert, M. J., Bhuiyan, N., Rahman, A., Ghosh, A., Hultenby, K., Weintraub, A., et al. (1996). Phage specific for Vibrio cholerae O139 Bengal. J. Clin. Microbiol. 34, 1843–1845.
Anany, H., Lingohr, E. J., Villegas, A., Ackermann, H. W., She, Y. M., Griffiths, M. W., et al. (2011). A Shigella boydii bacteriophage which resembles Salmonella phage viI. Virol. J. 8:242. doi: 10.1186/1743-422x-8-242
Anderson, E., and Williams, R. (1956). Bacteriophage typing of enteric pathogens and staphylococci and its use in epidemiology: a review. J. Clin. Pathol. 9, 94–127. doi: 10.1136/jcp.9.2.94
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available at: http://www.bioinformatics.babraham.ac.uk?/projects/fastqc/ (accessed October 6, 2011).
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Baker, K. S., Parkhill, J., and Thomson, N. R. (2015). Draft genome sequence of 24570, the type strain of Shigella flexneri. Genome Announc. 3:e393-15. doi: 10.1128/genomeA.00393-15
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Belcaid, M., Bergeron, A., and Poisson, G. (2010). Mosaic graphs and comparative genomics in phage communities. J. Comput. Biol. 17, 1315–1326. doi: 10.1089/cmb.2010.0108
Bratoeva, M. P., John, J., and Barg, N. (1992). Molecular epidemiology of trimethoprim-resistant Shigella boydii serotype 2 strains from Bulgaria. J. Clin. Microbiol. 30, 1428–1431.
Carver, T., Thomson, N., Bleasby, A., Berriman, M., and Parkhill, J. (2008). DNAPlotter: circular and linear interactive genome visualization. Bioinformatics 25, 119–120. doi: 10.1093/bioinformatics/btn578
Chattaway, M. A., Schaefer, U., Tewolde, R., Dallman, T. J., and Jenkins, C. (2017). Identification of Escherichia coli and Shigella species from whole-genome sequences. J. Clin. Microbiol. 55, 616–623. doi: 10.1128/jcm.01790-16
Chen, Y., and Young, R. (2016). The last r locus unveiled: T4 RIII is a cytoplasmic co-antiholin. J. Bacteriol. 198, 2448–2457. doi: 10.1128/JB.00294-16
Dressman, H. K., and Drake, J. W. (1999). Lysis and lysis inhibition in bacteriophage T4: rV mutations reside in the holin t gene. J. Bacteriol. 181, 4391–4396.
DuPont, H. L., Levine, M. M., Hornick, R. B., and Formal, S. B. (1989). Inoculum size in shigellosis and implications for expected mode of transmission. J. Infect. Dis. 159, 1126–1128. doi: 10.1093/infdis/159.6.1126
Faruque, S. M., Chowdhury, N., Khan, R., Hasan, M. R., Nahar, J., Islam, M. J., et al. (2003). Shigella dysenteriae type 1-specific bacteriophage from environmental waters in Bangladesh. Appl. Environ. Microbiol. 69, 7028–7031. doi: 10.1128/aem.69.12.7028-7031.2003
Fukushima, M., Kakinuma, K., and Kawaguchi, R. (2002). Phylogenetic analysis of Salmonella, Shigella, and Escherichia coli strains on the basis of the gyrB gene sequence. J. Clin. Microbiol. 40, 2779–2785. doi: 10.1128/jcm.40.8.2779-2785.2002
Ghosh, S., Dasgupta, S., Sen, A., and Sekhar Maiti, H. (2005). Low-temperature synthesis of nanosized bismuth ferrite by soft chemical route. J. Am. Ceram. Soc. 88, 1349–1352. doi: 10.1111/j.1551-2916.2005.00306.x
Goodridge, L. D. (2013). Bacteriophages for managing Shigella in various clinical and non-clinical settings. Bacteriophage 3:e25098. doi: 10.4161/bact.25098
Grimont, F., Lejay-Collin, M., Talukder, K. A., Carle, I., Issenhuth, S., Le Roux, K., et al. (2007). Identification of a group of shigella-like isolates as Shigella boydii 20. J. Med. Microbiol. 56(Pt 6), 749–754. doi: 10.1099/jmm.0.46818-0
Guy, L., Roat Kultima, J., and Andersson, S. G. (2010). genoPlotR: comparative gene and genome visualization in R. Bioinformatics 26, 2334–2335. doi: 10.1093/bioinformatics/btq413
Hamdi, S., Rousseau, G. M., Labrie, S. J., Tremblay, D. M., Kourda, R. S., Ben Slama, K., et al. (2017). Characterization of two polyvalent phages infecting Enterobacteriaceae. Sci. Rep. 7:40349. doi: 10.1038/srep40349
Hasman, H., Saputra, D., Sicheritz-Ponten, T., Lund, O., Svendsen, C. A., Frimodt-Moller, N., et al. (2014). Rapid whole-genome sequencing for detection and characterization of microorganisms directly from clinical samples. J. Clin. Microbiol. 52, 139–146. doi: 10.1128/jcm.02452-13
Hatfull, G. F. (2008). Bacteriophage genomics. Curr. Opin. Microbiol. 11, 447–453. doi: 10.1016/j.mib.2008.09.004
Hendrix, R. W. (2005). Bacteriophage HK97: assembly of the capsid and evolutionary connections. Adv. Virus Res. 64, 1–14. doi: 10.1016/S0065-3527(05)64001-8
Hillier, K. (2006). Babies and bacteria: phage typing, bacteriologists, and the birth of infection control. Bull. Hist. Med. 80, 733–761. doi: 10.1353/bhm.2006.0130
Houpt, E., Gratz, J., Kosek, M., Zaidi, A. K., Qureshi, S., Kang, G., et al. (2014). Microbiologic methods utilized in the MAL-ED cohort study. Clin. Infect. Dis. 59(Suppl._4), S225–S232. doi: 10.1093/cid/ciu413
Jiang, H., Lei, R., Ding, S.-W., and Zhu, S. (2014). Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics 15:182. doi: 10.1186/1471-2105-15-182
Jun, J. W., Giri, S. S., Kim, H. J., Yun, S. K., Chi, C., Chai, J. Y., et al. (2016). Bacteriophage application to control the contaminated water with Shigella. Sci. Rep. 6:22636. doi: 10.1038/srep22636
Jun, J. W., Yun, S. K., Kim, H. J., Chai, J. Y., and Park, S. C. (2014). Characterization and complete genome sequence of a novel N4-like bacteriophage, pSb-1 infecting Shigella boydii. Res. Microbiol. 165, 671–678. doi: 10.1016/j.resmic.2014.09.006
Kania, D. A., Hazen, T. H., Hossain, A., Nataro, J. P., and Rasko, D. A. (2016). Genome diversity of Shigella boydii. FEMS Pathog. Dis. 74:ftw027. doi: 10.1093/femspd/ftw027
Kotloff, K. L. (2017). The burden and etiology of diarrheal illness in developing countries. Pediatr. Clin. North Am. 64, 799–814. doi: 10.1016/j.pcl.2017.03.006
Kotloff, K. L., Nataro, J. P., Blackwelder, W. C., Nasrin, D., Farag, T. H., Panchalingam, S., et al. (2013). Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the global enteric multicenter study, GEMS): a prospective, case-control study. Lancet 382, 209–222. doi: 10.1016/s0140-6736(13)60844-2
Kotloff, K. L., Platts-Mills, J. A., Nasrin, D., Roose, A., Blackwelder, W. C., and Levine, M. M. (2017). Global burden of diarrheal diseases among children in developing countries: incidence, etiology, and insights from new molecular diagnostic techniques. Vaccine 35(49 Pt A), 6783–6789. doi: 10.1016/j.vaccine.2017.07.036
Kotloff, K. L., Winickoff, J. P., Ivanoff, B., Clemens, J. D., Swerdlow, D. L., Sansonetti, P. J., et al. (1999). Global burden of Shigella infections: implications for vaccine development and implementation of control strategies. Bull. World Health Organ. 77, 651–666.
Liu, B., Knirel, Y. A., Feng, L., Perepelov, A. V., Senchenkova, S. Y. N., Wang, Q., et al. (2008). Structure and genetics of Shigella O antigens. FEMS Microbiol. Rev. 32, 627–653. doi: 10.1111/j.1574-6976.2008.00114.x
Livio, S., Strockbine, N. A., Panchalingam, S., Tennant, S. M., Barry, E. M., Marohn, M. E., et al. (2014). Shigella isolates from the global enteric multicenter study inform vaccine development. Clin. Infect. Dis. 59, 933–941. doi: 10.1093/cid/ciu468
Mani, S., Wierzba, T., and Walker, R. I. (2016). Status of vaccine research and development for Shigella. Vaccine 34, 2887–2894. doi: 10.1016/j.vaccine.2016.02.075
Marchler-Bauer, A., Derbyshire, M. K., Gonzales, N. R., Lu, S., Chitsaz, F., Geer, L. Y., et al. (2015). CDD: NCBI’s conserved domain database. Nucleic Acids Res. 43, D222–D226. doi: 10.1093/nar/gku1221
Muthuirulandi Sethuvel, D. P., Devanga Ragupathi, N. K., Anandan, S., Walia, K., and Veeraraghavan, B. (2017). Molecular diagnosis of non-serotypeable Shigella spp.: problems and prospects. J. Med Microbiol. 66, 255–257. doi: 10.1099/jmm.0.000438
Olsen, J. E., Brown, D. J., Skov, M. N., and Christensen, J. P. (1993). Bacterial typing methods suitable for epidemiological analysis. applications in investigations of salmonellosis among livestock. Vet. Q. 15, 125–135. doi: 10.1080/01652176.1993.9694390
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Peng, Y., Jin, Y., Lin, H., Wang, J., and Khan, M. N. (2014). Application of the VPp1 bacteriophage combined with a coupled enzyme system in the rapid detection of vibrio parahaemolyticus. J. Microbiol. Methods 98, 99–104. doi: 10.1016/j.mimet.2014.01.005
Pettengill, E. A., Pettengill, J. B., and Binet, R. (2015). Phylogenetic analyses of Shigella and enteroinvasive Escherichia coli for the identification of molecular epidemiological markers: whole-genome comparative analysis does not support distinct genera designation. Front. Microbiol. 6:1573. doi: 10.3389/fmicb.2015.01573
R Core Team, (2013). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Ranjbar, R., Mammina, C., Pourshafie, M. R., and Soltan-Dallal, M. M. (2008). Characterization of endemic Shigella boydii strains isolated in Iran by serotyping, antimicrobial resistance, plasmid profile, ribotyping and pulsed-field gel electrophoresis. BMC Res. Notes 1:74. doi: 10.1186/1756-0500-1-74
Sambrook, J., Fritsch, E., and Maniatis, T. (1989). Molecular Cloning: A Laboratory Manual. New York, NY: Cold Spring Harbor Laboratory Press.
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shahnaij, M., Latif, H. A., Azmi, I. J., Amin, M. B., Luna, S. J., Islam, M. A., et al. (2018). Characterization of a serologically atypical Shigella flexneri Z isolated from diarrheal patients in Bangladesh and a proposed serological scheme for Shigella flexneri. PLoS One 13:e0202704. doi: 10.1371/journal.pone.0202704
Soffer, N., Woolston, J., Li, M., Das, C., and Sulakvelidze, A. (2017). Bacteriophage preparation lytic for Shigella significantly reduces Shigella sonnei contamination in various foods. PLoS One 12:e0175256. doi: 10.1371/journal.pone.0175256
Sullivan, M. J., Petty, N. K., and Beatson, S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. doi: 10.1093/bioinformatics/btr039
Talukder, K. A., and Azmi, I. J. (2012). “Population genetics and molecular epidemiology of Shigella species,” in Foodborne and Waterborne Bacterial Pathogens: Epidemiology, Evolution and Molecular Biology, ed. S. M. Faruque, (Poole: Caister Academic Press).
Talukder, K. A., Islam, M. A., Dutta, D. K., Hassan, F., Safa, A., Nair, G., et al. (2002). Phenotypic and genotypic characterization of serologically atypical strains of Shigella flexneri type 4 isolated in Dhaka, Bangladesh. J. Clin. Microbiol. 40, 2490–2497. doi: 10.1128/jcm.40.7.2490-2497.2002
Ud-Din, A., and Wahid, S. (2014). Relationship among Shigella spp. and enteroinvasive Escherichia coli (EIEC) and their differentiation. Braz. J. Microbiol. 45, 1131–1138. doi: 10.1590/s1517-83822014000400002
von Seidlein, L., Kim, D. R., Ali, M., Lee, H., Wang, X., Thiem, V. D., et al. (2006). A multicentre study of Shigella diarrhoea in six Asian countries: disease burden, clinical manifestations, and microbiology. PLoS Med. 3:e353. doi: 10.1371/journal.pmed.0030353
World Health Organization, (1987). Programme for Control of Diarrhoeal Diseases (CDD/83.3 Rev 1). MANUAL for Laboratory Investigations of Acute Enteric Infections. Geneva: World Health Organization, 9–20.
Keywords: shigellosis, Shigella boydii type 1, phage, diagnosis, low-cost
Citation: Akter M, Brown N, Clokie M, Yeasmin M, Tareq TM, Baddam R, Azad MAK, Ghosh AN, Ahmed N and Talukder KA (2019) Prevalence of Shigella boydii in Bangladesh: Isolation and Characterization of a Rare Phage MK-13 That Can Robustly Identify Shigellosis Caused by Shigella boydii Type 1. Front. Microbiol. 10:2461. doi: 10.3389/fmicb.2019.02461
Received: 16 May 2019; Accepted: 14 October 2019;
Published: 07 November 2019.
Edited by:
Robert Czajkowski, University of Gdańsk, PolandReviewed by:
Zoe Anne Dyson, University of Cambridge, United KingdomCopyright © 2019 Akter, Brown, Clokie, Yeasmin, Tareq, Baddam, Azad, Ghosh, Ahmed and Talukder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mahmuda Akter, bWFobXVkYWJtYi5kdWJkQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.