 Baoli Zhu
Baoli Zhu Jiaqi Wang
Jiaqi Wang Lauren M. Bradford
Lauren M. Bradford Katharina Ettwig2
Katharina Ettwig2 Baolan Hu
Baolan Hu Tillmann Lueders
Tillmann Lueders
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 10 July 2019
Sec. Microbiotechnology
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.01577
This article is part of the Research Topic New Microbiological Treatments for Removal of Oxyanions from Polluted Water and Soil View all 4 articles
Oxygenic denitrification represents a new route in reductive nitrogen turnover which differs from canonical denitrification in how nitric oxide (NO) is transformed into dinitrogen gas. Instead of NO reduction via N2O to N2, NO is proposed to be directly disproportionated into N2 and O2 in oxygenic denitrification, catalyzed by the putative NO dismutase (Nod). Although a high diversity of nod genes has been recovered from various environments, still little is known about the niche partitioning and ecophysiology of oxygenic denitrifiers. One constraint is that nod as a functional marker for oxygenic denitrifiers is not well established. To address this issue, we compared the diversity and phylogeny of nod, 16S rRNA and pmoA gene sequences of four NC10 enrichments that are capable of methane-driven oxygenic denitrification and one environmental sample. The phylogenies of nod, 16S rRNA and pmoA genes of these cultures were generally congruent. The diversity of NC10 bacteria inferred from different genes was also similar in each sample. A new set of NC10-specific nod primers was developed and used in qPCR. The abundance of NC10 bacteria inferred from nod genes was constantly lower than via 16S rRNA genes, but the difference was within one order of magnitude. These results suggest that nod is a suitable molecular marker for studying the diversity and phylogeny of methane-driven oxygenic denitrifiers, the further investigation of which may be of value to develop enhanced strategies for sustainable nitrogen or methane removal.
The defining step of oxygenic denitrification, the proposed nitric oxide (NO) dismutation, is conceived as an unusual oxygen-forming process, suggested to occur in nitrite-dependent anaerobic methanotrophs of the NC10 phylum (e.g., Candidatus Methylomirabilis oxyfera) and in a denitrifying alkane-degrading Gammaproteobacterium (Ettwig et al., 2010, 2012; Zedelius et al., 2011). In conventional denitrification, NO is reduced via N2O to N2 (Schreiber et al., 2012; Zhang et al., 2019). In contrast, in oxygenic denitrification, NO is suggested to be disproportionated directly into N2 and O2, thus bypassing the ozone-depleting potent greenhouse gas nitrous oxide (N2O). The released O2, in theory, could enable the use of aerobic catabolic routes in anoxic habitats, possibly entailing ecophysiological advantage for microbes to thrive on recalcitrant substrates in O2-limited environments (Zhu et al., 2017). So far, NO dismutation has been suggested to be involved in the oxidation of methane, alkanes (C6-C30) and benzene (Ettwig et al., 2010; Zedelius et al., 2011; Atashgahi et al., 2018). Recent molecular evidence, however, suggested that NO dismutation potentially occurs in only distantly related microorganisms with diverse catabolic capabilities (Zhu et al., 2017). Nonetheless, the current understanding of oxygenic denitrification, its key step of NO dismutation and the microbes hosting this capacity (referred to as oxygenic denitrifiers) is still very limited. So far, the NO dismutation step distinguishes, but not mutually excludes, oxygenic denitrifiers from conventional denitrifiers. It is known that the genes coding for enzymes involved in reductive N-cycling, e.g., nif, nir, and nor, are often subject to lateral gene transfer (Philippot, 2002; Jones et al., 2008; Andam et al., 2015). This complicates the inference of denitrifiers’ phylogeny via functional genes. Thus far, the possible evolutionary history of NO dismutase genes (nod) is unresolved. Although diverse nod gene pools have been recovered from the environment (Zhu et al., 2017; Zhang et al., 2018), their phylogenetic association often remains unclear.
Among all oxygenic denitrifiers, anaerobic methanotrophs of the NC10 phylum have been most intensively studied. They use CH4 as electron donor to fuel oxygenic denitrification (Ettwig et al., 2010). NC10 bacteria are present in various natural and engineered systems, such as wetlands, rivers, lakes, rice paddies, tidal zones, and wastewater treatment plants (Luesken et al., 2011b; Wang et al., 2012, 2015, 2018; Zhu et al., 2012; Shen et al., 2013, 2014; He et al., 2015). Due to their widespread occurrence and methane-oxidizing activity in nature, NC10 bacteria could actually represent an important methane sink in the environment (Zhu et al., 2012; Deutzmann et al., 2014; Hu et al., 2014). Still, the NC10 phylum has no pure culture representatives to date. Based on 16S rRNA sequence information, several groups within the phylum (group a, b, c, d, and e) have been classified (Ettwig et al., 2009; He et al., 2015). Of those, only members of the phylogroups a were found to be dominant in methane-oxidizing enrichment cultures obtained from various environments. The partial genome reconstruction of the clade d NC10 bacterium CSP1-5 from a subsurface sample suggested the capability for aerobic methanol oxidation and nitrate reduction, but not methane oxidation and NO dismutation (Hug et al., 2016). Thus, it is not likely that all members of the NC10 phylum are oxygenic methanotrophs.
It is known that 16S rRNA genes often do not allow inferring metabolic functions for environmental microbes. For aerobic methanotrophs, pmoA genes are a useful functional marker for diversity and phylogeny studies, and have been frequently used to target these populations in the environment (Bourne et al., 2001; Pacheco-Oliver et al., 2002). pmoA has also been used as a marker for methanotrophic NC10 bacteria, which also use particulate methane monooxygenase to activate methane. M. oxyfera-like pmoA sequences form a cluster distinct from pmoA of aerobic methanotrophs (Luesken et al., 2011c). When using a suggested OTU cut-off of 3 and 10% sequence similarity for 16S rRNA and pmoA (Luke and Frenzel, 2011), respectively, the diversity of NC10 pmoA genes recovered so far is much lower than the known NC10 16S rRNA gene diversity. Information about the presence of NC10 nod genes in different environments (Bhattacharjee et al., 2016; Padilla et al., 2016; Zhu et al., 2017) and in enrichment cultures is still scarce. This calls for a rigorous assessment of the utility and comparability of the different marker gene assays (nod, pmoA, and 16S) to recover the full spectrum of oxygenic methanotrophs within the NC10 phylum present in a given habitat.
In this study, the diversity and phylogeny of nod genes in four NC10 enrichment cultures and one aquifer sample were compared to the 16S rRNA and pmoA genes of the NC10 phylum detectable in the same samples. New NC10-specific nod primers were developed and validated, and NC10 abundance was evaluated by nod as well as 16S rRNA gene-targeted qPCR. Our results suggest that nod can be a functional and also a phylogenetic marker for oxygenic methanotrophs within the NC10 phylum. This will be useful for future studies seeking to address the ecology and ecophysiology of these enigmatic microbes in natural systems.
NC10 bacteria have been previously enriched under denitrifying, methane-oxidizing conditions, with nitrite and/or nitrate as electron acceptors. Four sediment-free NC10 enrichment cultures maintained under different conditions were analyzed here (Table 1). In cultures AAA-NC10, DAMO-VI and DAMO-CANON, M. oxyfera related bacteria were dominant (Ettwig et al., 2009, 2016). In DAMO-SC, a different NC10 species, M. sinica was dominant (He et al., 2016). Aquifer sediment samples from Siklós were collected in April 2015 from the bottom of a monitoring well in the center of a xylene plume in Siklós, Hungary (Tancsics et al., 2013), where diverse nod sequences have been previously reported (Zhu et al., 2017).

Table 1. NC10 enrichment cultures and aquifer sample analyzed in this study.
DNA of Siklós aquifer sediment was isolated as previously described (Pilloni et al., 2012) with minor modification, the final DNA precipitation was done at 4°C instead of 20°C. For DNA isolation from enrichment cultures, 0.5 – 1.0 ml homogenized culture was pipetted into 1.5 ml Eppendorf tubes, which were spun at 13,000 rpm for 2 min. The supernatant was then removed and the remaining biomass was weighed. DNA was isolated with the Power Soil DNA isolation kit (MoBio Laboratories Inc., United States) according to the manufacturer’s protocol. DNA concentration and quality were checked with the Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher, Waltham, United States) on a MX3000p cycler (Agilent, Santa Clara, United States) and by standard agarose gel electrophoresis.
First, the general nod primer pair nod684Fv2 and nod1706Rv2 (Zhu et al., 2017) was used to amplify nod gene fragments from NC10 enrichment cultures. The amplicons were cloned and sequenced as previously described (Winderl et al., 2008). The resulting nod sequences and previously reported NC10 cluster nod sequences (Zhu et al., 2017) were aligned with reference nod sequences in MEGA6 (Tamura et al., 2013). Based on the updated alignment, a new set of NC10-specific nod gene primers (nod840F and nod1012R) was developed, which should strictly target a 173 bp fragment of nod sequences within the NC10 phylum and be optimally suited for qPCR (Table 2). The specificity of this primer pair was verified by sequencing the short amplicons using environmental DNA extracted from Siklós aquifer sediment as template. The primer pair was used for qPCR analysis.
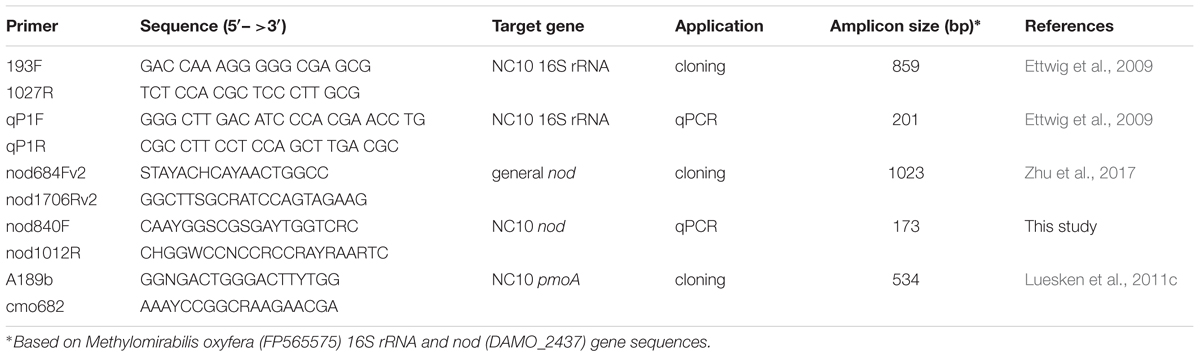
Table 2. primers used for cloning and qPCR analysis in this study.
DNA samples diluted by 10- or 100-fold were used as template for nod gene PCR analysis. Primer pairs used are listed in Table 2. To recover a potentially increased diversity for all analyzed nod, pmoA, and 16S rRNA genes, gradient PCRs with the following cycling conditions were performed: a 3 min initial dissociation at 96°C, followed by 30 cycles of amplification (45 s at 95°C, 60 s at 52–62°C, and 90 s at 72°C), and a final 5 min extension at 72°C. All PCRs were performed in 25 μl reactions containing nuclease-free H2O, 1 × PCR buffer, 1.5 mM MgCl2, 0.1 mM dNTPs, 0.5 U Taq polymerase (all Fermentas GmBH, Basel, Switzerland), 5 μg BSA (Roche Diagnostics GmbH, Basel, Switzerland), 0.5 μM of each primer, and 1 μl template DNA. PCR products were checked by standard agarose gel electrophoresis.
NC10 abundance in each sample was quantified by qPCR targeting nod as well as 16S rRNA genes, using the NC10-specific nod primers nod840F/nod1012R and the NC10-specific 16S rRNA primers qP1F/qP1R, respectively (Table 2). pGEM-T-easy vectors with respective inserts were used as standards. Sample DNA was quantified in 10- and 100-fold dilutions. Standards and samples were quantified in triplicate qPCR reactions. qPCR reactions (25 μl volume) were carried out with MX3000p cycler (Agilent, Santa Clara, United States). 2 × Takyon SYBR master mix (Eurogentec, Cologne, Germany) with Rox as the reference dye. The annealing temperatures used for nod and 16S rRNA qPCR were 57°C and 62°C, respectively. qPCR analyses with efficiencies of 100 ± 10% were used for calculating gene abundances. Absolute nod and 16S rRNA gene abundance of each sample were calculated per ng of genomic DNA.
nod, pmoA, and 16S rRNA PCR products of different annealing temperatures from each sample were pooled and purified with PCRextract spin columns (5Prime, Hamburg, Germany) according to the manufacturer’s protocol. Purified PCR products were ligated into the pGEM-T Easy Vector (Promega, Madison, United States) and then transformed into Escherichia coli JM109 competent cells. Selection of clones with correct insert size and Sanger sequencing was done as previously described (Winderl et al., 2008). High-quality sequences obtained were aligned with selected respective reference sequences with the ClustalW algorithm with default settings and were visually checked and adjusted before phylogeny constructions. nod and pmoA sequences were aligned with their translated protein sequences. Phylogenetic trees were constructed with MEGA6 using the neighbor-joining method. The robustness of each tree topology was tested by bootstrap analysis (1,000 replicates).
Representative 16S rRNA, pmoA and nod gene sequences obtained in this study were deposited at NCBI under the accession numbers MK909154 – MK909155, MK909156 – MK909157, and MK909158 – MK909164, respectively.
We recovered NC10-related marker genes from four enrichment cultures in chemostats (Table 1) that were active in methane and/or ammonia removal when samples were taken. No more than three different NC10 species-level OTUs were found in each reactor, with one OTU dominating over the others. This was inferred consistently via 16S rRNA, nod and pmoA gene diversities (Figures 1–3). The low diversity and uneven abundance of NC10 OTUs in each reactor allowed us to compare the phylogenies of 16S rRNA, nod and pmoA marker genes between reactors.
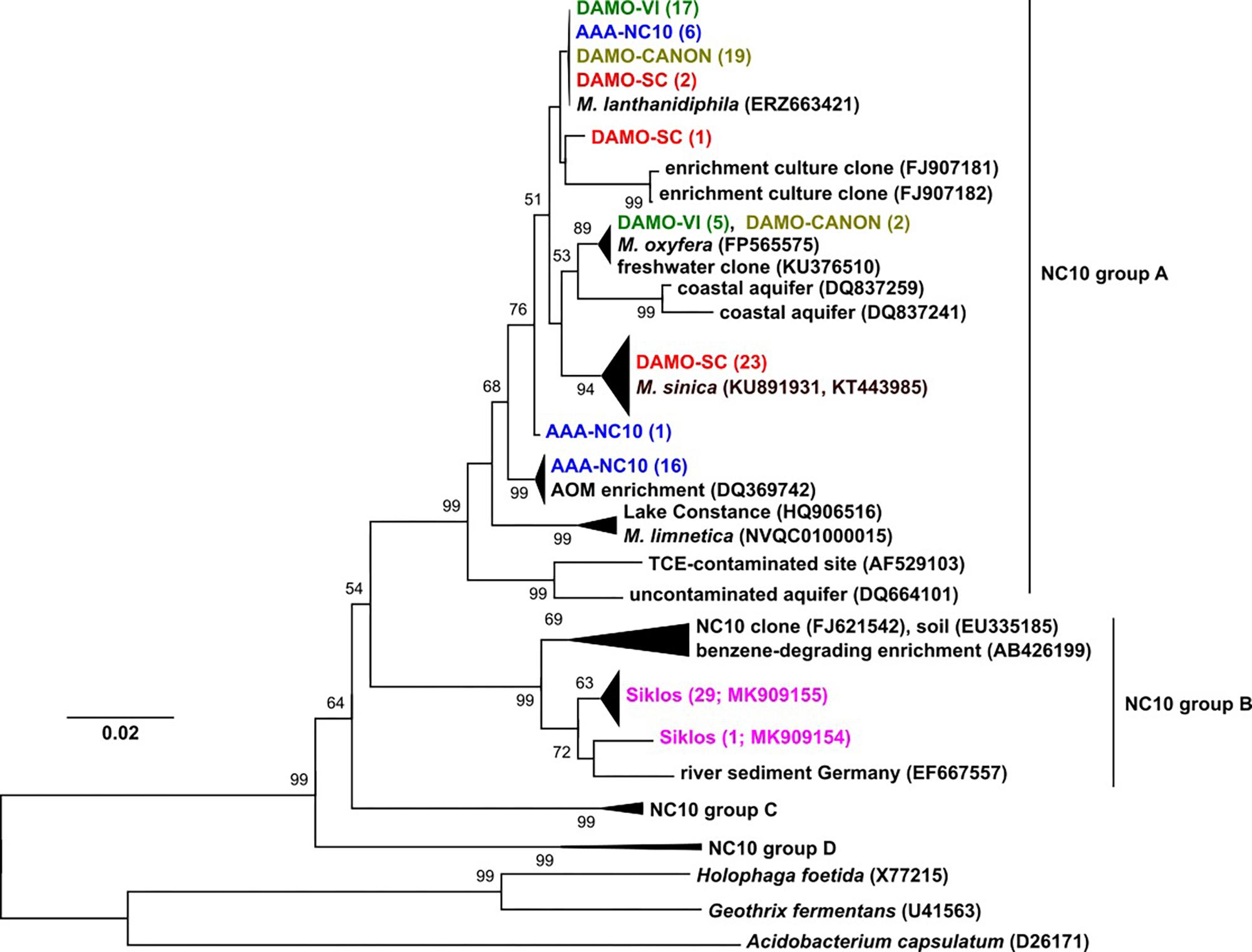
Figure 1. The 16S rRNA phylogeny of NC10 bacteria in the four reactors and Siklós aquifer. Sequences from different samples are labeled in different colors and are consistent in all figures. The number in bracket indicates number of clones in the branch or accession number of the reference and representative sequence.
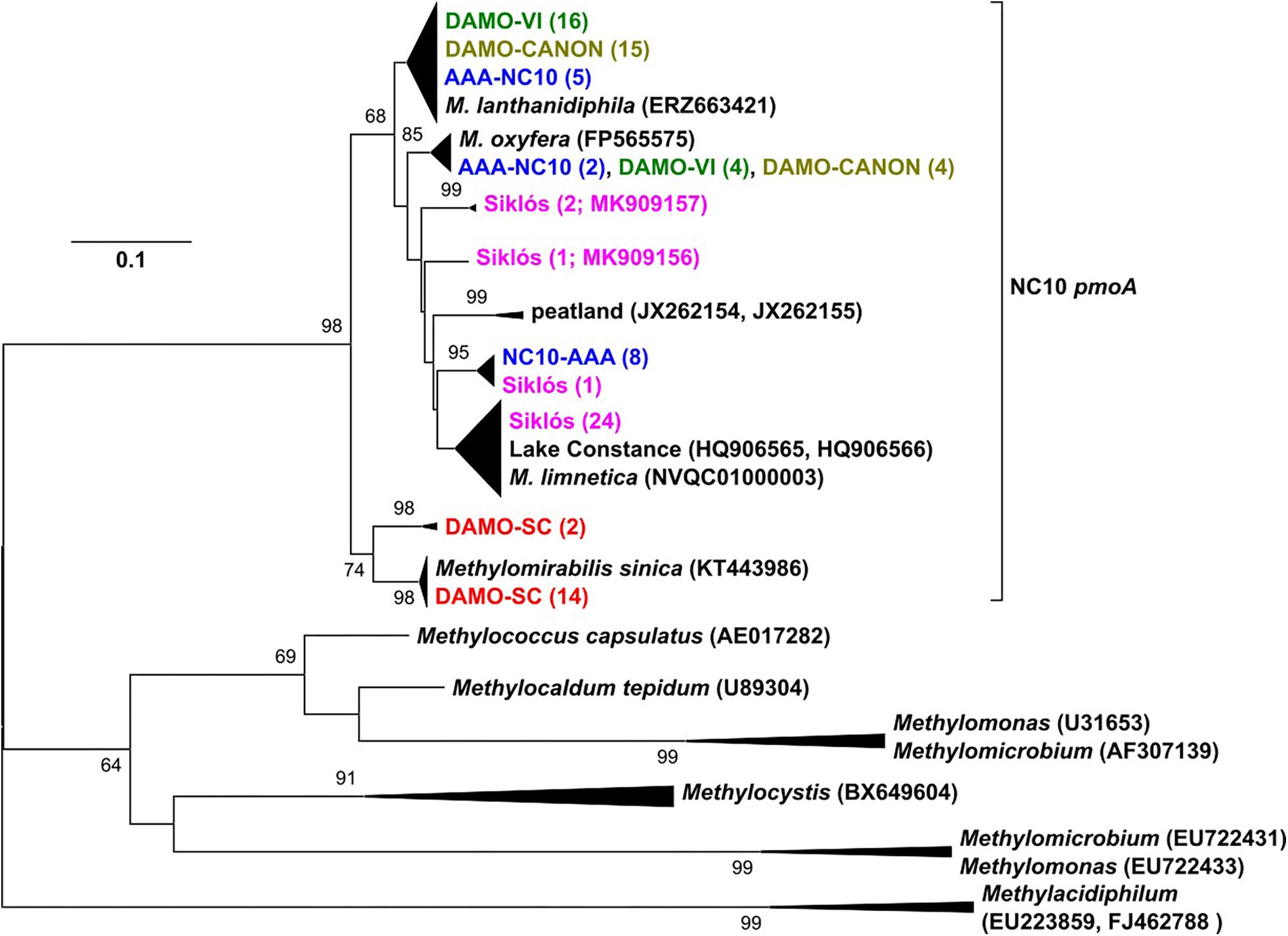
Figure 2. The pmoA phylogeny of NC10 bacteria in the four reactors and Siklós aquifer. Sequences from different samples are labeled in different colors and are consistent in all figures. The number in bracket indicates number of clones in the branch or accession number of the reference and representative sequence.
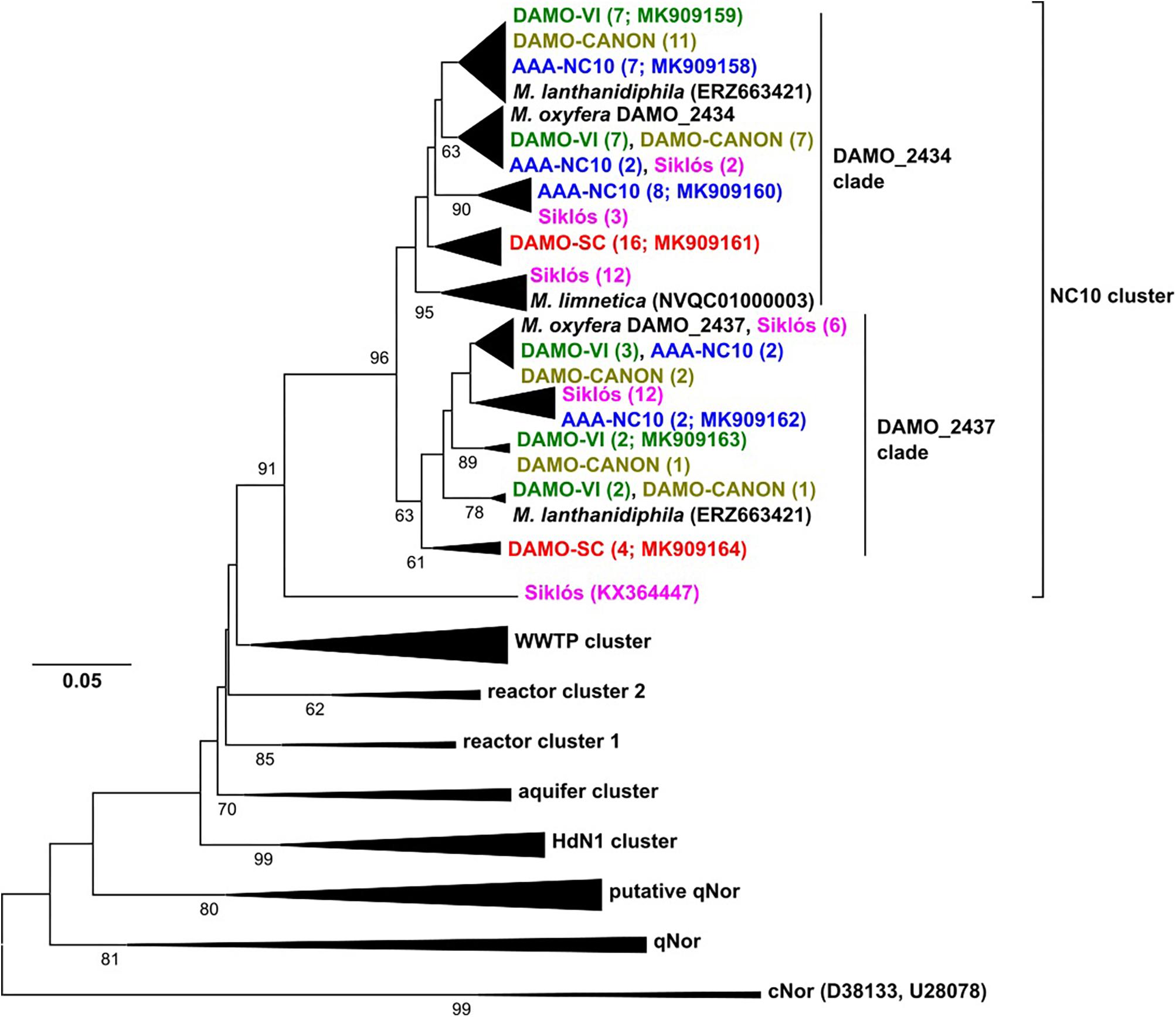
Figure 3. The nod gene phylogeny of NC10 bacteria in four reactors and Siklós aquifer. Sequences obtained from different samples are labeled in different color. Sequences from different samples are labeled in different colors and are consistent in all figures. The number in bracket indicates number of clones in the branch or accession number of the reference and representative sequence.
Phylogeny of 16S rRNA genes showed that in each of the four enrichment cultures, more than one NC10 species/subspecies-level OTU was detected. Five clones (out of 22) of enrichment DAMO-VI and two clones (out of 21) of DAMO-CANON cultures were highly similar (>99%) to the 16S rRNA gene of M. oxyfera, indicating these sequences belonged to M. oxyfera in a strict sense. Together with the recently sequenced M. lanthanidiphila (Versantvoort et al., 2018), the majority of DAMO-VI and DAMO-CANON reactor clones, and some clones from AAA-NC10 and DAMO-SC reactors formed a cluster distinct (97.5% identity) to M. oxyfera. The DAMO-CANON reactor had the same dominant NC10 species and a similar NC10 community composition as the DAMO-VI reactor (Figure 1). As the CANON reactor was inoculated with DAMO-VI and anammox biomass (Luesken et al., 2011a), this finding indicated that the co-culturing with anammox bacteria and the constant supply of ammonia did not induce a shift in NC10 population.
In DAMO-SC, nearly all NC10 16S rRNA sequences were closely related to that of M. sinica (KU891931 and KT443985), which has ∼97% sequence similarity to M. oxyfera. M. sinica, originally enriched from paddy soil, are reported as cocci (He et al., 2016), clearly distinct to the morphology of M. oxyfera. The dominant NC10 16S rRNA gene sequences in the AAA-NC10 reactor were very similar to the DQ369742 sequence, reported for the first enrichment culture of denitrifying anaerobic methane oxidizers (Raghoebarsing et al., 2006). Nitrate was the sole electron acceptor in the AAA-NC10 reactor, with only low (ca. 100 μM) transient nitrite accumulation (Ettwig et al., 2016). Similarly, the initial culture reported by Raghoebarsing et al. (2006) was obtained under high nitrate (>3 mM) and low nitrite (ca. 200 μM) availability. These conditions differed from high nitrite concentrations in the DAMO-VI and DAMO-CANON reactors (Luesken et al., 2011a). This suggests that nitrite concentrations could possibly play a role in selecting different NC10 strains. Thus, in addition to exploring diverse environments, varying nitrite and nitrate concentrations during enrichment may also help to cultivate divergent NC10 bacteria.
Interestingly, the dominant NC10 methanotroph M. limnetica in lake Zug (Graf et al., 2018) is closely related to lake Constance clones (Figure 1), suggesting some common lake conditions may favor this NC10 species over others. NC10 16S rRNA sequences obtained from the Siklós aquifer were all part of group B of NC10 bacteria (Figure 1). Although NC10 group B bacteria sequences were retrieved from different environments (e.g., Shen et al., 2016), they have not been enriched or isolated thus far, and their ecophysiology is unknown. Environments with only one dominant phylogroup of NC10 sequence types like the Siklós aquifer can be interesting for further elucidating their ecological functions.
pmoA genes of the NC10 phylum have been previously reported to form a cluster distinct to other aerobic methanotrophs, and have been used to detect NC10 methanotrophs in different ecosystems (Luesken et al., 2011c). In our reactor samples, the NC10 community revealed via pmoA was largely consistent to that inferred via 16S rRNA phylogeny, although pmoA sequences from the DAMO-SC reactor, instead of the Siklós aquifer, were placed in the basal region of the NC10 pmoA cluster. There were at least two different groups of pmoA sequences recovered from each reactor, again suggesting multiple NC10 (sub)species to exist in each community. Similar to 16S phylogeny, together with M. lanthanidiphila pmoA, the dominant pmoA type from the DAMO-VI and DAMO-CANON reactors, as well as five clones from AAA-NC10 reactor, clustered closely together (Figure 2). A minority of clones from the DAMO-VI and DAMO-CANON reactors clustered closely with M. oxyfera pmoA. This was congruent with 16S rRNA gene phylogeny (Figure 1). In contrast, pmoA from the DAMO-SC reactor and some of AAA-NC10 reactor clones formed distinct clusters (Figure 2). In 16S rRNA phylogeny, the branching of AAA-NC10 clones was deeper than that of DAMO-SC clones, while DAMO-SC clones branched close to the root in pmoA phylogeny.
Notably, although nearly all Siklós aquifer 16S rRNA sequences were highly similar and belonged to the group b of NC10 bacteria, Siklós pmoA seemed to be more diverse and dispersed among environmental and reactor pmoA sequences (Figure 2). Nevertheless, the majority of Siklós pmoA sequences formed a separate cluster and were close to NC10 pmoA sequences retrieved from Lake Constance (Deutzmann and Schink, 2011) and M. limnetica. This could indicate that group b NC10 bacteria are possibly capable of methane driven denitrification as well.
Primers (Table 2) used to recover pmoA gene fragments were initially intended to be NC10-specific (Luesken et al., 2011c). However, in reactor DAMO-SC, about 10% of the obtained sequences were affiliated to comammox amoA genes, which typically showed 79 and 90% sequence similarity on DNA and amino acid level, respectively, to that of Nitrospira inopinata (Daims et al., 2015). Comammox-related amoA sequences were not detected in other reactors and aquifer samples, though the same primers were applied. This indicates that the current NC10-specific pmoA primers may still need to be improved in specificity, as comammox bacteria are being discovered from more and more different environments (e.g., Xia et al., 2018). The competition of oxygenic denitrifiers and comammox bacteria for nitrite certainly warrants future investigations.
Besides sequenced NC10 bacteria genomes, environmental nod sequences have previously been reported from marine oxygen minimum zone (OMZ), aquifers, wastewater treatment systems, alpine wetland, and microaerobic pollutant-degrading sediment (Padilla et al., 2016; Zhu et al., 2017; Bradford et al., 2018; Zhang et al., 2018). Recently, nor sequences with nod characteristics were found in the genome of foraminifera, which, however, produced N2O (Woehle et al., 2018). In addition to the NC10 cluster, several unidentified nod clusters have also been reported from aquifer and wastewater systems (Zhu et al., 2017). Bhattacharjee et al. (2016) obtained several nearly identical nod sequences from a NC10 enrichment, although diverse NC10 bacteria were present in the culture.
The reactor nod gene sequences retrieved from the four reactors and Siklós aquifer expanded the known diversity of nod genes within the NC10 cluster. Compared to 16S rRNA and pmoA genes, the nod sequences in each reactor were more widely branching, suggesting that nod genes were less conserved than 16S rRNA and pmoA genes (Figure 3). The overall diversity of nod genes was higher than that of 16S rRNA and pmoA genes of the NC10 phylum. However, considering that the Methylomirabilis species sequenced so far host two distinct nod genes (Ettwig et al., 2010, 2012; Graf et al., 2018; Versantvoort et al., 2018), the diversity of NC10 bacteria inferred via nod gene was again similar to that of 16S rRNA and pmoA genes (Figure 3). Phylogenetically, NC10 cluster nod sequences can be divided into two major subgroups, named according to the nod homolog of M. oxyfera (DAMO_2434 or DAMO_2437) to which they are more closely related (Figure 3). The two nod sequences of M. lanthanidiphila fell into their respective clades. However, both of the M. limnetica nod copies clustered with some Siklós nod sequences, and were placed in the root of the DAMO_2434 clade.
The phylogenetic placements of nod genes within the DAMO_2434 clade was in good agreement with the phylogeny of NC10 16S rRNA genes: most sequences from DAMO-VI, DAMO-CANON and AAA-NC10 reactors were closely related, while sequences of the DAMO-SC reactor and Siklós were more deeply rooted. This general pattern was also seen amongst the sequences found within the DAMO-2437 clade, except Siküs nod sequences were clustered to DAMO-VI and AAA-NC10 reactor sequences, instead of in the basal region (Figure 3). Similar to pmoA, Siklós nod sequences also represented a higher NC10 diversity than Siklós 16S rRNA genes.
The general congruence among NC10 16S rRNA, pmoA and nod gene phylogenies suggested that nod can be used as a phylogenetic marker for these oxygenic methanotrophs. In order to validate if the abundance of methanotrophic NC10 bacteria in the environment can be directly quantified by targeting nod genes, a new qPCR primer set specific for NC10 nod-genes was developed and verified (Table 2).
In addition to the four reactor cultures, the primer pair was also applied to the Siklós aquifer sediment. Overall, the NC10 methanotroph abundance inferred from nod genes showed a strong linear correlation (R2 > 0.99) with that inferred from NC10 16S rRNA in all samples (Figure 4). The abundance of NC10 bacteria in all samples as quantified via nod genes was always lower than their abundance inferred from 16S rRNA genes. For the Siklós aquifer sediment, where NC10 bacteria were not enriched like in the reactor samples, the largest difference was observed: 3.0 × 103 16S rRNA and 8.3 × 102 nod (copy ng-1 DNA), but the difference was still within one order of magnitude (Table 1). This might suggest that not all NC10 bacteria are oxygenic denitrifiers. In fact, a methylotrophic member of NC10 without pmoA and nod genes was recently reported (Hug et al., 2016). Additionally, the difference may also be attributed to different PCR efficiencies when targeting the different genes. These results suggested that it is possible to directly quantify the abundance of oxygenic NC10 populations in enriched cultures as well as in environmental samples by using NC10-specific nod gene primers.
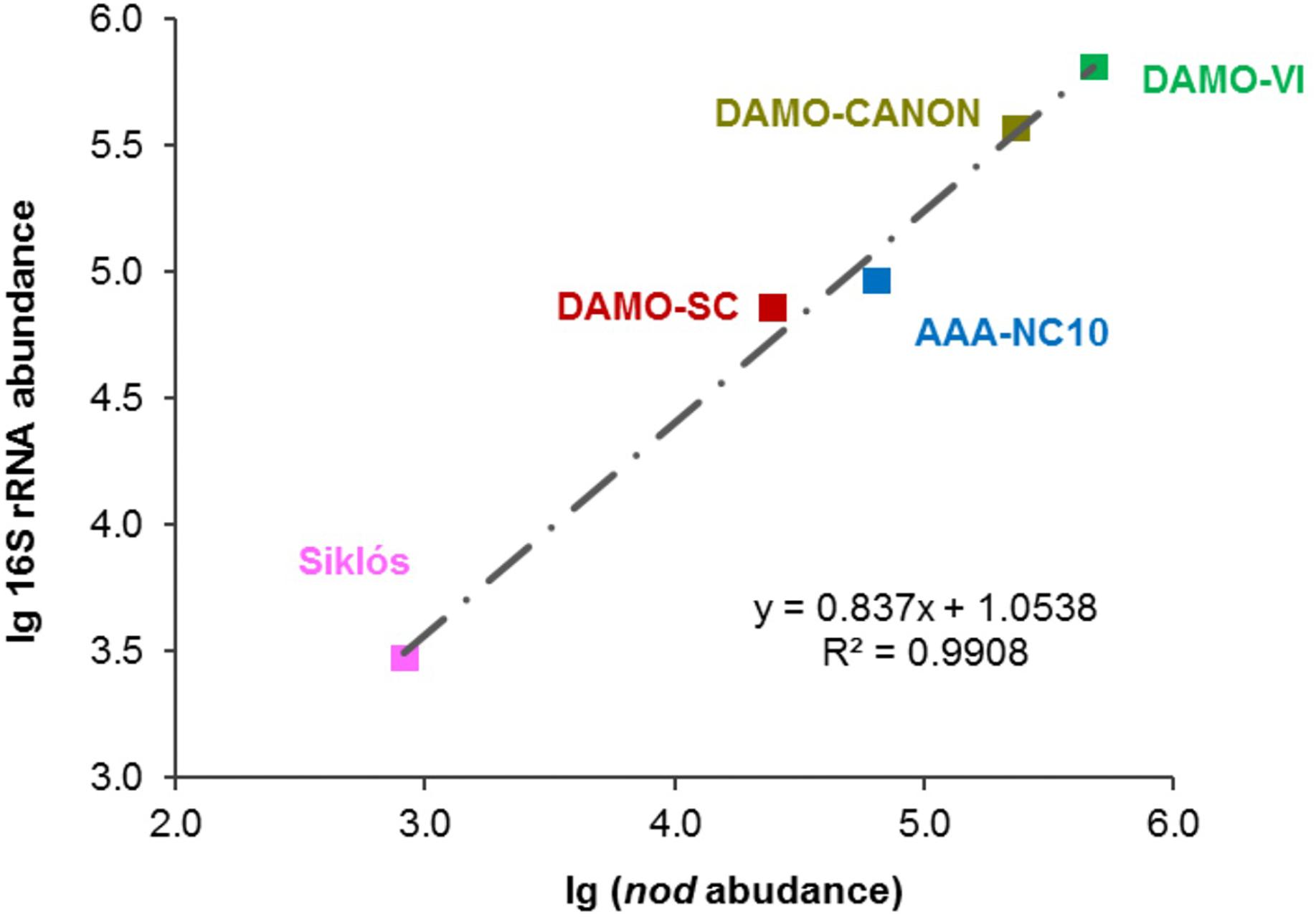
Figure 4. Strong linear relationship between 16S rRNA and nod gene abundances observed in these bioreactor and aquifer samples.
Highly diverse environmental nod gene pools in aquatic systems have suggested a high diversity of oxygenic denitrifiers (Zhu et al., 2017), among which NC10 bacteria use methane to drive oxygenic denitrification. Currently, the diversity and ecological importance of NC10 bacteria is not yet well understood. Here, we provide an integrated comparison of the detectability of NC10 populations using three different marker gene systems, and we found that the phylogeny of NC10 nod genes was generally congruent with those of 16S rRNA and pmoA genes. qPCR analysis targeting 16S rRNA and nod genes also resulted in different but still comparable NC10 bacteria abundance. Overall, the results suggest that nod can be used as a functional and phylogenetic marker for oxygenic NC10 methanotrophs. In future studies, researchers are suggested to include analyzing nod genes to better estimate the ecological importance of oxygenic NC10 bacteria in global methane cycling.
The datasets generated for this study can be found in the NCBI, MK909154 – MK909164.
BZ and TL designed the project. BZ, JW, and LB performed the experiment. BZ analyzed the data with help from KE, BH, and TL. BZ wrote the manuscript with inputs from all authors.
This research was funded by the European Research Council (ERC) under the European Union’s Seventh Framework Program (FP7/2007-2013), grant agreement 616644 (POLLOX) to TL. We acknowledge financial support from the Helmholtz Society.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Karin Stultiens (Radboud University Nijmegen, Netherlands) for providing bioreactor materials and András Táncsics (University of Gödöllö, Hungary) for access to the field site.
Andam, C. P., Carver, S. M., and Berthrong, S. T. (2015). Horizontal gene flow in managed ecosystems. Annu. Rev. Ecol. Evol. Syst. 46, 121–143. doi: 10.1146/annurev-ecolsys-112414-054126
Atashgahi, S., Hornung, B., van der Waals, M. J., da Rocha, U. N., Hugenholtz, F., Nijsse, B., et al. (2018). A benzene-degrading nitrate-reducing microbial consortium displays aerobic and anaerobic benzene degradation pathways. Sci. Rep. 8:4490. doi: 10.1038/s41598-018-22617-x
Bhattacharjee, A. S., Motlagh, A. M., Jetten, M. S., and Goel, R. (2016). Methane dependent denitrification- from ecosystem to laboratory-scale enrichment for engineering applications. Water Res. 99, 244–252. doi: 10.1016/j.watres.2016.04.070
Bourne, D. G., McDonald, I. R., and Murrell, J. C. (2001). Comparison of pmoA PCR primer sets as tools for investigating methanotroph diversity in three Danish soils. Appl. Environ. Microbiol. 67, 3802–3809. doi: 10.1128/aem.67.9.3802-3809.2001
Bradford, L. M., Vestergaard, G., Tancsics, A., Zhu, B. L., Schloter, M., and Lueders, T. (2018). Transcriptome-stable isotope probing provides targeted functional and taxonomic insights into microaerobic pollutant-degrading aquifer microbiota. Front. Microbiol. 9:2696. doi: 10.3389/fmicb.2018.02696
Daims, H., Lebedeva, E. V., Pjevac, P., Han, P., Herbold, C., Albertsen, M., et al. (2015). Complete nitrification by Nitrospira bacteria. Nature 528, 504–509. doi: 10.1038/nature16461
Deutzmann, J. S., and Schink, B. (2011). Anaerobic oxidation of methane in sediments of Lake Constance, an oligotrophic freshwater lake. Appl. Environ. Microbiol. 77, 4429–4436. doi: 10.1128/AEM.00340-11
Deutzmann, J. S., Stief, P., Brandes, J., and Schink, B. (2014). Anaerobic methane oxidation coupled to denitrification is the dominant methane sink in a deep lake. Proc. Natl. Acad. Sci. U.S.A. 111, 18273–18278. doi: 10.1073/pnas.1411617111
Ettwig, K. F., Butler, M. K., Le Paslier, D., Pelletier, E., Mangenot, S., Kuypers, M. M., et al. (2010). Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 464, 543–548. doi: 10.1038/nature08883
Ettwig, K. F., Speth, D. R., Reimann, J., Wu, M. L., Jetten, M. S., and Keltjens, J. T. (2012). Bacterial oxygen production in the dark. Front. Microbiol. 3:273. doi: 10.3389/fmicb.2012.00273
Ettwig, K. F., van Alen, T., van de Pas-Schoonen, K. T., Jetten, M. S., and Strous, M. (2009). Enrichment and molecular detection of denitrifying methanotrophic bacteria of the NC10 phylum. Appl. Environ. Microbiol. 75, 3656–3662. doi: 10.1128/AEM.00067-09
Ettwig, K. F., Zhu, B. L., Speth, D., Keltjens, J. T., Jetten, M. S. M., and Kartal, B. (2016). Archaea catalyze iron-dependent anaerobic oxidation of methane. Proc. Natl. Acad. Sci. U.S.A. 113, 12792–12796. doi: 10.1073/pnas.1609534113
Graf, J. S., Mayr, M. J., Marchant, H. K., Tienken, D., Hach, P. F., Brand, A., et al. (2018). Bloom of a denitrifying methanotroph, ’Candidatus Methylomirabilis limnetica’, in a deep stratified lake. Environ. Microbiol. 20, 2598–2614. doi: 10.1111/1462-2920.14285
He, Z., Cai, C., Wang, J., Xu, X., Zheng, P., Jetten, M. S., et al. (2016). A novel denitrifying methanotroph of the NC10 phylum and its microcolony. Sci. Rep. 6:32241. doi: 10.1038/srep32241
He, Z., Geng, S., Cai, C., Liu, S., Liu, Y., Pan, Y., et al. (2015). Anaerobic oxidation of methane coupled to nitrite reduction by halophilic marine NC10 bacteria. Appl. Environ. Microbiol. 81, 5538–5545. doi: 10.1128/AEM.00984-15
Hu, B. L., Shen, L. D., Lian, X., Zhu, Q., Liu, S., Huang, Q., et al. (2014). Evidence for nitrite-dependent anaerobic methane oxidation as a previously overlooked microbial methane sink in wetlands. Proc. Natl. Acad. Sci. U.S.A. 111, 4495–4500. doi: 10.1073/pnas.1318393111
Hug, L. A., Thomas, B. C., Sharon, I., Brown, C. T., Sharma, R., Hettich, R. L., et al. (2016). Critical biogeochemical functions in the subsurface are associated with bacteria from new phyla and little studied lineages. Environ. Microbiol. 18, 159–173. doi: 10.1111/1462-2920.12930
Jones, C. M., Stres, B., Rosenquist, M., and Hallin, S. (2008). Phylogenetic analysis of nitrite, nitric oxide, and nitrous oxide respiratory enzymes reveal a complex evolutionary history for denitrification. Mol. Biol. Evol. 25, 1955–1966. doi: 10.1093/molbev/msn146
Luesken, F. A., Sanchez, J., van Alen, T. A., Sanabria, J., Op den Camp, H. J., Jetten, M. S., et al. (2011a). Simultaneous nitrite-dependent anaerobic methane and ammonium oxidation. Appl. Environ. Microbiol. 77, 6802–6807. doi: 10.1128/aem.05539-11
Luesken, F. A., van Alen, T. A., van der Biezen, E., Frijters, C., Toonen, G., Kampman, C., et al. (2011b). Diversity and enrichment of nitrite-dependent anaerobic methane oxidizing bacteria from wastewater sludge. Appl. Microbiol. Biotechnol. 92, 845–854. doi: 10.1007/s00253-011-3361-9
Luesken, F. A., Zhu, B., van Alen, T. A., Butler, M. K., Diaz, M. R., Song, B., et al. (2011c). pmoA Primers for detection of anaerobic methanotrophs. Appl. Environ. Microbiol. 77, 3877–3880. doi: 10.1128/AEM.02960-10
Luke, C., and Frenzel, P. (2011). Potential of pmoA amplicon pyrosequencing for methanotroph diversity studies. Appl. Environ. Microbiol. 77, 6305–6309. doi: 10.1128/AEM.05355-11
Pacheco-Oliver, M., McDonald, I. R., Groleau, D., Murrell, J. C., and Miguez, C. B. (2002). Detection of methanotrophs with highly divergent pmoA genes from Arctic soils. FEMS Microbiol. Lett. 209, 313–319. doi: 10.1016/s0378-1097(02)00568-2
Padilla, C. C., Bristow, L. A., Sarode, N., Garcia-Robledo, E., Gomez Ramirez, E., Benson, C. R., et al. (2016). NC10 bacteria in marine oxygen minimum zones. ISME J. 10, 2067–2071. doi: 10.1038/ismej.2015.262
Philippot, L. (2002). Denitrifying genes in bacterial and Archaeal genomes. Biochim. Biophys. Acta 1577, 355–376. doi: 10.1016/s0167-4781(02)00420-7
Pilloni, G., Granitsiotis, M. S., Engel, M., and Lueders, T. (2012). Testing the limits of 454 pyrotag sequencing: reproducibility, quantitative assessment and comparison to T-RFLP fingerprinting of aquifer microbes. PLoS One 7:e40467. doi: 10.1371/journal.pone.0040467
Raghoebarsing, A. A., Pol, A., van de Pas-Schoonen, K. T., Smolders, A. J., Ettwig, K. F., Rijpstra, W. I., et al. (2006). A microbial consortium couples anaerobic methane oxidation to denitrification. Nature 440, 918–921. doi: 10.1038/nature04617
Schreiber, F., Wunderlin, P., Udert, K. M., and Wells, G. F. (2012). Nitric oxide and nitrous oxide turnover in natural and engineered microbial communities: biological pathways, chemical reactions, and novel technologies. Front. Microbiol. 3:372. doi: 10.3389/fmicb.2012.00372
Shen, L. D., Huang, Q., He, Z. F., Lian, X., Liu, S., He, Y. F., et al. (2014). Vertical distribution of nitrite-dependent anaerobic methane-oxidising bacteria in natural freshwater wetland soils. Appl. Microbiol. Biotechnol. 99, 349–357. doi: 10.1007/s00253-014-6031-x
Shen, L. D., Liu, S., Zhu, Q., Li, X. Y., Cai, C., Cheng, D. Q., et al. (2013). Distribution and diversity of nitrite-dependent anaerobic methane-oxidising bacteria in the sediments of the Qiantang river. Microb. Ecol. 67, 341–349. doi: 10.1007/s00248-013-0330-0
Shen, L. D., Wu, H. S., Gao, Z. Q., Liu, X., and Li, J. (2016). Comparison of community structures of Candidatus Methylomirabilis oxyfera-like bacteria of NC10 phylum in different freshwater habitats. Sci. Rep. 6:25647. doi: 10.1038/srep25647
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tancsics, A., Farkas, M., Szoboszlay, S., Szabo, I., Kukolya, J., Vajna, B., et al. (2013). One-year monitoring of meta-cleavage dioxygenase gene expression and microbial community dynamics reveals the relevance of subfamily I.2.C extradiol dioxygenases in hypoxic, BTEX-contaminated groundwater. Syst. Appl. Microbiol. 36, 339–350. doi: 10.1016/j.syapm.2013.03.008
Versantvoort, W., Guerrero-Cruz, S., Speth, D. R., Frank, J., Gambelli, L., Cremers, G., et al. (2018). Comparative genomics of Candidatus Methylomirabilis species and description of Ca. Methylomirabilis lanthanidiphila. Front. Microbiol. 9:1672. doi: 10.3389/fmicb.2018.01672
Wang, J., Cai, C., Li, Y., Hua, M., Wang, J., Yang, H., et al. (2018). Denitrifying anaerobic methane oxidation: a previously overlooked methane sink in Intertidal zone. Environ. Sci. Technol. 53, 203–212. doi: 10.1021/acs.est.8b05742
Wang, Y., Huang, P., Ye, F., Jiang, Y., Song, L., Op den Camp, H. M., et al. (2015). Nitrite-dependent anaerobic methane oxidizing bacteria along the water level fluctuation zone of the Three Gorges Reservoir. Appl. Microbiol. Biotechnol. 100, 1977–1986. doi: 10.1007/s00253-015-7083-2
Wang, Y., Zhu, G., Harhangi, H. R., Zhu, B., Jetten, M. S. M., Yin, C., et al. (2012). Co-occurrence and distribution of nitrite-dependent anaerobic ammonium and methane-oxidizing bacteria in a paddy soil. FEMS Microbiol. Lett. 336, 79–88. doi: 10.1111/j.1574-6968.2012.02654.x
Winderl, C., Anneser, B., Griebler, C., Meckenstock, R. U., and Lueders, T. (2008). Depth-resolved quantification of anaerobic toluene degraders and aquifer microbial community patterns in distinct redox zones of a tar oil contaminant plume. Appl. Environ. Microbiol. 74, 792–801. doi: 10.1128/aem.01951-07
Woehle, C., Roy, A. S., Glock, N., Wein, T., Weissenbach, J., Rosenstiel, P., et al. (2018). A novel eukaryotic denitrification pathway in foraminifera. Curr. Biol. 28, 2536–2543.e5. doi: 10.1016/j.cub.2018.06.027
Xia, F., Wang, J. G., Zhu, T., Zou, B., Rhee, S. K., and Quan, Z. X. (2018). Ubiquity and diversity of complete ammonia oxidizers (Comammox). Appl. Environ. Microbiol. 84:e01390-18. doi: 10.1128/AEM.01390-18
Zedelius, J., Rabus, R., Grundmann, O., Werner, I., Brodkorb, D., Schreiber, F., et al. (2011). Alkane degradation under anoxic conditions by a nitrate-reducing bacterium with possible involvement of the electron acceptor in substrate activation. Environ. Microbiol. Rep. 3, 125–135. doi: 10.1111/j.1758-2229.2010.00198.x
Zhang, M., Gu, J., and Liu, Y. (2019). Engineering feasibility, economic viability and environmental sustainability of energy recovery from nitrous oxide in biological wastewater treatment plant. Bioresour. Technol. 282, 514–519. doi: 10.1016/j.biortech.2019.03.040
Zhang, Y. F., Ma, A. Z., Liu, W. Z., Bai, Z. H., Zhuang, X. L., and Zhuang, G. Q. (2018). The occurrence of putative nitric oxide dismutase (n.d.) in an alpine wetland with a new dominant subcluster and the potential ability for a methane sink. Archaea 2018:6201541. doi: 10.1155/2018/6201541
Zhu, B., Bradford, L., Huang, S., Szalay, A., Leix, C., Weissbach, M., et al. (2017). Unexpected diversity and high abundance of putative nitric oxide dismutase (n.d.) genes in contaminated aquifers and wastewater treatment systems. Appl. Environ. Microbiol 83:e02750-16. doi: 10.1128/AEM.02750-16
Keywords: nitric oxide dismutation, oxygenic methanotrophs, oxygenic denitrification, NC10, nod, pmoA
Citation: Zhu B, Wang J, Bradford LM, Ettwig K, Hu B and Lueders T (2019) Nitric Oxide Dismutase (nod) Genes as a Functional Marker for the Diversity and Phylogeny of Methane-Driven Oxygenic Denitrifiers. Front. Microbiol. 10:1577. doi: 10.3389/fmicb.2019.01577
Received: 15 March 2019; Accepted: 25 June 2019;
Published: 10 July 2019.
Edited by:
Paul Hatzinger, Independent Researcher, United StatesReviewed by:
Meng Zhang, Nanyang Technological University, SingaporeCopyright © 2019 Zhu, Wang, Bradford, Ettwig, Hu and Lueders. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Baoli Zhu, YmFvbGkuemh1QHVuaS1iYXlyZXV0aC5kZQ==; YmFvbGl6aHVAZ21haWwuY29t; Tillmann Lueders, dGlsbG1hbm4ubHVlZGVyc0B1bmktYmF5cmV1dGguZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.