
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 28 June 2019
Sec. Evolutionary and Genomic Microbiology
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.01447
This article is part of the Research Topic Omics and Systems Approaches to Study the Biology and Applications of Lactic Acid Bacteria View all 20 articles
 Swaine L. Chen1,2*
Swaine L. Chen1,2*Streptococcus agalactiae, also known as Group B Streptococcus (GBS), is a bacteria with truly protean biology. It infects a variety of hosts, among which the most commonly studied are humans, cattle, and fish. GBS holds a singular position in the history of bacterial genomics, as it was the substrate used to describe one of the first major conceptual advances of comparative genomics, the idea of the pan-genome. In this review, I describe a brief history of GBS and the major contributions of genomics to understanding its genome plasticity and evolution as well as its molecular epidemiology, focusing on the three hosts mentioned above. I also discuss one of the major recent paradigm shifts in our understanding of GBS evolution and disease burden: foodborne GBS can cause invasive infections in humans.
Streptococcus agalactiae was first described in 1887 as a common bacteria infecting the udders of cattle, causing a disease termed mastitis (Nocard and Mollereau, 1887). This led to significantly lower milk production (Keefe, 1997; Ruegg, 2017), thus the species name agalactiae (from the Greek: a-, no; galactos, milk). In these cows, milk production can be reduced over 20% in infected cows, which, prior to the institution of active control measures, could conservatively affect 15–40% of all cows (Shaw and Beam, 1935). It came to be known by its other common name, Group B Streptococcus, with the Lancefield classification in 1933 (Lancefield, 1933), where the “Group B” references the species-specific carbohydrate “substance C” from Streptococci (Lancefield, 1928) recognized by rabbit immune sera (this substance C is associated with the cell wall and distinct from the polysaccharide capsule). Today, GBS is well known as the most common cause of neonatal meningitis, which is further classified into early onset (<7 days after birth) and late onset (7 days to 3 months after birth) (Berardi et al., 2007; Edwards et al., 2011; Nanduri et al., 2019). Transmission to the newborn can be vertical, through contact with mucus membranes, or through ingestion of infected amniotic fluid (Verani et al., 2010). Consistent with this, it is a frequent (20–40% of individuals) colonizer of the human gastrointestinal tract and the reproductive tract of women, based on two prevalence studies in a North American university (Bliss et al., 2002; Manning et al., 2004). However, GBS is also an increasingly common cause of severe invasive disease, typically in immunocompromised individuals and the elderly, since the 1960s (Farley et al., 1993; Schuchat, 1998; Farley, 2001; Francois Watkins et al., 2019).
These well-known facts about the history and medical importance of GBS parallel several deeper themes of GBS biology. The dual names by which we refer to this bacterium echo a split in how we have studied its biology in humans, cattle, and other animals. The rise of human infections, first in neonates and more recently in adults, matches a theme of ongoing evolution and niche expansion for the species. Finally, the shift in associated species from cattle to humans foreshadows additional potential species jumps that are apparently continuing to this day. GBS is now known to be widely distributed among diverse species of mammals, reptiles, amphibians, and fish (such as dogs, cats, goats, elephants, frogs, crocodiles, dolphins, seals, llamas, and camels) (Edelstein and Pegram, 1974; Bishop et al., 2007; Delannoy et al., 2013; Tavella et al., 2018), not only colonizing but in many cases also capable of causing severe invasive disease. Of particular note, besides its importance in human and bovine medicine, GBS is a significant pathogen for aquatic species, including those of importance for food production (Amal et al., 2011). Streptococcal infections are responsible for an estimated US$150 million in global losses in farmed tilapia in 2000 (close to 10% of the total value) (Amal et al., 2011). As with human and bovine infections, GBS infection in fish was first described relatively recently, with two seminal reports in 1958 and 1966 (Hoshina et al., 1958; Robinson and Meyer, 1966). Even from these first reports, GBS was noted to be a particular danger to farmed fish, being highly contagious and usually fatal (Hoshina et al., 1958; Robinson and Meyer, 1966).
GBS has therefore been studied in multiple contexts: human health, veterinary medicine, and agriculture. Research has thus been motivated by both health and economic goals, which naturally vary in importance across these different disciplines. Beyond the proximal questions of how GBS colonizes and causes specific diseases in specific hosts, GBS is an intriguing case study for the larger questions of how broad host range at the species level is maintained despite evidence of variation at the subspecies level. These larger evolutionary questions are again made more urgent by convincing evidence of ongoing adaptation and emergence of pathogenicity and resistance in GBS.
To tackle these specific questions about disease mechanisms as well as broader evolutionary questions, genomics is a natural fit. Recent years have seen an explosion of genomic data available for GBS, as is the case for all other bacteria. The transition to a post-genomic era for GBS holds an additional promise for a unified understanding across the medical and veterinary fields, which may lead to a fuller appreciation of the importance and impact of this versatile bacterium. In this review, I will focus on two primary topics: (1) GBS genome plasticity and evolution and (2) GBS molecular epidemiology related to geography and host range (focused on humans, cattle, and fish). I have included some contextual information drawn from non-genomic papers using other typing systems, but this review does not aspire to be complete in regard to the entire corpus of GBS studies.
GBS, like many other bacteria of medical importance, has long been recognized to have intraspecies variation that can be tracked with a variety of molecular methods. Early studies utilized immunological reactions, resulting in a (still commonly used) serotyping scheme consisting of 10 major serotypes (Ia, Ib, II-IX) (Edwards et al., 2011). Numerous other systems have been applied to GBS, including ribotyping (Huet et al., 1993), RAPD (Random Amplification of Polymorphic DNA) (Limansky et al., 1998), RFLP (Restriction Fragment Length Polymorphism) (Hauge et al., 1996), PFGE (Pulsed-Field Gel Electrophoresis) (Rolland et al., 1999), and MLEE (Multilocus Enzyme Electrophoresis) (Musser et al., 1989). The other major pre-genomic classification system (although, ironically, published after the first genome sequences became available) that still remains in common use today is MLST (Multilocus Sequence Typing) (Jones et al., 2003), due to its balance between ease of typing, portability between labs, and reasonable resolution (Maiden, 2006). From these initial pre-genomic studies, the general outlines of the population structure of GBS were inferred. The different major hosts (humans, cattle, and fish) have largely distinct populations of strains, with some notable exceptions that may be indicative of cross-species jumps. Furthermore, changes in the epidemiology of disease and responsible serotypes or MLST types have been noted (see Section “The Pan-Genome”). However, overall, prior to the genomic era, GBS could generally (though not exclusively) be classified based on its host species (Finch and Martin, 1984; Bohnsack et al., 2004; Sukhnanand et al., 2005; Evans et al., 2008; Pereira et al., 2010), which then could be subdivided into several (3–5) large groups of closely related strains (termed clonal complexes in the MLST nomenclature) that accounted for most diseases (Table 1).
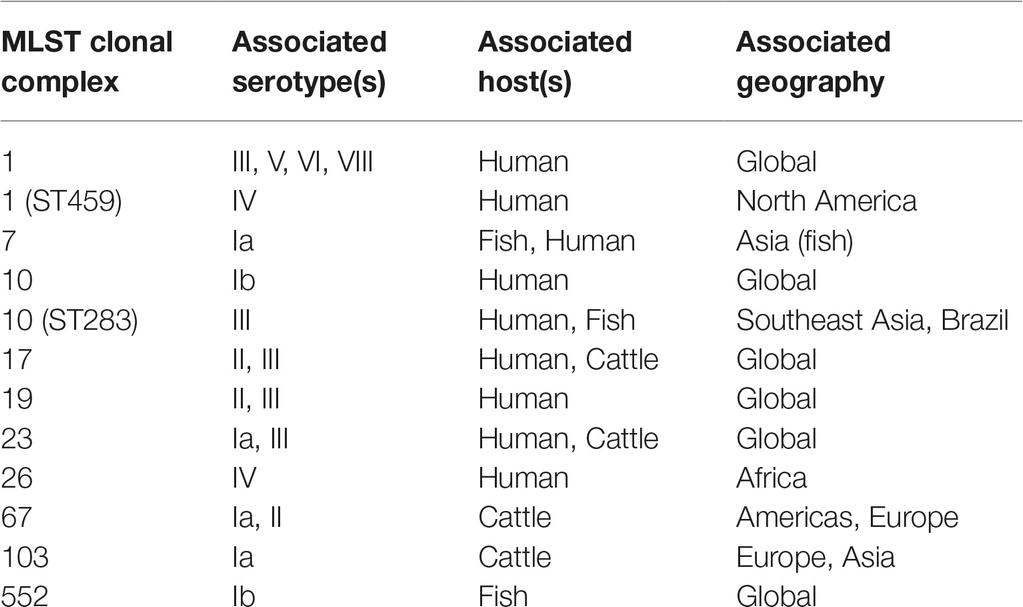
Table 1. Major MLST Clonal Complexes of GBS found in humans, cattle, and fish.
The first full genome sequences of GBS were of the human isolates NEM316 (ST23, Serotype III) (Glaser et al., 2002) and 2603V/R (ST110 (a single locus variant of ST19), Serotype V) (Tettelin et al., 2002), both published in late 2002. These two genomes provided a heretofore unprecedented view of the organization and potential evolution of the GBS genome, which was initially gleaned from comparisons with genomes of S. pyogenes and S. pneumoniae strains. Generally, the GBS genome consists of a conserved “backbone” that is punctuated by 14–15 genomic islands of variable gene content (and many smaller islets). GBS was remarkable for its high number of tRNAs (80), ABC transporters (62), and signal transduction systems (17–20 two component systems). In addition, there are multiple classes of mobile DNA elements that presumably contributed to disrupting, duplicating, and transferring genes. In particular, insertion sequences, prophages, and a triplicated integrated plasmid in NEM316 (denoted pNEM316–1) (Glaser et al., 2002) were major contributors to variation in gene content that was specific to GBS and that also varies among different GBS strains. With only a single full genome to analyze, neither of these initial genome papers were able to directly identify large-scale chromosomal recombination occurring in GBS, though it was clear from comparative genome hybridization experiments that genes within the species-specific GBS islands were also more likely to vary among GBS strains as well (Tettelin et al., 2002).
The structure of the GBS genomes, with a conserved backbone punctuated with islands of variable gene content, led directly to a simple hypothesis as to why different GBS strains may preferentially colonize or infect different hosts. GBS as a species might be defined by the conserved regions, while the variable islands, which often possessed features of genes important for virulence (Glaser et al., 2002; Tettelin et al., 2002), could carry genes that provided specific phenotypes important in different hosts or different disease settings. This structural stratification of gene conservation became more clear as more genomes were sequenced, not just for GBS but for other bacteria as well, most notably E. coli (Welch et al., 2002).
This represented, in a sense, a specific genomic extension to genetics. Clearly, different phenotypes for host specificity and disease would be traceable to genetic mechanisms; and now, perhaps, there was a structural genomic framework which would organize the adaptation and evolution of these traits. The first formalization of this idea was the pan-genome concept (though the potential structural aspect of genome organization was not noted). The seminal paper describing the concept of a pan-genome (the complete set of genes that is found in all individuals of a given species) was based on an analysis of eight GBS genome sequences (Tettelin et al., 2005). GBS therefore holds a special place in the early transition to the post-genomic era for bacteria. It was also the first organism described to have an “open” pan-genome; rarefaction analysis predicted that, even with an arbitrarily large number of genome sequences, every new genome sequence would contribute an extra 33 genes that had not previously been seen in any other GBS. The pan-genome concept was one of the first truly new results to arise from comparative genomics; it was a systematic rationalization of differing gene content that necessarily required the existence of multiple genome sequences, and it further gave rise to the companion concept of a core genome that consists of genes that are conserved across all individuals of a given species (Medini et al., 2005; Tettelin et al., 2005).
The initial ideas about core and pan-genomes were a remarkably useful organizational framework for thinking about a variety of issues relevant to bacteria and genomics, leading to their immense popularity. The pan-genome concept was closely related to ideas about genome plasticity, horizontal gene transfer, the concept of species for bacteria, and genome evolution (Medini et al., 2005). The conceptual simplicity of core (conserved) versus accessory (variable) genes in an organism was a natural fit for rationalizing differences in pathogenic potential, host range, and other variable phenotypes. Put simply, a core genome in some sense defined a species by providing conserved phenotypes and responses; of prime practical interest were those that medical microbiology leveraged to perform species identification in the lab. For an individual strain, the accessory genome (in other words, the subset of the pan-genome found in that individual above and beyond the core genome) could vary from other individuals, and would explain differences in observed phenotypes such as virulence; alternatively, as more genomes became sequenced, it became clear that sometimes gene set differences could also be related to different ecological niches, such as different geographical locations or host organisms. It was also further shown that large (up to 334 kb) chromosomal segments, including these islands, could be transferred horizontally between strains (Brochet et al., 2008).
The initial promise of the pan-genome concept for providing a holistic organizational framework for individual bacterial species, however, was difficult to fulfill. The initial eight GBS strains that were sequenced and analyzed were chosen for their representation of different serotypes and host organisms, both proxies for sampling the diversity of the species and its disease-relevant phenotypes (Tettelin et al., 2005). One implicit assumption embedded in this initial analysis was that new GBS genomes would sample similarly new subsets of the species diversity; returning to serotype or host organisms as proxies, larger data sets would inevitably begin to sample (at least some) very similar strains. In other words, the initial analysis looked at eight strains that were first chosen for sequencing because they were of different serotypes and MLSTs; they were chosen to represent the diversity of the species. With hundreds to thousands of genomes, however, an additional strain is unlikely to represent a divergent, previously unsampled clade or subclone. Thus the diversity modeled from eight relatively diverse strains may not be accurate when extrapolated to an arbitrary number of strains. Indeed, the high relatedness of many GBS isolates has become more clear in the observation of frequent clonal expansion of virulent or hypervirulent clones, especially among those causing disease (see examples below in the “Molecular Epidemiology” sections).
In addition, the original pan-genome paper inspired numerous similar analyses on other sets of genome sequences, and not all were limited to gene sequences (Lefébure and Stanhope, 2007; Liu et al., 2013; Kayansamruaj et al., 2015; Puymège et al., 2015; He et al., 2017; Wolf et al., 2018; Wang et al., 2018a); the result of an open pan-genome was consistently found. Beyond the use of genomics to calculate the sizes and “openness” of the core and pan-genomes, many studies performed additional analyses that provided several clear and general insights into overall GBS genome plasticity and evolution. GBS genomes have obviously evolved by recombination, likely driven by large-scale DNA transfers mediated by mobile elements (Bröker and Spellerberg, 2004; Brochet et al., 2008; Sørensen et al., 2010; Da Cunha et al., 2014). There is an interesting dichotomy between very clear evidence of large-scale recombination between different lineages of GBS (Springman et al., 2009; Da Cunha et al., 2014; Teatero et al., 2016; Campisi et al., 2016a) with very little recombination within expanding clones, which instead evolve largely by accumulation of mutations (Brochet et al., 2006; Flores et al., 2015; Almeida et al., 2017; Kalimuddin et al., 2017). Interestingly, there have been several noted instances of serotype switching, likely through recombination and after emergence of a successful lineage, which may confound earlier typing studies (Luan et al., 2005; Brochet et al., 2008; Martins et al., 2010; Bellais et al., 2012; Teatero et al., 2014; Neemuchwala et al., 2016; Wang et al., 2018a). These can occur through large-scale recombinations (most clearly seen in originally Serotype V ST1 strains that have converted to Serotype Ib, II, and IV through apparently single recombinations, ranging from 79 to 200Kbp, encompassing the capsule determining cps locus) (Neemuchwala et al., 2016). Notably, genomics provides the most clear view of this phenomenon, as previous studies using PFGE, MLST, lab-based serotyping, and sequencing of the cps locus estimated potential serotype switching events from 2 to 16% (Luan et al., 2005; Martins et al., 2010).
Therefore, the overall picture of GBS evolution is consistent with the hypothesis, most clearly articulated by the lab of Philippe Glaser, of a continuous generation of new lineages, through any mutational mechanism including mobile element activity, reductive evolution, or large-scale recombination, coupled with nearly clonal evolution of successful lineages, characterized by very little recombination and possibly by reductive evolution (Brochet et al., 2006, 2008; Lefébure and Stanhope, 2007; Sørensen et al., 2010; Rosinski-Chupin et al., 2013; Almeida et al., 2016). There are several additional strong results that provide insights into the mechanisms of subspecies adaptation. Notable examples are the consistent genome reduction in fish-adapted isolates (see Section “Group B Streptococcus Molecular Epidemiology in Fish”) (Liu et al., 2013; Rosinski-Chupin et al., 2013; Delannoy et al., 2016); the consistent presence of the scpA and lmb virulence factors in human isolates (though they are still found, at lower frequency, in animal isolates) (Franken et al., 2001; Sørensen et al., 2010); and the development of metabolic modifications matched to expected nutrient sources, exemplified by the acquisition of the Lac.2 operon enabling lactose fermentation in cow-associated strains (Richards et al., 2013).
As of this writing (March, 2019), there are over 7,000 GBS strains for which genome sequencing data are publicly available in the GenBank Sequence Read Archive (SRA). As with other microorganisms, the number of data sets has grown exponentially over the past few years, and the literature contains reports of several notable survey studies that together have contributed several thousand data sets (Metcalf et al., 2017), though several of these appear not to have been published in manuscripts yet (Table 2). Furthermore, the advent of journals like Genome Announcements, which publishes only genome sequences without analysis, has led to a large growth in manuscripts describing single or multiple genome sequences, and genome sequencing is being more routinely used as a tool (as opposed to the main endpoint of a study) (Table 3).
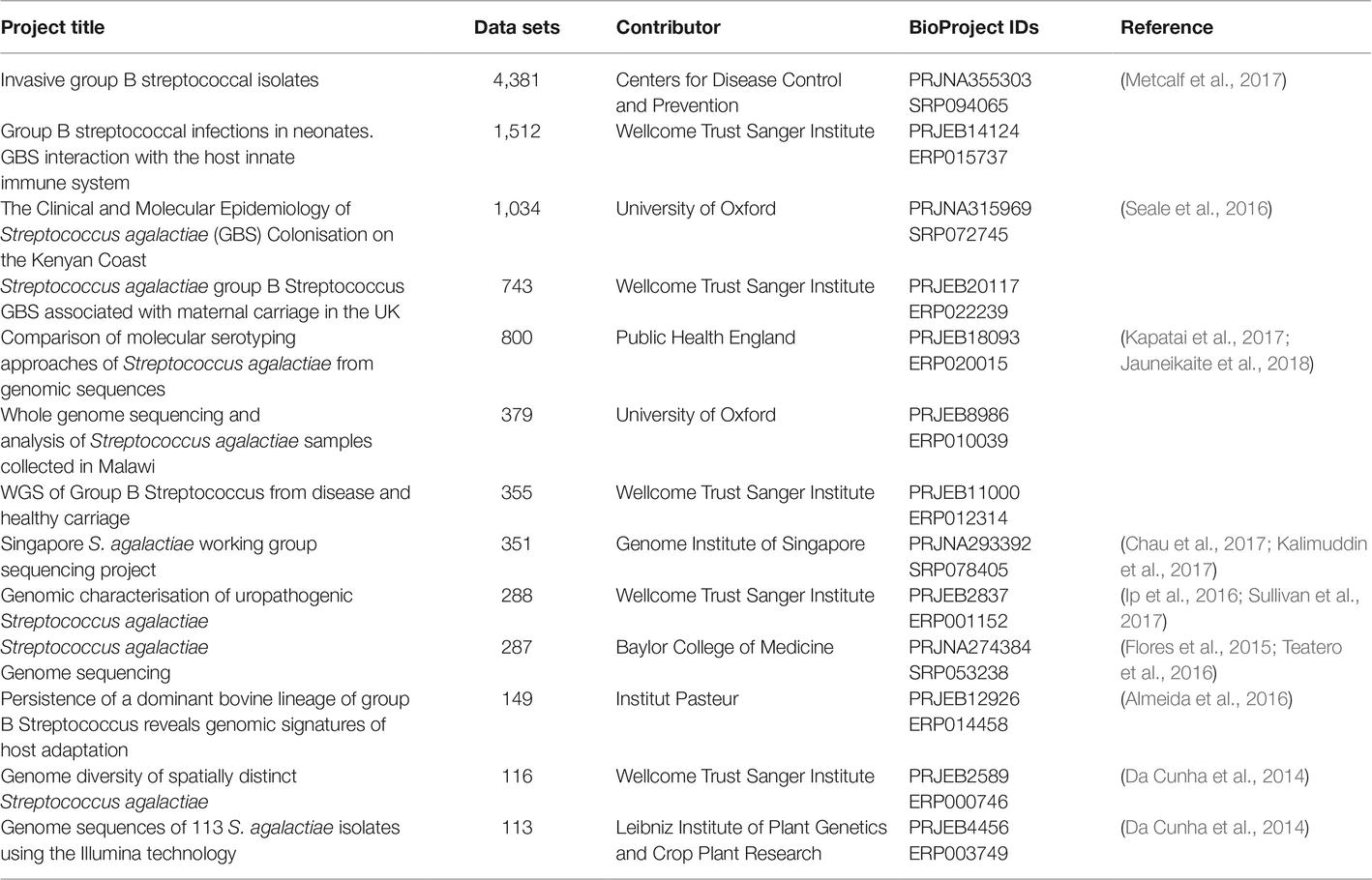
Table 2. Large BioProjects listed in GenBank with more than 100 Illumina whole genome sequencing data sets for GBS (As of March 7, 2019).
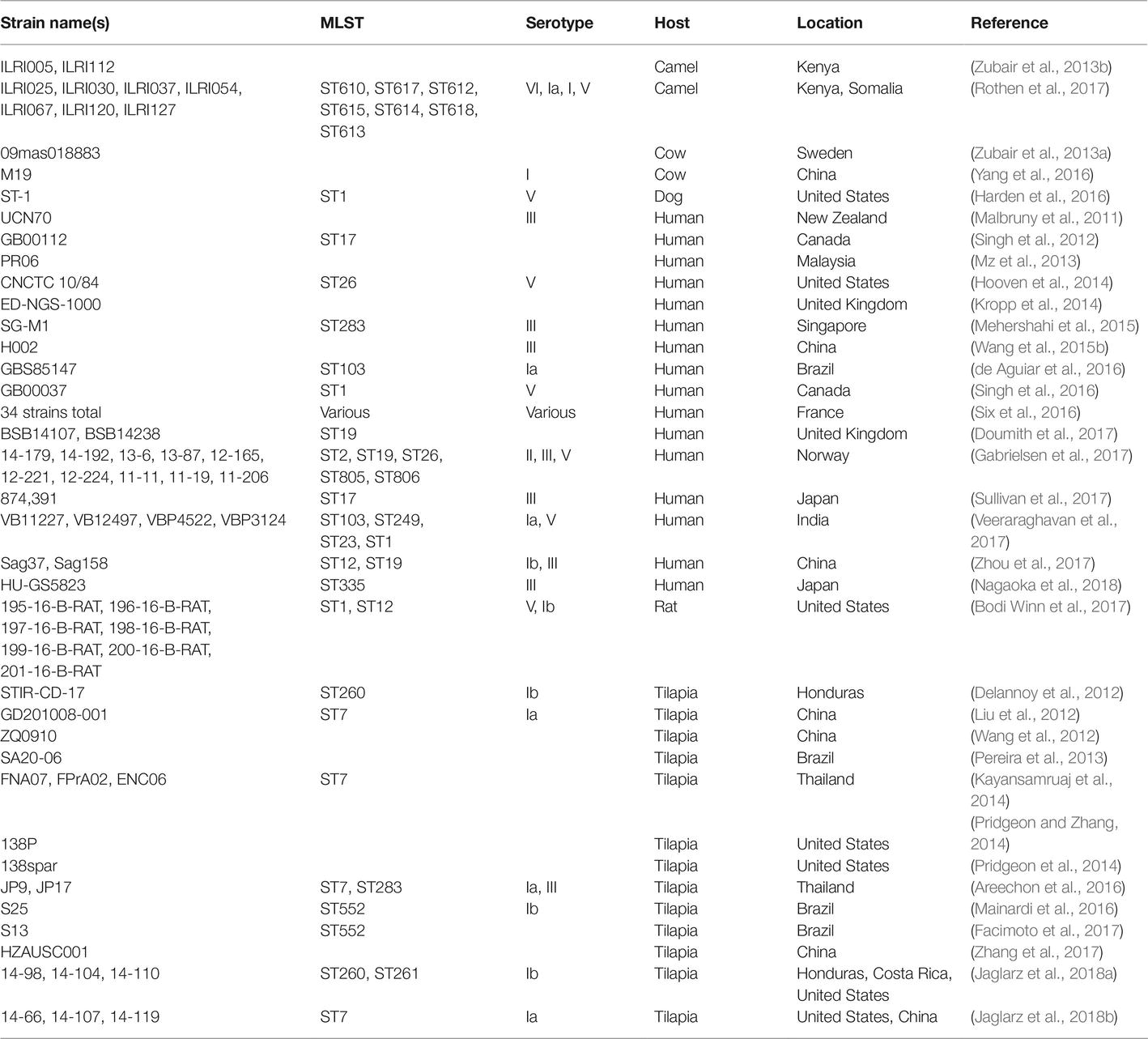
Table 3. GBS genome sequencing reports.
One of the major solutions that increases in genome sequencing throughput has delivered is definitive molecular epidemiology (Klemm and Dougan, 2016). The bacterial genomics field has variously described this also as phylogeography, phylogenomics, or global population structure studies; but the underlying concept of correlating strain relatedness with some other variable, such as isolation location, remains the same. The first major study to present enough sequences that were reasonably thought to represent nearly the full species diversity was published in 2014 (Da Cunha et al., 2014). This manuscript analyzed 229 strains, mostly (94%) isolated from humans, but also including 13 isolates from four other animals, all encompassing five different continents. This for the first time provided a global view of the species that could integrate the preceding MLST scheme. One prominent result from this analysis was the definitive conclusion that human GBS disease isolates arise from a limited number of clones. The clonal evolution and spread of individual clones was already strongly suggested by previous MLST studies, which had already identified clonal complexes that were variously associated with human and animal disease (Table 1). Interestingly, unlike several other bacterial pathogens, the distribution of GBS clones was not generally correlated with geography. There were some known exceptions [like the prevalence of CC26 in Africa (Brochet et al., 2009)], and the sampling of South America, Africa, and Asia was extremely low, both in this study and generally in the GBS literature (Dagnew et al., 2012; Johri et al., 2013; Kwatra et al., 2016). The overall conclusion from this first look, however, was that most of the major GBS CCs causing human disease had relatively low geographical stratification when compared with other surveyed bacteria.
A common theme for GBS, however, is that the general rules for the overall population are punctuated with notable exceptions. There has not been a similarly large-scale (i.e., species-wide) phylogeographic study of GBS since the Da Cunha 2014 publication. Instead, deeper analysis of individual clones, often with strong geographic biases, have added new perspectives to the distribution (and possibly evolution) of GBS in humans, and there has been encouraging recent progress in previously undersampled areas (not all using genomics) (Johri et al., 2013; Crespo-Ortiz et al., 2014; Dutra et al., 2014; Louthrenoo et al., 2014; Mitima et al., 2014; Belard et al., 2015; Bergal et al., 2015; Dangor et al., 2015; Eskandarian et al., 2015; Rivera et al., 2015; Villanueva-Uy et al., 2015; Wang et al., 2015, 2016; Le Doare et al., 2016; Seale et al., 2016, 2017; Sinha et al., 2016; Campisi et al., 2016b; Medugu et al., 2017; Slotved et al., 2017; Suhaimi et al., 2017; Veeraraghavan et al., 2017; Botelho et al., 2018; Emaneini et al., 2018; Guo et al., 2018; Li et al., 2018; Melo et al., 2018; Nkembe et al., 2018; Sigaúque et al., 2018; A’Hearn-Thomas et al., 2019; Lee et al., 2019; Mukesi et al., 2019; Nagano et al., 2019). However, as an example of the progress still needed, I could find only one study of human GBS each from Indonesia and the Philippines, and none from Vietnam, in Pubmed (Wibawan et al., 1992; Villanueva-Uy et al., 2015); Indonesia is the fourth most populous country in the world, and all three of these countries have populations that are larger than any individual European country.
There are several remarkable examples of what appears to be a single dominant clone of GBS causing the majority of disease in specific locations. From 1992 to 2013, more than 90% (210/229) of the invasive serotype V isolates were closely related isolates of an ST1 clone (however, this study did not examine other serotypes) (Flores et al., 2015). A similar serotype-restricted study found a rising incidence of serotype IV isolates in Minnesota from 2004 to 2008 [8.4% of 1,160 patients (Diedrick et al., 2010) compared to <1% of nearly 3,000 isolates from 1993 to 2002 from four cities including Minneapolis-St. Paul (Ferrieri et al., 2004)]. Subsequent genomic studies, again encompassing strains from Minnesota as well as Manitoba and Saskatchewan, Canada, determined that 89% of the serotype IV isolates from 2010 to 2014 were from the same clone of ST459 (Teatero et al., 2015a). Interestingly, in the geographically distant Toronto, where serotype V isolates were dominated by ST1, 81% of serotype IV isolates collected from 2009 to 2012 were comprised of just two STs, ST452 (CC23) and ST459 (CC1) (Teatero et al., 2015b), the same major STs previously found in Minneapolis (Diedrick et al., 2010). Additionally, there has recently been another dramatic reported expansion of a single clone causing human disease: ST283 (serotype III) in Southeast Asia (Ip et al., 2006; Kalimuddin et al., 2017) (see section “Emerging Group B Streptococcus Disease”).
The headline result from the Da Cunha et al. (2014) analysis was that resistance to tetracycline in GBS drove its increased importance for human disease (Da Cunha et al., 2014). All of the major clonal complexes infecting humans had a high (>90%) rate of tetracycline resistance, mostly mediated by the tetM gene. The high rate of tetracycline resistance was thought to be due to initial acquisition through a mobile genetic element (Tn916 or Tn5801 in all but one strain) then expansion of a subsequent clone. Importantly, the insertion position of the transposon was identical within all strains of the expanded clones causing human disease. A Bayesian analysis predicted that the divergence date of the expanded tetracycline-resistant clones corresponded well with the introduction of tetracycline for clinical use in 1948 (Da Cunha et al., 2014). This raised the possibility that the increasing virulence of GBS, or at least the rise in GBS cases, was caused by the simultaneous selection for more virulent and more tetracycline-resistant strains (Da Cunha et al., 2014).
Rising resistance is a nearly universal feature of medically important bacteria. For GBS, this has been reported not only for tetracycline, but for also for fluoroquinolones and aminoglycosides (Hays et al., 2016). Fortunately, beta-lactam antibiotics, particularly penicillins, which are first-line therapy for GBS, have remained highly effective, with large surveys documenting less than 1% of strains as resistant (Hays et al., 2016; Metcalf et al., 2017). Interestingly, vancomycin resistance has recently been reported for the first time in two GBS strains, through introduction of two different vanG elements found integrated at the same chromosomal locus in both strains (Srinivasan et al., 2014). Finally, macrolide resistance, most commonly measured for erythromycin, has also been rising, with rates measured since 2010 in the range of 14–59% (Lamagni et al., 2013; Da Cunha et al., 2014; Hays et al., 2016; Metcalf et al., 2017). Of great interest, a large survey in France found an exception to this trend, with rates of macrolide resistance falling from 47 to 30% between 2007 and 2014 (Hays et al., 2016). Many of these surveys leverage strong antibiotic susceptibility testing infrastructure in first-world countries; however, this is beginning to give way to genomic predictions that may eventually provide solutions for low resource settings. With regard to this, GBS-specific analyses for the prediction of antibiotic resistance and serotypes from genomic data were shown to provide high accuracy (Metcalf et al., 2017). Genomics currently cannot fully replace traditional antibiotic testing, as previously unknown (or rare) resistance mechanisms cannot be predicted from sequence data alone. However, genomics has the additional advantage of providing greater insight into the dynamics driving spread of resistance and changes in resistance rates. For example, antibiotic resistance in GBS is largely mediated by resistance gene acquisition for all of the major antibiotic classes except for fluoroquinolones, which instead arise mostly by mutations in the gyrA and parC genes and penicillins (Metcalf et al., 2017). In addition, there are multiple examples of rising resistance rates being associated with expansion of individual clones, as seen for tetracycline as described above (Da Cunha et al., 2014), erythromycin resistance in ST1 (Flores et al., 2015), and beta-lactam resistance in several examples of closely related isolates (Metcalf et al., 2017). Overall, therefore, genomics paints a general overall picture similar to that described for clonal emergence above; antibiotic resistance is initially acquired through horizontal transfer or possibly recombination (also enabling acquisition of fluoroquinolone or penicillin resistance), followed by clonal expansion of successful lineages that drive increases in antibiotic resistance rates.
As in other bacteria, mobile genetic elements are often associated with more than just antibiotic resistance genes. Many of these additional genes have features that suggest they may be involved in virulence, such as surface attachment signals (LPXTG), predicted surface localization, homology to adhesin proteins, novel metabolic activities, or predicted secretion and toxicity. One well-described example is the co-occurrence of AlpST-1, a predicted surface-exposed adhesin protein, that is encoded within the same mobile genetic element (denoted the RDF.2 MGE) as the tetM-carrying Tn916 in a collection of 202 ST1, serotype V strains from the US and Canada (Flores et al., 2015). In this clone, the authors argue that the close genetic linkage between tetM and the AlpST-1 virulence gene could account for the association between tetracycline resistance and virulence, so the virulence would not be due to the tetracycline resistance per se (Flores et al., 2015).
The concept that antibiotic resistance is associated with strains with high pathogenic potential for humans (and other animals) is not controversial. Indeed, therapeutic and agricultural antibiotic usage is perhaps one of the strongest influences that humans have exerted on the makeup of our microbial environment. However, we typically associate antibiotic resistance with a loss of fitness in bacteria, which is then overcome by the strong selection pressure of antibiotic administration (though numerous counterexamples exist). Antibiotic resistance is generally not considered a virulence determinant in and of itself. The observation of high tetracycline resistance rates, independently acquired in multiple lineages of common human GBS strains (CC1, 10, 17, 19, 23, and 26), is indeed an incontrovertibly strong demonstration that tetracycline use has dominated the evolution of GBS in humans (Da Cunha et al., 2014). As in the case of ST1, however, the underlying pathogenic mechanisms, which can lead to a stronger understanding of disease and novel strategies for prevention (Flores et al., 2015), remain the central unanswered question for essentially all GBS lineages.
In contrast to the idea that most of the GBS population was not strongly stratified by geography, it has long been known that GBS demonstrates relatively strong host specificity. In addition to human disease, the economic impact of lower milk production due to mastitis has driven substantial S. agalactiae research (Keefe, 1997; Ruegg, 2017). Substantial work, therefore, has also examined the potential commonality in the rise of human and cattle infections, most obviously mediated through milk and close contact with dairy farmers (Bliss et al., 2002; Bohnsack et al., 2004; Sukhnanand et al., 2005; Foxman et al., 2007; Manning et al., 2010).
The literature describing GBS that infects bovine hosts is extensive, in keeping with the initial S. agalactiae nomenclature (which refers to its effects on dairy cows). As with human isolates, pre-genomic techniques had already been used to sketch a general outline of the population structure. Bovine isolates generally fall into two main clusters, represented in the MLST scheme by CC67 and CC23. A significant minority of strains were found to fall into CCs that overlapped with human isolates, most prominently CC17 (Sørensen et al., 2010). As seen with human GBS, however, there are examples of recent changes in prevalent GBS clones which may differ based on geography. A recent rise in CC61 strains was noted in Portugal, with an origin estimated in the early 1990s (Almeida et al., 2016). From 84 bovine isolates collected from milk from 14 dairy farms in China between 2011 and 2016, all were either CC61 or CC103 (Pang et al., 2017). Interestingly, CC103 strains have also been noted in Denmark and Norway, but not the UK or US (Bisharat et al., 2004; Zadoks et al., 2011; Jørgensen et al., 2016).
The fact that GBS was known first as a bovine pathogen and only later noted to cause increasing rates of neonatal and adult disease led to a strong interest in exploring a potential zoonotic transmission from cows to humans (and vice versa). There was suggestive increase in GBS colonization rate among university students who drank milk (n = 150), but this was not statistically significant (Bliss et al., 2002). A larger, longitudinal study (3-week intervals over 3 months) in a similar population found no significant association between GBS colonization and beef or milk consumption; notably, there were positive associations with sexual activity and fish consumption (Foxman et al., 2007). Another epidemiological study examined the colonizing GBS in humans who had regular close contact with cattle. Of 68 matched human-cattle stool samples, one set had the same GBS as measured by MLST, capsule, RAPD pattern, and antibiotic resistance profile. Of note, the cattle sampled were not symptomatic, and stool is not a typical sampling location in cattle. Perhaps more interestingly, human colonization was significantly associated with exposure to cattle in the previous week (Manning et al., 2010). Overall, therefore, transmission between humans and cattle seemed rare if not nonexistent.
The conclusion that the human pathogenic CC17 lineage was derived from a bovine GBS ancestor was therefore a dramatic result (Bisharat et al., 2004). This initial report was based on an analysis of MLST gene sequences. In what has become a repeated testament to the value of whole genome analyses, this initial MLST result has been challenged by subsequent genomic studies (Brochet et al., 2006; Sørensen et al., 2010; Richards et al., 2013). Comparative whole genome hybridization on microarrays to assess gene content in a collection of 75 strains from humans and multiple animal hosts indicated that the common ancestor of the bovine ST61 and ST17 strains was likely more similar to a human ST17 strain, implying the reverse transmission direction (Brochet et al., 2006). An examination of 15 genes (including the seven MLST genes) in addition to several virulence-associated traits in a representative set of 55 strains (drawn from 238 in total) showed differences between the human and bovine CC17 strains that were not captured by the MLST genes. Combined with differences between human and bovine strains with respect to the ability to ferment lactose, the presence of the human-associated scpB virulence gene, and the presence of the PI-1 pilus island, the authors concluded that human CC17 strains evolved separately, perhaps from a common diverse pool, from the bovine CC17 isolates (Sørensen et al., 2010). Finally, whole genome sequencing data from 202 strains isolated from human and animal hosts was used as part of an analysis aimed at understanding the factors responsible for survival in the bovine mammary gland. Acquisition of the Lac.2 operon, which enables fermentation of lactose (the primary sugar in cow milk), was a consistent feature of bovine strains. In contrast, among ST17 strains of human and bovine origin, none of the human strains had the Lac.2 operon, which was interpreted as unlikely if a bovine strain was the ancestor of the human isolates (Richards et al., 2013). Notably, as with the association of human strains with presence of the scpA and lmb genes noted above, the association of the Lac.2 operon with bovine isolates, while strong, is not exclusive; 8/151 human strains in one survey carried Lac.2 (Richards et al., 2013), though for some strains, such as NEM316, the origin (possibly S. gordonii) may be distinct from the origin suspected in bovine strains (S. dysgalactiae subsp. dysgalactiae) (Richards et al., 2011). Therefore, with the benefit of larger scale genomic data, it still appears that bovine and human strains are largely (if not completely) separate, at least in terms of the severe disease-causing strains, though the possibility of overlap continues to be discussed (Lyhs et al., 2016; Wang et al., 2018b). There seem to be cases of colonization in both directions, but this appears to be transient in both cattle and humans (Jensen, 1982; Betsy Foxman et al., 2006). On a longer evolutionary scale, however, the genomic data are clear that some bovine- and human-adapted lineages do at least share common ancestors, most notably for CC17 and CC67 (Sørensen et al., 2010; Pang et al., 2017).
The next most extensively studied host organism for GBS is fish. Again, much of the motivation for this research has been economic interest, as Streptococcosis is a major disease affecting farmed fish (Amal et al., 2011). In fish, GBS are generally limited to certain MLST types, which leads to their occasional reference in this literature as biotypes, with a strong association with serotype. One early study proposed two new species, S. shiloi and S. difficile, as pathogens of fish in Israel starting in 1986 (Eldar et al., 1994). These were later resolved to S. iniae and S. agalactiae (serotype Ib), respectively (Eldar et al., 1995; Vandamme et al., 1997). Subsequently, the fish literature began to refer to biotypes of GBS, which persists today in the marketing for fish vaccines1. Biotype I corresponds to serotype Ia, ST7, and is mostly found in Asia. Biotype II corresponds with serotype Ib, ST260 or ST261 (and is referred to as CC552, though this has recently been refined), and is found generally throughout the globe (Delannoy et al., 2013; Paul, 2014; Munang’andu et al., 2016; Barony et al., 2017).
Similar to the situation with bovine GBS, one major topic of research has been the possibility of cross-species infections between humans and fish. Reliance on MLST again indicated that there was evidence for transmission: ST7 is found in both humans and fish (Evans et al., 2008; Delannoy et al., 2013), as well as other aquatic animals, and a large outbreak of fish infections in Kuwait Bay was thought to be due to human GBS isolates entering the water through sewage contamination (Jafar et al., 2008). Three genomic studies later examined human and fish ST7 strains more closely. In one study, the human and fish ST7 strains were very similar, with similar gene content and a uniformly low nucleotide divergence throughout the genome (Rosinski-Chupin et al., 2013). A second study noted that human and piscine ST7 strains were closely related based on genome content (CRISPR arrays, prophages, and virulence-associated genes) (Liu et al., 2013). The third study, which included fish strains from both CC7 and CC552, identified eight genomic regions, mostly located within genomic islands, containing genes associated with or specific to piscine strains, which were then confirmed by PCR screening across a larger collection of 43 isolates from various hosts (Delannoy et al., 2016). In addition, strains of ST23, which are common human pathogens, were unable to cause experimental infections in tilapia, while one of two human ST1 strains was equally pathogenic to tilapia compared a bona fide piscine ST7 isolate (Delannoy et al., 2016; Wang et al., 2017). Overall, therefore, it appears that much of the population of piscine and human isolates are largely separate, but at least some strains of CC7, may be able to infect both hosts. Another common theme is the idea that host specificity has evolved through genome reduction, where genes and pathways no longer needed in other environments (hosts such as humans) are lost once GBS specializes to colonize fish, most clearly demonstrated in CC7 and CC552 (Liu et al., 2013; Rosinski-Chupin et al., 2013; Delannoy et al., 2016). For example, several studies found multiple fish strains with genome sizes in the 1.7–1.8 Mbp range, compared with 2.0–2.2 Mbp for many human isolates (Liu et al., 2013; Rosinski-Chupin et al., 2013). This 200–300 Kbp accounts for over 100 otherwise “core” genes lost in fish isolates, which are enriched for carbohydrate transport and metabolism functions (Liu et al., 2013). Interestingly, another study of human ST19 strains found that at least some are able to cause experimental disease in tilapia, a phenotype that correlates with capsule type (Wang et al., 2018a).
One of the notable features of the research on piscine GBS is that the sampling covers a complementary geographic range to the human and much of the cattle GBS literature. Aquaculture is a rapidly growing business in South America, the Middle East, and Asia, particularly Southeast Asia; these are all areas that have been notably undersampled in human-focused GBS studies. In these areas, it appears that many of the major food fishes, found in both freshwater and saltwater, can be affected (including rainbow trout, seabream, tilapia, yellowtail, catfish, croaker, killfish, and pomfret) (Amal et al., 2011). Interestingly, our knowledge of the population structure of piscine GBS is actually stronger than that for human disease-causing isolates in many countries, for a variety of reasons including economics, infrastructure, and the assumption that seemingly globally distributed clones (for human disease) would be similarly dominant in unsampled regions. Accordingly, there are potentially emerging clones of piscine GBS that are being reported (such as Serotype III and IX in Southeast Asia and China, respectively) (Kalimuddin et al., 2017; Zhang et al., 2018).
The emerging GBS clone in Southeast Asia, ST283 (Serotype III), neatly intertwines the previously mentioned themes of ongoing GBS evolution (leading to potential cross-species host jumps), the potential for some clones to have strong geographical associations, and the value of genomics for enabling integration across different disciplines. Emerging clones such as ST283 therefore challenge our understanding of GBS ecology, evolution, disease, and management.
In 2015, Singapore experienced an outbreak of foodborne GBS infections. Associated with consumption of a local raw fish dish (魚生, yu sheng), over 200 patients suffered severe invasive disease, with bacteremia, meningitis, and septic arthritis (Rajendram et al., 2016; Tan et al., 2016; Kalimuddin et al., 2017)2. Prior to this outbreak, GBS had never been thought to be transmitted by the foodborne route. In retrospect, there had been indications that food consumption was associated with gastrointestinal colonization (Bliss et al., 2002; Foxman et al., 2007), and there had been an example of lizards contracting GBS sepsis through consuming contaminated mice (Hetzel et al., 2003). Furthermore, it seems clear that GI colonization by any organism in humans seems reasonable to assign to oral consumption until proven otherwise. Given that GBS is a common GI colonizer of both men and women, then, one interpretation is that even early onset neonatal meningitis is ultimately a foodborne disease (via vaginal colonization through the GI tract, then infecting the newborn during birth).
Interestingly, the outbreak organism, ST283, is a common MLST type that causes aquaculture-associated fish Streptococcosis. An ST283 strain and a single locus variant, ST491, had been identified in farmed fish in Vietnam and Thailand, isolated in the early 2000s (Delannoy et al., 2013). ST283 strains had also been reported to cause invasive infections similar to those seen in the 2015 Singapore outbreak in otherwise healthy humans from Hong Kong and Singapore as early as 1998 (Wilder-Smith et al., 2000; Ip et al., 2006, 2016; Barkham et al., 2018). Investigating after the 2015 Singapore outbreak, it was found that 71% of freshwater fish sold for raw consumption (which was associated with the outbreak) in Singapore carried GBS, with 14% carrying ST283; in contrast, 9–33% of saltwater fish carried GBS, with none being ST283 (Chau et al., 2017). Singapore imports the vast majority (>90%) of its food, including fish3; indeed, 4.6% of fish samples were already positive for GBS, with 1% of fish positive for ST283, at entry ports to Singapore (Chau et al., 2017), leading to a suspicion that regional aquaculture fish may also be colonized by ST283 (while ST283 is known to cause disease outbreaks in farmed fish, the fish being imported and sold appeared healthy). Troublingly, a recent report has identified ST283 as a cause of GBS outbreaks in at least five fish farms in 2016–2017 from four different states in Brazil, and this is suspected to be due to import of fish from Southeast Asia (Leal et al., 2019).
ST283 was originally described an emerging cause of invasive infections in Hong Kong and Singapore (Ip et al., 2006). It was only identified as a potential foodborne pathogen in 2015 (Rajendram et al., 2016; Tan et al., 2016; Kalimuddin et al., 2017). Epidemiological data for human infections are not consistently available in many countries in Southeast Asia, but recent data suggest that the endemic incidence of ST283 infections outside Singapore may be comparable to or higher than the incidence at the peak of the Singapore outbreak (Barkham et al., 2019; https://outbreakwatch.blogspot.com/2018/07/proahedr-strep-group-b-singapore.html). If ST283 is also a foodborne pathogen outside Singapore, as recently suspected in Thailand (Kayansamruaj et al., 2018), this suggests the remarkable interpretation that GBS infections in Southeast Asia are actually first and foremost a foodborne illness (in stark contrast to the current paradigm of neonatal and immunocompromised disease found in current medical teaching). This further raises the possibility that invasive GBS disease caused by non-ST283 strains in human adults may be at least partially foodborne, expanding our understanding of GBS in general as a possibly long unappreciated zoonotic pathogen, with attendant implications for food safety. As food consumption is associated with vaginal colonization in women (Foxman et al., 2007), a further implication is that neonatal infections may also ultimately be a late sequelae of foodborne consumption of GBS. Interestingly, there is a case report of a late-onset neonatal infection caused by a genomically indistinguishable strain that was consumed by the mother via placental pills (Buser et al., 2017).
GBS has a uniquely dynamic biology. At the overall species level, there is remarkably broad host range and global geographical reach. Standing in counterpoint to this broad generalism are numerous examples of strong host specificity and local geographical stratification. The source of this intraspecies heterogeneity likely traces back to similarly dynamic processes shaping the genome, with the capacity for large-scale chromosomal recombinations paired with highly independent, clonal evolution of individual successful lineages. GBS is not only an emerging pathogen in the traditional sense of rising incidence; a closer examination of its biology indicates that it is a continuously emerging pathogen that, in little more than a single human lifetime, has altered and continues to alter the tenets of epidemiology of human, bovine, and piscine colonization, infection, and economics. Since the first GBS genome sequences, genomics has been an ideal technology to capture the dynamism of this species. It therefore seems apt that GBS was the organism that birthed the concept of the pan-genome; its description as having an open pan-genome, with infinite possibility for evolution and adaptation, is a fitting metaphor for the recent history of GBS. GBS, in turn, provides a compelling argument for the continued progress of genomics and our need, as a community, to implement broad genomic monitoring in both developed and developing countries. The recent genomic history has demonstrated shifts in our understanding of GBS with respect to cattle, human neonates, human adults, aquacultured fish, and now the interface between fish and humans and food safety and economic development. Where else is GBS lurking? Further advances in genomics will hopefully enable us not only to reconstruct post hoc what GBS will do next, but to catch it in the early stages of its next evolutionary jump.
The author confirms being the sole contributor of this work and has approved it for publication.
The author was supported by the National Medical Research Council, Ministry of Health, Singapore (grant numbers NMRC/OFIRG/0009/2016 and NMRC/CIRG/1467/2017); the National Research Foundation, Singapore (NRF2018-ITS003-008); the Temasek Foundation Innovates through its Singapore Millennium Foundation Research Grant Programme; and the Genome Institute of Singapore (GIS)/Agency for Science, Technology and Research (A*STAR).
SC is a co-inventor on a patent application for a GBS ST283-specific test.
I would like to thank Timothy Barkham and Ruth Zadoks for their education on aspects of GBS microbiology and genomics; the members of the Chen lab for useful discussions about the ideas presented herein and for their constant dedication and support; and Hsu Li Yang and Koh Tse Hsien for their initial role, with Timothy Barkham, in convincing me to learn and care about GBS.
1. https://www.aquavac-vaccines.com/products/
2. http://10minus6cosm.tumblr.com/post/132590510556/serious-group-b-streptococcal-infection-in-adults
A’Hearn-Thomas, B., Khatami, A., Randis, T. M., Vurayai, M., Mokomane, M., Arscott-Mills, T., et al. (2019). High rate of serotype V Streptococcus agalactiae carriage in pregnant women in Botswana. Am. J. Trop. Med. Hyg. 100, 1115–1117. doi: 10.4269/ajtmh.18-0847
Almeida, A., Alves-Barroco, C., Sauvage, E., Bexiga, R., Albuquerque, P., Tavares, F., et al. (2016). Persistence of a dominant bovine lineage of group B Streptococcus reveals genomic signatures of host adaptation. Environ. Microbiol. 18, 4216–4229. doi: 10.1111/1462-2920.13550
Almeida, A., Rosinski-Chupin, I., Plainvert, C., Douarre, P.-E., Borrego, M. J., Poyart, C., et al. (2017). Parallel evolution of group B Streptococcus hypervirulent clonal complex 17 unveils new pathoadaptive mutations. mSystems 2, e00074–e00017. doi: 10.1128/mSystems.00074-17
Amal, M. N. A., Zamri-Saad, M., et al. (2011). Streptococcosis in Tilapia (Oreochromis niloticus): a review. Pert. J. Trop. Agri. Sci. 34, 195–206. http://www.pertanika.upm.edu.my/view_archives.php?journal=JTAS-34-2-8
Areechon, N., Kannika, K., Hirono, I., Kondo, H., and Unajak, S. (2016). Draft genome sequences of Streptococcus agalactiae serotype Ia and III isolates from tilapia farms in Thailand. Genome Announc. 4. doi: 10.1128/genomeA.00122-16
Barkham, T., Sheppard, A., Jones, N., and Chen, S. L. (2018). Streptococcus agalactiae that caused meningitis in healthy adults in 1998 are ST283, the same type that caused a food-borne outbreak of invasive sepsis in 2015: an observational molecular epidemiology study. Clin. Microbiol. Infect. 24, 923–925. doi: 10.1016/j.cmi.2018.04.006
Barkham, T., Zadoks, R. N., Azmai, M. N. A., Baker, S., Bich, V. T. N., Chalker, V., et al. (2019). One hypervirulent clone, sequence type 283, accounts for a large proportion of invasive Streptococcus agalactiae isolated from humans and diseased tilapia in Southeast Asia. PLoS Negl. Trop. Dis. (in press).
Barony, G. M., Tavares, G. C., Pereira, F. L., Carvalho, A. F., Dorella, F. A., Leal, C. A. G., et al. (2017). Large-scale genomic analyses reveal the population structure and evolutionary trends of Streptococcus agalactiae strains in Brazilian fish farms. Sci. Rep. 7:13538. doi: 10.1038/s41598-017-13228-z
Belard, S., Toepfner, N., Capan-Melser, M., Mombo-Ngoma, G., Zoleko-Manego, R., Groger, M., et al. (2015). Streptococcus agalactiae serotype distribution and antimicrobial susceptibility in pregnant women in Gabon, Central Africa. Sci. Rep. 5:17281. doi: 10.1038/srep17281
Bellais, S., Six, A., Fouet, A., Longo, M., Dmytruk, N., Glaser, P., et al. (2012). Capsular switching in group B Streptococcus CC17 hypervirulent clone: a future challenge for polysaccharide vaccine development. J. Infect. Dis. 206, 1745–1752. doi: 10.1093/infdis/jis605
Berardi, A., Tzialla, C., Riva, M., Cerbo, R. M., and Creti, R. (2007). Group B Streptococcus: early- and late-onset infections. J. Chemother. 19(Suppl. 2), 24–27.
Bergal, A., Loucif, L., Benouareth, D. E., Bentorki, A. A., Abat, C., and Rolain, J.-M. (2015). Molecular epidemiology and distribution of serotypes, genotypes, and antibiotic resistance genes of Streptococcus agalactiae clinical isolates from Guelma, Algeria and Marseille, France. Eur. J. Clin. Microbiol. Infect. Dis. 34, 2339–2348. doi: 10.1007/s10096-015-2487-6
Bisharat, N., Crook, D. W., Leigh, J., Harding, R. M., Ward, P. N., Coffey, T. J., et al. (2004). Hyperinvasive neonatal group B Streptococcus has arisen from a bovine ancestor. J. Clin. Microbiol. 42, 2161–2167. doi: 10.1128/JCM.42.5.2161-2167.2004
Bishop, E. J., Shilton, C., Benedict, S., Kong, F., Gilbert, G. L., Gal, D., et al. (2007). Necrotizing fasciitis in captive juvenile crocodylus porosus caused by Streptococcus agalactiae: an outbreak and review of the animal and human literature. Epidemiol. Infect. 135, 1248–1255. doi: 10.1017/S0950268807008515
Bliss, S. J., Manning, S. D., Tallman, P., Baker, C. J., Pearlman, M. D., Marrs, C. F., et al. (2002). Group B Streptococcus colonization in male and nonpregnant female university students: a cross-sectional prevalence study. Clini. Infect. Dis. 34, 184–190. doi: 10.1086/338258
Bodi Winn, C., Dzink-Fox, J., Feng, Y., Shen, Z., Bakthavatchalu, V., and Fox, J. G. (2017). Whole-genome sequences and classification of Streptococcus agalactiae strains isolated from laboratory-reared long-evans rats (Rattus norvegicus). Genome Announc. 5. doi: 10.1128/genomeA.01435-16
Bohnsack, J. F., Whiting, A. A., Martinez, G., Jones, N., Adderson, E. E., Detrick, S., et al. (2004). Serotype III Streptococcus agalactiae from bovine milk and human neonatal infections. Emerg. Infect. Dis. 10, 1412–1419. doi: 10.3201/eid1008.030917
Botelho, A. C. N., Oliveira, J. G., Damasco, A. P., Santos, K. T. B., Ferreira, A. F. M., Rocha, G. T., et al. (2018). Streptococcus agalactiae carriage among pregnant women living in Rio de Janeiro, Brazil, over a period of eight years. PLoS One 13:e0196925. doi: 10.1371/journal.pone.0196925
Brochet, M., Couvé, E., Bercion, R., Sire, J.-M., and Glaser, P. (2009). Population structure of human isolates of Streptococcus agalactiae from Dakar and Bangui. J. Clin. Microbiol. 47, 800–803. doi: 10.1128/JCM.01103-08
Brochet, M., Couvé, E., Zouine, M., Vallaeys, T., Rusniok, C., Lamy, M.-C., et al. (2006). Genomic diversity and evolution within the species Streptococcus agalactiae. Microbes Infect. 8, 1227–1243. doi: 10.1016/j.micinf.2005.11.010
Brochet, M., Rusniok, C., Couvé, E., Dramsi, S., Poyart, C., Trieu-Cuot, P., et al. (2008). Shaping a bacterial genome by large chromosomal replacements, the evolutionary history of Streptococcus agalactiae. Proc. Natl. Acad. Sci. USA 105, 15961–15966. doi: 10.1073/pnas.0803654105
Bröker, G., and Spellerberg, B. (2004). Surface proteins of Streptococcus agalactiae and horizontal gene transfer. Int. J. Med. Microbiol. 294, 169–175. doi: 10.1016/j.ijmm.2004.06.018
Buser, G. L., Mató, S., Zhang, A. Y., Metcalf, B. J., Beall, B., and Thomas, A. R. (2017). Notes from the field: late-onset infant group B Streptococcus infection associated with maternal consumption of capsules containing dehydrated placenta - Oregon, 2016. Morb. Mortal. Wkly Rep. 66, 677–678. doi: 10.15585/mmwr.mm6625a4
Campisi, E., Daniela Rinaudo, C., Donati, C., Barucco, M., Torricelli, G., Edwards, M. S., et al. (2016a). Serotype IV Streptococcus agalactiae ST-452 has arisen from large genomic recombination events between CC23 and the hypervirulent CC17 lineages. Sci. Rep. 6:29799. doi: 10.1038/srep29799
Campisi, E., Rosini, R., Ji, W., Guidotti, S., Rojas-López, M., Geng, G., et al. (2016b). Genomic analysis reveals multi-drug resistance clusters in group B Streptococcus CC17 hypervirulent isolates causing neonatal invasive disease in Southern Mainland China. Front. Microbiol. 7:1265. doi: 10.3389/fmicb.2016.01265
Chau, M. L., Chen, S. L., Yap, M., Hartantyo, S. H. P., Chiew, P. K. T., Fernandez, C. J., et al. (2017). Group B Streptococcus infections caused by improper sourcing and handling of fish for raw consumption, Singapore, 2015-2016. Emerg. Infect. Dis. 23, 2002–2010. doi: 10.3201/eid2312.170596
Crespo-Ortiz, M. d. P., Castañeda-Ramirez, C. R., Recalde-Bolaños, M., and Vélez-Londoño, J. D. (2014). Emerging trends in invasive and noninvasive isolates of Streptococcus agalactiae in a latin American hospital: a 17-year study. BMC Infect. Dis. 14:428. doi: 10.1186/1471-2334-14-428
Da Cunha, V., Davies, M. R., Douarre, P.-E., Rosinski-Chupin, I., Margarit, I., Spinali, S., et al. (2014). Streptococcus agalactiae clones infecting humans were selected and fixed through the extensive use of tetracycline. Nat. Commun. 5:4544. doi: 10.1038/ncomms5544
Dagnew, A. F., Cunnington, M. C., Dube, Q., Edwards, M. S., French, N., Heyderman, R. S., et al. (2012). Variation in reported neonatal group B streptococcal disease incidence in developing countries. Clini. Infect. Dis. 55, 91–102. doi: 10.1093/cid/cis395
Dangor, Z., Lala, S. G., Cutland, C. L., Koen, A., Jose, L., Nakwa, F., et al. (2015). Burden of invasive group B Streptococcus disease and early neurological sequelae in South African infants. PLoS One 10:e0123014. doi: 10.1371/journal.pone.0123014
de Aguiar, E. L., Mariano, D. C. B., Viana, M. V. C., Benevides, L. de J., de Souza Rocha, F., de Castro Oliveira, L., et al. (2016). Complete genome sequence of Streptococcus agalactiae strain GBS85147 serotype of type Ia isolated from human oropharynx. Stand. Genomic Sci. 11, 39. doi: 10.1186/s40793-016-0158-6
Delannoy, C. M. J., Crumlish, M., Fontaine, M. C., Pollock, J., Foster, G., Dagleish, M. P., et al. (2013). Human Streptococcus agalactiae strains in aquatic mammals and fish. BMC Microbiol. 13:41. doi: 10.1186/1471-2180-13-41
Delannoy, C. M. J., Zadoks, R. N., Crumlish, M., Rodgers, D., Lainson, F. A., Ferguson, H. W., et al. (2016). Genomic comparison of virulent and non-virulent streptococcus agalactiae in fish. J. Fish Dis. 39, 13–29. doi: 10.1111/jfd.12319
Delannoy, C. M. J., Zadoks, R. N., Lainson, F. A., Ferguson, H. W., Crumlish, M., Turnbull, J. F., et al. (2012). Draft genome sequence of a nonhemolytic fish-pathogenic Streptococcus agalactiae strain. J. Bacteriol. 194, 6341–6342. doi: 10.1128/JB.01552-12
Diedrick, M. J., Flores, A. E., Hillier, S. L., Creti, R., and Ferrieri, P. (2010). Clonal analysis of colonizing group B Streptococcus, serotype IV, an emerging pathogen in the United States. J. Clin. Microbiol. 48, 3100–3104. doi: 10.1128/JCM.00277-10
Doumith, M., Mushtaq, S., Martin, V., Chaudhry, A., Adkin, R., Coelho, J., et al. (2017). Genomic sequences of Streptococcus agalactiae with high-level gentamicin resistance, collected in the BSAC bacteraemia surveillance. J. Antimicrob. Chemother. 72, 2704–2707. doi: 10.1093/jac/dkx207
Dutra, V. G., Alves, V. M. N., Olendzki, A. N., Dias, C. A. G., de Bastos, A. F. A., Santos, G. O., et al. (2014). Streptococcus agalactiae in Brazil: serotype distribution, virulence determinants and antimicrobial susceptibility. BMC Infect. Dis. 14:323. doi: 10.1186/1471-2334-14-323
Edelstein, R. M., and Pegram, R. G. (1974). Contagious skin necrosis of Somali camels associated with Streptococcus agalactiae. Trop. Anim. Health Prod. 6, 255–256. doi: 10.1007/BF02383286
Edwards, M. S., Nizet, V., and Baker, C. J. (2011). “CHAPTER 12–group B streptococcal infections” in Infectious diseases of the fetus and newborn. 7th edn. eds. J. S. Remington, J. O. Klein, C. B. Wilson, V. Nizet, and Y. A. Maldonado (Philadelphia: W.B. Saunders), 419–469.
Eldar, A., Bejerano, Y., and Bercovier, H. (1994). Streptococcus shiloi and Streptococcus difficile: two new streptococcal species causing a meningoencephalitis in fish. Curr. Microbiol. 28, 139–143. doi: 10.1007/BF01571054
Eldar, A., Frelier, P. F., Assenta, L., Varner, P. W., Lawhon, S., and Bercovier, H. (1995). Streptococcus shiloi, the name for an agent causing septicemic infection in fish, is a junior synonym of Streptococcus iniae. Int. J. Syst. Bacteriol. 45, 840–842.
Emaneini, M., Jabalameli, F., van Leeuwen, W. B., and Beigverdi, R. (2018). Prevalence of group B Streptococcus in pregnant women in Iran: a systematic review and meta-analysis. Pediatr. Infect. Dis. J. 37, 186–190. doi: 10.1097/INF.0000000000001713
Eskandarian, N., Ismail, Z., Neela, V., van Belkum, A., Desa, M. N. M., and Amin Nordin, S. (2015). Antimicrobial susceptibility profiles, serotype distribution and virulence determinants among invasive, non-invasive and colonizing Streptococcus agalactiae (group B Streptococcus) from Malaysian patients. Eur. J. Clin. Microbiol. Infect. Dis. 34, 579–584. doi: 10.1007/s10096-014-2265-x
Evans, J. J., Bohnsack, J. F., Klesius, P. H., Whiting, A. A., Garcia, J. C., Shoemaker, C. A., et al. (2008). Phylogenetic relationships among Streptococcus agalactiae isolated from piscine, dolphin, bovine and human sources: a dolphin and piscine lineage associated with a fish epidemic in Kuwait is also associated with human neonatal infections in Japan. J. Med. Microbiol. 57, 1369–1376. doi: 10.1099/jmm.0.47815-0
Facimoto, C. T., Chideroli, R. T., Gonçalves, D. D., Carmo, A. O. do, Kalaphotakis, E., and Pereira, U. de P. (2017). Whole-genome sequence of Streptococcus agalactiae strain S13, isolated from a fish eye from a Nile Tilapia farm in Southern Brazil. Genome Announc. 5. doi: 10.1128/genomeA.00917-17
Farley, M. M. (2001). Group B streptococcal disease in nonpregnant adults. Clini. Infect. Dis. 33, 556–561. doi: 10.1086/322696
Farley, M. M., Harvey, R. C., Stull, T., Smith, J. D., Schuchat, A., Wenger, J. D., et al. (1993). A population-based assessment of invasive disease due to group B Streptococcus in nonpregnant adults. N. Engl. J. Med. 328, 1807–1811.
Ferrieri, P., Baker, C. J., Hillier, S. L., and Flores, A. E. (2004). Diversity of surface protein expression in Group B Streptococcal colonizing & invasive isolates. Indian J. Med. Res. 119(Suppl.), 191–196.
Finch, L. A., and Martin, D. R. (1984). Human and bovine group B streptococci: two distinct populations. J. Appl. Bacteriol. 57, 273–278.
Flores, A. R., Galloway-Peña, J., Sahasrabhojane, P., Saldaña, M., Yao, H., Xiaoping, S., et al. (2015). Sequence type 1 group B Streptococcus, an emerging cause of invasive disease in adults, evolves by small genetic changes. Proc. Natl. Acad. Sci. USA 112, 6431–6436. doi: 10.1073/pnas.1504725112
Foxman, B., Gillespie, B., Manning, S. D., Howard, L. J., Tallman, P., Zhang, L., et al. (2006). Incidence and duration of group B Streptococcus by serotype among male and female college students living in a single dormitory. Am. J. Epidemiol. 163, 544–551. doi: 10.1093/aje/kwj075
Foxman, B., Gillespie, B. W., Manning, S. D., and Marrs, C. F. (2007). Risk factors for group B streptococcal colonization: potential for different transmission systems by capsular type. Ann. Epidemiol. 17, 854–862. doi: 10.1016/j.annepidem.2007.05.014
Francois Watkins, L. K., McGee, L., Schrag, S. J., Beall, B., Jain, J. H., Pondo, T., et al. (2019). Epidemiology of invasive group B streptococcal infections among nonpregnant adults in the United States, 2008-2016. JAMA Intern. Med. 179, 479–488. doi: 10.1001/jamainternmed.2018.7269
Franken, C., Haase, G., Brandt, C., Weber-Heynemann, J., Martin, S., Lämmler, C., et al. (2001). Horizontal gene transfer and host specificity of beta-haemolytic streptococci: the role of a putative composite transposon containing scpB and Lmb. Mol. Microbiol. 41, 925–935. doi: 10.1046/j.1365-2958.2001.02563.x
Gabrielsen, C., Mæland, J. A., Lyng, R. V., Radtke, A., and Afset, J. E. (2017). Molecular characteristics of Streptococcus agalactiae strains deficient in alpha-like protein encoding genes. J. Med. Microbiol. 66, 26–33. doi: 10.1099/jmm.0.000412
Glaser, P., Rusniok, C., Buchrieser, C., Chevalier, F., Frangeul, L., Msadek, T., et al. (2002). Genome sequence of Streptococcus agalactiae, a pathogen causing invasive neonatal disease. Mol. Microbiol. 45, 1499–1513. doi: 10.1046/j.1365-2958.2002.03126.x
Guo, Y., Deng, X., Liang, Y., Liang, Z., Zhao, G.-P., and Zhou, Y. (2018). The draft genomes and investigation of serotype distribution, antimicrobial resistance of group B Streptococcus strains isolated from urine in Suzhou, China. Ann. Clin. Microbiol. Antimicrob. 17:28. doi: 10.1186/s12941-018-0280-y
Harden, L. K., Morales, K. M., and Hughey, J. R. (2016). Complete genome sequence of nonhemolytic Streptococcus agalactiae serotype V strain 1, isolated from the Buccal cavity of a Canine. Genome Announc. 4. doi: 10.1128/genomeA.01612-15
Hauge, M., Jespersgaard, C., Poulsen, K., and Kilian, M. (1996). Population structure of Streptococcus agalactiae reveals an association between specific evolutionary lineages and putative virulence factors but not disease. Infect. Immun. 64, 919–925.
Hays, C., Louis, M., Plainvert, C., Dmytruk, N., Touak, G., Trieu-Cuot, P., et al. (2016). Changing epidemiology of group B Streptococcus susceptibility to fluoroquinolones and aminoglycosides in France. Antimicrob. Agents Chemother. 60, 7424–7430. doi: 10.1128/AAC.01374-16
He, E.-M., Chen, C.-W., Guo, Y., Hsu, M.-H., Liang, Z., Chen, H.-L., et al. (2017). The genome of serotype VI Streptococcus agalactiae serotype VI and comparative analysis. Gene 597, 59–65. doi: 10.1016/j.gene.2016.10.030
Hetzel, U., König, A., Yildirim, A. O., Lämmler, C., and Kipar, A. (2003). Septicaemia in emerald monitors (Varanus Prasinus Schlegel 1839) caused by Streptococcus agalactiae acquired from mice. Vet. Microbiol. 95, 283–293. doi: 10.1016/S0378-1135(03)00184-6
Hooven, T. A., Randis, T. M., Daugherty, S. C., Narechania, A., Planet, P. J., Tettelin, H., et al. (2014). Complete genome sequence of Streptococcus agalactiae CNCTC 10/84, a hypervirulent sequence type 26 strain. Genome Announc. 2. doi: 10.1128/genomeA.01338-14
Hoshina, T., Sano, T., and Marimoto, Y. (1958). A Streptococcus pathogenic to fish. J. Tokyo Univ. Fisheries 44, 57–58.
Huet, H., Martin, C., Geslin, P., Grimont, F., and Quentin, R. (1993). Ribotyping of Streptococcus agalactiae strains isolated from vaginas of asymptomatic women. Res. Microbiol. 144, 457–465.
Ip, M., Ang, I., Fung, K., Liyanapathirana, V., Luo, M. J., and Lai, R. (2016). Hypervirulent clone of group B Streptococcus serotype III sequence type 283, Hong Kong, 1993-2012. Emerg. Infect. Dis. 22, 1800–1803. doi: 10.3201/eid2210.151436
Ip, M., Cheuk, E. S. C., Tsui, M. H. Y., Kong, F., Leung, T. N., and Gilbert, G. L. (2006). Identification of a Streptococcus agalactiae serotype III subtype 4 clone in association with adult invasive disease in Hong Kong. J. Clin. Microbiol. 44, 4252–4254. doi: 10.1128/JCM.01533-06
Jafar, Q. A., Sameer, A.-Z., Salwa, A.-M., Samee, A.-A., Ahmed, A.-M., and Al-Sharifi, F. (2008). Molecular investigation of Streptococcus agalactiae isolates from environmental samples and fish specimens during a massive fish kill in Kuwait bay. Pak. J. Biol. Sci. 11, 2500–2504. doi: 10.3923/pjbs.2008.2500.2504
Jaglarz, A., Gurgul, A., Leigh, W. J., Costa, J. Z., and Thompson, K. D. (2018a). Complete genome sequences of three fish-associated Streptococcus agalactiae isolates. Genome Announc. 6. doi: 10.1128/genomeA.00025-18
Jaglarz, A., Gurgul, A., Leigh, W. J., Costa, J. Z., and Thompson, K. D. (2018b). Complete genome sequences of three Streptococcus agalactiae serotype Ia isolates obtained from disease outbreaks in Nile Tilapia (Oreochromis niloticus). Genome Announc. 6. doi: 10.1128/genomeA.01432-17
Jauneikaite, E., Kapatai, G., Davies, F., Gozar, I., Coelho, J., Bamford, K. B., et al. (2018). Serial clustering of late-onset group B Streptococcal infections in the neonatal unit: a genomic re-evaluation of causality. Clin. Infect. Dis. 67, 854–860. doi: 10.1093/cid/ciy174
Jensen, N. E. (1982). Experimental bovine group-B streptococcal mastitis induced by strains of human and bovine origin. Nord. Vet. Med. 34, 441–450.
Johri, A. K., Lata, H., Yadav, P., Dua, M., Yang, Y., Xu, X., et al. (2013). Epidemiology of group B Streptococcus in developing countries. Vaccine 31(Suppl. 4), D43–D45. doi: 10.1016/j.vaccine.2013.05.094
Jones, N., Bohnsack, J. F., Takahashi, S., Oliver, K. A., Chan, M.-S., Kunst, F., et al. (2003). Multilocus sequence typing system for group B Streptococcus. J. Clin. Microbiol. 41, 2530–2536. doi: 10.1128/JCM.41.6.2530-2536.2003
Jørgensen, H. J., Nordstoga, A. B., Sviland, S., Zadoks, R. N., Sølverød, L., Kvitle, B., et al. (2016). Streptococcus agalactiae in the environment of bovine dairy herds–rewriting the textbooks? Vet. Microbiol. 184, 64–72. doi: 10.1016/j.vetmic.2015.12.014
Kalimuddin, S., Chen, S. L., Lim, C. T. K., Koh, T. H., Tan, T. Y., Kam, M., et al. (2017). 2015 Epidemic of severe Streptococcus agalactiae sequence type 283 infections in Singapore associated with the consumption of raw freshwater fish: a detailed analysis of clinical, epidemiological, and bacterial sequencing data. Clini. Infect. Dis. 64(Suppl. 2), S145–S152. doi: 10.1093/cid/cix021
Kapatai, G., Patel, D., Efstratiou, A., and Chalker, V. J. (2017). Comparison of molecular serotyping approaches of Streptococcus agalactiae from genomic sequences. BMC Genomics 18, 429. doi: 10.1186/s12864-017-3820-5
Kayansamruaj, P., Pirarat, N., Kondo, H., Hirono, I., and Rodkhum, C. (2014). Draft genome sequences of Streptococcus agalactiae strains isolated from Nile Tilapia (Oreochromis niloticus) farms in Thailand. Genome Announc. 2. doi: 10.1128/genomeA.01300-14
Kayansamruaj, P., Pirarat, N., Kondo, H., Hirono, I., and Rodkhum, C. (2015). Genomic comparison between pathogenic Streptococcus agalactiae isolated from Nile Tilapia in Thailand and fish-derived ST7 strains. Infect. Genet. Evol. 36, 307–314. doi: 10.1016/j.meegid.2015.10.009
Kayansamruaj, P., Soontara, C., Unajak, S., Dong, H. T., Rodkhum, C., Kondo, H., et al. (2018). Comparative genomics inferred two distinct populations of piscine pathogenic Streptococcus agalactiae, serotype Ia ST7 and serotype III ST283, in Thailand and Vietnam. Genomics. doi: 10.1016/j.ygeno.2018.11.016
Klemm, E., and Dougan, G. (2016). Advances in understanding bacterial pathogenesis gained from whole-genome sequencing and phylogenetics. Cell Host Microbe 19, 599–610. doi: 10.1016/j.chom.2016.04.015
Kropp, K. A., Lucid, A., Carroll, J., Belgrudov, V., Walsh, P., Kelly, B., et al. (2014). Draft genome sequence of a Streptococcus agalactiae strain isolated from a preterm neonate blood sepsis patient at the royal infirmary, Edinburgh, Scotland. Genome Announc. 2. doi: 10.1128/genomeA.00875-14
Kwatra, G., Cunnington, M. C., Merrall, E., Adrian, P. V., Ip, M., Klugman, K. P., et al. (2016). Prevalence of maternal colonisation with group B Streptococcus: a systematic review and meta-analysis. Lancet Infect. Dis. 16, 1076–1084. doi: 10.1016/S1473-3099(16)30055-X
Lamagni, T. L., Keshishian, C., Efstratiou, A., Guy, R., Henderson, K. L., Broughton, K., et al. (2013). Emerging trends in the epidemiology of invasive group B streptococcal disease in England and Wales, 1991-2010. Clini. Infect. Dis. 57, 682–688. doi: 10.1093/cid/cit337
Lancefield, R. C. (1928). THE antigenic complex of streptococcus haemolyticus: III. Chemical and immunological properties of the species-specific substance. J. Exp. Med. 47, 481–491.
Lancefield, R. C. (1933). A serological differentiation of human and other groups of hemolytic streptococci. J. Exp. Med. 57, 571–595.
Le Doare, K., Jarju, S., Darboe, S., Warburton, F., Gorringe, A., Heath, P. T., et al. (2016). Risk factors for group B Streptococcus colonisation and disease in Gambian women and their infants. J. Inf. Secur. 72, 283–294. doi: 10.1016/j.jinf.2015.12.014
Leal, C. A. G., Queiroz, G. A., Pereira, F. L., Tavares, G. C., and Figueiredo, H. C. P. (2019). Streptococcus agalactiae sequence type 283 in farmed fish, Brazil. Emerg. Infect. Dis. 25, 776–779.
Lee, H. T., Kim, S. Y., Park, P.-W., Ahn, J.-Y., Kim, K.-H., Seo, J. Y., et al. (2019). Detection and genomic analysis of genital group B Streptococcus in pregnant Korean women. J. Obstet. Gynaecol. Res. 45, 69–77. doi: 10.1111/jog.13810
Lefébure, T., and Stanhope, M. J. (2007). Evolution of the core and pan-genome of Streptococcus: positive selection, recombination, and genome composition. Genome Biol. 8:R71. doi: 10.1186/gb-2007-8-5-r71
Li, L., Wang, R., Huang, Y., Huang, T., Luo, F., Huang, W., et al. (2018). High incidence of pathogenic Streptococcus agalactiae ST485 strain in pregnant/puerperal women and isolation of hyper-virulent human CC67 strain. Front. Microbiol. 9:50. doi: 10.3389/fmicb.2018.03224
Limansky, A. S., Sutich, E. G., Guardati, M. C., Toresani, I. E., and Viale, A. M. (1998). Genomic diversity among Streptococcus agalactiae isolates detected by a degenerate oligonucleotide-primed amplification assay. J. Infect. Dis. 177, 1308–1313. doi: 10.1086/515275
Liu, G., Zhang, W., and Chengping, L. (2013). Comparative genomics analysis of Streptococcus agalactiae reveals that isolates from cultured Tilapia in China are closely related to the human strain A909. BMC Genomics 14:775. doi: 10.1186/1471-2164-14-775
Liu, G., Zhang, W., and Lu, C. (2012). Complete genome sequence of Streptococcus agalactiae GD201008-001, isolated in China from tilapia with meningoencephalitis. J. Bacteriol. 194, 6653. doi: 10.1128/JB.01788-12
Louthrenoo, W., Kasitanon, N., Wangkaew, S., Hongsongkiat, S., Sukitawut, W., and Wichainun, R. (2014). Streptococcus agalactiae: an emerging cause of septic arthritis. J. Clin. Rheumatol. 20, 74–78. doi: 10.1097/RHU.0000000000000071
Luan, S.-L., Granlund, M., Sellin, M., Lagergård, T., Spratt, B. G., and Norgren, M. (2005). Multilocus sequence typing of Swedish invasive group B Streptococcus isolates indicates a neonatally associated genetic lineage and capsule switching. J. Clin. Microbiol. 43, 3727–3733. doi: 10.1128/JCM.43.8.3727-3733.2005
Lyhs, U., Kulkas, L., Katholm, J., Waller, K. P., Saha, K., Tomusk, R. J., et al. (2016). Streptococcus agalactiae serotype IV in humans and cattle, northern Europe1. Emerg. Infect. Dis. 22, 2097–2103. doi: 10.3201/eid2212.151447
Maiden, M. C. J. (2006). Multilocus sequence typing of bacteria. Annu. Rev. Microbiol. 60, 561–588. doi: 10.1146/annurev.micro.59.030804.121325
Mainardi, R. M., Lima Júnior, E. A., Ribeiro Júnior, J. C., Beloti, V., Carmo, A. O., Kalapothakis, E., et al. (2016). Complete genome sequence of Streptococcus agalactiae strain S25 isolated from peritoneal liquid of Nile Tilapia. Genome Announc. 4. doi: 10.1128/genomeA.00784-16
Malbruny, B., Werno, A. M., Murdoch, D. R., Leclercq, R., and Cattoir, V. (2011). Cross-resistance to lincosamides, streptogramins A, and pleuromutilins due to the lsa(C) gene in Streptococcus agalactiae UCN70. Antimicrob. Agents Chemother. 55, 1470–1474. doi: 10.1128/AAC.01068-10
Manning, S. D., Cody Springman, A., Million, A. D., Milton, N. R., McNamara, S. E., Somsel, P. A., et al. (2010). Association of group B Streptococcus colonization and bovine exposure: a prospective cross-sectional cohort study. PLoS One 5:e8795. doi: 10.1371/journal.pone.0008795
Manning, S. D., Neighbors, K., Tallman, P. A., Gillespie, B., Marrs, C. F., Borchardt, S. M., et al. (2004). Prevalence of group B Streptococcus colonization and potential for transmission by casual contact in healthy young men and women. Clini. Infect. Dis. 39, 380–388. doi: 10.1086/422321
Martins, E. R., Melo-Cristino, J., and Ramirez, M. (2010). Evidence for rare capsular switching in Streptococcus agalactiae. J. Bacteriol. 192, 1361–1369. doi: 10.1128/JB.01130-09
Medini, D., Donati, C., Tettelin, H., Masignani, V., and Rappuoli, R. (2005). The microbial pan-genome. Curr. Opin. Genet. Dev. 15, 589–594. doi: 10.1016/j.gde.2005.09.006
Medugu, N., Iregbu, K. C., Parker, R. E., Plemmons, J., Singh, P., Audu, L. I., et al. (2017). Group B streptococcal colonization and transmission dynamics in pregnant women and their newborns in Nigeria: implications for prevention strategies. Clin. Microbiol. Infect. 23, 673.e9–673.e16. doi: 10.1016/j.cmi.2017.02.029
Mehershahi, K. S., Hsu, L. Y., Koh, T. H., and Chen, S. L. (2015). Complete genome sequence of Streptococcus agalactiae serotype III, multilocus sequence type 283 strain SG-M1. Genome Announc. 3. doi: 10.1128/genomeA.01188-15
Melo, S. C. C. S. d., Costa, A. B., da Silva, F. T. R., Silva, N. M. M. G., Tashima, C. M., Cardoso, R. F., et al. (2018). Prevalence of Streptococcus agalactiae colonization in pregnant women from the 18th health region of Paraná state. Rev. Inst. Med. Trop. Sao Paulo 60:e2. doi: 10.1590/s1678-9946201860002
Metcalf, B. J., Chochua, S., Gertz, R. E. Jr., Hawkins, P. A., Ricaldi, J., Li, Z., et al. (2017). Short-read whole genome sequencing for determination of antimicrobial resistance mechanisms and capsular serotypes of current invasive Streptococcus agalactiae recovered in the USA. Clin. Microbiol. Infect. 23, 574.e7–574.e14. doi: 10.1016/j.cmi.2017.02.021
Mitima, K. T., Ntamako, S., Birindwa, A. M., Mukanire, N., Kivukuto, J. M., Tsongo, K., et al. (2014). Prevalence of colonization by Streptococcus agalactiae among pregnant women in Bukavu, Democratic Republic of the Congo. J. Infect. Dev. Ctries. 8, 1195–1200. doi: 10.3855/jidc.5030
Mukesi, M., Iweriebor, B. C., Obi, L. C., Nwodo, U. U., Moyo, S. R., and Okoh, A. I. (2019). Prevalence and capsular type distribution of Streptococcus agalactiae isolated from pregnant women in Namibia and South Africa. BMC Infect. Dis. 19:179. doi: 10.1186/s12879-019-3809-6
Munang’andu, H. M., Paul, J., and Evensen, Ø. (2016). An overview of vaccination strategies and antigen delivery systems for Streptococcus agalactiae vaccines in Nile Tilapia (Oreochromis niloticus). Vaccine 4:48. doi: 10.3390/vaccines4040048
Musser, J. M., Mattingly, S. J., Quentin, R., Goudeau, A., and Selander, R. K. (1989). Identification of a high-virulence clone of type III Streptococcus agalactiae (group B Streptococcus) causing invasive neonatal disease. Proc. Natl. Acad. Sci. USA 86, 4731–4735. doi: 10.1073/pnas.86.12.4731
Mz, I. S., Teh, L. K., and Salleh, M. Z. (2013). Draft genome sequence of Streptococcus agalactiae PR06. Genome Announc. 1. doi: 10.1128/genomeA.00351-13
Nagano, N., Koide, S., Hayashi, W., Taniguchi, Y., Tanaka, H., Maeyama, Y., et al. (2019). Population-level transition of capsular polysaccharide types among sequence type 1 group b Streptococcus isolates with reduced penicillin susceptibility during a long-term hospital epidemic. Int. J. Antimicrob. Agents 53, 203–210. doi: 10.1016/j.ijantimicag.2018.11.003
Nagaoka, K., Konno, S., Murase, K., Kikuchi, T., and Nakagawa, I. (2018). Complete genome sequence of Streptococcus agalactiae serotype III, Multilocus sequence type 335 strain HU-GS5823, isolated from a human patient in Japan with severe invasive infection. Microbiol. Resour. Announc. 7. doi: 10.1128/MRA.01303-18
Nanduri, S. A., Petit, S., Smelser, C., Apostol, M., Alden, N. B., Harrison, L. H., et al. (2019). Epidemiology of invasive early-onset and late-onset group b streptococcal disease in the United States, 2006 to 2015: multistate laboratory and population-based surveillance. JAMA Pediatr. 173, 224–233. doi: 10.1001/jamapediatrics.2018.4826
Neemuchwala, A., Teatero, S., Athey, T. B. T., McGeer, A., and Fittipaldi, N. (2016). Capsular switching and other large-scale recombination events in invasive sequence type 1 group B Streptococcus. Emerg. Infect. Dis. 22, 1941–1944. doi: 10.3201//eid2211.152064
Nkembe, N. M., Kamga, H. G., Baiye, W. A., Chafa, A. B., and Njotang, P. N. (2018). Streptococcus agalactiae prevalence and antimicrobial susceptibility pattern in vaginal and anorectal swabs of pregnant women at a tertiary hospital in Cameroon. BMC. Res. Notes 11:480. doi: 10.1186/s13104-018-3589-x
Nocard, M., and Mollereau, R. (1887). Sur Une Mammite Contagieuse Des Vaches Laitieres. Ann. Inst. Pasteur 1:109.
Pang, M., Sun, L., He, T., Bao, H., Zhang, L., Zhou, Y., et al. (2017). Molecular and virulence characterization of highly prevalent Streptococcus agalactiae circulated in bovine dairy herds. Vet. Res. 48:65. doi: 10.1186/s13567-017-0461-2
Paul, J. (2014). Characterization of immunogenic properties of Streptococcus agalactiae biotype I and II. November. doi: 10.1016/j.gdata.2014.06.010
Pereira, U. P., Mian, G. F., Oliveira, I. C. M., Benchetrit, L. C., Costa, G. M., and Figueiredo, H. C. P. (2010). Genotyping of Streptococcus agalactiae strains isolated from fish, human and cattle and their virulence potential in Nile Tilapia. Vet. Microbiol. 140, 186–192. doi: 10.1016/j.vetmic.2009.07.025
Pereira, U. de P., Rodrigues Dos Santos, A., Hassan, S. S., Aburjaile, F. F., Soares, S. de C., Ramos, R. T. J., et al. (2013). Complete genome sequence of Streptococcus agalactiae strain SA20-06, a fish pathogen associated to meningoencephalitis outbreaks. Stand. Genomic Sci. 8, 188–197. doi: 10.4056/sigs.3687314
Pridgeon, J. W., and Zhang, D. (2014). Complete genome sequence of a virulent Streptococcus agalactiae strain, 138P, isolated from diseased Nile Tilapia. Genome Announc. 2. doi: 10.1128/genomeA.00295-14
Pridgeon, J. W., Zhang, D., and Zhang, L. (2014). Complete genome of the attenuated sparfloxacin-resistant Streptococcus agalactiae strain 138spar. Genome Announc. 2. doi: 10.1128/genomeA.00431-14
Puymège, A., Bertin, S., Guédon, G., and Payot, S. (2015). Analysis of Streptococcus agalactiae pan-genome for prevalence, diversity and functionality of integrative and conjugative or mobilizable elements integrated in the tRNA(Lys CTT) gene. Mol. Gen. Genomics. 290, 1727–1740. doi: 10.1007/s00438-015-1031-9
Rajendram, P., Kyaw, W. M., Leo, Y. S., Ho, H., Chen, W. K., Lin, R., et al. (2016). Group B Streptococcus sequence type 283 disease linked to consumption of raw fish, Singapore. Emerg. Infect. Dis. 22, 1974–1977. doi: 10.3201/eid2211.160252
Richards, V. P., Choi, S. C., Pavinski Bitar, P. D., Gurjar, A. A., and Stanhope, M. J. (2013). Transcriptomic and genomic evidence for Streptococcus agalactiae adaptation to the bovine environment. BMC Genomics 14:920. doi: 10.1186/1471-2164-14-920
Richards, V. P., Lang, P., Pavinski, P. D., Bitar, T. L., Schukken, Y. H., Zadoks, R. N., et al. (2011). Comparative genomics and the role of lateral gene transfer in the evolution of bovine adapted Streptococcus agalactiae. Infect. Genet. Evol. 11, 1263–1275. doi: 10.1016/j.meegid.2011.04.019
Rivera, L., Sáez-Llorens, X., Feris-Iglesias, J., Ip, M., Saha, S., Adrian, P. V., et al. (2015). Incidence and serotype distribution of invasive group B streptococcal disease in young infants: a multi-country observational study. BMC Pediatr. 15:143. doi: 10.1186/s12887-015-0460-2
Rolland, K., Marois, C., Siquier, V., Cattier, B., and Quentin, R. (1999). Genetic features of Streptococcus agalactiae strains causing severe neonatal infections, as revealed by pulsed-field gel electrophoresis and hylB gene analysis. J. Clin. Microbiol. 37, 1892–1898.
Rothen, J., Schindler, T., Pothier, J. F., Younan, M., Certa, U., Daubenberger, C., et al. (2017). Draft genome sequences of seven Streptococcus agalactiae strains isolated from Camelus dromedarius at the Horn of Africa. Genome Announc. 5. doi: 10.1128/genomeA.00525-17
Rosinski-Chupin, I., Sauvage, E., Mairey, B., Mangenot, S., Ma, L., Da Cunha, V., et al. (2013). Reductive evolution in Streptococcus agalactiae and the emergence of a host adapted lineage. BMC Genomics 14:252. doi: 10.1186/1471-2164-14-252
Ruegg, P. L. (2017). A 100-year review: mastitis detection, management, and prevention. J. Dairy Sci. 100, 10381–10397. doi: 10.3168/jds.2017-13023
Schuchat, A. (1998). Epidemiology of group B streptococcal disease in the United States: shifting paradigms. Clin. Microbiol. Rev. 11, 497–513. doi: 10.1128/CMR.11.3.497
Seale, A. C., Bianchi-Jassir, F., Russell, N. J., Kohli-Lynch, M., Tann, C. J., Hall, J., et al. (2017). Estimates of the burden of group B Streptococcal disease worldwide for pregnant women, stillbirths, and children. Clini. Infect. Dis. 65(Suppl. 2), S200–S219. doi: 10.1093/cid/cix664
Seale, A. C., Koech, A. C., Sheppard, A. E., Barsosio, H. C., Langat, J., Anyango, E., et al. (2016). Maternal colonization with Streptococcus agalactiae and associated stillbirth and neonatal disease in coastal Kenya. Nat. Microbiol. 1:16067. doi: 10.1038/nmicrobiol.2016.67
Shaw, A. O., and Beam, A. L. (1935). The effect of mastitis upon milk production. J. Dairy Sci. 18, 353–357.
Sigaúque, B., Kobayashi, M., Vubil, D., Nhacolo, A., Chaúque, A., Moaine, B., et al. (2018). Invasive bacterial disease trends and characterization of group B Streptococcal isolates among young infants in Southern Mozambique, 2001-2015. PLoS One 13:e0191193. doi: 10.1371/journal.pone.0191193
Singh, P., Aronoff, D. M., Davies, H. D., and Manning, S. D. (2016). Draft genome sequence of an invasive Streptococcus agalactiae isolate lacking pigmentation. Genome Announc. 4. doi: 10.1128/genomeA.00015-16
Singh, P., Springman, A. C., Davies, H. D., and Manning, S. D. (2012). Whole-genome shotgun sequencing of a colonizing multilocus sequence type 17 Streptococcus agalactiae strain. J. Bacteriol. 194, 6005. doi: 10.1128/JB.01378-12
Sinha, A., Russell, L. B., Tomczyk, S., Verani, J. R., Schrag, S. J., Berkley, J. A., et al. (2016). Disease burden of group B Streptococcus among infants in sub-Saharan Africa: a systematic literature review and meta-analysis. Pediatr. Infect. Dis. J. 35, 933–942. doi: 10.1097/INF.0000000000001233
Six, A., Firon, A., Plainvert, C., Caplain, C., Bouaboud, A., Touak, G., et al. (2016). Molecular characterization of nonhemolytic and nonpigmented group B Streptococci responsible for human invasive infections. J. Clin. Microbiol. 54, 75–82. doi: 10.1128/JCM.02177-15
Slotved, H.-C., Dayie, N. T. K. D., Banini, J. A. N., and Frimodt-Møller, N. (2017). Carriage and serotype distribution of Streptococcus agalactiae in third trimester pregnancy in Southern Ghana. BMC Preg. Childbirth 17:238. doi: 10.1186/s12884-017-1419-0
Sørensen, U. B. S., Poulsen, K., Ghezzo, C., Margarit, I., and Kilian, M. (2010). Emergence and global dissemination of host-specific Streptococcus agalactiae clones. MBio 1. doi: 10.1128/mBio.00178-10
Springman, A. C., Lacher, D. W., Wu, G., Milton, N., Whittam, T. S., Dele Davies, H., et al. (2009). Selection, recombination, and virulence gene diversity among group B Streptococcal genotypes. J. Bacteriol. 191, 5419–5427. doi: 10.1128/JB.00369-09
Srinivasan, V., Metcalf, B. J., Knipe, K. M., Ouattara, M., McGee, L., Shewmaker, P. L., et al. (2014). vanG element insertions within a conserved chromosomal site conferring vancomycin resistance to Streptococcus agalactiae and Streptococcus anginosus. MBio 5, e01386–e01314. doi: 10.1128/mBio.01386-14
Suhaimi, M. E. S., Desa, M. N. M., Eskandarian, N., Pillay, S. G., Ismail, Z., Neela, V. K., et al. (2017). Characterization of a group B Streptococcus infection based on the demographics, serotypes, antimicrobial susceptibility and genotypes of selected isolates from sterile and non-sterile isolation sites in three major hospitals in Malaysia. J. Infect. Public Health 10, 14–21. doi: 10.1016/j.jiph.2016.01.009
Sukhnanand, S., Dogan, B., Ayodele, M. O., Zadoks, R. N., Craver, M. P. J., Dumas, N. B., et al. (2005). Molecular subtyping and characterization of bovine and human Streptococcus agalactiae isolates. J. Clin. Microbiol. 43, 1177–1186. doi: 10.1128/JCM.43.3.1177-1186.2005
Sullivan, M. J., Forde, B. M., Prince, D. W., Ipe, D. S., Ben Zakour, N. L., Davies, M. R., et al. (2017). Complete genome sequence of serotype III Streptococcus agalactiae sequence type 17 strain 874391. Genome Announc. 5. doi: 10.1128/genomeA.01107-17
Tan, S., Lin, Y., Foo, K., Koh, H. F., Tow, C., Zhang, Y., et al. (2016). Group B Streptococcus serotype III sequence type 283 bacteremia associated with consumption of raw fish, Singapore. Emerg. Infect. Dis. 22, 1970–1973. doi: 10.3201/eid2211.160210
Tavella, A., Bettini, A., Cocchi, M., Idrizi, I., Colorio, S., Viel, L., et al. (2018). Isolation of Streptococcus agalactiae in a female Llama (Lama glama) in South Tyrol (Italy). BMC Vet. Res. 14:343. doi: 10.1186/s12917-018-1676-9
Teatero, S., Athey, T. B. T., Van Caeseele, P., Horsman, G., Alexander, D. C., Melano, R. G., et al. (2015a). Emergence of serotype IV group B Streptococcus adult invasive disease in Manitoba and Saskatchewan, Canada, is driven by clonal sequence type 459 strains. J. Clin. Microbiol. 53, 2919–2926. doi: 10.1128/JCM.01128-15
Teatero, S., Lemire, P., Dewar, K., Wasserscheid, J., Calzas, C., Mallo, G. V., et al. (2016). Genomic recombination leading to decreased virulence of group B Streptococcus in a mouse model of adult invasive disease. Pathogens 5:54. doi: 10.3390/pathogens5030054
Teatero, S., McGeer, A., Li, A., Gomes, J., Seah, C., Demczuk, W., et al. (2015b). Population structure and antimicrobial resistance of invasive serotype IV group B Streptococcus, Toronto, Ontario, Canada. Emerg. Infect. Dis. 21, 585–591. doi: 10.3201/eid2014.140759
Teatero, S., McGeer, A., Low, D. E., Li, A., Demczuk, W., Martin, I., et al. (2014). Characterization of invasive group B Streptococcus strains from the greater Toronto area, Canada. J. Clin. Microbiol. 52, 1441–1447. doi: 10.1128/JCM.03554-13
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial ‘pan-genome. Proc. Natl. Acad. Sci. USA 102, 13950–13955. doi: 10.1073/pnas.0506758102
Tettelin, H., Masignani, V., Cieslewicz, M. J., Eisen, J. A., Peterson, S., Wessels, M. R., et al. (2002). Complete genome sequence and comparative genomic analysis of an emerging human pathogen, serotype V Streptococcus agalactiae. Proc. Natl. Acad. Sci. USA 99, 12391–12396. doi: 10.1073/pnas.182380799
Vandamme, P., Devriese, L. A., Pot, B., Kersters, K., and Melin, P. (1997). Streptococcus difficile is a nonhemolytic group B, type Ib Streptococcus. Int. J. Syst. Bacteriol. 47, 81–85. doi: 10.1099/00207713-47-1-81
Veeraraghavan, B., Ragupathi, N. K. D., Santhanam, S., Verghese, V. P., Inbanathan, F. Y., and Livingston, C. (2017). Whole genome shotgun sequencing of Indian strains of Streptococcus agalactiae. Genom. Data 14, 63–65. doi: 10.1016/j.gdata.2017.10.001
Verani, J. R., McGee, L., and Schrag, S. J. Division of Bacterial Diseases National Center for Immunization and Respiratory Diseases, Centers for Disease Control and Prevention (CDC) (2010). “Prevention of perinatal group B Streptococcal disease–revised guidelines from CDC, 2010.” MMWR 59, 1–36.
Villanueva-Uy, M. E., Wongsiridej, P., Sangtawesin, V., Chiu, V., Tallo, V., Nazaire-Bermal, N., et al. (2015). THE burden of invasive neonatal group B streptococcal (GBS) disease in Thailand and the Philippines. Southeast Asian J. Trop. Med. Public Health 46, 728–737.
Wang, B., Jian, J., Lu, Y., Cai, S., Huang, Y., Tang, J., et al. (2012). Complete genome sequence of Streptococcus agalactiae ZQ0910, a pathogen causing meningoencephalitis in the GIFT strain of Nile tilapia (Oreochromis niloticus). J. Bacteriol. 194, 5132–5133. doi: 10.1128/JB.01080-12
Wang, R., Li, L., Huang, T., Huang, Y., Huang, W., Yang, X., et al. (2018b). Phylogenetic, comparative genomic and structural analyses of human Streptococcus agalactiae ST485 in China. BMC Genomics 19:716. doi: 10.1186/s12864-018-5084-0
Wang, R., Li, L., Huang, T., Huang, W., Lei, A., and Chen, M. (2018a). Capsular switching and ICE transformation occurred in human Streptococcus agalactiae ST19 with high pathogenicity to fish. Front. Veter. Sci. 5:281. doi: 10.3389/fvets.2018.00281
Wang, R., Li, L.-P., Huang, T., Lei, A.-Y., Huang, Y., Luo, F.-G., et al. (2017). Genomic comparison of virulent and non-virulent serotype V ST1 Streptococcus agalactiae in fish. Vet. Microbiol. 207, 164–169. doi: 10.1016/j.vetmic.2017.06.007
Wang, R., Li, L., Luo, F., Liang, W., Gan, X., and Chen, M. (2015b). Genome sequence of Streptococcus agalactiae strain H002, serotype III, isolated in China from a pregnant woman. Genome Announc. 3. doi: 10.1128/genomeA.01109-15
Wang, Y.-H., Lu, C.-C., Chiu, C.-H., Wang, M.-H., Yang, T.-H., and Chu, C. (2016). Genetically diverse serotypes III and VI substitute major clonal disseminated serotypes Ib and V as prevalent serotypes of Streptococcus agalactiae from 2007 to 2012. J. Microbiol. Immunol. Infect. 49, 672–678. doi: 10.1016/j.jmii.2015.05.022
Wang, P., Ma, Z., Tong, J., Zhao, R., Shi, W., Yu, S., et al. (2015). Serotype distribution, antimicrobial resistance, and molecular characterization of invasive group B Streptococcus isolates recovered from Chinese neonates. Int. J. Infect. Dis. 37, 115–118. doi: 10.1016/j.ijid.2015.06.019
Welch, R. A., Burland, V., Plunkett, G. 3rd., Redford, P., Roesch, P., Rasko, D., et al. (2002). Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. USA 99, 17020–17024. doi: 10.1073/pnas.252529799
Wibawan, I. W., Lämmler, C., Lautrou, Y., and Warsa, U. C. (1992). Serotyping and further characterization of group B streptococcal isolates from Indonesia. Zentralbl. Bakteriol. 277, 260–266. doi: 10.1016/S0934-8840(11)80621-3
Wilder-Smith, E., Chow, K. M., Kay, R., Ip, M., and Tee, N. (2000). Group B Streptococcal meningitis in adults: recent increase in Southeast Asia. Aust. NZ J. Med. 30, 462–465.
Wolf, I. R., Paschoal, A. R., Quiroga, C., Domingues, D. S., de Souza, R. F., Pretto-Giordano, L. G., et al. (2018). Functional annotation and distribution overview of RNA families in 27 Streptococcus agalactiae genomes. BMC Genomics 19:556. doi: 10.1186/s12864-018-4951-z
Yang, F., Li, H., Zhang, S., and Wang, X. (2016). Draft genome sequence of Streptococcus agalactiae serotype Ia strain M19, a multidrug-resistant isolate from a cow with bovine mastitis. Genome Announc. 4. doi: 10.1128/genomeA.01093-16
Zadoks, R. N., Middleton, J. R., McDougall, S., Katholm, J., and Schukken, Y. H. (2011). Molecular epidemiology of mastitis pathogens of dairy cattle and comparative relevance to humans. J. Mammary Gland Biol. Neoplasia 16, 357–372. doi: 10.1007/s10911-011-9236-y
Zhang, Z., Lan, J., Li, Y., Hu, M., Yu, A., Zhang, J., et al. (2018). The pathogenic and antimicrobial characteristics of an emerging Streptococcus agalactiae serotype IX in Tilapia. Microb. Pathog. 122, 39–45. doi: 10.1016/j.micpath.2018.05.053
Zhang, Z., Yu, A., Lan, J., Zhang, Y., Zhang, H., Li, Y., et al. (2017). Draft genome sequence of an attenuated Streptococcus agalactiae strain isolated from the Gut of a Nile Tilapia (Oreochromis niloticus). Genome Announc. 5. doi: 10.1128/genomeA.01627-16
Zhou, K., Xie, L., Han, L., Guo, X., Wang, Y., and Sun, J. (2017). ICESag37, a novel integrative and conjugative element carrying antimicrobial resistance genes and potential virulence factors in Streptococcus agalactiae. Front. Microbiol. 8, 1921. doi: 10.3389/fmicb.2017.01921
Zubair, S., de Villiers, E. P., Fuxelius, H. H., Andersson, G., Johansson, K.-E., Bishop, R. P., et al. (2013a). Genome sequence of Streptococcus agalactiae strain 09mas018883, isolated from a Swedish cow. Genome Announc. 1. doi: 10.1128/genomeA.00456-13
Keywords: group B Streptococcus, Streptococcus agalactiae, foodborne disease, review, genomics, molecular epidemiology, whole genome sequencing
Citation: Chen SL (2019) Genomic Insights Into the Distribution and Evolution of Group B Streptococcus. Front. Microbiol. 10:1447. doi: 10.3389/fmicb.2019.01447
Edited by:
Pierre Renault, Institut National de la Recherche Agronomique (INRA), FranceReviewed by:
Bernard Beall, Centers for Disease Control and Prevention (CDC), United StatesCopyright © 2019 Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Swaine L. Chen, c2xjaGVuQGdpcy5hLXN0YXIuZWR1LnNn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.