 Xiangyuan Mei
Xiangyuan Mei Chunhui Wu
Chunhui Wu Jin Zhao
Jin Zhao Tian Yan
Tian Yan Peng Jiang
Peng Jiang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 28 May 2019
Sec. Aquatic Microbiology
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.01192
Golden tides dominated by Sargassum spp. are occurring at an accelerated rate worldwide. In China, Sargassum has started to bloom in the Yellow Sea and led to tremendous economic losses, but the underlying biological causes and mechanisms are still unclear. Although algae-associated bacteria were suggested to play crucial roles in algal blooms, the profiles of bacteria associated with drifting Sargassum remain unexplored. In this study, the community structures and functions of Sargassum-associated bacteria were analyzed using the high-throughput sequencing data of the V5–V7 hypervariable region of the 16S rRNA gene. Molecular identification revealed that the golden tide analyzed in the Yellow Sea was dominated by a single species, Sargassum horneri. They were a healthy brown color nearshore but were yellow offshore with significantly decreased chlorophyll contents (P < 0.01), which indicates that yellow S. horneri was under physiological stress. The structural and functional analyses of bacterial communities indicated that the drifting S. horneri had an obvious selectivity on their associated bacteria against surrounding seawater. Although the bacterial communities phylogenetically differed between brown and yellow S. horneri (P < 0.01), their dominant functions were all nitrogen and iron transporters, which strongly indicates microbial contribution to blooming of the algal host. For the first time, potential epiphytic and endophytic bacteria associated with Sargassum were independently analyzed by a modified co-vortex method with silica sand. We showed that the composition of dominant endophytes, mainly Bacillus and Propionibacterium, was relatively consistent regardless of host status, whereas the epiphytic operational taxonomic units (OTUs) greatly varied in response to weakness of host status; however, dominant functions were consistent at elevated intensities, which might protect the host from stress related to nitrogen or iron deficiency. Thus, we propose that host physiological status at different intensities of functional demands, which were related to variable environmental conditions, may be a critical factor that influences the assembly of epiphytic bacterial communities. This study provided new insight into the structure and potential functions of associated bacteria with golden tide blooms.
Golden tides are harmful algal blooms that are occurring at an accelerated rate worldwide and are caused by rapid proliferation of drifting brown seaweed Sargassum (Smetacek and Zingone, 2013; Sissini et al., 2017). For several decades, drifting Sargassum horneri aggregations have been reported in the East China Sea, but its origin remains unknown (Komatsu et al., 2014b; Qi et al., 2017). However, at the end of 2016, a large-scale golden tide bloomed in the adjacent Yellow Sea for the first time and destroyed nearshore Pyropia yezoensis cultivation, which resulted in substantial economic losses of as much as 500 million CNY (approximately 73 million United States dollars) (Xing et al., 2017). It was suggested that golden tides along coastal China might undergo notable variation, and the mechanism of blooming needs to be clarified for future disaster management.
Although up to 17 local Sargassum species are distributed along the coast of the Yellow Sea and East China Sea, which are fixed to substrate (Lu and Tseng, 2004; Huang et al., 2017), S. horneri was identified as the only dominant species of drifting Sargassum in the Yellow Sea (Su L. et al., 2018). This fact indicated that, except environmental factors such as nutrient content or ocean current, the distinct biological causes of S. horneri were also worthy of special attention. To date, the floating mechanism of S. horneri has been preliminarily studied, and it was found that the vesicles provide the floating force (Komatsu et al., 2014b), and the degree of maturity affects the detachment of the thallus from substrate (Xu et al., 2016); however, the cause of golden tides is still unclear.
Algae–bacteria interactions have been widely reported. As a host, algae provide habitat, oxygen, and carbohydrates such as algal polysaccharides for associated bacteria. In turn, bacteria provide hormones, vitamins, minerals, and carbon dioxide to algae, thus playing important roles in algal morphogenesis, growth, immune defense, and even spore release and germination (Matsuo et al., 2003; Croft et al., 2005; Singh and Reddy, 2014; Cho et al., 2015; Kouzuma and Watanabe, 2015). In addition, the associated bacteria also greatly contribute to algal bloom initiation and maintenance by secreting growth-promoting substances (Park et al., 2016), participating in nitrogen metabolism through nitrogen fixation (Falcon et al., 2002), or dissimilatory nitrate reduction to ammonium (DNRA), which would benefit the blooming of algal host from nutrient competition (An and Gardner, 2002). Therefore, it was suggested that the analyzing on associated bacteria may provide new insights into the causes and dynamics of seaweed blooming (Fu et al., 2018).
However, almost all related studies were performed on blooming microalgae, such as diatoms, dinoflagellates, or cyanobacteria (Sison-Mangus et al., 2016; Berry et al., 2017; Parulekar et al., 2017), and only a few studies have examined blooming macroalgae. Additionally, only epiphytic bacterial communities were preliminarily analyzed by denaturing gradient gel electrophoresis (DGGE) in green tide seaweed (Liu et al., 2011). To clarify the community structure and functions of bacteria associated with drifting Sargassum, three issues needed to be addressed.
Bacterial communities associated with attached macroalgae are obviously distinct from those in ambient seawater (Tujula et al., 2010; Burke et al., 2011b; Michelou et al., 2013) and also various among macroalgae species (Longford et al., 2007; Lachnit et al., 2009). This strong selectivity indicated specific functional requirements that seaweed imposed on associated bacteria, especially in relatively stable environments (Burke et al., 2011a; Zhang et al., 2018). However, drifting Sargassum experience rapid changes of both ambient conditions and physiological status (Komatsu et al., 2014a); therefore, whether the selectivity on associated bacteria still exists and varies under these circumstances still needs to be illustrated.
Preliminary tissue differentiation occurs in brown seaweed, and variation in bacterial communities among tissues has been noticed (Goecke et al., 2010). Sargassum possesses highly specialized structures, such as vesicles and blades with distinct adaptive functions for floating, but it was unknown whether the associated bacteria differed among tissues. In addition, almost all previous studies analyzed epiphytes rather than endophytes. To date, endophytic bacteria were mainly reported in red and green seaweed (Hollants et al., 2011b; Singh et al., 2011; Aires et al., 2015), and usually have a simple and stable composition (Hollants et al., 2011a). A comparative genomic analysis of an endophytic isolate from Ulva prolifera implied that endophytes might benefit hosts from nitrogen competition during blooming (Fu et al., 2018). Alternatively, similar research has not been carried out on brown seaweed, because of a lack of available methods for obtaining endophytic bacteria.
Studies on attached green macroalgae demonstrated that the bacterial communities were essentially assembled based on function (Burke et al., 2011a; Florez et al., 2017; Roth-Schulze et al., 2018). Blooming seaweeds usually have a higher metabolic level and are extremely sensitive to environmental stresses (Wang et al., 2012). To better understand the roles that the associated bacteria play in the blooming process, it is necessary to determine the relationships between bacterial communities and functions, environmental conditions and host status.
To address these issues, high-throughput sequencing of 16S ribosomal RNA (rRNA) genes was employed to analyze the community structures of bacteria associated with drifting Sargassum in the Yellow Sea. Moreover, a method for removal of epiphytic bacteria from the Sargassum surface was optimized to determine bacterial spatial distribution characteristics. In addition, the functions of bacterial communities were predicted to analyze their potential contributions to blooming.
At the end of December in 2016, a large-scale of floating Sargassum was reported to drift southward from Shandong Peninsula to nearshore of Jiangsu (Xing et al., 2017). Then some biomass stayed in the nearshore area intertwined by Pyropia cultivation rafts, and others kept drifting and finally entered the offshore area. Until April when south wind occurred, these two parts of biomass all started to shift to drift northward (unpublished data). A cruise carried out by research vessel R/V Ke Xue III was conducted in early June to investigate the Sargassum bloom in the Yellow Sea. The floating Sargassum were sampled from both nearshore (A, C) and offshore (B, D) sites (Figure 1 and Table 1), in which the seawater transparency and seaweed color significantly varied. The dark brown seaweed was distributed nearshore on the surface of muddy seawater. Alternatively, the light yellow seaweed was always drifting in offshore seawater that was more transparent (Figures 2A,B). Approximately 400-ml surface water samples were also collected using CTD (SBE911, Sea-Bird Electronics, Inc., United States) from each site as environmental controls. The seawater transparency was measured using a secchi disk.
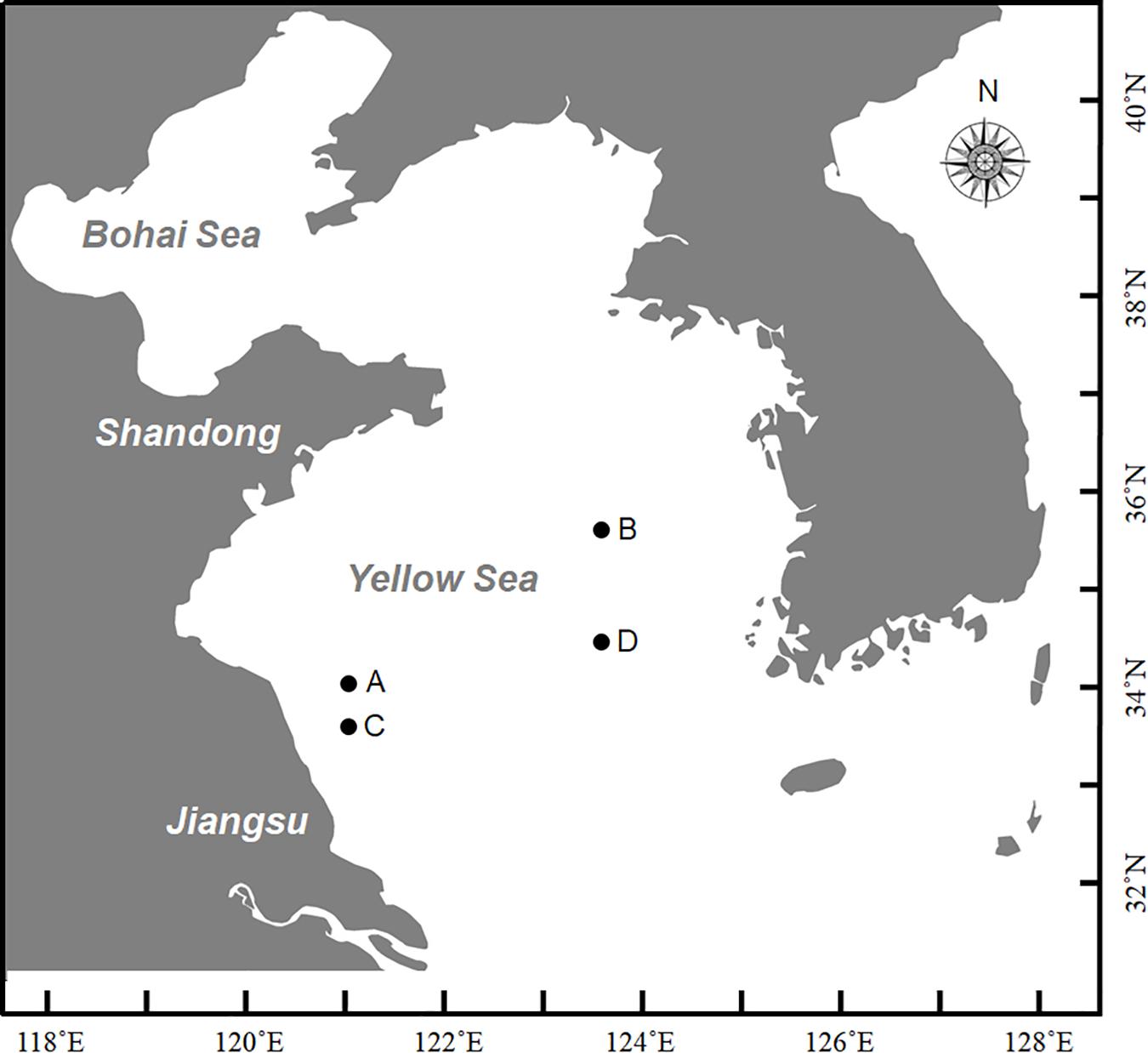
Figure 1. Sites for sampling in this study during Ke Xue III cruise. A and C represented nearshore sites with lower transparency where floating seaweeds were in brown color; B and D represented offshore sites with higher transparency where floating seaweeds were in yellow color.
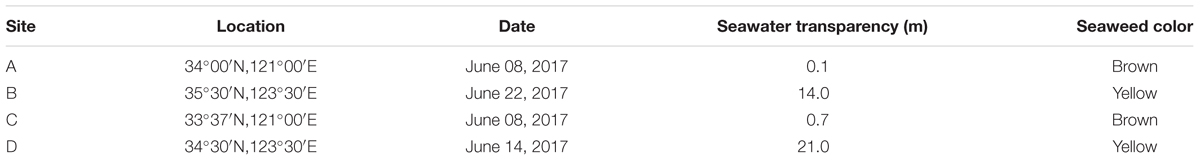
Table 1. Information on sampling sites surveyed in this study.

Figure 2. The status in field, morphology and tissues of drifting Sargassum. (A) In light yellow from offshore site. (B) In dark brown from nearshore site. (C) Thallus. (D) Blades. (E) Stalks. (F) Vesicles. Scale bars = 1 cm.
Five individuals from each sampling site were selected for molecular identification. Blades were washed with sterile seawater twice to remove any epiphytes and then homogenized by an electric tissue grinder (OSE-Y10, Tiangen, China). Total DNA was extracted with a Plant Genomic DNA Extraction Kit (Tiangen, China) and measured by spectrophotometer (Titertek Berthold, Germany). Nuclear-encoded ribosomal DNA internal transcribed spacer (ITS) 2 was amplified with a gradient thermal cycler (T960, Heal Force, China), following a previously described protocol (Yoshida et al., 2000). All PCR products were sub-cloned into pGEM-T cloning vector (Promega, United States) and sequenced by Sangon Biotech Co., Ltd. (China). Based on ITS2 sequence alignments from samples and representative taxa of brown seaweed using Clustal X 1.83, a phylogenetic neighbor-joining (NJ) tree was constructed by MEGA 5.0 using the Kimura 2-parameter model with 1000 bootstrap replications. To determine seaweed chlorophyll contents, 0.1 g of a segment from each individual was used for chlorophyll extraction and spectrophotometric analysis according to a previously described protocol (Jeffrey and Humphrey, 1975), and this was repeated in triplicate. After Levene test for the homogeneity samples variances via SPSS (v. 20), the difference of chlorophyll contents between brown and yellow samples were analyzed by One-way ANOVA using Microsoft Excel (2017).
A previous study on the green macroalgae Ulva showed that co-vortex of seaweed segments with silica sand was effective for removing and collecting most epiphytic bacteria; simultaneously, endophytic bacteria are prepared by grinding the remaining seaweed segments (Liu, 2015). Compared with Ulva, Sargassum has apparent tissue differentiation and a much tougher thallus surface. To test the effects on three kinds of Sargassum tissues (i.e., blade, vesicle, and stalk), the number of 15-min vortex replications were optimized from 1 to 10. The vortex suspension was composed of a piece of tissue (Figures 2C–F), silica sand with grain sizes of 125–250 μm, and sterilized seawater in a 1.5-ml centrifuge tube. After each round of vortexing for 15 min, the suspension was entirely renewed for the next round. From pipetted suspension, 10 μl was transferred to 2 ml of 2216E liquid media for culture at 28°C for 72 h; then, 200 μl of culture was transferred into a 96-well plate to measure the OD600 absorbance with a multi-functional microplate detector (M1000 Pro, Tecan, China). Because culturable bacteria account for a low percentage of environmental colonial samples (Amann et al., 1995), the removal effects were further detected by scanning electron microscopy (S-3400N, Hitachi, Japan) for tissues vortexed for 0, 5, or 10 replications. All tissues were fixed by 5% glutaraldehyde for 12 h, then gradient dehydration was carried out with 30, 50, 80, 90, and 100% ethanol at room temperature for 15 min, respectively. Observation was conducted at 5.00 kV and 1.50 K magnification. All treatments were conducted in triplicate.
Isolation of both epiphytic and endophytic bacteria from Sargassum was conducted on the research vessel just after seaweed collection. Individuals of floating Sargassum were selected from nearshore and offshore sites, and washed with autoclaved seawater. Each of the three kinds of tissues (i.e., blades, vesicles, and stalks), were cut by autoclaved dissecting knives and then co-vortexed with sterilized silica sand five times, which was determined to be the optimal number of replications by the optimization procedure. For each sample, all vortex suspensions produced over the five rounds were combined and pre-filtrated by mixed cellulose ester filter membranes (Merck Millipore, United States) with 5.0-μm pores to remove silica sand and other contamination. Then, epiphytic bacteria were collected by a filter membrane with 0.22-μm pores. The surface-cleaned Sargassum tissue was homogenized by an electric tissue grinder (OSE-Y10, Tiangen, China), and sterilized seawater was added for resuspension. Using the same multi-grade filtration as for epiphytic bacteria, the potential endophytic bacteria and environmental bacteria were prepared for nearshore and offshore seawater samples. All filter membranes with 0.22-μm pores were kept in liquid nitrogen until return to the laboratory for subsequent molecular analysis.
The microbial genomic DNA was isolated by a DNeasy PowerSoil Kit (QIAGEN, Netherlands) following the manufacturer’s instructions. To prevent potential amplification of 16S rRNA genes from organelle genomes of Sargassum, evaluations were conducted by aligning sequences of several pairs of universal primers with those of S. horneri mitochondrial (KJ938300.1) or chloroplast (KP881334.1) genomes. Finally, PCR amplifications of the V5–V7 region of the bacterial 16S rRNA gene were performed with primers 799F (5′-AACMGGATTAGATACCCKG-3′) and 1193R (5′-ACGTCATCCCCACCTTCC-3′) (Beckers et al., 2016). Sample-specific 7-bp barcodes were incorporated into the primers for multiplex sequencing. The PCR components contained 5 μl of Q5 reaction buffer (5×), 5 μl of Q5 High-Fidelity GC buffer (5×), 0.25 μl of Q5 High-Fidelity DNA Polymerase (5 U/μl), 2 μl (2.5 mM) of dNTPs, 1 μl (10 μM) of each forward and reverse primer, 2 μl of DNA template, and ddH2O added to 25 μl. Thermal cycling consisted of initial denaturation at 98°C for 2 min, followed by 25 cycles of denaturation at 98°C for 15 s, annealing at 55°C for 30 s, and extension at 72°C for 30 s, with a final extension at 72°C for 5 min. PCR amplicons were purified with Agencourt AMPure Beads (Beckman Coulter, United States) and quantified using the PicoGreen dsDNA Assay Kit (Invitrogen, United States). Amplicons were pooled in equal amounts, and paired-end (2 × 300 bp) sequencing was performed using the Illumina MiSeq platform with MiSeq Reagent Kit v3 at Shanghai Personal Biotechnology Co., Ltd. (China).
The Quantitative Insights into Microbial Ecology (QIIME, v1.8.0) pipeline was employed to process the sequencing data (Caporaso et al., 2010). The raw data of each sample were merged using Fast Length Adjustment of SHort reads (FLASH) to obtain raw tags (Magoc and Salzberg, 2011). After identifying and removing chimeric sequences, the effective tags were obtained and clustered to operational taxonomic units (OTUs) with a 97% similarity threshold. Taxonomic assignment of OTUs was performed by comparing sequences to the SILVA database.
Sequence data analyses were mainly performed using QIIME (v1.8.0) and R packages (v3.2.0). OTU-level alpha diversity analyses, including Chao1, Abundance-based Coverage Estimator (ACE), the Shannon index, and the Simpson index, were calculated using the OTU table in QIIME. The differences of the Chao1, ACE, Shannon index, and Simpson index between brown and yellow seaweed samples were calculated by One-way ANOVA after Bartlett test of homogeneity samples variances in R packages. OTU-level ranked abundance curves were generated to compare the richness and evenness of OTUs among samples. To investigate the structural variation of microbial communities across samples, beta diversity was estimated using UniFrac distance metrics (Lozupone and Knight, 2005; Lozupone et al., 2007) and visualized via Unweighted Pair Group Method with Arithmetic Means (UPGMA) hierarchical clustering using unweighted or weighted Unifrac Matrices (Ramette, 2007). Differences in Unifrac distances for pairwise comparisons among groups were determined using the Student’s t-test and the Monte Carlo permutation test with 1000 permutations via QIIME. Principal Component Analysis (PCA) using Euclidean distances was also conducted by R package “vegan” based on RDA function using the genus-level compositional profiles (Ramette, 2007). The significance of differentiation of microbiota structure among groups was assessed by Permutational Multivariate Analysis of Variance (PERMANOVA) (McArdle and Anderson, 2001) and Analysis of Similarities (ANOSIM) (Clarke, 1993; Warton et al., 2012) using the R package “vegan”. Taxa abundances at the phylum, class, order, family, genus, and species levels were statistically compared among samples or groups by Metastats (White et al., 2009) and visualized as column plots. The generalization error was estimated using 10-fold cross-validation. The expected “baseline” error was also included, which was obtained by a classifier that simply predicts the most common category label. Based on OTU data, microbial functions were predicted by Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) (Langille et al., 2013) and then categorized by Kyoto Encyclopedia of Genes and Genomes (KEGG) classification, and visualized in heatmap clustered by Hierarchical cluster via R package.
All ITS2 sequences from 20 individuals were determined as two genotypes G1 (accession number MK182418.1) and G2 (accession number MK182419.1), which were distinguished by a single SNP and both deposited in GenBank. These two genotypes were found to represent intra-individual polymorphism in both yellow and brown samples. Phylogenetic analysis with G1, G2, and homologous sequences from other Sargassum species showed that all drifting seaweed belonged to a single species, S. horneri (Figure 3), which was consistent with the findings of a previous report (Su L. et al., 2018).
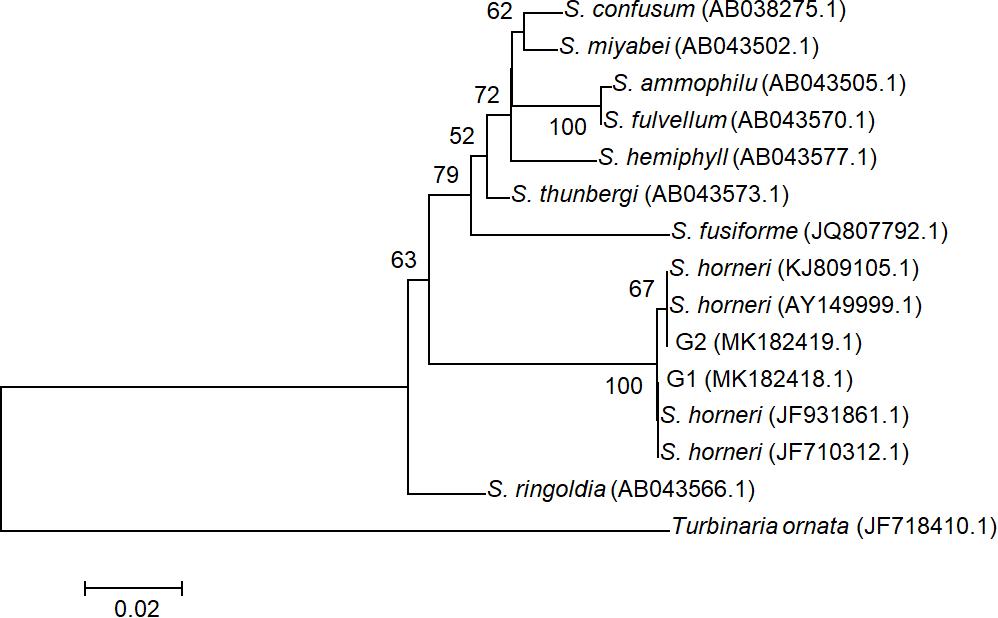
Figure 3. Phylogenetic NJ tree based on ITS2 alignment. Numbers at internal nodes indicate support values obtained using the bootstrap method with NJ. Scale indicates substitutions per site. G1 and G2 represented two genotypes of ITS2 sequences in S. horneri, representing intra-individual polymorphism in both yellow and brown samples.
However, the chlorophyll contents in brown and yellow S. horneri were significantly different (P = 1.41072E-05 for chlorophyll a, P < 0.01; P = 1.27422E-05 for chlorophyll c1 + c2, P < 0.01). As shown in Figure 4, the contents of chlorophyll a and c1 + c2 in brown S. horneri (147.30 μg/g ± 6.51 and 41.97 μg/g ± 1.559) were 5.93 and 4.5 times higher than those in yellow S. horneri (24.85 μg/g ± 5.195 and 9.33 μg/g ± 1.500), respectively, which indicates that the brown and yellow samples were in different physiological status.
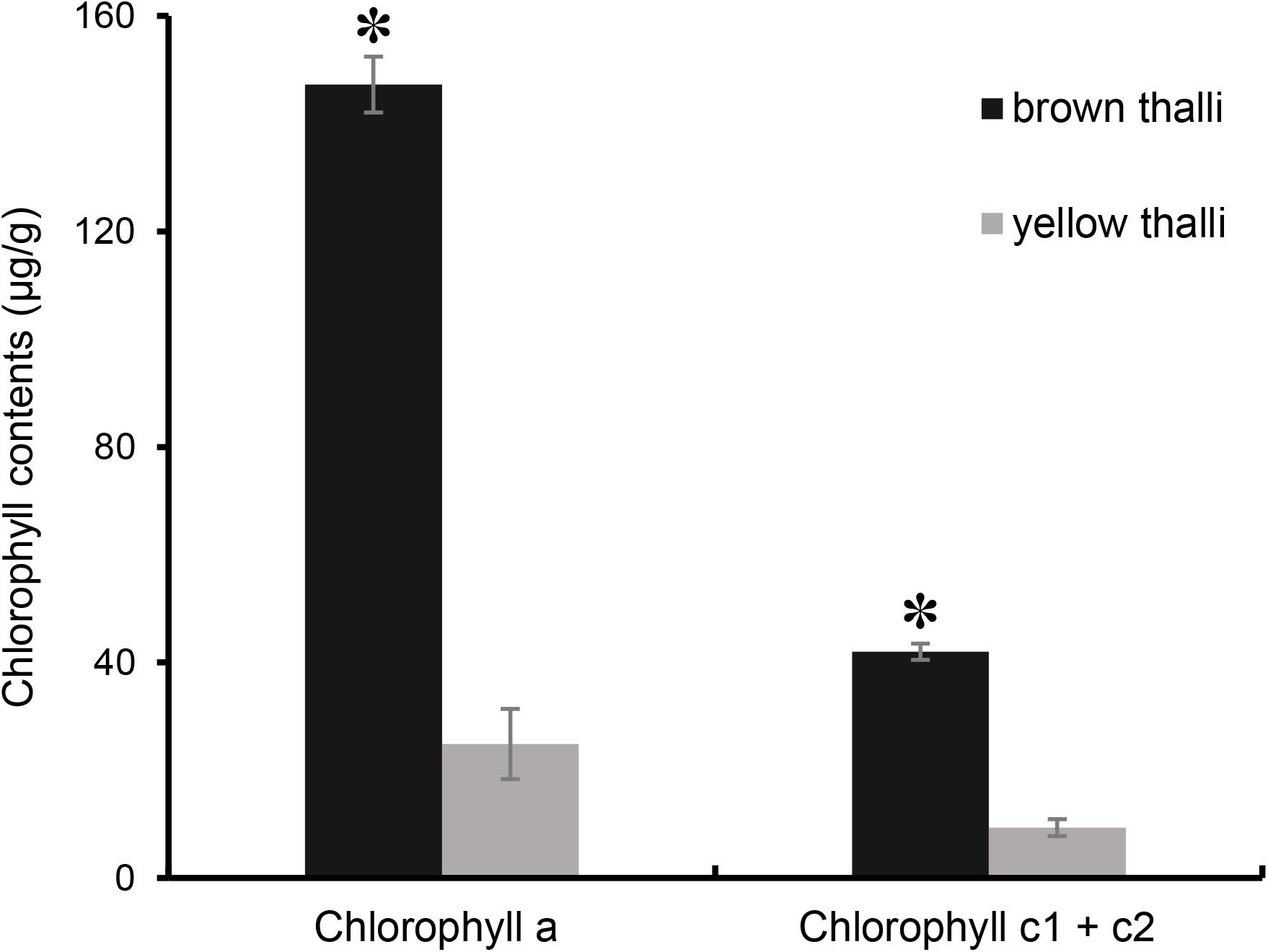
Figure 4. Comparison of chlorophyll contents between brown and yellow S. horneri thalli. The error bar represented the standard deviation, the asterisk above represented the significant differences.
As shown in Figure 5, an increase in number of co-vortex replications of seaweed tissues and silica sand was associated with a decrease in OD600 absorbance values in all kinds of tissues, which reflected the abundance of bacteria removed from each round of vortexing, and nearly reached to zero after five vortex replications, which indicates possible removal of all epiphytes. Subsequently, the abundance of removed bacteria slightly increased, especially in stalk groups, which indicates a potential release of endophytic bacteria from damaged Sargassum cells due to overtreatment. These results were consistent with those revealed by scanning electron microscopy, in which five rounds of vortexing could eliminate almost all epiphytic bacteria while retaining intact Sargassum cells, whereas 10 rounds of vortexing resulted in obvious damage to the seaweed surface, especially the stalk (Figure 6). Therefore, five replicates was determined to be optimal for subsequent vortex treatments.
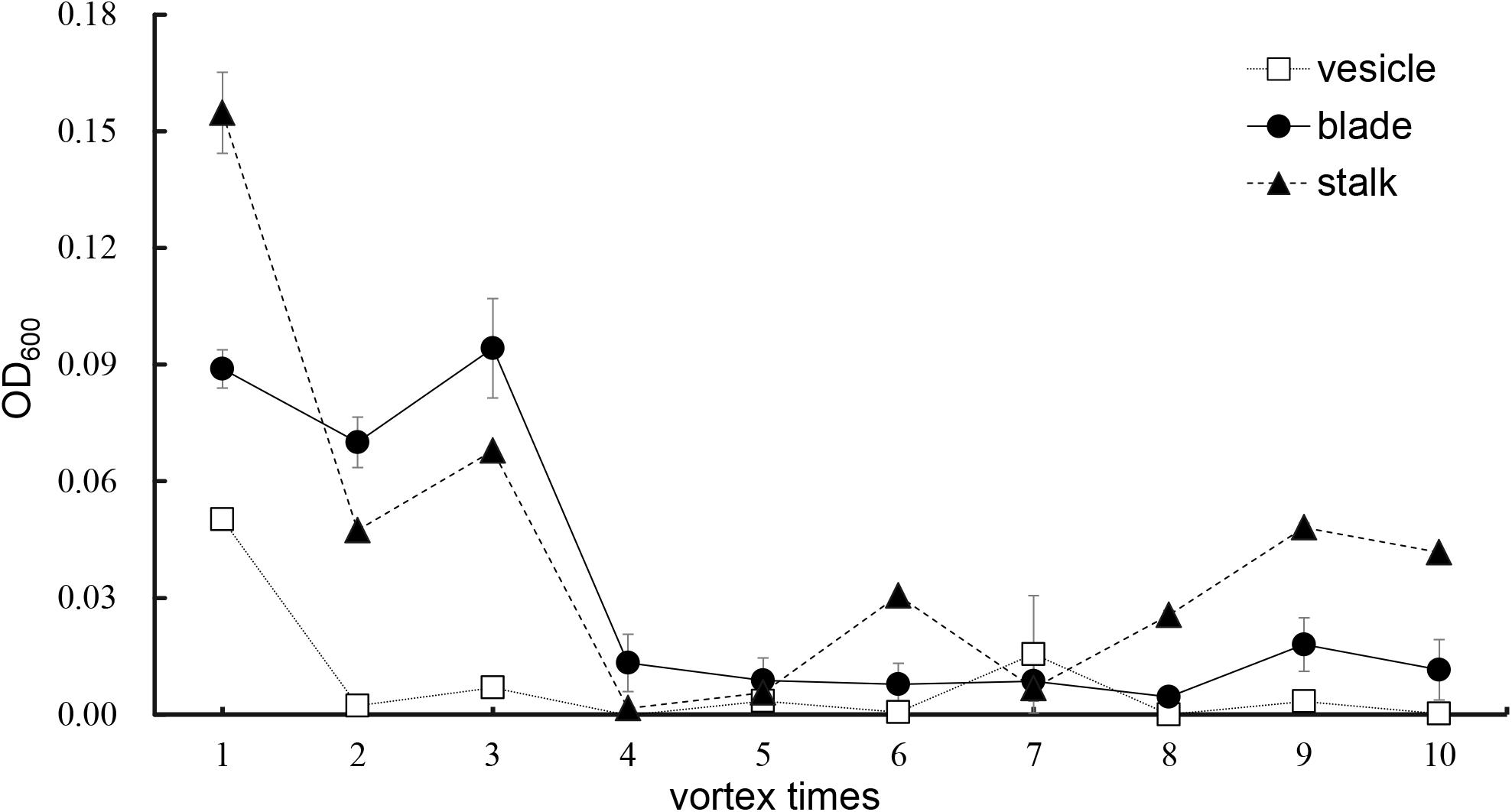
Figure 5. Culture method detection for removal effects of epiphytic bacteria from three kinds of S. horneri tissues.
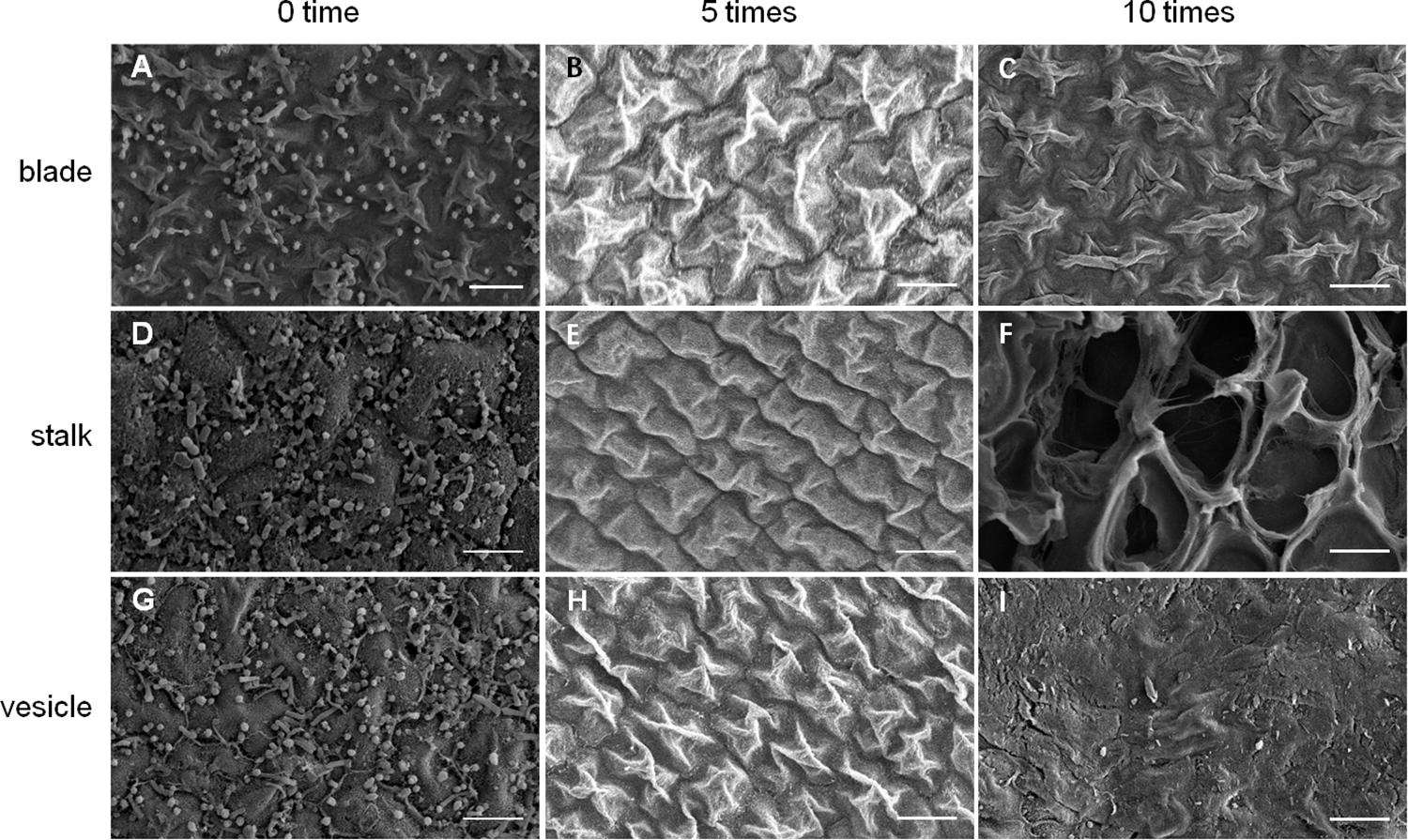
Figure 6. SEM detection for removal effects of epiphytic bacteria from three kinds of S. horneri tissues. (A) Blade blank control. (B) Blade vortexed for 5 times. (C) Blade vortexed for 10 times. (D) Stalk blank control. (E) Stalk vortexed for 5 times. (F) Stalk vortexed for 10 times. (G) Vesicle blank control. (H) Vesicle vortexed for 5 times. (I) Vesicle vortexed for 10 times. Scale bars = 5 μm.
Annotation of sample names was based on the numbers and characters: 1, nearshore sites; 2, offshore sites; B, blade; S, stalk; V, vesicle; en, endophyte; ep, epiphyte; W, water sample. And this annotation was used throughout this paper including figure legends.
Based on Illumina MiSeq sequencing data, a total of 306,784 high-quality 16S rRNA gene sequences were obtained from 14 groups of samples, in which the percentage of sequences generated from organelle genomes of S. horneri were lower than 0.08%, which indicated that there were no notable interference to further analyses. All raw sequences were deposited in the NCBI Sequence Read Archive (SRA) under the accession number of PRJNA504763. According to the amplicon analyses, all sequences clustered into 19,867 OTUs at a 97% identity threshold, which were further classified into 525 bacterial groups at the genus level. Rarefaction curves and species accumulation curves demonstrated that the sequencing depth was sufficient to profile bacterial richness and diversity in all samples (Supplementary Figures S1, S2). The Chao1 and ACE estimators in Supplementary Table S1 indicated that the bacterial richness from the nearshore samples was much higher than that from offshore samples (Chao1: P = 0.013; ACE: P = 0.014). However, the Shannon and Simpson indexes exhibited similar bacterial diversity between these two types of sampling locations (Shannon: P = 0.144; Simpson: P = 0.140). It was shown that all samples were similarly dominated by sequences from Proteobacteria, Bacteroidetes, Actinobacteria, and Firmicutes at the phylum level (Supplementary Figure S3), but they were much more divergent at the genus level (Supplementary Figure S4).
High-throughput sequencing showed that the bacterial communities of drifting seaweed were clearly distinct from those of seawater regardless of if they were from nearshore or offshore habitats. As shown in Figure 7, the UPGMA trees constructed using weighted or unweighted Unifrac distance were similar, in which almost all samples of brown S. horneri were roughly separated from those of yellow S. horneri, whereas two ambient seawater samples, 1W and 2W, closely clustered together, which indicated obvious selectivity of seaweed host on their associated bacteria against the surrounding seawater. This was also shown clearly in the bar plot of Figure 8. In nearshore sites, the top five dominant bacterial genera from dark brown S. horneri were Bacillus (12.2%), Propionibacterium (9.48%), Kocuria (8.01%), Pseudomonas (7.65%), and Bacteroides (5.21%); however, they were present in extremely low amounts in seawater (0.299, 0.516, 0.179, 0.598, and 0.560%, respectively) (Figure 8A). A similar situation occurred in offshore sites, in which Flavobacterium (16.5%), Paracoccus (11.5%), Bacillus (6.97%), and Propionibacterium (5.27%) were mainly selected by light yellow S. horneri, whereas their percentages in seawater were 0.851, 0.483, 0.278, and 0.859%, respectively (Figure 8B). It was notable that, except for Bacillus, all dominant genera in seaweed were mainly composed of a single OTU.
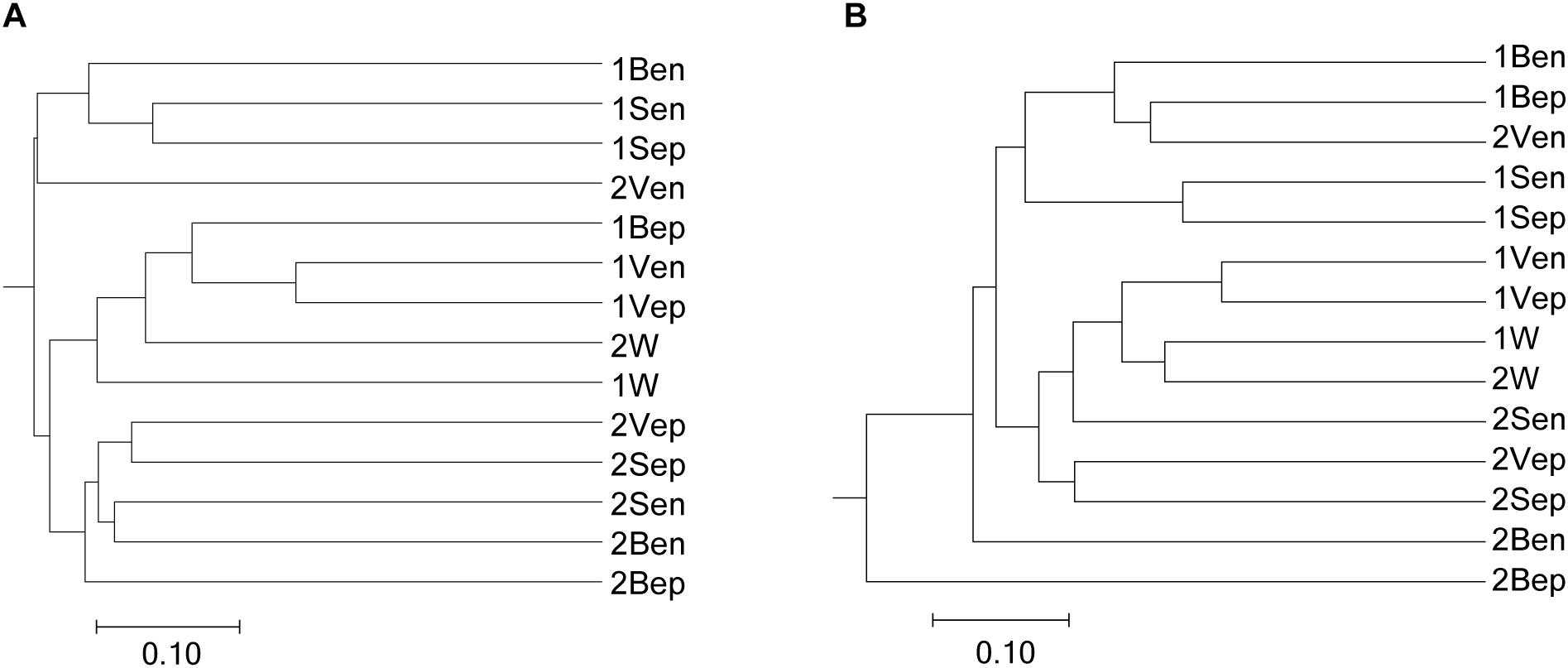
Figure 7. Dendogram of similarity of all samples clustered by the UPGMA method. (A) Unweighted. (B) Weighted. The scale bar shows the distance between clusters in UniFrac units.
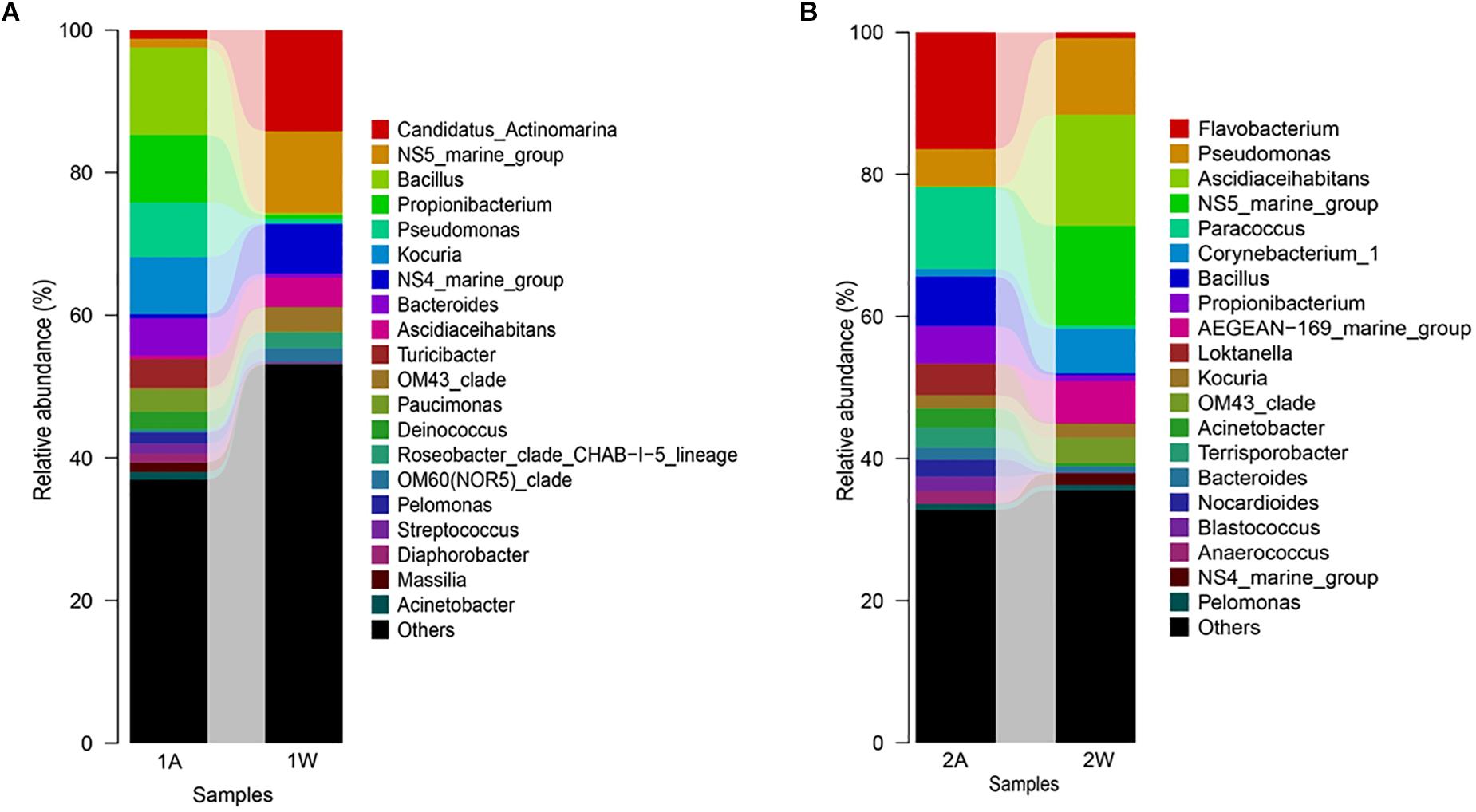
Figure 8. Bar-plot of bacterial composition of S. horneri and surrounding seawater at genus levels. (A) Nearshore. (B) Offshore. 1A: total bacteria isolated from brown S. horneri, 1W: total bacteria isolated from nearshore seawater, 2A: total bacteria isolated from yellow S. horneri, 2W: total bacteria isolated from offshore seawater.
Nevertheless, the selectivity of floating S. horneri on bacterial communities was not consistent between brown and yellow thalli. The Adonis/PERMANOVA results revealed that the bacteria associated with brown S. horneri were significantly different from those associated with yellow S. horneri (P = 0.006, unweighted UniFrac; P = 0.009, weighted UniFrac), which was visualized via PCA plot in which these two groups were distributed in different clusters (Figure 9). These two types of seaweed were sampled in different habitats; however, the two ambient seawater samples had similar bacterial structures (Figures 7, 9). Because brown and yellow S. horneri significantly varied in chlorophyll contents (Figure 4), we suggest that the seaweed selectivity on associated bacteria was highly affected by host physiological status.
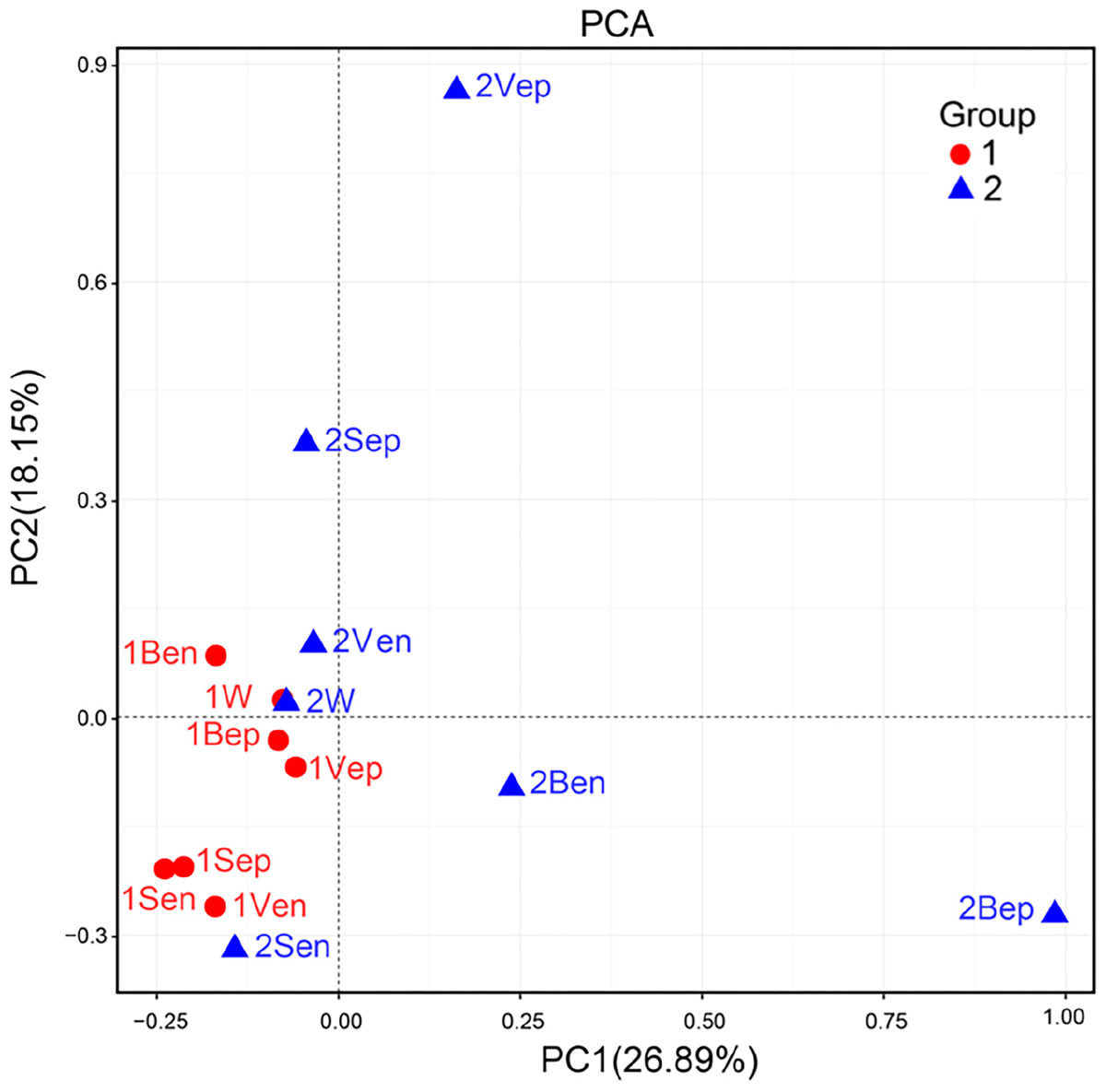
Figure 9. Principal Component Analysis (PCA) of the bacterial communities from all samples based on Euclidean distance at genus level.
As shown in Figure 10, the bacterial community structures of floating S. horneri seemed to be shaped by seaweed tissues, but in an algal physiological status-dependent manner. In brown S. horneri, samples from each of the three tissues (i.e., blade, vesicle, and stalk) clustered into a separate group (Figure 10A), and the Adonis/PERMANOVA results showed that the differences among the three tissues were close to significant (P = 0.067, unweighted UniFrac; P = 0.067, weighted UniFrac). By contrast, in yellow S. horneri, no distinct bacterial communities were detected among tissues (Figure 10B) and P-values were much higher (P = 0.600, unweighted UniFrac; P = 0.267, weighted UniFrac).
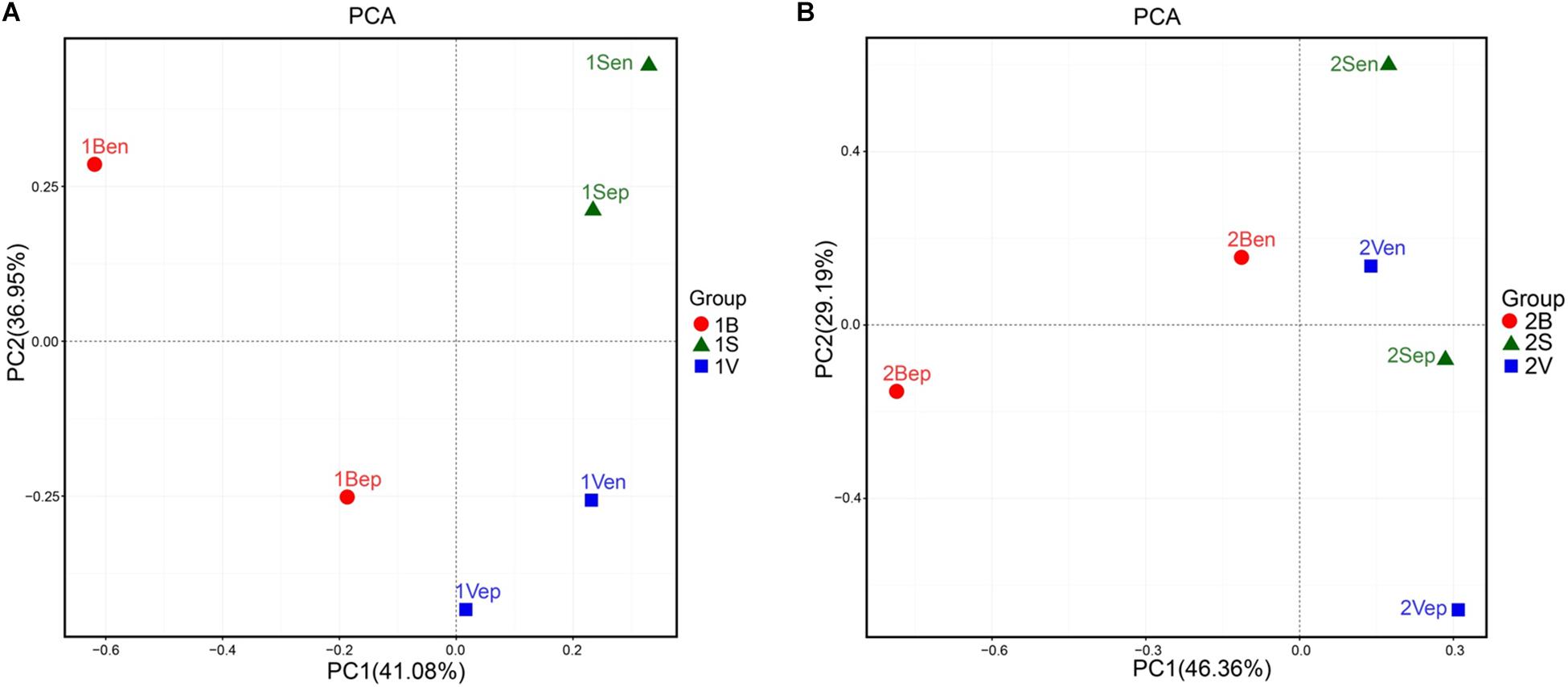
Figure 10. Principal Component Analysis (PCA) of the bacterial communities from blade, stalk, and vesicle of S. horneri based on Euclidean distance at genus level. (A) Brown S. horneri. (B) Yellow S. horneri.
To further distinguish potential epiphytic and endophytic bacteria, the abundance and ranks of each dominant genus (abundance greater than 5% in any sample) were compared based on the data collected before and after grinding with the same types of seaweed samples. As shown in Figure 11, the abundances of Flavobacterium and Paracoccus, the two most dominant genera from yellow S. horneri, significantly decreased by 63.6 and 93.6% after grinding, respectively. Likewise, in brown S. horneri, the abundances of the three main dominant genera, Kociria, Bacteroides, and Pseudomonas, were reduced by 60.8, 60.2, and 49.5%, respectively (Figure 11A). Because violent friction effectively removed epiphytes, we suggest that bacteria from the above genera were most likely located on the surface of the seaweed thallus. Alternatively, some bacteria became dramatically much more predominant after grinding. Interestingly, no matter in yellow or brown S. horneri, Bacillus, and Propionibacterium significantly increased and became extremely dominant after removing epiphytes. In total, their abundances were increased by 60.6 and 64.8%, which indicates that they were the two main types of potential endophytes. Moreover, some genera were suspected to be specific endophytes of yellow or brown S. horneri. For example, Turicibacter was only detected in brown S. horneri, and their abundance increased by 82.8% after grinding; similarly, Loktanella abundance increased by 98.2% in yellow S. horneri (Figure 11B).
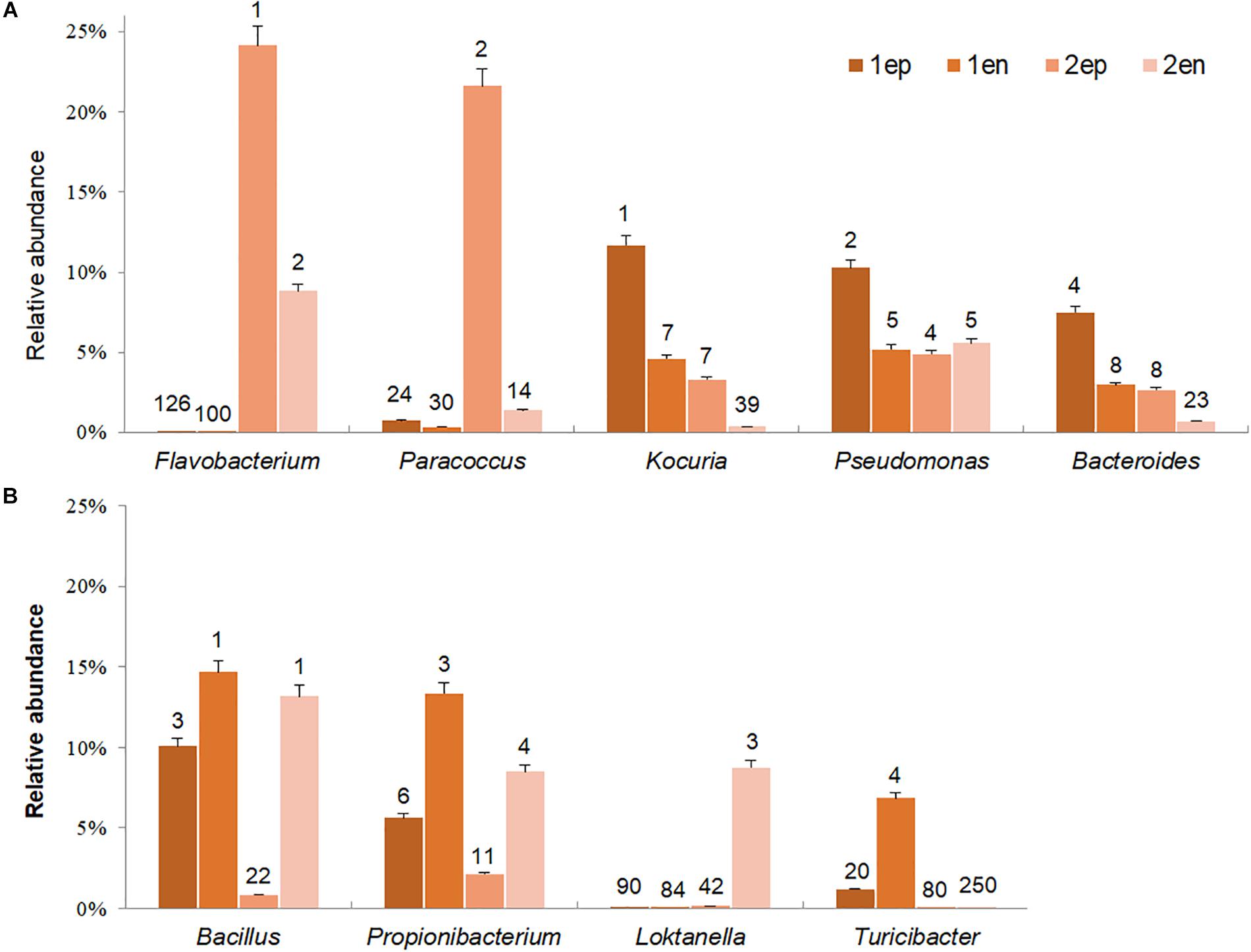
Figure 11. Potential dominant epiphytic and endophytic bacterial genera in brown and yellow S. horneri. (A) Epiphytic bacterial genera. (B) Endophytic bacterial genera. The number above column indicated the rank in abundance. 1ep: all epiphytic bacteria associated with brown thallus, 1en: all endophytic bacteria associated with brown thallus, 2ep: all epiphytic bacteria associated with yellow thallus, 2en: all endophytic bacteria associated with yellow thallus.
The endophytic bacteria dominated by Bacillus and Propionibacterium appeared to be much more consistent than epiphytes, regardless of host physiological status. Bacillus was mainly located inside vesicles (1Ven, 27.9%) or stalks (1Sen, 15.8%; 2Sen, 33.4%), whereas Propionibacterium occurred inside blades (1Ben, 36.9%) or vesicles (2Ven, 17.8%). In contrast, a sharp variation in composition of dominant epiphytes was observed between yellow and brown S. horneri. In yellow S. horneri, Flavobacterium, and Paracoccus occupied the surface of blades (2Bep, 63.0%) and vesicles (2Vep, 49.1%), respectively, with extremely high dominance. Alternatively, in brown S. horneri, Kocuria mainly occurred on the surface of vesicles (1Vep, 22.1%), and Bacteroides shared the blade surface (1Bep, 18.9%) with Pseudomonas (1Bep, 18.5%).
After assessing the metabolic functions of bacterial communities by PICRUSt via the KEGG database, a total of 6,909 KEGG orthology (KO) categories were obtained and were assigned to 328 subsystems of 41 metabolic classes. As shown in Figure 12, the vertical topological structure illustrated the phylogeny of all samples based on the abundance of 50 most dominant functional proteins (Supplementary Table S2), which indicated that epiphytes were predicted to potentially have more functions than endophytes. Interestingly, the horizontal topological structure revealed that the nitrogen and iron metabolism-related proteins were enriched in both yellow and brown seaweed relative to seawater, which indicated that they might be crucial for golden tide blooms. These proteins were mainly classified into (1) nitrogen transport: peptide and nickel transporters (K02032–K02035), sulfate/nitrate/taurine transport system (K02049–K02051), and polar amino acid transport system (K02029 and K02030); and (2) iron transport: iron complex transport system (K06147 and K02013–K02016). Notably, these proteins were more dominant in yellow than brown seaweed. Alternatively, the sugar transport-related proteins, namely monosaccharide or oligosaccharide transporters (K02025–K02027), were only abundant in brown seaweed and nearshore seawater bacteria.
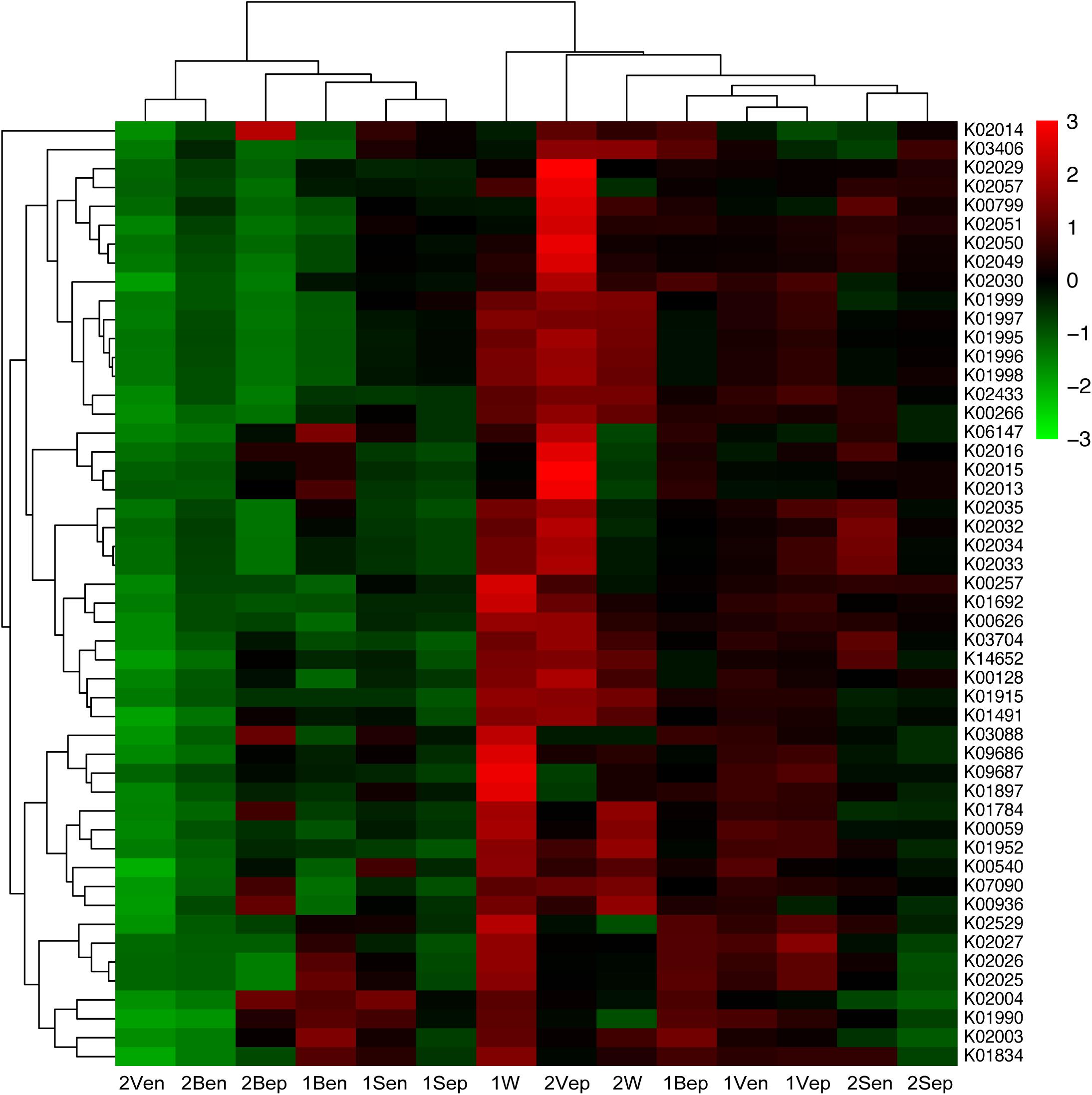
Figure 12. Heat-map of top 50 abundant functional proteins combined with the phylogenetic distribution of samples clustered by Hierarchical cluster. The color key scale represented the abundance of corresponding proteins. Each row represented the abundance distribution of right KEGG orthologous category (KO) among samples, and each column represented the abundance distribution of various KO in each sample whose name was shown at bottom. Lateral topology was contributed by the similarity of KO abundance distribution among different samples, and longitudinal topology was contributed by similarity of different KO species distribution in each sample.
Analysis of spatial distribution of function proteins revealed that the nitrogen transport system was mainly dominant in 2Vep, 2Sen, 1Bep, and 1Vep; the iron transport system was mostly dominant in 2Vep, 2Bep, 1Ben, and 1Bep; and the sugar transport system was mainly distributed in 1Bep, 1Vep, 1Ben, 1Ven, and 1W. Overall, nitrogen and iron transport were predicted to be dominant functions in the epiphytic bacteria of vesicles and blades, especially in yellow S. horneri, and sugar transport mainly occurred on the surface of vesicles and blades in brown S. horneri and in nearshore seawater.
In this study, the community structures of bacteria associated with blooming S. horneri and ambient seawater were analyzed and compared. We found that the seaweed host exhibited obvious selectivity on associated bacteria, as the bacterial communities of drifting S. horneri were clearly distinct from those of seawater in both nearshore and offshore habitats (Figure 8). Algal–bacterial specificity has been widely reported in red seaweed (Longford et al., 2007; de Oliveira et al., 2012), green seaweed (Burke et al., 2011b; Hollants et al., 2013), brown seaweed (Lachnit et al., 2009; Michelou et al., 2013), and microalgae (Jasti et al., 2005); therefore, it is thought that the substance exchange between algae and associated bacteria may shape and maintain the algal–bacterial communities (Egan et al., 2013; Hollants et al., 2013).
However, seaweed selectivity on associated bacteria was not always stable and was affected by various environmental factors, such as season (Bengtsson et al., 2010; Tujula et al., 2010), salinity (Dittami et al., 2016), nutrients (Piggott et al., 2015), and even desiccation during low tide (Zhang et al., 2018). For blooming algae, such as U. prolifera (Liu et al., 2011), Alexandrium tamarense (Doucette, 1995), Alexandrium catenella (Zhou et al., 2018), and Cochlodinium polykrikoides (Park B.S. et al., 2015), the community structures of associated bacteria also varied at different bloom stages. In our two types of sampling locations with distinct physicochemical properties (Pan et al., 2018), the floating S. horneri obviously differed in color, chlorophyll contents (Figure 4), and bacterial communities. The seaweed chlorophylls contents have been proved to be related to their physiological status such as photosynthetic activity or growth rate (Zhang et al., 2013; Costa et al., 2017). Therefore, because the nearshore and offshore seawater microbiota were similar (Figures 7, 9), it was strongly indicated that host physiological status might shape the assembly of associated bacteria to meet the altered functional demands of the host (Lekunberri et al., 2012; Grottoli et al., 2018).
Studies on terrestrial plants showed that different parts of plants, like roots and leaves, may shape their specific bacterial compositions because of the substantial amount of variation in their respective local habitats (Sarria-Guzmán et al., 2016). For seaweed, although all parts of fronds live in the same seawater environment, the composition of associated bacteria among different parts or tissues also varied, which was also reported in Chara vulgaris (Ariosa et al., 2004), Saccharina latissima (Staufenberger et al., 2008), and Ascophyllum nodosum (Cundell et al., 1977). The lack of vascular connections was considered the key that might limit nutrient transport among tissues, thus leading to variant supplies of substances to associated bacteria (Goecke et al., 2010). In this study, such a tissue-specific distribution pattern of bacterial communities was also roughly shown in brown floating S. horneri, but was not apparent in yellow S. horneri (Figure 10). Further study with larger sample size is needed to confirm whether this pattern is affected by seaweed host physiological status.
The scanning electron microscopy and culture analysis results (Figures 5, 6) showed that co-vortex of S. horneri segments with silica sand might effectively remove most epiphytic bacteria. In particular, compared with other chemical methods involving strong oxidant (Kientz et al., 2011; Aires et al., 2012), this method provides a feasible approach for studying the community structure of endophytes with minimal interference (Liu, 2015). Moreover, we found that for each specific seaweed material, the protocol for parameter optimization was necessary, since overtreatment would result in the release of endophytes by damaging the seaweed cell walls. Using this new method, the potential dominant endophytic and epiphytic bacteria from golden tide-forming drifting Sargassum were suggested. As shown in Figure 11, most dominant endophytic bacteria of S. horneri belonged to Bacillus and Propionibacterium. Four dominate endophytic OTUs were identified in Bacillus, which shared high sequence identity with those endophytic Bacillus strains reported in the brown seaweed Sargassum sabrepandum (Ahmed et al., 2016), green seaweed Ulva lactuca (Nair et al., 2016), and red seaweed Gracilaria dura (Singh et al., 2011). Alternatively, only one endophytic strain was detected in Propionibacterium, and exhibited extremely high identity (99%) with Propionibacterium acnes, which lives inside higher plants (Tan et al., 2011; Sánchez-López et al., 2018). In addition, two other endophytic genera, Loktanella and Turicibacter, both of which only consisted of one OTU, were also found to be the main endo-symbionts in cnidaria (Doepke et al., 2012) and animal guts (Auchtung et al., 2016), respectively. Moreover, we found that epiphytic bacteria from Flavobacterium, Paracoccus, Kocuria, Pseudomonas, and Bacteroides mainly occupied the surface of S. horneri, and all of these genera are common among brown seaweed-associated bacteria (Staufenberger et al., 2008; Hollants et al., 2013; Park S.H. et al., 2015; Uzair et al., 2018). Interestingly, Bacteroides is known to be strictly anaerobic, we speculate that the presence of abundant polysaccharides at the surface of healthy seaweed thallus might likely protect Bacteroides from exposure to oxygen.
It is worth noting that, following a change in environment and host status, the composition of dominant endophytes was quite consistent, whereas that of major epiphytes substantially changed; this could be main reason for the significant variation between the bacterial communities of yellow and brown S. horneri. Several studies reported that the composition of algal endophytes was very stable and unique (Hollants et al., 2011a), and has been used to reveal the invasion route of algal hosts (Aires et al., 2013). The identification of endophytes in drifting S. horneri may inspire additional approach, because the origin of golden tides in the Yellow Sea is still unknown (Komatsu et al., 2014b; Qi et al., 2017).
Metabolic protein prediction (Figure 12) revealed that the bacteria associated with seaweed differed from those in seawater in both phylogeny and function. Moreover, their functions in nitrogen and iron transport were dominant in seaweed regardless of host status, which indicated that they might play crucial roles both in golden tide blooms and in the survivals of seaweed under stress. Because the bacterial communities in yellow and brown S. horneri were significantly different in phylogeny, we suggest that algae-associated bacteria tended to be assembled by function, which was consistent with the competitive lottery theory (Burke et al., 2011b). Interestingly, the abundances of nitrogen and iron transport proteins were much higher in yellow S. horneri, whereas sugar transport proteins were dominant in brown S. horneri; this demonstrated an obvious correlation among environmental characteristics, host demands, and bacterial community functions.
Nitrate accounted for 80–100% of dissolved inorganic nitrogen (DIN) in the South Yellow Sea (Gao et al., 2012). According to the data collected from the same cruise, the nitrate concentration was approximately 30 μM in the nearshore locations but close to zero in the offshore locations (Pan et al., 2018), which indicated that yellow S. horneri was under nitrogen deficiency stress compared with brown S. horneri. We found that the nitrogen transport functions were extremely dominant on the surface of vesicles in yellow S. horneri, and this appeared to be associated with a single OTU from Paracoccus, which was extremely dominant (49%) among epiphytes. Active nitrate transport was reported in Paracoccus denitrificans, a closely related strain capable of nitrate assimilation (Goddard et al., 2017), and several closely related strains of Paracoccus had urease activity (Liu et al., 2008; Shen et al., 2012), which could be influenced by the peptide and nickel transporters (Hiron et al., 2010). Therefore, we speculated that nitrogen transport enhancement by the epiphytes might improve host growth nearshore, and help protect the host from nitrogen deficiency-based stress offshore by enriching some unique OTUs with enhanced functions. However, the potential mechanism of nitrogen transmission between seaweed and bacteria was still unclear.
As predicted, the iron complex (siderophore iron) outer membrane receptor protein (OMP) and iron complex transport system proteins were abundantly distributed in epiphytic bacteria of blades and vesicles, respectively, especially in yellow S. horneri. Iron is essential for chlorophyll synthesis in seaweed. According to the data collected in the Yellow Sea in spring 2015, the concentration of dissolved iron was approximately 70 nM nearshore, but as low as approximately 12 nM in offshore seawater (Su et al., 2015; Su H. et al., 2018). Studies showed that S. horneri was yellow in 14.4 nM of dissolved iron, and turned brown above 43 nM, which indicates increased chlorophyll content (Miki et al., 2016). Thus, we speculate that the yellow S. horneri were also under iron deficiency stress compared with brown S. horneri. Approaches on iron concentration mechanism in brown seaweed showed that, the carboxylate group of alginate on the thallus surface could absorb iron or possibly exchange iron with bacterial siderophores (Miller et al., 2016). We found that Flavobacterium highly dominated the surface of blades and Paracoccus dominated the surface of vesicles, and both were prominent in iron transport. TonB-dependent OMP, which was crucial for transport of the siderophore–iron complex, has been recognized in several closely related strains of Flavobacterium (Møller et al., 2005; Guan et al., 2013) and several closely related Paracoccus strains had strong abilities to produce siderophores (Bergeron et al., 1985; Bergeron and Weimar, 1990; Sedlácek et al., 2009). These two dominant OTUs might play key roles in supplying iron to hosts under stress. However, further study is needed to determine whether siderophore-mediated iron transmission occurs between seaweed and bacteria.
In addition, sugar transport was predicted to be abundant on both the surface of brown S. horneri and in nearshore seawater, which has been proved in other seaweed (Burke et al., 2011b; Zhang and Wang, 2017). A study indicated that the seaweed surface continued to secrete polysaccharides into surrounding seawater, which provides a carbon source for bacteria (Goecke et al., 2010). Consistent with the function prediction, we found that the surface of brown S. horneri was much more slippery than that of yellow S. horneri, which might arise from a low metabolic level and deficient polysaccharide secretion in yellow S. horneri.
It should be emphasized that the metabolic function analysis was only based on bioinformatics prediction by PICRUSt, the accuracy of PICRUSt relies on the availability of completely sequenced genomes for the representative organisms (Wilkinson et al., 2018), and this method might lead to low accuracy for function prediction of endophytes (Langille et al., 2013). In this study, Bacillus and Propionibacterium OTUs were identified as dominant endophytic bacteria in drifting S. horneri, but no strong functions were predicted. However, Bacillus has been widely reported to be plant endophytes with various abilities that enhance the host’s competitive advantage, including promotion of host growth (Li et al., 2018), bioleaching of iron (Sun et al., 2017), producing IAA and fixing nitrogen to promote sprouting for seaweed (Singh et al., 2011), and releasing antibacterial substances to protect hosts against disease (Pathak et al., 2012). Therefore, endophytes should be investigated further, because several Bacillus strains in this study have been successfully isolated and cultured. In addition, nitrogen and iron metabolism still need to be verified in the field, by RT-PCR of target genes, and metagenomics analysis, because of the high proportion of unknown OTUs observed in bacterial communities.
In this study, the community structure and functions of bacteria associated with drifting S. horneri were analyzed. We showed that the seaweed host had obvious selectivity on associated bacteria, which were assembled via functions rather than phylogeny. The proteins involved in nitrogen and iron transporters dominated the predicted functions, which indicated that the microbial community may greatly contribute to algal host blooms. In response to weakness of host status, the epiphytic OTUs extensively varied, whereas the dominant functions were consistent but at elevated intensities, which might protect the host from nitrogen or iron deficiency-based stress. Alternatively, the composition of endophytic bacteria remained consistent, and they potentially play key roles in rapid growth of the host. We propose that host physiological status, with different intensities of functional demands related to variable environmental conditions, may be a critical factor that influences epiphytic bacterial community assembly. This study provided new insights into the structure and potential functions of associated bacteria with golden tide blooms.
PJ and XM conceived and designed the experiments. XM and CW performed the experiments. JZ, XM, and PJ analyzed the data. CW and TY collected the samples. XM and PJ wrote the original draft. PJ revised the manuscript. All authors reviewed and approved the final manuscript.
This work was supported by Strategic Priority Research Program of the Chinese Academy of Sciences (XDA23050302), National Natural Science Foundation of China (41776153), Open Fund of Laboratory for Marine Ecology and Environmental Science, Qingdao National Laboratory for Marine Science and Technology (LMEES201801), Open Fund of Qingdao National Laboratory for Marine Science and Technology (QNLM2016ORP0303), Marine S&T Fund of Shandong Province for Pilot National Laboratory for Marine Science and Technology (Qingdao) (2018SDKJ050404), and Nantong Municipal Science and Technology Project (MS12017019-4).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01192/full#supplementary-material
Ahmed, E. F., Hassan, H. M., Rateb, M. E., Abdel-Wahab, N., Sameer, S., Aly Taie, H. A., et al. (2016). A comparative biochemical study on two marine endophytes, bacterium SRCnm and Bacillus sp. JS, isolated from red sea algae. Pak. J. Pharm. Sci. 29, 17–26.
Aires, T., Marba, N., Serrão, E. A., Duarte, C. M., and Arnaud-Haond, S. (2012). Selective elimination of chloroplastidial DNA for metagenomics of bacteria associated with the green alga Caulerpa taxifolia (Bryopsidophyceae). J. Phycol. 48, 483–490. doi: 10.1111/j.1529-8817.2012.01124.x
Aires, T., Moalic, Y., Serrao, E. A., and Arnaud-Haond, S. (2015). Hologenome theory supported by cooccurrence networks of species-specific bacterial communities in siphonous algae (Caulerpa). FEMS Microbiol. Ecol. 91:fiv067. doi: 10.1093/femsec/fiv067
Aires, T., Serrão, E. A., Kendrick, G., Duarte, C. M., and Arnaud-Haond, S. (2013). Invasion is a community affair: clandestine followers in the bacterial community associated to green algae, Caulerpa racemosa, track the invasion source. PLoS One 8:e68429. doi: 10.1371/journal.pone.0068429
Amann, R. I., Ludwig, W., and Schleifer, K. H. (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59, 143–169. doi: 10.1016/S0882-4010(95)90076-4
An, S., and Gardner, W. S. (2002). Dissimilatory nitrate reduction to ammonium (DNRA) as a nitrogen link, versus denitrification as a sink in a shallow estuary (laguna madre/baffin bay, texas). Mar. Ecol. Prog. Ser. 237, 41–50. doi: 10.3354/meps237041
Ariosa, Y., Quesada, A., Aburto, J., Carrasco, D., Carreres, R., Leganés, F., et al. (2004). Epiphytic cyanobacteria on Chara vulgaris are the main contributors to N2 fixation in rice fields. Appl. Environ. Microbiol. 70, 5391–5397. doi: 10.1128/aem.70.9.5391-5397.2004
Auchtung, T. A., Holder, M. E., Gesell, J. R., Ajami, N. J., Duarte, R. T., Itoh, K., et al. (2016). Complete genome sequence of Turicibacter sp. strain H121, isolated from the feces of a contaminated germ-free mouse. Genome Announc. 4, e00114–e00116. doi: 10.1128/genomeA.00114-16
Beckers, B., Op De Beeck, M., Thijs, S., Truyens, S., Weyens, N., Boerjan, W., et al. (2016). Performance of 16S rDNA primer pairs in the study of rhizosphere and endosphere bacterial microbiomes in metabarcoding studies. Front. Microbiol. 7:650. doi: 10.3389/fmicb.2016.00650
Bengtsson, M. M., Sjøtun, K., and Øvreås, L. (2010). Seasonal dynamics of bacterial biofilms on the kelp Laminaria hyperborea. Aquat. Microb. Ecol. 60, 71–83. doi: 10.3354/ame01409
Bergeron, R. J., Dionis, J. B., Elliott, G. T., and Kline, S. J. (1985). Mechanism and stereospecificity of the parabactin-mediated iron-transport system in Paracoccus denitrificans. J. Biol. Chem. 260, 7936–7944. doi: 10.1016/0165-022X(85)90070-3
Bergeron, R. J., and Weimar, W. R. (1990). Kinetics of iron acquisition from ferric siderophores by Paracoccus denitrificans. J. Bacteriol. 172, 2650–2657. doi: 10.1128/jb.172.5.2650-2657.1990
Berry, M. A., Davis, T. W., Cory, R. M., Duhaime, M. B., Johengen, T. H., Kling, G. W., et al. (2017). Cyanobacterial harmful algal blooms are a biological disturbance to western lake erie bacterial communities. Environ. Microbiol. 19, 1149–1162. doi: 10.1111/1462-2920.13640
Burke, C., Steinberg, P., Rusch, D., Kjelleberg, S., and Thomas, T. (2011a). Bacterial community assembly based on functional genes rather than species. Proc. Natl. Acad. Sci. U.S.A. 108, 14288–14293. doi: 10.1073/pnas.1101591108
Burke, C., Thomas, T., Lewis, M., Steinberg, P., and Kjelleberg, S. (2011b). Composition, uniqueness and variability of the epiphytic bacterial community of the green alga Ulva australis. ISME J. 5, 590–600. doi: 10.1038/ismej.2010.164
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Cho, D. H., Ramanan, R., Heo, J., Lee, J., Kim, B. H., Oh, H. M., et al. (2015). Enhancing microalgal biomass productivity by engineering a microalgal-bacterial community. Bioresour. Technol. 175, 578–585. doi: 10.1016/j.biortech.2014.10.159
Clarke, K. R. (1993). Non-parametric multivariate analyses of changes in community structure. Austral J. Ecol. 18, 117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x
Costa, G. B., Simioni, C., Pereira, D. T., Ramlov, F., Maraschin, M., Chow, F., et al. (2017). The brown seaweed Sargassum cymosum: changes in metabolism and cellular organization after long-term exposure to cadmium. Protoplasma 254, 817–837. doi: 10.1007/s00709-016-0992-9
Croft, M. T., Lawrence, A. D., Raux-Deery, E., Warren, M. J., and Smith, A. G. (2005). Algae acquire vitamin B12 through a symbiotic relationship with bacteria. Nature 438, 90–93. doi: 10.1038/nature04056
Cundell, A. M., Sleeter, T. D., and Mitchell, R. (1977). Microbial populations associated with the surface of the brown alga Algaascophyllum nodosum. Microb. Ecol. 4, 81–91. doi: 10.1007/BF02010431
de Oliveira, L. S., Gregoracci, G. B., Silva, G. G., Salgado, L. T., Filho, G. A., Alves-Ferreira, M., et al. (2012). Transcriptomic analysis of the red seaweed Laurencia dendroidea (Florideophyceae, Rhodophyta) and its microbiome. BMC Genomics 13:487. doi: 10.1186/1471-2164-13-487
Dittami, S. M., Duboscq-Bidot, L., Perennou, M., Gobet, A., Corre, E., Boyen, C., et al. (2016). Host-microbe interactions as a driver of acclimation to salinity gradients in brown algal cultures. ISME J. 10, 51–63. doi: 10.1038/ismej.2015.104
Doepke, H., Herrmann, K., and Schuett, C. (2012). Endobacteria in the tentacles of selected cnidarian species and in the cerata of their nudibranch predators. Helgol. Mar. Res. 66, 43–50. doi: 10.1007/s10152-011-0245-4
Doucette, G. J. (1995). Interactions between bacteria and harmful algae: a review. Nat. Toxins 3, 65–74. doi: 10.1002/nt.2620030202
Egan, S., Harder, T., Burke, C., Steinberg, P., Kjelleberg, S., and Thomas, T. (2013). The seaweed holobiont: understanding seaweed–bacteria interactions. FEMS Microbiol. Rev. 37, 462–476. doi: 10.1111/1574-6976.12011
Falcon, L. I., Escobar-Briones, E., and Romero, D. (2002). Nitrogen fixation patterns displayed by cyanobacterial consortia in alchichica crater-lake. Hydrobiologia 467, 71–78. doi: 10.1023/A:1014984629985
Florez, J. Z., Camus, C., Hengst, M. B., and Buschmann, A. H. (2017). A functional perspective analysis of macroalgae and epiphytic bacterial community interaction. Front. Microbiol. 8:2561. doi: 10.3389/fmicb.2017.02561
Fu, H. H., Jiang, P., Zhao, J., and Wu, C. H. (2018). Comparative genomics of Pseudomonas sp. strain SI-3 associated with macroalga Ulva prolifera, the causative species for green tide in the yellow sea. Front. Microbiol. 9:1458. doi: 10.3389/fmicb.2018.01458
Gao, S., Shi, X. Y., and Wang, T. (2012). Variation of nutrient concentrations at the inshore coastal area of northern Jiangsu province and the occurrence of green tide caused by Enteromorpha prolifera. Environ. Sci. 33, 2204–2209. doi: 10.13227/j.hjkx.2012.07.004
Goddard, A. D., Bali, S., Mavridou, D. A. I., Luque-Almagro, V. M., Gates, A. J., Roldán, M. D., et al. (2017). The Paracoccus denitrificans NarK-like nitrate and nitrite transporters-probing nitrate uptake and nitrate/nitrite exchange mechanisms. Mol. Microbiol. 103, 117–133. doi: 10.1111/mmi.13546
Goecke, F., Labes, A., Wiese, J., and Imhoff, J. F. (2010). Chemical interactions between marine macroalgae and bacteria. Mar. Ecol. Prog. Ser. 409, 267–299. doi: 10.3354/meps08607
Grottoli, P. D., Wilkins, M. J., Johnston, M. D., Warner, M. E., Cai, W. J., et al. (2018). Coral physiology and microbiome dynamics under combined warming and ocean acidification. PLoS One 13:e0191156. doi: 10.1371/journal.pone.0191156
Guan, L., Santander, J., Mellata, M., Zhang, Y., and Curtiss, R. (2013). Identification of an iron acquisition machinery in Flavobacterium columnare. Dis. Aquat. Organ. 106, 129–138. doi: 10.3354/dao02635
Hiron, A., Posteraro, B., Carrière, M., Remy, L., Delporte, C., La Sorda, M., et al. (2010). A nickel ABC-transporter of Staphylococcus aureus is involved in urinary tract infection. Mol. Microbiol. 77, 1246–1260. doi: 10.1111/j.1365-2958.2010.07287.x
Hollants, J., Decleyre, H., Leliaert, F., De Clerck, O., and Willems, A. (2011a). Life without a cell membrane: challenging the specificity of bacterial endophytes within Bryopsis (Bryopsidales, Chlorophyta). BMC Microbiol. 11:255. doi: 10.1186/1471-2180-11-255
Hollants, J., Leliaert, F., De Clerck, O., and Willems, A. (2013). What we can learn from sushi: a review on seaweed-bacterial associations. FEMS Microbiol. Ecol. 83, 1–16. doi: 10.1111/j.1574-6941.2012.01446.x
Hollants, J., Leroux, O., Leliaert, F., Decleyre, H., De Clerck, O., and Willems, A. (2011b). Who is in there? Exploration of endophytic bacteria within the siphonous green seaweed Bryopsis (Bryopsidales, Chlorophyta). PLoS One 6:e26458. doi: 10.1371/journal.pone.0026458
Huang, C. H., Sun, Z. M., Gao, D. H., Yao, J. T., Hu, Z. M., Li, Y. H., et al. (2017). Molecular analysis of Sargassum from the northern China seas. Phytotaxa 319, 71–83. doi: 10.11646/phytotaxa.319.1.3
Jasti, S., Sieracki, M. E., Poulton, N. J., Giewat, M. W., and Rooney-Varga, J. N. (2005). Phylogenetic diversity and specificity of bacteria closely associated with Alexandrium spp. and other phytoplankton. Appl. Environ. Microbiol. 71, 3483–3494. doi: 10.1128/aem.71.7.3483-3494.2005
Jeffrey, S. W., and Humphrey, G. F. (1975). New spectrophotometric equations for determining chlorophylls a, b, c1 and c2 in higher plants, algae and natural phytoplankton. Biochem. Physiol. Pflanz. 167, 191–194. doi: 10.1016/s0015-3796(17)30778-3
Kientz, B., Thabard, M., Cragg, S. M., Pope, J., and Hellio, C. (2011). A new method for removing microflora from macroalgal surfaces: an important step for natural product discovery. Bot. Mar. 54, 457–469. doi: 10.1515/bot.2011.053
Komatsu, T., Fukuda, M., Mikami, A., Mizuno, S., Kantachumpoo, A., Tanoue, H., et al. (2014a). Possible change in distribution of seaweed, Sargassum horneri, in northeast Asia under A2 scenario of global warming and consequent effect on some fish. Mar. Pollut. Bull. 85, 317–324. doi: 10.1016/j.marpolbul.2014.04.032
Komatsu, T., Mizuno, S., Natheer, A., Kantachumpoo, A., Tanaka, K., Morimoto, A., et al. (2014b). Unusual distribution of floating seaweeds in the East China Sea in the early spring of 2012. J. Appl. Phycol. 26, 1169–1179. doi: 10.1007/s10811-013-0152-y
Kouzuma, A., and Watanabe, K. (2015). Exploring the potential of algae/bacteria interactions. Curr. Opin. Biotech. 33, 125–129. doi: 10.1016/j.copbio.2015.02.007
Lachnit, T., Blumel, M., Imhoff, J. F., and Wahl, M. (2009). Specific epibacterial communities on macroalgae: phylogeny matters more than habitat. Aquat. Biol. 5, 181–186. doi: 10.3354/ab00149
Langille, M. G. I., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Lekunberri, I., Lefort, T., Romera-Castillo, C., Cardelus, C., Coll-Llado, M., Ruiz-Gonzalez, C., et al. (2012). Relationship between induced phytoplankton blooms and the structure and dynamics of the free-living heterotrophic bacterial community. Mar. Ecol. Prog. Ser. 448, 23–37. doi: 10.3354/meps09480
Li, L., Mohamad, O. A. A., Ma, J., Friel, A. D., Su, Y., Wang, Y., et al. (2018). Synergistic plant-microbe interactions between endophytic bacterial communities and the medicinal plant Glycyrrhiza uralensis F. Antonie Van Leeuwenhoek 111, 1735–1748. doi: 10.1007/s10482-018-1062-4
Liu, M., Dong, Y., Zhao, Y., Zhang, G. T., Zhang, W. C., and Xiao, T. (2011). Structures of bacterial communities on the surface of Ulva prolifera and in seawaters in an Ulva blooming region in Jiaozhou Bay. China World J. Microbiol. Biotechnol. 27, 1703–1712. doi: 10.1007/s11274-010-0627-9
Liu, X. J. (2015). Methodological Innovation for Analysis of the Bacterial Community Structure and the Study of CFB Group Associated with Ulva prolifera. Master thesis, Institute of Oceaonology Chinese Academy of Sciences, China.
Liu, Z. P., Wang, B. J., Liu, X. Y., Dai, X., Liu, Y. H., and Liu, S. J. (2008). Paracoccus halophilus sp. nov., isolated from marine sediment of the South China sea, China, and emended description of genus paracoccus davis 1969. Int. J. Syst. Evol. Microbiol. 58, 257–261. doi: 10.1099/ijs.0.65237-0
Longford, S. R., Tujula, N. A., Crocetti, G. R., Holmes, A. J., Holmstroem, C., Kjelleberg, S., et al. (2007). Comparisons of diversity of bacterial communities associated with three marine eukaryotes. Aquat. Microb. Ecol. 48, 217–229. doi: 10.3354/ame048217
Lozupone, C., and Knight, R. (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235. doi: 10.1128/aem.71.12.8228-8235.2005
Lozupone, C. A., Hamady, M., Kelley, S. T., and Knight, R. (2007). Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73, 1576–1585. doi: 10.1128/aem.01996-06
Lu, B. R., and Tseng, C. K. (2004). Studies on four new species of the malacocarpic Sargassum (Sargassaceae. Heterokontophyta) in China. Hydrobiologia 512, 193–199. doi: 10.1023/b:hydr.0000020327.28056.fa
Magoc, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Matsuo, Y., Suzuki, M., Kasai, H., Shizuri, Y., and Harayama, S. (2003). Isolation and phylogenetic characterization of bacteria capable of inducing differentiation in the green alga Monostroma oxyspermum. Environ. Microbiol. 5, 25–35. doi: 10.1046/j.1462-2920.2003.00382.x
McArdle, B. H., and Anderson, M. J. (2001). Fitting multivariate models to community data: a comment on distance-based redundancy analysis. Ecology 82, 290–297. doi: 10.1890/0012-9658(2001)082
Michelou, V. K., Caporaso, J. G., Knight, R., and Palumbi, S. R. (2013). The ecology of microbial communities associated with Macrocystis pyrifera. PLoS One 8:e67480. doi: 10.1371/journal.pone.0067480
Miki, O., Nagai, T., Marzuki, M., Okumura, C., Kosugi, C., and Kato, T. (2016). Effects of Fe fertilizer eluate on the growth of Sargassum horneri, at the germling and immature stages. J. Appl. Phycol. 28, 1775–1782. doi: 10.1007/s10811-015-0729-8
Miller, E. P., Auerbach, H., Schünemann, V., Tymon, T., and Carrano, C. J. (2016). Correction: surface binding, localization and storage of iron in the giant kelp Macrocystis pyrifera. Metallomics 8, 403–411. doi: 10.1039/c6mt90018f
Møller, J. D., Ellis, A. E., Barnes, A. C., and Dalsgaard, I. (2005). Iron acquisition mechanisms of Flavobacterium psychrophilum. J. Fish. Dis. 28, 391–398. doi: 10.1111/j.1365-2761.2005.00639.x
Nair, D., Vanuopadath, M., Nair, B. G., Pai, J. G., and Nair, S. S. (2016). Identification and characterization of a library of surfactins and fengycins from a marine endophytic Bacillus sp. J. Basic. Microbiol. 56, 1159–1172. doi: 10.1002/jobm.201600029
Pan, J., Yu, F., Wei, C. J., Ren, Q., Sun, S. H., and Li, J. (2018). Relationship between green tide outbreak and nitrate and hydrological environmental factors in the South Yellow Sea. Oceanologia et Limnologia Sinica 49, 1031–1037. doi: 10.11693/hyhz20171200327
Park, B. S., Joo, J. H., Baek, K. D., and Han, M. S. (2016). A mutualistic interaction between the bacterium Pseudomonas asplenii and the harmful algal species Chattonella marina (Raphidophyceae). Harmful Algae 56, 29–36. doi: 10.1016/j.hal.2016.04.006
Park, B. S., Kim, J. H., Kim, J. H., Gobler, C. J., Baek, S. H., and Han, M. S. (2015). Dynamics of bacterial community structure during blooms of Cochlodinium polykrikoides (Gymnodiniales, Dinophyceae) in Korean coastal waters. Harmful Algae 48, 44–54. doi: 10.1016/j.hal.2015.07.004
Park, S. H., Kim, J. Y., Kim, Y. J., and Heo, M. S. (2015). Flavobacterium jejuensis sp. nov., isolated from marine brown alga Ecklonia cava. J. Microbiol. 53, 756–761. doi: 10.1007/s12275-015-5280-4
Parulekar, N. N., Kolekar, P., Jenkins, A., Kleiven, S., Utkilen, H., Johansen, A., et al. (2017). Characterization of bacterial community associated with phytoplankton bloom in a eutrophic lake in South Norway using 16S rRNA gene amplicon sequence analysis. PLoS One 12:e0173408. doi: 10.1371/journal.pone.0173408
Pathak, K. V., Keharia, H., Gupta, K., Thakur, S. S., and Balaram, P. (2012). Lipopeptides from the banyan endophyte, Bacillus subtilis K1: mass spectrometric characterization of a library of fengycins. J. Am. Soc. Mass Spectrom. 23, 1716–1728. doi: 10.1007/s13361-012-0437-4
Piggott, J. J., Salis, R. K., Lear, G., Townsend, C. R., and Matthaei, C. D. (2015). Climate warming and agricultural stressors interact to determine stream periphyton community composition. Glob. Chang. Biol. 21, 206–222. doi: 10.1111/gcb.12661
Qi, L., Hu, C. M., Wang, M. Q., Shang, S. L., and Wilson, C. (2017). Floating algae blooms in the East China Sea. Geophys. Res. Lett. 44, 11501–11509. doi: 10.1002/2017GL075525
Ramette, A. (2007). Multivariate analyses in microbial ecology. FEMS Microbiol. Ecol. 62, 142–160. doi: 10.1111/j.1574-6941.2007.00375.x
Roth-Schulze, A. J., Pintado, J., Zozaya-Valdes, E., Cremades, J., Ruiz, P., Kjelleberg, S., et al. (2018). Functional biogeography and host specificity of bacterial communities associated with the marine green alga Ulva spp. Mol. Ecol. 27, 1952–1965. doi: 10.1111/mec.14529
Sánchez-López, A. S., Thijs, S., Beckers, B., Gonzalez-Chavez, M. C., Weyens, N., Carrillo-Gonzalez, R., et al. (2018). Community structure and diversity of endophytic bacteria in seeds of three consecutive generations of Crotalaria pumila growing on metal mine residues. Plant Soil 422, 51–65. doi: 10.1007/s11104-017-3176-2
Sarria-Guzmán, Y., Chávez-Romero, Y., Gómez-Acata, S., Montes-Molina, J. A., Morales-Salazar, E., Dendooven, L., et al. (2016). Bacterial communities associated with different Anthurium andraeanum L. plant tissues. Microbes Environ. 31, 321–328. doi: 10.1264/jsme2.ME16099
Sedlácek, V., van Spanning, R. J., and Kucera, I. (2009). Ferric reductase A is essential for effective iron acquisition in Paracoccus denitrificans. Microbiology 155, 1294–1301. doi: 10.1099/mic.0.022715-0
Shen, W. L., Chen, H. H., Jia, K. Z., Ni, J., Yan, X., and Li, S. P. (2012). Cloning and characterization of a novel amidase from Paracoccus sp. M-1, showing aryl acylamidase and acyl transferase activities. Appl. Microbiol. Biotechnol. 94, 1007–1018. doi: 10.1007/s00253-011-3704-6
Singh, R. P., Bijo, A. J., Baghel, R. S., Reddy, C. R. K., and Jha, B. (2011). Role of bacterial isolates in enhancing the bud induction in the industrially important red alga Gracilaria dura. FEMS Microbiol. Ecol. 76, 381–392. doi: 10.1111/j.1574-6941.2011.01057.x
Singh, R. P., and Reddy, C. R. K. (2014). Seaweed-microbial interactions: key functions of seaweed-associated bacteria. FEMS Microbiol. Ecol. 88, 213–230. doi: 10.1111/1574-6941.12297
Sison-Mangus, M. P., Jiang, S., Kudela, R. M., and Mehic, S. (2016). Phytoplankton-associated bacterial community composition and succession during toxic diatom bloom and non-bloom events. Front. Microbiol. 7:1433. doi: 10.3389/fmicb.2016.01433
Sissini, M. N., Barreto, M. B. B. D., Szechy, M. T. M., de Lucena, M. B., Oliveira, M. C., Gower, J., et al. (2017). The floating Sargassum (Phaeophyceae) of the south atlantic ocean-likely scenarios. Phycologia 56, 321–328. doi: 10.2216/16-92.1
Smetacek, V., and Zingone, A. (2013). Green and golden seaweed tides on the rise. Nature 504, 84–88. doi: 10.1038/nature12860
Staufenberger, T., Thiel, V., Wiese, J., and Imhoff, J. F. (2008). Phylogenetic analysis of bacteria associated with Laminaria saccharina. FEMS Microbiol. Ecol. 64, 65–77. doi: 10.1111/j.1574-6941.2008.00445.x
Su, H., Yang, R. J., Li, Y., and Wang, X. C. (2018). Influence of humic substances on iron distribution in the East China Sea. Chemosphere 204, 450–462. doi: 10.1016/j.chemosphere.2018.04.018
Su, L., Shan, T. F., Pang, S. J., and Li, J. (2018). Analyses of the genetic structure of Sargassum horneri in the yellow sea: implications of the temporal and spatial relations among floating and benthic populations. J. Appl. Phycol. 30, 1417–1424. doi: 10.1007/s10811-017-1296-y
Su, H., Yang, R. J., Zhang, A. B., and Li, Y. (2015). Dissolved iron distribution and organic complexation in the coastal waters of the East China Sea. Mar. Chem. 173, 208–221. doi: 10.1016/j.marchem.2015.03.007
Sun, Z. K., Liu, K., Zhang, J., Zhang, Y., Xu, K. D., Yu, D. S., et al. (2017). IAA producing Bacillus altitudinis alleviates iron stress in Triticum aestivum L. seedling by both bioleaching of iron and up-regulation of genes encoding ferritins. Plant Soil 419, 1–11. doi: 10.1007/s11104-017-3218-9
Tan, H. M., Zhou, S. N., Deng, Z. J., He, M., and Cao, L. X. (2011). Ribosomal-sequence-directed selection for endophytic streptomycete strains antagonistic to Ralstonia solanacearum to control tomato bacterial wilt. Biol. Control 59, 245–254. doi: 10.1016/j.biocontrol.2011.07.018
Tujula, N. A., Crocetti, G. R., Burke, C., Thomas, T., Holmstrom, C., Holmström, C., et al. (2010). Variability and abundance of the epiphytic bacterial community associated with a green marine Ulvacean alga. ISME J. 4, 301–311. doi: 10.1038/ismej.2009.107
Uzair, B., Menaa, F., Khan, B. A., Mohammad, F. V., Ahmad, V. U., Djeribi, R., et al. (2018). Isolation, purification, structural elucidation and antimicrobial activities of kocumarin, a novel antibiotic isolated from actinobacterium Kocuria marina CMG S2 associated with the brown seaweed Pelvetia canaliculata. Microbiol. Res. 206, 186–197. doi: 10.1016/j.micres.2017.10.007
Wang, Y., Wang, Y., Zhu, L., Zhou, B., and Tang, X. X. (2012). Comparative studies on the ecophysiological differences of two green tide macroalgae under controlled laboratory conditions. PLoS One 7:e38245. doi: 10.1371/journal.pone.0038245
Warton, D. I., Wright, S. T., and Wang, Y. (2012). Distance-based multivariate analyses confound location and dispersion effects. Methods Ecol. Evol. 3, 89–101. doi: 10.1111/j.2041-210X.2011.00127.x
White, J. R., Nagarajan, N., and Pop, M. (2009). Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 5:e1000352. doi: 10.1371/journal.pcbi.1000352
Wilkinson, T. J., Huws, S. A., Edwards, J. E., Kingston-Smith, A. H., Siu-Ting, K., Hughes, M., et al. (2018). CowPI: a rumen microbiome focussed version of the PICRUSt functional inference software. Front. Microbiol. 9:1095. doi: 10.3389/fmicb.2018.01095
Xing, Q. G., Guo, R. H., Wu, L. L., An, D. Y., Cong, M., Qin, S., et al. (2017). High-resolution satellite observations of a new hazard of golden tides caused by floating Sargassum in winter in the Yellow Sea. IEEE Geosci. Remote Sens. Lett. 14, 1815–1819. doi: 10.1109/lgsr.2017.2737079
Xu, M., Sakamoto, S., and Komatsu, T. (2016). Attachment strength of the subtidal seaweed Sargassum horneri, (Turner) C. Agardh varies among development stages and depths. J. Appl. Phycol. 28, 3679–3687. doi: 10.1007/s10811-016-0869-5
Yoshida, T., Stiger, V., and Horiguchi, T. (2000). Sargassum boreale sp. nov. (Fucales, Phaeophyceae) from hokkaido, Japan. Phycol. Res. 48, 125–131. doi: 10.1111/j.1440-1835.2000.tb00207.x
Zhang, J. H., Huo, Y. Z., Zhang, Z. L., Yu, K. F., He, Q., Zhang, L. H., et al. (2013). Variations of morphology and photosynthetic performances of Ulva prolifera during the whole green tide blooming process in the yellow sea. Mar. Environ. Res. 92, 35–42. doi: 10.1016/j.marenvres.2013.08.009
Zhang, T., and Wang, X. (2017). Release and microbial degradation of dissolved organic matter (DOM) from the macroalgae Ulva prolifera. Mar. Pollut. Bull. 125, 192–198. doi: 10.1016/j.marpolbul.2017.08.029
Zhang, X., Hu, B. X., Ren, H., and Zhang, J. (2018). Composition and functional diversity of microbial community across a mangrove-inhabited mudflat as revealed by 16S rDNA gene sequences. Sci. Total Environ. 633, 518–528. doi: 10.1016/j.scitotenv.2018.03.158
Keywords: 16S rRNA gene, high-throughput sequencing, bacterial community, golden tide, Sargassum horneri, the Yellow Sea
Citation: Mei X, Wu C, Zhao J, Yan T and Jiang P (2019) Community Structure of Bacteria Associated With Drifting Sargassum horneri, the Causative Species of Golden Tide in the Yellow Sea. Front. Microbiol. 10:1192. doi: 10.3389/fmicb.2019.01192
Received: 01 February 2019; Accepted: 10 May 2019;
Published: 28 May 2019.
Edited by:
David A. Walsh, Concordia University, CanadaReviewed by:
Yvonne Vallès, The University of the West Indies, Cave Hill, BarbadosCopyright © 2019 Mei, Wu, Zhao, Yan and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peng Jiang, amlhbmdwZW5nQHFkaW8uYWMuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.