 Kelsey R. Moore
Kelsey R. Moore Cara Magnabosco2
Cara Magnabosco2 Lily Momper
Lily Momper Tanja Bosak
Tanja Bosak Gregory P. Fournier
Gregory P. Fournier- 1Department of Earth, Atmospheric and Planetary Sciences, Massachusetts Institute of Technology, Cambridge, MA, United States
- 2Center for Computational Biology, Flatiron Institute, Simons Foundation, New York, NY, United States
- 3Department of Earth and Planetary Sciences, University of California, Davis, Davis, CA, United States
The phylum Cyanobacteria includes free-living bacteria and plastids, the descendants of cyanobacteria that were engulfed by the ancestral lineage of the major photosynthetic eukaryotic group Archaeplastida. Endosymbiotic events that followed this primary endosymbiosis spread plastids across diverse eukaryotic groups. The remnants of the ancestral cyanobacterial genome present in all modern plastids, enable the placement of plastids within Cyanobacteria using sequence-based phylogenetic analyses. To date, such phylogenetic studies have produced conflicting results and two competing hypotheses: (1) plastids diverge relatively recently in cyanobacterial evolution and are most closely related to nitrogen-fixing cyanobacteria, or (2) plastids diverge early in the evolutionary history of cyanobacteria, before the divergence of most cyanobacterial lineages. Here, we use phylogenetic analysis of ribosomal proteins from an expanded data set of cyanobacterial and representative plastid genomes to infer a deep placement for the divergence of the plastid ancestor lineage. We recover plastids as sister to Gloeomargarita and show that the group diverges from other cyanobacterial groups before Pseudanabaena, a previously unreported placement. The tree topologies and phylogenetic distances in our study have implications for future molecular clock studies that aim to model accurate divergence times, especially with respect to groups containing fossil calibrations. The newly sequenced cyanobacterial groups included here will also enable the use of novel cyanobacterial microfossil calibrations.
Introduction
Two major groups of organisms produce oxygen by oxygenic photosynthesis: Cyanobacteria, the bacterial group in which this metabolism first evolved, and photosynthetic eukaryotes. Photosynthesis in eukaryotes is carried out by plastids, specialized organelles capable of capturing and converting light energy using Photosystems I and II. Following the first suggestion of a cyanobacterial ancestor of the plastids (Schimper, 1885), studies have attempted to characterize the commonalities and relationships between different groups of cyanobacteria and plastids. Subsequent work firmly established that plastids originated in an endosymbiotic event in which an early eukaryote engulfed a cyanobacterium (Whatley et al., 1979; Whatley and Whatley, 1981; Cavalier-Smith, 1982; Douglas, 1998). A major component of this work has attempted to determine the sequence of evolutionary events leading from the single endosymbiotic event in the ancestor of Archaeplastida to the subsequent diversification of the three primary photosynthetic eukaryote lineages (Glaucophyta, Rhodophyta, and Viridiplantae) (Whatley and Whatley, 1981; Cavalier-Smith, 1982; McFadden, 2001). However, the nature of the direct cyanobacterial ancestor of the major plastid lineages and its relationship to other cyanobacteria remain poorly understood (Sato, 2007). Cyanobacteria and plastids share several key features such as thylakoid membranes containing Photosystems I and II, but the ancestral plastid lineage is highly derived and altered from its cyanobacterial ancestor. Additionally, the ultrastructure and pigments of plastids differ among the various groups of photosynthetic eukaryotes. Consequently, structural and morphological comparisons alone cannot be used to determine the closest cyanobacterial relative to the plastid ancestor.
Phylogenetic reconstructions based on nucleotide or amino acid sequence data provide an alternative strategy for determining the evolutionary relationships and history of plastids and Cyanobacteria. However, these approaches have an independent set of challenges. Studies to date have attempted to uncover the placement of plastids within the cyanobacterial tree using a range of sequence datasets and taxonomic sampling. The topologies for the major cyanobacterial groups produced by these different analyses are in general agreement (e.g., Shih et al., 2013; Schirrmeister et al., 2015). However, the placement of plastids – and therefore the divergence of their ancestral cyanobacterial lineage – has proven contentious. Two main hypotheses emerge from these studies. Hypothesis 1 places the divergence of the plastid lineage relatively recently, close to the nitrogen fixing cyanobacteria (Douglas and Turner, 1991; Douglas, 1998; Ochoa de Alda et al., 2014). This topology was recovered using 16S rRNA sequences (Douglas and Turner, 1991), a concatenation of 16S rRNA and 23S rRNA sequences (Ochoa de Alda et al., 2014), genes such as tufA, atpB, rpoC1, and psbA (Douglas, 1998), a concatenated dataset of 16S rRNA and the rbcL genes (Falcón et al., 2010), and photosynthetic eukaryotic nuclear encoded protein sequences and their cyanobacterial homologs (Dagan et al., 2012). An additional study by Deusch et al. (2008) used nuclear encoded proteins in a group of photosynthetic eukaryotic lineages with cyanobacterial homologs and found that plastids are most closely related to heterocystous nitrogen-fixing cyanobacteria. Hypothesis 2 places plastids much deeper in the tree. This topology was recovered by studies that used aligned amino acid sequences from a wide range of concatenated protein and rRNA datasets (Rodríguez-Ezpeleta et al., 2005; Reyes-Prieto et al., 2010; Criscuolo and Gribaldo, 2011; Li et al., 2014). In general, many of these studies use a limited set of cyanobacteria and do not always recover the phylogenetic placement of plastids with high confidence. A more recent analysis included a newly sequenced cyanobacterium (Gloeomargarita lithophora) and used a concatenation of 97 proteins chosen from plastid genomes to suggest a deep placement of plastids with G. lithophora as a sister group to their cyanobacterial ancestor (Ponce-Toledo et al., 2017). This result was further supported by phylogenies of concatenated 16S and 23S rRNA datasets, although Bayesian consensus trees generated from these datasets did not resolve the deep relationship between Pseudanabaena, G. lithophora+plastids, and other more derived clades of cyanobacteria. Another recent study similarly recovered deep divergences of both plastids and G. lithophora using a concatenation of proteins for Cyanobacteria and a concatenation of 26 nucleotide sequences for the plastids (Sánchez-Baracaldo et al., 2017). Both analyses recovered a topology in which plastids diverged after Pseudanabaena and the clade containing Thermosynechococcus (Sánchez-Baracaldo et al., 2017). Overall, the range of data sets and taxon sampling used in previous studies, and the absence or differing placement of some key groups like Pseudanabaena – a possible sister group to the plastids (Shih et al., 2013) – suggest that establishing the placement of plastids within the cyanobacterial tree requires further investigation.
Resolving the placement of plastids is complicated by several factors that may influence phylogenetic reconstructions. For example, the transition from endosymbiont to organelle is accompanied by gene loss, endosymbiotic gene transfer, amino acid and nucleotide compositional changes, and even rearrangement of the genetic code itself (Kurland, 1992). These processes decrease the amount of phylogenetic information available for evolutionary inference and increase the chance of tree reconstruction artifacts such as long branch attraction. Such factors have been noted in debates regarding the placement of the alphaproteobacterial ancestor lineage of mitochondria (Fitzpatrick et al., 2006; Ferla et al., 2013; Wang and Wu, 2015; Martijn et al., 2018), and may impact the nucleotide or amino acid sequences used in any given phylogenetics study to varying degrees. This may, in part, explain the conflicting topologies recovered for the placement of plastids within Cyanobacteria. Another complicating factor is horizontal gene transfer (HGT), as the cyanobacterial lineage ancestral to plastids may have acquired genes from other cyanobacterial groups. This is a frequent occurrence, preferentially involving cyanobacteria-specific genes, including photosynthesis-associated genes subsequently inherited by plastids (Yerrapragada et al., 2009).
Resolving the phylogenomic history of cyanobacterial genes, including plastid genes, is a complex problem. It is entirely plausible that many plastid genes trace different deep evolutionary histories through cyanobacterial evolution. This is a well-known critique and challenge to Tree of Life studies in general (Bapteste et al., 2009). One approach, favored by several of the studies above, and consistent with many Tree of Life studies (e.g., Williams et al., 2012; Hug et al., 2016) is to use a standard “core” set of conserved and ubiquitous genes that are expected to infrequently experience HGT, and thus are more likely to trace the underlying history of cellular descent. While such a tree may not capture the totality of the complex evolution of plastid genomes, it is more likely to represent the tree of cellular descent that necessarily links the plastid to its cyanobacterial origins.
Here, we elaborate on ribosomal protein studies addressing the placement of plastids within the cyanobacterial tree. We use an updated and expanded taxonomic sampling, which includes Pseudanabaena and 20 previously unsequenced cyanobacterial species, to generate a plastid/cyanobacteria species tree. This expanded diversity provides a means to potentially better resolve the deep divergences within Cyanobacteria, including the ancestor lineage to plastids. The resulting phylogenies show long branches among plastids relative to crown Cyanobacteria, and place the plastids deep within the cyanobacterial tree, between Synechococcus sp. JA-2-3B a 2-13 and Pseudanabaena. These results are especially relevant to molecular clock studies that use cyanobacterial/plastid phylogenetic trees calibrated by fossil evidence, as accurate tree topologies are important for estimation of divergence times.
Materials and Methods
Selection of Available Sequences
We selected a core set of 36 cyanobacterial species, 32 plastid lineages, and 45 bacterial outgroups from genomes that were available on the NCBI database1 (Table 1). We selected between one and eleven representative taxa from each of the major cyanobacterial groups described in previous work (Schirrmeister et al., 2011; Shih et al., 2013), depending on how many genomes are currently available from those groups. We additionally selected representatives within groups that had the most complete set of ribosomal proteins used in our analyses in an effort to maximize the effectiveness of our data set and fairly compare across all groups without introducing bias in lack of sequence information. All protein analyses used a concatenation of 30 conserved large and small subunit ribosomal protein sequences (Table 2) as initially selected in Magnabosco et al. (2018), with orthologs from additional cyanobacterial and plastid genomes identified in Genbank using BLASTp (Altschul et al., 1990), taking the top reciprocal hit in each case, when present. In some cases, a homolog to the query ribosomal protein was not detected. These were left as missing data in the concatenated alignment and are noted in section “rRNA Sequence Trimming” in Supplementary Methods. Taxa were selected to provide representative coverage of the major cyanobacterial groups, and to avoid oversampling of heavily sequenced cyanobacteria such as Prochlorococcus.
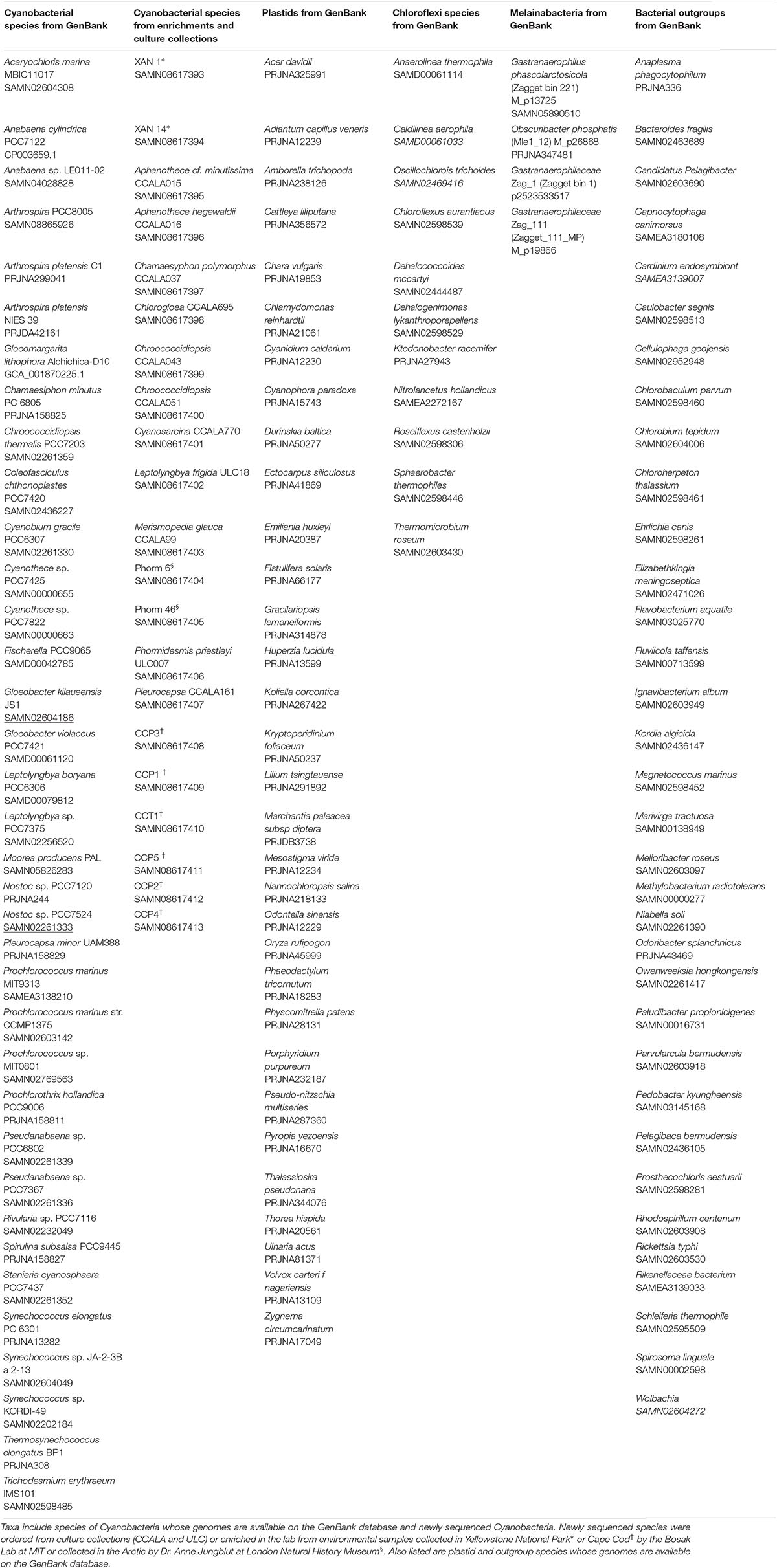
Table 1. List of taxa included in phylogenetic trees.

Table 2. Large and small subunit proteins used in this study.
For ribosomal RNA datasets, available 16S rRNA sequences were obtained from each of the selected genomes through the Silva high quality ribosomal RNA database (Quast et al., 2013). 23S rRNA was additionally obtained from genomes where a 16S sequence was found. Complete rRNA gene sequences were identified in the newly sequenced cyanobacterial genomes using BLASTn (Altschul et al., 1990). In the case of rRNA gene duplicates within genomes, only one sequence was selected.
Selection and Sequencing of Additional Cyanobacteria
In addition to cyanobacterial species with ribosomal protein sequences that are currently available on the GenBank database, our analyses included 20 previously unsequenced cyanobacteria and one genome previously sequenced and described by Lara et al. (2017) and Cornet et al. (2018) (Table 1). These were ordered from culture collections or enriched from environmental samples (See Supplementary Methods). We sequenced the genomes of these species both to increase the coverage of cyanobacteria represented in phylogenetic trees, and to improve our understanding of the phylogenetic placement of organisms with distinct morphologies, behaviors and ecological niches.
DNA was extracted using a PowerSoil DNA Isolation Kit (Mo Bio Laboratories, Inc., San Diego, CA, United States), and DNA concentrations were measured using a Qubit 2.0 Fluorometer (ThermoFischer Scientific, Waltham, MA, United States). The extracted DNA was sent to the MIT BioMicro Center Core Facility for sequencing. Libraries were prepared using Nextera DNA Library Prep and DNA was sequenced on an Illumina MiSeq or HiSeq 2000 platform at the MIT Center for Environmental Health Sciences (CEHS) Genomics Facilities Core. The sequencing yielded an average of 250 base pair (bp) paired end reads. Quality control was performed using Trimmomatic 0.36 with default parameters and a minimum sequence length of 50 base pairs (Bolger et al., 2014). Reads were assembled using SPAdes 3.9.0 (Bankevich et al., 2012) with a minimum contig length of 1,000 base pairs. Target Cyanobacteria genomes were extracted and binned using sequence composition and read-pair linkage through the CONCOCT algorithm within the Anvi’o software (Alneberg et al., 2014; Eren et al., 2015). MAGs were manually refined and curated using the interactive interface in the Anvi’o program (Eren et al., 2015). After refinement, genome completeness and contamination were assessed using the CheckM workflow (Parks et al., 2015) and output is listed in Supplementary Table 2. Following successful sequencing and reconstruction of cyanobacterial genomes, small and large subunit ribosomal proteins were identified and extracted from the resulting genomic assemblies using BLAST+ with a blastx search of whole genomes against an index of ribosomal proteins.
Data Deposition
Sequence data for newly sequenced cyanobacteria (Table 1) are available on the NCBI database under accession numbers PVWP00000000, PXOH00000000, PVWE00000000, PVWD00000000, PYGV00000000, PYFY00000000, PYEQ 00000000, PYER00000000, PVWO00000000, PVWN00000000, PVWM00000000, PYCI00000000, PVWL00000000, PVWK 00000000, PVWJ00000000, PVWH00000000, PVWI00000000, PVWG00000000, PVWF00000000, SAMN08828726, and SAMN08828728. All alignment and tree files are available on Figshare at doi: 10.6084/m9.figshare.7629383.
Phylogenetic Tree Reconstruction
Sequences of large and small subunit ribosomal proteins for cyanobacteria, plastids and bacterial outgroups were aligned with MUSCLE v3.8.31 (Edgar, 2004) and concatenated using FASconCAT v1.0 (Kück and Meusemann, 2010). Substitution model analyses were carried out using ProtTest v. 3.4.2 (Abascal et al., 2005; Darriba et al., 2011).
The monophyly of Cyanobacteria+plastids was tested for each ribosomal gene family by producing maximum-likelihood trees using IQtree (LG model with four gamma distributed rate categories). The resulting trees show that the concatenated alignment does not contain extensive HGT from outside this group that could potentially confound phylogenetic inference. A small number of sequences were identified as placing outside of Cyanobacteria+plastids in their respective gene trees: L15, L18 (Chlamydomonas reinhardtii, Volvox carteri f. nagariensis); S11 (Ectocarpus siliculosus, Arthrospira, Nannochloropsis salina). It is unclear if these exceptions were due to legitimate HGT detection, or lack of phylogenetic signal within such short individual proteins. The impact of these potentially transferred sequences was assessed by removing them from the concatenated protein alignment and generating a maximum-likelihood tree (IQTree, LG model, 4 gamma distributed rate categories). The resulting placement of plastids within the deep cyanobacterial tree topology was identical to that observed in maximum-likelihood and Bayesian inference consensus trees generated from the full concatenated protein dataset, and so these were retained in subsequent analyses.
Phylogenetic trees were made using both maximum likelihood (ML) criteria with RAxML v8.1.9 (Stamatakis, 2006) [four gamma distributed site rate categories with estimated shape parameter (α = 0.827794) and an LG substitution model], and Bayesian inference using PhyloBayes 3.3 (Lartillot et al., 2013) (C20 site specific substitution models, convergence criterion cutoff of 0.3 for all parameters). Bayesian inference phylogenies were also generated from Dayhoff-recoded alignments to test the potential impact of nucleotide compositional bias on non-synonymous substitutions. Three recordings were performed: (A) the dayhoff6 recoding as included in the PhyloBayes 3.3 package (recoded amino acid sets: AGPST, DENQ, HKR, FYW,ILVM); (B) a more conservative recoding that synonymized only physiochemically similar amino acids with varying G+C content between synonymous codons (recoded amino acid sets: ILVM, FYW, KR, NQ); (C) a highly conservative recoding based on the specific prediction of a high rate of substitution between K and R within plastids driven by nucleotide bias (recoded amino acid sets KR) (Li et al., 2014).
Ribosomal RNA sequence alignments were performed using the Silva SINA (v1.2.11) global aligner service (Quast et al., 2013). The subsequent alignments were manually curated, as several gap-adjacent regions showed clearly misaligned bases. The resulting edited alignments were then further curated by the removal of “gappy” regions that contained ambiguously aligned residues. The unedited, edited, and trimmed alignments, including a list of the removed regions, are available as supporting materials (section “rRNA Sequence Trimming” in Supplementary Methods and Supplementary Table 1). Maximum-likelihood trees for 16S, 23S, and concatenated 16S/23S alignments were generated in IQ-TREE (Nguyen et al., 2015), with best-fitting models determined by ModelFinder (Kalyaanamoorthy et al., 2017). 100 bootstrap replicates were performed in each case. Best-fitting models were determined by Bayesian Information Criterion (BIC). For the 16S and concatenated rRNA alignments, the best fitting model was GTR, with empirical base frequencies and five free rate parameters. For 23S, the best fitting model was SYM with five free rate parameters. Bayesian consensus trees were generated in Phylobayes3.3f (Lartillot et al., 2013), under a CAT-GTR model including site-specific base exchangeabilities, with consensus trees generated from tree samples following convergence in two chains across all parameters to a variance of <0.3.
Results and Discussion
Cyanobacterial Tree Topology
The analyses presented here combine the sequences of 37 cyanobacterial and 32 plastid genomes with 20 newly sequenced species to expand cyanobacterial phylogeny and constrain the divergence of plastids with better resolution (Table 1). Of the 20 genomes that our group sequenced for this study, 17 were high quality, and 4 were of medium quality, according to the current accepted standards (Bowers et al., 2017). We conducted both ML and Bayesian analyses that used a concatenation of 30 large and small subunit ribosomal protein sequences (Table 2). We chose to use amino acid sequences rather than nucleotide sequences because the latter are more strongly affected by saturation over long time scales (Li et al., 2014). While Cyanobacteria and plastids share additional homologous proteins, such as those associated with the photosynthetic machinery, ribosomal sequence data remain a standard for species-tree phylogenetic inference for three main reasons. Firstly, ribosomal proteins are generally conserved, and provide many thousands of well-aligned, informative sites. Secondly, ribosomal proteins are infrequently transferred and are usually present as orthologs encoded by single gene loci within genomes. This makes reticulate scenarios that can confound species tree inferences (gene transfers, duplications, and losses) unlikely. Thirdly, the use of ribosomal protein sequences shared by many bacterial phyla enables the rooting of Cyanobacteria by outgroup sequences. This is critical for inferring deep cyanobacterial phylogenetic topology and the inferred placement of plastids (Lyons-Weiler et al., 1998), and cannot be done with sequences that are absent from the bacterial species tree outgroups.
All iterations of our models and tree reconstruction methods recovered congruent sets of bipartitions between deeply branching cyanobacterial taxa including G. lithophora, and the plastid donor/ancestor lineage. For non-recoded analyses that included bacterial outgroups for rooting, Gloeobacter was the deepest branching group (assigned here as clade 1, including G. kilaueensis and G. violaceus), followed by a deeply branching strain of Synechococcus (Synechococcus sp. JA-2-3B a 2-13; clade 2). In all analyses, plastids and G. lithophora diverged immediately after Synechococcus sp. JA-2-3B a 2-13. These placements were statistically supported with 100% bootstrap support (Figure 1) and posterior probability (Supplementary Figure 1). A clade comprised of two strains of Pseudanabaena (sp. PCC7367 and sp. PCC6820, clade 3) did not diverge until after plastids, followed by clade 4, comprised of Acaryochloris marina MBIC11017, Thermosynechococcus elongatus BP1, and Cyanothece sp. PCC7425 (Figure 1 and Supplementary Figure 1). An additional analysis that excluded Pseudanabaena recovered a similarly deeply branching plastid clade, although the removal of Pseudanabaena altered the cyanobacterial topology slightly with lower bootstrap support (Figure 2) and posterior probability (Supplementary Figure 2) relative to trees that included Pseudanabaena (e.g., nodes that had bootstrap support values of 90–99 decreased to well below 70, and those that had posterior probabilities of 1 decreased to 0.8–0.9). Specifically, the removal of this group altered the position of clade 4, placing it between clades 6 and 7 with very low bootstrap support (<30). Because of the low support for this topology, and its inconsistency with previously published trees, we favor the topology that includes Pseudanabaena. Notably, this test confirms that the deep placement of plastids in our tree is not driven by the inclusion or exclusion of Pseudanabaena.
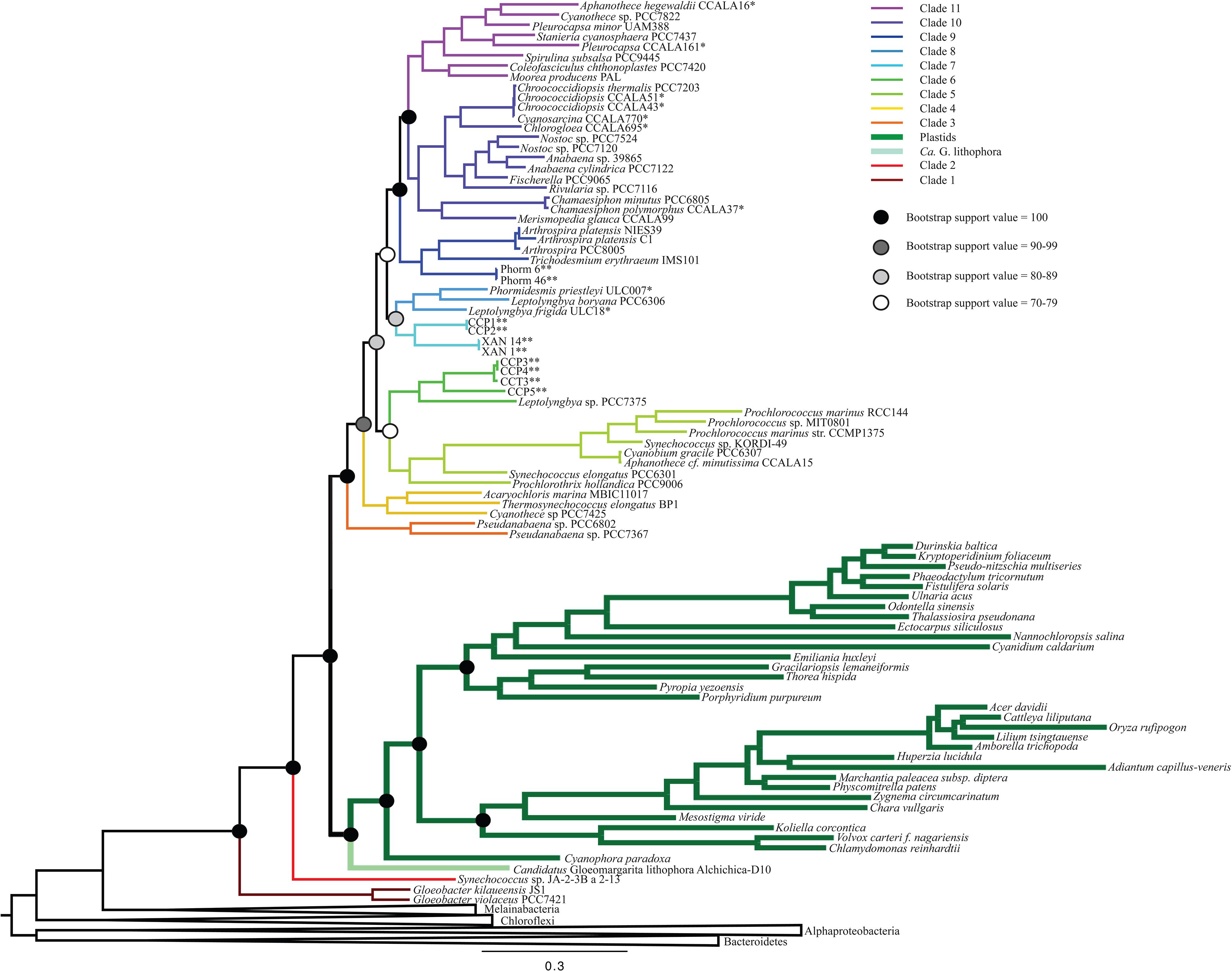
Figure 1. A maximum likelihood (ML) phylogenetic tree made using a concatenation of 30 large and small subunit ribosomal proteins (Table 2) and RAxML. This tree supports a deep placement of the plastid clade with high bootstrap support. Bootstrap values are denoted for major divergences of cyanobacterial and plastid clades. All internal nodes have bootstrap values of >90.
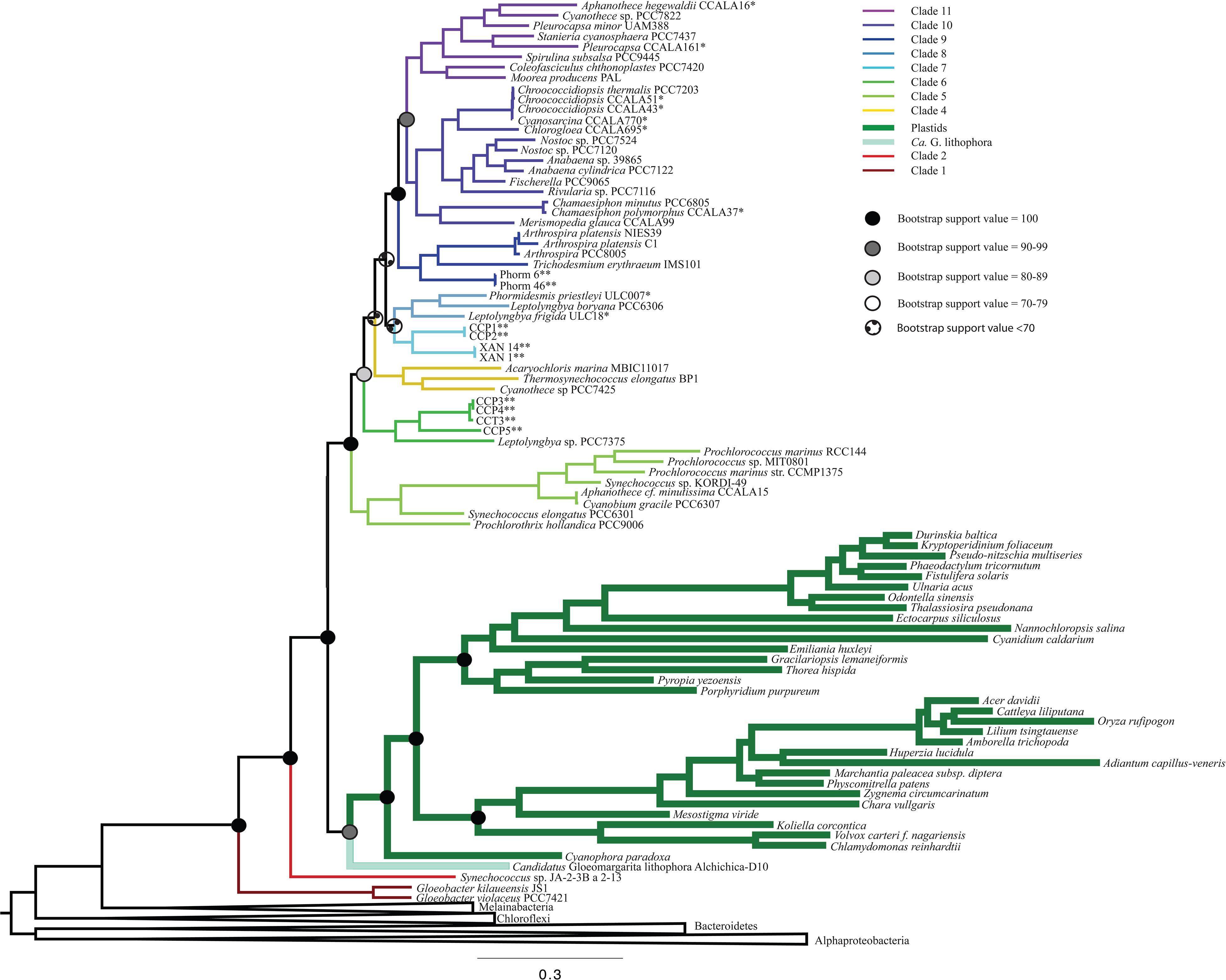
Figure 2. A ML phylogenetic tree made without the inclusion of Pseudanabaena using RAxML. The placement of plastids in this tree is consistent with Figure 1, but with lower bootstrap support values. Additionally, the placement of clade 4 is shifted to be more recently branching in this tree than in trees that contain Pseudanabaena.
The remaining cyanobacterial species grouped into seven clades (clades 5 through 11). Two of these clades were new, and five corresponded to clades recovered and described in previously published cyanobacterial phylogenies. Clade 5 includes Prochlorothrix hollandica as well as several species of marine Synechococcus and Prochlorococcus, and corresponds to clade 3 in Schirrmeister et al. (2015) and clade C in Shih et al. (2013). Clades 9, 10, and 11, respectively, correspond to clades 4, 5, and 6 in Schirrmeister et al. (2015) and A, B1, and B2 in Shih et al. (2013). Clades 6 and 7 in our study contain newly sequenced species of benthic cyanobacteria from the Arctic, Cape Cod and Yellowstone National Park. Some of these strains group with Leptolyngbya boryana (clade 6) that corresponds to clade D in Shih et al. (2013), while the strains from the Arctic group together into a new clade (clade 8). Our analyses also identified one additional clade that was composed entirely of newly sequenced cyanobacteria, and therefore is not directly comparable to any previously described clades. This clade (clade 7) included four newly sequenced filamentous cyanobacteria enriched from microbial mats (Momper et al., 2019). Other newly sequenced species grouped within the remaining clades in a predictable fashion (i.e., species from culture collections generally grouped with other organisms of the same assigned genus; Figure 1 and Supplementary Figure 1).
To determine the influence of newly sequenced cyanobacterial genomes on the overall topology, we carried out additional analyses that excluded these genomes (Figure 3 and Supplementary Figure 3). These analyses produced a topology consistent with those that include new sequences (Figure 1). However, the trees that did not include the new ribosomal protein sequences had lower bootstrap support values (Figure 3) and posterior probabilities (Supplementary Figure 3) compared to analyses that included newly sequenced cyanobacteria (e.g., some nodes that previously had bootstrap support values of 100 decreased to 70–79, and some nodes that previously had posterior probabilities of 1 decreased to 0.7–0.79). These results support our tree topology and demonstrate the value of including newly sequenced strains in our phylogenetic analyses.
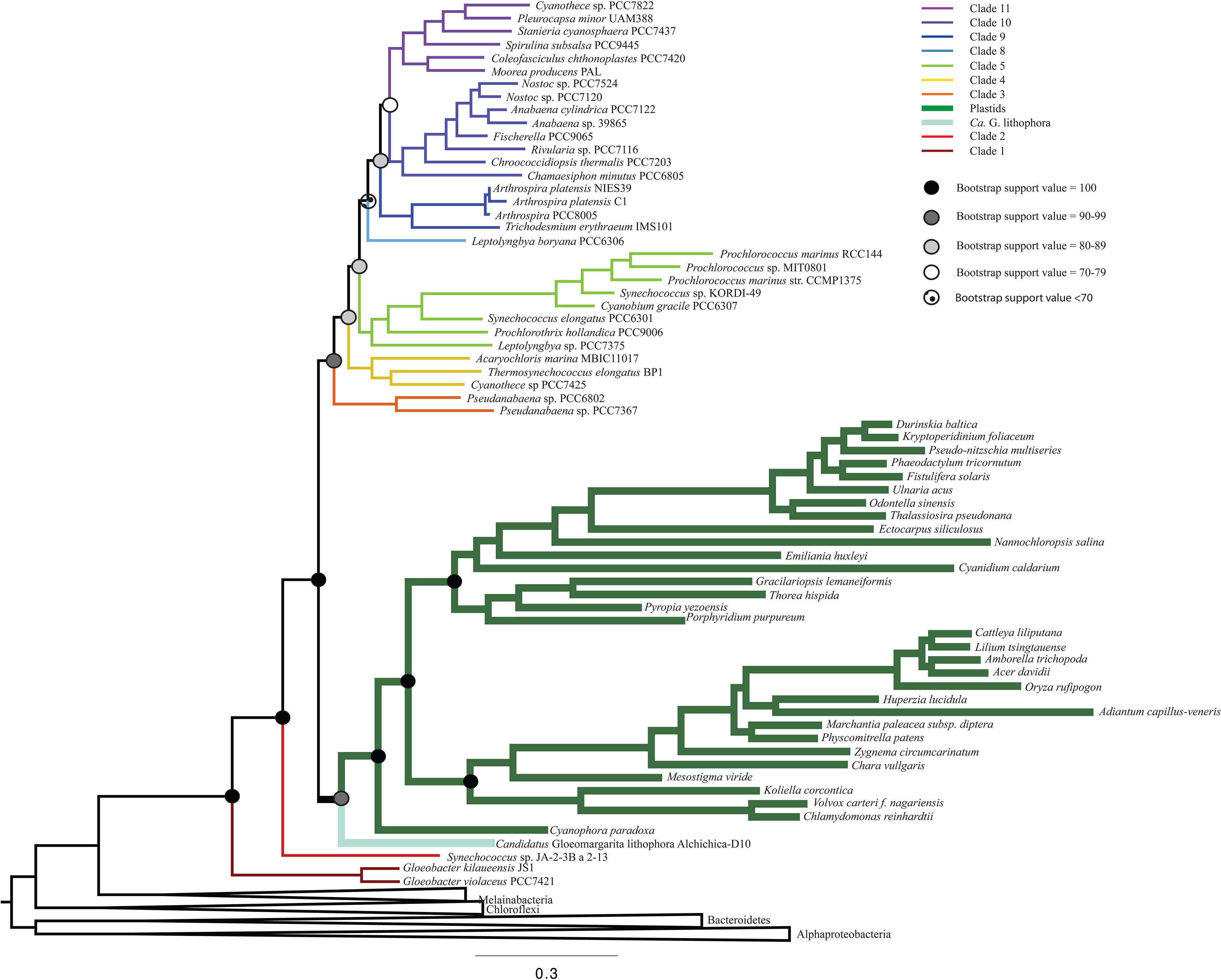
Figure 3. A ML phylogenetic tree that does not include any newly sequenced genomes, made using RAxML. This tree recovers a topology consistent with Figure 1, but with lower bootstrap support values.
To extend comparisons to previous analyses and further investigate the ribosomal evolutionary signal within plastids, trees were also generated for ribosomal RNA sequences, including 16S, 23S, concatenated 16S and 23S alignment datasets, and ribosomal protein trees for Cyanobacteria alone, without outgroups (Supplementary Figures 6–12). For each dataset, maximum-likelihood and Bayesian inference consensus trees produced similar topologies and placements for plastids and G. lithophora. For the 16S dataset, both trees recovered a deep placement of plastids within Cyanobacteria, deeper than Pseudanabaena, albeit with low bootstrap and posterior probability support (38/100 and 0.53, respectively). In contrast to trees generated from concatenated proteins, G. lithophora grouped more deeply, together with Synechococcus sp. JA-2-3B’a(2-13). Support values across internal nodes for shallower cyanobacterial clades were extremely low, and most cyanobacterial groups shared a major multifurcation in the Bayesian inference consensus tree. 23S alignments produced substantially different tree topologies. While plastids grouped together with G. lithophora with high bootstrap and posterior probability values (83/100 and 1.00, respectively) similar to the protein trees, this group has a shallow placement within the tree, similar to the position observed in Ochoa de Alda et al. (2014), and with very low bootstrap and posterior probability values (25/100 and 0.67, respectively). Interestingly, in the 23S trees Pseudanabaena also branches shallowly in contrast to 16S and ribosomal protein tree results, although still relatively close to G. lithophora and plastids. Concatenated 16S and 23S alignments generate trees similar to the 23S tree, with somewhat higher but still relatively low support values for placements of major groups, and a major multifurcation persisting in the Bayesian inference consensus tree.
Ribosomal RNA appears to contain a relatively weak evolutionary signal for resolving the placement of many major groups of cyanobacteria, including plastids and G. lithophora. This is similar to the result obtained in previous analyses (e.g., Ponce-Toledo et al., 2017), which also recovered low supports and multifurcations for these clades. However, we also note that the 16S rRNA tree’s “deep” placement of plastids, while poorly supported, does recover as a consensus signal two of the key bipartitions observed in trees generated from ribosomal protein datasets. Clades 1–2, Gloeomargarita, and plastids group together to the exclusion of other cyanobacteria, and cyanobacterial clades 4–11 subsequently group together to the exclusion of other cyanobacteria and plastids, including Pseudanabaena (Clade 3). The generally low support values and surprisingly inconsistent tree topologies between 16S and 23S datasets suggest that a focus on protein sequence analysis is the potentially more fruitful approach for investigating of plastid origins.
Testing the Position of Plastids
The placement of plastids within our trees is deeper than the placements that have been suggested by most analyses to date. Our analyses consistently show a deep divergence of the plastid/G. lithophora clade after the deeply branching Synechococcus sp. JA-2-3B a 2-13, but before clade 3 (Pseudanabaena; Figure 1 and Supplementary Figure 1). This bifurcation, supported by high bootstrap values and high posterior probabilities, indicates an ancient ancestry of plastids. Previous studies that place plastids within the nitrogen-fixing cyanobacteria (clades 9 and 10 in this study) recover a shallow placement using predominantly nucleotide sequences or protein sequences from nuclear encoded proteins in photosynthetic eukaryotes and their homologs in cyanobacteria (Table 3). These studies use data sets that range from small subunit rRNA sequences to genes such as rbcl (Douglas and Turner, 1991; Falcón et al., 2010) and nuclear encoded proteins from photosynthetic eukaryotes with cyanobacterial homologs (Dagan et al., 2012), and many include only a limited dataset of cyanobacteria. A few analyses using nucleotide data have produced topologies with a deeper placement of plastids (Nelissen et al., 1995; Turner et al., 1999), but these also used a limited set of cyanobacterial sequences and had low bootstrap support values. In contrast, studies based on amino acid sequences generally recover a deeper placement of plastids (Rodríguez-Ezpeleta et al., 2005; Reyes-Prieto et al., 2010; Criscuolo and Gribaldo, 2011; Shih et al., 2013). However, analyses that recovered a deep placement either showed a divergence of plastids after the Pseudanabaena (Shih et al., 2013) or did not include deeply branching groups like Pseudanabaena and Synechococcus sp. JA-2-3B a 2-13 at all (Rodríguez-Ezpeleta et al., 2005; Reyes-Prieto et al., 2010; Criscuolo and Gribaldo, 2011). A recent analysis (Sánchez-Baracaldo et al., 2017) used a large set of concatenated proteins including parts of the photosynthetic machinery, resulting in a placement of plastids after both the filamentous Pseudanabaena and the clade containing unicellular Thermosynechococcus, Cyanothece, and Acaryochloris (clade 4 in this study). This study used a technique in which the cyanobacterial topology was produced using concatenated proteins as a fixed “backbone” onto which the plastids were placed based on their aligned gene sequences (Sánchez-Baracaldo et al., 2017). The same study also recovered a broadly similar topology using a concatenation technique without the “backbone” (Sánchez-Baracaldo et al., 2017). In contrast, our analyses use a consistent dataset across taxa and include bacterial outgroups to allow for a true root to be placed on the tree. Using this approach, we recovered a deeper placement than previous analyses (Table 3), before Pseudanabaena, with high resolution and support. The same deep cyanobacterial and plastid phylogeny was recovered in the absence of outgroups (Supplementary Figure 12). Support values were slightly lower in these cases, but still high. This is expected, as the presence of outgroups polarizes characters within the tree.
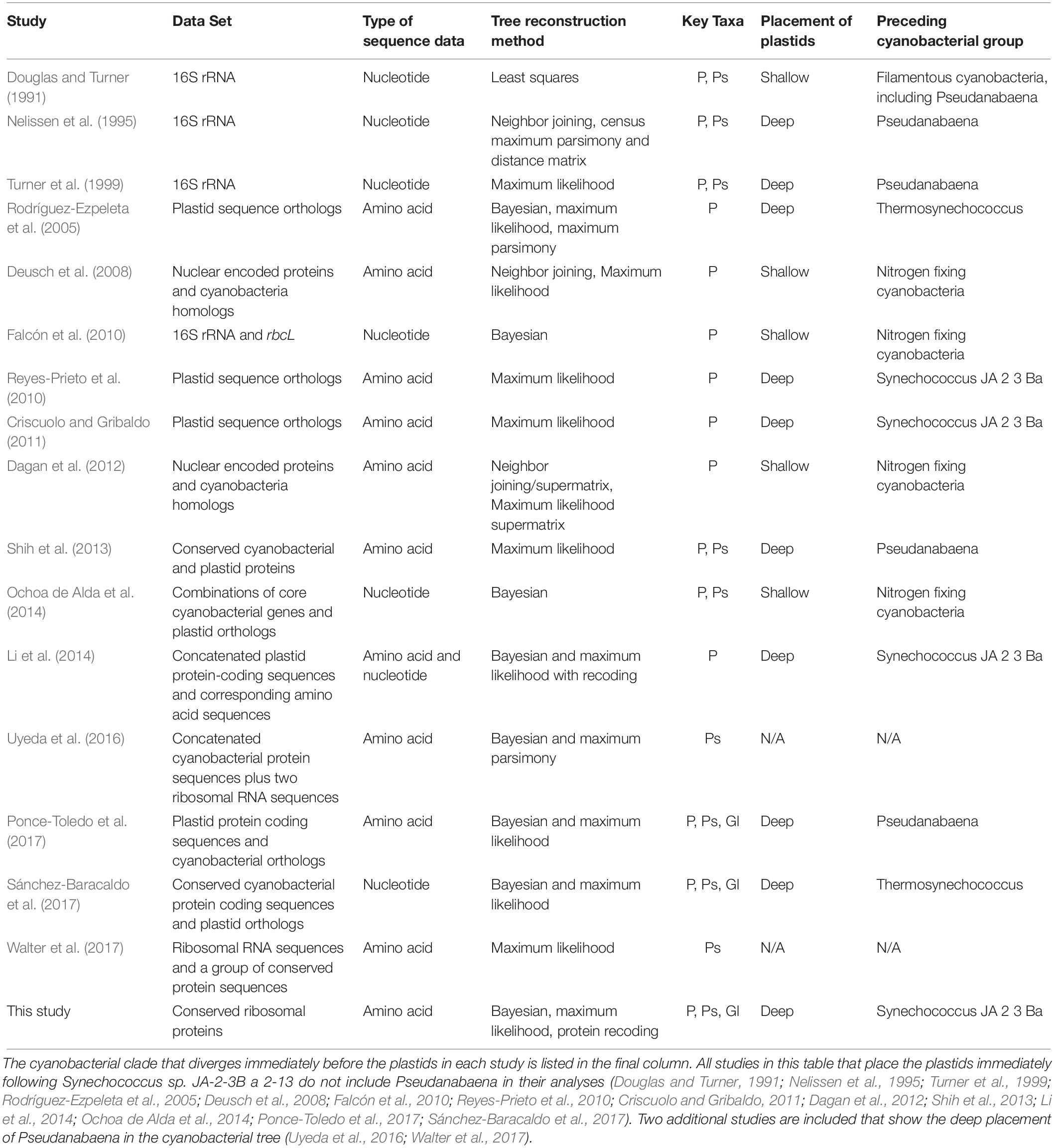
Table 3. Previous phylogenetic studies of plastid placement within the cyanobacterial tree.
A recent study that described a newly sequenced cyanobacterium (G. lithophora) suggested that this group is a sister group to the plastids and also recovered a deep divergence of both G. lithophora and plastids, after the divergence of Pseudanabaena (Ponce-Toledo et al., 2017). However, it is possible that the sister relationship between plastids and G. lithophora may reflect a long branch attraction artifact (LBA), or otherwise influence the placement of plastids with respect to other cyanobacterial groups. To test the effect of G. lithophora on the placement of the plastids in the cyanobacterial tree, we ran two sets of analyses; one that included G. lithophora (Analysis 1, Figure 1 and Supplementary Figure 1) and one that did not (Analysis 2, Supplementary Figure 4). The results confirmed that the placement of the plastids in our topology was not affected by the exclusion or inclusion of G. lithophora. Analysis 1 placed G. lithophora as a sister group to the plastids, and the rest of the tree topology was identical to the analysis that did not include G. lithophora (Analysis 2). Both analyses also recovered identical deep branching of the plastids immediately after clade 2 (Figure 1 and Supplementary Figures 1, 4). Therefore, our analyses support G. lithophora as a sister group to the cyanobacterial plastid ancestor, but the inclusion of this group does not impact the placement of plastids within our phylogenetic analyses. Furthermore, the persistent placement of this group on a short internal branch of the tree shows that the observed placement of plastids is not an artifact arising from LBA.
It has been suggested that the reason for the large discrepancy in the placement of plastids may result from the type of sequences used (Li et al., 2014; Ochoa de Alda et al., 2014). Ochoa de Alda et al. (2014) explored this hypothesis by testing multiple datasets and phylogenetic approaches including (1) large and small subunit rRNA sequences with and without removal of saturated sites; (2) a consensus tree of gene trees including both cyanobacteria and plastids; and (3) phylogenetic analysis of concatenated protein alignments. Their results showed a clear difference between the topologies produced by the gene sequences, which recovered a shallow branching of the plastids, and those produced by amino acid sequences, which show a deeper placement (Ochoa de Alda et al., 2014). The authors attributed this discrepancy to differences in model selection rather than the datasets used, and proposed that the CAT model implemented in nucleotide analyses is a better predictor of the evolutionary relationships than the LG + discrete gamma model used in amino acid analyses (Ochoa de Alda et al., 2014). Because of this, the authors favored the shallow placement produced by analyses using core cyanobacterial and plastid genes with a CAT model (Ochoa de Alda et al., 2014). In contrast, our alignment generates the same tree topology under both an LG substitution model and a CAT site-specific substitution model, showing that our result is not sensitive to this particular increase in model complexity.
Li et al. (2014) tested the difference in plastid placement between phylogenies based on nucleotide sequences and those based on the proteins that these nucleotide sequences encode. These authors suggest that the reason for this discrepancy may be related to compositional biases in the first and third codon positions of the nucleotide sequences, which supports the use of amino acid sequences over nucleotide sequences for these types of analyses (Li et al., 2014). The use of non-homogeneous composition models and a codon-degeneracy recoding technique to reconstruct the cyanobacteria/plastid phylogeny in their study results in a tree that is similar to our results, and places the root of the plastids deep within the tree, after Synechococcus sp. JA-2-3B a 2-13 (Li et al., 2014). However, this study did not include Pseudanabaena. Our analyses are the first to support a deep placement of the plastids between Synechococcus sp. JA-2-3B a 2-13 and Pseudanabaena, and provide an independent support for the findings of Li et al. (2014) using highly conserved concatenated proteins from an expanded set of cyanobacterial taxa.
Given that nucleotide composition may also impact non-synonymous amino acid substitutions (Li et al., 2014), we performed additional Bayesian phylogenetic reconstructions using Dayhoff-recoded alignments designed to mitigate the impact of this effect. All three implemented Dayhoff recoding schemes returned phylogenies with deep cyanobacterial topologies identical to non-recoded analyses, including the monophyly of G.lithophora and plastids, and their placement with respect to other cyanobacteria. Furthermore, with one exception, all of these bipartitions were recovered with posterior probabilities of 100%. The exception to this was the monophyly of plastids and G. lithophora, with a posterior probability of only 51% in coding scheme [B]. Coding scheme [B] still placed plastids deeper in the tree than all included cyanobacteria except Gloeobacter, Synechococcus, and G.lithophora with 100% posterior probability. Of non-consensus bipartitions, 40% rooted plastids one node shallower than G. lithophora.
Phylogeny of Plastids
Our analyses consistently recover the monophly of plastids. In this clade, the cyanelle of Cyanophora paradoxa is the deepest lineage, followed by two additional major clades: clade A includes the green algae and land plants and clade B is comprised of the red algae, dinoflagellates, coccolithophores, and diatoms. Even when the deeply diverging Cyanophora paradoxa is removed, our analyses recover the sister relationship between clades A and B (see Supplementary Figure 5). This relationship is in agreement with previously published results that support the monophyly of plastids despite the secondary plastid acquisition that led to groups like dinoflagellates (Cavalier-Smith, 1982; Douglas and Turner, 1991; Turner et al., 1999; Moreira et al., 2000; Nozaki et al., 2003; Palmer, 2003; Rodríguez-Ezpeleta et al., 2005). Some uncertainty remains as to the exact relationships between these nucleocytoplasmic lineages and their secondary (and tertiary) endosymbionts (Keeling, 2009), but these much more recent divergences are not the primary focus of this study.
The phylogenetic trees produced by our analyses also show that the plastid lineages have very long branches compared to other cyanobacteria, with the exception of the long branches within the clade containing Synechococcus/Prochlorococcus and their close relatives. The long branches of Prochlorococcus have been attributed to their reduced genome sizes and accelerated rates of evolution (Sun and Blanchard, 2014), a potentially analogous situation to that within plastids. The transfer of genes from cyanobacteria into ancestral plastid bearing lineages has been noted and investigated by Martin et al. (2002). These authors identified homologous proteins in Arabidopsis and three cyanobacterial genomes to determine the amount of gene transfer that likely occurred, and found that roughly 18% of the total Arabidopsis genome was transferred from a chloroplast (Martin et al., 2002). This study underlines the amount of gene transfer that likely occurred through this endosymbiotic event (Martin et al., 2002), an occurrence which may have similarly diminished the genome of the endosymbiont as these genes were transferred to the host.
Conclusion
The placement of plastids within a cyanobacterial phylogeny is key to understanding the evolutionary timing and relationship of these groups, but this placement remains in question. Our study generates phylogenies using a concatenation of highly conserved protein sequences from an expanded set of cyanobacterial and plastid lineages. We recover a deeper placement of the plastid/G. lithophora clade than has been suggested by most previous studies. This topology is the first to demonstrate a divergence of plastids before the group containing Pseudanabaena, after the deeply branching Synechococcus sp. JA-2-3B a 2-13. Our results are consistent with the findings of Li et al. (2014) using a highly conserved set of concatenated proteins and expanded set of taxa, and are statistically supported in both maximum-likelihood and Bayesian phylogenetic reconstructions, as well as multiple sequence recoding schemes. Additionally, the trees in this study show very long branches among plastid lineages, consistent with a faster rate of evolution of plastids relative to crown group cyanobacteria. These results are of particular importance for future molecular clock studies of cyanobacterial lineages that include sequences from plastid-containing eukaryotes and fossil calibrations based on these organisms. An accurate tree topology, appropriate modeling of rates of evolution, and well-informed fossil calibrations are all crucial in building molecular clock models to depict the evolutionary history of cyanobacteria and plastids.
Author Contributions
KM carried out the laboratory culturing and enrichment of environmental samples, data analysis, sequence alignment, phylogenetic tree reconstruction, simplified molecular clock models, and drafted the manuscript. CM participated in sequence collection and analysis. CM, LM, DG, TB, and GF contributed to the manuscript revision and editing. LM carried out the DNA extraction and genome reconstruction of newly sequenced species. DG carried out the statistical analyses. TB and GF conceived and coordinated the study, and provided funding. All authors gave final approval for the manuscript.
Funding
This work was supported by the Simons Foundation through a Simons Early Career Investigator in Marine Microbial Ecology and Evolution grant to TB (#344707) and Simons Collaboration on Origins of Life grants to GF and TB (#339603, #327126).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Anne D. Jungblut (Natural History Museum, London, United Kingdom) and Warwick F. Vincent [Centre for Northern Studies (CEN), Laval University, Canada] for cyanobacteria strain isolation and field sampling for strains Phorm 6 and Phorm 46 through the program “Northern Ellesmere Island in the Global Environment” (NEIGE). Additional thanks to the Bosak and Fournier labs for support and training. LM would like to thank the Massachusetts Institute of Technology for the award of the W. O. Crosby Postdoctoral Fellowship.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01612/full#supplementary-material
Footnotes
References
Abascal, F., Zardoya, R., and Posada, D. (2005). ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21, 2104–2105. doi: 10.1093/bioinformatics/bti263
Alneberg, J., Bjarnason, B. S., De Bruijn, I., Schirmer, M., Quick, J., Ijaz, U. Z., et al. (2014). Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146. doi: 10.1038/nmeth.3103
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1006/jmbi.1990.9999
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bapteste, E., O’Malley, M. A., Beiko, R. G., Ereshefsky, M., Gogarten, J. P., Franklin-Hall, L., et al. (2009). Prokaryotic evolution and the tree of life are two different things. Biol. Direct 4:34. doi: 10.1186/1745-6150-4-34
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bowers, R. M., Kyrpides, N. C., Stepanauskas, R., Harmon-Smith, M., Doud, D., Reddy, T. B. K., et al. (2017). Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35, 725–731. doi: 10.1038/nbt.3893
Cornet, L., Bertrand, A. R., Hanikenne, M., Javaux, E. J., Wilmotte, A., and Baurain, D. (2018). Metagenomic assembly of new (sub)polar cyanobacteria and their associated microbiome from non-axenic cultures. Microb. Genomics 4:e000212. doi: 10.1099/mgen.0.000212
Criscuolo, A., and Gribaldo, S. (2011). Large-scale phylogenomic analyses indicate a deep origin of primary plastids within Cyanobacteria. Mol. Biol. Evol. 28, 3019–3032. doi: 10.1093/molbev/msr108
Dagan, T., Roettger, M., Stucken, K., Landan, G., Koch, R., Major, P., et al. (2012). Genomes of stigonematalean cyanobacteria (Subsection V) and the evolution of oxygenic photosynthesis from prokaryotes to plastids. Genome Biol. Evol. 5, 31–44. doi: 10.1093/gbe/evs117
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2011). ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165. doi: 10.1007/978-3-642-21878-1_22
Deusch, O., Landan, G., Roettger, M., Gruenheit, N., Kowallik, K. V., Allen, J. F., et al. (2008). Genes of cyanobacterial origin in plant nuclear genomes point to a heterocyst-forming plastid ancestor. Mol. Biol. Evol. 25, 748–761. doi: 10.1093/molbev/msn022
Douglas, S. E. (1998). Plastid evolution: origins, diversity, trends. Curr. Opin. Genet. Dev. 8, 655–661. doi: 10.1016/s0959-437x(98)80033-6
Douglas, S. E., and Turner, S. (1991). Molecular evolution for the origin of plastids from a cyanobacterium-like ancestor. J. Mol. Evol. 33, 267–273. doi: 10.1007/bf02100678
Edgar, R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. doi: 10.1186/1471-2105-5-113
Eren, A. M., Esen, Ö. C., Quince, C., Vineis, J. H., Morrison, H. G., Sogin, M. L., et al. (2015). Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3:e1319. doi: 10.7717/peerj.1319
Falcón, L. I., Magallón, S., and Castillo, A. (2010). Dating the cyanobacterial ancestor of the chloroplast. ISME J. 4, 777–783. doi: 10.1038/ismej.2010.2
Ferla, M. P., Thrash, J. C., Giovannoni, S. J., and Patrick, W. M. (2013). New rRNA gene-based phylogenies of the Alphaproteobacteria provide perspective on major groups, mitochondrial ancestry and phylogenetic instability. PLoS One 8:e83383. doi: 10.1371/journal.pone.0083383
Fitzpatrick, D. A., Creevey, C. J., and McInerney, J. O. (2006). Genome phylogenies indicate a meaningful α-proteobacterial phylogeny and support a grouping of the mitochondria with the Rickettsiales. Mol. Biol. Evol. 23, 74–85. doi: 10.1093/molbev/msj009
Hug, L. A., Baker, B. J., Anantharaman, K., Brown, C. T., Probst, A. J., Castelle, C. J., et al. (2016). A new view of the tree of life. Nat. Microbiol. 1:16048. doi: 10.1038/nmicrobiol.2016.48
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., Von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Keeling, P. J. (2009). Chromalveolates and the evolution of plastids by secondary endosymbiosis. J. Eukaryot. Microbiol. 56, 1–8. doi: 10.1111/j.1550-7408.2008.00371.x
Kück, P., and Meusemann, K. (2010). FASconCAT: convenient handling of data matrices. Mol. Phylogenet. Evol. 56, 1115–1118. doi: 10.1016/j.ympev.2010.04.024
Kurland, C. G. (1992). Evolution of mitochondrial genomes and the genetic code. BioEssays 14, 709–714. doi: 10.1002/bies.950141013
Lara, Y., Durieu, B., Cornet, L., Verlaine, O., Rippka, R., Pessi, I. S., et al. (2017). Draft genome sequence of the axenic strain Phormidesmis priestleyi ULC007, a cyanobacterium isolated from Lake Bruehwiler (Larsemann Hills, Antarctica). Genome Announc. 5:e01546-16. doi: 10.1128/genomea.01546-16
Lartillot, N., Rodrigue, N., Stubbs, D., and Richer, J. (2013). PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Softw. Syst. Evol. 62, 611–615. doi: 10.5061/dryad.c459h
Li, B., Lopes, J. S., Foster, P. G., Embley, T. M., and Cox, C. J. (2014). Compositional biases among synonymous substitutions cause conflict between gene and protein trees for plastid origins. Mol. Biol. Evol. 31, 1697–1709. doi: 10.1093/molbev/msu105
Lyons-Weiler, J., Hoelzer, G. U. Y. A., and Tausch, R. J. (1998). Optimal outgroup analysis. Biol. J. Linn. Soc. 64, 493–511. doi: 10.1006/bijl.1998.0229
Magnabosco, C., Moore, K. R., Wolfe, J. M., and Fournier, G. P. (2018). Dating phototrophic microbial lineages with reticulate gene histories. Geobiology 16, 179–189. doi: 10.1111/gbi.12273
Martijn, J., Vosseberg, J., Guy, L., Offre, P., and Ettema, T. J. G. (2018). Deep mitochondrial orign outside the sampled alphaproteobacteria. Nature 557, 101–109. doi: 10.1111/ijfs.12827
Martin, W., Rujan, T., Richly, E., Hansen, A., Cornelsen, S., Lins, T., et al. (2002). Evolutionary analysis of Arabidopsis, cyanobacterial, and chloroplast genomes reveals plastid phylogeny and thousands of cyanobacterial genes in the nucleus. Proc. Natl. Acad. Sci. U.S.A. 99, 12246–12251. doi: 10.1073/pnas.182432999
McFadden, G. I. (2001). Chloroplast origin and integration. Plant Physiol. 125, 50–53. doi: 10.1104/pp.125.1.50
Momper, L., Hu, E., Moore, K. R., Skoog, E. J., Tyler, M., Evans, A. J., et al. (2019). Metabolic versatility in a modern lineage of cyanobacteria from terrestrial hot springs. Free Radic. Biol. Med. 1–9. doi: 10.1016/j.freeradbiomed.2019.05.036 [Epub ahead of print].
Moreira, D., Le Guyader, H., and Philippe, H. (2000). The origin of red algae and the evolution of chloroplasts. Nature 405, 69–72. doi: 10.1038/35011054
Nelissen, B., Van de Peer, Y., Wilmotte, A., and De Wachter, R. (1995). An early origin of plastids within the cyanobacterial divergence is suggested by evolutionary trees based on complete 16S rRNA sequences. Mol. Biol. Evol. 12, 1116–1173. doi: 10.1093/oxfordjournals.molbev.a040289
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nozaki, H., Ohta, N., Matsuzaki, M., Misumi, O., and Kuroiwa, T. (2003). Phylogeny of plastids based on cladistic analysis of gene loss inferred from complete plastid genome sequences. J. Mol. Evol. 57, 377–382. doi: 10.1007/s00239-003-2486-6
Ochoa de Alda, J. A. G., Esteban, R., Diago, M. L., and Houmard, J. (2014). The plastid ancestor originated among one of the major cyanobacterial lineages. Nat. Commun. 5:4937. doi: 10.1038/ncomms5937
Palmer, J. D. (2003). The symbiotic birth and spread of plastids: how many times and whodunit? J. Phycol. 39, 4–11.
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Ponce-Toledo, R. I., Deschamps, P., López-García, P., Zivanovic, Y., Benzerara, K., and Moreira, D. (2017). An early-branching freshwater cyanobacterium at the origin of plastids. Curr. Biol. 27, 386–391. doi: 10.1016/j.cub.2016.11.056
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, 590–596. doi: 10.1093/nar/gks1219
Reyes-Prieto, A., Yoon, H. S., Moustafa, A., Yang, E. C., Andersen, R. A., Boo, S. M., et al. (2010). Differential gene retention in plastids of common recent origin. Mol. Biol. Evol. 27, 1530–1537. doi: 10.1093/molbev/msq032
Rodríguez-Ezpeleta, N., Brinkmann, H., Burey, S. C., Roure, B., Burger, G., Löffelhardt, W., et al. (2005). Monophyly of primary photosynthetic eukaryotes: green plants, red algae, and glaucophytes. Curr. Biol. 15, 1325–1330. doi: 10.1016/j.cub.2005.06.040
Sánchez-Baracaldo, P., Raven, J. A., Pisani, D., and Knoll, A. H. (2017). Early photosynthetic eukaryotes inhabited low-salinity habitats. Proc. Natl. Acad. Sci. U.S.A. 114, E7737–E7745. doi: 10.1073/pnas.1620089114
Sato, N. (2007). “Origin and evolution of plastids: Genomic view on the unification and diversity of plastids,” in The Structure and Function of Plastids, eds R. R. Wise and K. J. Hoober (Dordrecht: Springer Netherlands), 76–93.
Schimper, A. F. (1885). Untersuchungen über die chlorophyllkörner und die ihnen homologen gebilde. Jahrb. Wiss. Bot. 16, 1–247.
Schirrmeister, B. E., Anisimova, M., Antonelli, A., and Homayoun, C. (2011). Evolution of cyanobacterial morphotypes: taxa required for improved phylogenomic approaches. Commun. Integr. Biol. 4, 1–4. doi: 10.4161/cib.16183
Schirrmeister, B. E., Gugger, M., and Donoghue, P. C. J. (2015). Cyanobacteria and the great oxidation event: evidence from genes and fossils. Palaeontology 58, 769–785. doi: 10.1111/pala.12178
Shih, P. M., Wu, D., Latifi, A., Axen, S. D., Fewer, D. P., Talla, E., et al. (2013). Improving the coverage of the cyanobacterial phylum using diversity-driven genome sequencing. Proc. Natl. Acad. Sci. U.S.A. 110, 1053–1058. doi: 10.1073/pnas.1217107110
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Sun, Z., and Blanchard, J. L. (2014). Strong genome-wide selection early in the evolution of Prochlorococcus resulted in a reduced genome through the loss of a large number of small effect genes. PLoS One 9:e88837. doi: 10.1371/journal.pone.0088837
Turner, S., Pryer, K. M., Miao, V. P. W., and Palmer, J. D. (1999). Investigating deep phylogenetic relationships among Cyanobacteria and plastids by small subunit rRNA sequence analysis. J. Eukaryot. Microbiol. 46, 327–338. doi: 10.1111/j.1550-7408.1999.tb04612.x
Uyeda, J. C., Harmon, L. J., and Blank, C. E. (2016). A comprehensive study of cyanobacterial morphological and ecological evolutionary dynamics through deep geologic time. PLoS One 11:e0162539. doi: 10.1371/journal.pone.0162539
Walter, J. M., Coutinho, F. H., Dutilh, B. E., Swings, J., Thompson, F. L., and Thompson, C. C. (2017). Ecogenomics and taxonomy of Cyanobacteria phylum. Front. Microbiol. 8:2132. doi: 10.3389/fmicb.2017.02132
Wang, Z., and Wu, M. (2015). An integrated phylogenomic approach toward pinpointing the origin of mitochondria. Sci. Rep. 5:7949. doi: 10.1038/srep07949
Whatley, J. M., John, P., and Whatley, F. R. (1979). From extracellular to intracellular: the establishment of mitochondria and chloroplasts. Proc. R. Soc. B 204, 165–187. doi: 10.1098/rspb.1979.0020
Williams, T. A., Foster, P. G., Nye, T. M. W., Cox, C. J., and Embley, T. M. (2012). A congruent phylogenomic signal places eukaryotes within the Archaea. Proc. R. Soc. B Biol. Sci. 279, 4870–4879. doi: 10.1098/rspb.2012.1795
Keywords: cyanobacteria, Archaeplastida, chloroplast, evolution, phylogenetic tree
Citation: Moore KR, Magnabosco C, Momper L, Gold DA, Bosak T and Fournier GP (2019) An Expanded Ribosomal Phylogeny of Cyanobacteria Supports a Deep Placement of Plastids. Front. Microbiol. 10:1612. doi: 10.3389/fmicb.2019.01612
Received: 25 January 2019; Accepted: 27 June 2019;
Published: 12 July 2019.
Edited by:
Rekha Seshadri, Lawrence Berkeley National Laboratory, United StatesReviewed by:
Steven Graham Ball, Université de Lille, FranceJonathan Badger, National Cancer Institute (NCI), United States
Denis Baurain, University of Liège, Belgium
Copyright © 2019 Moore, Magnabosco, Momper, Gold, Bosak and Fournier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kelsey R. Moore, a3Jtb29yZUBtaXQuZWR1