 Lin Zhu
Lin Zhu Jie Song
Jie Song Jing Si
Jing Si Bao-Kai Cui
Bao-Kai Cui
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 17 April 2019
Sec. Fungi and Their Interactions
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.00812
This article is part of the Research Topic Fungal Molecular Systematics, Ranking, and Evolution Using Divergence Time View all 7 articles
“Sanghuang” is a popular fungus used as a Chinese traditional medicine. In fact, it represents a group of fungi belonging to the genus Sanghuangporus, but little is known about its origin and biogeography. The aim of this study was to characterize the molecular relationships, origin and biogeographical distribution of Sanghuangporus. The multi-locus phylogenetic analyses were used to infer the phylogenetic relationships. In addition, based on Bayesian evolutionary analysis using sequences from the internal transcribed spacer (ITS), nuclear large subunit rDNA (nLSU), translation elongation factor 1-α (EF1-α), and the largest and second largest subunits of RNA polymerase II (RPB1 and RPB2), we used a fungus fossil-based approach to gain insight into the divergence time of species in Sanghuangporus. The molecular phylogeny strongly supports the monophyly of Sanghuangporus (MP = 100%, ML = 100%, and BPP = 1.00), and 13 species are recognized in this genus. The Bayesian uncorrelated lognormal relaxed molecular clock using BEAST and reconstructed ancestral areas indicate that the maximum crown age of Sanghuangporus is approximately 30.85 million years. East Asia is the likely ancestral area (38%). Dispersal and differentiation to other continents then occurred during the late Middle Miocene and Pliocene. The ancestor of Sanghuangporus probably originated in palaeotropical Northeast Asia and covered Northeast Asia and East Africa during the Oligocene-Miocene, hosted by plants that expanded via the “Gomphotherium Landbridge.” Six kinds of dispersal routes are proposed, including intercontinental dispersal events of three clades between Northeast Asia and East Africa, between East Asia and North America, and between Northeast Asia and Europe.
The genus Sanghuangporus Sheng H. Wu, L.W. Zhou, and Y.C. Dai belongs to the order Hymenochaetales, and its species are prized as medicinal fungi. The medicinal functions of Sanghuangporus are antitumor, antioxidation, anti-inflammatory, hypoglycemic effect, hepatoprotective effect, and improving immunity (Dai et al., 2009; Lee et al., 2015; Zhu and Cui, 2016; Lin et al., 2017). Species in this genus form parasitic relationships with Alnus, Juglans, Lonicera, Morus, Populus, Quercus, Syringa, and Weigela (Dai, 2010; Wu et al., 2012). In the past few years, several studies have examined the phylogenetic relationships within Sanghuangporus (Dai, 2010; Wu et al., 2012; Vlasák et al., 2013; Tian et al., 2013; Ghobad-Nejhad, 2015; Tomsovsky, 2015; Zhou et al., 2016; Zhu et al., 2017). Thirteen species are currently accepted in the genus: S. alpinus (Y.C. Dai and X.M. Tian) L.W. Zhou and Y.C. Dai, S. baumii (Pilát) L.W. Zhou and Y.C. Dai, S. ligneus Ghobad-Nejhad, S. lonicericola (Parmasto) L.W. Zhou and Y.C. Dai, S. lonicerinus (Bondartsev) Sheng H. Wu, L.W. Zhou and Y.C. Dai, S. microcystideus (Har. and Pat.) L.W. Zhou and Y.C. Dai, S. pilatii (Cerný) Tomšovský, S. quercicola L. Zhu and B.K. Cui, S. sanghuang (Sheng H. Wu, T. Hatt., and Y.C. Dai) Sheng H. Wu, L.W. Zhou, and Y.C. Dai, S. vaninii (Ljub.) L.W. Zhou and Y.C. Dai, S. weigelae (T. Hatt. and Sheng H. Wu) Sheng H. Wu, L.W. Zhou and Y.C. Dai, S. weirianus (Bres.) L.W. Zhou and Y.C. Dai and S. zonatus (Y.C. Dai and X.M. Tian) L.W. Zhou and Y.C. Dai (Dai, 2010; Wu et al., 2012; Tian et al., 2013; Vlasák et al., 2013; Ghobad-Nejhad, 2015; Tomsovsky, 2015; Zhou et al., 2016; Zhu et al., 2017).
Previous phylogenetic analyses of Sanghuangporus used the sequences of the internal transcribed spacer (ITS) (Wu et al., 2012; Tian et al., 2013; Vlasák et al., 2013; Ghobad-Nejhad, 2015; Tomsovsky, 2015), and these studies were mainly focused on the descriptions of new species. Recently, Zhou et al. (2016) used the ITS and the nuclear large subunit rDNA (nrLSU) to analyze Sanghuangporus and Tropicoporus L.W. Zhou and Y.C. Dai. However, a comprehensive multilocus phylogenetic analysis of the taxa in Sanghuangporus has not been performed.
Sanghuangporus exhibits a wide intercontinental disjunct distribution, and the majority of species are in the Northern Hemisphere (Zhou et al., 2016; Zhu and Cui, 2016). Molecular biogeography studies have provided important insights into the histories of “species” range changes (Eastwood et al., 2011; Feng et al., 2012), and several origin and biogeography studies of fungi have focused on biogeographic distribution and geological events (Floudas et al., 2012; Cai et al., 2014; Chen et al., 2015; Song and Cui, 2017). However, there has been no the estimation of divergence time and no examination of the biogeography of the Sanghuangporus mushrooms.
In this study, the sequences of the ITS, nrLSU, translation elongation factor 1-α (EF1-α), and the largest and second largest subunits of RNA polymerase II (RPB1 and RPB2) were used to study the phylogeny of Sanghuangporus. Furthermore, the divergence time between Ascomycota and Basidiomycota (582 Mya), as estimated based on fossil evidence of Paleopyrenomycites devonicus by Taylor et al. (2004), was used as a calibration point to estimate divergence time and explore the biogeography of Sanghuangporus.
Sixty two samples of Sanghuangporus species from Europe, North America, East Asia, Central Asia, West Asia, and East Africa were studied for their morphological characters. The specimens and cultures were obtained from the herbaria of the Institute of Microbiology, Beijing Forestry University (BJFC) and the Institute of Applied Ecology, Chinese Academy of Sciences (IFP). The detailed information and GenBank accession numbers of the samples are given in Table 1.
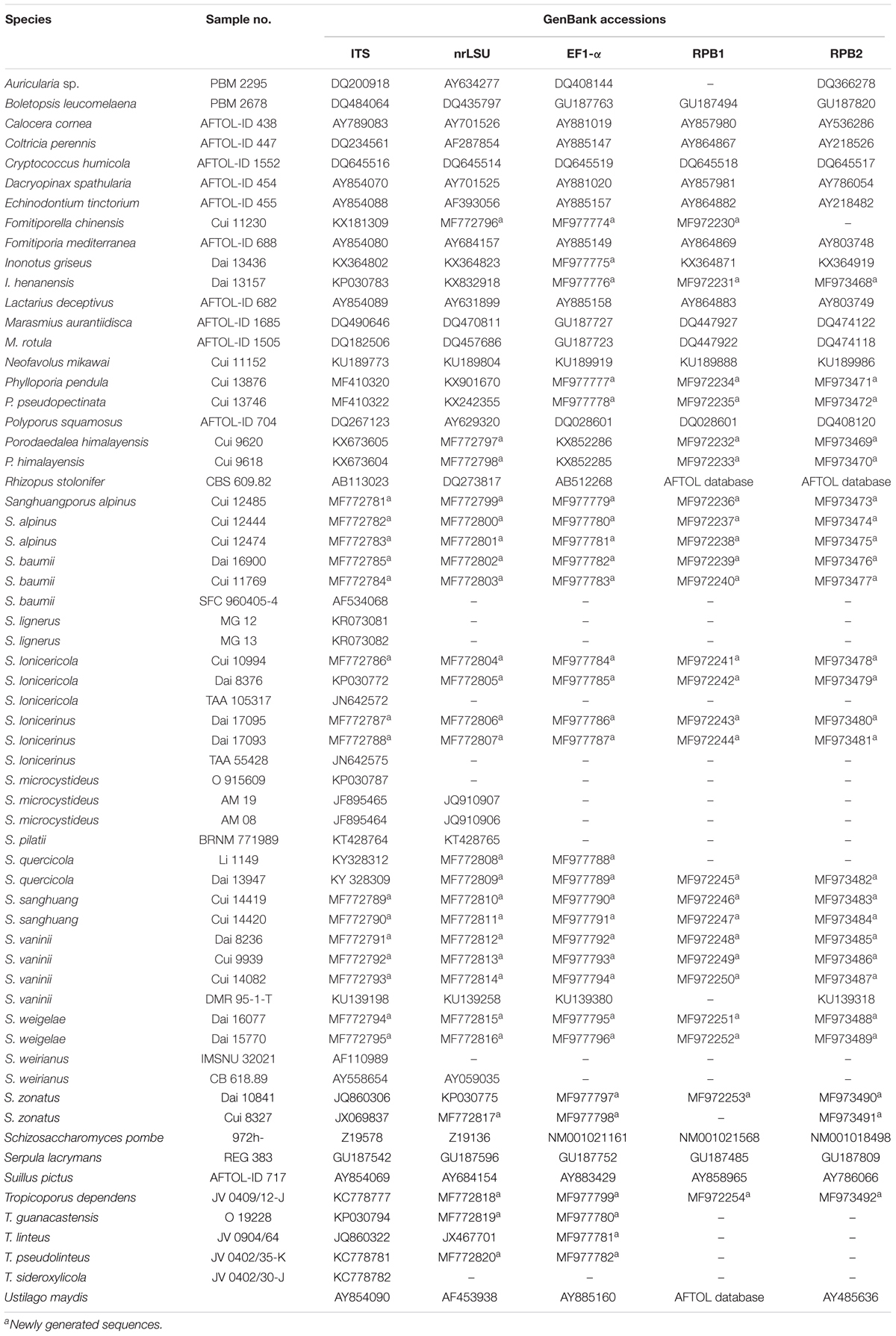
Table 1. Information of sequences used in this study.
Macro-morphological descriptions were based on the field notes. Special color terms followed Petersen (1996). Micro-morphological data were obtained from the dried specimens, and observed under a light microscope following Han et al. (2016). Sections were studied at a magnification of up to ×1000 using a Nikon Eclipse 80i microscope and phase contrast illumination. Spores were measured from sections cut from the tubes. In presenting the variation of spore size, 5% of measurements were excluded from each end of the range, and were given in parentheses. The following abbreviations were used: KOH = 5% potassium hydroxide, CB = Cotton Blue, CB+ = cyanophilous, CB- = acyanophilous, IKI = Melzer’s reagent, IKI- = neither amyloid nor dextrinoid, L = mean spore length (arithmetic average of all spores).
Genomic DNA was extracted from dried specimens and cultures using CTAB rapid plant genome extraction kit (Demeter Biotech Co., Ltd, Beijing) according to the manufacturer’s instructions with modifications. Five DNA gene fragments were analyzed, including those coding for RPB1, RPB2 and EF1-α, along with two non-protein coding regions: ITS and nrLSU. The primer pairs ITS5/4, LR0R /LR7, 983F/1567R, Af/Cf, and 5F/7CR listed in Table 2 were used to amplify ITS, nrLSU, EF1-α, RPB1, and RPB2, respectively (Vilgalys and Hester, 1990; Liu et al., 1999; Matheny et al., 2002; Rehner and Buckley, 2005; Binder et al., 2010). To improve the success rate of RPB2 amplification, a new primer pair, R2s-1 (CCTCGTTACGGGCTTGTT) and R2a-1 (AGCATTTGGAAGTGCCTTG), was designed based on eleven obtained sequences using Primer-Premier 5 (Premier Biosoft International, Palo Alto, CA, United States). PCR was performed in a reaction mixture containing 25 μl of 2 × EasyTaq® PCR SuperMix, 2 μl of Forward Primer (10 μM), 2 μl of Reverse Primer (10 μM), and 2 μl of Template DNA. The total volume was adjusted to 50 μl with sterile deionized H2O. The PCR amplifications were conducted using an Eppendorf Master Cycler (Eppendorf, Netheler-Hinz, Hamburg, Germany). The PCR procedure for ITS was: initial denaturation at 95°C for 2 min, followed by 35 cycles of denaturation at 94°C for 45 s, annealing at 53°C for 45 s and extension at 72°C for 2 min, and a final extension at 72°C for 10 min. The PCR procedure for nrLSU was: initial denaturation at 94°C for 5 min, followed by 35 cycles of denaturation at 94°C for 1 min, annealing at 50°C for 1 min and extension at 72°C for 90 min, and a final extension at 72°C for 10 min. The PCR procedure for EF1-α was: initial denaturation at 95°C for 2 min, followed by 35 cycles of denaturation at 94°C for 45 s, annealing at 56°C for 45 s and extension at 72°C for 2 min, and a final extension at 72°C for 10 min. The PCR procedure for RPB1 and RPB2 was: initial denaturation at 94°C for 2 min, followed by 36 cycles of denaturation at 94°C for 45 s, annealing at 53°C for 90 s and extension at 72°C for 90 s, and a final extension at 72°C for 10 min. The PCR products were visualized by agarose gel electrophoresis and stored at -20°C after visualization. The PCR products were purified and sequenced at the Beijing Genomics Institute (China) using the same primers. 116 sequences of Sanghuangporus used in this paper, and 90 sequences of Sanghuangporus were newly generated, including 14 ITS (44% new), 19 nrLSU (76% new), 20 EF1-α (95% new), 18 RPB1 (100% new), and 19 RPB2 (95% new). All newly generated sequences were deposited in GenBank1.
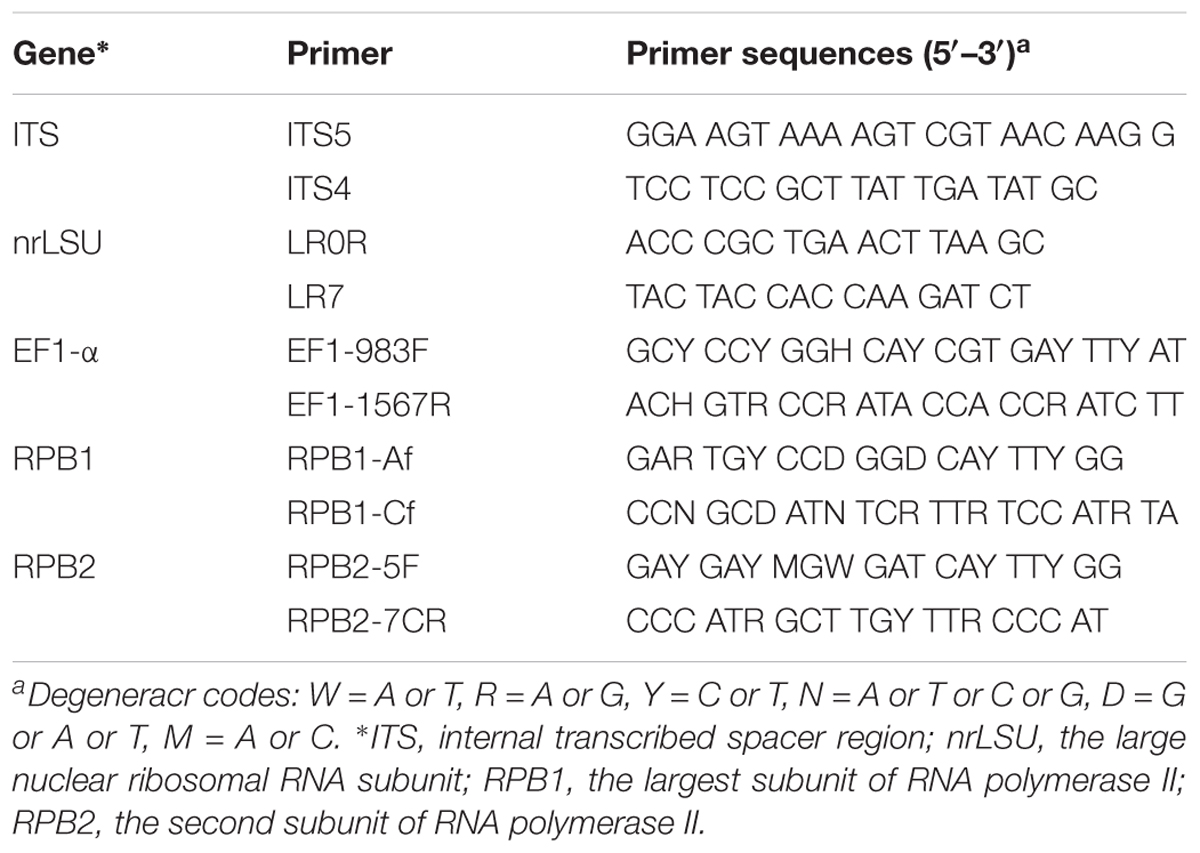
Table 2. PCR primers used in this study.
Phylogenetic analyses were applied to the dataset that contained the ITS+nrLSU+EF1-α+RPB1+RPB2 sequences. Settings for phylogenetic analyses followed Shen et al. (2019). The five genes were initially aligned separately using Clustal Omega (Sievers et al., 2011) and then manually optimized in BioEdit (Hall, 1999). The missing sequences were coded as “N.” Ambiguous nucleotides were coded as “N.” Ambiguous sequences at the start and the end were deleted and gaps were adjusted to optimize the alignment. Ambiguously aligned regions were excluded from subsequent analyses. Finally, the five gene fragments were concatenated with SEAVIEW 4. One thousand partition homogeneity test (PHT) replicates of the ITS, nrLSU, EF1-α, RPB1, and RPB2 sequences were tested by PAUP∗ version 4.0 beta 10 (Swofford, 2002) to determine whether the partitions were homogeneous. The PHT results indicated all the DNA sequences display a congruent phylogenetic signal (P-value = 1). The sequences of Inonotus griseus L.W. Zhou and I. henanensis Juan Li and Y.C. Dai were used as outgroups (Zhou and Wang, 2015). Sequence alignments were deposited at TreeBase (submission ID 215692).
Maximum parsimony (MP) analysis was performed in PAUP∗ version 4.0 beta 10 (Swofford, 2002). All characters were equally weighted, and gaps were treated as missing data. Trees were inferred using the heuristic search option with TBR branch swapping and 1000 random sequence additions. Max-trees was set to 5000, branches of zero length were collapsed, and all parsimonious trees were saved. Descriptive tree statistics tree length (TL), consistency index (CI), retention index (RI), rescaled consistency index (RC), and homoplasy index (HI) were calculated for each maximum parsimonious tree (MPT) generated.
For the maximum likelihood (ML) and Bayesian phylogenetic inference (BI) analyses, the optimal substitution models for the combined dataset were determined using the Akaike Information Criterion (AIC) implemented in MrModeltest 2.2 (Nylander, 2004) after scoring 24 models of evolution by PAUP∗ version 4.0 beta 10 (Swofford, 2002). The selected substitution models for both the combined dataset were general time reversible+proportion invariant+gamma (GTR+I+G).
The ML analysis was conducted on RAxmlGUI 1.31 (Michalak, 2012), the concatenated dataset was partitioned into five parts by sequence region, and 1000 ML searches under the GTR+GAMMA model with all model parameters estimated using the RAxmlGUI 1.31 program. The best fit maximum likelihood tree from all searches was kept. In addition, 1000 rapid bootstrap replicates were run with the GTR+CAT model to assess the reliability of the nodes.
The BI analysis was performed with MrBayes 3.2 (Ronquist and Huelsenbeck, 2003). Four Markov chains were run for 20 million generations and trees were sampled every 1000 generations. The first 25% of the sampled trees were discarded as burn-in, and the remaining ones were used to reconstruct a majority rule consensus and calculate Bayesian posterior probabilities (BPP) of the clades. Branches that received bootstrap support values for MP and ML greater than or equal to 60% and BPP greater than or equal to 0.95 were considered as significantly supported.
Given that fossil records of fungi had been used to calibration point to estimate the divergence time for any fungal groups (Song and Cui, 2017). We used internal calibration point by placing P. devonicus in the subphylum Pezizomycotina, 582 Mya, divergence time between Ascomycota and Basidiomycota in the Figure 3. The estimated divergence time was constrained by the following value: the estimated divergence time between Ascomycota and Basidiomycota is at least 400 Mya (the divergence time of P. devonicus) (Taylor et al., 2004). A normal distribution was applied by setting the mean and the standard deviation to 582.5 and 50.15, respectively. The parameter settings for the calibrations was the same as those used in several studies (Chen et al., 2015; Song and Cui, 2017). We retrieved the sequences of four additional species – Marasmius rotula (Scop.) Fr., Mycena amabilissima Peck, Fomitiporia mediterranea M. Fisch., and Coltricia perennis (L.) Murrill – as representative taxa of the initial diversification of mushroom-forming fungi (based on the 90-million-year-old fossil, Archaeomarasmius leggetti Hibbett, D. Grimaldi and Donoghue) (Hibbett et al., 1997), and the divergence of the Hymenochaetaceae (based on the 125-million-year-old fossil, Q. cranhamii S.Y. Sm., Currah, and Stockey) (Smith et al., 2004).
The origin time of Sanghuangporus was estimated in BEAST v1.8.0 (Drummond and Rambaut, 2007) with the molecular clock and substitution models unlinked but with the trees linked for each gene partition. Two nuclear ribosomal RNA genes (ITS and nrLSU) and three protein coding genes (EF1-α, RPB1, and RPB2), were concatenated for molecular dating. ITS1, ITS2, and the introns in EF1-α, RPB1, and RPB2 were excluded for a conservation analysis. MrModeltest v2.2 was used to select the best models of evolution using the hierarchical likelihood ratio test (Nylander, 2004). The GTR+I+G model was used for the EF1-α+RPB1+RPB2 and the HKY+I+G model for the ITS+nrLSU data, based on the results from the Modeltest. The uncorrelated lognormal relaxed molecular clock and the Yule speciation prior set were set were used to estimate the divergence time and the corresponding credibility intervals. We approximated the posterior distributions of parameters using MCMC analysis for 50 million generations with a burn-in percentage of 10%. The BEAST input files were constructed using BEAUti (within BEAST). The convergence of the chains was confirmed using Tracer v1.63, and samples from the posterior distributions were summarized on a maximum clade credibility tree with the maximum sum of posterior probabilities listed on its internal nodes using the program TreeAnnotator v1.8.0 (Drummond and Rambaut, 2007) with the posterior probability limits set to 0.8 to summarize the mean node heights. FigTree v1.4.2 (Rambaut, 2012) was used to visualize the resulting tree and to obtain the means and 95% HPD (Drummond and Rambaut, 2007). A 95% HPD marks the shortest interval that contains 95% of the values sampled.
We also estimated the divergence time of the main nodes in Sanghuangporus using the ITS dataset containing representatives of all 13 species. The estimated crown age of this genus inferred by the combined ITS+nrLSU and EF1-α+RPB1+RPB2 data was used as the calibration point to date the ITS phylogeny by setting the prior to a normal distribution. The other procedures were the same as the ones applied in the estimation using the combined dataset.
Reconstruction makes it possible to infer the original location and dispersal routes of the organisms (Song and Cui, 2017). The geographic distributions of ancestor lineages were defined based on plate-tectonic and dispersal paths histories (Esseghir et al., 2000; Meulenkamp and Sissingh, 2003). To infer ancestral areas, we performed Bayesian binary Markov chain Monte Carlo (BBM) analysis implemented in RASP 3.2 (Yu et al., 2015) by setting the generations to 10 million and by discarding the first 10% of samples as burn-ins; the other parameters used were the default settings. The geographic distributions for the Sanghuangporus were delimited into seven areas: (A) Northeast Asia, including Northeast China, Russian Far-East, Korea, and Japan, (B) South and Central China, located to south of the Qinling Mountains, (C) Central Asia, (D) West Asia, (E) North America, (F) Europe, and (G) East Africa. ArcGIS v10.1 (ArcGIS Platform4) was used to visualize the geographic distribution and possible dispersal routes of Sanghuangporus.
Thirteen species of Sanghuangporus have been accepted. They are widely distributed across temperate to subtropical and tropical regions. Basidiomata of several Sanghuangporus species are shown in Figure 1. The main morphological characters and distribution details of Sanghuangporus are listed in Table 3.

Figure 1. Basidiomata of Sanghuangporus species. (A) S. alpinus. (B) S. lonicerinus. (C) S. sanghuang. (D) S. baumii. (E) S. vaninii. (F) S. lonicericola. Bars = 1 cm.
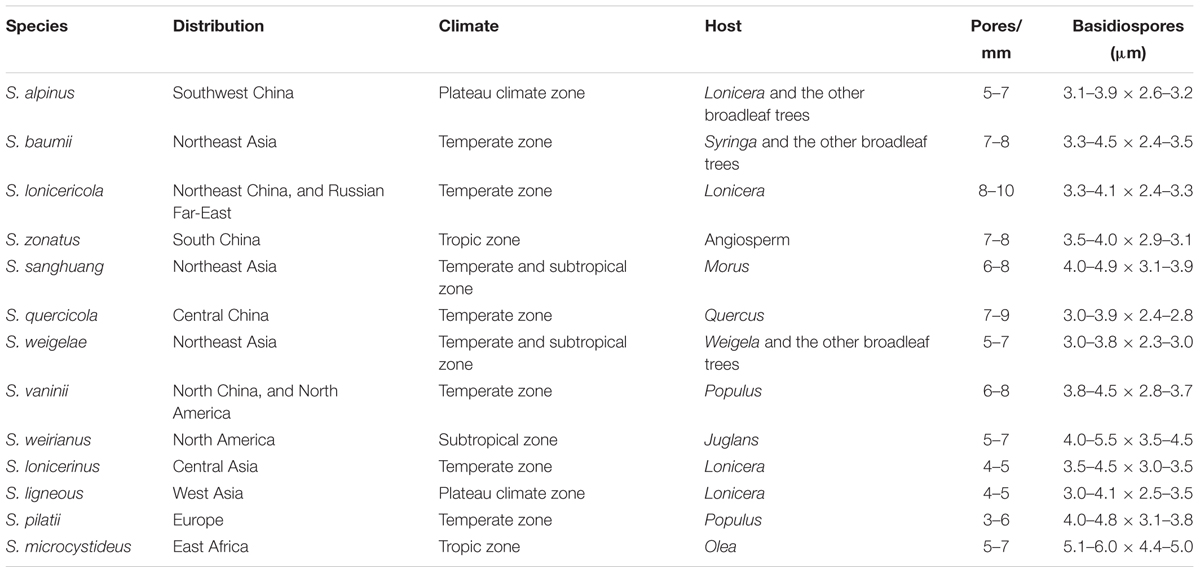
Table 3. Main characteristics and distribution information of Sanghuangporus.
The combined dataset (ITS+nrLSU+EF1-α+RPB1+RPB2) has an aligned length of 3867 characters, of which 2520 are constant, 372 are variable and parsimony-uninformative, and 975 are parsimony-informative characters. In the MP analysis, 18,609,864 rearrangements were attempted, and two equal maximally parsimonious trees were retained (TL = 2268, CI = 0.770, RI = 0.806, RC = 0.621, HI = 0.230). The best model for this alignment set used for estimation and applied in the Bayesian phylogenetic inference (BI) was general time reversible+proportion invariant+gamma (GTR+I+G). The Bayesian analysis resulted in a similar topology, with an average standard deviation of split frequencies = 0.002972.
The trees obtained from the maximum likelihood (ML), MP, and Bayesian posterior probability (BPP) analyses are shown in Figure 2.
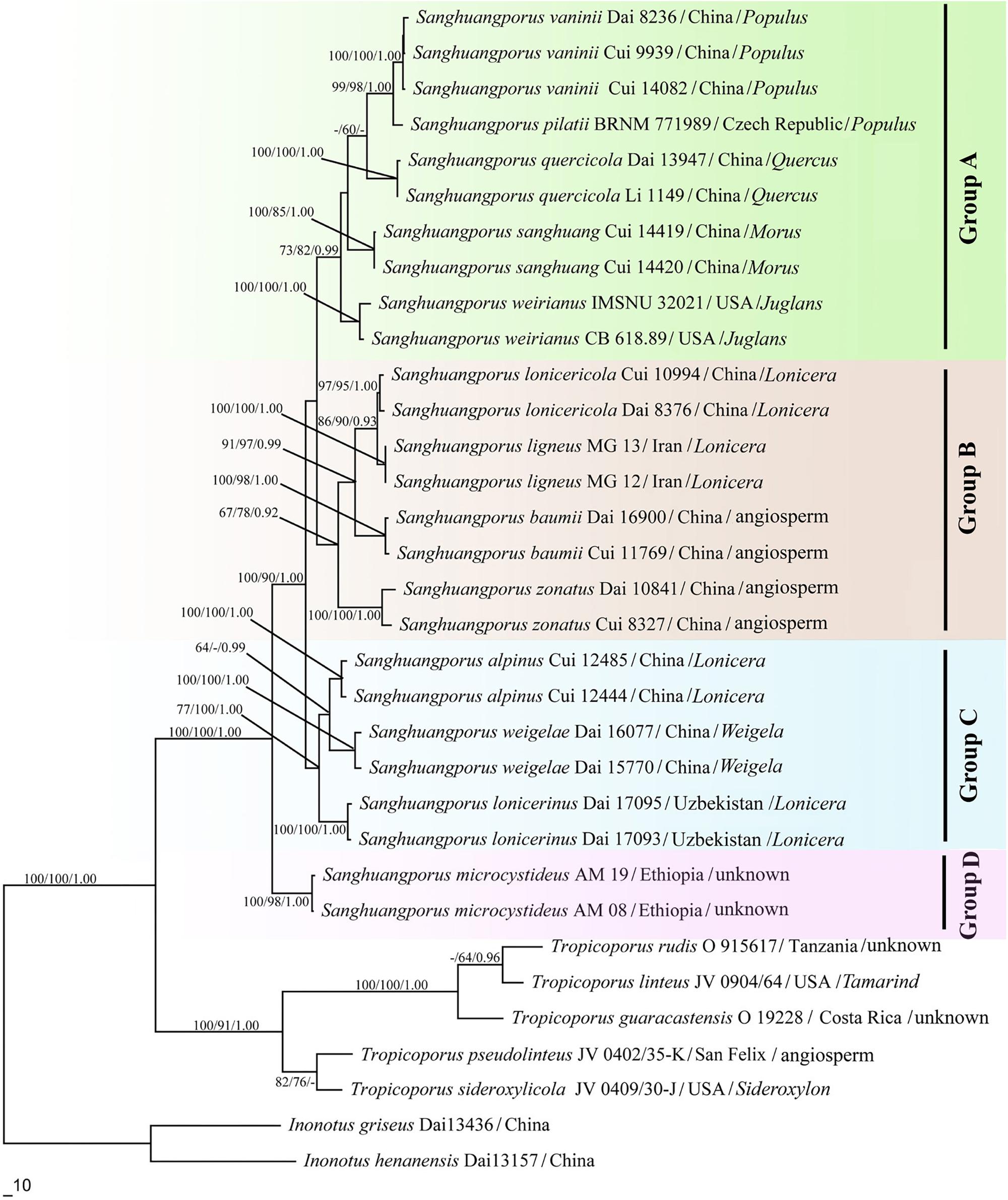
Figure 2. Phylogenetic consensus tree inferred from the maximum likelihood (ML) analysis based on a concatenated, multi-locus dataset (ITS+nrLSU+EF1-α+RPB1+RPB2). Branches are labeled where MP/ML support is greater than 60% and collapsed below that support threshold. BPP is labeled where greater than 0.95.
The BEAST-derived chronogram of Sanghuangporus (Figure 3) was based on the alignment of the two concatenated datasets (ITS+nrLSU and EF1-α+RPB1+RPB2). There were 36 taxa recovered, which were 814 and 1267 bp in length, respectively, for the two datasets. The aligned ITS dataset was 598 bp in length and was used to estimate the divergence time and biogeographical history of Sanghuangporus. Using the divergence time between Ascomycota and Basidiomycota (582 Mya) as a calibration point, we estimated the divergence time of Hymenochaetales as 207.21 ± 0.56 Mya (146.74–265.46 Mya, 95% higher posterior density (HPD), which was more recent than the estimate by Feng et al. (2012) (divergence time at 220.81 Mya). The divergence time of Agaricales at 123.57 ± 0.45 Mya (80.52–170.72, 95% HPD) is consistent with a previous estimate (Eastwood et al., 2011). The initial diversification of Sanghuangporus occurred during the Late Oligocene, 30.85 ± 0.19 Mya (15.65–37.18 Mya, 95% HPD). The divergences of the major clades within Sanghuangporus occurred mainly during the Late Miocene, and the divergence times of the main nodes are shown in Figure 3 and summarized in Table 4.
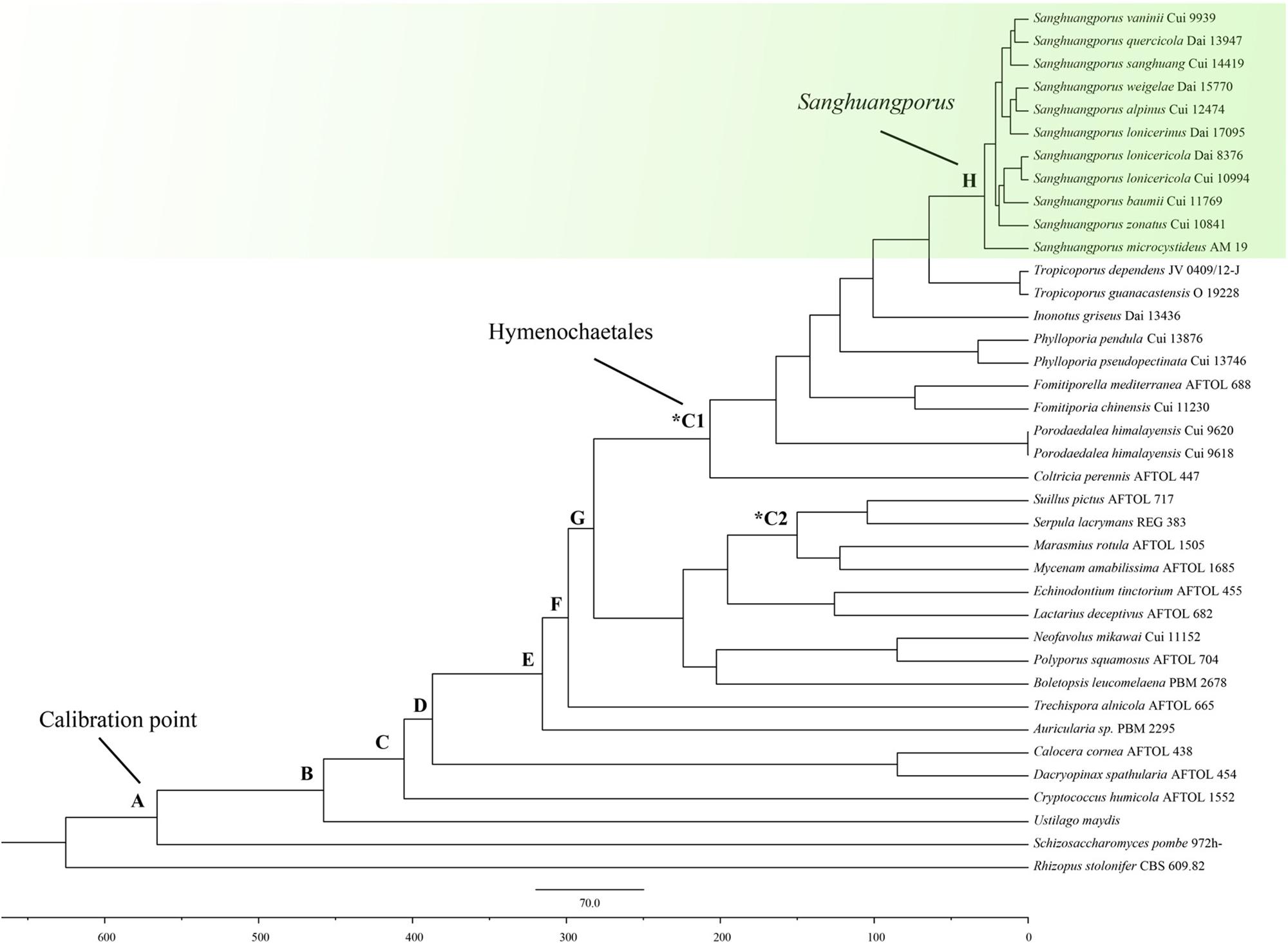
Figure 3. Chronogram and estimated divergence times of Sanghuangporus generated by molecular clock analysis using the two concatenated datasets (ITS+nrLSU and EF1-α+RPB1+RPB2) dataset. The chronogram was obtained using the Ascomycota–Basidiomycota divergence time of 582 Mya as the calibration point. The calibration point and objects of this study are marked in the chronogram. The geological time scale is in millions of years ago (Mya).
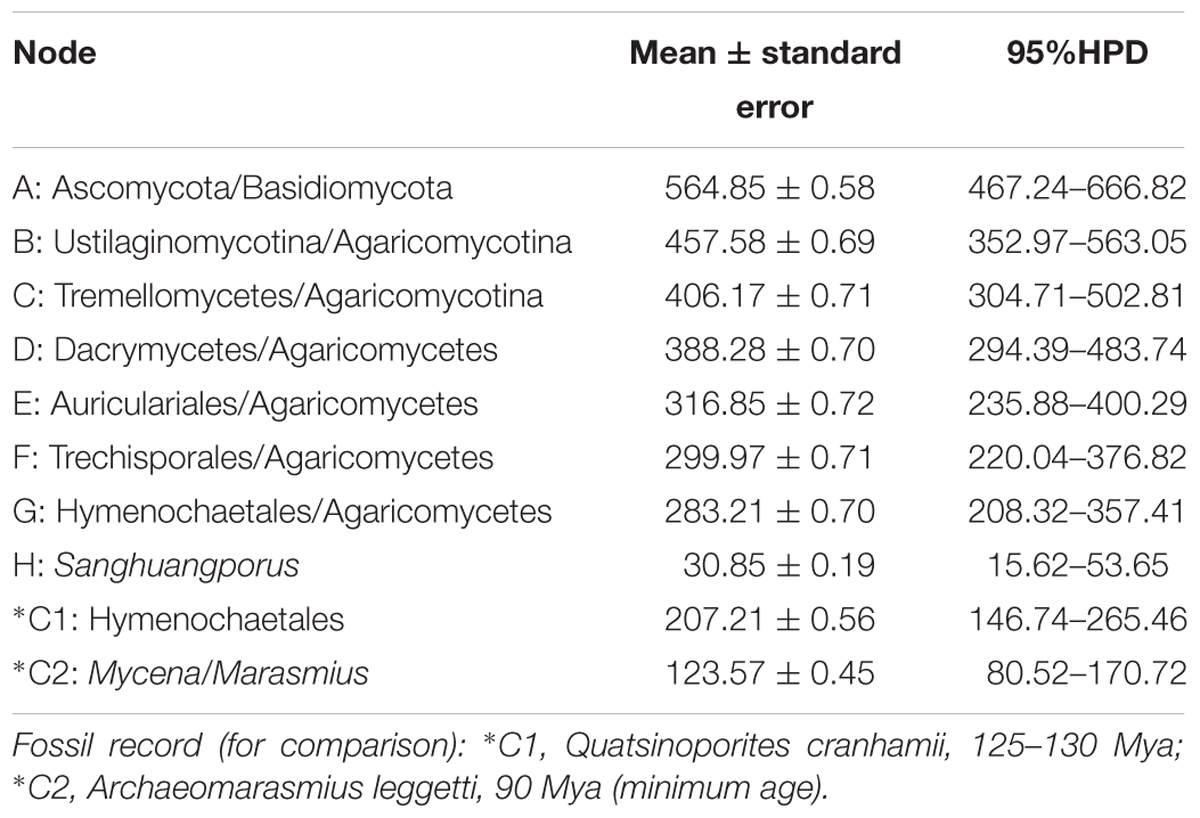
Table 4. Estimated divergence times of the main nodes.
The inferred historical biogeographic scenarios from the analyses using RASP are shown in Figure 4 and summarized in Table 5. The Bayesian binary Markov chain Monte Carlo analysis shows that East Asia has the highest probability (38%) of being the ancestral area of Sanghuangporus. Northeast Asia was the most likely ancestral area for Group 1 ∖∗ ROMAN and Group 2 ∖∗ ROMAN 4 ∖∗ ROMAN, at 70 and 69%. The most likely ancestral area for Group 3 ∖∗ ROMAN (51%) and Group 4 ∖∗ ROMAN (61%) was East Asia. Ten dispersal events and six vicariance events are needed to explain the current distribution of the genus. The basal species (S. microcystideus) exhibited a tropical distribution pattern (Zhou and Wang, 2015). We estimated the crown node of Sanghuangporus to correspond to 30.85 ± 0.19 Mya. The Northeast Asian–East Africa lineages split during the end of the Mid-Miocene, which is consistent with the previous biogeographic studies of Tertiary relics (Zhou et al., 2012; Yang et al., 2016). Additionally, six kinds of dispersal routes were inferred: Northeast Asia–East Africa, Northeast Asia–Southern China, Northeast Asia–Western Asia, Northeast Asia–Europe, East Asia–North America, and East Asia–Central Asia (Figure 5).
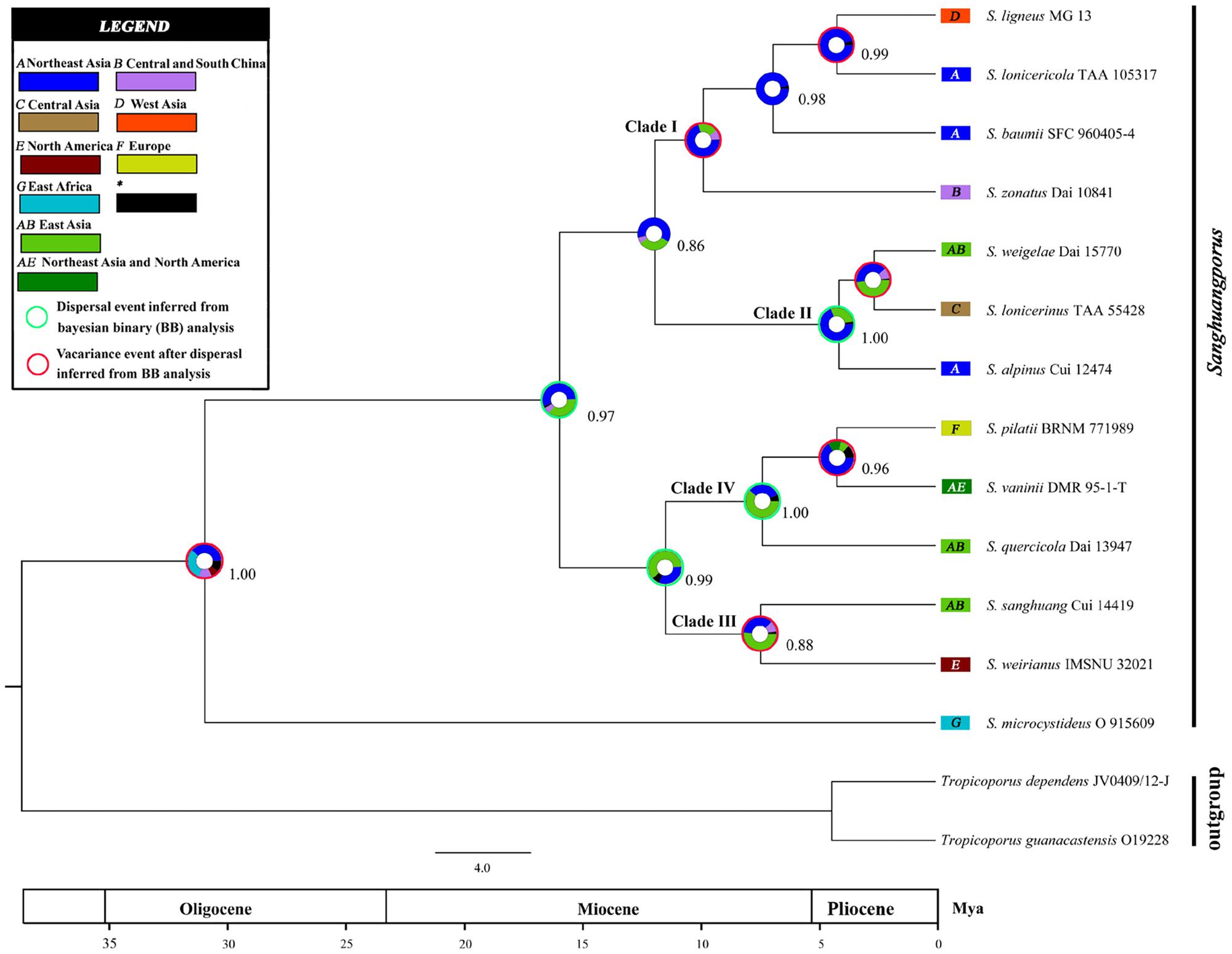
Figure 4. Divergence time estimation and ancestral area reconstruction of Sanghuangporus using the ITS dataset. The chronogram was obtained by molecular clock analysis using BEAST. The pie chart in each node indicates the possible ancestral distributions inferred from Bayesian Binary MCMC analysis (BBM) implemented in RASP. Bayesian credibility values (PP) over 0.85 are indicated near the pie chart of the tree. Green circle around pie charts indicate possible dispersal events, red circle indicate dispersal and vicariance events as suggested by BBM analysis.
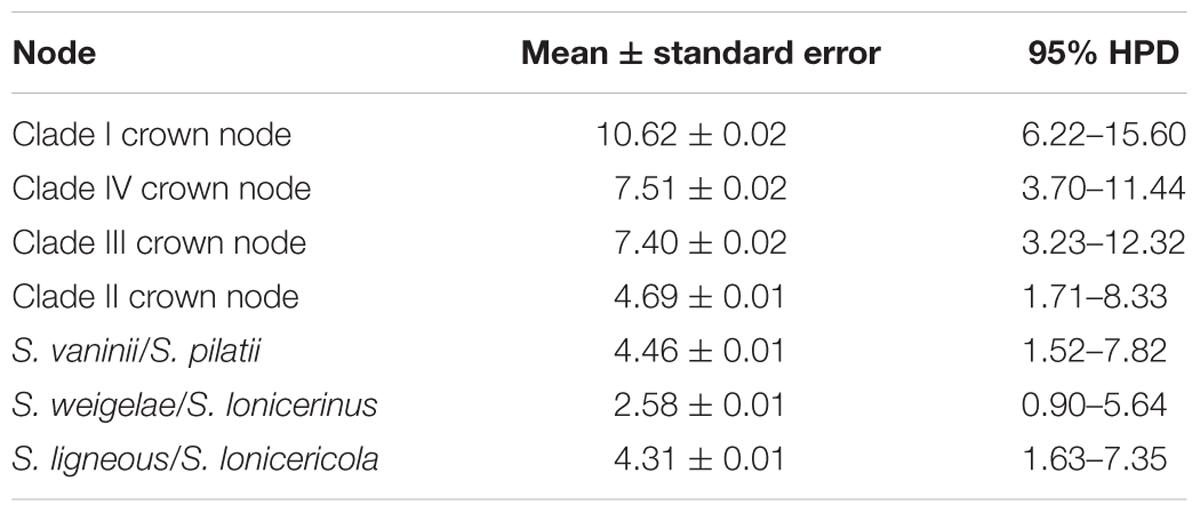
Table 5. Estimated divergence times of the main groups correspond with the dating analysis of ITS datasets.
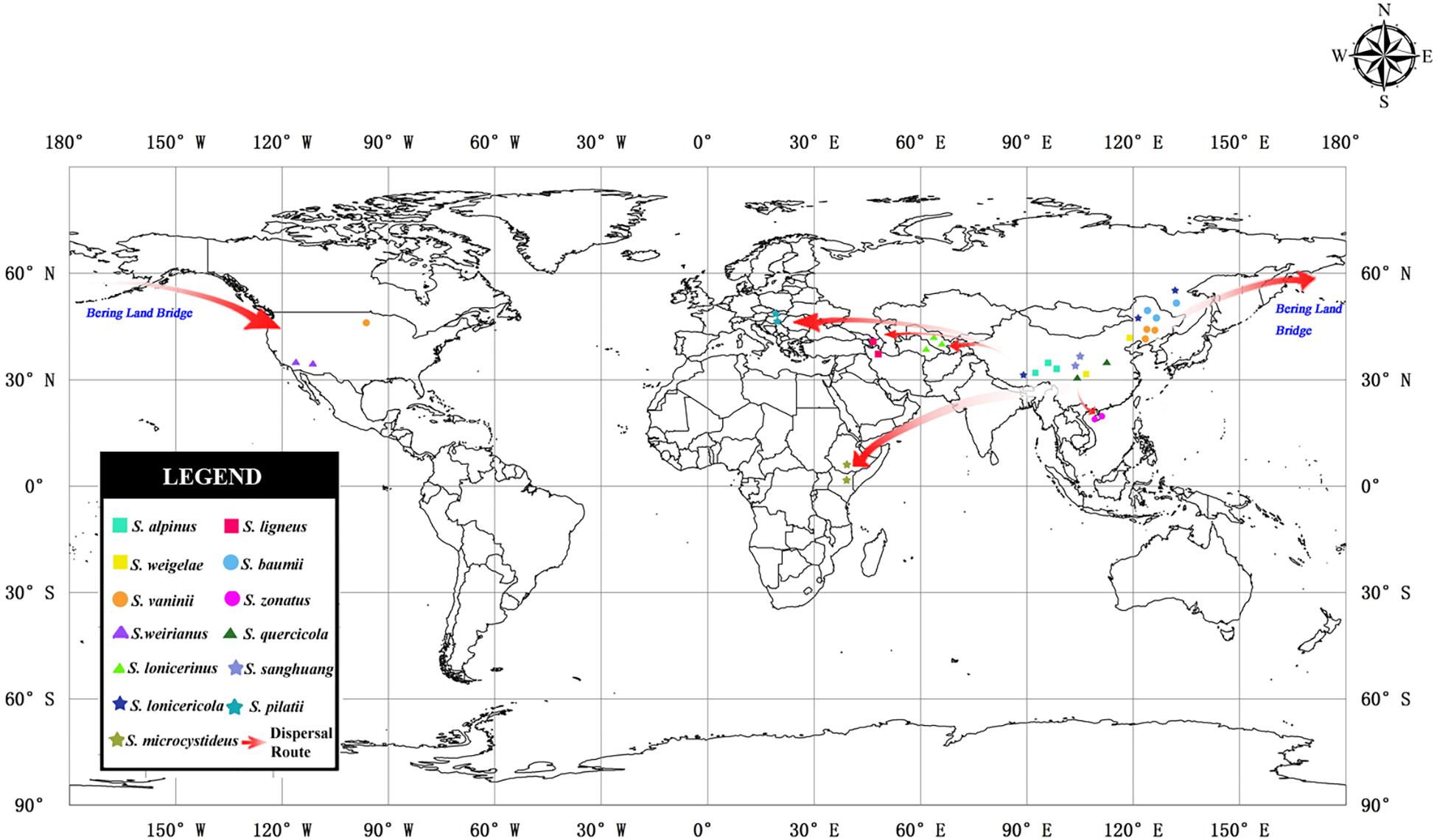
Figure 5. Map of the geographic distribution of Sanghuangporus and possible dispersal routes generated by ArcGIS v10.1. A hypothetical schematic depiction of the original locations, the migration routes the speciation of Sanghuangporus.
The genus Sanghuangporus is one of the most important medicinal fungal genera. Currently, 13 species are accepted in Sanghuangporus. These species form four major clades (Figure 2). This study also shows that Tropicoporus is as a sister genus to Sanghuangporus (MP = 100%, ML = 100%, and BPP = 1.00).
Group A contains five Sanghuangporus species from Europe, East Asia, and North America. In the combined dataset topology (Figure 2), S. vaninii, S. pilatii, and S. quercicola are grouped together, with significant supports from the MP/ML/BI analyses (MP = 99%, ML = 98%, BPP = 1.00). These three species share several morphological synapomorphies including (1) a dark brown pileal surface and a buff-yellow pore surface; (2) similar dimitic hyphal structure; (3) basidiospores ovoid to broadly ellipsoid (Wu et al., 2012; Tomsovsky, 2015; Zhu et al., 2017). Nevertheless, S. quercicola grows on Quercus, but S. pilatii and S. vaninii mostly grow on Populus (Wu et al., 2012; Tomsovsky, 2015; Zhu et al., 2017). Therefore, these species have close relationships to each other except for differences in their host plants, suggesting a gene exchange in this clade. In addition, S. weirianus is restricted to living walnut substrata in Arizona, New Mexico, and Mexico (Gilbertson, 1979). Sanghuangporus sanghuang has brownish yellow basidiospores, and this species grows exclusively on Morus in East Asia (Wu et al., 2012). Moreover, the latter differs from the former in its shallower yellow basidiospores.
In Group B, Sanghuangporus lonicericola, S. baumii, S. zonatus, and S. ligneous form a weakly supported clade in our phylogenic analysis (MP = 67%, ML = 78%, BPP = 0.92) in Figure 2. To date, S. zonatus was only recorded from the tropics (Tian et al., 2013). It is characterized by an applanate pileus with an acute margin (Tian et al., 2013). The molecular data suggest a closer relationship between the East Asian species S. lonicericola and the West Asian species S. ligneous. Both have similar basidiomes and grow on Lonicera. This similarity may result from morphological stasis caused by similar refuges, hosts and habitats in the Quaternary Ice Age (Berbee and Taylor, 1992; Percudani et al., 1999; Djamali et al., 2012). In the East Asian species, S. baumii is often mistaken for S. lonicericola, but, S. baumii has slightly larger basidiospores (3.5–4.5 × 3.2–3.5 μm vs. 3.3–4.1 × 2.4–3.3 μm for S. lonicericola) and larger pores (5–7 per mm vs. 8–10 per mm for S. lonicericola) and grows mostly on Syringa (Wu et al., 2012).
Group C contains two East Asian species, Sanghuangporus alpinus and S. weigelae, and a Central Asia species, S. lonicerinus, and this group is well supported by the ML and BI analyses (ML = 100%, BPP = 1.00) and moderately supported by the MP analysis (MP = 77%). The two East Asian species form a strongly supported clade in the BI analysis (BPP = 0.99), but this was not supported in the ML analysis. S. alpinus is distinguished by its homogeneous context, but S. weigelae shows duplex context (Wu et al., 2012). In addition, the former species is presently only known from the plateau region, but the latter has a warm temperate to subtropical distribution (Tian et al., 2013). S. lonicerinus covers most parts of Central Asia and grows mostly on Lonicera (Wu et al., 2012). These species are distinct from those of the other clades in the phylogenetic tree (Figure 2).
Group D consists of only Sanghuangporus microcystideus. It resembles S. alpinus in having a dark brown pileal surface, but the basidiospores of S. microcystideus are slightly larger (5.1–6.0 × 4.1–5.0 μm). This East African species was collected on Olea (Zhou et al., 2016). In the phylogenetic analysis (Figure 2), Group D formed a significantly supported terminal lineage (MP = 100%, ML = 98%, BPP = 1.00).
The maximum crown age of Sanghuangporus was estimated to be around the Oligocene (30.85 ± 0.19 Mya) (Figures 3, 4), and Northeast Asia was inferred as the most likely (38%) ancestral area (Figure 4). The changes caused by tectonic activity and dramatic climate change since the collision between the Arabian plate and the Eurasian closed the Tethys Sea. Asia and Africa were connected via the “Gomphotherium Landbridge” during the late Oligocene (Rögl, 1999, 2001). Expanded tropical forests associated with the Miocene warming trend may be an important factor for species exchanges between Asia and Africa (Yuan et al., 2005). It is probable that the common ancestor between Asia and Africa migrated via the expanded rainforests at the end of the Mid-Miocene. Later, global cooling and increasing aridification (16–2.8 Mya) replaced the Mid-Miocene climate optimum (approximately 15 Mya) after the temperature began to fluctuate in the Miocene–Oligocene boundary (approximately 24 Mya) (Zachos et al., 2001; Morley, 2003; Guo et al., 2008; Besnard et al., 2009; Chen et al., 2014). As a consequence, the expansion of the grassland vegetation gradually replaced tropical forests during the Quaternary ice ages. Climatic shifts would exacerbate the depth of divergence between sister lineages (Godfrey, 2000; Fleagle and Gilbert, 2006). This biogeographic distribution pattern was also identified in Searsia, Lychnis, Uvaria, and it is associated with the “out-of-Africa” dispersal of primates (Fleagle and Gilbert, 2006; Popp et al., 2008; Zhou et al., 2012; Yang et al., 2016). With repeated climatic oscillations and the major growth of the Arctic Ice Sheet, there was climate deterioration in the late Pliocene and Pleistocene in Europe, Asia, and North America (Godfrey, 2000; Shawn and John, 2006). These severe climatic oscillations affected environmental heterogeneity and produced great changes in Sanghuangporus species distributions in the Northern Hemisphere.
According to the molecular clock results, the separation of Clade I and Clade II ∖∗ ROMAN initially occurred at 12.47 Mya (Figure 4). The estimated divergence time coincides with the climate changes in the middle Miocene, probably following the uplift of the Himalaya-Tibetan Plateau (Zhang and Fritsch, 2010). As a consequence, increasingly East Asian monsoons served as a key trigger of the aridification of central China (Molnar et al., 2010; Li et al., 2011; Zhao et al., 2012; Chatterjee et al., 2013; Zhang et al., 2015). Two sister clades within this lineage exhibited independent east-to-west dispersal in the ancestral area reconstruction analyses (Figures 4, 5). In Clade I, the divergence between the West Asia species Sanghuangporus ligneous and its East Asian counterpart (S. lonicericola) is estimated to have occurred approximately 4.31 Mya in the Pliocene, and East Asia is inferred to be the common ancestral area. A complex sequence of tectonic setting deformation occurred, leading to the uplift of the mountain ranges starting in the Late Miocene (Axen et al., 2001; Eronen et al., 2010; Djamali et al., 2012). The variation event might indicate a relatively stable climate during the late Miocene-Pliocene compared to the Quaternary period that caused small species range shifts and gene flow in the Iranian plateau (Sherkatia and Letouzeyb, 2004; Ritz et al., 2006). In Clade 2 ∖∗ ROMAN, three Sanghuangporus species from East Asia and Central Asia are closely related (Figures 2, 4). We inferred vicariance events within Central Asia during the Pliocene and early Pleistocene (2.58 Mya). Palaeogeographical evidence suggests a dramatic change from a region with a humid climate to a semi-arid climate in the western part of central Asia (Manafzadeh et al., 2014; Wang et al., 2016; Lauer et al., 2017). This change is thought to have spurred speciation as a result of habitat shifts promoted by progressive aridification and the Quaternary glacial-interglacial cycles, which started approximately 2.6 Mya at the beginning of the Quaternary (Gibbard et al., 2009; Zhang and Zhang, 2012). Hence, the climatic shifts caused range contraction and speciation in the region.
Our results indicate that two independent intercontinental distribution patterns (Clade 3 ∖∗ ROMAN and Clade 4 ∖∗ ROMAN) started to diversify during the Miocene (Figure 4). Clade 3 ∖∗ ROMAN is divided into two sister species, Sanghuangporus sanghuang and S. weirianus (Figure 4), and this clade had an intercontinental distribution between East Asian and North America. The divergence of Clade III and Clade IV occurred before the Bering Land Bridge (BLB) was detached approximately 5.5 Mya (Milne, 2006). We speculate that intercontinental transfers of the Sanghuangporus ancestor occurred between East Asia and North America via the BLB. Estimated vicariance events between North America and Asia in the late Miocene (7.51 Mya) are also consistent with biogeographic patterns supported by the bidirectional floristic exchange in the temperate taxa (Manos and Stanford, 2001). The vicariance due to reduced precipitation may be responsible for the endemism of Sanghuangporus in America. Dispersal-vicariance theory is used to understand the intercontinental migration patterns (Donoghue and Smith, 2004; Cai et al., 2014). In addition, in Clade 4 ∖∗ ROMAN, the estimated divergence time of S. vaninii is consistent with the known dispersal of the host plant (Populus) from Asia to North America (Godfrey, 2000; Du et al., 2015). Palaeobotanical data indicate that the BLB route played a crucial role in plant dispersal for Populus, Lonicera, Leibnitzia, and Linnaea (Baird et al., 2010; Smith and Donoghue, 2010; Du et al., 2015; Wang et al., 2015). The notable finding is that the biogeographical pattern of the European species (S. pilatii) and the Northeast Asian and North American species (S. pilatii) nested in the East Asia lineage, which would be expected given the probable direction of ancestor migration. The estimated crown age is approximately 4.46 Mya between S. pilatii and S. vaninii. Pliocene climatic events caused range contraction, fragmentation, and the extinction of numerous species, which accelerated the intra-phylogroup diversification processes in Sanghuangporus. Long-distance dispersal has also been detected in other fungi, such as Bondarzewia Singer (Bondarzewiaceae, Russulales) (Song et al., 2016). Our data suggest that the other East Asian species in this clade, S. quercicola, diversified approximately 7.44 Mya. Afterward, the ice sheets expanded in the Northern Hemisphere (Chang et al., 2017). Divergence due to habitat fragmentation in the late Miocene is thought to be responsible for the endemism of S. quercicola. The divergence events exhibited by the plant taxa (Quercus glauca and Pinus armandii) confirm the geological events associated with species diversification in subtropical Asia (Andrew and Hall, 2006; Xu et al., 2015).
We found that the samples of Sanghuangporus are scant in some parts of the world, such as South America, the Indian Subcontinent and Australia. Therefore, the further sampling is required to clarify the population history of these areas.
B-KC and LZ designed the experiments. LZ, JSi, and BK-C prepared the samples. LZ, JSo, and JL-Z conducted the molecular experiments and analyzed the data. LZ, JSi, JL-Z, JSo, and BK-C drafted the manuscript. All of the authors approved the manuscript.
This research was financed by the Fundamental Research Funds for the Central Universities (No. 2016ZCQ04) and the National Natural Science Foundation of China (Project No. 31670016).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to Prof. Yu-Cheng Dai (Beijing Forestry University, China) for improving the manuscript and help in field collections. We extend special thanks to Drs. Xiao-Lan He (Soil and Fertilizer Institute, Sichuan Academy of Agricultural Sciences, China) and Juan Li (Institute of Applied Ecology, Chinese Academy of Sciences, China) for collecting specimens, as well as to Drs. Hai-Sheng Yuan and Li-Wei Zhou (Institute of Applied Ecology, Chinese Academy of Sciences, China) for the loaning of specimens. Mr. Li-Yan Yang and Feng Qiu (Beijing Forestry University, China) are thanked for help in preparing the figures. IMC 11 is acknowledged for providing a poster presentation for the authors to show the results of this study.
BBM, Bayesian binary Markov chain Monte Carlo; BI, Bayesian inference; BJFC, Herbarium of the Institute of Microbiology, Beijing Forestry University; BPP, Bayesian posterior probabilities; CI, consistency index; EF1-α, translation elongation factor 1-α; GTR-I+G, general time reversible+proportion invariant+gamma; HI, homoplasy index; HPD, higher posterior densities; IFP, Herbarium of the Institute of Institute of Applied Ecology, Chinese Academy of Sciences; ITS, internal transcribed spacer; ML, maximum likelihood; MP, maximum parsimony; Mya, million years ago; nrLSU, nuclear large subunit rDNA; RC, rescaled consistency index; RI, retention index; RPB1, DNA-directed RNA polymerase II subunit 1; RPB2, DNA-directed RNA polymerase II subunit 2; TL, tree length.
Andrew, J., and Hall, B. D. (2006). Phylogeny, historical biogeography, and patterns of diversification for Pinus (Pinaceae): phylogenetic tests of fossil-based hypotheses. Mol. Phylogenet. Evol. 40, 166–182. doi: 10.1016/j.ympev.2006.03.009
Axen, G. J., Lam, P. S., Grove, M., Stockli, D. F., and Hassanzadeh, J. (2001). Exhumation of the west-central Alborz Mountains, Iran, Caspian subsidence, and collision-related tectonics. Geology 29, 559–562.
Baird, K. E., Funk, V. A., Wem, J., and Weeks, A. (2010). Molecular phylogenetic analysis of Leibnitzia Cass. (Asteraceae: Mutisieae: Gerbera-complex), an Asian-North American disjunct genus. J. Syst. Evol. 48, 161–174. doi: 10.1111/j.1759-6831.2010.00077.x
Berbee, M. L., and Taylor, J. W. (1992). Detecting morphological convergence in true fungi, using 18S rRNA gene sequence data. Biosystems 28, 117–125. doi: 10.1016/0303-2647(92)90014-p
Besnard, G., Casas, R. R., Christin, P., and Vargas, P. (2009). Phylogenetics of Olea (Oleaceae) based on plastid and nuclear ribosomal DNA sequences: tertiary climatic shifts and lineage differentiation times. Ann. Bot. 104, 143–160. doi: 10.1093/aob/mcp105
Binder, M., Larsson, K. H., Matheny, P. B., and Hibbett, D. S. (2010). Amylocorticiales ord. nov. and Jaapiales ord. nov.: early diverging clades of Agaricomycetidae dominated by corticioid forms. Mycologia 102, 865–880. doi: 10.3852/09-288
Cai, Q., Tulloss, R. E., Tang, L. P., Tolgor, B., Zhang, P., Chen, Z. H., et al. (2014). Multi-locus phylogeny of lethal amanitas: implications for species diversity and historical biogeography. BMC Evol. Biol. 14:143. doi: 10.1186/1471-2148-14-143
Chang, H., An, Z. S., Wu, F., Song, Y. G., Qiang, X. K., and Li, L. Y. (2017). Late Miocene – early Pleistocene climate change in the mid-latitude westerlies and their influence on Asian monsoon as constrained by the K/Al ratio record from drill core Ls2 in the Tarim Basin. Catena 53, 75–82. doi: 10.1016/j.catena.2017.02.002
Chatterjee, S., Goswami, A., and Scptese, C. R. (2013). The longest voyage: tectonic, magmatic, and paleoclimatic evolution of the Indian plate during its northward flight from Gondwana to Asia. Gondwana Res. 23, 238–267. doi: 10.1016/j.gr.2012.07.001
Chen, C., Qi, Z. C., Xu, X. H., Comes, H. P., Koch, M. A., Fu, C. X., et al. (2014). Understanding the formation of Mediterranean–African–Asian disjunctions: evidence for Miocene climate-driven vicariance and recent long-distance dispersal in the Tertiary relict Smilax aspera (Smilacaceae). New Phytol. 204, 243–255. doi: 10.1111/nph.12910
Chen, J. J., Cui, B. K., Zhou, L. W., Korhonen, K., and Dai, Y. C. (2015). Phylogeny, divergence time estimation, and biogeography of the genus Heterobasidion (Basidiomycota, Russulales). Fungal Divers. 71, 185–200. doi: 10.1007/s13225-014-0317-2
Dai, Y. C. (2010). Hymenochaetaceae (Basidiomycota) in China. Fungal Divers. 45, 131–343. doi: 10.1007/s13225-010-0066-9
Dai, Y. C., Yang, Z. L., Cui, B. K., Yu, C. J., and Zhou, L. W. (2009). Species diversity and utilization of medicinal mushrooms and fungi in China. Int. J. Med. Mushrooms 11, 287–302. doi: 10.1615/IntJMedMushr.v11.i3.80
Djamali, M., Baume, A., Brewer, S., Jackson, S. T., Kadereit, J. W., López-Vinyallonga, S., et al. (2012). Ecological implications of Cousinia Cass. (Asteraceae) persistence through the last two glacial-interglacial cycles in the continental Middle East for the Irano-Turanian flora. Rev. Palaeobot. Palynol. 172, 10–20. doi: 10.1016/j.revpalbo.2012.01.005
Donoghue, M. J., and Smith, S. A. (2004). Patterns in the assembly of temperate forests around the Northern Hemisphere. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359, 1633–1644. doi: 10.1098/rstb.2004.1538
Drummond, A. J., and Rambaut, A. (2007). BEAST: bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. doi: 10.1186/1471-2148-7-214
Du, S. H., Wang, Z. S., Ingvarsson, P. K., Wang, D. S., Wang, J. H., Wu, Z. Q., et al. (2015). Multilocus analysis of nucleotide variation and speciation in three closely related Populus (Salicaceae) species. Mol. Ecol. 24, 4994–5005. doi: 10.1111/mec.13368
Eastwood, D. C., Floudas, D., Binder, M., Majcherczyk, A., Schneider, P., Aerts, A., et al. (2011). The plant cell wall-decomposing machinery underlies the functional diversity of forest fungi. Science 333, 762–765. doi: 10.1126/science.1205411
Eronen, J. T., Puolamäki, K., Liu, L., Lintulaakso, K., Damuth, J., Janis, C., et al. (2010). Precipitation and large herbivorous mammals II: application to fossil data. Evol. Ecol. Res. 12, 235–248.
Esseghir, S., Ready, P. D., and Ben-Ismsil, R. (2000). Speciation of Phlebotomus sandflies of the subgenus Larroussius coincided with the late Miocene-Pliocene aridification of the Mediterranean subregion. Biol. J. Linn. Soc. 70, 189–219. doi: 10.1006/bijl.1999.0393
Feng, B., Xu, J. P., Wu, G., Zeng, N. K., Li, Y. C., Tolgor, B., et al. (2012). DNA sequences analyses reveal abundant diversity, endemism and evidence for Asian origin of the porcini. PLoS One 7:e37567. doi: 10.1371/journal.pone.0037567
Fleagle, J. G., and Gilbert, C. C. (2006). “The biogeography of primate evolution: the role of plate tectonics, climate and chance,” in Primate Biogeography, eds S. M. Lehman and J. G. Fleagle (New York, NY: Springer), 375–418. doi: 10.1007/0-387-31710-4_13
Floudas, D., Binder, M., Riley, R., Barry, K., Blanchette, R. A., Henrissat, B., et al. (2012). The Paleozonic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 336, 1715–1719. doi: 10.1126/science.1221748
Ghobad-Nejhad, M. (2015). Collections on Lonicera in Northwest Iran represent an undescribed species in the Inonotus linteus complex (Hymenochaetales). Mycol. Prog. 14:90.
Gibbard, P. L., Head, M. J., and Walker, M. J. C. (2009). Formal ratification of the Quaternary system/period and the Pleistocene series/epoch with a base at 2.588 Ma. J. Quat. Sci. 25, 96–102. doi: 10.1002/jqs.1338
Gilbertson, R. L. (1979). The genus Phellinus (Aphyllophorales: Hymenochaetaceae) in western North America. Mycotaxon 9, 51–89.
Godfrey, H. (2000). The genetic legacy of the Quaternary ice ages. Nature 405, 907–913. doi: 10.1038/35016000
Guo, Z. T., Sun, B., Zhang, Z. S., Peng, S. Z., Xiao, G. Q., Ge, J. Y., et al. (2008). A major reorganization of Asian climate by the early Miocene. Clim. Past 4, 153–174. doi: 10.5194/cp-4-153-2008
Hall, T. A. (1999). Bioedit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 41, 95–98. doi: 10.1021/bk-1999-0734.ch008
Han, M. L., Chen, Y. Y., Shen, L. L., Song, J., Vlasák, J., Dai, Y. C., et al. (2016). Taxonomy and phylogeny of the brown-rot fungi: Fomitopsis and its related genera. Fungal Divers. 80, 343–373. doi: 10.1007/s13225-016-0364-y
Hibbett, D. S., Grimaldi, D., and Donoghue, M. J. (1997). Fossil mushrooms from Miocene and Cretaceous ambers and the evolution of homobasidiomycetes. Am. J. Bot. 84, 981–991. doi: 10.2307/2446289
Lauer, T., Vlaminck, S., Frechen, M., Rolf, C., Kehl, M., Sharifi, J., et al. (2017). The Agh Band loess-palaeosol sequence – a terrestrial archive for climatic shifts during the last and penultimate glacial-interglacial cycles in a semiarid region in northern Iran. Quatern. Int. 429, 13–30. doi: 10.1016/j.quaint.2016.01.062
Lee, M. S., Hwang, B. S., Lee, I. K., Seo, G. S., and Yun, B. S. (2015). Chemical constituents of the culture broth of Phellinus linteus and their antioxidant activity. Mycobiology 44, 43–48. doi: 10.5941/MYCO.2015.43.1.43
Li, G. J., Pettke, T., and Chen, J. (2011). Increasing Nd isotopic ratio of Asian dust indicates progressive uplift of the north Tibetan Plateau since the middle Miocene. Geology 39, 199–202. doi: 10.1130/g31734.1
Lin, W. C., Deng, J. S., Huang, S. S., Lin, W. R., Wu, S. H., Lin, H. Y., et al. (2017). Anti-inflammatory activity of Sanghuangporus sanghuang by suppressing the TLR4-mediated PI3K/AKT/mTOR/IKKβ signaling pathway. RSC Adv. 7, 21234–21251. doi: 10.1039/C7RA01000A
Liu, Y. J., Wheelen, S., and Hall, B. D. (1999). Phylogenetic relationships among ascomycetes: evidence from an RNA polymerase II subunit. Mol. Biol. Evol. 16, 1799–1808. doi: 10.1093/oxfordjournals.molbev.a026092
Manafzadeh, S., Salvo, G., Conti, E., and Ebach, M. (2014). A tale of migrations from east to west: the Irano-Turanian floristic region as a source of Mediterranean xerophytes. J. Biogeogr. 41, 366–379. doi: 10.1111/jbi.12185
Manos, P. S., and Stanford, A. M. (2001). The historical biogeography of Fagaceae: tracking the Tertiary history of temperate and subtropical forests of the Northern hemisphere. Int. J. Plant Sci. 162, 18–27. doi: 10.1086/323280
Matheny, P. B., Liu, Y. J., Ammirati, J. F., and Hall, B. D. (2002). Using RPB1 sequences to improve phylogenetic inference among mushrooms (Inocybe, Agaricales). Am. J. Bot. 89, 688–698. doi: 10.3732/ajb.89.4.688
Meulenkamp, J. E., and Sissingh, W. (2003). Tertiary palaeogeography and tectonostratigraphic evolution of the Northern and Southern Peri-Tethys platforms and the intermediate domains of the African-Eurasian convergent plate boundary zone. Palaeogeogr. Palaeocl. 196, 209–228. doi: 10.1016/s0031-0182(03)00319-5
Michalak, S. (2012). RaxmlGUI: a graphical front-end for RAxML.Org. Divers. Evol. 12, 335–337. doi: 10.1007/s13127-011-0056-0
Milne, R. I. (2006). Northern hemisphere plant disjunctions: a window on Tertiary Land Bridges and climate change? Ann. Bot. 98, 465–472. doi: 10.1093/aob/mcl148
Molnar, P., Boos, W. R., and Battisti, D. S. (2010). Orographic controls on climate and paleoclimate of Asia: thermal and mechanical roles for the Tibetan Plateau. Ann. Rev. Ear. Planet. Sci. 38, 77–102. doi: 10.1146/annurev-earth-040809-152456
Morley, R. J. (2003). Interplate dispersal paths for megathermal angiosperms. Perspect. Plant Ecol. 6, 5–20. doi: 10.1078/1433-8319-00039
Nylander, J. A. A. (2004). MrModeltest v2. Program Distributed by the Author. Uppsala: Uppsala University. doi: 10.1078/1433-8319-00039
Percudani, R., Trevisi, A., Zambonelli, A., and Ottonello, S. (1999). Molecular phylogeny of truffles (Pezizales: Terfeziaceae, Tuberaceae) derived from nuclear rDNA sequence analysis. Mol. Phylogenet. Evol. 13, 169–180. doi: 10.1006/mpev.1999.0638
Petersen, J. H. (1996). Farvekort. The Danish Mycological Society’s Colour-Chart. Greve: Foreningen til Svampekundskabens Fremme, 1–6.
Popp, M., Gizaw, A., Nemomissa, S., Suda, J., and Brochmann, C. (2008). Colonization and diversification in the African sky ‘islands’ by Eurasian Lychnis L. (Caryophyllaceae). J. Biogeogr. 35, 1016–1029. doi: 10.1111/nph.13937
Rambaut, A. (2012). Molecular Evolution, Phylogenetics and Epidemiology. FigTree ver. 1.4 Software. Available at: http://tree.bio.ed.ac.uk/software/figtree/ (accessed May 1, 2017).
Rehner, S. A., and Buckley, E. (2005). A Beauveria phylogeny inferred from nuclear ITS and EF-1α sequences: evidence for cryptic diversification and links to Cordyceps teleomorphs. Mycologia 97, 84–98. doi: 10.1080/15572536.2006.11832842
Ritz, J. F., Nazari, H., Ghassemi, A., Salamati, R., Shafei, A., Solaymani, S., et al. (2006). Active transtension inside central Alborz: a new insight into northern Iran-southern Caspian geodynamics. Geology 36, 477–480. doi: 10.1130/g22319.1
Rögl, F. (1999). Mediterranean and Paratethys. facts and hypotheses of an Oligocene to Miocene Paleogeography (short overview). Geol. Carpath. 50, 339–349.
Rögl, F. (2001). Mid-Miocene circum-mediterranean paleogeography. Ber. Inst. Geol. Paläont. K. F. Univ. Graz 4, 49–59.
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Shawn, M. L., and John, G. F. (2006). Biogeography and Primates: A Review. Primate Biogeography. New York, NY: Springer, 1–58.
Shen, L. L., Wang, M., Zhou, J. L., Xing, J. H., Cui, B. K., and Dai, Y. C. (2019). Taxonomy and phylogeny of Postia. Multi-gene phylogeny and taxonomy of the brown-rot fungi: Postia (Polyporales, Basidiomycota) and related genera. Persoonia 42, 101–126. doi: 10.3767/persoonia.2019.42.05
Sherkatia, S., and Letouzeyb, J. (2004). Variation of structural style and basin evolution in the central Zagros (Izeh zone and Dezful Embayment), Iran. Mar. Petrol. Geol. 21, 535–554. doi: 10.1016/j.marpetgeo.2004.01.007
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W. Z., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Smith, S. A., and Donoghue, M. J. (2010). Combining historical biogeography with niche modeling in the Caprifolium Clade of Lonicera (Caprifoliaceae, Dipsacales). Syst. Biol. 59, 322–341. doi: 10.1093/sysbio/syq011
Smith, S. Y., Currah, R. S., and Stockey, R. A. (2004). Cretaceous and Eocene poroid hymenophores from Vancouver Island, British Columbia. Mycologia 96, 180–186. doi: 10.2307/3762001
Song, J., Chen, J. J., Wang, M., Chen, Y. Y., and Cui, B. K. (2016). Phylogeny and biogeography of the remarkable genus Bondarzewia (Basidiomycota, Russulales). Sci. Rep. 6:34568. doi: 10.1038/srep34568
Song, J., and Cui, B. K. (2017). Phylogeny, divergence time and historical biogeography of Laetiporus (Basidiomycota, Polyporales). BMC Evol. Biol. 17:102. doi: 10.1186/s12862-017-0948-5
Swofford, D. L. (2002). PAUP∗. Phylogenetic Analysis Using Parsimony (∗ and Other Methods). Version 4.0b10. Sunderland, MA: Sinauer Associates, doi: 10.1111/j.0014-3820.2002.tb00191.x
Taylor, T. N., Hass, H., Kerp, H., Krings, M., and Hanlin, R. T. (2004). Perithecial ascomycetes from the 400 million year old Rhynie chert: an example of ancestral polymorphism. Mycologia 96, 1403–1419. doi: 10.2307/3762157
Tian, X. M., Yu, H. Y., Decock, C., Vlasák, J., and Dai, Y. C. (2013). Phylogeny and taxonomy of the Inonotus linteus complex. Fungal Divers. 58, 159–160. doi: 10.1007/s12275-016-5520-2
Tomsovsky, M. (2015). Sanghuangporus pilatii, a new combination, revealed as European relative of Asian medicinal fungi. Phytotaxa 239, 82–88. doi: 10.11646/phytotaxa.239.1.8
Vilgalys, R., and Hester, M. (1990). Rapid genetic identification and mapping of enzymatically amplified ribosomal DNA from several Cryptococcus species. J. Bacteriol. 172, 4238–4246. doi: 10.1128/jb.172.8.4238-4246.1990
Vlasák, J., Li, H. J., Zhou, L. W., and Dai, Y. C. (2013). A further study on Inonotus linteus complex (Hymenochaetales, Basidiomycota) in tropical America. Phytotaxa 124, 25–36. doi: 10.11646/phytotaxa.124.1.3
Wang, H. F., Landrein, S., Dong, W. P., Nie, Z. L., Kondo, K., Funamoto, T., et al. (2015). Molecular phylogeny and biogeographic diversification of Linnaeoideae (Caprifoliaceae s. l.) disjunctly distributed in Eurasia, North America and Mexico. PLoS One 10:e0116485. doi: 10.1371/journal.pone.0116485
Wang, X., Wei, H. T., Taheri, M., Khormali, F., Danukalova, G., and Chen, F. H. (2016). Early Pleistocene climate in western arid central Asia inferred from loess-palaeosol sequences. Sci. Rep. 6:20556. doi: 10.1038/srep20560
Wu, S. H., Dai, Y. C., Hattori, T., Yu, T. W., Wang, D. M., Parmasto, E., et al. (2012). Species clarification for the medicinally valuable ‘sanghuang’ mushroom. Bot. Stud. 53, 135–149.
Xu, J., Deng, M., Jiang, X. L., Westwood, M., Song, Y. G., and Turkington, R. (2015). Phylogeography of Quercus glauca (Fagaceae), a dominant tree of East Asian subtropical evergreen forests, based on three chloroplast DNA interspace sequences. Tree Genet. Genomes 11:805.
Yang, Y. Y., Meng, Y., Wen, J., Sun, H., and Nie, Z. H. (2016). Phylogenetic analyses of Searsia (Anacardiaceae) from eastern Asia and its biogeographic disjunction with its African relatives. S. Afr. J. Bot. 106, 129–136. doi: 10.1016/j.sajb.2016.05.021
Yu, Y., Harris, A. J., Blair, C., and He, X. J. (2015). RASP (reconstruct ancestral state in phylogenies): a tool for historical biogeography. Mol. Phylogenet. Evol. 87, 46–49. doi: 10.1016/j.ympev.2015.03.008
Yuan, Y. M., Wohlhauser, S., Moller, M., Klackenberg, J., Callmander, M. W., and Kupfer, P. (2005). Phylogeny and biogeography of Exacum (Gentianaceae): a disjunctive distribution in the Indian Ocean basin resulted from long distance dispersal and extensive radiation. Syst. Biol. 54, 21–34. doi: 10.1080/10635150590905867
Zachos, J., Pagani, M., Sloan, L., Thomas, E., and Billups, K. (2001). Trends, rhythms, and aberrations in global climate 65 Ma to present. Science 292, 686–693. doi: 10.1126/science.1059412
Zhang, H. X., and Zhang, M. L. (2012). Genetic structure of the Delphinium naviculare species group tracks Pleistocene climatic oscillations in the Tianshan Mountains, arid Central Asia. Palaeogeogr. Palaeocl. 353, 93–103. doi: 10.1016/j.palaeo.2012.07.013
Zhang, M. L., and Fritsch, P. W. (2010). Evolutionary response of Caragana (Fabaceae) to Qinghai–Tibetan Plateau uplift and Asian interior aridification. Plant Syst. Evol. 288, 191–199. doi: 10.1007/s00606-010-0324-z
Zhang, M. L., Temirbayeva, K., Sanderson, S. C., and Chen, X. (2015). Young dispersal of xerophil Nitraria lineages in intercontinental disjunctions of the Old World. Sci. Rep. 5:13840. doi: 10.1038/srep13840
Zhao, N., Dai, C. Y., Wang, W. J., Zhang, R. Y., Qu, Y. H., Song, G., et al. (2012). Pleistocene climate changes shaped the divergence and demography of Asian populations of the great tit Parus major: evidence from phylogeographic analysis and ecological niche models. J. Avian Biol. 43, 297–310. doi: 10.1111/j.1600-048x.2012.05474.x
Zhou, L. L., Su, Y., Thomas, D. C., and Richard, S. (2012). ‘Out’-of-Africa dispersal of floras during the Miocene climatic optimum: evidence from Uvaria (Annonaceae). J. Biogeogr. 39, 322–335. doi: 10.1111/j.1365-2699.2011.02598.x
Zhou, L. W., Vlasák, J., Decock, C., Asefa, A., Stelid, J., Abate, D., et al. (2016). Global diversity and taxonomy of the Inonotus linteus complex (Hymenochaetales, Basidiomycota): Sanghuangporus gen. nov., Tropicoporus excentrodendri and T. guanacastensis gen. et spp. nov., and 17 new combinations. Fungal Divers. 77, 335–347. doi: 10.1007/s13225-015-0335-8
Zhou, L. W., and Wang, X. Y. (2015). Inonotus griseus sp. nov. from eastern China. Mycotaxon 130, 661–669. doi: 10.5248/130.661
Zhu, L., and Cui, B. K. (2016). Progress on the studies of medicinal mushrooms “Sanghuang” group. J. Fungal Res. 14, 201–209.
Keywords: biogeography, Hymenochaetaceae, medicinal mushroom, molecular clock, phylogeny
Citation: Zhu L, Song J, Zhou J-L, Si J and Cui B-K (2019) Species Diversity, Phylogeny, Divergence Time, and Biogeography of the Genus Sanghuangporus (Basidiomycota). Front. Microbiol. 10:812. doi: 10.3389/fmicb.2019.00812
Received: 17 December 2018; Accepted: 01 April 2019;
Published: 17 April 2019.
Edited by:
Rui-Lin Zhao, Institute of Microbiology (CAS), ChinaReviewed by:
Somayeh Dolatabadi, Westerdijk Fungal Biodiversity Institute, NetherlandsCopyright © 2019 Zhu, Song, Zhou, Si and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bao-Kai Cui, Y3VpYmFva2FpQHlhaG9vLmNvbQ==; YmFva2FpY3VpMjAxM0BnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.