 Gaia Piazza
Gaia Piazza Laura Ercoli
Laura Ercoli Marco Nuti
Marco Nuti Elisa Pellegrino
Elisa Pellegrino- Institute of Life Sciences, Scuola Superiore Sant’Anna, Pisa, Italy
Soil biodiversity accomplishes key roles in agro-ecosystem services consisting in preserving and enhancing soil fertility and nutrient cycling, crop productivity and environmental protection. Thus, the improvement of knowledge on the effect of conservation practices, related to tillage and N fertilization, on soil microbial communities is critical to better understand the role and function of microorganisms in regulating agro-ecosystems. In the Mediterranean area, vulnerable to climate change and suffering for management-induced losses of soil fertility, the impact of conservation practices on soil microbial communities is of special interest for building mitigation and adaptation strategies to climate change. A long-term experiment, originally designed to investigate the effect of tillage and N fertilization on crop yield and soil organic carbon, was utilized to understand the effect of these management practices on soil prokaryotic and fungal community diversity. The majority of prokaryotic and fungal taxa were common to all treatments at both soil depths, whereas few bacterial taxa (Cloacimonates, Spirochaetia and Berkelbacteria) and a larger number of fungal taxa (i.e., Coniphoraceae, Debaryomycetaceae, Geastraceae, Cordicypitaceae and Steccherinaceae) were unique to specific management practices. Soil prokaryotic and fungal structure was heavily influenced by the interaction of tillage and N fertilization: the prokaryotic community structure of the fertilized conventional tillage system was remarkably different respect to the unfertilized conservation and conventional systems in the surface layer. In addition, the effect of N fertilization in shaping the fungal community structure of the surface layer was higher under conservation tillage systems than under conventional tillage systems. Soil microbial community was shaped by soil depth irrespective of the effect of plowing and N addition. Finally, chemical and enzymatic parameters of soil and crop yields were significantly related to fungal community structure along the soil profile. The findings of this study gave new insights on the identification of management practices supporting and suppressing beneficial and detrimental taxa, respectively. This highlights the importance of managing soil microbial diversity through agro-ecological intensified systems in the Mediterranean area.
Introduction
Agricultural systems are essential sources of provision services, such as food, forage, fiber, and bioenergy, as well as for regulating services, such as climate and disease regulation (Power, 2010). High intensity of management practices has potential negative environmental consequences, which may determine the reduction of agricultural incomes and the increase of production costs (Zhang et al., 2007). Intensive plow-based systems along with inefficient use of chemical inputs are known to accelerate the oxidation of soil organic matter (SOM), disrupt soil physical structure, decrease soil moisture retention and modify soil biological diversity, with strong consequences on biogeochemical cycles (Peng et al., 2013; Degrune et al., 2017; Wang et al., 2018). Moreover, agricultural activities have been recognized responsible for 30% of global anthropogenic greenhouse gas emissions that are one of the causes of global warming and climate change (IPCC, 2015).
Conservation agriculture (CA) is a systemic approach aiming to a sustainable intensification of production, coping with the ecosystem disservices determined by conventional management practices (Pretty, 2008; Friedrich, 2013). Conservation agriculture is based on the application of the following linked principles: minimum or no soil disturbance; maintenance of permanent soil mulch cover; adoption of crop rotations and an appropriate use of fertilizers (Lal, 2015; Thierfelder et al., 2018). Conservation agriculture covers worldwide about 180 million ha across different agro-ecosystems, representing 13% of the global cropland, with an increase of about 69% from 2015–2016 to 2008–2009 (Kassam et al., 2019).
Soil biodiversity accomplishes key roles in ecosystem services, consisting in preserving and enhancing soil fertility and nutrient cycling, crop productivity and environmental protection (Yang et al., 2017; Mommer et al., 2018; Wang et al., 2019). Thus, the improvement of knowledge on patterns and drivers of soil microbial community composition is critical to better understand the role and function of soil microorganisms in regulating agro-ecosystem structure, process and functioning. Linkages between above- and belowground communities of living organisms have been largely investigated: plant community composition can shape the diversity of belowground organisms, such as decomposers, symbionts or pathogens, whose activity affect plant metabolism and shoot/root biomass production (Bezemer and van Dam, 2005; De Deyn and Van der Putten, 2005; Bardgett and Wardle, 2010; van Der Heijden et al., 2016). In this context, bacteria and fungi represent the dominant microorganisms in soil, consisting in 102–104 μg biomass carbon (C) g–1 soil (Fierer, 2017) and carrying out 80–90% of soil processes (Nannipieri and Badalucco, 2003).
Several studies found increases of microbial biomass under long-term conservation tillage systems (i.e., no tillage, NT; minimum tillage, MT) (e.g., Mathew et al., 2012; Zhang et al., 2012; Murugan et al., 2014). Moreover, recent studies, addressing the molecular diversity of microbial communities, have shown a higher abundance of Acidobacteria and Bacteroidetes and a lower abundance of Proteobacteria and Actinobacteria under MT systems respect to conventional tillage (CT) (Dorr de Quadros et al., 2012; Degrune et al., 2017; Babin et al., 2019). These evidences were explained through the different nutrient status and aerobic/anaerobic conditions of soil. According to the oligotrophy-copiotrophy theory, fast-growing r-strategist microorganisms (or copiotrophics, e.g., classes Alpha- and Beta-proteobacteria), showing a preference to use labile C compounds, are highly present in CT (Fontaine et al., 2003; Fierer et al., 2007; Trivedi et al., 2015, 2017). Conversely, slow-growing K-strategists (or oligotrophics, e.g., phylum Acidobacteria), mainly feeding on recalcitrant C, are highly present in MT. Moreover, under NT or MT, a shift from soil bacterial- to fungal-dominated communities was associated to SOC accumulation, due to the improvement of soil structural stability through mycelium development and deposition of fungal-derived C (Simpson et al., 2004; Kong et al., 2011). Minimum soil disturbance could also allow suitable conditions for the promotion of the abundance and diversity of symbiotic fungi, such as arbuscular mycorrhizal fungi (AMF), as well as shifts of their community structures (Alguacil et al., 2008; Brito et al., 2012). Arbuscular mycorrhizal fungi can improve soil aggregation and structure on one side, and enhance plant growth, crop production, plant nutrient status and health on the other side (Ercoli et al., 2017; Caruso, 2018; Powell and Rillig, 2018; Zhang et al., 2018; Coccina et al., 2019). However, according to a recent meta-analysis, the response of soil microbial diversity to tillage is highly variable depending on climatic conditions and soil properties (de Graaff et al., 2019). Overall, MT systems increase bacterial diversity by 7% and do not affect either soil total fungal diversity or AMF diversity respect to CT. The Mediterranean area is characterized by summer drought and erratic distribution of rainfall and thus is vulnerable to climate change and suffers for losses of soil fertility. In these conditions, the strong decrease of AMF diversity following CT and the large difference in the responses of functional bacterial and fungal taxa need to be better clarified (Ceja-Navarro et al., 2010; Brito et al., 2012; Pastorelli et al., 2013; Ciccolini et al., 2015, 2016a,b; Pellegrino et al., 2015b).
Bacterial diversity was increased by mineral N fertilization at low input (<150 kg N ha–1 y–1) and with applications over more than 5 years, while fungal diversity was increased only for specific taxonomic groups (e.g., yeasts), having a copiotrophic lifestyle (de Graaff et al., 2019). On the other hand, AMF diversity was reduced by N fertilization and this was supposedly due to a less investment in mycorrhizal symbioses by host plants (Treseder, 2004) and to a reduction in pH induced by N addition that improves phosphorus (P) availability (Egerton-Warburton et al., 2007; Marklein and Houlton, 2012). In the Mediterranean cropping systems, also suffering from leaching N and S losses, the effect of N fertilization on the diversity and structure of functional soil prokaryotes and fungi was poorly investigated (Dias et al., 2000; Ercoli et al., 2012; Alguacil et al., 2014; Mariotti et al., 2015). Therefore, in this area the impact of agricultural management (i.e., tillage and N fertilization) on soil microbial communities needs to be better clarified in order to reduce environmental pollution and water deficit and improve crop yield and SOC accumulation.
Gradient of soil nutrients and environmental factors throughout the soil profile have been shown to affect abundance, composition and structure of soil prokaryotic and fungal communities (Fierer et al., 2003; Oehl et al., 2005; Eilers et al., 2012; Somenahally et al., 2018; Yao et al., 2018). In the surface layer, where the availability of nutrients is high, bacteria prevail over fungi, whereas in the lower layer the opposite occurs, and fungi prevail since they are more competitive for the uptake of N and P. By contrast, if a large amount of plant material input and recalcitrant organic matter is in the surface layer, the fungal biomass prevails over bacteria, since fungi are able to decompose more complex organic matter (Jumpponen et al., 2010). Therefore, there is still a need to clarify the composition and structure of soil microbial community shifts along the soil depth profile.
Long-term experiments in the Mediterranean area, such as the one located in Pisa (Italy) and utilized for this study, originally designed to investigate the effect of tillage and N fertilization on crop yield and SOC, provide a great opportunity for improving our understanding on the effect of management practices on soil prokaryotic and fungal diversity. In this study, we tested the following hypotheses: (1) the interaction of long-term conservation tillage and N fertilization shifts molecular community diversity, composition and structure of soil prokaryotes and fungi; (2) soil depth plays a role in shaping the diversity, composition and structure of soil prokaryotes and fungi; (3) soil physico-chemical and biochemical parameters affect soil microbial community structure and in reverse microbes affect soil parameters, with specific taxa playing major roles in nutrient cycling.
Materials and Methods
Field Experiment
The long-term field experiment was set up in 1993 comparing two tillage intensities to a bread wheat (Triticum aestivum L.) - soybean (Glycine max L. Merr.) rotation: conventional tillage (CT), mouldboard plowing at 25-cm depth, disking and harrowing at 15-cm depth; MT, disk harrowing at 15-cm depth. Two N fertilization levels were also applied on bread wheat: 0 and 200 kg N ha–1 (N0 and N200, respectively). These treatments were applied arranged following a split-plot design with tillage as main-plot factor and N fertilization as subplot factor, with three replicate plots (dimension: 11.5 × 14.5 m). The experiment was conducted at the Centro Interdipartimentale di Ricerche Agro-Ambientali “Enrico Avanzi” (San Piero a Grado, Pisa, Italy; 43°40’ latitude N; 10°19’ longitude E; 1 m above sea level) of the University of Pisa in an alluvial silt loam soil (131, 613 and 256 g kg–1 of sand, silt and clay, respectively, in the 0–30 cm soil layer). The soil is classified as Typic Xerofluvent by USDA system (Soil Survey Staff, 1975) and as Fluvisol by FAO (IUSS working group WRB, 2006). Climate of the site is cold, humid Mediterranean (Csa), according to the Köppen–Geiger climate classification (Kottek et al., 2006). Nitrogen fertilizer treatment was applied to wheat as urea and the rate was splitted into three applications before seeding (60 kg N ha–1), at first node detectable (70 kg N ha–1), and 15 days after this stage (70 kg N ha–1). Under CT, almost 100% of the residues were incorporated in the 0–25 cm soil layer, whereas under MT approximately 50% of the crop residues were incorporated at 0–15-cm depth. Crops were managed following the common agronomical technique applied in the area, comprising pre-emergence herbicide application for weed control, whereas no disease or insect treatment was applied.
Soil Sampling and Analysis of Soil Physico-Chemical and Enzymatic Parameters
For the evaluation of stable systems after long-term changes, soil samples were collected in Spring 2016, before soybean sowing in order to avoid sampling close to the main management practices (Picci and Nannipieri, 2002; Pellegrino et al., 2011). Spring is considered the best time to assess soil microbial diversity in the study area, since the land is dry enough to access and soil temperature is optimal for the growth of microbes. This depends to the fact that in the area the risk of high and low rainfall events is the lowest in Spring compared with other seasons and air temperature does not show low and high extreme values (Vallebona et al., 2015). In each replicate plot, a homogenized sample was taken by mixing the content of four soil cores collected at two soil depths (0–15 and 15–30 cm). Once in the laboratory, each sample was air-dried, gently broken apart and then passed through a 2-mm sieve.
Soil samples were analyzed for soil bulk density (BD), soil organic carbon (SOC), total nitrogen (Total N), available P (Avail P), ammonium (NH4-N) and nitrate (NO3-N). Moreover, soil enzyme potential activities were measured as: cellulose (Cell), chitinase (NAG), α-glucosidase (α-gluc), β-glucosidase (β-gluc), xylosidase (Xylos), phosphatase (Phosph), arylsulphatase (Aryls) and leucine (L.AP). The synthetic enzymatic index (SEI), synthetic enzyme index for the C-cycle (SEIc) were calculated, as well as the microbial functional diversity (Shannon diversity index, H). Finally, the ecoenzymatic C/N and N/P acquisition activities were measured by the ratios of β-glucosidase/(chitinase + leucine) [β-gluc/(NAG + L.AP)] and (chitinase + leucine)/phosphatase activities [(NAG + L.AP)/Phosph], respectively. Details about the analytical methods are given in Supplementary Materials and Methods.
Molecular Analyses
DNA was extracted from 0.25 g of soil samples using the DNeasy PowerSoil Kit (QIAGEN, Venlo, Netherlands), following the instructions of the manufacturer. The extracted DNA was quantified by a spectrophotometer (NanoDrop Technology, Wilmington, DE) and then stored at −20°C for further analyses. PCRs were generated from 10 ng μL–1 genomic DNA in volumes of 25 μL with 0.125 U μL–1 of GoTaq® Hot Start Polymerase (Promega Corporation, WA, United States), 0.5 μM of each primer set for prokaryotes and fungi, 0.2 mM of each dNTP, 1 mM of MgCl2 and 1x reaction buffer, using a S1000 Thermal CyclerTM (BioRad, Hercules, CA, United States). The primer sets used for the amplification of prokaryotes and fungi, relative sequences, targeted genes and PCR reaction conditions are given in Supplementary Table S1. All PCR amplifications were carried out using the primer pairs that have Illumina sequencing tags attached, and in the case of the forward primers a 13 bp random sequence was included to improve cluster definition on the MiSeq slide. All PCR products were examined by electrophoresis through a 1% agarose gel in 0.5 × TBE buffer, then purified with magnetic beads (Agencourt® AMPure® XP, Beckman Coulter, United States) and freshly prepared 80% ethanol, and quantified by a fluorimetry with the Quant-iTTM dsDNA High-Sensitivity Assay Kit (Invitrogen by Thermo Fisher Scientific, CA, United States), following the instructions of the manufacturer. Cleaned and quantified amplicons of each library were adjusted in an equimolar ratio (10 ng/μL) for dual-index barcodes addition using Nextera® Index kit (Illumina Inc., CA, United States), and the resulting metabarcoding libraries were sequenced on an Illumina MiSeq sequencer (2 × 300 bp paired-end reads) at the Eurofins MWG Operon, Ebersberg (Germany).
Bioinformatic and Statistical Analyses
Raw data generated from the Illumina MiSeq sequencing run were processed and analyzed following the pipelines of QIIME 2 (2018.4) and USEARCH (v10.0.240) (Caporaso et al., 2010; Edgar, 2010). Forward and reverse paired-end sequences were assembled independently for each sample using -fastq_mergepairs USEARCH command. Primer sequences were then trimmed off by employing cutadapt plugin (2018.4) with default settings. After optimizing the sequences, there were 1,417,762 and 1,173,836 valid sequences for prokaryotes and fungi, respectively. The average length distribution was approximately 260 and 290 for prokaryotes and fungi, respectively. To avoid potential errors in sequencing data, quality of sequence reads was checked by -fastq_eestats2 USEARCH command, using the expected number of errors in a read as a measure of quality for filtering (Edgar and Flyvbjerg, 2015). Reads were then trimmed at the length where the “drop-off” point for the maximum expected error value occurred (250 bp). Quality filtered reads were de-replicated by -fastx_uniques USEARCH command, then operational taxonomic units (OTUs) of prokaryotes and fungi were generated using USEARCH by clustering sequence reads at the 97% similarity threshold. Chimeric sequences and singletons were removed from the dataset during the process.
The OTUs were phylogenetically assigned using two distinct sequence reference databases: the taxonomic identity of prokaryotes was identified by the 16S SSU SILVA database by clustering sequence reads at the 97% similarity threshold (version 132, release date 13.12.2017; Quast et al., 2012; Yilmaz et al., 2013), whereas the identity of fungi by the ITS UNITE database and a dynamic threshold values of clustering (version 7.2, release date 01.12.2017; Kõljalg et al., 2013). For the curation, the sequences were aligned using the algorithms ClustalW and MUSCLE for prokaryotes and fungi, respectively. Neighbor Joining (NJ) phylogenetic trees were built in MEGA71 (Kumar et al., 2016) and then the most abundant sequence of each prokaryotic and fungal OTU was selected, after branch collapsing, and used as representative sequence for that OTU. Then, the representative sequences were re-aligned using the same algorithms and phylogenetic trees were inferred in MEGA7 using the NJ analysis with 1,000 boostrap replicates and the Kimura 2-parameter model (uniform rates) for prokaryotes, and using the maximum likelihood (ML) phylogenetic analysis with 1,000 boostrap replicates and the general time reverse (GTR) evolutionary model (gamma distributed) for Ascomycota, Basidiomycota, Chytridiomycota and the phyla of Glomeromycota and Mortierellomycota, separately. The phylograms were drawn by the interactive tree of life (ITOL) (Letunic and Bork, 2006) and edited by Adobe Illustrator CS4. All representative sequences were deposited in the NCBI sequence read (SRA) database (prokaryotic accession numbers MK903871-MK904485; fungal accession numbers MK881790-MK881896).
Because there was a high variability in the number of reads per sample, sequencing depth per sample was standardized to the median number of reads across the samples in each data matrix (prokaryotes and fungi) using the package Vegan in R (de Cárcer et al., 2011; Oksanen et al., 2013). Applying this approach, bias due to differences in sample size is reduced by randomly choosing in each sample a number of reads equal to the median number of reads across all samples. Samples that had fewer reads than the median were left unchanged. These reads were used for data input in ITOL for building the pie charts, describing the community structure of Prokaryota, Ascomycota and Basidiomycota among treatments at 0–15 and 15–30 cm soil depth.
For testing the hypothesis 1, for each soil depth, richness and diversity indexes (Shannon index, H’ and Simpson index, λ) were calculated at phylum level and class/family levels (class level: for prokaryotes; family level: for fungi), while the relative abundances for prokaryotes and fungi were calculated at phylum level, and the relative abundances of Ascomycota and Basidiomycota at class level. Richness and diversity indexes were analyzed by a two-way ANOVA following the split-plot experimental design. Data were ln- and arcsine-transformed when needed to fulfill the assumptions of the ANOVA. Post hoc Tukey-B significant difference test was used for comparisons among treatments. Means and standard errors given are for untransformed data. All the analyses were performed using the SPSS software package version 21.0 (SPSS Inc., Chicago, IL, United States). Then, the permutational analysis of variance (PERMANOVA; Anderson, 2001) was used to test the effect of tillage (MT and CT) and N fertilization (N0 and N200) on the community structure (relative abundance) of prokaryotes and fungi at phylum and at class/family levels. Response data matrices were square-root transformed prior to the analyses in order to down-weight the importance of dominant taxa and the Bray-Curtis index of dissimilarity was calculated to measure ecological distance. P-values were calculated using the Monte-Carlo test (Anderson and Braak, 2003). Since PERMANOVA is sensitive to differences in multivariate location (average community composition of a group) and dispersion (within-group variability), the analysis of homogeneity of multivariate dispersion (PERMDISP; Anderson, 2006) was performed to check the homogeneity of dispersion among groups (beta-diversity) (Anderson et al., 2006). When PERMANOVA indicated a significant effect, the principal coordinate analysis (PCO) was performed (Torgerson, 1958) for visualizing the most relevant patterns in the data. In each PCO biplot, we displayed only the phyla, classes and families with a strong correlation (r ≥ 0.70) with the ordination scores indicated on each PCO axis. The circle in each plot, whose diameter is 1.0, allows the reader to understand the scale of the vectors in the vector plot. Analyses were performed using PRIMER 6 and PERMANOVA + software (Clarke and Gorley, 2006; Anderson et al., 2008).
The standardized datasets were also used to generate the Venn diagrams, representing the community composition and unique OTUs to each tillage system and N fertilization or shared among treatments for each soil depth. The Venn diagrams were generated using Venny version 2.1 software2 (Oliveros, 2007).
For testing the hypothesis 2, richness and diversity indexes were analyzed by one-way ANOVA using soil depth as fixed factor, and tillage and N fertilization as covariates. Then, the PERMANOVA was used to test the effect of soil depth on the community structure (relative abundances) of prokaryotes and fungi at phylum and at class/family levels. Finally, the standardized datasets were used to generate the Venn diagrams at each soil depth. PERMDISPs and the PCOs were also performed in order to visualize the most relevant patterns and only the phyla, classes and families with a strong correlation (r ≥ 0.60) were displayed.
We applied co-inertia analysis (Dolédec and Chessel, 1994) to test the relationship between soil physico-chemical and enzymatic parameters and prokaryotic and fungal community structures (hypothesis 3). The co-inertia approach was utilized because it lacks assumptions on the metrics of the datasets. The first step of the co-inertia analysis was based on a detrended correspondence analysis on the soil microbial community structure using as supplementary variables soil parameters, whereas the second step was based on a principal component analysis on soil parameters. Since co-inertia analysis does not allow testing the significance of the relationship, the analysis was combined with the Mantel test (Mantel and Valand, 1970). Co-inertia analyses can handle a large number of species/variables in both dataset, and no dataset takes the response or predictor role. Co-inertia analyses were performed by CANOCO 5 (ter Braak and Šmilauer, 2012), whereas Mantel test was performed by PC-ORD 5 (Grandin, 2006).
Results
Illumina Sequencing Information
A total of 1,341,713 and 1,100,704 quality-filtered 16S SSU rRNA and ITS1 sequences were obtained for prokaryotes and fungi, respectively, from 24 soil samples. For prokaryotes, after BLAST against the 16S SSU SILVA database, we found 1,189,331 reads, ranging from 10 to 103,317 reads per sample that were assigned to a total of 2,875 OTUs. For fungi, after BLAST against the ITS UNITE database we found 1,065,465 reads, ranging from 8 to 174,386 reads per sample that were assigned to a total of 590 OTUs. After the curation of prokaryotic sequences, 1,168,953 reads, ranging from 10 to 94,874 reads per sample, were retrieved and assigned to 615 OTUs (Figure 1A). After the curation of fungal sequences, 1,023,727 reads, ranging from to 8 to 164,728 reads per sample, were retrieved and assigned to 107 OTUs (Figures 1B,C and Supplementary Table S2). Following the standardization to the median number per sample, a total of 1,165,667 reads belonging to 615 OTUs, 52 classes and 24 phyla were retrieved for prokaryotes (Figures 1A, 2A), while for the fungal data, a total of 830,329 reads belonging to 107 OTUs, 68 families, 8 and 6 classes for Ascomycota and Basidiomycota, respectively, were retrieved (Figures 1B,C,2B–D and Supplementary Table S2). Overall, five fungal phyla were retrieved: Ascomycota, Basidiomycota, Chytridiomycota, Glomeromycota and Mortierellomycota (Figure 2B).
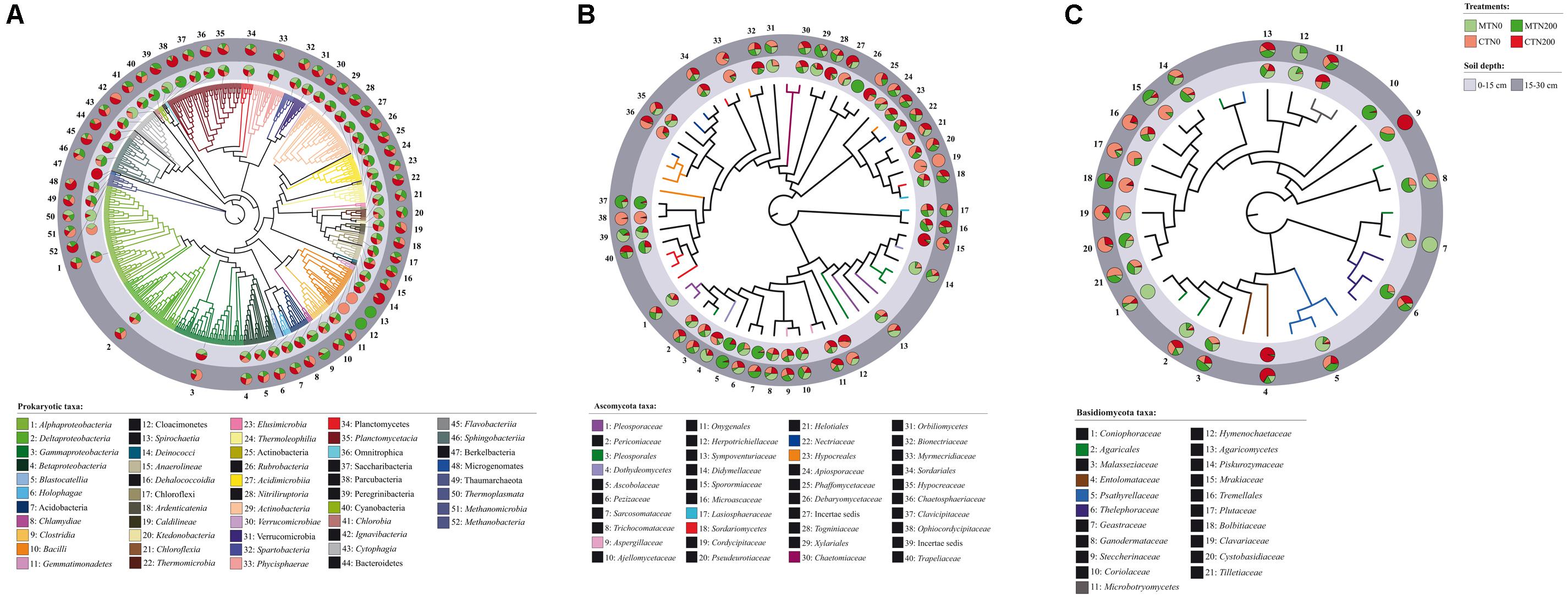
Figure 1. Neighbor-joining (NJ) tree of 52 prokaryotic taxon representative sequences (A) and Maximum Likelihood (ML) trees of 40 Ascomycota (B) and 21 Basidiomycota taxon representative sequences (C) found in soil under long-term tillage and nitrogen fertilization. Treatments are: MTN0 (minimum tillage and 0 kg N ha–1), MTN200 (minimum tillage and 200 kg N ha–1), CTN0 (conventional tillage and 0 kg N ha–1) and CTN200 (conventional tillage and 200 kg N ha–1). Soil depths are: 0–15 and 15–30 cm soil depths. NJ tree of prokaryotes is based on the sequences obtained from the amplification of the V4 region (16 SSU rRNA gene) and ML trees of fungi are based on the sequences obtained from the amplification of the ITS1 region (for details see Supplementary Table S1). The prokaryotic taxa were assigned to Operational Taxonomic Unit (OTU) by BLAST against the 16S SSU SILVA database by clustering sequence reads at the 97% similarity threshold. The fungal taxa were assigned by BLAST against the ITS UNITE database and were selected by dynamic threshold values of clustering. For each OTU, the proportion of sequences retrieved from treatment (MTN0, light green; MTN200, dark green; CTN0, light red; CTN200, dark red) and soil depths (0–15 cm: light gray; 15–30 cm: dark gray) are shown in the pie charts. The name of each OTU is composed by the name of the class/phylum phylogenetic resolution and a serial number that is reported also in the trees.
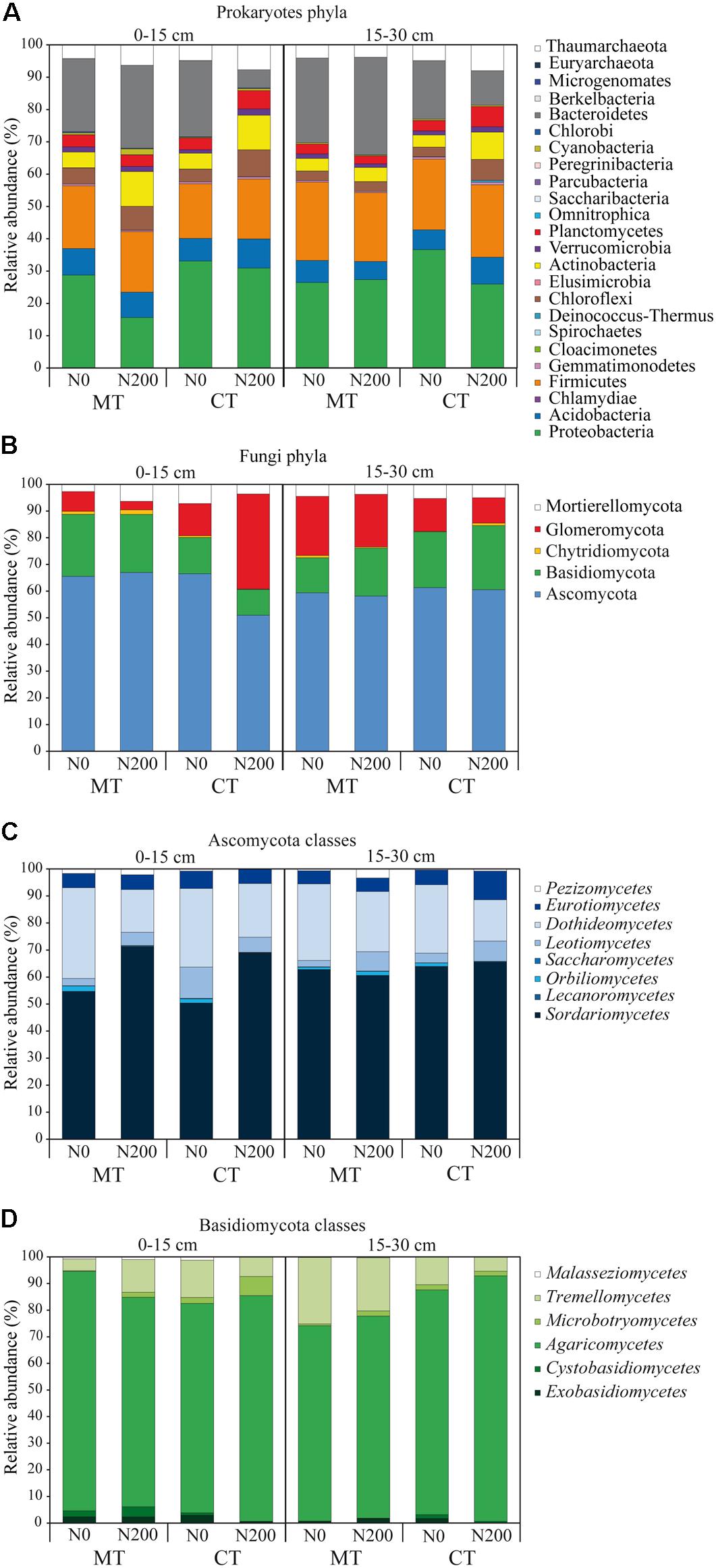
Figure 2. Community diversity of prokaryotic phyla (A), fungal phyla (B), Ascomycota classes (C), Basidiomycota classes (D) shown as relative abundances of operational taxonomic units found in soil under long-term tillage and nitrogen fertilization. Treatments are: MTN0 (minimum tillage and 0 kg N ha–1), MTN200 (minimum tillage and 200 kg N ha–1), CTN0 (conventional tillage and 0 kg N ha–1) and CTN200 (conventional tillage and 200 kg N ha–1). Soil depths are: 0–15 and 15–30 cm soil depths.
Long-Term Effect of Conservation Tillage and N Fertilization on Community Diversity, Composition and Structure of Soil Prokaryotes (Hypothesis 1)
Prokaryotic richness at class and phylum level at 0–15 cm and at class level at 15–30 cm were not modified either by tillage or N fertilization, whereas at phylum level at 15–30 cm it was modified by tillage (MT < CT: 17.34 vs. 19.34 number of OTUs) (Supplementary Table S3). Shannon and Simpson indexes (H’ and λ, respectively) were significantly increased by N fertilization at both class and phylum level at 0–15 cm (N200 > N0), whereas at 15–30 cm only H’ at phylum level was increased by tillage (CT > MT).
The total number of prokaryotic classes at 0–15 cm was 47, 44, 49 and 47 under MTN0, MTN200, CTN0 and CTN200, respectively, while at 15–30 was 47, 48, 48 and 49, respectively (Supplementary Figures S1A,B). Looking at a higher phylogenetic resolution level, the total number of prokaryotic phyla at 0–15 cm was 20, 20, 23, 21 under MTN0, MTN200, CTN0 and CTN200, respectively, while at 15–30 was 21 for all treatments (Supplementary Figures S1D,E). Proteobacteria (28%), Bacteroidetes (20%), Firmicutes (20%), Acidobacteria (7%), Actinobacteria (6%), Thaumarchaeota (5%), Chloroflexi (5%), Plantomycetes (4%), Verrucomicrobia (2%), were the dominant phyla, whereas the other phyla occurred with relative abundances below 1% (Figure 2A). Although the majority of those taxa were common to all treatments at both soil depths (0–15 cm: 43 classes and 19 phyla; 15–30 cm: 41 classes and 19 phyla), some were exclusively found in each treatment (Supplementary Figures S1A,B,D,E). Specifically, at 0–15 cm soil depth, the phylum Cloacimonetes (#12) and the class Spirochaetia (#13) within the phylum Spirochaetes were retrieved in CTN0, whereas the phylum Berkelbacteria (#47) was retrieved in CTN200 (Figure 1A). By contrast, at 15–30 cm soil depth, the phylum Cloacimonetes and the class Spirochaetia were exclusively retrieved in MTN200. However, these unique taxa occurred at very low relative abundances (data not shown).
The relative abundance of the other taxa at both soil depths was largely different among treatments, as shown in the pie/bar charts of Figures 1A, 2A. PERMANOVA showed that at 0–15 cm prokaryotic community structure at class level was significantly affected by N fertilization and by the interaction between tillage and N fertilization (Supplementary Table S4). In the PCO biplot, the first two principal coordinates explained 82.4% of the total variance (Figure 3A), and the variation partitioning analysis highlighted that N fertilization explained 22% of total variance (Table 1), in agreement with the fact that samples clearly clustered in the biplot in two groups along the first axis. However, while the community structure of soil prokaryotes under MTN0, MTN200 and CTN0 were almost similar (e.g., Alphaproteobacteria, Sphingobacteriia, Betaproteobacteria, Deltaproteobacteria), CTN200 showed a different prokaryotic community structure composed by many taxa (e.g., Chloroflexi, Verrucomicrobia, Actinobacteria, Acidimicrobiia, Bacilli, Thermoleophilia, Thermomicrobia, Thaumarchaeota) (Figure 3A). This was supported by the large amount of total variance explained by the interaction between tillage and N fertilization (50%) (Table 1). PERMANOVA showed that, at the same soil depth, prokaryotic community structure at phylum level was significantly affected only by N fertilization, which explained 24% of total variance (Table 1 and Supplementary Table S4), according to the total variance explained by the first principal coordinate (78.2% of the total variance) and to the sample clustering along the first axis (N0: Cloacimonetes; N200: Acidobacteria, Actinobacteria, Berkelbacteria, Chloroflexi, Plantomycetes, Thaumarchaeota, Verrucomicrobia etc.) (Figure 3B). Moreover, the output of PERMDISP for both analyses confirmed the differences in community dispersion between N fertilization treatments (Supplementary Table S4). By contrast, at 15–30 cm, no effect of tillage and N fertilization was reported on prokaryotic community structure at both class and phylum level.
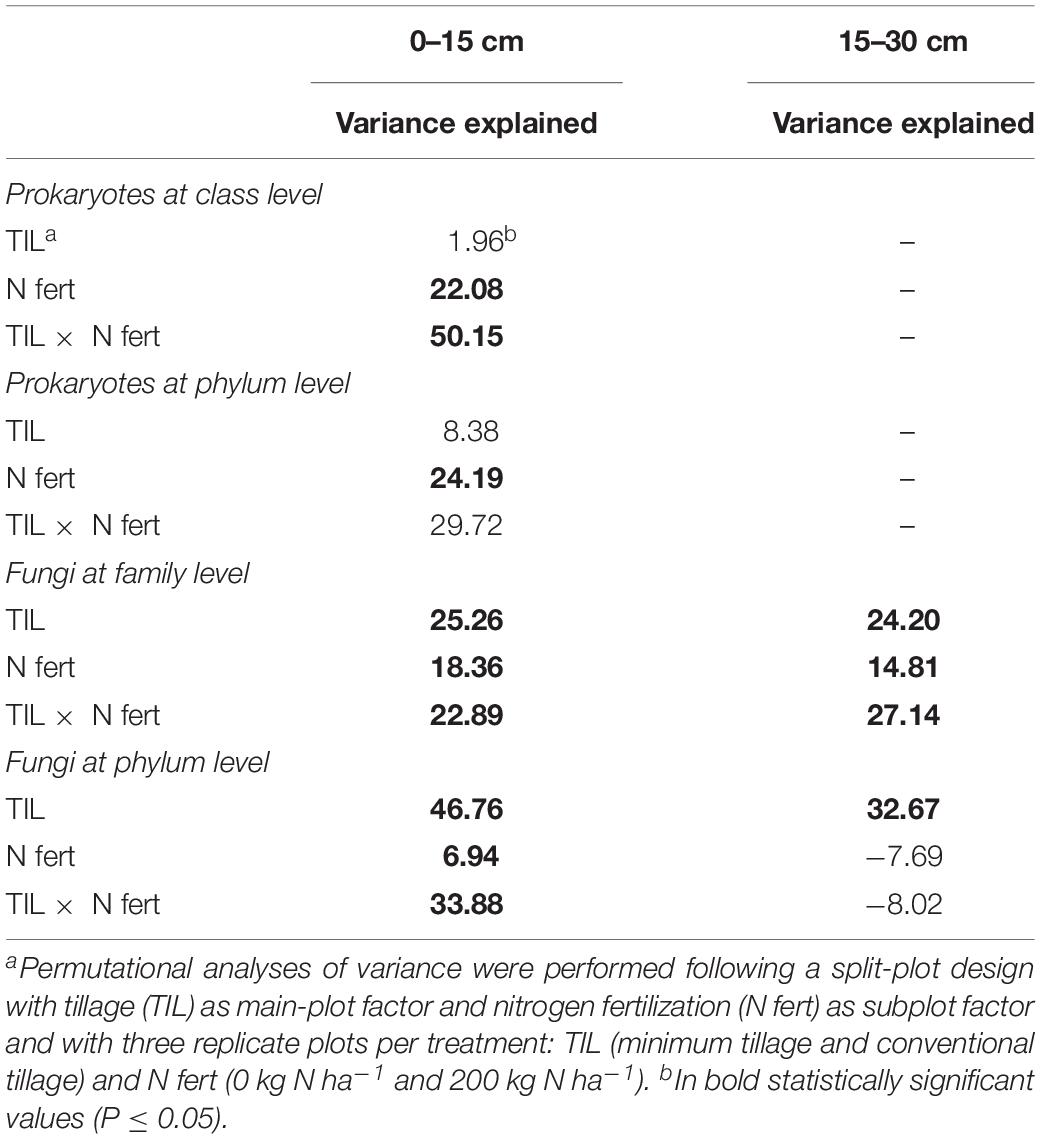
Table 1. Variation partitioning of the long-term effect of tillage and nitrogen fertilization on soil prokaryotic and fungal diversity at different phylogenetic resolution and two soil depths, in a wheat-soybean rotation in the Mediterranean area.
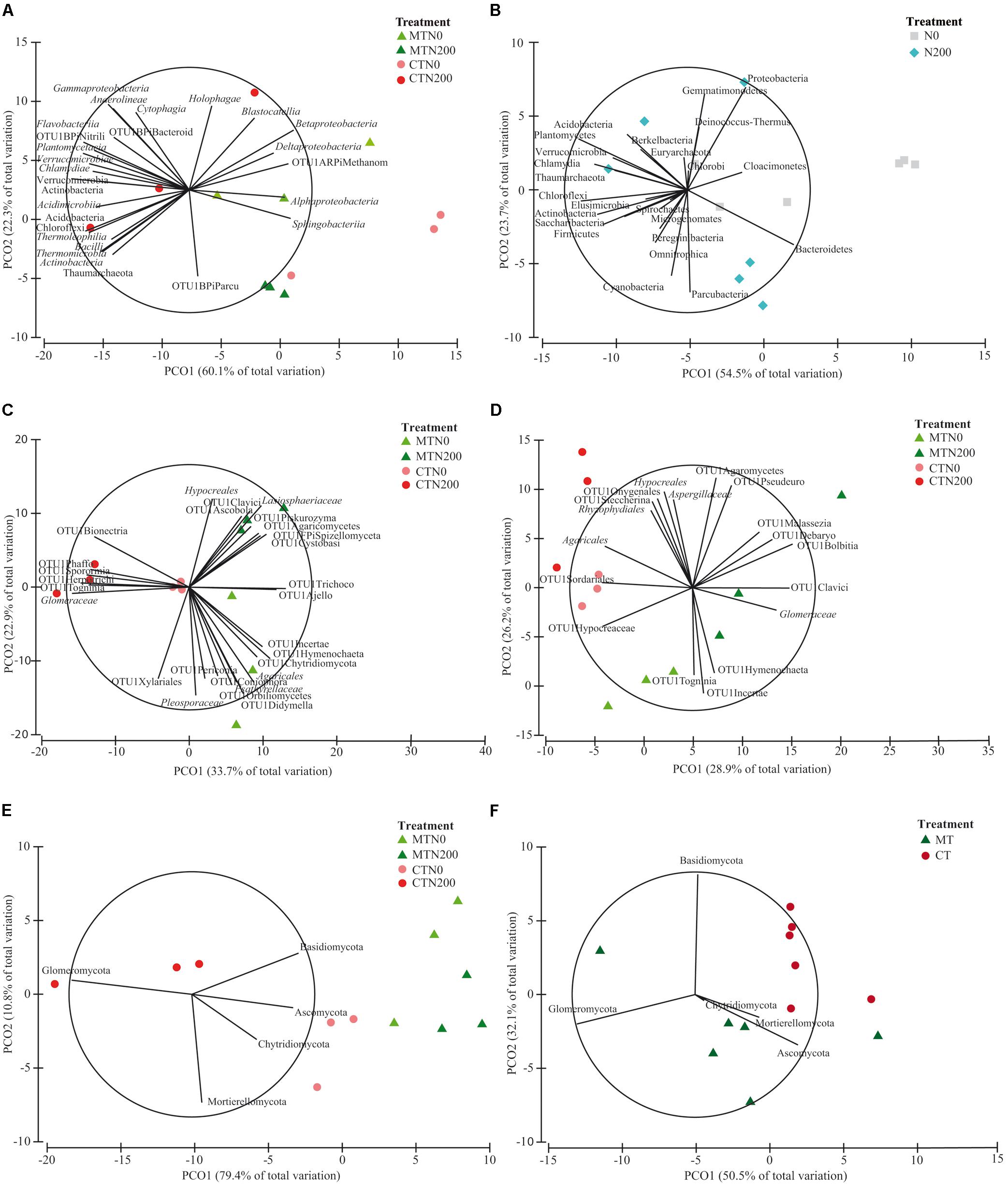
Figure 3. Principal Coordinates Analysis (PCO) biplots on the long-term effect of tillage and nitrogen fertilization on community diversity of prokaryotic classes at 0–15 cm soil depth (A), prokaryotic phyla at 0–15 cm (B), fungal families at 0–15 cm (C), fungal families at 15–30 cm (D), fungal phyla at 0–15 cm (E), fungal families at 15–30 cm (F). The output of the subfigures is based on the significant effect of treatments following the permutational analysis of variance. In each PCO biplot, we displayed only the phyla, classes and families with a strong correlation (r ≥ 0.70) with the ordination scores on each PCO axis. Data standardized to the median were square-root transformed and Bray-Curtis coefficients of similarity were calculated between samples.
Long-Term Effect of Conservation Tillage and N Fertilization on Community Diversity, Composition and Structure of Soil Fungi (Hypothesis 1)
As regard soil fungi, a lower variability due to treatments was observed respect to prokaryotes (Supplementary Table S3). In MT, N fertilization increased fungal richness at family level at 0–15 cm, whereas in CT it determined a decrease. Moreover, at the same phylogenetic resolution and soil depth, N fertilization did not modify the diversity indexes (H’ and λ) under MT, whereas it decreased both indexes under CT. At 15–30 cm, tillage affected only λ at family level (MT < CT).
The total numbers of fungal families at 0–15 cm was 60, 67, 63, and 60 under MTN0, MTN200, CTN0 and CTN200, respectively, while at 15–30 was 64, 63, 63, and 62, respectively (Supplementary Figures S1G,H). At a higher phylogenetic resolution level, the number of fungal phyla at 0–15 and 15–30 cm was five for all treatments (Figure 2B). The relative abundance of the five phyla was as follows: Ascomycota (61%), Basidiomycota (18%), Glomeromycota (15%), Mortierellomycota (5%), and Chytridiomycota (1%). Looking at a higher phylogenetic resolution, the increasingly predominant classes of Ascomycota were Leotiomycetes (6%), Eurotiomycetes (6%), Dothideomycetes (24%), Sordariomycetes (62%), while those of the Basiodiomycota were Exobasidiomycetes (2%), Microbotryomycetes (2%), Tremellomycetes (12%) and Agaricomycetes (82%) (Figures 2C,D). By contrast, each of the other classes accounted for less than 1%. Although the majority of fungal taxa were common to all treatments at both soil depths (0–15 cm: 53 families; 15–30 cm: 59 families), some were exclusively found in each treatment (Supplementary Figures S1G,H). Specifically, at 0–15 cm, the families Coniophoraceae (#1; Basidiomycota) and Debaryomycetaceae (#26; Ascomycota) were retrieved in MTN0 and in MTN200, respectively. At 15–30 cm, the families Geastraceae (#7; Basidiomycota), Cordycipitaceae (#19; Ascomycota) and Steccherinaceae (#9; Basidiomycota) were exclusively retrieved in MTN0, CTN0 and CTN200, respectively. However, these unique taxa occurred at very low relative abundances (data not shown).
The relative abundance of the other taxa at both soil depths was largely different among treatments as shown in the pie/bar charts of Figures 1B,C, 2B–D. PERMANOVA showed that fungal community structure at family and phylum level was significantly affected by tillage, N fertilization and the interaction between tillage and N fertilization at 0–15 cm (Supplementary Table S4). Looking at corresponding PCO biplots, the first two principal coordinates explained 56.6 and 90.2% of the total variance, respectively (Figures 3C,E). At family level, tillage, N fertilization and their interaction explained 25, 18 and 23% of total variance, respectively, while at phylum level, the same treatments explained 47, 7, 34% of total variance, respectively (Table 1). At family level, samples belonging to MT and CT clearly clustered into two separated groups along the first axis (PCO1: 33.7% of total variance) (Figure 3C). Within MT, samples belonging to N0 and N200 were separated in two clusters along the second axis (PCO2: 22.9% of total variance), showing a strong difference of their fungal community structure (e.g., MTN0: OTU1Chytridiomycota, OTU1Coniophora, Pleosporaceae, OTU1Didymella; MTN200: Lasiophaeriaceae). Within CT, many fungal families were commonly found in N0 and N200, but their relative abundances were consistently higher in N200 than in N0 (e.g., Glomeraceae, OTU1Herpotrichi and OTU1Phaffo). At phylum level, samples belonging to MT and CT clearly clustered into two separated groups along the first axis (PCO1: 79.4% of total variance), while samples belonging to N0 and N200 clustered into two separated groups along the second axis (PCO2: 10.8% of total variance) (Figure 3E). Basidiomycota, Ascomycota and Chytridiomycota were highly abundant in MTN0, MTN200 and CTN0, whereas Glomeromycota in CTN200.
PERMANOVA showed that fungal community structure at family level was significantly affected by tillage, N fertilization and the interaction between tillage and N fertilization at 15–30 cm, whereas at phylum level only by tillage (Supplementary Table S4). At family level, tillage, N fertilization and their interaction explained 24, 15, 27% of total variance, respectively, whereas at phylum level tillage explained 33% of total variance (Table 1). At family level, MT and CT samples clustered into two separated groups along the first axis (PCO1: 28.9% of total variance) (Figure 3D). Within MT, samples under N0 and N200 were separated in two clusters along the second axis (PCO2: 26.2% of total variance) (e.g., MTN0: OTU1Hymenochaeta, OTU1 Togninia; MTN200: Glomeraceae, OTU1Malassezia and OTU1Debaryo). Within CT, samples under N0 and N200 were more similar than in MT, although in N0 some fungal families were more abundant than in N200 (e.g., Agaricales, OTU1Sordariales, OTU1Steccherina). At phylum level, MT and CT samples clustered into two groups along the first axis (PCO1: 50.5% of total variance), with Glomeromycota highly abundant in MT (Figure 3F). PERMDISP for all the significant analyses generally confirmed the differences in fungal community dispersion (Supplementary Table S4).
Role of Soil Depth in Shaping Diversity, Composition and Structure of Soil Prokaryotes and Fungi (Hypothesis 2)
Richness, H’ and λ of prokaryotes at both class and phylum level were higher at 0–15 than at 15–30 cm (Supplementary Table S5). As regard fungi, no differences were observed according to soil depth at both family and phylum levels.
The majority of prokaryotic and fungal taxa were common to both soil depths (prokaryotes: 51 classes and 22 phyla; fungi: 66 families) (Supplementary Figures S1C,F,I). The prokaryotic taxa exclusively found at 0–15 cm and 15–30 cm were the phylum Berkelbacteria (#47) and the class Methanobacteria (#52; Euryarchaeota), respectively (Figure 1A). The fungal taxa exclusively found at 0–15 cm were the families Coriolaceae (#10; Basidiomycota) and Debarymycetaceae (#26; Ascomycota). These unique taxa occurred at very low relative abundances (data not shown).
PERMANOVA showed that soil depth had a significant effect on prokaryotic community structure at class and phylum level, whereas it affected fungal structure only at family level (Supplementary Table S6). In the corresponding PCO biplots, the first two principal coordinates explained 70.3, 78.6 and 34.0% of the total variance (Figure 4), and the variation partitioning analysis highlighted that soil tillage depth explained 37, 10, 21% of total variance, respectively (Table 2), in agreement with the fact that samples clearly and consistently clustered in the biplots in two groups along the second axis. For prokaryotes, the biplots highlighted that almost all the families and phyla were more abundant at 0–15 cm than at 15–30 cm (Figures 4A,B). For fungi, the community structure at 0–15 cm was characterized by Glomeraceae (Glomeromycota), OTU1Sordariales (Ascomycota), OTU1 Phaffo (Ascomycota), whereas the community structure at 15–30 cm was characterized by Agaricales (Basidiomycota), and OTU1Mrakia (Basidiomycota). PERMDISP for all the analyses confirmed the differences in community dispersion between the two soil depths (Supplementary Table S6).
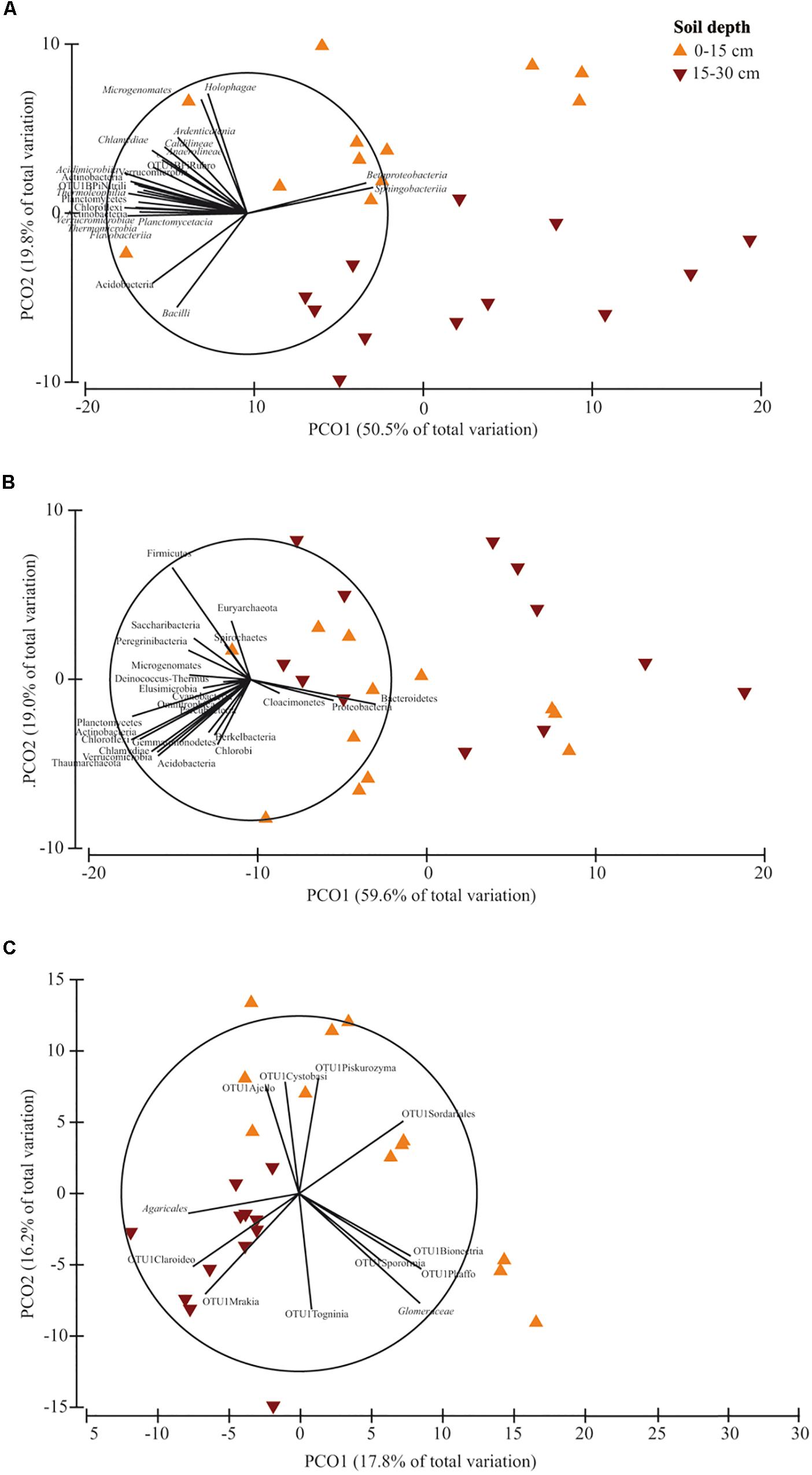
Figure 4. Principal Coordinates Analysis (PCO) biplots on the effect of soil depth (0–15 vs. 15–30 cm) on community diversity of prokaryotic classes (A), prokaryotic phyla (B) and fungal families (C). Tillage and nitrogen fertilization were used as covariates. The output of the subfigures is based on the significant effect of soil depth following the permutational analysis of variance. In each PCO biplot, we displayed only the phyla, classes and families with a strong correlation (r ≥ 0.70) with the ordination scores on each PCO axis. Data standardized to the median were square-root transformed and Bray-Curtis coefficients of similarity were calculated between samples.

Table 2. Variation partitioning of the effect of soil depth (0–15 vs. 15–30 cm) on soil prokaryotic and fungal diversity at different phylogenetic resolution in a wheat-soybean rotation in the Mediterranean area.
Linking Soil/Yield Parameters to Microbial Community Structure (Hypothesis 3)
Co-inertia analyses highlighted significant relationships between soil/yield parameters and fungal community structure at family level at both soil depths (Figure 5). Co-inertia values were 0.253 and 0.209 at 0–15 and 15–30 cm, respectively. The significance of the relationships between data sets was verified by the Mantel test (0–15 cm: r = 0.314, P = 0.031; 15–30 cm: r = 0.304; P = 0.050). At 0–15 cm soil depth, the axis 1 and axis 2 loadings of the first step account for 25.9% and 18.6% of the total variance, respectively (Figure 5A) and at 15–30 cm for 22.2 and 14.4% of the total variance, respectively (Figure 5B). Moreover, at 0–15 cm, the axis 1 and axis 2 loadings of the second step account for 57.0 and 28.8% of the total variance, respectively (Figure 5A) and at 15–30 cm for 57.7 and 27.6% of the total variance, respectively (Figure 5B).
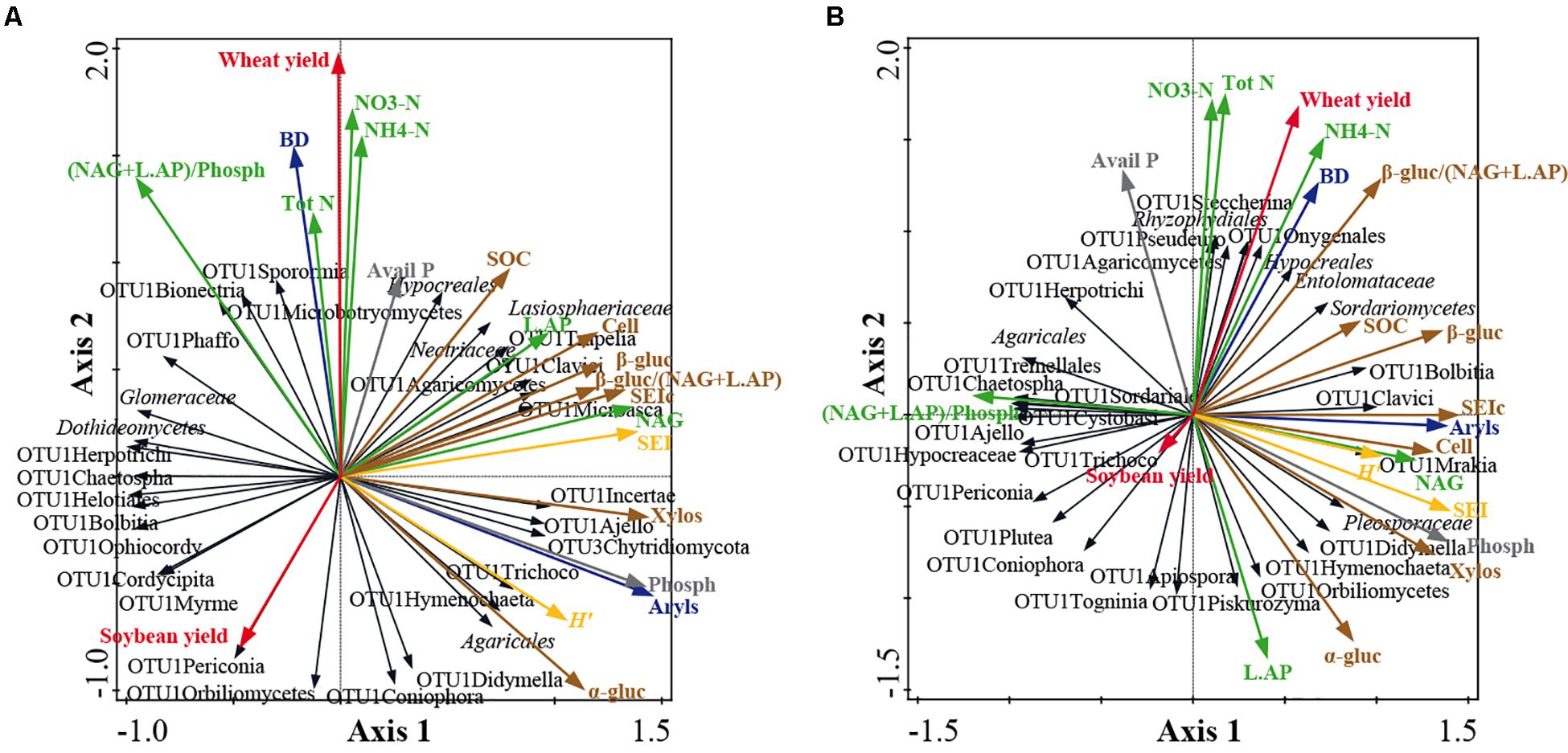
Figure 5. Co-inertia biplots on fungal community structure at family level and soil physico-chemical and enzymatic parameters at 0–15 cm soil depth (A) and at 15–30 cm soil depth (B). See Figures 1B,C and Supplementary Table S2 for fungal taxon assignment. The colored arrows represent the soil parameters: bulk density (BD) and arylsulphatase (Aryls) (blue arrows); wheat and soybean yields (red arrows); P cycle parameters: available P, Avail P; phosphatase, Phosph (gray arrows); N cycle parameters: total N, Tot N; ammonium, NH4-N; nitrate, NO3-N; chitinase, NAG; Leucine, AP, L.AP; ecoenzymatic N/P acquisition activity, (NAG + L.AP)/Phosph (green arrows); C cycle parameters: soil organic carbon, SOC; cellulose, Cell; α-glucosidase, α-gluc; β-glucosidase, β-gluc; xylosidase, Xylos; synthetic enzyme index of C cycle, SEIc; ecoenzymatic C/N acquisition activity, β-gluc/(NAG + L.AP) (brown arrows); enzyme indexes: synthetic enzyme index, SEI; Shannon index, H’ (yellow arrows). The black arrows represent fungal taxa. The lengths of the vector arrows indicate the influence of the parameters on the co-inertia co-structure.
At both soil depths, the C parameters discriminated the fungal community structure along axis 1, whereas N parameters and crop yields mostly discriminated along axis 2 (Figure 5). Moreover, at both soil depths, all C-cycling enzymes were positively related to SOC. At 0–15 cm, the fungal taxa positively associated to C parameters were: Lasiospheraceae, OTU1Clavici, OTU1Agaricomycetes, OTU1Microasca and OTU1Hymenochaeta (Figure 5A). At 15–30 cm, the fungal taxa positively associated to C parameters were: Sordariomycetes, Pleosporaceae, OTU1Bolbitia, OTU1Clavici, OTU1Didymella and OTU1Hymenochaeta (Figure 5B).
At 0–15 cm, all N enzymes were positively related to N in soil (N tot, NO3-N and NH4-N), whereas at 15–30 cm they were negatively related (Figure 5). At 0–15 cm, the fungal taxa positively associated to N parameters were: Nectriaceae, OTU1Trapelia, OTU1Sporormia, and OTU1Microbotryomycetes (Figure 5A). At 15–30 cm, the fungal taxa associated to N parameters were: OTU1Steccherina, Rhyzophydiales and OTU1Agiospora (Figure 5B). At 0–15 cm soil depth, N in soil was positively related to wheat yield and negatively with soybean yield, whereas at 15–30 cm N in soil was positively related only to wheat yield (Figure 5).
Several taxa were negatively related to C and P parameters at 0–15 cm (e.g., Glomeraceae, Dothideomycetes, OTU1Herpotrichi and OTU1Bolbitia) and at 15–30 cm (e.g., OTU1Periconia, OTU1Hypocreaceae, OTU1Ajello and Agaricales) (Figure 5). Moreover, some other taxa were negatively related to N in soil at 0–15 cm (e.g., OTU1Coniophora, OTU1Orbiliomycetes and OTU1Didymella) (Figure 5A). By contrast, no significant relationships were observed along the soil profile for prokaryotes at both phylogenetic resolutions and for fungi at phylum level (data not shown).
Discussion
Here, using the high-throughput resolution of Illumina sequencing, we found that (i) the majority of prokaryotic and fungal taxa were common to all treatments at both soil depths; (ii) few bacterial taxa (Cloacimonates, Spirochaetia and Berkelbacteria) and a larger number of fungal taxa (i.e., Coniphoraceae, Debaryomycetaceae, Geastraceae, Cordicypitaceae and Steccherinaceae) were unique to specific management practices; (iii) soil prokaryotic and fungal structure was heavily influenced by the interaction of tillage and N fertilization; (iv) the prokaryotic community structure of the fertilized conventional tillage system was remarkably different respect to the unfertilized conservation and conventional systems in the surface layer; (v) the effect of N fertilization in shaping the fungal community structure of the surface layer was higher under conservation tillage systems than under conventional tillage systems; (vi) soil microbial community was shaped by soil depth irrespective of the effect of plowing and N addition; (vii) chemical and enzymatic parameters of soil and crop yields were significantly related to fungal community structure along the soil profile.
Long-Term Effect of Conservation Tillage and N Fertilization on Diversity of Soil Prokaryotes and Fungi
The diversity of soil prokaryotes at class and phylum level, as measured by Shannon and Simpson index, at 0–15 cm soil depth was increased by N fertilization (+2%). It is possible to infer that in the surface layer, where tillage disturbs soil, N fertilization increases the homogeneity among taxa as well as the occurrence of the most common taxa. Accordingly, de Graaff et al. (2019) found in several field studies across the world, including the Mediterranean area, that N fertilization increased soil bacterial diversity by 3%. This result was explained by the addition of N organic fertilizers, alone or in combination with mineral fertilizers. It is interesting to highlight that, in line with the length of our experiment, the duration of N mineral fertilizer addition (≥5 years) was pointed out as a major determinant for the increase of soil bacterial diversity.
By contrast, N fertilizer addition decreased soil fungal diversity up to 5% (richness, H’ and λ) in the surface layer under the plowed system (CT) and not in the disk-harrowed system (MT), where only fungal richness was promoted (+7%). To date, the effects of N enrichment on fungal diversity was less studied in agricultural soil compared to forests (e.g., Wallenda and Kottke, 1998; Eom et al., 1999; Egerton-Warburton and Allen, 2000; Peter et al., 2001; Avis et al., 2003; Frey et al., 2004; Jumpponen and Johnson, 2005; Antoninka et al., 2011; Lin et al., 2012). Many of those studies were based on the assessment of ecto- and endo-mycorrhizal diversity and not on total fungi. Recently, Leff et al. (2015), by analyzing the soil fungal diversity of 25 globally distributed grasslands, observed that Shannon index responds weakly to N addition, although the diversity of Glomeromycota was consistently reduced. In our study, the reduction of total fungal diversity may be explained by a toxic effect of urea due to its hydrolyzation after application, leading to a net decrease of soil pH (Omar and Ismail, 1999). In addition, the different response of fungal diversity indexes (H’ and λ) to N fertilization between minimum and conventional tillage may be driven by the increase of fungal richness observed under N fertilization in MT respect to the reduction in CT.
In the subsurface layer (15–30 cm soil depth), the richness and H’ of prokaryotes at phylum level was increased in CT compared to MT, supporting the idea that plowing is not detrimental for the diversity of soil prokaryotes. The higher BD found in deeper soil layers in MT systems results in a reduction of pore size and soil aeration, causing local anoxic conditions and therefore the reduction of the diversity of prokaryotes (Berisso et al., 2012).
Long-Term Effect of Conservation Tillage and N Fertilization on Community Composition of Soil Prokaryotes and Fungi
A conserved core community of soil prokaryotic and fungal taxa was retrieved across treatments at both soil depths and at different phylogenetic resolutions. Indeed, averaged over soil depths, 85% and 81% of total prokaryotes at class and phylum resolution, respectively, were shared among treatments, whereas 85% of total fungi were shared at family level. The fact that soil microbes are strongly conserved throughout these long-term treatments is surprising and noticeable.
Interestingly, at 0–15 cm soil depth, the bacterial phylum Cloacimonetes and the class Spirochaetia were the taxa uniquely found in unfertilized conventional tillage (CTN0), whereas the newly recognized phylum Berkelbacteria (Wrighton et al., 2014) was uniquely found in conventional fertilized tillage (CTN200). Cloacimonetes is a mesophylic phylum able to degrade cellulose and produce methane, while the members of Spirochaetia degrade cellulose and chitin (Limam et al., 2014; Chojnacka et al., 2015; Dai et al., 2016; Deng et al., 2018) and anaerobically decompose organic matter (Baba et al., 2014). Moreover, the phylum Berkelbacteria is an ATP binding and a putative transcriptional regulator with helix-turn-helix (HTH)-like protein. By contrast, at 15–30 cm soil depth, the phylum Cloacimonetes and the class Spirochaetia were the taxa uniquely found in fertilized minimum tillage (MTN200). These results point at the overall metabolic flexibility of these groups of bacteria or at the presence of different members belonging to the same group, but possessing diverse metabolic potential.
As regard fungi, the Basidiomycota family Coniophoraceae, exclusively found under MTN0 in the surface layer, includes saprophytic brown-rot fungi (Thorn et al., 1996; Lee et al., 2004; Utobo and Tewari, 2015), as well as the mycorrhizal fungi Boletales spp., and other members known to be cellulolytic. Moreover, the Ascomycota family Debaryomycetaceae, exclusively found under MTN200 in the surface layer, includes xylose-fermenting yeasts (Steindorff et al., 2016) Cordycipitaceae, exclusively found in the subsurface layer of CTN0 and CTN200, is a family belonging to Ascomycota including entomophatogenic fungi (Vega et al., 2009), whereas the Steccherinaceae, found in the same conditions, is a family belonging to Basidiomycota, includes fungi that produce proteinase (Kudryavtseva et al., 2008). Finally, Geastraceae, exclusively found in MTN0, are a family belonging to Basidiomycota and including fungi that decay SOM and recycle C and N, also acting as biofertilizers (Eriksson et al., 1990; Gadd, 2001).
Long-Term Effect of Conservation Tillage and N Fertilization on Community Structure of Soil Prokaryotes and Fungi
In recent years, using molecular tools, such as Sanger or next-generation sequencing, the effect of tillage or N fertilization were explored to give indications on management practices that support beneficial taxa and suppress detrimental ones (Hartmann et al., 2015; Francioli et al., 2016). These studies assessed soil bacteria and fungi (e.g., tillage and bacteria/fungi: Mirás-Avalos et al., 2011; Bevivino et al., 2014; Sharma-Poudyal et al., 2017; N fertilization and bacteria/fungi: Lienhard et al., 2014; Ding et al., 2017; Li et al., 2017; Wang et al., 2018). Other studies focused on the interactions of tillage or N fertilization with crop rotation (Degrune et al., 2017; Ai et al., 2018). Only one study, carried out in a long-term vineyard experiment in Germany, found the interaction between soil tillage and N fertilization as major explanatory variable for the shift of soil bacterial and fungal communities (Pingel et al., 2019). In our study, despite the high degree of similarity in term of microbial composition among treatments, we firstly found that the prokaryotic and fungal community structure is strongly shaped by the interaction of tillage and N fertilization. However, N fertilization played a crucial role in shaping prokaryotic and fungal assemblages, whereas for soil fungi tillage was the additional main driver. In our study, Alpha-, Beta-, and Delta-proteobacteria showed higher relative abundances at the surface layer in the minimum soil-disturbed system irrespective of N fertilization as well as in the plowed unfertilized one. Alphaproteobacteria is a class important for cellulose hydrolyzation, not affected by the addition of N/P and known to actively fix N2 (e.g., Rhizobia) (Pankratov et al., 2006). As regard the fertilized conventional tillage, we found a high number of abundant taxa compared to the other treatments, such as Acidimicrobiia, Actinobacteria, Bacilli, Chloroflexi, Gammaproteobacteria and Verrucomicrobia. Among these taxa, the high abundance of the class Acidimicrobiia and of the whole phylum Acidobacteria may have been determined by a reduction of pH with urea application (Omar and Ismail, 1999). Acidobacteria were described as low-growing oligotrophics (or K-selected) and less present with high concentration of labile C (Fierer et al., 2007), and were found highly present at low N fertilization rates in several studies (Fierer et al., 2007; Ramirez et al., 2010; Eo and Park, 2016; Francioli et al., 2016; Wang et al., 2018). This inconsistency can be explained by the fact that not all the members of the phylum can be considered distinctly copiotrophic or oligrotrophic and therefore their functional behavior can be variable. The members of the phylum Actinobacteria are well-known decomposers, chitin utilizers, rhizosphere colonizers and strongly responding to root exudates (DeAngelis et al., 2008, 2009). Similar to results of our study, Actinobacteria were shown to increase with high N input (Ramirez et al., 2010). The high presence of Bacilli is positive since they cohabit in the rhizosphere with AMF and promote plant growth by producing phytohormones (e.g., auxins and gibberellins) (Gutiérrez-Manero et al., 1996; Gutiérrez-Mañero et al., 2001; Medina et al., 2003; Ramesh et al., 2014). In agreement with our results, Chlroflexi were found abundant under fertilized systems (Francioli et al., 2016; Jiménez-Bueno et al., 2016). This phylum includes filamentous bacteria playing a role in anaerobic ammonium oxidation, degradation of SOM, using sucrose, glucose and N-acetyl-glucosamine (Kragelund et al., 2007; Kindaichi et al., 2012). Gammaproteobacteria is a class generally found under high N level following manure application, playing a crucial role in the decomposition of labile C compounds and in the control of bacterial pathogens (Fierer et al., 2007; Eo and Park, 2016; Francioli et al., 2016). Finally, the phylum Verrucomicrobia was found to be influenced by soil management and nutritional regimes (Buckley and Schmidt, 2001; Kielak et al., 2009).
By looking at the prokaryotic shifts at a lower level of resolution (phylum level), we could detect only the effect N fertilization, supporting the fact that it is important to utilize molecular tools at high resolution to allow a deeper discrimination.
As regards soil fungi, we found a strong interaction between tillage and N fertilization not only in the surface layer, as for prokaryotes, but also in the subsurface layer, confirming that this eukaryotic community could be more sensitive to management practices respect to prokaryotes (Degrune et al., 2017; Sharma-Poudyal et al., 2017). At the surface layer, the phylum Chytridiomycota and the families Coniophoraceae and Pleosporaceae were highly abundant in MTN0. Chytridiomycota comprises either saprophytic or parasitic fungi that degrade cellulose and secrete many extracellular enzymes (Gulis et al., 2008; Kameshwar and Qin, 2016). In line with our results, the family Pleosporaceae, belonging to the class of Dothideomycetes, which comprises many saprophytic and plant pathogens (e.g., Alternaria spp.), was not affected or decreased by mineral fertilization (Freedman et al., 2015; Francioli et al., 2016; Ding et al., 2017). On the other hand, we found the family Lasiosphaeraceae, which includes wood-decaying fungi (Nordeìn and Paltto, 2001; Cannon and Kirk, 2007), highly abundant under MTN200, and this is in line with previous results (Hartmann et al., 2015; Ding et al., 2017). Moreover, N fertilization under conventional tillage increased the abundance of the Ascomycota families Phaffomycetaceae, including yeasts, utilizing different N sources (Linder, 2019), and Herpotrichiellaceae highly abundant under N fertilized conditions (Weber et al., 2013; Ding et al., 2017). Although it was expected that reduced soil disturbance and low N availability would be more beneficial for AMF (Liu et al., 2015; Mbuthia et al., 2015; Bowles et al., 2017), we surprisingly found a strong effect of the interaction between tillage and N fertilization. Indeed, Glomeraceae at the surface layer were more abundant under CTN200 compared to the other treatments and at the subsurface layer they were more abundant under MTN200. The positive effect of N fertilization on Glomeraceae is likely due not to the direct effect of increase of N availability, but to the indirect effect of promotion of plant growth and root size. In the surface layers, where the majority of roots are concentrated, loosed and aerated soil promote highly competitive taxa able to rapidly recover following tillage, while in the subsurface hard soil, prevail taxa able to tolerate low oxygen availability, but highly sensitive to the disturbance caused by tillage (Jansa et al., 2002; Avio et al., 2006; Voets et al., 2006).
Role of Soil Depth in Shaping Diversity, Composition and Structure of Soil Prokaryotes and Fungi
Although it is widely acknowledged that soil microbes affect nutrient cycling along the soil profile, to date the understanding of the diversity, composition and structure of soil prokaryotic and fungal communities is mainly limited to the surface layer, with the majority of studies targeting the up 15 cm. The higher diversity of prokaryotes found in this study in the surface layer respect to the deeper layer was previously reported by Eilers et al. (2012). By contrast, the similarity in the fungal diversity we found along the soil profile is in disagreement with previous studies that reported a declining trend in the whole fungal and AMF diversity with increasing soil depth (Oehl et al., 2005; Jumpponen et al., 2010).
Moreover, the fact that the fungal family Coriolaceae was exclusively found at 0–15 cm is positive since this group of wood-decaying white-rot fungi are able to secrete hydrolytic (e.g., endoglucanase and endoxylanase) and oxidative (e.g., manganese peroxidase and laccase) extracellular enzymes (Hofrichter, 2002; Levin et al., 2007). In addition, the prokaryotic class Methanobacteria, belonging to the largely less explored Archaea phylum Euryarchaeota, was uniquely found at 15–30 cm, in agreement with their role to reduce nitrate (Cabello et al., 2004).
Looking at prokaryotic and fungal structure at class/family resolution, we found strong differences between the two depths, irrespective of tillage and N fertilization. This is in agreement with previous studies (Blume et al., 2002; Griffiths et al., 2003; Hartmann et al., 2009). Root morphology and therefore exudation deeply vary along the soil profile (Wen et al., 2019) and this can be considered a major factor shaping microbial structure. Another factor can be the indirect effect of AMF in soil and within roots along the soil profile (Kabir et al., 1998; Oehl et al., 2005; Jones et al., 2018). This is also supported by our data showing a high relative abundance of Glomeraceae at 0–15 cm and of Claroideoglomeraceae at 15–30 cm.
Linking Soil/Yield Parameters to Microbial Community Structure
Several attempts were done in order to associate soil microbial community structures and soil parameters and/or plant species (i.e., Johnson et al., 2004; Berg and Smalla, 2009; Zhu et al., 2012; Ciccolini et al., 2015, 2016b). However, these attempts were based on the implicit assumption that microorganisms are unilaterally affected by soil parameters, crop residues or species identity/communities. Actually, microorganisms are also the main players in nutrient cycling, determining nutrient accumulation, and soil structure (Li et al., 2017; Sahu et al., 2017; Powell and Rillig, 2018). Indeed, the co-inertia analyses we performed aimed to disentangling the roles of microbial taxa in soil nutrient cycling and crop yield and to clarify how these roles vary accordingly to soil conditions. The analyses highlighted meaningful bilateral interactions with soil parameters and crop yields only for soil fungi at low taxonomic level (i.e., family level). At higher taxonomic resolution (i.e., order, class, phylum), it was not possible to detect the fungal/bacterial taxa playing functional roles in nutrient cycling and yield production. This is due to the heterogeneous composition of taxa at high taxonomic level, in terms of roles and ecosystem services (Fra̧c et al., 2018).
At surface layer, Lasiospheraceae, the fungal family belonging to Ascomycota that we found positively associated to C-cycling parameters, was previously described as saprobes only in soil of temperate forests (Cannon and Kirk, 2007). Other Ascomycota, positively related to C, were the families Clavicipitaceae and Microascaceae that were previously described as biotrophics/necrotrophs of other fungi and having a high affinity for materials of animal origin (Mycology Web pages of the New Brunswick Museum, 2019). Moreover, also members of the phylum Basidiomycota, such the class Agaricomycetes and the family Hymenochaetaceae, were highly and positively associated to SOC and enzymes linked to C-cycling. The members of Agaricomycetes are symbionts (ectomycorrhizal fungi) and saprobes playing a key role in lignocellulose decomposition (Kersten and Cullen, 2013), while the members of Hymenochaetaceae are described as parasites or saprobes (Mycology Web pages of the New Brunswick Museum, 2019). By contrast, at the subsurface layer, Pleosporaceae and Didymellaceae (phylum Ascomycota), previously described as saprobes as well as plant pathogens (Rodriguez et al., 2009), were the fungal families positively associated to the C cycle. In such a layer, main players of the C-cycling were also members of the family Bolbitiaceae (phylum Basidiomycota) that were already described as saprobes and symbionts (ectomycorrhizal fungi) (Cannon and Kirk, 2007). Moreover, the occurrence of members of the families Clavicipitaceae and Hymenochaetaceae also in the subsurface layer supports their key role in C-cycling along the profile.
At surface layer, the families Nectriaceae and Trapeliaceae, belonging to the phylum Ascomycota, and previously shown to play important roles in organic matter decomposition and being associated with green algae, respectively (Zhou et al., 2017), were positively related to N-cycling parameters, together with the ascribed saprobes Sporormiaceae (phylum Ascomycota, Pleosporales) and Microbotryomycetes (phylum Basidiomycota) (Cannon and Kirk, 2007). By contrast, at the subsurface layer, the main players in N cycle were members of the family Steccherinaceae (phylum Basiomycota) that were previously described as soil saprobes producing protolithic enzyme, such as protease (Kudryavtseva et al., 2008), and members of the order Rhizophydiales (phylum Chytridiomycota), parasites of plants, algae, protists and invertebrates. Therefore, our results support the fact that different fungal taxa play key roles in N and C cycles along the soil profile. This can be well explained taking into consideration the soil physical conditions, such as porosity, moisture, temperature and air diffusion that provide different microhabitats for soil microbes (Horn et al., 1994). The differential production of exudates from the root system mainly concentrated in the surface layer may therefore modify the patterns of accumulation of SOM and the concentration of ammonium and nitrate. Our results confirm previous conspicuous data reporting that AMF biomass in roots and soil is markedly decreased by high P availability and C rich substrates (Abbott et al., 1984; Smith et al., 2011). In addition, the quality of residues (wheat and soybean), highly discriminating the fungal structures at both soil depths, may be strongly related to the quality of crop residues, as previously found (Pellegrino et al., 2011, 2015a).
Conclusion
This study circumvented the problem of the highly complexity of microbial diversity in soil for evaluating the influence of agricultural management by an appropriate plot size, homogenizing soil samples and using a long-term field trial. A conserved core of prokaryotic and fungal taxa across tillage and N fertilizer treatments was identified. However, the interaction between tillage systems and N fertilizion strongly shifted soil prokaryotic and fungal community structure toward a higher presence of functional taxa linked to nutrient cycling, with positive implications on wheat yield. Indeed, the increase of wheat yield was associated to the soil parameters linked to N cycle and to specific fungal taxa (e.g., Nectriaceae, Trapeliaceae, Steccherinaceae), whereas the increase of SOC was associated to the other fungal taxa (e.g., C cycle: Lasiospheraceae, Microascacea, Plesoporaceae). The families Clavicipitaceae and Hymenochaetaceae were consistently associated to C cycle along the soil profile. These results, only detectable for fungi at low taxonomic level, are important advances for supporting cropping systems based on minimum tillage and adequate N fertilization in the Mediterranean area. However, whether these results are of general validity in different soil types across the Mediterranean area is still a matter for further investigation based on the identification of taxa at a lower taxonomic resolution (i.e., species) or ‘omics approaches that allow to measure gene expression linked to nutrient cycling.
Data Availability
The datasets generated for this study can be found in the NCBI Sequence Read (SRA) database (prokaryotic accession numbers MK903871–MK904485; fungal accession numbers MK881790–MK881896).
Author Contributions
EP and GP contributed to designing the experiments, collecting the data, analyzing the data, and writing the manuscript. LE contributed to designing the experiments and discussing the data. MN contributed to discussing the data. All authors reviewed and approved the manuscript before its submission.
Funding
This work was funded by the European Agricultural Fund for Rural Development 2007–2013 for Tuscany (Italy), measure 16.1 for GO groups (FERTIBIO project), project leader EP. GP was supported by a Ph.D. scholarship from Scuola Superiore Sant’Anna.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the Emeritus Professor Enrico Bonari for allowing to sample in the long-term field experiment at the Centro Interdipartimentale di Ricerche Agro-Ambientali Enrico Avanzi, San Piero a Grado, Pisa, Italy.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02047/full#supplementary-material
Footnotes
References
Abbott, L. K., Robson, A. D., and De Boer, G. (1984). The effect of phosphorus on the formation of hyphae in soil by the vesicular-arbuscular mycorrhizal fungus. Glomus fasciculatum. New Phytol. 97, 437–446. doi: 10.1111/j.1469-8137.1984.tb03609.x
Ai, C., Zhang, S., Zhang, X., Guo, D., Zhou, W., and Huang, S. (2018). Distinct responses of soil bacterial and fungal communities to changes in fertilization regime and crop rotation. Geoderma 319, 156–166. doi: 10.1016/j.geoderma.2018.01.010
Alguacil, M. M., Lumini, E., Roldan, A., Salinas-Garcia, J. R., Bonfante, P., and Bianciotto, V. (2008). The impact of tillage practices on arbuscular mycorrhizal fungal diversity in subtropical crops. Ecol. Appl. 18, 527–536. doi: 10.1890/07-0521.1
Alguacil, M. M., Torrecillas, E., García-Orenes, F., and Roldán, A. (2014). Changes in the composition and diversity of AMF communities mediated by management practices in a Mediterranean soil are related with increases in soil biological activity. Soil Biol. Biochem. 76, 34–44. doi: 10.1016/j.soilbio.2014.05.002
Anderson, M. J. (2001). A new method for non-parametric multivariate analysis of variance. Austral Ecol. 26, 32–46. doi: 10.1111/j.1442-9993.2001.01070.pp.x
Anderson, M. J. (2006). Distance-based tests for homogeneity of multivariate dispersions. Biometrics 62, 245–253. doi: 10.1111/j.1541-0420.2005.00440.x
Anderson, M. J., and Braak, C. T. (2003). Permutation tests for multi-factorial analysis of variance. J. Stat. Comput. Simul. 73, 85–113. doi: 10.1080/00949650215733
Anderson, M. J., Ellingsen, K. E., and McArdle, B. H. (2006). Multivariate dispersion as a measure of beta diversity. Ecol. Lett. 9, 683–693. doi: 10.1111/j.1461-0248.2006.00926.x
Anderson, M. J., Gorley, R. N., and Clarke, R. K. (2008). Permanova+ for Primer: Guide to Software and Statistical Methods. Plymouth: Primer-E Limited.
Antoninka, A., Reich, P. B., and Johnson, N. C. (2011). Seven years of carbon dioxide enrichment, nitrogen fertilization and plant diversity influence arbuscular mycorrhizal fungi in a grassland ecosystem. New Phytol. 192, 200–214. doi: 10.1111/j.1469-8137.2011.03776.x
Avio, L., Pellegrino, E., Bonari, E., and Giovannetti, M. (2006). Functional diversity of arbuscular mycorrhizal fungal isolates in relation to extraradical mycelial networks. New Phytol. 172, 347–357. doi: 10.1111/j.1469-8137.2006.01839.x
Avis, P. G., McLaughlin, D. J., Dentinger, B. C., and Reich, P. B. (2003). Long-term increase in nitrogen supply alters above- and below-ground ectomycorrhizal communities and increases the dominance of Russula spp. in a temperate oak savanna. New Phytol. 160, 239–253. doi: 10.1046/j.1469-8137.2003.00865.x
Baba, R., Kimura, M., Asakawa, S., and Watanabe, T. (2014). Analysis of [FeFe]-hydrogenase genes for the elucidation of a hydrogen-producing bacterial community in paddy field soil. FEMS Microbiol. Lett. 350, 249–256. doi: 10.1111/1574-6968.12335
Babin, D., Deubel, A., Jacquiod, S., Sørensen, S. J., Geistlinger, J., Grosch, R., et al. (2019). Impact of long-term agricultural management practices on soil prokaryotic communities. Soil Biol. Biochem. 129, 17–28. doi: 10.1016/j.soilbio.2018.11.002
Bardgett, R. D., and Wardle, D. A. (2010). Aboveground-Belowground Linkages: Biotic Interactions, Ecosystem Processes, and Global Change. Oxford: Oxford University Press.
Berg, G., and Smalla, K. (2009). Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. Fems Microbiol. Ecol. 68, 1–13. doi: 10.1111/j.1574-6941.2009.00654.x
Berisso, F. E., Schjønning, P., Keller, T., Lamandeì, M., Etana, A., de Jonge, L. W., et al. (2012). Persistent effects of subsoil compaction on pore size distribution and gas transport in a loamy soil. Soil Tillage Res. 122, 42–51. doi: 10.1016/j.still.2012.02.005
Bevivino, A., Paganin, P., Bacci, G., Florio, A., Pellicer, M. S., Papaleo, M. C., et al. (2014). Soil bacterial community response to differences in agricultural management along with seasonal changes in a Mediterranean region. PLoS One 9:e105515. doi: 10.1371/journal.pone.0105515
Bezemer, T. M., and van Dam, N. M. (2005). Linking aboveground and belowground interactions via induced plant defenses. Trends Ecol. Evol. 20, 617–624. doi: 10.1016/j.tree.2005.08.006
Blume, E., Bischoff, M., Reichert, J. M., Moorman, T., Konopka, A., and Turco, R. F. (2002). Surface and subsurface microbial biomass, community structure and metabolic activity as a function of soil depth and season. Appl. Soil Ecol. 20, 171–181. doi: 10.1016/S0929-1393(02)00025-2
Bowles, T. M., Jackson, L. E., Loher, M., and Cavagnaro, T. R. (2017). Ecological intensification and arbuscular mycorrhizas: a meta-analysis of tillage and cover crop effects. J .Appl. Ecol. 54, 1785–1793. doi: 10.1111/1365-2664.12815
Brito, I., Goss, M. J., de Carvalho, M., Chatagnier, O., and van Tuinen, D. (2012). Impact of tillage system on arbuscular mycorrhiza fungal communities in the soil under Mediterranean conditions. Soil Tillage Res. 121, 63–67. doi: 10.1016/j.still.2012.01.012
Buckley, D. H., and Schmidt, T. M. (2001). Environmental factors influencing the distribution of rRNA from Verrucomicrobia in soil. FEMS Microbiol. Ecol. 35, 105–112. doi: 10.1111/j.1574-6941.2001.tb00793.x
Cabello, P., Roldan, M. D., and Moreno-Vivian, C. (2004). Nitrate reduction and the nitrogen cycle in archaea. Microbiology 150, 3527–3546. doi: 10.1099/mic.0.27303-0
Cannon, P. F., and Kirk, P. M. (2007). Fungal Families of the World. Wallingford: CAB International.
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Caruso, T. (2018). Disentangling the factors shaping arbuscular mycorrhizal fungal communities across multiple spatial scales. New Phytol. 220, 954–956. doi: 10.1111/nph.15212
Ceja-Navarro, J. A., Rivera-Orduna, F. N., Patino-Zúniga, L., Vila-Sanjurjo, A., Crossa, J., Govaerts, B., et al. (2010). Phylogenetic and multivariate analyses to determine the effects of different tillage and residue management practices on soil bacterial communities. Appl. Environ. Microbiol. 76, 3685–3691. doi: 10.1128/AEM.02726-2729
Chojnacka, A., Szczêsny, P., Błaszczyk, M. K., Zielenkiewicz, U., Detman, A., Salamon, A., et al. (2015). Noteworthy facts about a methane-producing microbial community processing acidic effluent from sugar beet molasses fermentation. PLoS One 10:e0128008. doi: 10.1371/journal.pone.0128008
Ciccolini, V., Bonari, E., Ercoli, L., and Pellegrino, E. (2016a). Phylogenetic and multivariate analyses to determine the effect of agricultural land-use intensification and soil physico-chemical properties on N-cycling microbial communities in drained Mediterranean peaty soils. Biol. Fertil. Soils 52, 811–824. doi: 10.1007/s00374-016-1121-1129
Ciccolini, V., Ercoli, L., Davison, J., Vasar, M., Opik, M., and Pellegrino, E. (2016b). Land-use intensity and host plant simultaneously shape the composition of arbuscular mycorrhizal fungal communities in a Mediterranean peatland. FEMS Microbiol. Ecol. 92, 1–13. doi: 10.1093/femsec/fiw186
Ciccolini, V., Bonari, E., and Pellegrino, E. (2015). Land-use intensity and soil properties shape the composition of fungal communities in Mediterranean peaty soils drained for agricultural purposes. Biol. Fertil. Soils 51, 719–731. doi: 10.1007/s00374-015-1013-1014
Coccina, A., Cavagnaro, T. R., Pellegrino, E., Ercoli, L., McLaughlin, M. J., and Watts-Williams, S. J. (2019). The mycorrhizal pathway of zinc uptake contributes to zinc accumulation in barley and wheat grain. BMC Plant Biol. 19:133. doi: 10.1186/s12870-019-1741-y
Dai, Y., Yan, Z., Jia, L., Zhang, S., Gao, L., Wei, X., et al. (2016). The composition, localization and function of low-temperature-adapted microbial communities involved in methanogenic degradations of cellulose and chitin from Qinghai–Tibetan Plateau wetland soils. J. Appl. Microbiol. 121, 163–176. doi: 10.1111/jam.13164
de Cárcer, D. A., Denman, S. E., McSweeney, C., and Morrison, M. (2011). Evaluation of subsampling-based nor- malization strategies for tagged high-throughput sequencing data sets from gut microbiomes. Appl. Environ. Microbiol. 77, 8795–8798. doi: 10.1128/AEM.05491-5411
De Deyn, G. B., and Van der Putten, W. H. (2005). Linking aboveground and belowground diversity. Trends Ecol. Evol. 20, 625–633. doi: 10.1016/j.tree.2005.08.009
de Graaff, M. A., Hornslein, N., Throop, H., Kardol, P., and van Diepen, L. T. (2019). Effects of agricultural intensification on soil biodiversity and implications for ecosystem functioning: a meta-analysis. Adv. Agron. 155, 1–44. doi: 10.1016/bs.agron.2019.01.001
DeAngelis, K. M., Brodie, E. L., DeSantis, T. Z., Andersen, G. L., Lindow, S. E., and Firestone, M. K. (2009). Selective progressive response of soil microbial community to wild oat roots. ISME J. 3, 168–178. doi: 10.1038/ismej.2008.103
DeAngelis, K. M., Lindow, S. E., and Firestone, M. K. (2008). Bacterial quorum sensing and nitrogen cycling in rhizosphere soil. FEMS Microbiol. Ecol. 66, 197–207. doi: 10.1111/j.1574-6941.2008.00550.x
Degrune, F., Theodorakopoulos, N., Colinet, G., Hiel, M. P., Bodson, B., Taminiau, B., et al. (2017). Temporal dynamics of soil microbial communities below the seedbed under two contrasting tillage regimes. Front. Microbiol. 8:1127. doi: 10.3389/fmicb.2017.01127
Deng, Y., Huang, Z., Ruan, W., Miao, H., Shi, W., and Zhao, M. (2018). Enriching ruminal polysaccharide-degrading consortia via co-inoculation with methanogenic sludge and microbial mechanisms of acidification across lignocellulose loading gradients. Appl. Microbiol. Biotechnol. 102, 3819–3830. doi: 10.1007/s00253-018-8877-8879
Dias, T., Malveiro, S., Chaves, S., Tenreiro, R., Branquinho, C., Martins-Loução, M. A., et al. (2000). “Effects of increased N availability on biodiversity of Mediterranean-type ecosystems: A case study in a Natura 2000 site in Portugal,” in Nitrogen Deposition and Natura 2000Book of Proceedings of the COST729 Mid-term workshop, eds W. K. Hicks, C. P. Whitfield, W. J. Bealey, and M. A. Sutton (Laxenburg), 173–181.
Ding, J., Jiang, X., Guan, D., Zhao, B., Ma, M., Zhou, B., et al. (2017). Influence of inorganic fertilizer and organic manure application on fungal communities in a long-term field experiment of Chinese Mollisols. Appl. Soil Ecol. 111, 114–122. doi: 10.1016/j.apsoil.2016.12.003
Dolédec, S., and Chessel, D. (1994). Co-inertia analysis: an alternative method for studying species-environment relationships. Freshwater Biol. 31, 277–294. doi: 10.1111/j.1365-2427.1994.tb01741.x
Dorr de Quadros, P., Zhalnina, K., Davis-Richardson, A., Fagen, J. R., Drew, J., Bayer, C., et al. (2012). The effect of tillage system and crop rotation on soil microbial diversity and composition in a subtropical acrisol. Diversity 4, 375–395. doi: 10.3390/d4040375
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C., and Flyvbjerg, H. (2015). Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics 31, 3476–3482. doi: 10.1093/bioinformatics/btv401
Egerton-Warburton, L. M., and Allen, E. B. (2000). Shifts in arbuscular mycorrhizal communities along an anthropogenic nitrogen deposition gradient. Ecol Appl. 10, 484–496. doi: 10.1890/1051-0761(2000)010[0484:siamca]2.0.co;2
Egerton-Warburton, L. M., Johnson, N. C., and Allen, E. B. (2007). Mycorrhizal community dynamics following nitrogen fertilization: a cross-site test in five grasslands. Ecol. Monograph 77, 527–544. doi: 10.1890/06-1772.1
Eilers, K. G., Debenport, S., Anderson, S., and Fierer, N. (2012). Digging deeper to find unique microbial communities: the strong effect of depth on the structure of bacterial and archaeal communities in soil. Soil Biol. Biochem. 50, 58–65. doi: 10.1016/j.soilbio.2012.03.011
Eo, J., and Park, K. C. (2016). Long-term effects of imbalanced fertilization on the composition and diversity of soil bacterial community. Agric. Ecosyst. Environ. 231, 176–182. doi: 10.1016/j.agee.2016.06.039
Eom, A. H., Hartnett, D. C., Wilson, G. W., and Figge, D. A. (1999). The effect of fire, mowing and fertilizer amendment on arbuscular mycorrhizas in tallgrass prairie. Am. Midl. Nat. 142, 55–71.
Ercoli, L., Arduini, I., Mariotti, M., Lulli, L., and Masoni, A. (2012). Management of sulphur fertiliser to improve durum wheat production and minimise S leaching. Eur. J. Agron. 38, 74–82. doi: 10.1016/j.eja.2011.12.004
Ercoli, L., Arduini, I., Schüßler, A., and Pellegrino, E. (2017). Strong increase of durum wheat iron and zinc content by field-inoculation with arbuscular mycorrhizal fungi at different soil nitrogen availabilities. Plant Soil 19, 153–167. doi: 10.1007/s11104-017-3319-3315
Eriksson, K. E. L., Blanchette, R. A., and Ander, P. (1990). Microbial and Enzymatic Degradation of Wood and Wood Components. Berlin: Springer Science & Business Media.
Fierer, N. (2017). Embracing the unknown: disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 15, 579–590. doi: 10.1038/nrmicro.2017.87
Fierer, N., Bradford, M. A., and Jackson, R. B. (2007). Toward an ecological classification of soil bacteria. Ecology 88, 1354–1364. doi: 10.1890/05-1839
Fierer, N., Schimel, J. P., and Holden, P. A. (2003). Variations in microbial community composition through two soil depth profiles. Soil Biol. Biochem. 35, 167–176. doi: 10.1016/S0038-0717(02)00251-251
Fontaine, S., Mariotti, A., and Abbadie, L. (2003). The priming effect of organic matter: a question of microbial competition? Soil Biol. Biochem. 35, 837–843. doi: 10.1016/S0038-0717(03)00123-128
Fra̧c, M., Hannula, S. E., Bełka, M., and Jêdryczka, M. (2018). Fungal biodiversity and their role in soil health. Front. Microbiol. 9:707. doi: 10.3389/fmicb.2018.00707
Francioli, D., Schulz, E., Lentendu, G., Wubet, T., Buscot, F., and Reitz, T. (2016). Mineral vs organic amendments: microbial community structure, activity and abundance of agriculturally relevant microbes are driven by long-term fertilization strategies. Front. Microbiol. 7:1446. doi: 10.3389/fmicb.2016.01446
Freedman, Z. B., Romanowicz, K. J., Upchurch, R. A., and Zak, D. R. (2015). Differential responses of total and active soil microbial communities to long-term experimental N deposition. Soil Biol. Biochem. 90, 275–282. doi: 10.1016/j.soilbio.2015.08.014
Frey, S. D., Knorr, M., Parrent, J. L., and Simpson, R. T. (2004). Chronic nitrogen enrichment affects the structure and function of the soil microbial community in temperate hardwood and pine forests. Forest Ecol. Manag. 196, 159–171. doi: 10.1016/j.foreco.2004.03.018
Friedrich, T. (2013). Conservation agriculture as a means of achieving sustainable intensification of crop production. Agr. Develop. 19, 7–11.
Grandin, U. (2006). PC-ORD version 5: a user-friendly toolbox for ecologists. J. Veg. Sci. 17, 843–844. doi: 10.1111/j.1654-1103.2006.tb02508.x
Griffiths, R. I., Whiteley, A. S., O’donnell, A. G., and Bailey, M. J. (2003). Influence of depth and sampling time on bacterial community structure in an upland grassland soil. FEMS Microbiol. Ecol. 43, 35–43. doi: 10.1111/j.1574-6941.2003.tb01043.x
Gulis, V., Suberkropp, K., and Rosemond, A. D. (2008). Comparison of fungal activities on wood and leaf litter in unaltered and nutrient-enriched headwater streams. Appl. Environ. Microbiol. 74, 1094–1101. doi: 10.1007/s00027-010-0133-z
Gutiérrez-Manero, F. J., Acero, N., Lucas, J. A., and Probanza, A. (1996). The influence of native rhizobacteria on European alder (Alnus glutinosa L. Plant Soil 182, 67–74. doi: 10.1007/bf00010996
Gutiérrez-Mañero, F. J., Ramos-Solano, B., Probanza, A. N., Mehouachi, J. R., Tadeo, F., and Talon, M. (2001). The plant-growth-promoting rhizobacteria Bacillus pumilus and Bacillus licheniformis produce high amounts of physiologically active gibberellins. Physiol. Plant 111, 206–211. doi: 10.1034/j.1399-3054.2001.1110211.x
Hartmann, M., Frey, B., Mayer, J., Mäder, P., and Widmer, F. (2015). Distinct soil microbial diversity under long-term organic and conventional farming. ISME J. 9, 1177–1194. doi: 10.1038/ismej.2014.210
Hartmann, M., Lee, S., Hallam, S. J., and Mohn, W. W. (2009). Bacterial, archaeal and eukaryal community structures throughout soil horizons of harvested and naturally disturbed forest stands. Environ. Microbiol. 11, 3045–3062. doi: 10.1111/j.1462-2920.2009.02008.x
Hofrichter, M. (2002). Review: lignin conversion by manganese peroxidase (MnP). Enzyme Microb. Technol. 30, 454–466. doi: 10.1016/S0141-0229(01)00528-522
Horn, R., Taubner, H., Wuttke, M., and Baumgartl, T. (1994). Soil physical properties related to soil structure. Soil Till. Res. 30, 187–216. doi: 10.1016/0167-1987(94)90005-1
IPCC (2015). Climate Change 2014: Synthesis Report. Geneva: Intergovernmental Panel on Climate Change.
IUSS working group WRB, (2006). World reference base for soil resources 2006 - A framework for international classification, correlation and communication. Rome: Food and Agriculture Organization of the United Nations.
Jansa, J., Mozafar, A., Anken, T., Ruh, R., Sanders, I., and Frossard, E. (2002). Diversity and structure of AMF communities as affected by tillage in a temperate soil. Mycorrhiza 12, 225–234. doi: 10.1007/s00572-002-0163-z
Jiménez-Bueno, N. G., Valenzuela-Encinas, C., Marsch, R., Ortiz-Gutiérrez, D., Verhulst, N., Govaerts, B., et al. (2016). Bacterial indicator taxa in soils under different long-term agricultural management. J. Appl. Microbiol. 120, 921–933. doi: 10.1111/jam.13072
Johnson, D., Vandenkoornhuyse, P. J., Leake, J. R., Gilbert, L., Booth, R. E., Grime, J. P., et al. (2004). Plant communities affect arbuscular mycorrhizal fungal diversity and community composition in grassland microcosms. New Phytol. 161, 503–515. doi: 10.1046/j.1469-8137.2003.00938.x
Jones, D. L., Magthab, E. A., Gleeson, D. B., Hill, P. W., Sánchez-Rodríguez, A. R., Roberts, P., et al. (2018). Microbial competition for nitrogen and carbon is as intense in the subsoil as in the topsoil. Soil Biol. Biochem. 117, 72–82. doi: 10.1016/j.soilbio.2017.10.024
Jumpponen, A., and Johnson, L. C. (2005). Can rDNA analyses of diverse fungal communities in soil and roots detect effects of environmental manipulations-a case study from tallgrass prairie. Mycologia 97, 1177–1194. doi: 10.1080/15572536.2006.11832728
Jumpponen, A., Jones, K. L., and Blair, J. (2010). Vertical distribution of fungal communities in tallgrass prairie soil. Mycologia 102, 1027–1041. doi: 10.3852/09-316
Kabir, Z., O’Halloran, I. P., Fyles, J. W., and Hamel, C. (1998). Dynamics of the mycorrhizal symbiosis of corn (Zea mays L.): effects of host physiology, tillage practice and fertilization on spatial distribution of extra-radical mycorrhizal hyphae in the field. Agric. Ecosyst. Environ. 68, 151–163. doi: 10.1016/S0167-8809(97)00155-152
Kameshwar, A. K., and Qin, W. (2016). Recent developments in using advanced sequencing technologies for the genomic studies of lignin and cellulose degrading microorganisms. Int. J. Biol. Sci. 12, 156–171. doi: 10.7150/ijbs.13537
Kassam, A., Friedrich, T., and Derpsch, R. (2019). Global spread of conservation agriculture. Int. J. Environ. Stud. 76, 29–51. doi: 10.1080/00207233.2018.1494927
Kersten, P., and Cullen, D. (2013). “Recent advances on the genomics of litter-and soil-inhabiting Agaricomycetes,” in Genomics of Soil-and Plant-Associated Fungi, eds B. A. Horwitz, P. K. Mukherjee, M. Mukherjee and C. P. Kubice Berlin: Springer, 311–332. doi: 10.1007/978-3-642-39339-6_13
Kielak, A., Rodrigues, J. L., Kuramae, E. E., Chain, P. S., Van Veen, J. A., and Kowalchuk, G. A. (2009). Phylogenetic and metagenomic analysis of Verrucomicrobia in former agricultural grassland soil. FEMS Microbiol. Ecol. 71, 23–33. doi: 10.1111/j.1574-6941.2009.00785.x
Kindaichi, T., Yuri, S., Ozaki, N., and Ohashi, A. (2012). Ecophysiological role and function of uncultured Chloroflexi in an anammox reactor. Wat. Sci. Tech. 66, 2556–2561. doi: 10.2166/wst.2012.479
Kõljalg, U., Nilsson, R. H., Abarenkov, K., Tedersoo, L., Taylor, A. F., Bahram, M., et al. (2013). Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 22, 5271–5277. doi: 10.1111/mec.12481
Kong, A. Y., Scow, K. M., Córdova-Kreylos, A. L., Holmes, W. E., and Six, J. (2011). Microbial community composition and carbon cycling within soil microenvironments of conventional, low-input, and organic cropping systems. Soil Biol. Biochem. 43, 20–30. doi: 10.1016/j.soilbio.2010.09.005
Kottek, M., Grieser, J., Beck, C., Rudolf, B., and Rubel, F. (2006). World map of the Köppen-Geiger climate classification updated. Meteorol. Z. 15, 259–263. doi: 10.1127/0941-2948/2006/0130
Kragelund, C., Caterina, L., Borger, A., Thelen, K., Eikelboom, D., Tandoi, V., et al. (2007). Identity, abundance and ecophysiology of filamentous Chloroflexi species present in activated sludge treatment plants. FEMS Microbiol. Ecol. 59, 671–682. doi: 10.1111/j.1574-6941.2006.00251.x
Kudryavtseva, O. A., Dunaevsky, Y. E., Kamzolkina, O. V., and Belozersky, M. A. (2008). Fungal proteolytic enzymes: features of the extracellular proteases of xylotrophic basidiomycetes. Microbiology 77, 643–653. doi: 10.1134/S0026261708060015
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lal, R. (2015). Sequestering carbon and increasing productivity by conservation agriculture. J. Soil Water Conserv. 70, 55A–62A. doi: 10.2489/jswc.70.3.55A
Lee, K. H., Wi, S. G., Singh, A. P., and Kim, Y. S. (2004). Micromorphological characteristics of decayed wood and laccase produced by the brown-rot fungus Coniophora puteana. J. Wood Sci. 50, 281–284. doi: 10.1007/s10086-003-0558-552
Leff, J. W., Jones, S. E., Prober, S. M., Barberán, A., Borer, E. T., Firn, J. L., et al. (2015). Consistent responses of soil microbial communities to elevated nutrient inputs in grasslands across the globe. Proc. Natl. Acad. Sci. U.S.A. 112, 10967–10972. doi: 10.1073/pnas.1508382112
Letunic, I., and Bork, P. (2006). Interactive tree of life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128. doi: 10.1093/bioinformatics/btl529
Levin, L., Villalba, L., Da Re, V., Forchiassin, F., and Papinutti, L. (2007). Comparative studies of loblolly pine biodegradation and enzyme production by Argentinean white rot fungi focused on biopulping processes. Process Biochem. 42, 995–1002. doi: 10.1016/j.procbio.2007.03.008
Li, F., Chen, L., Zhang, J., Yin, J., and Huang, S. (2017). Bacterial community structure after long-term organic and inorganic fertilization reveals important associations between soil nutrients and specific taxa involved in nutrient transformations. Front. Microbiol. 8:187. doi: 10.3389/fmicb.2017.00187
Lienhard, P., Terrat, S., Prévost-Bouré, N. C., Nowak, V., Régnier, T., Sayphoummie, S., et al. (2014). Pyrosequencing evidences the impact of cropping on soil bacterial and fungal diversity in Laos tropical grassland. Agron. Sustain. Dev. 34, 525–533. doi: 10.1007/s13593-013-0162-169
Limam, R. D., Chouari, R., Mazeas, L., Wu, T. D., Li, T., Grossin-Debattista, J., et al. (2014). Members of the uncultured bacterial candidate division WWE 1 are implicated in anaerobic digestion of cellulose. Microbiologyopen 3, 157–167. doi: 10.1002/mbo3.144
Lin, X., Feng, Y., Zhang, H., Chen, R., Wang, J., Zhang, J., et al. (2012). Long-term balanced fertilization decreases arbuscular mycorrhizal fungal diversity in an arable soil in North China revealed by 454 pyrosequencing. Environ. Sci. Technol. 46, 5764–5771. doi: 10.1021/es3001695
Linder, T. (2019). Cyanase-independent utilization of cyanate as a nitrogen source in ascomycete yeasts. World J. Microbiol. Biotechnol. 35:3. doi: 10.1007/s11274-018-2579-2574
Liu, W., Wang, Q., Wang, B., Wang, X., Franks, A. E., Teng, Y., et al. (2015). Changes in the abundance and structure of bacterial communities under long-term fertilization treatments in a peanut monocropping system. Plant Soil 395, 415–427. doi: 10.1007/s11104-015-2569-2563
Mantel, N., and Valand, R. S. (1970). A technique of nonparametric multi-variate analysis. Biometrics 26, 547–558. doi: 10.1371/journal.pone.0050332
Mariotti, M., Masoni, A., Ercoli, L., and Arduini, I. (2015). Nitrogen leaching and residual effect of barley/field bean intercropping. Plant Soil Environ. 61, 60–65. doi: 10.17221/832/2014-PSE
Marklein, A. R., and Houlton, B. Z. (2012). Nitrogen inputs accelerate phosphorus cycling rates across a wide variety of terrestrial ecosystems. New Phytol. 193, 696–704. doi: 10.1111/j.1469-8137.2011.03967.x
Mathew, R. P., Feng, Y., Githinji, L., Ankumah, R., and Balkcom, K. S. (2012). Impact of no-tillage and conventional tillage systems on soil microbial communities. Appl. Environ. Soil Sci. 2012, 1–10. doi: 10.1155/2012/548620
Mbuthia, L. W., Acosta-Martínez, V., DeBruyn, J., Schaeffer, S., Tyler, D., Odoi, E., et al. (2015). Long term tillage, cover crop, and fertilization effects on microbial community structure, activity: implications for soil quality. Soil Biol. Biochem. 89, 24–34. doi: 10.1016/j.soilbio.2015.06.016
Medina, A., Probanza, A., Gutiérrez-Mañero, F. J., and Azcón, R. (2003). Interactions of arbuscular-mycorrhizal fungi and Bacillus strains and their effects on plant growth, microbial rhizosphere activity (thymidine and leucine incorporation) and fungal biomass (ergosterol and chitin). Appl. Soil Ecol. 22, 15–28. doi: 10.1016/S0929-1393(02)00112-119
Mirás-Avalos, J. M., Antunes, P. M., Koch, A., Khosla, K., Klironomos, J. N., and Dunfield, K. E. (2011). The influence of tillage on the structure of rhizosphere and root-associated arbuscular mycorrhizal fungal communities. Pedobiologia 54, 235–241. doi: 10.1016/j.pedobi.2011.03.005
Mommer, L., Cotton, T. A., Raaijmakers, J. M., Termorshuizen, A. J., van Ruijven, J., Hendriks, M., et al. (2018). Lost in diversity: the interactions between soil-borne fungi, biodiversity and plant productivity. New Phytol. 218, 542–553. doi: 10.1111/nph.15036
Murugan, R., Koch, H. J., and Joergensen, R. G. (2014). Long-term influence of different tillage intensities on soil microbial biomass, residues and community structure at different depths. Biol. Fertil. Soils 50, 487–498. doi: 10.1007/s00374-013-0871-x
Mycology Web pages of the New Brunswick Museum (2019). Mycology Web pages of the New Brunswick Museum. Available at: http://website.nbm-mnb.ca/mycologywebpages/NaturalHistoryOfFungi/Home.html (accessed on 25 July 2019).
Nannipieri, P., and Badalucco, L. (2003). “Biological processes,” in Handbook of Processes and Modeling in the Soil Plant System, eds D. K. Benbi and R. Niedere (New York, NY: Haworth Binghamton), 57–82.
Nordeìn, B., and Paltto, H. (2001). Wood-decay fungi in hazel wood: species richness correlated to stand age and dead wood features. Biol. Cons. 101, 1–8. doi: 10.1016/S0006-3207(01)00049-40
Oehl, F., Sieverding, E., Ineichen, K., Ris, E. A., Boller, T., and Wiemken, A. (2005). Community structure of arbuscular mycorrhizal fungi at different soil depths in extensively and intensively managed agroecosystems. New Phytol. 165, 273–283. doi: 10.1111/j.1469-8137.2004.01235.x
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O’hara, R. B., et al. (2013). Package ‘vegan’. Community Ecology Package, version 2.9.
Oliveros, J. C. (2007). Venny. An interactive tool for comparing lists with Venn’s diagrams. Available at: https://bioinfogp.cnb.csic.es/tools/venny/index.html
Omar, S. A., and Ismail, M. A. (1999). Microbial populations, ammonification and nitrification in soil treated with urea and inorganic salts. Folia Microbiol. 44, 205–212. doi: 10.1007/BF02816244
Pankratov, T. A., Dedysh, S. N., and Zavarzin, G. A. (2006). The leading role of actinobacteria in aerobic cellulose degradation in Sphagnum peat bogs. Dokl. Biol. Sci. 410, 428–430. doi: 10.1134/s0012496606050243
Pastorelli, R., Vignozzi, N., Landi, S., Piccolo, R., Orsini, R., Seddaiu, G., et al. (2013). Consequences on macroporosity and bacterial diversity of adopting a no-tillage farming system in a clayish soil of Central Italy. Soil Biol. Biochem. 66, 78–93. doi: 10.1016/j.soilbio.2013.06.015
Pellegrino, E., Bosco, S., Ciccolini, V., Pistocchi, C., Sabbatini, T., Silvestri, N., et al. (2015a). Agricultural abandonment in Mediterranean reclaimed peaty soils: long-term effects on soil chemical properties, arbuscular mycorrhizas and CO2 flux. Agric. Ecosyst. Environ. 199, 164–175. doi: 10.1016/j.agee.2014.09.004
Pellegrino, E., Öpik, M., Bonari, E., and Ercoli, L. (2015b). Responses of wheat to arbuscular mycorrhizal fungi: a meta-analysis of field studies from 1975 to 2013. Soil Biol. Biochem. 84, 210–217. doi: 10.1016/j.soilbio.2015.02.020
Pellegrino, E., Di Bene, C., Tozzini, C., and Bonari, E. (2011). Impact on soil quality of a 10-year-old short-rotation coppice poplar stand compared with intensive agricultural and uncultivated systems in a Mediterranean area. Agric. Ecosyst. Environ. 140, 245–254. doi: 10.1016/j.agee.2010.12.011
Peng, G., Bing, W., Guangpo, G., and Guangcan, Z. (2013). Spatial distribution of soil organic carbon and total nitrogen based on GIS and geostatistics in a small watershed in a hilly area of northern China. PloS One 8:e83592. doi: 10.1371/journal.pone.0083592
Peter, M., Ayer, F., and Egli, S. (2001). Nitrogen addition in a Norway spruce stand altered macromycete sporocarp production and belowground ectomycorrhizal species composition. New Phytol. 149, 311–325. doi: 10.1046/j.1469-8137.2001.00030.x
Picci, G., and Nannipieri, P. (2002). Metodi di Analisi Microbiologica del suolo. Roma: Franco Angeli.
Pingel, M., Reineke, A., and Leyer, I. (2019). A 30-years vineyard trial: plant communities, soil microbial communities and litter decomposition respond more to soil treatment than to N fertilization. Agric. Ecosyst. Environ. 272, 114–125. doi: 10.1016/j.agee.2018.11.005
Powell, J. R., and Rillig, M. C. (2018). Biodiversity of arbuscular mycorrhizal fungi and ecosystem function. New Phytol. 220, 1059–1075. doi: 10.1111/nph.15119
Power, A. G. (2010). Ecosystem services and agriculture: tradeoffs and synergies. Philos. Trans. R. Soc. Lond. B Biol. Sci. 365, 2959–2971. doi: 10.1098/rstb.2010.0143
Pretty, J. (2008). Agricultural sustainability: concepts, principles and evidence. Philos. Trans. R. Soc. Lond. B 363, 447–466. doi: 10.1098/rstb.2007.2163
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2012). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucl. Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Ramesh, A., Sharma, S. K., Sharma, M. P., Yadav, N., and Joshi, O. P. (2014). Inoculation of zinc solubilizing Bacillus aryabhattai strains for improved growth, mobilization and biofortification of zinc in soybean and wheat cultivated in Vertisols of central India. Appl. Soil Ecol. 73, 87–96. doi: 10.1016/j.apsoil.2013.08.009
Ramirez, K. S., Lauber, C. L., Knight, R., Bradford, M. A., and Fierer, N. (2010). Consistent effects of nitrogen fertilization on soil bacterial communities in contrasting systems. Ecology 91, 3463–3470. doi: 10.1890/10-0426.1
Rodriguez, R. J., White, J. F. Jr., Arnold, A. E., and Redman, A. R. A. (2009). Fungal endophytes: diversity and functional roles. New Phytol. 182, 314–330. doi: 10.1111/j.1469-8137.2009.02773.x
Sahu, N., Vasu, D., Sahu, A., Lal, N., and Singh, S. K. (2017). :Strength of Microbes in Nutrient Cycling: A Key to Soil Health,” in Agriculturally Important Microbes for Sustainable Agriculture, eds V. Meena, P. Mishra, J. Bisht and A. Pattanayak Singapore: Springer, 69–86. doi: 10.1007/978-981-10-5589-8_4
Sharma-Poudyal, D., Schlatter, D., Yin, C., Hulbert, S., and Paulitz, T. (2017). Long-term no-till: a major driver of fungal communities in dryland wheat cropping systems. PloS One 12:e0184611. doi: 10.1371/journal.pone.0184611
Simpson, R. T., Frey, S. D., Six, J., and Thiet, R. K. (2004). Preferential accumulation of microbial carbon in aggregate structures of no-tillage soils. Soil Sci. Soc. Am. J. 68, 1249–1255. doi: 10.2136/sssaj2004.1249
Smith, S. E., Jakobsen, I., Grønlund, M., and Smith, F. A. (2011). Roles of arbuscular mycorrhizas in plant phosphorus nutrition: interactions between pathways of phosphorus uptake in arbuscular mycorrhizal roots have important implications for understanding and manipulating plant phosphorus acquisition. Plant Physiol. 156, 1050–1057. doi: 10.1104/pp.111.174581
Soil Survey Staff (1975). Soil taxonomy. A Basic System of Soil Classification for Making and Interpreting Soil Surveys. USDA-SCS Agrjic. Handb. Washington, DC: U.S. Government Printing Office, 436.
Somenahally, A., DuPont, J. I., Brady, J., McLawrence, J., Northup, B., and Gowda, P. (2018). Microbial communities in soil profile are more responsive to legacy effects of wheat-cover crop rotations than tillage systems. Soil Biol. Biochem. 123, 126–135. doi: 10.1016/j.soilbio.2018.04.025
Steindorff, A., Souza, M., de Almeida, J. R. M., and Formighieri, E. (2016). Genomic and mitochondrial assembly of xylose-fermenting yeasts from Debaryomycetaceae family. Brasília, DF: Embrapa Agroenergia, 48–54.
ter Braak, C. J. F., and Šmilauer, P. (2012). Canoco Reference Manual and user’s guide: Software for Ordination (version 50). Itacha: Microcomputer power.
Thierfelder, C., Baudron, F., Setimela, P., Nyagumbo, I., Mupangwa, W., Mhlanga, B., et al. (2018). Complementary practices supporting conservation agriculture in southern Africa. A review. Agron. Sustain. Dev. 38:16. doi: 10.1007/s13593-018-0492-498
Thorn, R. G., Reddy, C. A., Harris, D., and Paul, E. A. (1996). Isolation of saprophytic basidiomycetes from soil. Appl. Environ. Microbiol. 62, 4288–4292.
Treseder, K. K. (2004). A meta-analysis of mycorrhizal responses to nitrogen, phosphorus, and atmospheric CO2 in field studies. New Phytol. 164, 347–355. doi: 10.1111/j.1469-8137.2004.01159.x
Trivedi, P., Delgado Baquerizo, M., Jeffries, T. C., Trivedi, C., Anderson, I. C., Lai, K., et al. (2017). Soil aggregation and associated microbial communities modify the impact of agricultural management on carbon content. Environ. Microbiol. 19, 3070–3086. doi: 10.1111/1462-2920.13779
Trivedi, P., Rochester, I. J., Trivedi, C., Van Nostrand, J. D., Zhou, J., Karunaratne, S., et al. (2015). Soil aggregate size mediates the impacts of cropping regimes on soil carbon and microbial communities. Soil Biol. Biochem. 91, 169–181. doi: 10.1016/j.soilbio.2015.08.034
Utobo, E. B., and Tewari, L. (2015). Soil enzymes as bioindicators of soil ecosystem status. Appl. Ecol. Env. Res. 13, 147–169. doi: 10.15666/aeer/1301_147169
Vallebona, C., Pellegrino, E., Frumento, P., and Bonari, E. (2015). Temporal trends in extreme rainfall intensity and erosivity in the Mediterranean region: a case study in southern Tuscany. Italy. Clim. Change 128, 139–151. doi: 10.1007/s10584-014-1287-1289
van Der Heijden, M. G., De Bruin, S., Luckerhoff, L., Van Logtestijn, R. S., and Schlaeppi, K. (2016). A widespread plant-fungal-bacterial symbiosis promotes plant biodiversity, plant nutrition and seedling recruitment. ISME J. 10, 389. doi: 10.1038/ismej.2015.120
Vega, F. E., Goettel, M. S., Blackwell, M., Chandler, D., Jackson, M. A., Keller, S., et al. (2009). Fungal entomopathogens: new insights on their ecology. Fungal Ecol. 2, 149–159. doi: 10.1016/j.funeco.2009.05.001
Voets, L., De La Providencia, I. E., and Declerck, S. (2006). Glomeraceae and Gigasporaceae differ in their ability to form hyphal networks. New Phytol. 172, 185–188. doi: 10.1111/j.1469-8137.2006.01873.x
Wallenda, T., and Kottke, I. (1998). Nitrogen deposition and ectomycorrhizas. New Phytol. 139, 169–187. doi: 10.1046/j.1469-8137.1998.00176.x
Wang, M., Ruan, W., Kostenko, O., Carvalho, S., Hannula, S. E., Mulder, P. P., et al. (2019). Removal of soil biota alters soil feedback effects on plant growth and defense chemistry. New Phytol. 223, 1478–1491. doi: 10.1111/nph.15485
Wang, Y., Wang, Z. L., Zhang, Q., Hu, N., Li, Z., Lou, Y., et al. (2018). Long-term effects of nitrogen fertilization on aggregation and localization of carbon, nitrogen and microbial activities in soil. Sci. Total Environ. 624, 1131–1139. doi: 10.1016/j.scitotenv.2017.12.113
Weber, C. F., Vilgalys, R., and Kuske, C. R. (2013). Changes in fungal community composition in response to elevated atmospheric CO2 and nitrogen fertilization varies with soil horizon. Front. Microbiol. 4:78. doi: 10.3389/fmicb.2013.00078
Wen, Z., Li, H., Shen, Q., Tang, X., Xiong, C., Li, H., et al. (2019). Trade-offs among root morphology, exudation and mycorrhizal symbioses for phosphorus-acquisition strategies of 16 crop species. New Phytol 223, 882–895. doi: 10.1111/nph.15833
Wrighton, K. C., Castelle, C. J., Wilkins, M. J., Hug, L. A., Sharon, I., Thomas, B. C., et al. (2014). Metabolic interdependencies between phylogenetically novel fermenters and respiratory organisms in an unconfined aquifer. ISME J. 8, 1452–1463. doi: 10.1038/ismej.2013.249
Yang, T., Adams, J. M., Shi, Y., He, J. S., Jing, X., Chen, L., et al. (2017). Soil fungal diversity in natural grasslands of the Tibetan Plateau: associations with plant diversity and productivity. New Phytol. 215, 756–765. doi: 10.1111/nph.14606
Yao, X., Zhang, N., Zeng, H., and Wang, W. (2018). Effects of soil depth and plant-soil interaction on microbial community in temperate grasslands of northern China. Sci. Total Environ. 630, 96–102. doi: 10.1016/j.scitotenv.2018.02.155
Yilmaz, P., Parfrey, L. W., Yarza, P., Gerken, J., Pruesse, E., Quast, C., et al. (2013). The SILVA and “all-species living tree project (LTP)” taxonomic frameworks. Nucl. Acids Res. 42, D643–D648. doi: 10.1093/nar/gkt1209
Zhang, B., He, H. B., Ding, X. L., Zhang, X. D., Zhang, X. P., Yang, X. M., et al. (2012). Soil microbial community dynamics over a maize (Zea mays L.) growing season under conventional- and no-tillage practices in a rainfed agroecosystem. Soil Tillage Res. 124, 153–160. doi: 10.1016/j.still.2012.05.011
Zhang, W., Ricketts, T. H., Kremen, C., Carney, K., and Swinton, S. M. (2007). Ecosystem services and dis-services to agriculture. Ecol. Econ. 64, 253–260. doi: 10.1016/j.ecolecon.2007.02.024
Zhang, X., Xin, X., Zhu, A., Yang, W., Zhang, J., Ding, S., et al. (2018). Linking macroaggregation to soil microbial community and organic carbon accumulation under different tillage and residue managements. Soil Tillage Res. 178, 99–107. doi: 10.1016/j.still.2017.12.020
Zhou, X., Zhang, J., Gao, D., Gao, H., Guo, M., Li, L., et al. (2017). Conversion from long-term cultivated wheat field to Jerusalem artichoke plantation changed soil fungal communities. Sci. Rep. 7:41502. doi: 10.1038/srep41502
Keywords: soil bacteria, soil fungi, soil archaea, arbuscular mycorrhizal fungi, Glomeromycota, Illumina sequencing, 16S rRNA, ITS1 region
Citation: Piazza G, Ercoli L, Nuti M and Pellegrino E (2019) Interaction Between Conservation Tillage and Nitrogen Fertilization Shapes Prokaryotic and Fungal Diversity at Different Soil Depths: Evidence From a 23-Year Field Experiment in the Mediterranean Area. Front. Microbiol. 10:2047. doi: 10.3389/fmicb.2019.02047
Received: 22 May 2019; Accepted: 20 August 2019;
Published: 04 September 2019.
Edited by:
Per Bengtson, Lund University, SwedenReviewed by:
Engracia Madejon, Institute of Natural Resources and Agrobiology of Seville (CSIC), SpainNobuhiro Kaneko, Fukushima University, Japan
Copyright © 2019 Piazza, Ercoli, Nuti and Pellegrino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gaia Piazza, Z2FpYS5waWF6emFAc2FudGFubmFwaXNhLml0
†These authors have contributed equally to this work