 Ruchira Sharma1
Ruchira Sharma1 Brittany A. Pielstick1
Brittany A. Pielstick1 Kimberly A. Bell1
Kimberly A. Bell1 Tanner B. Nieman1
Tanner B. Nieman1 Olivia A. Stubbs1
Olivia A. Stubbs1 Edward L. Yeates1
Edward L. Yeates1 David A. Baltrus2
David A. Baltrus2 Julianne H. Grose1*
Julianne H. Grose1*- 1Department of Microbiology and Molecular Biology, Brigham Young University, Provo, UT, United States
- 2School of Plant Sciences, The University of Arizona, Tucson, AZ, United States
Erwinia amylovora is a plant pathogen from the Erwiniaceae family and a causative agent of the devastating agricultural disease fire blight. Here we characterize eight lytic bacteriophages of E. amylovora that we isolated from the Wasatch front (Utah, United States) that are highly similar to vB_EamM_Ea35-70 which was isolated in Ontario, Canada. With the genome size ranging from 271 to 275 kb, this is a novel jumbo family of bacteriophages. These jumbo bacteriophages were further characterized through genomic and proteomic comparison, mass spectrometry, host range and burst size. Their proteomes are highly unstudied, with over 200 putative proteins with no known homologs. The production of 27 of these putative proteins was confirmed by mass spectrometry analysis. These bacteriophages appear to be most similar to bacteriophages that infect Pseudomonas and Ralstonia rather than Enterobacteriales bacteria by protein similarity, however, we were only able to detect infection of Erwinia and the closely related strains of Pantoea.
Introduction
Whitman et al. (1998) estimated that there are approximately 5 × 1030 bacteria on earth, which is more than the number of plants and animals combined. Most, or likely all, bacteria are subject to infection by one or more viruses or “bacteriophages,” making bacteriophages the most common and diverse biological entity at an estimated 1032 (Bergh et al., 1989; Wommack and Colwell, 2000; Hambly and Suttle, 2005). Bacteriophages were likely first reported in 1896, when Ernest Hanbury Hankin discovered antibacterial activity against cholera in the waters of two large rivers in India, the Ganges and Yamuna (Abedon et al., 2011). They were independently characterized and named in the 1900s by bacteriologist Twort (1915, 1936) and microbiologist D’Herelle (1917, 2007), Keen (2015). During the infection process, bacteriophages can transfer foreign DNA to their host (including virulence factors), integrate into the host genome, and/or kill their host through cell lysis (Chen and Novick, 2009). The sheer number of bacteriophages combined with their clear evolutionary influence makes them an important target for understanding the ecology and evolution of bacteria, including pathogenic strains (Bollback and Huelsenbeck, 2009; Boyd, 2012). In addition, their specificity, genomic plasticity, and rapid multiplication rates make them a potential weapon to treat bacterial infections (Sulakvelidze, 2005; Shivaswamy et al., 2015).
One such bacterial infection caused by a phytopathogen Erwinia amylovora (Zwet and Keil, 1979) is called fire blight and mainly affects ornamental plants of the Rosaceae family. The symptoms of the infected tissues include wilting, ooze production and death of blossoms, shoots branches and entire trees (Thomson, 2000). We have recently isolated and characterized twenty eight bacteriophages that infect E. amylovora (Esplin et al., 2017; Sharma et al., 2018). Out of these 28, there is a distinct group of eight highly related bacteriophages: vB_EamM_Special G (Special G), vB_EamM_Simmy50 (Simmy50), vB_EamM_RAY (RAY), vB_EamM_Deimos-Minion (Deimos-Minion or DM), vB_EamM_Bosolaphorus (Bosolaphorus), vB_EamM_Desertfox (Desertfox), vB_EamM_MadMel (MadMel), and vB_EamM_Mortimer (Mortimer) very similar to Erwinia bacteriophage Ea35-70 which was isolated in Ontario, Canada (Yagubi et al., 2014). These nine bacteriophages were recently added as the Agrican357virus genus of bacteriophages by the ICTV (Kuhn et al., 2013; Adams et al., 2017) and are considered jumbo bacteriophages due to their large genome (>200 kb) and particle size (Yuan and Gao, 2017).
As reviewed in 2017, jumbo bacteriophages have diverse genome sizes (ranging from 208 to 497 kb) as well as diverse virion morphology and complex virion structure (Yuan and Gao, 2017). They often encode greater than 60 structural proteins with some displaying complex head structures composed of more than five proteins (Effantin et al., 2013) or long, wavy, curly tail fibers (Yuan and Gao, 2016). Jumbo bacteriophages were also found to be highly diverse, with over 11 clusters and five singleton bacteriophages suggested from 52 complete jumbo bacteriophage genomes analyzed in 2017, many of which are uncharacterized (Yuan and Gao, 2017). Only a few jumbo bacteriophage families have been characterized beyond sequence analysis and EM, including the phiKZ-like bacteriophages 201phi2-1 (Thomas et al., 2008), KTN4 (Danis-Wlodarczyk et al., 2016), phiPA3 (Monson et al., 2011), phiRSL2 (Bhunchoth et al., 2016), phiRSF1 (Bhunchoth et al., 2016), OBP (Shaburova et al., 2006), EL (Sokolova et al., 2014), and phiKZ (Lecoutere et al., 2009), related bacteriophages phiRSL1 (Yamada et al., 2010) and PaBG (Kurochkina et al., 2018), Cronobacter bacteriophage CR5 (Lee et al., 2016), Prochlorococcus bacteriophage P-SSM2 (Sullivan et al., 2005), related bacteriophages KVP40 (Miller et al., 2003) and Aeh1 (Gibb and Edgell, 2007), Aeromonas bacteriophage phiAS5 (Kim et al., 2012), Pectobacterium bacteriophage CBB (Buttimer et al., 2017), Caulobacter bacteriophage phiCbK (Gill et al., 2012), related Erwinia bacteriophages Joad and RisingSun (Arens et al., 2018), related bacteriophage RaK2 (Simoliunas et al., 2013) and GAP32 (Abbasifar et al., 2014), Bacillus bacteriophage 0305phi8-36 (Thomas et al., 2007), related Bacillus bacteriophages BpSp (Yuan and Gao, 2016) and AR9 (Lavysh et al., 2016, 2017). Herein we further analyze the genome, proteome, and host range of our eight Agrican357virus jumbo bacteriophages. Their lytic nature and plethora of novel genes makes them a unique entity to be studied further and analyzed. As a close relative of the animal pathogens Escherichia coli and Salmonella (Zhao and Qi, 2011), viruses that infect E. amylovora may help us understand the evolution of pathogenic strains in this family.
Materials and Methods
Bacteriophage Isolation and Genome Sequencing
Environmental samples of leaves, branches and soil surrounding infected trees were collected from around the state of Utah (United States) and used to create enrichment cultures with the host E. amylovora. To test the presence of amplified bacteriophages, the enrichment cultures were spun at 4000 rpm and 4°C for 20 min and the supernatant was removed and used without filtering. 50 μL of this supernatant was incubated at room temperature with 500 μL of E. amylovora ATCC 29780 bacteria for 30–45 min, mixed with 5 ml NBDYE top agar (at half concentration agar), plated on NBSYE agar plate, and incubated at 25°C overnight. Plaque presence on the plates was the primary indicator of bacteriophage presence in the environmental sample. Using a sterile needle or pipette tip, we picked a plaque from the initial bacteriophage identification plate and performed three rounds of plaque purification. All eight isolated bacteriophages: Special G (KU886222), Simmy50 (KU886223), RAY (KU886224), Deimos-Minion (KU886225), Bosolaphorus (MG655267), Desertfox (MG655268), MadMel (MG655269), and Mortimer (MG655270) were able to infect E. amylovora ATCC 29780 (Esplin et al., 2017; Sharma et al., 2018). Bacteriophage DNA was extracted using the Phage DNA isolation kit (Norgen Biotek Corporation), and was sequenced, assembled and annotated as previously described (Esplin et al., 2017; Sharma et al., 2018).
Electron Microscopy
Electron microscopy was performed at Brigham Young University in the Life Sciences Microscopy Lab using a FEI Helios NATOCAB 600i DualBeam FIB/SEM with STEM detector. The samples for SEM analysis were prepared by placing 15 μL of high-titer bacteriophage lysate on a 200-mesh copper carbon type-B electron microscope grid for one-two minutes. The lysate was wicked away and the grids were stained for 2 min using 15 μL of 2% phosphotungstic acid (pH = 7). Residual liquid was wicked away using Kimtech wipes and the grid was allowed to dry before being imaged. Bacteriophage structures in electron micrographs were measured using ImageJ (Abramoff et al., 2004). The average and standard deviation for each measurement was calculated from a minimum of four separate measurements.
Burst Size
Burst size was calculated by performing single-infection assay as described by Delbruck (1945). The bacteria-bacteriophage mixture was allowed to adsorb for 10 min at a multiplicity of infection (MOI) of 100. The lysate was then removed at different time-intervals ranging from 1 to 6 h and diluted to avoid secondary infection. Soft agar plaque method was used to determine titers and a graph of 10 separate readings was plotted with their average titers and time.
Host Range
Host range of all eight bacteriophages was determined using the soft agar plaque method (Hockett and Baltrus, 2017). For this, 50 μL of bacteriophage lysate dilutions were incubated with 500 μL of bacteria grown overnight for 30 min before plating in top agar. The plates were incubated with the top agar facing up at 25°C overnight for this assay. Seventeen bacterial strains including E. amylovora ATCC 29780 (Ritchie and Klos, 1976) as control were used including five other E. amylovora strains [Ea110 (Ritchie and Klos, 1976), GH9 (McGhee and Sundin, 2012), EaBH (McGhee and Sundin, 2012), RB02 (McGhee and Sundin, 2012), Ea273 (Sebaihia et al., 2010)], Pantoea agglomerans E325 (Pusey, 1997), Pantoea vagans C-91 (Ishimaru et al., 1987; Smits et al., 2010), E. coli K-12 BW 25113 (Datsenko and Wanner, 2000), Salmonella enterica LT2 (generously donated by John Roth lab), Yersinia pestis KIM6 (Fetherston et al., 1992; Gong et al., 2001), Enterobacter cloacae ATCC 13047 (Ren et al., 2010), Klebsiella pneumoniae ATCC 10031 (Otman et al., 2007), Bacillus subtilis ATCC 6033 (Nakamura et al., 1999), Cronobacter sakazakii ATCC 29544 (Iversen et al., 2007; Gicova et al., 2014), standard clinical isolate Pseudomonas aeruginosa PA100 (Cartagena et al., 2007) and Pseudomonas chlororaphis ATCC 13985 (Conway et al., 1956). An average of two readings was taken to obtain bacteriophage titers post-infection.
Computational Analysis and Genomic Comparison
Bacteriophages with any similarities to Agrican357virus genus were identified using a blastx analysis of their putative major capsid and terminase proteins, and the corresponding bacteriophage for all retrieved hits with a cutoff e-value of less than 1.00E-04 and 33% similarity were downloaded from GenBank. In addition, any bacteriophages that showed up in at least three qblast hits while annotating were also retrieved. These sequences were then used in Gepard (Krumsiek et al., 2007) to generate the dot plots of nucleic acid and protein sequences. PhamDB, a web interface (Lamine et al., 2016) was used for creating databases and Phamerator, (Cresawn et al., 2011) an open-source program was used to generate pham map of bacteriophage genomes. PhamDB uses kClust (Hauser et al., 2013) to cluster large protein sequence databases. The default settings of PhamDB were used in this comparison. Splitstree (Huson and Bryant, 2006) protein analysis was produced from the exported pham table of conserved proteins converted to a Nexus file using Janus1. The Average Nucleotide Identity (ANI) percentages comparing each of the E. amylovora bacteriophage genomes were calculated using MAFFT (Katoh et al., 2002) plugin in Geneious R8.1 (Kearse et al., 2012).
The evolutionary history was inferred by using the Maximum Likelihood method and Poisson correction model (Zuckerkandl and Pauling, 1965). The bootstrap consensus tree inferred from 100 replicates (Felsenstein, 1985) is taken to represent the evolutionary history of the taxa analyzed (Felsenstein, 1985). Branches corresponding to partitions reproduced in less than 50% bootstrap replicates are collapsed. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (100 replicates) are shown next to the branches (Felsenstein, 1985). Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model, and then selecting the topology with superior log likelihood value. This analysis involved 59 amino acid sequences. There were a total of 1302 positions in the final dataset. Evolutionary analyses were conducted in MEGA X (Kumar et al., 2018).
Mass Spectrometry
Sample preparation was performed (Guttman et al., 2009) by diluting crude lysates of RAY and Deimos-Minion in TNE (50 mM Tris pH 8.0, 100 mM NaCl, 1 mM EDTA) buffer and adding RapiGest SF reagent (Waters Corp.) to a final concentration of 0.1%. Samples were then boiled for 5 min followed by addition of 1 mM (final concentration) of TCEP [Tris (2-carboxyethyl) phosphine] and incubated at 37°C for 30 min. Afterward, carboxymethylation of samples was done with 0.5 mg/ml of iodoacetamide for 30 min at 37°C followed by neutralization with 2 mM TCEP (final concentration). Trypsin (trypsin: protein ratio – 1:50) was used overnight at 37°C to digest the crude lysates prepared as above. The samples were treated with 250 mM HCl at 37°C for 1 h followed by centrifugation at 14000 rpm for 30 min at 4°C to degrade and remove RapiGest. The soluble fraction was then added to a new tube and Aspire RP30 desalting columns (Thermo Fisher Scientific) were used for extraction and desalting of the peptides.
High pressure liquid chromatography (HPLC) coupled with tandem mass spectroscopy (LC-MS/MS) using nano spray ionization was used to analyze Trypsin-digested peptides (McCormack et al., 1997). A TripleT of 5600 hybrid mass spectrometer (ABSCIEX) interfaced with nano-scale reversed-phase HPLC (Tempo) using a 10 cm-100-micron ID glass capillary packed with 5-μm C18 ZorbaxTM beads (Agilent Technologies, Santa Clara, CA) was used to perform the nano-spray ionization experiments. By using a linear gradient (5–60%) of ACN (Acetonitrile) at a flow rate of 250 μl/min for 1 h, peptides were eluted from the C18 column into the mass spectrometer The ACN gradient was created using these buffers: buffer A (98% H2O, 2% ACN, 0.2% formic acid, and 0.005% TFA) and buffer B (100% ACN, 0.2% formic acid, and 0.005% TFA). In a data-dependent manner MS/MS data were acquired in which the MS1 data was acquired for 250 ms at m/z of 400 to 1250 Da and the MS/MS data was acquired from m/z of 50 to 2,000 Da. For Independent data acquisition (IDA) parameters MS1-TOF 250 ms, followed by 50 MS2 events of 25 ms each. The IDA criteria; over 200 counts threshold, charge state of plus 2–4 with 4 s exclusion window. Finally, MASCOT® (Matrix Sciences) was used to analyze the collected data and Protein Pilot 4.0 (ABSCIEX) was used for peptide identifications.
Extracellular Polymeric Substance (EPS) Depolymerase Mediated Biofilm Degradation Assay
Soft agar plaque method (Hockett and Baltrus, 2017), as described previously in host range method, was used to detect the presence of halo zone on P. vagans strain C9-1 and E. amylovora ATCC 29780. The putative EPS-depolymerase from bacteriophage RAY was PCR amplified from lysate using primers designed to amplify the full length gp76. It was cloned by digesting with enzymes NdeI/SalI into a similarly digested pET15b. The resulting plasmid (JG1700) was amplified by transforming into E. coli DH5α and plated on LB-amp. Resulting colonies were PCR checked and were used to start overnight cultures and DH5α without plasmid pJG1700 was grown as a control. The protein was induced using IPTG and extracted by lysing cells via sonication. Post-sonication, cell debris was removed from both cultures by centrifuging at 12000 rpm and 4°C for 2 × 20 min. 10 μl of resulting supernatant was spotted on bacterial lawns of P. vagans strain C9-1 and E. amylovora ATCC 29780 embedded in top agar after plating for 2 h.
Motif Identification and Analysis
MEME (Bailey et al., 2009) and FIMO (Grant et al., 2011) tools at public phage galaxy2 were used to scan bacteriophage genome of Agrican357virus for statistically significant motifs. Motifs found by MEME (Bailey et al., 2009) with e-value less than 1.00E-02 were selected by FIMO (Grant et al., 2011) to be searched for their coordinates and iterations in their respective genomes. User defined cut-off values (P-value <1.00E-03, Q-value <0.05), as described in Berg et al. (2016) were used to maximize the results. The location of the motifs within bacteriophage genomes was determined from the annotated GenBank files (Esplin et al., 2017).
Results and Discussion
Isolation and Characterization of Eight Closely Related Large Bacteriophages Infecting E. amylovora
Eight novel bacteriophages (Deimos-Minion, Special G, RAY, Simmy50, Bosolaphorus, Desertfox, Mortimer, and MadMel) that infect E. amylovora were plaque isolated and their genomes were subsequently sequenced and annotated as previously described (Esplin et al., 2017; Sharma et al., 2018). All eight bacteriophages have relatively large genomes with genome sizes of 271 to 275 kb (Table 1), which are comparable to the related bacteriophage Ea35-70 (271084 bp). These bacteriophages have correspondingly large putative proteomes, with 317 to 324 predicted ORFs. A search for tRNA’s using tRNA ScanSE (Lowe and Chan, 2016) suggests that RAY, Simmy50, Bosolaphorus, and Mortimer have 1 tRNA each coding for Asparagine, whereas no tRNA’s were detected for Deimos-Minion, Special G, MadMel, and Desertfox. No lysogeny related genes were identified (including integrase, excisionase or repressors). Their clear plaque morphology and ease in obtaining higher titers (∼108–1010 pfu/ml) suggest they may be lytic bacteriophages, however, rigorous testing for bacterial lysogeny has not been performed.
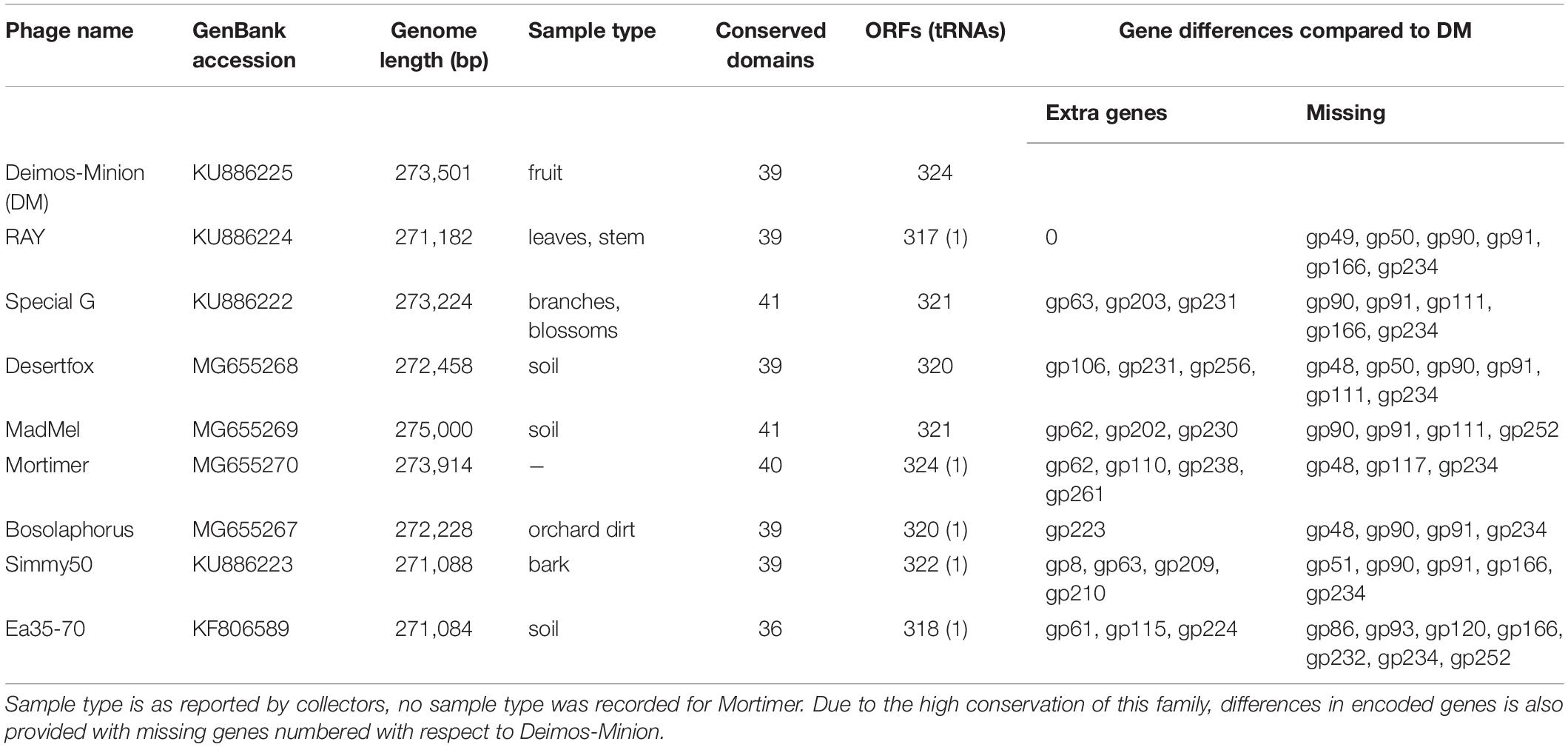
Table 1. General characteristics of related bacteriophages Deimos-Minion (DM), RAY, Special G, Desertfox, MadMel, Mortimer, Bosolaphorus, Simmy50, and Ea35-70 that infect E. amylovora ATCC 29780.
Electron Microscopy Reveals Myovirus Structure for Five E. amylovora Bacteriophages
Deimos-Minion, Special G, RAY, Simmy50, Mortimer, MadMel, Desertfox, and Bosolaphorus were all found to be similarly sized Myoviridae, having contractile tails (average size 159 + −11.4 nm), a tail sheath (average size 78.5 + −9.28 nm), visible tail fibers, and large capsids (average size 128 + −5.96 nm) (Figure 1). This morphology is supported by their protein-based relationships to other jumbo Myoviridae discussed below. Due to apparent similarity within these bacteriophages, only RAY’s morphological calculations are listed but all eight of these bacteriophages were imaged extensively.
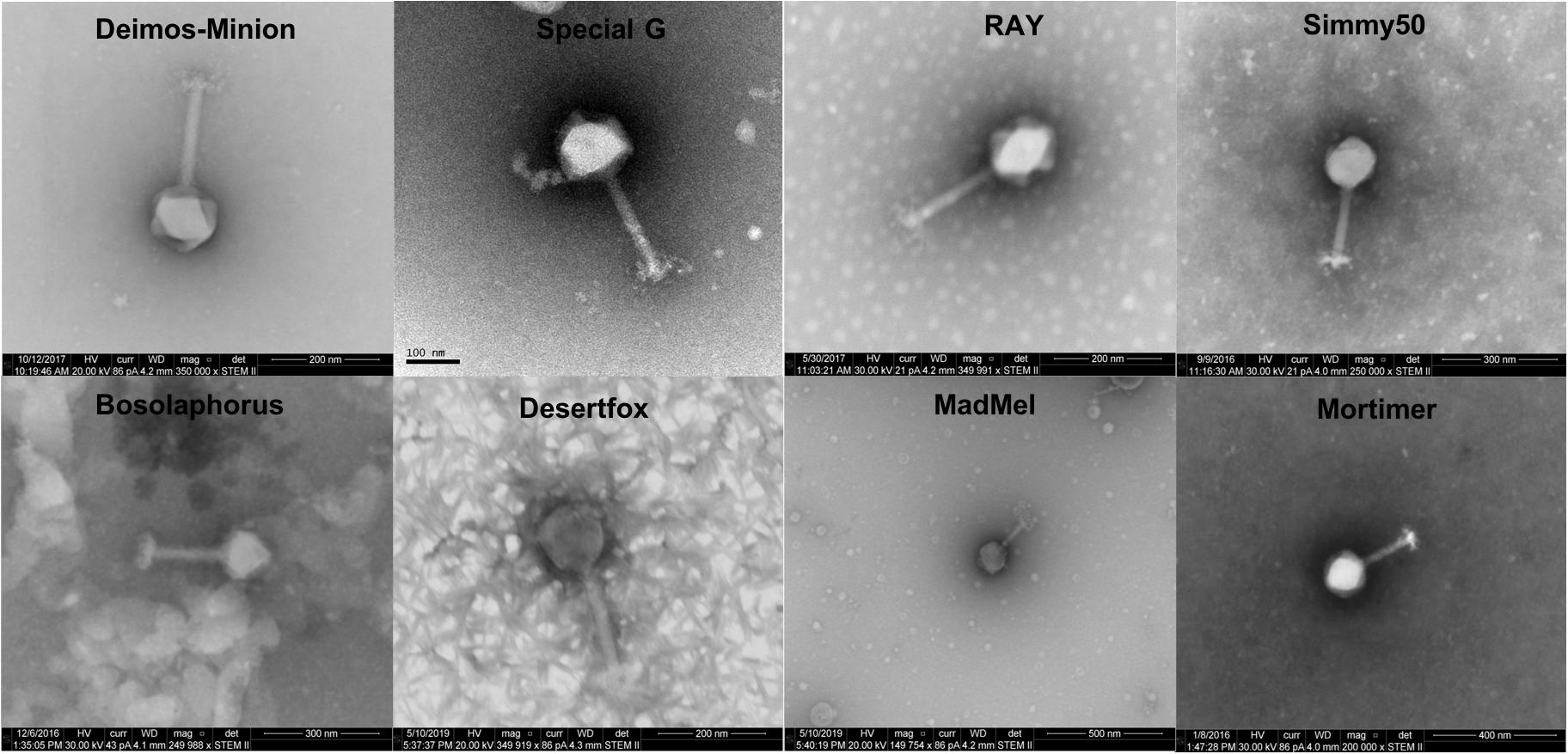
Figure 1. Electron microscopy STEM images of Deimos-Minion, Special G, RAY, Simmy50, Bosolaphorus, Desertfox, MadMel, and Mortimer revealed Myoviruses with long contractile tails.
Host Range and Burst Size
Bacteriophages of the Agrican357virus family were tested for activity against seventeen different bacterial strains. Out of these, fifteen were from the Enterobacteriales- P. agglomerans E325 (Pusey, 1997), P. vagans C-91 (Ishimaru et al., 1987; Smits et al., 2010), E. coli K-12 BW 25113 (Datsenko and Wanner, 2000), S. enterica (generous donation by roth lab), Y. pestis KIM6 (Fetherston et al., 1992; Gong et al., 2001), E. cloacae ATCC 13047 (Ren et al., 2010), K. pneumoniae ATCC 10031 (Otman et al., 2007), B. subtilis ATCC 6033 (Nakamura et al., 1999), C. sakazakii ATCC 29544 (Iversen et al., 2007; Gicova et al., 2014), E. amylovora Ea110 (Ritchie and Klos, 1976), E. amylovora GH9 (McGhee and Sundin, 2012), E. amylovora EaBH (McGhee and Sundin, 2012), E. amylovora RB02 (McGhee and Sundin, 2012), E. amylovora Ea273 (Sebaihia et al., 2010), E. amylovora ATCC 29780 (control) (Ritchie and Klos, 1976), and two from Pseudomonadaceae- P. aeruginosa PA100 (Cartagena et al., 2007) and P. chlororaphis ATCC 13985 (Conway et al., 1956). Enterobacteriales strains were chosen due to being members of the same bacterial order as Erwinia, whereas Pseudomonadaceae strains were the hosts of bacteriophages related to the Agrican357virus bacteriophages based on protein BLAST. Our current analyses displayed that Agrican357virus bacteriophages infect all Erwinia strains (with the exception of Special G and Mortimer that failed to infect GH9 and EaBH, respectively) as well as closely related genera also commonly found on plants– P. agglomerans (Deletoile et al., 2009) and P. vagans (Palmer et al., 2016; Table 2).
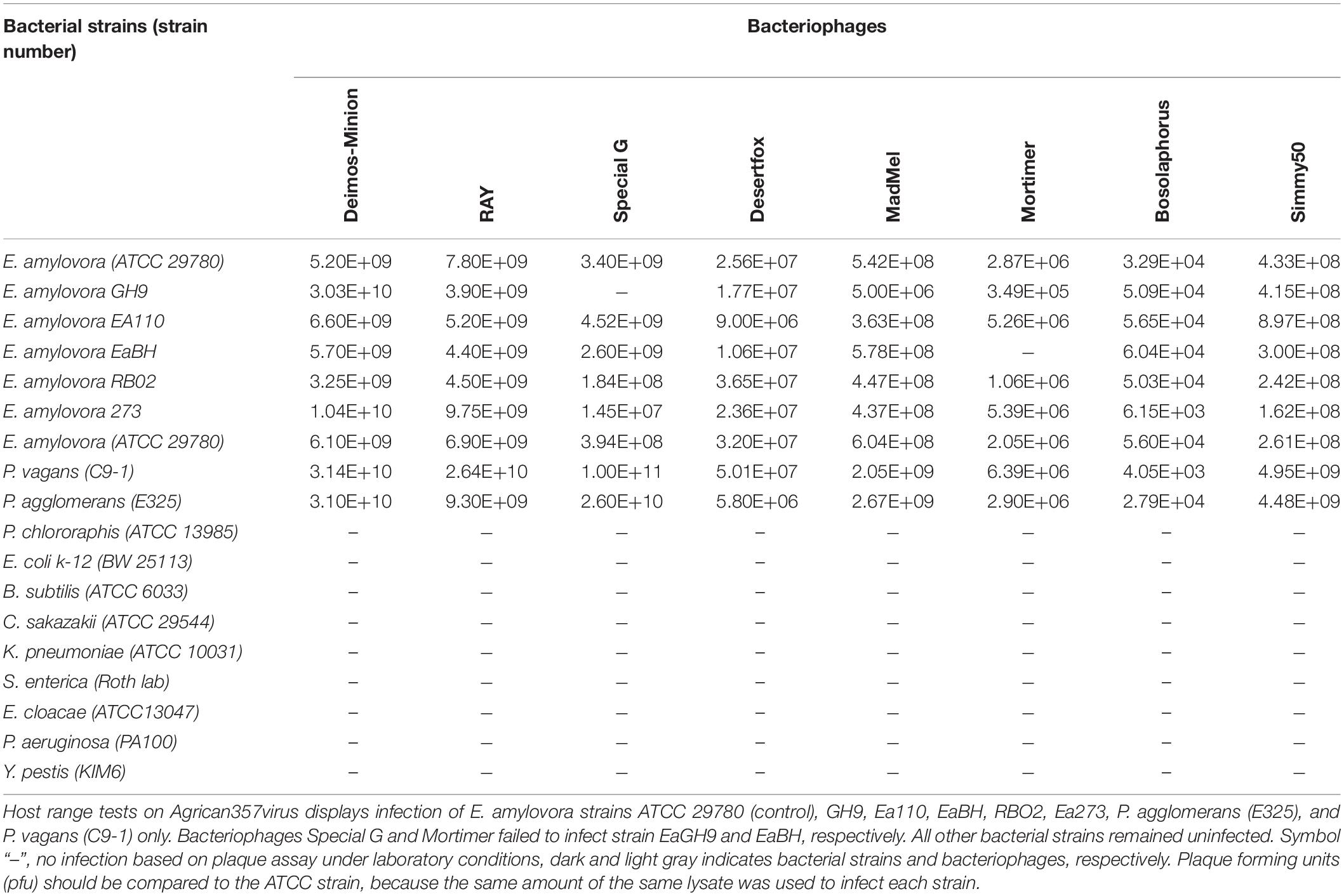
Table 2. Host range analysis of eight Agrican357virus bacteriophages.
Owing to the large nature of Agrican357virus bacteriophages, we investigated the burst size of bacteriophage Deimos-Minion on E. amylovora strain ATCC 29780. Burst size studies suggested that when infected at MOI of 100 Deimos-Minion has burst size of 4.6–4.9 with latent period of 3–4 h before the first burst (Figure 2) under the laboratory growth conditions used herein, consistent with their large size. As seen in Figure 2, a second burst is appearing at the end of this 6 h period. The observed burst size (∼5) was confirmed with bacteriophage RAY (data not shown) and is consistent with other large Myoviridae in that P. aeruginosa bacteriophage KTN4 has a reported burst of 6–8 and may be due to the need to build internal cellular structures for the Jumbo viruses to be built (Danis-Wlodarczyk et al., 2016), or due to sub-optimal assay conditions for proliferation.
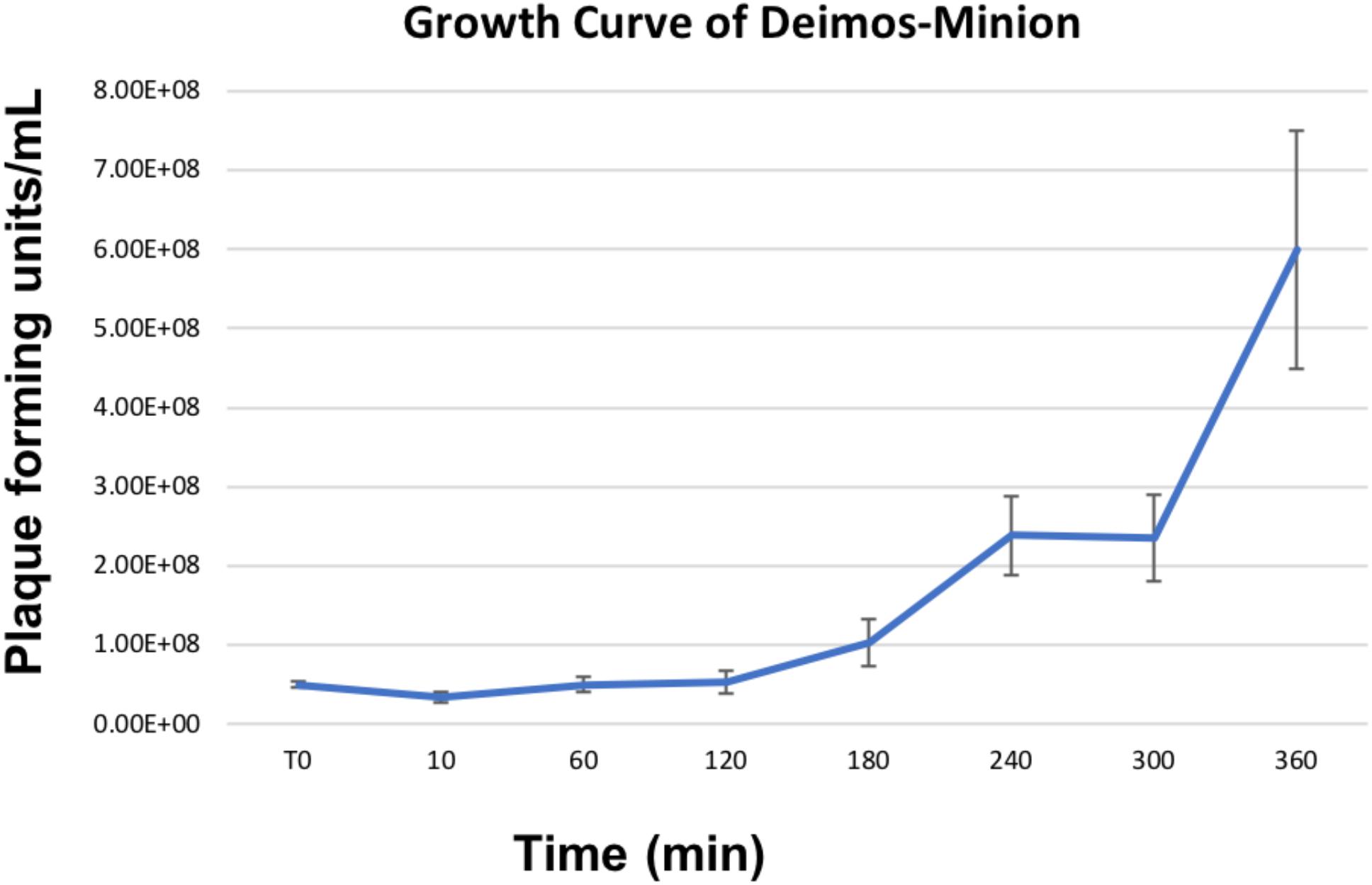
Figure 2. Growth Curve of Deimos-Minion with host E. amylovora ATCC 29780 by plaque assays shows first burst at ∼4 h and second burst at ∼6 h.
Genomic and Evolutionary Characteristics
To determine the overall genomic and proteomic similarity of our eight novel bacteriophages to available bacteriophages in GenBank, related bacteriophages were identified by BLAST (qblast) using each of the putative gene products encoded by RAY. The bacteriophages with e-values below 1.00E-04 and above 33% identity that were identified in three or more BLAST searches were then compared using Gepard dot plot (Krumsiek et al., 2007), average nucleotide identity (ANI analysis) (Lassmann and Sonnhammer, 2005), and BLAST alignment (Altschul et al., 1990). Dot plots were constructed using whole genome sequences, major capsid protein amino acid, and terminase amino acid sequences (Figures 3A–C, respectively). While looking at the results of the whole genome dot plot, all eight of our bacteriophages show no similarity to any other bacteriophages used in the dot plot except for very close similarity to Ea35-70 (KF806589) (Yagubi et al., 2014), an Erwinia bacteriophage isolated in Canada in 2014 (see Figure 3A). In addition, their average nucleotide identity (ANI) using Geneious (Kearse et al., 2012) was remarkably high >94% (see Supplementary Table S1). These results indicate that these eight bacteriophages Deimos-Minion, Simmy50, RAY, Special G, Bosolaphorus, Desertfox, MadMel, and Mortimer along with Ea35-70 make a distinct family of bacteriophages, consistent with the International committee on taxonomy of viruses’ classification as new species of a new genus Agrican357virus in the family Myoviridae of order Caudovirales (Adams et al., 2017).
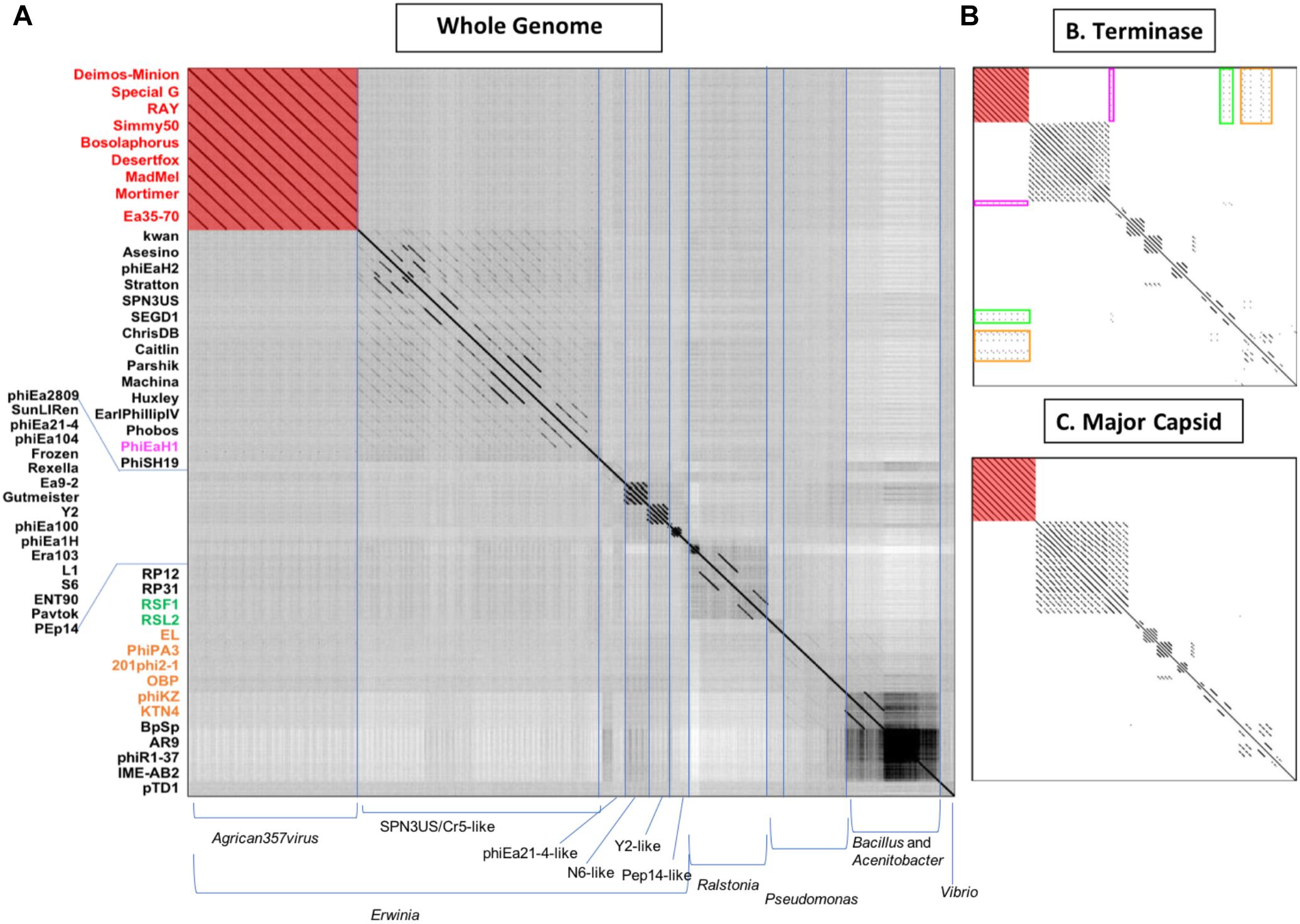
Figure 3. Whole-genome nucleotide (A) and protein terminase (B) or major capsid protein (C) dot plot analysis reveals a fairly isolated cluster of bacteriophages that includes Deimos-Minion, Special G, RAY, Simmy50, Bosolaphorus, Desertfox, MadMel, Mortimer and Ea35-70. Dot plots were constructed using Gepard.
The major capsid protein (MCP) and terminase proteins are two of the most conserved proteins in bacteriophage genomes and have been used to group bacteriophages in families by single gene analysis (Smith et al., 2013). In order to identify distant bacteriophage relatives, a proteomic comparison of these bacteriophages was performed using terminases (see Figure 3B) and MCPs (see Figure 3C) by Gepard dot plot (Krumsiek et al., 2007). The same bacteriophage order from the whole genome dot plot (Figure 3A) was used in these dot plots. Whole genome and terminase dot plots both displayed limited synteny between Agrican357virus bacteriophages and Erwinia bacteriophage phiEaH1 (4.00E-155 from blastp of terminase) indicating this bacteriophage as the closest known relative from Erwiniaceae. In contrast, little similarity to Pseudomonas bacteriophages phiKZ (8.00E-156), KTN4 (8.00E-156), phiPA3 (2.00E-149), 201phi2-1 (5.00E-140), OBP (2.00E-101), EL (3.00E-77), and Ralstonia bacteriophages RSF1 (9.00E-122) and RSL2 (3.00E-120) can be seen in the terminase dot plot which was not apparent in the whole genome and major capsid protein dot plots. All of these bacteriophages are distantly related jumbo Myoviridae.
The two subunits of terminase protein; large and small, are an essential part of DNA packaging (Kuebler and Rao, 1998; Mesyanzhinov et al., 2002). All eight of our Agrican357virus bacteriophages have a putative terminase gene with identical amino acid sequences: Deimos-Minion gp189, Special G gp185, RAY gp183, Simmy50 gp186, Desertfox gp184, Bosolaphorus gp185, MadMel gp185, and Mortimer gp188. This protein is also present in Ea35-70 gp181. This indicates that it is a highly conserved protein for this family. Considering the similarity between these bacteriophages, it can be inferred that all nine bacteriophages of Agrican357virus may have headful packaging (Figure 4). In support of this conclusion, blastp results demonstrated a match with Pseudomonas bacteriophage phiKZ with an e-value of 8.00E-156, a terminase large subunit from Erwinia bacteriophage PhiEaH2 with an e-value of 6.00E-122 and a terminase large subunit of Pseudomonas bacteriophage 201phi2-1 with an e-value of 5.00E-140. Bacteriophages phiKZ, phiEaH2 and 201phi2-1 are all known to have headful packaging (Merrill et al., 2016). In addition to blastp, bacteriophage termini and packaging mode for six bacteriophages (excluding Deimos-Minion and Special G) was also determined using randomly fragmented next-generation sequencing (NGS) data with the help of software PhageTerm (Garneau et al., 2017)3. PhageTerm analysis indicated that RAY, MadMel, Desertfox, Bosolaphorus, Simmy50, and Mortimer have headful packaging without a pac site. Thus, the headful packaging strategy is supported by terminase homology and NGS sequencing data.
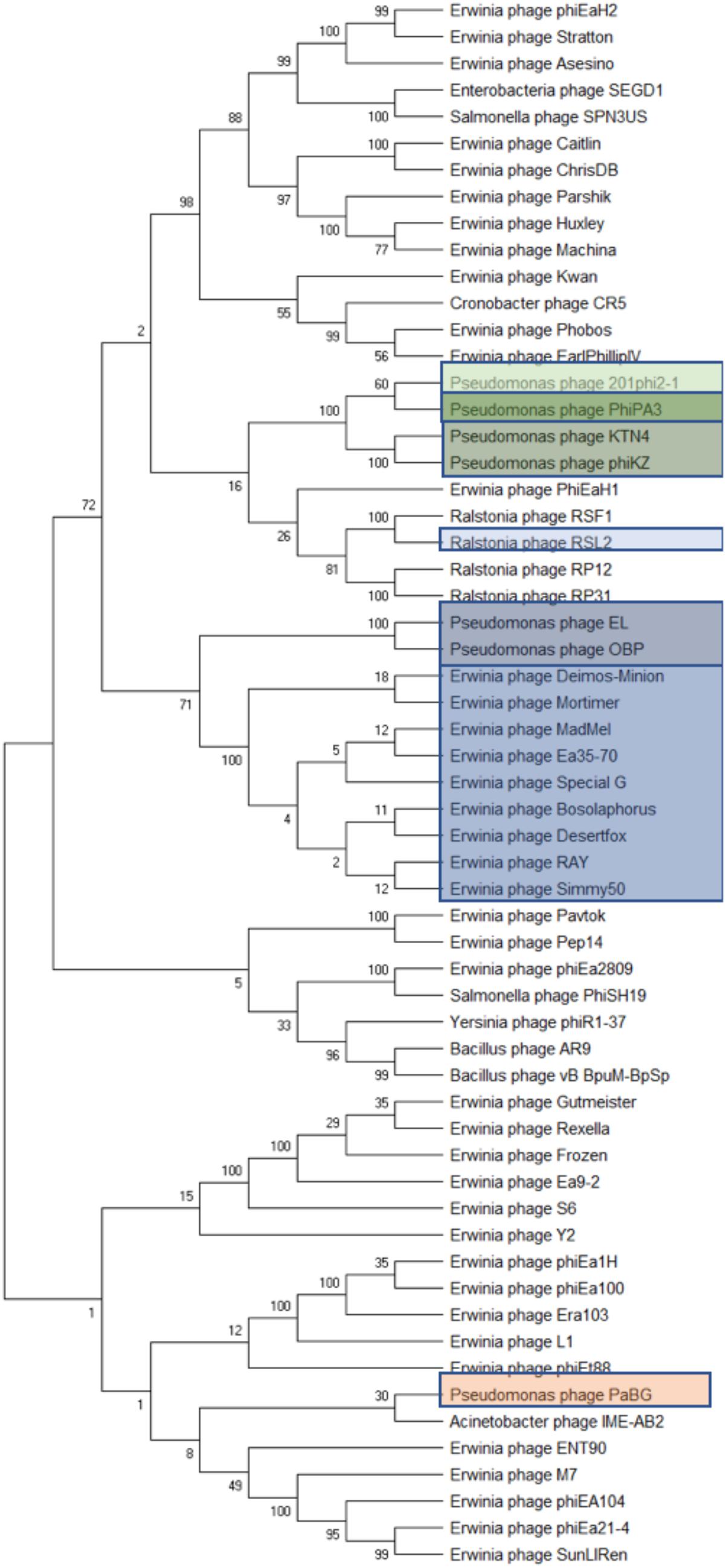
Figure 4. Phylogenetic analyses of phage terminase proteins supports the relationships depicted by dot plot analysis of the Agrican357virus bacteriophages. The evolutionary history was inferred using the Maximum Likelihood method and Poisson correction model in MEGA X. Branches corresponding to partitions reproduced in less than 50% bootstrap replicates are collapsed where 100 was set to be initial bootstrapping value.
Proteomic Analysis of the Agrican357virus Family
Due to great similarity between these bacteriophages we randomly chose RAY as a representative for the protein classification. Proteomic analysis of RAY reveals the novel nature of these bacteriophages in that of 318 proteins, 202 proteins were considered to be novel with no BLAST hit (the e-value cutoff was <1.00E-04), 50 were hypothetical proteins with BLAST hits, and 67 were proteins with a putative function based on their BLAST hit (Supplementary Figure S1A). Thus, over half of the proteins had no BLAST hit outside of the Agrican357virus bacteriophages. These proteins represent a considerable proteomic “dark matter” (Hatfull, 2015), and underscore the vast biological richness harbored in bacteriophages. Of the 67 proteins with predicted function, a majority appear to be structural proteins (∼41%), and DNA metabolism proteins (approximately 41%) (Supplementary Figure S1B).
The computer program Phamerator (Cresawn et al., 2011) was used to compare the entire genomes of the nine Agrican357virus bacteriophages that infect E. amylovora: Deimos-Minion, Special G, RAY, Simmy50, Bosolaphorus, Desertfox, MadMel, Mortimer, and Ea35-70 (Figure 5). Despite their large size, these genomes display remarkable nucleotide sequence and proteomic conservation (>94% ANI, see Supplementary Table S1). The genomes encode recognizable structural and enzymatic bacteriophage proteins vital to the bacteriophage life cycle, including terminase proteins, major capsid proteins, and tail fiber proteins as well as proteins involved in DNA transcription and translation, such as helicase proteins, DNA polymerase, and RNA polymerase. Though the genomes of these nine bacteriophages are virtually identical, a few genes are differentially present across these bacteriophage genomes. Most of these are hypothetical proteins, however, HNH endonucleases also differed consistently between the Agrican357virus bacteriophages. HNH endonucleases are proteins that splice DNA and assist in the movement of introns and other intron-like sequences (Chevalier and Stoddard, 2001). Deimos-Minion has two such HNH endonucleases, gp93 and gp234 that do not appear to be homologs based on protein similarity. Protein BLAST results of gp93 show that the HNH endonuclease is also found in bacteriophages Bosolaphorus, Desertfox, MadMel, RAY, Simmy50, Special G and Ea35-70, and is similar to those found in some Pseudomonas bacteriophages (phiKZ and KTN4) as well as both Gram-negative and Gram-positive strains of bacteria. However, only the HNH endonuclease domain (∼amino acid 58-109 of bp93) is primarily conserved, the remaining 278 amino acid protein is not conserved in bacteria. On the other hand, homologs of HNH endonuclease gp234 are only found in Deimos-Minion and MadMel, as well as several Gram-positive and Gram-negative bacteria. Genomes of Deimos-Minion, Desertfox and MadMel also displayed a reversed order of two proteins (gp93-gp94 in Deimos-Minion, gp88-gp89 in Desertfox and gp90-gp91 in MadMel) when compared to similar proteins in other bacteriophages of this family. The proteins involved are HNH endonuclease and ribonucleotide reductase. To search for repetitive sequences in the genome which may be involved in recombination, MEME (Bailey et al., 2009) and FIMO (Grant et al., 2011) were used to locate motifs in the genomes of all eight of our Agrican357virus bacteriophages. Several common and unique motifs were discovered, however, they had poor e-values with little or no significance and were not followed further.

Figure 5. Whole genome Phamerator map of E. amylovora bacteriophages illustrates the high similarity of bacteriophages Mortimer, MadMel, Desertfox, Bosolaphorus, Deimos-Minion, Special G, RAY, Simmy50, and Ea35-70. Bacteriophages were mapped using Phamerator and arranged based on highest protein similarity. Violet shading between genomes indicates genome nucleotide homology (with standard e-value cutoff of 1.00E-04) and the ruler indicates genome base pairs, while white spaces indicate areas without significant nucleotide similarity. Boxes above and below the genome ruler indicate ORFs going in the forward and reverse direction, respectively. They are labeled with predicted function, occasionally numbered, and colored to indicate protein homologs between the bacteriophages.
Due to the large size of these bacteriophages, and their terminase similarity to bacteriophage phiKZ, these bacteriophages likely belong to the jumbo bacteriophages (Hertveldt et al., 2005; Yuan and Gao, 2017) making it no surprise that the structural proteins are found in other bacteriophages. Along with hypothetical proteins, the proteins that are conserved with other phiKZ-like jumbo bacteriophages include: RNA polymerase beta subunit, nuclease RtcB-like, SbcC like, helicase, virion structural proteins, tail fiber, tail sheath, lysozyme domain, terminase, and major capsid protein. A splitstree analysis showing the relationship of the related jumbo bacteriophages by protein conservation is displayed in Figure 6. This protein-based tree suggests seven groups of related jumbo Myoviridae bacteriophages, with the Agrican357virus group as the most distant group. It further confirms that proteins of Agrican357virus family are more similar to proteins from Pseudomonas bacteriophages EL and OBP and Ralstonia bacteriophage RSL2 than to other Enterobacteriales bacteriophages.
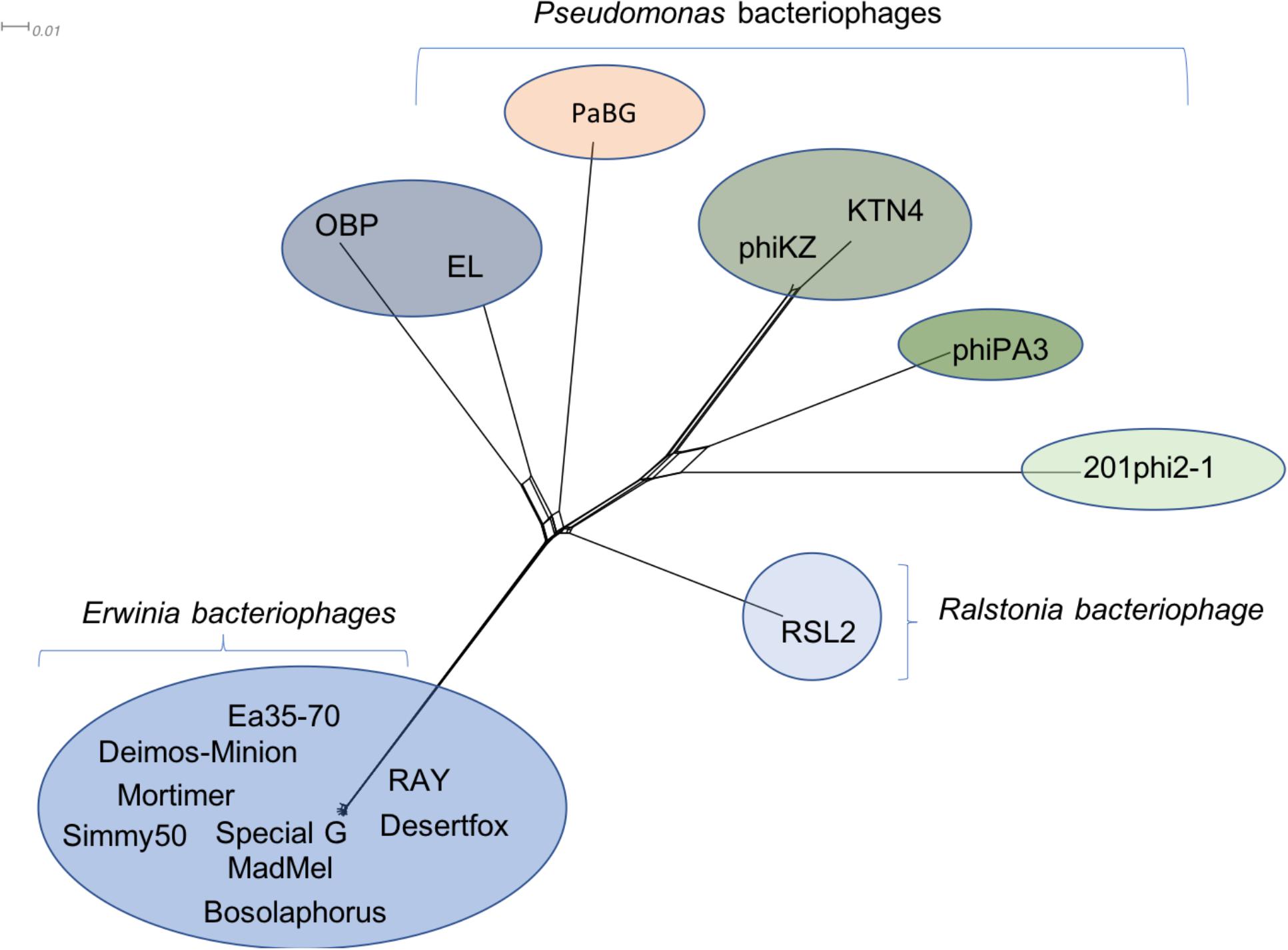
Figure 6. Protein-conservation analysis displayed by Splitstree of the Agrican357virus genus with related jumbo Myoviridae bacteriophages reveals Agrican357virus as a distant evolutionary group.
Mass Spectrometry Validates 27 Hypothetical Proteins as Proteins of Unknown Function
Further analysis of Deimos-Minion and RAY genomes via mass spectrometry (MS) detected several novel proteins, promoting the status of 27 proteins from hypothetical proteins to proteins of unknown function. In RAY and Deimos-Minion genomes collectively, MS analysis identified seventeen proteins with a putative function, eighteen novel hypothetical proteins specific to this bacteriophage family and nine hypothetical proteins (seven known bacteriophage proteins and two other) with blastp hits to other bacteriophages (Table 3). The majority of proteins found through MS are novel hypothetical proteins found only in this family, followed by putative bacteriophage structural proteins, hypothetical bacteriophage proteins, proteins with putative functions and other hypothetical proteins (see Table 3). This analysis agrees with our predicted conservation of proteins depicted through Phamerator analyses.
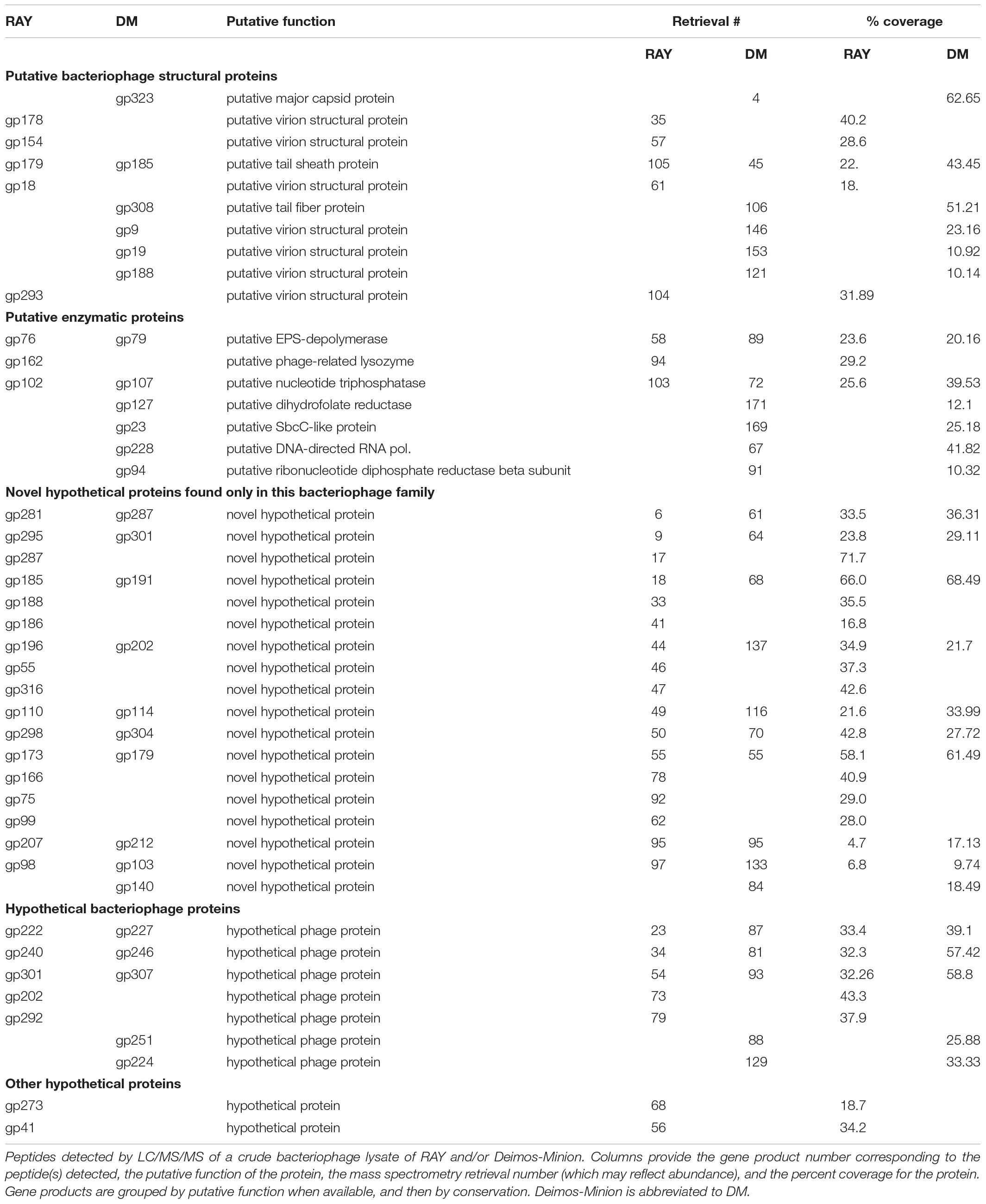
Table 3. Mass Spectrometry reveals 27 Hypothetical Proteins as Proteins of Unknown Function.
Biofilm Degradation (EPS) Assays Suggest Specificity for Pantoea
Enzymatic proteins like extracellular polysaccharide (EPS) depolymerase and phage-related lysozyme are few of the annotated proteins with putative functions which were also predicted via mass spectrometry. EPS depolymerase (Kim and Geider, 2000) is an enzyme that degrades EPS and phage-related lysozyme is shown to lyse the bacterial cell wall (Emrich and Streisinger, 1968). It has been shown that halo formation on the host could be a result of biofilm degradation activity (Cornelissen et al., 2012; Majkowska-Skrobek et al., 2016). The presence of halo zone after in infections of Agrican357virus family was first observed on P. vagans strain C9-1 (Figure 7A). To investigate further the EPS- depolymerase gene was cloned into a plasmid pJG1700, amplified using E. coli DH5α, and spotted on P. vagans stain C-91 and E. amylovora strain ATCC 29780 (Figure 7B). Lysate from a similarly grown and prepared DH5α culture was used as a control. The clearing is indicative of EPS depolymerase activity on P. vagans. This activity was not seen on E. amylovora ATCC 29780.
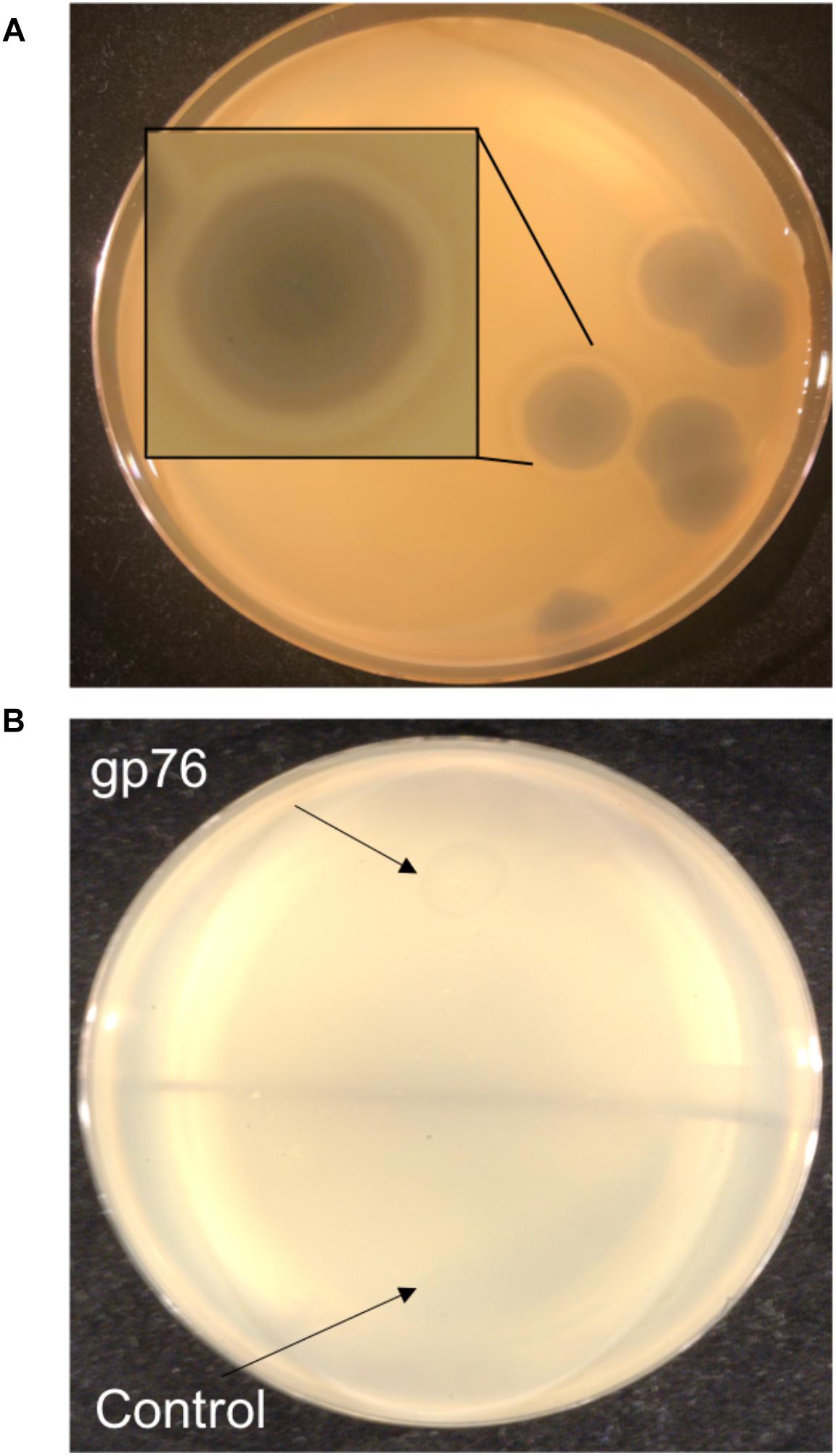
Figure 7. Halo formation on P. vagans by RAY (A) and bio-film degradation activity of gp76 on P. vagans (B).
Structural Prediction Supports the Putative Function of Several Proteins
To further understand Agrican357virus and verify their protein functions, we studied proteins involved in DNA metabolism (∼45%), the largest group of functional proteins conserved in the Agrican357virus. Multiple mechanisms for DNA regulation and repair are evident with the presence of proteins that are hypothesized to aid DNA synthesis, repair, and recombination. These proteins may increase the stability and survival of these jumbo bacteriophages (Supplementary Table S2 and Supplementary Figure S2). In order to proliferate in host cells, bacteriophages need to be equipped with proteins that allow them to reproduce effectively. Although many bacteriophages harbor proteins for DNA damage repair and DNA reproduction inside a host bacteria cell, these large bacteriophages may require extremely viable progeny due to lower burst sizes (∼4.6 functional virions compared to thousands reported for other bacteriophages).
Two proteins with a conserved domain found in the nine Agrican357virus bacteriophages are SbcC and a SbcCD nuclease (see Supplementary Figure S2A). The ability of SbcC and SbcCD to regulate and repair DNA has been shown to be essential for the stability and proliferation of some bacteriophages (Connelly et al., 1998). During DNA replication, palindromic sequences will create hairpin-like structures that can inhibit the progression of DNA polymerase (Leach et al., 1997). SbcC and SbcCD proteins work together to cleave both double- stranded and single-stranded DNA, and have been shown to recognize and specifically cleave hairpin structures. This breaks down the replication fork, allowing the genome to be repaired through recombination, so replication can proceed (Leach et al., 1997; Connelly et al., 1998). The proteins SbcC and SbcCD nucleases preserve the viability of the genome by allowing replication without excising the palindromic sequences (Leach et al., 1997). There are many types of DNA damage that may occur within a genome, making recombination and repair of DNA important, such as mutations due to UV damage. UV damage creates kinks or abnormalities within a genome, and prohibits proliferation. Exodeoxyribonuclease VIII breaks double stranded DNA, and degrades a genome on both 5′ ends (Joseph and Kolodner, 1983; Kolodner et al., 1994). This allows the kinked and abnormal portions of a genome to be straightened and repaired through homologous recombination. Additionally, exodeoxyribonuclease VIII does not require ATP to perform DNA repair, enabling repair of the genome even in low-energy environments where the bacteriophage does not have access to ATP (Kolodner et al., 1994). We hypothesize that exodeoxyribonuclease VIII enables the bacteriophages to remain stable despite mutations from UV damage. However, unique from our other predicted structure alignments, the protein from RAY does not match up well with other exodeoxyribonuclease VIII homologs (see Supplementary Figure S2B). It is possible that since these proteins do not have the same protein folding and alignment, they may not have the same function but a related, adapted function.
In the Agrican357virus bacteriophages, there are several encoded proteins with conserved domains of the thymidylate kinase and thymidine kinase (see Supplementary Table S2). Structural prediction and alignment confirm these proteins as likely thymidine kinases (see Supplementary Figures S2C,D), a necessary step due to the distant relationship (low e-values) of Agrican357virus bacteriophage proteins when compare to other biological entities. Thymidine kinase is an enzyme that catalyzes the phosphorylation of thymidine monophosphate (Kokoris and Black, 2002). Thymidylate kinase then catalyzes the phosphorylation of thymidine diphosphate (Doharey et al., 2016), which is an essential precursor for DNA (Chaudhary et al., 2013). Therefore, these proteins are regulatory enzymes that make bacteriophage cell growth and survival possible by aiding proliferation through the synthesis of DNA (Chaudhary et al., 2013; Cui et al., 2013; Doharey et al., 2016). Other proteins shown in Supplementary Figures S2E,F are putative UvsX recombinase and a putative SF2 helicase with conserved helicase domain known as UvsW, which finishes the recombination (Madej et al., 1995; Gajewski et al., 2011). UvsX and UvsW are proteins that have been known to work together to repair broken replication forks through homologous recombination (Kadyrov and Drake, 2004; Maher and Morrical, 2013). Homologous recombination is one of the most efficient ways to have error free DNA repair, and is beneficial to bacteriophages to have this repair mechanism. These repair mechanisms would be important to the bacteriophages because it would not only help repair broken replication forks but it would also help repair damaged or broken DNA (Kadyrov and Drake, 2004; Maher and Morrical, 2013). It has been shown that the absence of UvsX increases UV sensitivity (Kadyrov and Drake, 2004).
Conclusion
Agrican357virus genus of bacteriophages are Myoviridae with dsDNA, large capsids, long contractile tails and high GC content. Their genomes are nearly identical (>94% ANI). All three dot plots (whole genome, major capsid protein, and terminase protein) show no close similarity between the Agrican357virus family and any of the other bacteriophages on NCBI (see Figures 3A–C). We have also found that the Agrican357virus cluster is more closely related to bacteriophages infecting Pseudomonas and Ralstonia, than those infecting E. amylovora. The contrast that we observe between this cluster of bacteriophages and the distantly related bacteriophage analyzed by dot plot contributes valuable information about evolutionary relationships between these other clusters (see Figure 3), suggesting the distant relationship may emphasize the importance of ecological niche, since most other Enterobacteriales bacteriophages isolated infect animal pathogens rather than plant pathogens. It may also, however, simply indicate the abundance of unstudied bacteriophages. The Agrican357virus family of bacteriophages is a novel family, with very low similarity to any other viruses, providing approximately 250 novel proteins to add to the viral dark matter that have no homolog by blastp (Hatfull, 2015). To understand a bacteriophage, it is vital to understand the encoded proteome. A bacteriophage’s proteins determine how effectively it can infect bacteria, and how stable and safe it would be to use in a phage cocktail (a mixture of bacteriophages used together for phage therapy). Of the proteins with predicted function, this family encodes primarily DNA metabolism and repair proteins. Since the bacteriophage host, E. amylovora, is found primarily on the blossoms of fruit trees of the Rosaceae family, these proteins may be particularly vital due to the onset of UV radiation including putative thymidine and thymidylate kinases which aid the production of the nucleotide thymine for DNA synthesis (Doharey et al., 2016), putative SbcC and SbcCD proteins which protect against DNA damage by cleaving harmful hairpin structures during replication (Connelly et al., 1998), putative exodeoxyribonuclease VIII which makes double stranded DNA breaks to help repair DNA damage at low energy (Joseph and Kolodner, 1983), and putative UvsX recombinase and putative SF2 helicase which aid in repair and recombination of DNA (Maher and Morrical, 2013). The small burst size we report herein for these jumbo bacteriophages (∼4.6 functional virions), may require a high level of fidelity to ensure success in the environment.
A paper published in 2003 on evolutionary pathways of P. aeruginosa bacteria demonstrated that phiKZ-like bacteriophages have a very broad host range (Krylov et al., 2003). In 1995, Campbell et al. (Campbell et al., 1995) isolated bacteriophages from barley rhizosphere that infected Pseudomonas spp. other than P. aeruginosa. These bacteriophages displayed great morphological similarity to phiKZ-like bacteriophages despite low genomic similarity (Mesyanzhinov et al., 2002; Krylov et al., 2003; Sokolova et al., 2014). Similarly, Agrican357virus bacteriophages display proteomic similarity to phiKZ-like bacteriophages, particularly with their structural proteins, with little genomic synteny. These results suggest the phiKZ-like bacteriophages are highly divergent, derived from a common ancestor and successful in a wide range of ecological niches. It is highly likely that Agrican357virus family evolved through both mutational divergence and modular evolution (acquisition of larger regions of DNA, or modules), which is a common phenomenon in bacteriophages (Botstein, 1980), and yet there is extremely low variance in all isolates thus far (>94% ANI). Such high conservation in these large genomes may reflect selective forces on a majority of the genome, which is for the most part uncharacterized. The great challenge ahead is both the abundance of bacteriophages that are completely uncharacterized, and the abundance of novel proteins harbored in their genomes.
Data Availability
All datasets generated for this study are included in the manuscript and the Supplementary Files.
Author Contributions
RS designed and wrote the manuscript, performed genomic and proteomic analysis, executed host range, EPS depolymerase cloning, halo zone and burst size experiments, prepared samples for electron microscopy and mass spectrometry, and drew all the figures and tables. KB, BP, and OS performed the structural prediction of proteins and contributed to writing. TN and EY helped with Dotplots. DB contributed with funding and ideas and also edited the manuscript. JG is the corresponding author and principal investigator of this study and as such contributed to the design and execution of experiments, oversaw all students, edited the manuscript, and contributed with funding. All authors read and approved the submitted version.
Funding
DB was awarded a grant by the United States Dairy Association. Financial support by The University of Arizona, Brigham Young University, Department of Microbiology and Molecular Biology, and the College of Life Sciences in the form of student wage funds, support, and publication fees is gratefully acknowledged.
Conflict of Interest Statement
JG is working on licensing a phage-based therapy for fire blight.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors extend a special thanks to the Science Education Alliance – Phage Hunters Advancing Genomics and Evolutionary Science (SEA-PHAGES) for support and training on bacteriophage analysis, Steven Cresawn for training on Phamerator, Majid Ghassemian and BMPMSF at UCSD for carrying out mass spectrometry, George W. Sundin for donating E. amylovora strains, Edward Wilcox from the BYU Sequencing Center, Michael Standing from the BYU Microscopy Lab and Daniel Thompson for helping with phage microscopy, David Parady and Brandon Ekins from Life Science IT at BYU for hosting DNAmaster and PhamDB on BYU server, Tsz Ching Tam for media preparation, Daniel Arens for helping with cloning and Yomesh Sharma for helping with the formatting of the manuscript. The authors also like to thank the following undergraduates from the BYU Phage Hunters’ program for their contributions to isolation of the bacteriophages: Garrett Jensen, Jared Kruger, Madison Melville, Trevon Galbraith, Savannah Grossarth, Hannah Ferguson, and Austin Simister.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01533/full#supplementary-material
Footnotes
References
Abbasifar, R., Griffiths, M. W., Sabour, P. M., Ackermann, H. W., Vandersteegen, K., Lavigne, R., et al. (2014). Supersize me: Cronobacter sakazakii phage Gap32. Virology 460-461, 138–146. doi: 10.1016/j.virol.2014.05.003
Abedon, S. T., Thomas-Abedon, C., Thomas, A., and Mazure, H. (2011). Bacteriophage prehistory: is or is not Hankin, 1896, a phage reference? Bacteriophage 3, 174–178. doi: 10.4161/bact.1.3.16591
Abramoff, M. D., Magalhaes, P. J., and Ram, S. J. (2004). Image processing with imageJ. Biophoton. Int. 11, 36–42.
Adams, M. J., Lefkowitz, E. J., King, A. M. Q., Harrach, B., Harrison, R. L., Knowles, N. J., et al. (2017). Changes to taxonomy and the international code of virus classification and nomenclature ratified by the international committee on taxonomy of viruses. Arch. Virol. 162, 2505–2538.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1006/jmbi.1990.9999
Arens, D. K., Brady, T. S., Carter, J. L., Pape, J. A., Robinson, D. M., Russell, K. A., et al. (2018). Characterization of two related erwinia myoviruses that are distant relatives of the phikz-like jumbo phages. PLoS One 13:e0200202. doi: 10.1371/journal.pone.0200202
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). Meme suite: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. doi: 10.1093/nar/gkp335
Berg, J. A., Merrill, B. D., Crockett, J. T., Esplin, K. P., Evans, M. R., Heaton, K. E., et al. (2016). Characterization of five novel brevibacillus bacteriophages and genomic comparison of Brevibacillus phages. PLoS One 11:e0156838. doi: 10.1371/journal.pone.0156838
Bergh, O., Borsheim, K. Y., Bratbak, G., and Heldal, M. (1989). High abundance of viruses found in aquatic environments. Nature 340, 467–468. doi: 10.1038/340467a0
Bhunchoth, A., Blanc-Mathieu, R., Mihara, T., Nishimura, Y., Askora, A., Phironrit, N., et al. (2016). Two Asian jumbo phages, varphirsl2 and varphirsf1, infect ralstonia solanacearum and show common features of varphikz-related phages. Virology 494, 56–66. doi: 10.1016/j.virol.2016.03.028
Bollback, J. P., and Huelsenbeck, J. P. (2009). Parallel genetic evolution within and between bacteriophage species of varying degrees of divergence. Genetics 181, 225–234. doi: 10.1534/genetics.107.085225
Botstein, D. (1980). A theory of modular evolution for bacteriophages. Ann. N. Y. Acad. Sci. 354, 484–490.
Boyd, E. F. (2012). Bacteriophage-encoded bacterial virulence factors and phage-pathogenicity island interactions. Adv. Virus Res. 82, 91–118. doi: 10.1016/B978-0-12-394621-8.00014-5
Buttimer, C., Hendrix, H., Oliveira, H., Casey, A., Neve, H., McAuliffe, O., et al. (2017). Things are getting hairy: Enterobacteria bacteriophage Vb_Pcam_Cbb. Front. Microbiol. 8:44. doi: 10.3389/fmicb.2017.00044
Campbell, J. I. A., Albrechtsen, M., and Sorensen, J. (1995). Large Pseudomonas phages isolated from barley rhizosphere. FEMS Microbiol. Ecol. 18, 63–74. doi: 10.1016/0168-6496(95)00043-a
Cartagena, E., Colom, O. A., Neske, A., Valdez, J. C., and Bardon, A. (2007). Effects of plant lactones on the production of biofilm of Pseudomonas Aeruginosa. Chem. Pharm. Bull. 55, 22–25. doi: 10.1248/cpb.55.22
Chaudhary, S. K., Jeyakanthan, J., and Sekar, K. (2013). Cloning, expression, purification, crystallization and preliminary X-Ray crystallographic study of thymidylate kinase (Ttha1607) from thermus thermophilus Hb8. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 69(Pt 2), 118–121. doi: 10.1107/S1744309112050208
Chen, J., and Novick, R. P. (2009). Phage-mediated intergeneric transfer of toxin genes. Science 323, 139–141. doi: 10.1126/science.1164783
Chevalier, B. S., and Stoddard, B. L. (2001). Homing endonucleases: structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res. 29, 3757–3774. doi: 10.1093/nar/29.18.3757
Connelly, J. C., Kirkham, L. A., and Leach, D. R. (1998). The sbccd nuclease of Escherichia Coli is a structural maintenance of chromosomes (Smc) family protein that cleaves hairpin DNA. Proc. Natl. Acad. Sci. U.S.A. 95, 7969–7974. doi: 10.1073/pnas.95.14.7969
Conway, H. F., Haynes, W. C., Jackson, R. W., Locke, J. M., Pridham, T. G., Sohns, V. E., et al. (1956). Pseudomonas aureofaciens kluyver and phenazine alpha-carboxylic acid, its characteristic pigment. J. Bacteriol. 72, 412–417.
Cornelissen, A., Ceyssens, P. J., Krylov, V. N., Noben, J. P., Volckaert, G., and Lavigne, R. (2012). Identification of Eps-degrading activity within the tail spikes of the novel Pseudomonas putida phage Af. Virology 434, 251–256. doi: 10.1016/j.virol.2012.09.030
Cresawn, S. G., Bogel, M., Day, N., Jacobs-Sera, D., Hendrix, R. W., and Hatfull, G. F. (2011). Phamerator: a bioinformatic tool for comparative bacteriophage genomics. BMC Bioinformatics 12:395. doi: 10.1186/1471-2105-12-395
Cui, Q., Shin, W. S., Luo, Y., Tian, J., Cui, H., and Yin, D. (2013). Thymidylate kinase: an old topic brings new perspectives. Curr. Med. Chem. 20, 1286–1305. doi: 10.2174/0929867311320100006
Danis-Wlodarczyk, K., Vandenheuvel, D., Jang, H. B., Briers, Y., Olszak, T., Arabski, M., et al. (2016). A proposed integrated approach for the preclinical evaluation of phage therapy in Pseudomonas infections. Sci. Rep. 6:28115. doi: 10.1038/srep28115
Datsenko, K. A., and Wanner, B. L. (2000). “One-step inactivation of chromosomal genes in Escherichia Coli K-12 using Pcr products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645. doi: 10.1073/pnas.120163297
Delbruck, M. (1945). The burst size distribution in the growth of bacterial viruses (Bacteriophages). J. Bacteriol. 50, 131–135.
Deletoile, A., Decre, D., Courant, S., Passet, V., Audo, J., Grimont, P., et al. (2009). Phylogeny and Identification of Pantoea species and typing of pantoea agglomerans strains by multilocus gene sequencing. J. Clin. Microbiol. 47, 300–310. doi: 10.1128/JCM.01916-08
D’Herelle, F. (1917). Sur un microbe invisible antagoniste des bacilles dysentériques. CR Acad. 165, 373–375.
D’Herelle, F. (2007). On an invisible microbe antagonistic toward dysenteric bacilli: brief note by Mr. F. D’herelle, presented by Mr. Roux. 1917. Res. Microbiol. 158, 553–554. doi: 10.1016/j.resmic.2007.07.005
Doharey, P. K., Singh, S. K., Verma, P., Verma, A., Rathaur, S., and Saxena, J. K. (2016). Insights into the structure-function relationship of Brugia Malayi Thymidylate Kinase (Bmtmk). Int. J. Biol. Macromol. 88, 565–571. doi: 10.1016/j.ijbiomac.2016.04.004
Effantin, G., Hamasaki, R., Kawasaki, T., Bacia, M., Moriscot, C., Weissenhorn, W., et al. (2013). Cryo-electron microscopy three-dimensional structure of the jumbo phage phirsl1 infecting the phytopathogen Ralstonia solanacearum. Structure 21, 298–305. doi: 10.1016/j.str.2012.12.017
Emrich, J., and Streisinger, G. (1968). The role of phage lysozyme in the life cycle of phage T4. Virology 36, 387–391. doi: 10.1016/0042-6822(68)90163-3
Esplin, I. N. D., Berg, J. A., Sharma, R., Allen, R. C., Arens, D. K., Ashcroft, C. R., et al. (2017). Genome sequences of 19 novel erwinia amylovora bacteriophages. Genome Announc. 5:e00931-17. doi: 10.1128/genomeA.00931-17
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x
Fetherston, J. D., Schuetze, P., and Perry, R. D. (1992). Loss of the pigmentation phenotype in Yersinia pestis is due to the spontaneous deletion of 102 Kb of chromosomal DNA which is flanked by a repetitive element. Mol. Microbiol. 6, 2693–2704. doi: 10.1111/j.1365-2958.1992.tb01446.x
Gajewski, S., Webb, M. R., Galkin, V., Egelman, E. H., Kreuzer, K. N., and White, S. W. (2011). “Crystal structure of the phage T4 recombinase Uvsx and its functional interaction with the T4 Sf2 helicase Uvsw. J. Mol. Biol. 405, 65–76. doi: 10.1016/j.jmb.2010.10.004
Garneau, J. R., Florence, D., Louis-Charles, F., David, B., and Marc, M. (2017). Phageterm: a fast and user-friendly software to determine bacteriophage termini and packaging mode using randomly fragmented Ngs data. bioRxiv
Gibb, E. A., and Edgell, D. R. (2007). Multiple controls regulate the expression of mobe, an hnh homing endonuclease gene embedded within a ribonucleotide reductase gene of phage Aeh1. J. Bacteriol. 189, 4648–4661. doi: 10.1128/jb.00321-07
Gicova, A., Orieskova, M., Oslanecova, L., Drahovska, H., and Kaclikova, E. (2014). Identification and characterization of cronobacter strains isolated from powdered infant foods. Lett. Appl. Microbiol. 58, 242–247. doi: 10.1111/lam.12179
Gill, J. J., Berry, J. D., Russell, W. K., Lessor, L., Escobar-Garcia, D. A., Hernandez, D., et al. (2012). The Caulobacter crescentus phage phicbk: genomics of a canonical phage. BMC Genomics 13:542. doi: 10.1186/1471-2164-13-542
Gong, S., Bearden, S. W., Geoffroy, V. A., Fetherston, J. D., and Perry, R. D. (2001). Characterization of the Yersinia pestis Yfu Abc inorganic iron transport system. Infect. Immun. 69, 2829–2837. doi: 10.1128/iai.67.5.2829-2837.2001
Grant, C. E., Bailey, T. L., and Noble, W. S. (2011). “Fimo: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. doi: 10.1093/bioinformatics/btr064
Guttman, M., Betts, G. N., Barnes, H., Ghassemian, M., van der Geer, P., and Komives, E. A. (2009). Interactions of the Npxy microdomains of the low density lipoprotein receptor-related protein 1. Proteomics 9, 5016–5028. doi: 10.1002/pmic.200900457
Hambly, E., and Suttle, C. A. (2005). The viriosphere, diversity, and genetic exchange within phage communities. Curr. Opin. Microbiol. 8, 444–450. doi: 10.1016/j.mib.2005.06.005
Hatfull, G. F. (2015). Dark matter of the biosphere: the amazing world of bacteriophage diversity. J. Virol. 89, 8107–8110. doi: 10.1128/JVI.01340-15
Hauser, M., Mayer, C. E., and Soding, J. (2013). Kclust: fast and sensitive clustering of large protein sequence databases. BMC Bioinformatics 14:248. doi: 10.1186/1471-2105-14-248
Hertveldt, K., Lavigne, R., Pleteneva, E., Sernova, N., Kurochkina, L., Korchevskii, R., et al. (2005). “Genome comparison of Pseudomonas Aeruginosa large phages. J. Mol. Biol. 354, 536–545. doi: 10.1016/j.jmb.2005.08.075
Hockett, K. L., and Baltrus, D. A. (2017). Use of the soft-agar overlay technique to screen for bacterially produced inhibitory compounds. J. Vis. Exp. 119:55064. doi: 10.3791/55064
Huson, D. H., and Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
Ishimaru, C. A., Klos, E. J., and Brubaker, R. R. (1987). Multiple antibiotic production by Erwinia herbicola. Phytopathology 78, 746–750.
Iversen, C., Lehner, A., Mullane, N., Bidlas, E., Cleenwerck, I., Marugg, J., et al. (2007). The taxonomy of Enterobacter sakazakii: proposal of a new genus cronobacter gen. nov. and descriptions of Cronobacter sakazakii comb. nov. Cronobacter sakazakii subsp. sakazakii, comb. nov., Cronobacter sakazakii subsp. Malonaticus subsp. nov., Cronobacter turicensis sp. nov., Cronobacter muytjensii sp. nov., Cronobacter dublinensis sp. nov. and cronobacter genomospecies 1. BMC Evol. Biol. 7:64. doi: 10.1186/1471-2148-7-64
Joseph, J. W., and Kolodner, R. (1983). Exonuclease Viii of Escherichia Coli. I. purification and physical properties. J. Biol. Chem. 258, 10411–10417.
Kadyrov, F. A., and Drake, J. W. (2004). Uvsx recombinase and dda helicase rescue stalled bacteriophage T4 DNA replication forks in vitro. J. Biol. Chem. 279, 35735–35740. doi: 10.1074/jbc.m403942200
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). Mafft: a novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Keen, E. C. (2015). A century of phage research: bacteriophages and the shaping of modern biology. Bioessays 37, 6–9. doi: 10.1002/bies.201400152
Kim, J. H., Son, J. S., Choi, Y. J., Choresca, C. H. Jr., Shin, S. P., Han, J. E., et al. (2012). Complete genome sequence and characterization of a broad-host range T4-Like bacteriophage phias5 infecting Aeromonas salmonicida Subsp. Salmonicida. Vet. Microbiol. 157, 164–171. doi: 10.1016/j.vetmic.2011.12.016
Kim, W. S., and Geider, K. (2000). Characterization of a viral eps-depolymerase, a potential tool for control of fire blight. Phytopathology 90, 1263–1268. doi: 10.1094/PHYTO.2000.90.11.1263
Kokoris, M. S., and Black, M. E. (2002). Characterization of herpes simplex virus type 1 thymidine kinase mutants engineered for improved ganciclovir or acyclovir activity. Protein Sci. 11, 2267–2272. doi: 10.1110/ps.2460102
Kolodner, R., Hall, S. D., and Luisi-DeLuca, C. (1994). Homologous pairing proteins encoded by the Escherichia Coli Rece and Rect Genes. Mol. Microbiol. 11, 23–30. doi: 10.1111/j.1365-2958.1994.tb00286.x
Krumsiek, J., Arnold, R., and Rattei, T. (2007). Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 23, 1026–1028. doi: 10.1093/bioinformatics/btm039
Krylov, V., Pleteneva, E., Bourkaltseva, M., Shaburova, O., Volckaert, G., Sykilinda, N., et al. (2003). Myoviridae bacteriophages of Pseudomonas Aeruginosa: a long and complex evolutionary pathway. Res. Microbiol. 154, 269–275. doi: 10.1016/s0923-2508(03)00070-6
Kuebler, D., and Rao, V. B. (1998). Functional analysis of the DNA-packaging/terminase protein Gp17 from bacteriophage T4. J. Mol. Biol. 281, 803–814. doi: 10.1006/jmbi.1998.1952
Kuhn, J. H., Radoshitzky, S. R., Bavari, S., and Jahrling, P. B. (2013). The international code of virus classification and nomenclature (Icvcn): proposal for text changes for improved differentiation of viral taxa and viruses. Arch. Virol. 158, 1621–1629. doi: 10.1007/s00705-012-1582-6
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). Mega X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kurochkina, L. P., Semenyuk, P. I., Sykilinda, N. N., and Miroshnikov, K. A. (2018). The unique two-component tail sheath of giant Pseudomonas phage pabg. Virology 515, 46–51. doi: 10.1016/j.virol.2017.12.010
Lamine, J. G., DeJong, R. J., and Nelesen, S. M. (2016). Phamdb: a web-based application for building phamerator databases. Bioinformatics 32, 2026–2028. doi: 10.1093/bioinformatics/btw106
Lassmann, T., and Sonnhammer, E. L. (2005). Kalign–an accurate and fast multiple sequence alignment algorithm. BMC Bioinform. 6:298.
Lavysh, D., Sokolova, M., Minakhin, L., Yakunina, M., Artamonova, T., Kozyavkin, S., et al. (2016). The genome of Ar9, a giant transducing bacillus phage encoding two multisubunit RNA polymerases. Virology 495, 185–196. doi: 10.1016/j.virol.2016.04.030
Lavysh, D., Sokolova, M., Slashcheva, M., Forstner, K. U., and Severinov, K. (2017). Transcription profiling of Bacillus subtilis cells infected with Ar9, a giant phage encoding two multisubunit RNA polymerases. MBio 8:e02041-16. doi: 10.1128/mBio.02041-16
Leach, D. R., Okely, E. A., and Pinder, D. J. (1997). Repair by recombination of DNA containing a palindromic sequence. Mol. Microbiol. 26, 597–606. doi: 10.1046/j.1365-2958.1997.6071957.x
Lecoutere, E., Ceyssens, P. J., Miroshnikov, K. A., Mesyanzhinov, V. V., Krylov, V. N., Noben, J. P., et al. (2009). Identification and comparative analysis of the structural proteomes of phikz and El, two giant Pseudomonas Aeruginosa bacteriophages. Proteomics 9, 3215–3219. doi: 10.1002/pmic.200800727
Lee, J. H., Bai, J., Shin, H., Kim, Y., Park, B., Heu, S., et al. (2016). “A novel bacteriophage targeting Cronobacter sakazakii is a potential biocontrol agent in foods. Appl. Environ. Microbiol. 82, 192–201. doi: 10.1128/AEM.01827-15
Lowe, T. M., and Chan, P. P. (2016). Trnascan-Se on-line: integrating search and context for analysis of transfer rna genes. Nucleic Acids Res. 44, W54–W57. doi: 10.1093/nar/gkw413
Madej, T., Gibrat, J. F., and Bryant, S. H. (1995). Threading a database of protein cores. Proteins 23, 356–369. doi: 10.1002/prot.340230309
Maher, R. L., and Morrical, S. W. (2013). Coordinated binding of single-stranded and double-stranded DNA by Uvsx recombinase. PLoS One 8:e66654. doi: 10.1371/journal.pone.0066654
Majkowska-Skrobek, G., Latka, A., Berisio, R., Maciejewska, B., Squeglia, F., Romano, M., et al. (2016). Capsule-targeting depolymerase, derived from klebsiella Kp36 phage, as a tool for the development of anti-virulent strategy. Viruses 8:E324.
McCormack, A. L., Schieltz, D. M., Goode, B., Yang, S., Barnes, G., Drubin, D., et al. (1997). Direct analysis and identification of proteins in mixtures by Lc/Ms/Ms and database searching at the low-femtomole level. Anal. Chem. 69, 767–776. doi: 10.1021/ac960799q
McGhee, G. C., and Sundin, G. W. (2012). Erwinia amylovora crispr elements provide new tools for evaluating strain diversity and for microbial source tracking. PLoS One 7:e41706. doi: 10.1371/journal.pone.0041706
Merrill, B. D., Ward, A. T., Grose, J. H., and Hope, S. (2016). Software-based analysis of bacteriophage genomes, physical ends, and packaging strategies. BMC Genomics 17:679. doi: 10.1186/s12864-016-3018-2
Mesyanzhinov, V. V., Robben, J., Grymonprez, B., Kostyuchenko, V. A., Bourkaltseva, M. V., Sykilinda, N. N., et al. (2002). The genome of bacteriophage phikz of Pseudomonas Aeruginosa. J. Mol. Biol. 317, 1–19.
Miller, E. S., Heidelberg, J. F., Eisen, J. A., Nelson, W. C., Durkin, A. S., Ciecko, A., et al. (2003). Complete genome sequence of the broad-host-range vibriophage Kvp40: comparative genomics of a T4-related bacteriophage. J. Bacteriol. 185, 5220–5233. doi: 10.1128/jb.185.17.5220-5233.2003
Monson, R., Foulds, I., Foweraker, J., Welch, M., and Salmond, G. P. (2011). The Pseudomonas Aeruginosa generalized transducing phage phipa3 is a new member of the phikz-like group of ‘jumbo’ phages, and infects model laboratory strains and clinical isolates from cystic fibrosis patients. Microbiology 157(Pt 3), 859–867. doi: 10.1099/mic.0.044701-0
Nakamura, L. K., Roberts, M. S., and Cohan, F. M. (1999). Relationship of Bacillus subtilis clades associated with strains 168 and W23: a proposal for Bacillus Subtilis Subsp. Subtilis subsp. nov. and Bacillus Subtilis Subsp. Spizizenii Subsp. Nov. Int. J. Syst. Bacteriol. 49(Pt 3), 1211–1215. doi: 10.1099/00207713-49-3-1211
Otman, J., Perugini, M. E., Tognim, M. C. B., and Vidotto, M. C. (2007). Atypical phenotypic characteristics of Klebsiella Pneumoniae isolates from an outbreak in a neonatal intensive care unit in Brazil. Brazil. J. Microbiol. 38, 273–277. doi: 10.1590/s1517-83822007000200016
Palmer, M., de Maayer, P., Poulsen, M., Steenkamp, E. T., van Zyl, E., Coutinho, T. A., et al. (2016). Draft genome sequences of pantoea agglomerans and pantoea vagans isolates associated with termites. Stand Genomic Sci. 11:23. doi: 10.1186/s40793-016-0144-z
Pusey, P. L. (1997). Crab apple blossoms as a model for research on biological control of fire blight. Phytopathology 87, 1096–1102. doi: 10.1094/PHYTO.1997.87.11.1096
Ren, Y., Ren, Y., Zhou, Z., Guo, X., Li, Y., Feng, L., et al. (2010). Complete genome sequence of Enterobacter Cloacae subsp. cloacae type strain Atcc 13047. J. Bacteriol. 192, 2463–2464. doi: 10.1128/JB.00067-10
Ritchie, D. F., and Klos, E. J. (1976). Isolation of Erwinia amylovora bacteriophage from aerial parts of apple trees. Phytopathology 67, 101–104.
Sebaihia, M., Bocsanczy, A. M., Biehl, B. S., Quail, M. A., Perna, N. T., Glasner, J. D., et al. (2010). Complete genome sequence of the plant pathogen Erwinia amylovora Strain Atcc 49946. J. Bacteriol. 192, 2020–2021. doi: 10.1128/JB.00022-10
Shaburova, O. V., Hertveldt, K., de la Crus, D. M., Krylov, S. V., Pleteneva, E. A., Burkaltseva, M. V., et al. (2006). Comparison of new giant bacteriophages Obp and Lu11 of soil pseudomonads with bacteriophages of phikz-supergroup of Pseudomonas Aeruginosa. Genetika 42, 1065–1074.
Sharma, R., Berg, J. A., Beatty, N. J., Choi, M. C., Cowger, A. E., and Cozzens, B. J. R. (2018). Genome sequences of nine Erwinia amylovora bacteriophages. Microbiol. Resour. Announc. 7:e00944-18. doi: 10.1128/MRA.00944-18
Shivaswamy, V. C., Kalasuramath, S. B., Sadanand, C. K., Basavaraju, A. K., Ginnavaram, V., Bille, S., et al. (2015). Ability of bacteriophage in resolving wound infection caused by multidrug-resistant Acinetobacter Baumannii in uncontrolled diabetic rats. Microb. Drug Resist. 21, 171–177. doi: 10.1089/mdr.2014.0120
Simoliunas, E., Kaliniene, L., Truncaite, L., Zajanckauskaite, A., Staniulis, J., Kaupinis, A., et al. (2013). Klebsiella phage Vb_Klem-Rak2 - a giant singleton virus of the family myoviridae. PLoS One 8:e60717. doi: 10.1371/journal.pone.0060717
Smith, K. C., Castro-Nallar, E., Fisher, J. N., Breakwell, D. P., Grose, J. H., and Burnett, S. H. (2013). Phage cluster relationships identified through single gene analysis. BMC Genomics 14:410. doi: 10.1186/1471-2164-14-410
Smits, T. H., Rezzonico, F., Kamber, T., Goesmann, A., Ishimaru, C. A., Stockwell, V. O., et al. (2010). Genome sequence of the biocontrol agent pantoea vagans strain C9-1. J. Bacteriol. 192, 6486–6487. doi: 10.1128/JB.01122-10
Sokolova, O. S., Shaburova, O. V., Pechnikova, E. V., Shaytan, A. K., Krylov, S. V., Kiselev, N. A., et al. (2014). Genome packaging in El and Lin68, two giant phikz-like bacteriophages of P. Aeruginosa. Virology 468-470, 472–478. doi: 10.1016/j.virol.2014.09.002
Sulakvelidze, A. (2005). Phage therapy: an attractive option for dealing with antibiotic-resistant bacterial infections. Drug Discov. Today 10, 807–809. doi: 10.1016/s1359-6446(05)03441-0
Sullivan, M. B., Coleman, M. L., Weigele, P., Rohwer, F., and Chisholm, S. W. (2005). Three Prochlorococcus cyanophage genomes: signature features and ecological interpretations. PLoS Biol. 3:e144. doi: 10.1371/journal.pbio.0030144
Thomas, J. A., Hardies, S. C., Rolando, M., Hayes, S. J., Lieman, K., Carroll, C. A., et al. (2007). Complete genomic sequence and mass spectrometric analysis of highly diverse, atypical bacillus thuringiensis phage 0305phi8-36. Virology 368, 405–421. doi: 10.1016/j.virol.2007.06.043
Thomas, J. A., Rolando, M. R., Carroll, C. A., Shen, P. S., Belnap, D. M., Weintraub, S. T., et al. (2008). Characterization of Pseudomonas Chlororaphis myovirus 201varphi2-1 via genomic sequencing, mass spectrometry, and electron microscopy. Virology 376, 330–338. doi: 10.1016/j.virol.2008.04.004
Thomson, S. V. (2000). Epidemiology of Fire Blight. Fire Blight; the Disease and Its Causative Agent, Erwinia Amylovora, ed. J. Vanneste (Wallingford: CABI Publishing).
Twort, F. W. (1915). An investigation on the nature of ultra-microscopic viruses. Lancet 186, 1241–1243. doi: 10.1016/s0140-6736(01)20383-3
Twort, F. W. (1936). Further investigations on the nature of ultra-microscopic viruses and their cultivation. J. Hyg. 36, 204–235. doi: 10.1017/s0022172400043606
Whitman, W. B., Coleman, D. C., and Wiebe, W. J. (1998). Prokaryotes: the unseen majority. Proc. Natl. Acad. Sci. U.S.A. 95, 6578–6583. doi: 10.1073/pnas.95.12.6578
Wommack, K. E., and Colwell, R. R. (2000). Virioplankton: viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 64, 69–114. doi: 10.1128/mmbr.64.1.69-114.2000
Yagubi, A. I., Castle, A. J., Kropinski, A. M., Banks, T. W., and Svircev, A. M. (2014). Complete genome sequence of Erwinia amylovora bacteriophage Vb_Eamm_Ea35-70. Genome Announc.2:e00413-14. doi: 10.1128/genomeA.00413-14
Yamada, T., Satoh, S., Ishikawa, H., Fujiwara, A., Kawasaki, T., Fujie, M., et al. (2010). A jumbo phage infecting the phytopathogen Ralstonia solanacearum defines a new lineage of the Myoviridae family. Virology 398, 135–147. doi: 10.1016/j.virol.2009.11.043
Yuan, Y., and Gao, M. (2016). Proteomic analysis of a novel bacillus jumbo phage revealing glycoside hydrolase as structural component. Front. Microbiol. 7:745. doi: 10.3389/fmicb.2016.00745
Yuan, Y., and Gao, M. (2017). Jumbo bacteriophages: an overview. Front. Microbiol. 8:403. doi: 10.3389/fmicb.2017.00403
Zhao, Y., and Qi, M. (2011). Comparative genomics of Erwinia amylovora and related Erwinia species-what do we learn? Genes 2, 627–639. doi: 10.3390/genes2030627
Zuckerkandl, E., and Pauling, L. (1965). “Evolutionary Divergence and Convergence in Proteins,” in Evolving Genes and Proteins, eds V. Bryson and H. J. Vogel (New York, NY: Academic Press).
Keywords: jumbo bacteriophage, Agrican357virus, burst size, novel, Erwinia, Pantoea, genome, proteome
Citation: Sharma R, Pielstick BA, Bell KA, Nieman TB, Stubbs OA, Yeates EL, Baltrus DA and Grose JH (2019) A Novel, Highly Related Jumbo Family of Bacteriophages That Were Isolated Against Erwinia. Front. Microbiol. 10:1533. doi: 10.3389/fmicb.2019.01533
Received: 08 March 2019; Accepted: 19 June 2019;
Published: 23 July 2019.
Edited by:
Manuel Martinez Garcia, University of Alicante, SpainReviewed by:
Jeroen Wagemans, KU Leuven, BelgiumAna-Belen Martin-Cuadrado, University of Alicante, Spain
Lars Fieseler, Zurich University of Applied Sciences, Switzerland
Copyright © 2019 Sharma, Pielstick, Bell, Nieman, Stubbs, Yeates, Baltrus and Grose. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julianne H. Grose, Z3Jvc2VqdWxpYW5uZUBnbWFpbC5jb20=