 Lei Zhou1,2†
Lei Zhou1,2† Ruibai Wang
Ruibai Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 13 August 2019
Sec. Infectious Agents and Disease
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.01887
This article is part of the Research Topic Innovative Approaches in Diagnosis of Emerging/re-emerging Infectious Diseases View all 20 articles
Background: Tuberculosis (TB) and non-tuberculous mycobacteriosis are serious threats to health worldwide. A simple non-sequencing method is needed for rapid diagnosis, especially in less experienced hospitals, but there is no specific biomarker commonly used for all mycobacteria. The ku gene of the prokaryotic error-prone non-homologous end joining system (NHEJ) has the potential to be a highly specific detection biomarker for mycobacteria.
Methods: A total of 7294 mycobacterial genomes and 14 complete genomes of other families belonging to Corynebacteriales with Mycobacteriaceae were downloaded and analyzed for the existence and variation of the ku gene. Mycobacterium tuberculosis complex (MTBC) and non-tuberculosis mycobacteria (NTM)- specific primers were designed and the actual amplification and identification efficiencies were tested with 150 strains of 40 Mycobacterium species and 10 kinds of common respiratory pathogenic bacteria.
Results: The ku gene of the NHEJ system was ubiquitous in all genome sequenced Mycobacterium species and absent in other families of Corynebacteriales. On the one hand, as a single gene non-sequencing biomarker, its specific primers could effectively distinguish mycobacteria from other bacteria, MTBC from NTM, which would make the clinical detection of mycobacteria easy and have great clinical practical value. On the other hand, the sequence of ku gene can effectively distinguish NTM to species level with high resolution.
Conclusion: The Ku protein existed before the differentiation of Mycobacterium species, which was an important protein involved in maintaining of the genome’s integrity and related to the special growth stage of mycobacteria. It was rare in prokaryotes. These features made it a highly special differential biomarker for Mycobacterium.
Mycobacterium is a genus of over 190 species and 13 subspecies. Apart from the causative agents, the Mycobacterium tuberculosis complex (MTBC) and Mycobacterium leprae, the other members of this genus are grouped together and termed non-tuberculosis mycobacteria (NTM). Both tuberculosis (TB) and non-tuberculous mycobacteriosis pose serious threats to health worldwide, especially with the increase of multi-drug and pan-drug-resistant strains.
Mycobacteria have completely different culture characteristics and therapeutic antibiotics from other bacteria. A preliminary discrimination of the infection as mycobacteriosis, TB or NTM, or a mixed infection of these two kinds of mycobacteria, will make the subsequent culture more directed and help improve the isolation/culture rate and appropriately administer clinical medication to reduce transmission more effectively, especially for less-experienced hospitals. This method should be simple, fast, accurate and culture-free.
Compared with the relatively easy diagnosis of tuberculosis, the diagnosis of non-tuberculosis is more dependent on culture and biochemical tests or by the exclusion that negative TB detection in smear/culture of acid-positive samples. Because of the cumbersome procedure, isolation, cultivation and identification of NTM is not actually done in many hospitals in China. The incidence and disease burden of NTM are continuously increasing in many regions, and the prevalence of NTM in aged people, human immunodeficiency virus (HIV)-infected patients and those with severely damaged immune systems is significant, even more than that of TB (Wang et al., 2014; Halstrom et al., 2015; Mortaz et al., 2018). Therefore, the rapid diagnosis of NTM is also a prominent problem. It is necessary to find a better single-gene biomarker, which can be used for both of MTBC and NTM identification.
Mycobacteria have three DNA double-strand break repair pathways, which include the NHEJ system required for the CRISPR/Cas9 system in the second step (Pitcher et al., 2006). The NHEJ system is absent in most prokaryotic cells. To date, eukaryotic NHEJ homologs have only been identified in M. smegmatis, M. tuberculosis, and Bacillus subtilis (Bs). Furthermore, prokaryotic NHEJ is a much simpler system that needs only two key proteins, Ku and ligase D (LigD) (Gong et al., 2005; Korycka-Machala et al., 2006; Shuman and Glickman, 2007; Gupta et al., 2011; Zheng et al., 2017). The Ku protein exists as a homodimer and preferentially binds to dsDNA ends (Weller et al., 2002). LigD is an adenosine triphosphate (ATP)-dependent DNA ligase that contains polymerase and nuclease domains, which facilitates the joining of long linear DNA molecules with different incompatible ends (Della et al., 2004). The rarity of the NHEJ system in bacteria hints that it may be developed into a Mycobacterium specific detection biomarker. Although, the NHEJ system has been confirmed existing in the M. tuberculosis strain H37Rv (Rv0937c and Rv0938 encoded) (Doherty et al., 2001; Weller et al., 2002), its distribution, especially the distribution of the Ku protein that could specifically stimulate LigD and suppress homologous recombination (Della et al., 2004) in other Mycobacterium species has not yet been elucidated. In this study, we analyzed Mycobacterium genome data submitted in GenBank before September 2018 to explore the existence of the ku gene in the Mycobacterium genus and determine its applicability for Mycobacterium identification.
We downloaded a total of 7294 genomes from 139 definite species, seven subspecies and five variants of Mycobacterium from the NCBI’s FTP site submitted before September 2018, including 5245 genomes of M. tuberculosis, 1376 genomes of M. abscessus, 152 genomes of M. avium, and 70 M. tuberculosis variant bovis (Supplementary Table 1).
All the regions annotated as ku or mku and/or homologous to Rv0937c of M. tuberculosis were extracted from the genomes. Sequence alignments and comparisons were performed using the MEGA program version 6.0 (Tamura et al., 2013). Sequences were aligned on ClustalW using a gap opening penalty of 15 and a gap extension penalty of 6.66. Maximum likelihood trees were drawn. In each Mycobacterium species/variant, every ku sequence with even a nucleotide difference was defined as a genotype and listed in Supplementary Table 1 with a representative sequence. The IS6110 and rpoB genes were analyzed much as the ku gene was, and Supplementary Table 2 lists the genotypes of the rpoB gene.
Primers were designed using Oligo 6.0 and followed the general design principle of PCR primer. Simulated PCRs were performed using the Analyze Mix Wizard of Clone Manager Professional 9.0. The 379 ku genotype sequences were added as molecules in the mix.
In actual PCR amplification, the boiled DNA temples of M. tuberculosis H37Rv, M. bovis 93006 and 43 American Type Culture Collection (ATCC) NTM strains belonging to 40 species were used (Table 1). In addition, clinical isolates including 42 M. tuberculosis strains, 10 M. abscessus strains, 10 M. marseillense strains, 10 M. avium strains, 10 M. kansasii strains and 23 strains of 10 kinds of common respiratory pathogenic bacteria (Table 1) were also tested. PCR amplification condition was 5 min at 95°C followed by 30 cycles of 95°C 30 s, 58°C 30 s, and 72°C 1 min, with a final extension step at 72°C for 5 min. Four primer sets 16S 27f/16S907r (16SrRNA) (28), Tb11/Tb12 (hsp65) (Telenti et al., 1993), Myco-F/Myco-R (rpoB) (Adekambi and Drancourt, 2004), and 16S-1511f/23S-23r (ITS) (Harmsen et al., 2003) were used as control.
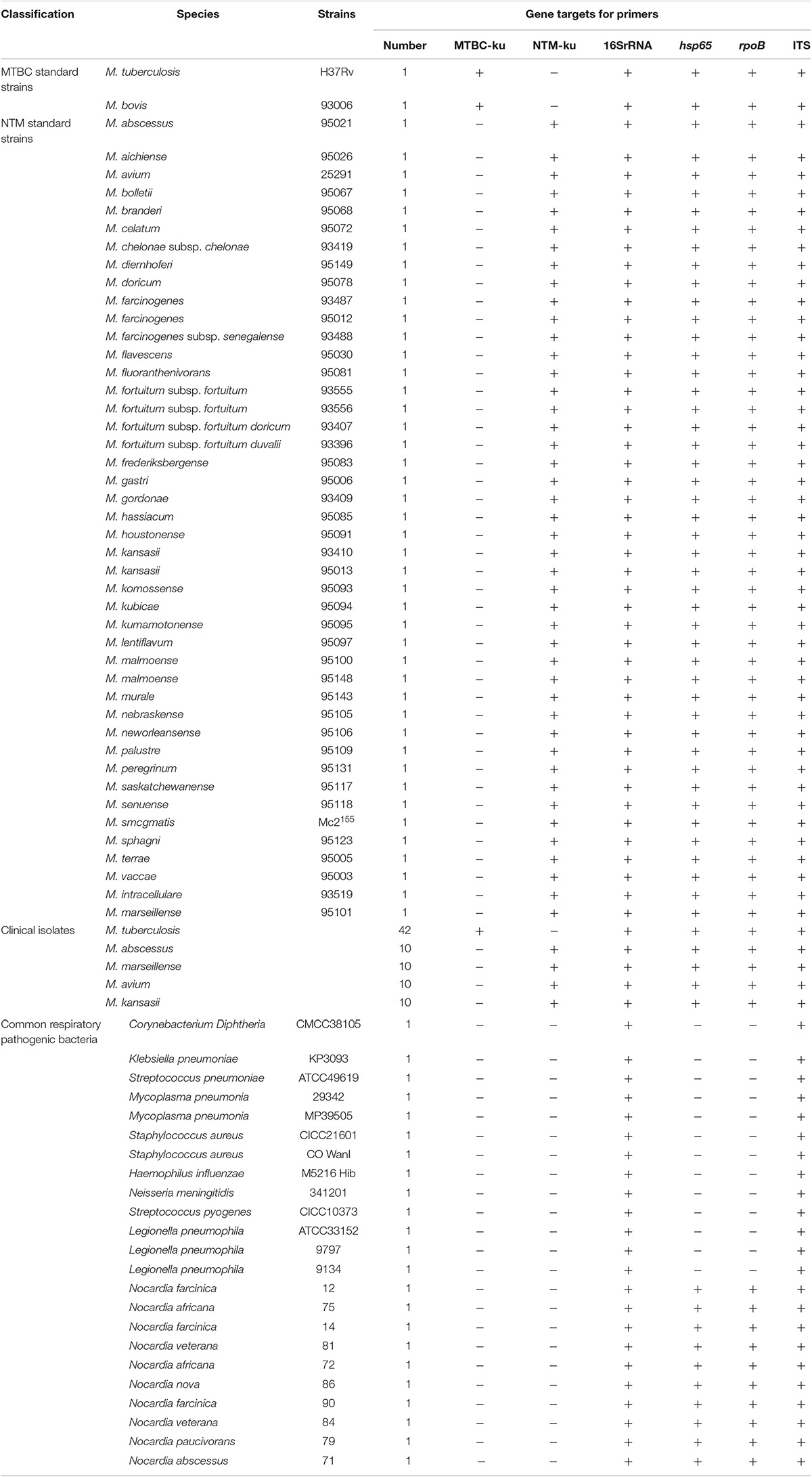
Table 1. Strains and the amplification in the actual PCR tests.
Fourfold table Chi-square test was used to test the differences in variant rates between the ku and rpoB genes.
The ku gene was found to be distributed in almost all of the 7294 Mycobacterium genomes with three exceptions: two incomplete genomes, M. setense strain 852014-10208_SCH5295773 and M. tuberculosis strain 0109V, without sequence analogous to the ku gene, and an incomplete ku gene in M. tuberculosis strain AH26_28866, with the first 290 bp in contig NZ_LKMH01000091.1 and 304–822 bp in contig NZ_LKMH01000168.1.
In M. tuberculosis, the ku gene was highly conserved. Of the 5243 M. tuberculosis genomes, there were 39 ku genotypes, of which 5149 (98.17%) genomes harbored the Rv0937c genotype, while 25 genomes were one genome with one genotype (Supplementary Table 1). The similarity of the ku gene sequence of the 39 genotypes was also very high. Only 37 sites on the 822 bp of the ku gene had variants, and sites 287, 449, and 451 had the highest rate of variation, at 5.13% (2/39).
In MTBC, except for M. tuberculosis, 116 genomes of four species (M. africanum, M. bovis, M. caprae, M. microti, and M. pinnipedii) and five variants (M. canettii, M. decipiens, M. mungi, and M. orygis) had 14 genotypes and 17 variant sites, among which the sequences of RN09_1148 genotype of M. africanum (28/29), LH58_05105 of M. bovis (68/70), BBG46_05065 of M. caprae (2/2), MORY_05401 of M. orygis (1/1), MPS_4136 of M. pinnipedii (2/2), B8W66_10645 of M. decipiens (1/1) and RN08_1045 of M. microti (1/1) were completely identical to that of the Rv0937c genotype, as a result of which Rv0937c was the dominant genotype in the MTBC (98%, 5253/5359). On the other hand, nine M. canettii genomes were found to have six genotypes and two variant sites, A210G and A487G, which appeared only in all M. canettii, indicating obvious species specificity. In addition, from the high conservation of the ku gene in MTBC, we inferred that M. tuberculosis strain 0109V and M. setense strain 852014-10208_SCH5295773 might also carry the ku gene, and the missing of the ku gene was caused by the incompletion of the genomic data.
In NTM, 278 sites of the ku gene were conserved in all NTM genotypes, but the conservation of the ku gene in each species varied greatly (Supplementary Table 1). In some species, the ku gene was highly conserved and had few genotypes. For example, 91.4% (139/152) of M. avium were concentrated in three types (MAV_1050, IQU_02120, and O982_17680), 94.1% (16/17) of M. immunogenum in ABG82_05475, and 61.9% (13/21) of M. kansasii in MKSMC1_52900. However, most species are genotype polymorphic. There were nine genotypes among 10 genomes in M. asiaticum and 14 genotypes among 42 genomes in M. chelonae. Furthermore, each genome had a genotype in some species, such as M. heckeshornense, M. llatzerense, and M. mageritense.
Overall, 32.4% sites (266/822) of the ku gene were conserved in all Mycobacterium genotypes, but none of the NTM genotype were 100% identical to the genotypes of MTBC, including Rv0937c. On phylogeny tree based on the ku gene, MTBC could be clearly separated from NTM without any exception (Figure 1). The ku genotypes in different species but having identical sequences are listed in Table 2. For MTBC, only M. canettii, M. decipiens, and M. mungi had completely special genotypes, other species of MTBC could not be separated by the ku gene sequence. For NTM, the genotypes of M. avium and M. intracellulare, M. abscessus, and M. chelonae only accounted for a very small proportion. Therefore, except the other 18 NTM species in Table 2, most of the NTMs could be identified to species level by the ku gene sequence.
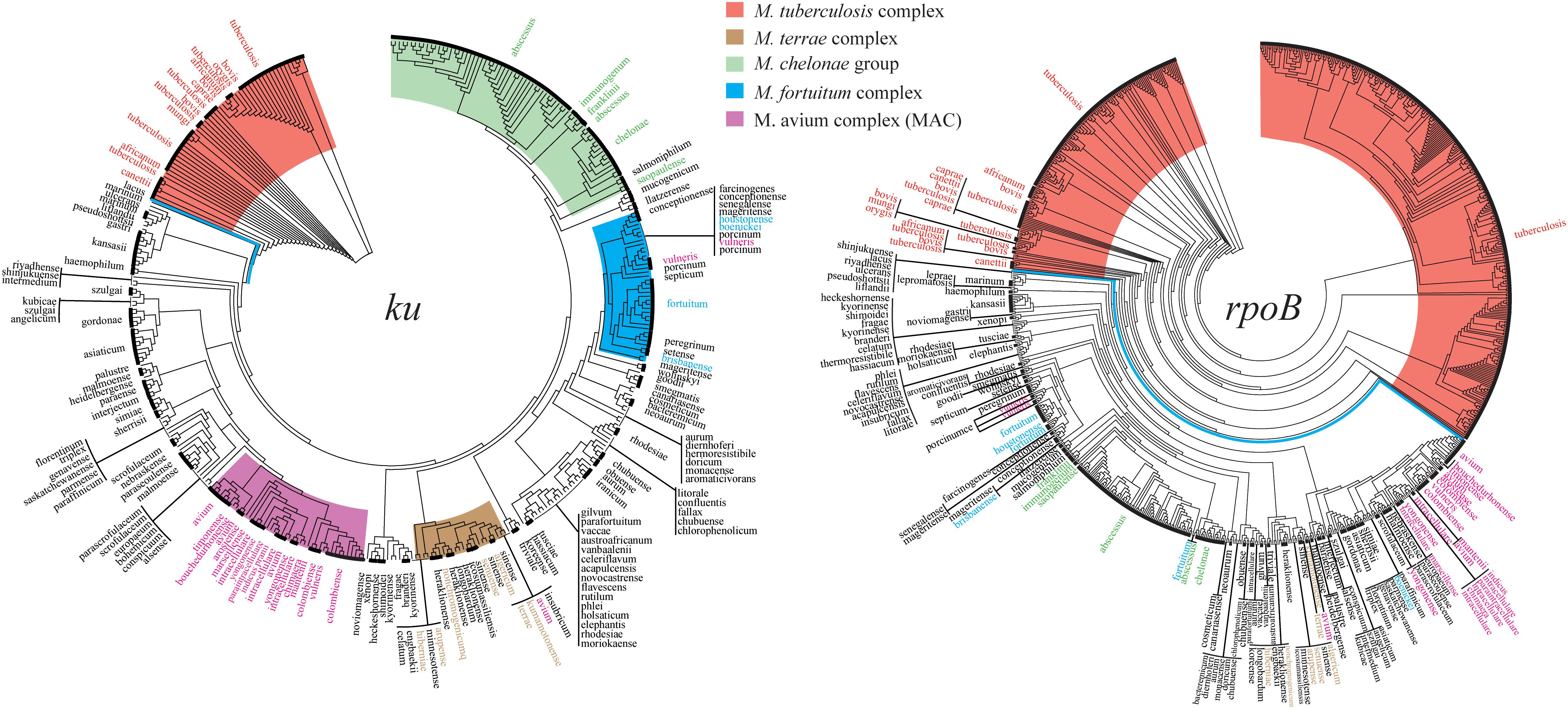
Figure 1. Phylogenetic trees drawn on the basis of the ku and rpoB genes. The Mycobacterium tuberculosis complex (MTBC) and the four complex groups of non-tuberculous mycobacteria (NTM) are highlighted by different colors.
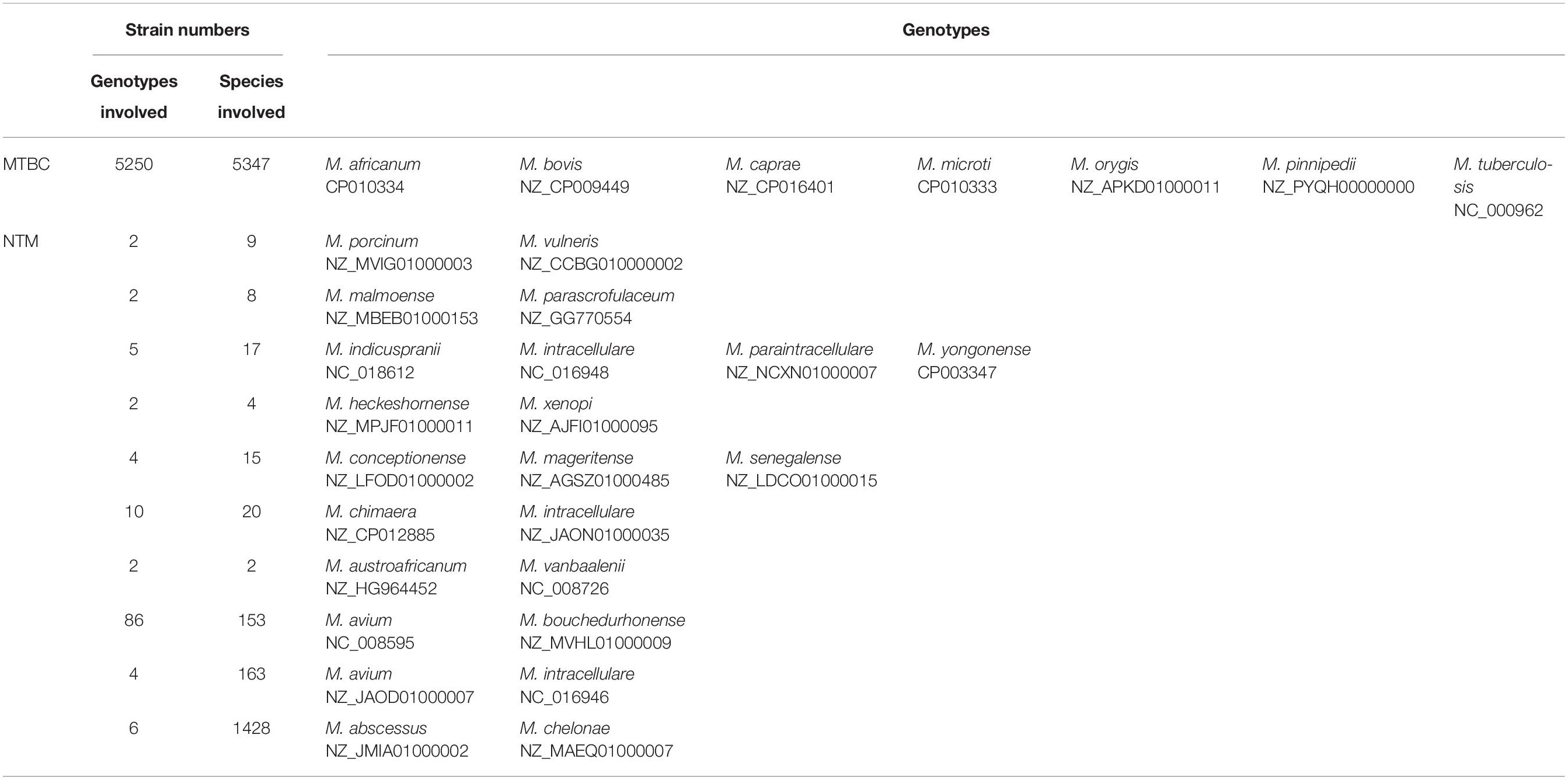
Table 2. ku genotypes in different species but with identical sequences.
The distribution of the two most used identification loci, IS6110 and rpoB, in the whole Mycobacterium genus had also been analyzed in this study.
The IS6110 element has 16 completely identical copies in the M. tuberculosis strain H37Rv genome. There was at least one IS6110 copy in 4450 M. tuberculosis genomes with identical sequence to the IS6110 element of H37Rv. The IS6110 element in 428 genomes were only partially identical to that of H37Rv. The IS6110 of 306 M. tuberculosis genomes were located at the end of contig and were incomplete. The remaining 61 genomes, including the complete genome of M. tuberculosis UT205, had no sequences homologous to that of IS6110. In the other MTBC species/variants, the complete genomes of M. canettii CIPT 140070008 and 140070017 also had no sequences similar to that of IS6110. Therefore, even though it has been used as an important diagnostic marker to identify MTBC species (Coros et al., 2008; Guernier et al., 2017), the IS6110 element was not common to all the MTBC strains, even M. tuberculosis. Tests based on it have false negatives, which is consistent with previous studies (Viana-Niero et al., 2006; Freidlin et al., 2017).
The rpoB gene was widely distributed in Mycobacterium and had a total of 861 genotypes, excluding 57 incomplete rpoB sequences from analysis (Supplementary Table 2). The rpoB gene had 454 variant sites in M. tuberculosis alone, compared to only 37 variant sites of the ku gene in M. tuberculosis. Even after standardization by gene length, the variant rate of rpoB gene in M. tuberculosis was 14.4% (454/3519 bp), which was far greater than that of the ku gene at 4.5% (37/822 bp) (Σ2 = 46.872, P < 0.01). The rpoB gene could set apart the MTBC from all NTM without any exception on its phylogenetic tree, but the four complex groups of NTM could not be separated as distinctly as the tree drawn on the ku gene (Figure 1).
To confirm the results of Weller et al. (2002), we downloaded 14 whole genomic sequences of other families belonging to Corynebacteriales with Mycobacteriaceae, Corynebacteriaceae (CP008913.1, CP017639.1, CP026947.1, and CP026948.1), Dietziaceae (CP027238.1, and CP024869.1), Gordoniaceae (CP002907.1, NZ_CP025435.1, and CP023405.1), Segniliparaceae (CP001958.1), Tsukamurellaceae (CP001966.1), and Nocardiaceae (CP018082.1, CP032568.1, and CP016819.1) and blasted Rv0937c (NC_000962) with these genomes. The results showed that there was no region homologous to Rv0937c in these genomes.
For the rarity of the ku gene in bacteria and the distinct clusters of MTBC and NTM on the phylogenetic tree, we inferred that mycobacteria might be specially identified from other bacteria and MTBC from NTM only by PCR without sequencing. Afterward, the PCR system was developed.
Mycobacterium tuberculosis complex-specific primers were designed in the conservative regions that were identical in the 55 ku genotypes of MTBC, but different to all ku genotypes of NTM. Candidate primer sets with different product lengths were designed and screened. The pair with the most sufficient difference between MTBC and NTM was selected. ku-MTBC-U: GGT GGT CGA CTA CCG CGA TCT T and ku-MTBC-L: TCT TCG GGC TCG TCC AGC AAC C were located at 159–180 bp and 719–740 bp of the reference sequence of Rv0937c genotype, respectively. Figure 2 shows that the design region of this pair of primers has a high degree of variation in NTM. Especially, the first base of the 3′ end of the upstream primer and the first, third and fourth base of the 3′ end of the downstream primer were single nucleotide polymorphic loci that completely distinguished MTBC from NTM. Simulated PCR revealed that 55 MTBC genotypes could be amplified by this primer set, but 324 NTM genotypes could not be amplified.
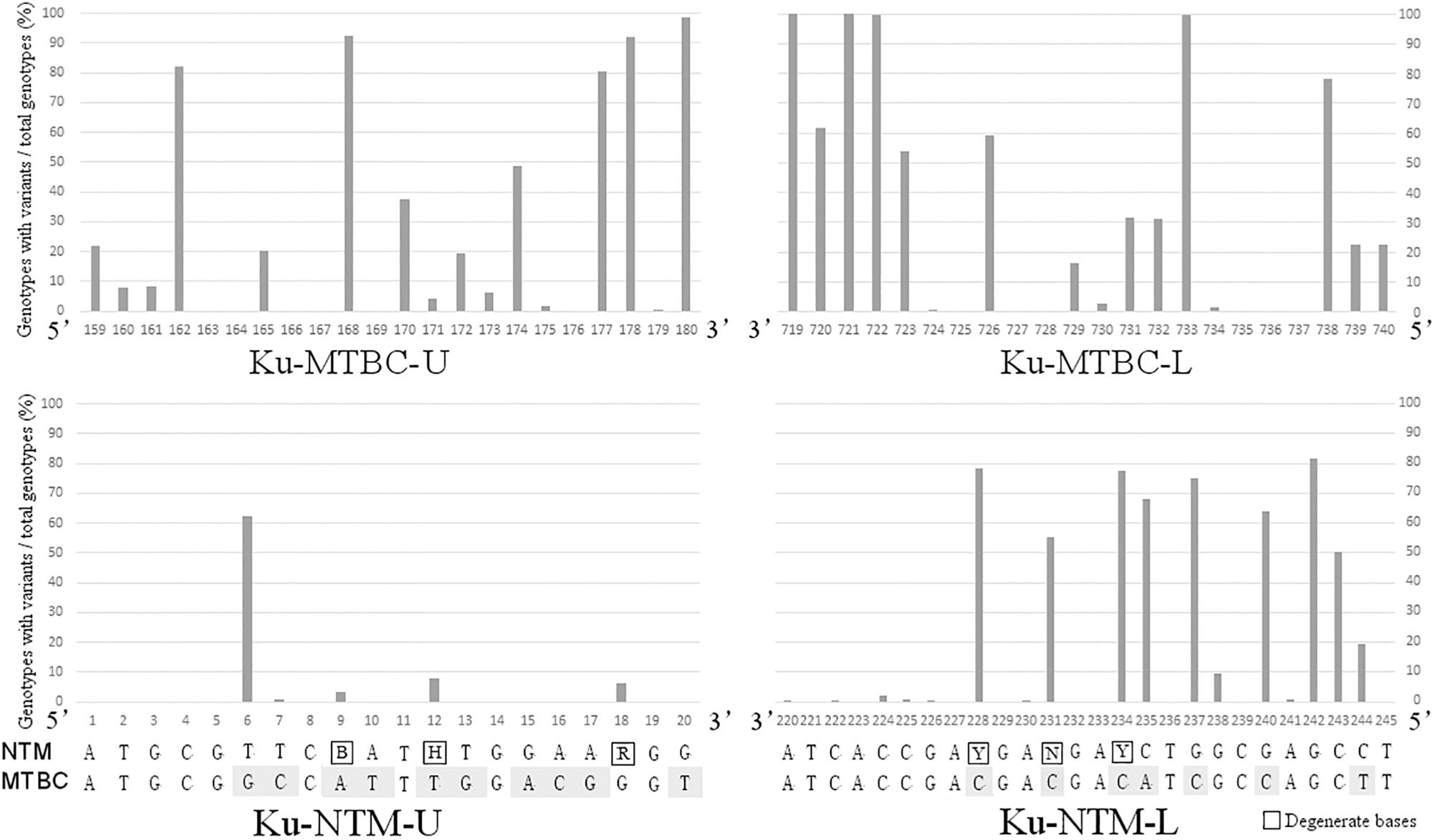
Figure 2. The variation rates of the primer design sites in NTM ku genotypes.
NZ_CP009616.seq of M. abscessus was used as reference of NTM for primer design. By marking the completely conserved sites in NTM on the reference sequence, it was found that the initial 1–20 bp of ku gene was the best region for the design of upstream primer ku-NTM-U with the most conserved sites in NTM and nine base difference to MTBC (Figure 2). The downstream primer Ku-NTM-L was an NTM/MTBC universal primer at 220–245 bp with eight conserved bases at the 3′ end. To improve the amplification efficiency of all NTMs, degenerate base were used. The primer set was ku-NTM-U: ATG CGT TCB ATH TGG AAR GG and ku-NTM-L: AGG CTC GCC AGR TCN TCR TCG GTG AT, and its specificity for NTM depends on the upstream primer.
In the actual PCR amplification of the 148 Mycobacterium and common respiratory pathogenic bacteria, all of the MTBC strains were positive for the amplification of ku-MTBC-U/L and negative for ku-NTM-U/L. The amplifications of NTM strains were opposite, and all of the 23 respiratory pathogenic bacteria were negative for both of them. The sensitivity and specificity of the two pairs of primers were 100%. Among the control primers, primers of 16SrRNA and ITS were universal and almost all of the tested strains, including the 23 respiratory pathogenic strains, were positive for them. Primers for hsp60 and rpoB were specific for Mycobacterium. But all of the 10 Nocardia strains belonging to six Nocardia species were positive, although other respiratory pathogenic strains were negative (Table 1). Without sequencing, none of the four pairs of primers would be suitable for the identification of Mycobacterium, let alone the distinction between MTBC and NTM.
The GenBank database contains plenty of Mycobacterium genome data. The sensitivity and specificity of molecular detection methods can be predicted and compared before actual use. In this work, approximately 151 definite species/variants of Mycobacterium have been included, accounting for 79.7% of all known mycobacterial species and all submitted genomes. This work has the same coverage as the phylogenomics and comparative genomics studies of Gupta et al. (2018). Although it does not cover all the Mycobacterium species, it may be the most comprehensive analysis that can be done so far and includes all of the clinically common species.
The similarity between the eukaryotic and bacterial Ku proteins suggested that they were evolved from a common ancestor and very ancient process (Weller et al., 2002). The ku gene should be present in the Mycobacterium genome before the differentiation of Mycobacterium species. Additionally, The Ku-based NHEJ system participates in the repairing of DSBs of Mycobacterium, which maintains the genome integrity and is pivotal for cell survival. The ku gene is less likely to be lost in the evolution of Mycobacterium. Thus, it is reasonable that the ku gene is distributed and conserved in all sequenced Mycobacterium genomes. Moreover, the research of Weller et al. (2002) has speculated that the Ku ligase system might be related to the special growth stage of mycobacteria, especially the bacterial sporulation and the long stationary phase of life cycle. Different growth rates and culture and biochemical test characteristics are just important indicators for distinguishing Mycobacterium species, especially NTM. Therefore, in theory, it is not unexpected that the ku gene can completely distinguish MTBC from NTM and distinguish species in NTM.
Among the detection methods of Mycobacterium, acid-fast (AF) staining, also known as Ziehl–Neelsen stain, is currently the most widely used preliminary diagnostic method. The sensitivity of AF staining compared with culture ranges from 22 to 78%, and its limit of detection ranges from 5 × 103 to 1 × 104 bacilli/mL. However, AF staining is not specific for Mycobacterium detection, and Mycobacterium spp. cannot be distinguished from other AF bacteria, such as Nocardia, Rhodococcus, Tsukamurella, Gordona, Dietzia, Legionella micdadei, Cryptosporidium, Isospora belli, and Cyclospora cayetanensis and parasites such as Sarcocystis and Taenia saginata. Moreover, non-AF tuberculosis bacilli (Vilcheze and Kremer, 2017) also exist.
Thus, nucleic acid detection methods (NADMs) are becoming more and more important in the diagnosis and identification of mycobacteria (Niemann et al., 2000; Brunello et al., 2001). Loci of developed NADM include species-specific insertion sequences, such as IS6110 for members of the M. tuberculosis complex (Thierry et al., 1990), IS900 and F57 for M. avium subsp. paratuberculosis (Slana et al., 2008), IS901 for M. avium subsp. avium (Slana et al., 2010), IS2404 and IS2606 for M. ulcerans, M. liflandii, M. pseudoshottsii, and M. shottsii (mycolactone-producing mycobacteria) (Fyfe et al., 2007), and common shared bacterial genes, for example 16S rRNA, hsp65, rpoB genes and the internal transcribed spacer (ITS) of broad-range sequencing approaches. Multi-genes analysis (Homolka et al., 2012; Perez-Lago et al., 2014; Gupta et al., 2018) and whole genome sequencing (Vissa et al., 2009; Fedrizzi et al., 2017; Trofimov et al., 2018) have also been used in Mycobacterium and because of their high resolution, they can identify mycobacteria to the species level. Methods based on sequencing and homology comparison, especially on sequences of hundreds of core genes or whole genome are more available for research purposes, and they are promising tools in the identification of new mycobacteria species, new molecules for bacterial typing and new candidate genes for multidrug resistance. But they are less practical for rapid screening of large samples and for primary hospitals without the ability of bioinformatics analysis. This is also the reason that single locus, IS6110 and rpoB, are the most commonly used biomarkers. But IS6110 cannot be applied to NTM. Although rpoB is used not only for identification but also for antibiotic resistance prediction, and GeneXpert assay which based on it requires little technical training and can obtained from unprocessed sputum samples in 90 min, with minimal biohazard. GeneXpert is replacing AF and endorsed by the World Health Organization (WHO) (Moure et al., 2011; Li et al., 2017), but it is also only applicable to MTBC and requires special instruments. In fact, until now, there has been no specific biomarker that is commonly used for all mycobacteria without sequencing.
The analysis in this study showed the value of the ku gene as a diagnostic biomarker. The ku gene is not a common gene of bacteria. Its rarity in prokaryotes (Weller et al., 2002), especially its absence in bacteria closely related to Mycobacterium (such as Nocardia), endows it with high specificity. Its wide distribution in all sequenced Mycobacterium makes it widely applicable for MTBC and NTM. Both features actualize the greatest application value of the ku gene, that is, they can directly distinguish mycobacteria, MTBC and NTM by PCR, and achieve the purpose of rapid clinical diagnosis. This actual application value has been confirmed by the MTBC/NTM- specific primers we designed and the testing of the standard and clinical strains. In conclusion, Ku gene is a new single-gene biomarker of Mycobacterium that differentiates MTBC and NTM and makes the identification of MTBC and NTM simpler and more accurate. However, it does not define the resistance of Mycobacterium, so it can’t take into account the identification of strains and the prediction of drug resistance simultaneously as ropB gene does. Its application value lies in that it can be used as a single indicator for the primary screening and identification of mycobacteria, or can be combined with rpoB to complement its deficiencies for accurate identification of Mycobacterium. More sensitive detection methods based on this gene and application for the detection of different samples besides pure cultures will be explored in our further study to help the diagnosis of Tb worldwide.
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
RW conceived the project, analyzed and interpreted the data, designed the primers, and prepared the figures and manuscript. LZ, CM, and TX did the PCR testing. ML and XZ collected the strains. HL downloaded the Mycobacterium genomes from the GenBank database. KW revised the manuscript. All authors had full access to all the data in the study and approved the final version of the manuscript for submission.
The authors have applied for a pending Chinese patent titled “The application of the Ku protein in Mycobacterium,” Number: 201910154948.5.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01887/full#supplementary-material
Adekambi, T., and Drancourt, M. (2004). Dissection of phylogenetic relationships among 19 rapidly growing Mycobacterium species by 16S rRNA, hsp65, sodA, recA and rpoB gene sequencing. Int. J. Syst. Evol. Microbiol. 54, 2095–2105. doi: 10.1099/ijs.0.63094-0
Brunello, F., Ligozzi, M., Cristelli, E., Bonora, S., Tortoli, E., and Fontana, R. (2001). Identification of 54 mycobacterial species by PCR-restriction fragment length polymorphism analysis of the hsp65 gene. J. Clin. Microbiol. 39, 2799–2806. doi: 10.1128/jcm.39.8.2799-2806.2001
Coros, A., Deconno, E., and Derbyshire, K. M. (2008). IS6110, a Mycobacterium tuberculosis complex-specific insertion sequence, is also present in the genome of Mycobacterium smegmatis, suggestive of lateral gene transfer among Mycobacterial species. J. Bacteriol. 190, 3408–3410. doi: 10.1128/JB.00009-08
Della, M., Palmbos, P. L., Tseng, H. M., Tonkin, L. M., Daley, J. M., Topper, L. M., et al. (2004). Mycobacterial Ku and ligase proteins constitute a two-component NHEJ repair machine. Science 306, 683–685. doi: 10.1126/science.1099824
Doherty, A. J., Jackson, S. P., and Weller, G. R. (2001). Identification of bacterial homologues of the Ku DNA repair proteins. FEBS Lett. 500, 186–188. doi: 10.1016/s0014-5793(01)02589-3
Fedrizzi, T., Meehan, C. J., Grottola, A., Giacobazzi, E., Fregni Serpini, G., Tagliazucchi, S., et al. (2017). Genomic characterization of nontuberculous Mycobacteria. Sci. Rep. 7:45258. doi: 10.1038/srep45258
Freidlin, P. J., Nissan, I., Luria, A., Goldblatt, D., Schaffer, L., Kaidar-Shwartz, H., et al. (2017). Structure and variation of CRISPR and CRISPR-flanking regions in deleted-direct repeat region Mycobacterium tuberculosis complex strains. BMC Genomics 18:168. doi: 10.1186/s12864-017-3560-6
Fyfe, J. A., Lavender, C. J., Johnson, P. D., Globan, M., Sievers, A., Azuolas, J., et al. (2007). Development and application of two multiplex real-time PCR assays for the detection of Mycobacterium ulcerans in clinical and environmental samples. Appl. Environ. Microbiol. 73, 4733–4740. doi: 10.1128/aem.02971-06
Gong, C., Bongiorno, P., Martins, A., Stephanou, N. C., Zhu, H., Shuman, S., et al. (2005). Mechanism of nonhomologous end-joining in mycobacteria: a low-fidelity repair system driven by Ku, ligase D and ligase C. Nat. Struct. Mol. Biol. 12, 304–312. doi: 10.1038/nsmb915
Guernier, V., Diefenbach-Elstob, T., Pelowa, D., Pollard, S., Burgess, G., Mcbryde, E. S., et al. (2017). Molecular diagnosis of suspected tuberculosis from archived smear slides from the Balimo region, Papua New Guinea. Int. J. Infect. Dis. 67, 75–81. doi: 10.1016/j.ijid.2017.12.004
Gupta, R., Barkan, D., Redelman-Sidi, G., Shuman, S., and Glickman, M. S. (2011). Mycobacteria exploit three genetically distinct DNA double-strand break repair pathways. Mol. Microbiol. 79, 316–330. doi: 10.1111/j.1365-2958.2010.07463.x
Gupta, R. S., Lo, B., and Son, J. (2018). Phylogenomics and comparative genomic studies robustly support division of the genus Mycobacterium into an emended genus Mycobacterium and four novel genera. Front. Microbiol. 9:67. doi: 10.3389/fmicb.2018.00067
Halstrom, S., Price, P., and Thomson, R. (2015). Review: environmental Mycobacteria as a cause of human infection. Int. J. Mycobacteriol. 4, 81–91. doi: 10.1016/j.ijmyco.2015.03.002
Harmsen, D., Dostal, S., Roth, A., Niemann, S., Rothganger, J., Sammeth, M., et al. (2003). RIDOM: comprehensive and public sequence database for identification of Mycobacterium species. BMC Infect. Dis. 3:26.
Homolka, S., Projahn, M., Feuerriegel, S., Ubben, T., Diel, R., Nubel, U., et al. (2012). High resolution discrimination of clinical Mycobacterium tuberculosis complex strains based on single nucleotide polymorphisms. PLoS One 7:e39855. doi: 10.1371/journal.pone.0039855
Korycka-Machala, M., Brzostek, A., Rozalska, S., Rumijowska-Galewicz, A., Dziedzic, R., Bowater, R., et al. (2006). Distinct DNA repair pathways involving RecA and nonhomologous end joining in Mycobacterium smegmatis. FEMS Microbiol. Lett. 258, 83–91. doi: 10.1111/j.1574-6968.2006.00199.x
Li, Y., Pang, Y., Zhang, T., Xian, X., Wang, X., Yang, J., et al. (2017). Rapid diagnosis of extrapulmonary tuberculosis with xpert Mycobacterium tuberculosis/rifampicin assay. J. Med. Microbiol. 66, 910–914. doi: 10.1099/jmm.0.000522
Mortaz, E., Moloudizargari, M., Varahram, M., Movassaghi, M., Garssen, J., Kazempour Dizagie, M., et al. (2018). What immunological defects predispose to non-tuberculosis Mycobacterial infections? Iran J. Allergy Asthma Immunol. 17, 100–109.
Moure, R., Munoz, L., Torres, M., Santin, M., Martin, R., and Alcaide, F. (2011). Rapid detection of Mycobacterium tuberculosis complex and rifampin resistance in smear-negative clinical samples by use of an integrated real-time PCR method. J. Clin. Microbiol. 49, 1137–1139. doi: 10.1128/JCM.01831-10
Niemann, S., Harmsen, D., Rusch-Gerdes, S., and Richter, E. (2000). Differentiation of clinical Mycobacterium tuberculosis complex isolates by gyrB DNA sequence polymorphism analysis. J. Clin. Microbiol. 38, 3231–3234.
Perez-Lago, L., Navarro, Y., and Garcia-De-Viedma, D. (2014). Current knowledge and pending challenges in zoonosis caused by Mycobacterium bovis: a review. Res. Vet. Sci. 97(Suppl.), S94–S100. doi: 10.1016/j.rvsc.2013.11.008
Pitcher, R. S., Tonkin, L. M., Daley, J. M., Palmbos, P. L., Green, A. J., Velting, T. L., et al. (2006). Mycobacteriophage exploit NHEJ to facilitate genome circularization. Mol. Cell. 23, 743–748. doi: 10.1016/j.molcel.2006.07.009
Shuman, S., and Glickman, M. S. (2007). Bacterial DNA repair by non-homologous end joining. Nat. Rev. Microbiol. 5, 852–861.
Slana, I., Kaevska, M., Kralik, P., Horvathova, A., and Pavlik, I. (2010). Distribution of Mycobacterium avium subsp. avium and M. a. hominissuis in artificially infected pigs studied by culture and IS901 and IS1245 quantitative real time PCR. Vet. Microbiol. 144, 437–443. doi: 10.1016/j.vetmic.2010.02.024
Slana, I., Kralik, P., Kralova, A., and Pavlik, I. (2008). On-farm spread of Mycobacterium avium subsp. paratuberculosis in raw milk studied by IS900 and F57 competitive real time quantitative PCR and culture examination. Int. J. Food Microbiol. 128, 250–257. doi: 10.1016/j.ijfoodmicro.2008.08.013
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Telenti, A., Marchesi, F., Balz, M., Bally, F., Bottger, E. C., and Bodmer, T. (1993). Rapid identification of mycobacteria to the species level by polymerase chain reaction and restriction enzyme analysis. J. Clin. Microbiol. 31, 175–178.
Thierry, D., Cave, M. D., Eisenach, K. D., Crawford, J. T., Bates, J. H., Gicquel, B., et al. (1990). IS6110, an IS-like element of Mycobacterium tuberculosis complex. Nucleic Acids Res. 18:188.
Trofimov, V., Kicka, S., Mucaria, S., Hanna, N., Ramon-Olayo, F., Del Peral, L. V., et al. (2018). Antimycobacterial drug discovery using mycobacteria-infected amoebae identifies anti-infectives and new molecular targets. Sci. Rep. 8:3939. doi: 10.1038/s41598-018-22228-6
Viana-Niero, C., Rodriguez, C. A., Bigi, F., Zanini, M. S., Ferreira-Neto, J. S., Cataldi, A., et al. (2006). Identification of an IS6110 insertion site in plcD, the unique phospholipase C gene of Mycobacterium bovis. J. Med. Microbiol. 55, 451–457. doi: 10.1099/jmm.0.46364-0
Vilcheze, C., and Kremer, L. (2017). Acid-fast positive and acid-fast negative Mycobacterium tuberculosis: the koch paradox. Microbiol Spectr 5:TBTB2-TBTB0003. doi: 10.1128/microbiolspec.TBTB2-0003-2015
Vissa, V. D., Sakamuri, R. M., Li, W., and Brennan, P. J. (2009). Defining mycobacteria: shared and specific genome features for different lifestyles. Indian J. Microbiol. 49, 11–47. doi: 10.1007/s12088-009-0006-0
Wang, L., Zhang, H., Ruan, Y., Chin, D. P., Xia, Y., Cheng, S., et al. (2014). Tuberculosis prevalence in China, 1990-2010; a longitudinal analysis of national survey data. Lancet 383, 2057–2064. doi: 10.1016/S0140-6736(13)62639-2
Weller, G. R., Kysela, B., Roy, R., Tonkin, L. M., Scanlan, E., Della, M., et al. (2002). Identification of a DNA nonhomologous end-joining complex in bacteria. Science 297, 1686–1689. doi: 10.1126/science.1074584
Zheng, X., Li, S. Y., Zhao, G. P., and Wang, J. (2017). An efficient system for deletion of large DNA fragments in Escherichia coli via introduction of both Cas9 and the non-homologous end joining system from Mycobacterium smegmatis. Biochem. Biophys. Res. Commun. 485, 768–774. doi: 10.1016/j.bbrc.2017.02.129
Keywords: Mycobacterium, non-homologous DNA end-joining, polymorphism, biomarker, non-tuberculosis mycobacteria, non-sequencing
Citation: Zhou L, Ma C, Xiao T, Li M, Liu H, Zhao X, Wan K and Wang R (2019) A New Single Gene Differential Biomarker for Mycobacterium tuberculosis Complex and Non-tuberculosis Mycobacteria. Front. Microbiol. 10:1887. doi: 10.3389/fmicb.2019.01887
Received: 03 May 2019; Accepted: 30 July 2019;
Published: 13 August 2019.
Edited by:
Aleksandra Barac, University of Belgrade, SerbiaReviewed by:
Muge Cevik, University of St Andrews, United KingdomCopyright © 2019 Zhou, Ma, Xiao, Li, Liu, Zhao, Wan and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruibai Wang, d2FuZ3J1aWJhaUBpY2RjLmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.