 Soon-Hwan Oh1
Soon-Hwan Oh1 Brooke Smith2
Brooke Smith2 Andrew N. Miller3Bart Staker4Christopher Fields5
Andrew N. Miller3Bart Staker4Christopher Fields5 Alvaro Hernandez5
Alvaro Hernandez5 Lois L. Hoyer1*
Lois L. Hoyer1*- 1Department of Pathobiology, University of Illinois at Urbana-Champaign, Urbana, IL, United States
- 2Department of Biology, Millikin University, Decatur, IL, United States
- 3Illinois Natural History Survey, Urbana, IL, United States
- 4Seattle Structural Genomics Center for Infectious Disease, Seattle Children’s Hospital, Seattle, WA, United States
- 5Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign, Urbana, IL, United States
The agglutinin-like sequence (Als) proteins are best-characterized in Candida albicans and known for their role in adhesion of the fungal cell to host and abiotic surfaces. ALS sequences are often misassembled in whole-genome sequence data because each species has multiple ALS loci that contain similar sequences, most notably tandem copies of highly conserved repeated sequences. The Candida parapsilosis species complex includes Candida parapsilosis, Candida orthopsilosis, and Candida metapsilosis, three distinct but closely related species. Using publicly available genome resources, de novo genome assemblies, and laboratory experimentation including Sanger sequencing, five ALS genes were characterized in C. parapsilosis strain CDC317, three in C. orthopsilosis strain 90–125, and four in C. metapsilosis strain ATCC 96143. The newly characterized ALS genes shared similar features with the well-known C. albicans ALS family, but also displayed unique attributes such as novel short, imperfect repeat sequences that were found in other genes encoding fungal cell-wall proteins. Evidence of recombination between ALS sequences and other genes was most obvious in CmALS2265, which had the 5′ end of an ALS gene and the repeated sequences and 3′ end from the IFF/HYR family. Together, these results blur the boundaries between the fungal cell-wall families that were defined in C. albicans. TaqMan assays were used to quantify relative expression for each ALS gene. Some measurements were complicated by the assay location within the ALS gene. Considerable variation was noted in relative gene expression for isolates of the same species. Overall, however, there was a trend toward higher relative gene expression in saturated cultures rather than younger cultures. This work provides a complete description of the ALS genes in the C. parapsilosis species complex and a toolkit that promotes further investigations into the role of the Als proteins in host-fungal interactions.
Introduction
The study of adhesive mechanisms is pivotal in pathogenic microbiology because adhesion promotes host colonization, which is a foothold in the process of causing disease. The agglutinin-like sequence (ALS) gene family encodes cell-surface glycoproteins that are involved in adhesion of fungal cells to host and abiotic surfaces (Hoyer and Cota, 2016). ALS genes and their encoded proteins are best characterized in C. albicans. A binding cavity located within the N-terminal Als domain is responsible for adhesion to peptide ligands (Lin et al., 2014). Another hallmark of C. albicans Als proteins is an often-extensive central domain of tandemly repeated sequences that are rich in serine, threonine, and sometimes proline.
The rapid pace at which draft genome sequences are developed is a benefit to the research community. At this time, however, most available draft genomes were assembled from short-read DNA sequences of no more than a few hundred nucleotides (e.g., Riccombeni et al., 2012; Pryszcz et al., 2015). Considering that the repeated DNA unit in ALS genes is 108 bp and that the tandemly repeated copies of highly similar sequences may extend for several kilobases, it is no surprise that many genome assemblies fail within the ALS coding regions. In some published fungal genomes, ALS genes may be assembled accurately, while in others, it is not possible to calculate basic information such as the number of loci present in the gene family. Long-read DNA sequence technology provides the opportunity to create a larger template upon which the more-accurate short-read sequences can be assembled (Madoui et al., 2015), providing a new approach to characterizing the ALS genes in pathogenic fungi.
The focus of this work is the Candida parapsilosis species complex: Candida parapsilosis, Candida orthopsilosis, and Candida metapsilosis. The three species are closely related; recognition as separate species is a relatively recent event (Tavanti et al., 2005). The species are gaining importance as pathogens, with adhesion and colonization mechanisms playing a central role in the disease process (Bliss, 2015). As such, it is important to understand the nature of the ALS family, its variability across alleles within and among various strains, and the relative gene expression patterns that may reveal which of the genes could play the largest role in the adhesion process.
Using a combination of published sequence data and de novo approaches, we establish the nature of the ALS genes in the C. parapsilosis species complex, their allelic variability, and expression patterns. The resulting data reveal novel sequences and recombination between genes that demonstrate the blurring of gene family definitions that were developed in C. albicans. The work provides a toolkit that can be used to further knowledge of the ALS genes, as well as highlights mechanisms for maintaining the diversity of cell-surface adhesins in the pathogenic fungi.
Materials and Methods
Microbial Strains
Table 1 shows the microbial strains used in this study. Fungal identifications were validated using RAPD methodology (Tavanti et al., 2005). Products were separated on 1% agarose/Tris-Acetate-EDTA gels and visualized by ethidium bromide staining. Routine PCR analyses were conducted as described previously (Green et al., 2004).
Table 1. C. parapsilosis, C. orthopsilosis, and C. metapsilosis strains and sources.
Publicly Available Genome Sequences
Previously published genome sequences were used for this study. These included C. parapsilosis strains CDC317 (Butler et al., 2009; GenBank accession number ASM18276v2; GCA_000182765.2), CBS6318 (GCA_0000982555.2), and GA1 (GCA_0000982675.1). Strain CDC317 was used as the representative genome sequence. A genome sequence was available for C. orthopsilosis strain 90-125 (GCA_000315875.1). A new 90–125 sequence was derived from short-read and long-read sequence data as described previously (Lombardi et al., 2019; PQBP00000000). Sequences for strains MCO456 (GCA_900002835.2) and AY2 (GCA_000304155.1) were also examined. The C. metapsilosis genome sequence under accession number CBZN0200000000 was included in the analysis although it was compiled from more than one isolate (Pryszcz et al., 2015). A new C. metapsilosis genome sequence was generated from short-read and long-read sequence data as described below.
Genome Sequence Data Collection, Assembly, and Validation
A new genome sequence was generated for C. metapsilosis strain ATCC 96143. Genomic DNA initially was extracted from cells that were grown to saturation (16 h) at 37°C in YPD medium (per liter: 10 g yeast extract, 20 g peptone, 20 g glucose) with 200 rpm shaking. The genome assembly from this preparation was of lower quality than desired, so the method was repeated, this time with a culture that was grown to mid-log phase. Mid-log-phase cells appeared to lack an extracellular polysaccharide that was present in the saturated culture, presumably leading to a cleaner DNA preparation that did not clog the pores of the MinION sequencer (see below). The mid-log phase sequence data replaced those from the saturated culture prep in the GenBank deposit (PQNC00000000) and were used in the analysis presented here.
DNA was extracted using the method of Sherman et al. (1986). Briefly, cells were treated with zymolyase (MP Biomedicals) to form spheroplasts that were lysed with sodium dodecyl sulfate. Gentle mixing by inversion was used to handle the spheroplasts, and during subsequent phenol extraction and isopropyl alcohol precipitation of the DNA. Verification of the quality of the high-molecular-weight DNA was assessed using agarose gel electrophoresis.
C. metapsilosis DNA libraries were constructed and sequenced at the Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign. Data were derived using Illumina (short-read) and Oxford Nanopore (long-read) methods. MiSeq shotgun genomic libraries were prepared with the Hyper Library construction kit (Kapa Biosystems). The library was quantitated by qPCR and sequenced on one MiSeq flowcell for 151 cycles from each end of the fragment using a MiSeq 300-cycle sequencing kit (version 2). FASTQ files were generated and demultiplexed with the bcl2fastq Conversion Software (Illumina, version 2.17.1.14). MiSeq reads were quality trimmed using Trimmomatic (Bolger et al., 2014) with the parameters “LEADING:30 TRAILING:30” prior to assembly.
For Oxford Nanopore long-read sequencing, 1 μg of genomic DNA was sheared in a gTube (Covaris, Woburn, MA, United States) for 1 min at 6,000 rpm in a MiniSpin plus microcentrifuge (Eppendorf, Hauppauge, NY, United States). The sheared DNA was converted into a shotgun library with the LSK-108 kit from Oxford Nanopore, following their manual. The library was sequenced on a SpotON R9.4 RevC flowcell for 48 h using a MinION MK 1B sequencer.
Basecalling was performed with the software Albacore version 2.3.1 from Oxford Nanopore. Sixty nucleotides were removed from both ends of each Oxford Nanopore read, followed by additional trimming using a Github checkout (commit 92c0b65f) of Porechop (Wick et al., 2017a) to remove reads with potential internal barcodes which were likely chimeric. Only reads longer than 800 nt were used in the final assembly. Canu v1.7 (Koren et al., 2017) was used for assembly with the following parameters: ‘canu -p asm -d C_meta_default genomeSize = 14m useGrid = false -nanopore-raw c_metapsilosis.qualtrim.clean.fastq.gz.’
Oxford Nanopore reads were then aligned against the assembly using minimap v2.8 (Li, 2018), and the alignment was then used to polish the assembly using nanopolish v 0.9.0 (Senol Cali et al., 2018). Quality-trimmed MiSeq data were used to iteratively polish the assembly using a Python script based on the iterative polishing method utilized during assembly with the hydrid assembler Unicycler (Wick et al., 2017b) using Pilon v1.22 for error correction (Walker et al., 2014). Haplomerger2 (release 20180603; Huang et al., 2017) was then used to both locate and resolve potential haplotypes, generating a haploid reference genome with a separate assembly with alternate haplotypes.
Ambiguities in the genome sequence data were corrected by PCR amplification of the region and Sanger sequencing of the product. Supplementary Table S1 lists PCR primer sequences. Supplementary File S1 includes details regarding the computational analyses and characteristics of the resulting C. metapsilosis genome sequence.
Tools for Identification of ALS Genes and Features of Predicted Als Proteins
BLAST1 was used to identify potential ALS genes and Als proteins in the various genome sequences. C. albicans sequences used as queries included CaALS1 (L25902), CaALS2 (AH006927), CaALS3 (AY223552), CaALS4 (AH006929), CaALS5 (AY227440), CaALS6 (AY225310), CaALS7 (AF201684), CaALS9-1 (AY269423), and CaALS9-2 (AY269422). As additional ALS genes were located, those sequences were also used as BLAST queries until no additional new sequences were detected.
Secretory signal peptides were detected in putative Als proteins using the SignalP 4.1 Server2 (Nielsen, 2017). Putative GPI anchor addition sites were identified using the big-PI Predictor3 (Eisenhaber et al., 1999). Nucleotide sequence translation, multiple sequence alignments, and other general bioinformatic processes used the European Bioinformatics Institute (EMBL-EBI) tools and data resources4 (Cook et al., 2017).
Phylogenetics Analysis
Nucleotide sequences from 21 concatenated genes from the ALS family were analyzed using maximum likelihood (ML) and Bayesian methods. Alignment of genes was conducted in SeaView v4.6.1 (Gouy et al., 2010) using Muscle v3.8 (Edgar, 2004). Ambiguous regions were removed using Gblocks v0.91b (Castresana, 2000), allowing for less strict flanking regions, gap positions within the final blocks, and smaller final blocks. Maximum likelihood analysis was conducted using PhyML (Guindon and Gascuel, 2003) under the GTR substitution model with four rate classes and optimized invariable sites based on the results from jModelTest 2.0 (Posada, 2008). The best nearest neighbor interchange and subtree pruning and regrafting tree improvement was implemented on the unrooted BioNJ starting tree and branch support was determined with 1,000 non-parametric bootstrap replicates. Bayesian analysis was conducted using MrBayes 3.2.2 on XSEDE (3.2.6) through the CIPRES Science Gateway (Miller et al., 2010). Bayesian analysis was conducted using the GTR + I + G model with four rate classes and four independent chains and ran for 10 million generations sampling every 1,000 trees with the first 25% of trees discarded as burn-in. A consensus tree with the remaining 7,500 trees was produced using PAUP 4.0a (Swofford, 2002). Nodes with ≥70% bootstrap support and ≥95% Bayesian posterior probability were considered significantly supported (Alfaro et al., 2003).
Structural Analysis
Structural homology models were calculated using Phyre2 (Kelly et al., 2015). Sequence alignments were created to identify structurally conserved residues and models were built using the Threading option to maintain the integrity of the peptide-binding cavity (PBC) and allow for comparison of side-chain residue changes between sequence variants. Structures were compared and analyzed using Coot (Emsley et al., 2010) and Maestro (Schrodinger LLC, Portland, OR, United States).
Growth Conditions for Gene Expression Analysis
All fungal isolates were stored as pure cultures in 30% glycerol at -80°C. Isolates were streaked to YPD agar as needed. YPD plates were stored at 4°C for no more than 1 week prior to use. Some growth conditions were used for assessing ALS gene expression because they mimicked conditions used in another publication. Other selected conditions were intended to detect large trends in differential gene expression that are characteristic of the C. albicans ALS family (e.g., assessing gene expression at early or late time points in different growth media; Hoyer et al., 2008). In each experiment, cells were collected from the specified growth condition, pelleted by centrifugation, and the pellet flash frozen in a dry ice-ethanol bath. Frozen pellets were stored at -80°C until RNA was extracted.
C. orthopsilosis strain 90-125 was grown to mimic the conditions described by Lombardi et al. (2019). A single colony from a YPD plate was inoculated into 10 ml YPD liquid medium and incubated for 24 h at 30°C and 200 rpm shaking. 0.5 ml of the culture was inoculated into 20 ml fresh YPD medium and incubated for either 1 h or 24 h under the same temperature and agitation conditions. Cells were harvested by centrifugation, flash frozen in dry ice/ethanol, and stored at -80°C. Duplicate cultures were grown on three separate occasions.
A similar approach was used for gene expression analysis of the C. parapsilosis genome isolate (CDC317) and the type strain (ATCC 22019). A single colony from a YPD plate was inoculated into 10 ml YPD medium and grown for 24 h at 30°C and 200 rpm shaking. C. parapsilosis cells were also grown using the liquid filament induction conditions of Lackey et al. (2013). In preliminary experiments, growth of strain ATCC 22019 for 24 h produced slightly elongated cells while strain CDC317 appeared as a typical budding yeast. One colony was inoculated into 20 ml YPD at 30°C and 200 rpm shaking for 24 h. Cells were pelleted by centrifugation and washed twice in sterile water. A culture of SC medium [6.7 g/liter yeast nitrogen base without amino acids, supplemented 2% glucose and 2 g of complete amino acid mixture as defined by Lackey et al. (2013)] containing 20% fetal bovine serum (Gibco; 26140-079) was inoculated at an OD600 of 0.6. The culture was incubated for 2 h at 37°C and 200 rpm shaking. Duplicate cultures were grown on three separate occasions.
For C. metapsilosis, genome strain (ATCC 96143) and strains 61, 397, and 482 were studied. A single colony from the YPD plate was inoculated into 10 ml YPD liquid medium and incubated for 24 h at 37°C and 200 rpm shaking. Cells were harvested from the 24-h culture, replicate 2.5-ml aliquots were removed from the culture; cells were collected, frozen, and stored as described above. The remaining 5 ml of culture was washed in Dulbecco’s Phosphate Buffered Saline (BioWhittaker; 17-512Q), diluted, and counted using a hemocytometer. The total cell number was resuspended in RPMI 1640 tissue culture medium (Gibco; 11875-135) at a density of 5 × 107 cells/ml. The culture was incubated for 1 h at 37°C and 200 rpm shaking. Cells were collected by filtration across a 0.45-micron membrane (GVS Life Sciences; 1213776). The filter was placed into a 50-ml conical centrifuge tube, flash frozen, and stored at -80°C.
Design and Validation of TaqMan Assays to Quantify Relative ALS Gene Expression Levels
All steps for developing, validating, and running the TaqMan assays followed the Guide to Performing Relative Quantitation of Gene Expression Using Real-Time Quantitative PCR (Applied Biosystems Inc., 2008). TaqMan primers and probes were designed using PrimerQuest (Integrated DNA Technologies). Primers had an optimal Tm of 61°C, GC content of 40% and 25-nt length. Probes had an optimal Tm of 70°C, GC content of 40% and 28-nt length. To identify divergent sequences that could be exploited for TaqMan assay design, Clustal Omega5 was used to align the nucleotide sequences from the 5′ end of each ALS gene. An amplicon size of 140 bp was sought; some products were larger or smaller, depending on the availability of unique sequences among the genes. PCR efficiencies were measured for each TaqMan assay; each was between 90 and 110% (Supplementary Table S2).
Because some genes were so similar within a given species, specificity of the TaqMan assays was demonstrated experimentally (Supplementary Table S3). Fragments were PCR amplified and cloned into the pJet1.2/blunt vector and transformed into E. coli INVαF’ (Thermo Fisher). The accuracy of cloned fragments was verified using Sanger sequencing (Roy J. Carver Biotechnology Center, Urbana, IL, United States). Dilutions of purified, quantified plasmid DNA were used as templates to demonstrate specificity of TaqMan assays (see section “Results”).
Control TaqMan assays featured the ACT1 and TEF1 genes from each species. Primers (Supplementary Table S1) were used to amplify the genes from each species and validate its sequence using the Sanger method. Some TaqMan control assays were designed to recognize a single species (Supplementary Table S2). Control assays that recognize multiple species were also designed with an eye on cost savings, as use of a common control would eliminate the need to order one control assay per species.
Real-Time PCR Analysis of Gene Expression
RNA was extracted using the hot acidic phenol method of Collart and Oliviero (1993) with the addition of approximately 100 μl volume of 0.5 mm sterile, baked, acid-washed glass beads to each tube. Nucleic acid concentration was measured using a NanoDrop spectrophotometer (Thermo Scientific). The presence of contaminating genomic DNA in RNA preparations was detected by PCR using ITS4 and ITS5 primers6 (White et al., 1990). The reaction template was 25 ng of RNA in a 25 μl reaction containing PCR buffer with 2.5 mM of MgCl2, 1 μM of each primer, 0.5 mM of dNTP, and Taq polymerase (Invitrogen; 10342-020). PCR conditions were 93°C for 5 min followed by 40 cycles of 93°C for 30 s, 53°C for 30 s, 72°C for 1 min with a 7-min final extension at 72°C. DNase (Invitrogen; AM2224) was used according to manufacturer’s instructions to digest contaminating genomic DNA. Often, multiple rounds of DNase digestion were required to eliminate DNA below the detection limit of the PCR assay. Each RNA sample was cleaned using an RNeasy Mini spin column (Qiagen; 74104). RNA preparations were quantified and their quality assessed using Qubit (Invitrogen; Q10210) and/or the 2100 Bioanalyzer (Agilent Technologies). Aliquots were stored at -80°C until used for real-time gene expression assays.
The Superscript III cDNA synthesis kit (Invitrogen; 18080-051) was used to make cDNA according to manufacturer’s instructions. The amount of RNA added to the cDNA synthesis reaction was titrated by adding RNA dilutions to the cDNA synthesis reaction, then using the reaction products as the basis for real-time PCR quantification of a control gene. The Ct value for 0.2 μg RNA was in the linear portion of the graph of RNA amount vs. Ct, so 0.2 μg RNA was used for all cDNA synthesis reactions.
The TaqMan assay reaction mixture was 15 μl in PrimeTime Gene Expression Master Mix (Integrated DNA Technologies) with 500 nM of each primer and 250 nM of probe. Synthesized cDNA was diluted 1:5 and 5 μl added to each reaction. PCR amplification used an ABI 7500 Real-Time PCR System (Applied Biosystems) with 95°C for 3 min followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Triplicate reactions were run for the C. metapsilosis RNA samples while duplicate reactions were run for the other species to fit as many reactions from the same species onto a single PCR plate.
Combining data across plates relied on an identical set of controls that were present on each: CmACT1 and CmTEF1 for C. metapsilosis RNA, and CmCoCpACT1 and CmCoCpTEF1 for C. parapsilosis and C. orthopsilosis samples. TaqMan analyses for comparison to SYBR Green data used only the CmCoCpACT1 control since the published SYBR Green assay relied only on ACT1 as a control (Lombardi et al., 2019). The default threshold for each primer was applied and ΔCt calculated using Expression Suite software v1.1 (Thermo Fisher Scientific).
A control sample of RNA without reverse transcriptase was analyzed using the CmCoCpACT1 TaqMan assay to monitor background amplification caused by residual genomic DNA. In all reactions but one (C. metapsilosis 1-Y-61; Ct = 38.7), no detectable product was generated during 40 amplification cycles.
Simple calculation of means and standard errors of the mean used Prism7 software (GraphPad). The statistical significance of results was assessed using a mixed-model analysis of variance (PROC MIXED in SAS 9.4; SAS Institute Inc., Cary, NC, United States). Separation of means used the LSMEANS option.
Results
Number and Nature of ALS Genes in Each Species of the C. parapsilosis Complex
Three C. parapsilosis genome sequences (strains CDC317, CBS6318, GA1) were found in the National Center for Biotechnology Information (NCBI) database7. The annotated CDC317 sequence was also located in the Candida Genome Database8. BLAST using C. albicans ALS gene sequences as queries revealed five ALS genes in strain CDC317 (Figure 1). Four of the ALS genes were located on contig 006372 and transcribed in the same direction; the fifth gene was located on contig 006139 (Figure 1). The same five ALS genes were detected in strain CBS6318 and their arrangement appeared similar to strain CDC317. Only two ALS genes were apparent in the GA1 genome sequence: one was most similar to CpALS4800 while the other most closely resembled CpALS660.
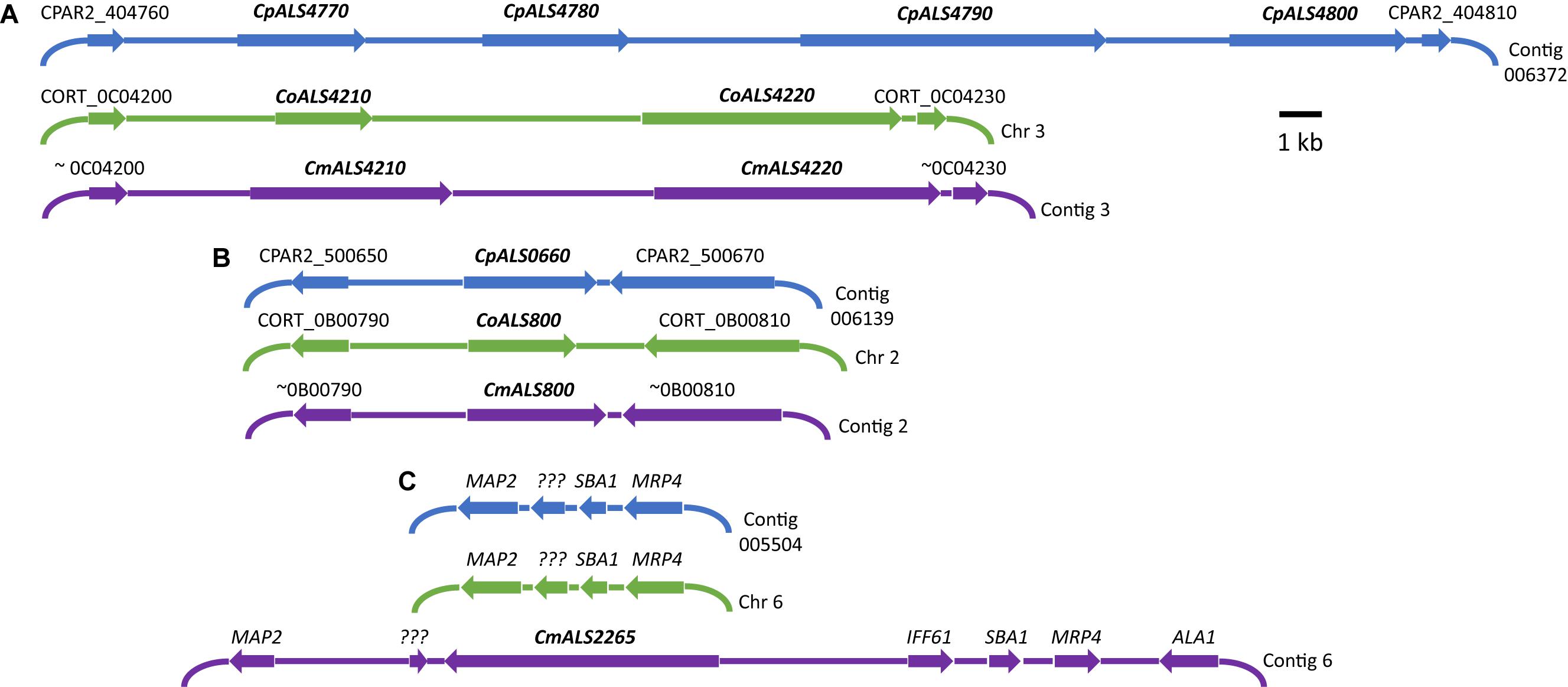
Figure 1. Schematic of chromosomal locations of ALS genes in C. parapsilosis, C. orthopsilosis, and C. metapsilosis, drawn to scale. (A) Location of contiguous ALS genes on C. parapsilosis contig 006372, C. orthopsilosis chromosome 3, and contig 3 from the C. metapsilosis genome assembly PQNC00000000. ALS gene names are in bold italics type. Arrows show the direction of transcription for each gene. Flanking sequences were identified to indicate gene order conservation among the three species. Names of the flanking ORFs were taken from the Candida Genome Database (www.candidagenome.org) and/or GenBank. (B) Location of genes CpALS0660 (C. parapsilosis contig 006139), CoALS800 (C. orthopsilosis chromosome 2), and CmALS800 (C. metapsilosis contig 2). Features were drawn similarly to (A). (C) Location of the fourth C. metapsilosis ALS gene (CmALS2265) on contig 6. This region was orthologous to C. parapsilosis contig 005504 and C. orthopsilosis chromosome 6 but contained approximately 16 kb of extra sequence. The region contained the reading frame for CmIff61 (GenBank accession number MK205443) that encoded a 341-amino acid protein not present in the other species. Gene names were shown above other coding regions. ??? encoded a hypothetical protein (CPAR2_601270; CORT_0F02270) that was conserved in all three species, but shorter in C. metapsilosis. MAP2 = CPAR2_601280, CORT_0F02280; SBA1 = CPAR2_601260, CORT_0F02260; MRP4 = CPAR2_601250, CORT_0F02250. Some genes in this region (ALA1, MRP4, SBA1) were reminiscent of those that are located near CaALS5, CaALS1, and CaALS9 on C. albicans chromosome 6 (Zhao et al., 2003).
ALS genes in C. orthopsilosis were noted originally in the sequence of strain 90-125 that was available on the Candida Gene Order Browser (Maguire et al., 2013) and in the NCBI database. Because these sequences were incomplete due primarily to misassembled repeated sequence regions, we generated a new genome sequence using both Oxford Nanopore (long-read) and Illumina data (Lombardi et al., 2019). In collaboration with the laboratory of Arianna Tavanti (Pisa, Italy), we deduced the ALS genes and validated their sequence and variability. Strain 90-125 encoded three C. orthopsilosis genes (Figure 1). Two were contiguous on chromosome 3 and transcribed in the same direction while the third gene was on chromosome 2, reminiscent of the ALS gene arrangement in C. parapsilosis. Flanking regions of the genome were orthologous in each species. Genome sequences for two more C. orthopsilosis strains were available on the NCBI database: MCO456 and AY2. BLAST searches with C. albicans and C. parapsilosis ALS queries revealed multiple “hits,” however, the sequences were not assembled completely enough to determine an accurate number of ALS loci.
A genome sequence for C. metapsilosis was available in the NCBI database (accession CBZN0200000000) and was reported to encode a single ALS gene (Pryszcz et al., 2015). We generated a novel genome assembly for strain ATCC 96143 using the long-read and Illumina technologies. The Oxford Nanopore flowcell produced over 4.3 Gb of sequence from 670,717 reads. The minimum read length was 1,000 bp while the maximum was 89,846 bp. The mean read length was 6,476 bp. The majority of reads were 6–30 kb in length, with the longest at 89 kb. The genome assembly separated into diploid chromosomes and was deposited in GenBank as assembly PQNC00000000. A summary of computational analyses and characteristics of the resulting genome sequences was provided in Supplementary File S1.
BLAST using previously identified ALS genes (including those from C. albicans, C. parapsilosis, and C. orthopsilosis) revealed four ALS genes in C. metapsilosis (Figure 1). Three were orthologs of the C. orthopsilosis ALS genes, reflecting the close relationship between the species (Tavanti et al., 2005). The fourth C. metapsilosis ALS gene did not have an ortholog in either C. parapsilosis or C. orthopsilosis. Figure 1 shows the corresponding genome region where the fourth C. metapsilosis ALS gene was located and insertion of a 16-kb DNA fragment that was not present in either C. parapsilosis or C. orthopsilosis. BLAST of the published C. metapsilosis genome assembly (CBZN0200000000) showed the same four genes as detected in the new ATCC 96143 assembly.
The 20 diploid contigs for the new C. metapsilosis ATCC 96143 genome assembly were arranged in descending order by size; similar data were examined for the C. orthopsilosis and C. parapsilosis genomes (Table 2). Color coding indicated the location of ALS genes from Figure 1A (red), Figure 1B (blue), and Figure 1C (purple). Variable chromosomal localizations for the ALS genes suggested the potential for large karyotypic differences between these three closely related species. For example, the ALS gene cluster was located on chromosome 3 in C. orthopsilosis, and on the fourth-largest contig in C. parapsilosis. Although CpALS0660 and CoALS800 appear to be orthologs, one is found on the sixth-largest contig in C. parapsilosis and on the second-largest in C. orthopsilosis. Karyotypes for these species were not apparent in the literature, nor were tools such as a physical map that was essential for guiding assembly of the C. albicans genome (Chu et al., 1993).
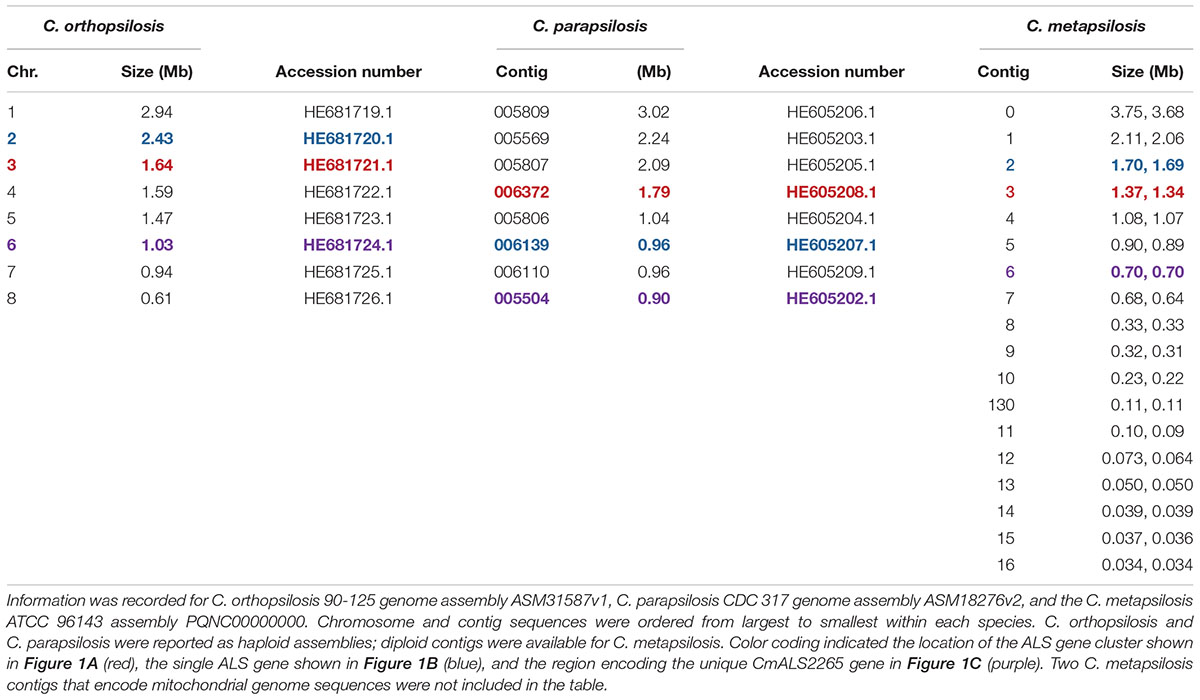
Table 2. Chromosomes and contigs for the C. parapsilosis, C. orthopsilosis, and C. metapsilosis genome sequences.
Identification of new ALS genes raised the question of how they should be named. ALS genes in C. albicans are numbered starting with “1” and include ALS1 through ALS7, and ALS9 (Hoyer et al., 2008). Because the new genes presented here were different from the C. albicans ALS genes, we developed a new naming scheme based on Figure 1. The gene names (Table 3) reflected the close relationships between ALS genes in the C. parapsilosis species complex. The names accurately reflected the arrangement of genes on the various contigs, and the annotation available in the first genome assembly deposited in the NCBI database. For example, the names CpALS4770 and CpALS4780 suggested that the ORFs were ALS genes from C. parapsilosis. The consecutive numbers (4770 and 4780) were taken directly from the genome annotation and indicated that the genes were contiguous. Literature precedent for these ideas was already present (Table 3).
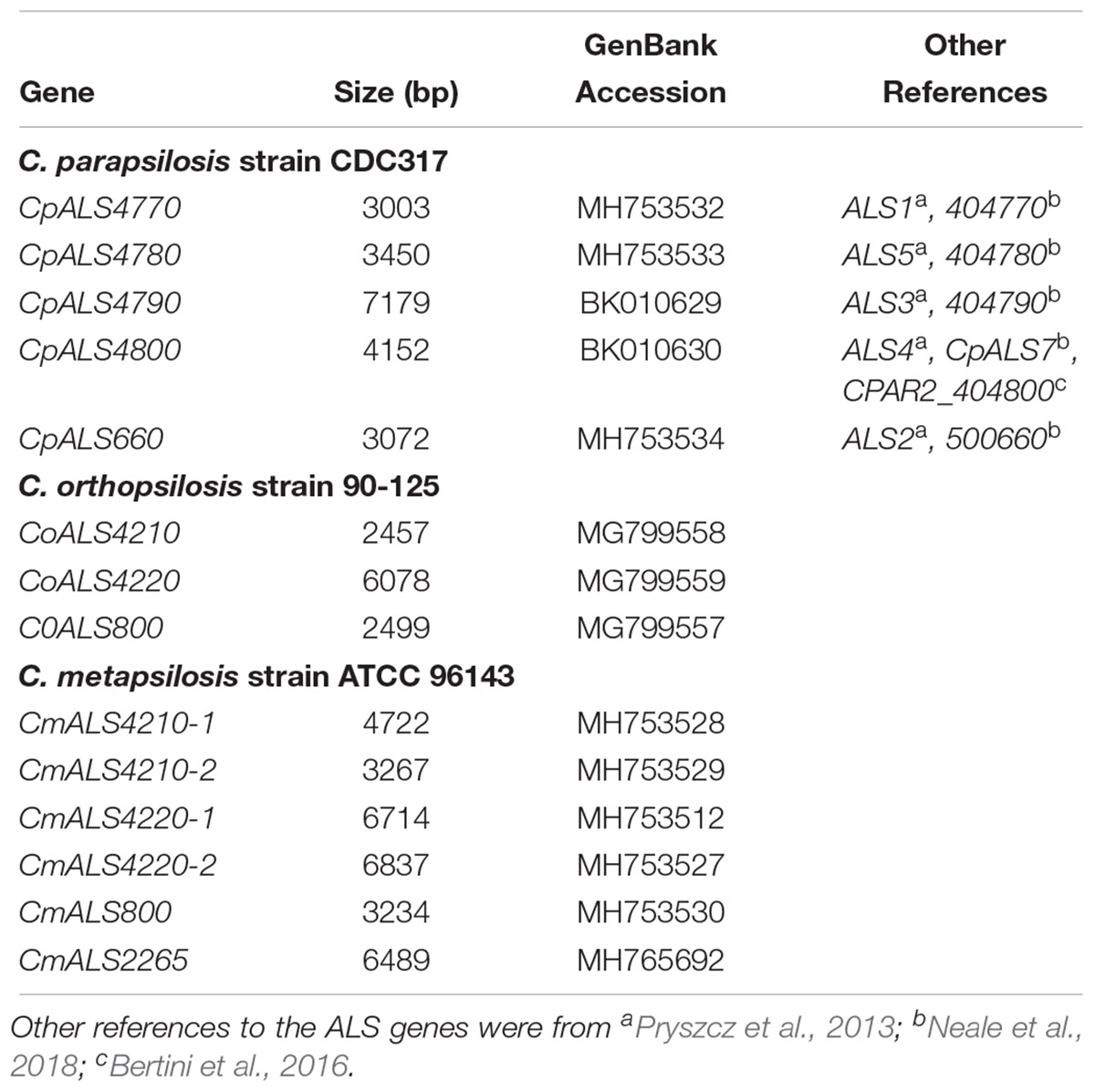
Table 3. ALS genes from the Candida parapsilosis species complex and their GenBank accession numbers.
Annotation of the C. orthopsilosis assembly proceeded somewhat differently than for C. parapsilosis (Riccombeni et al., 2012), leading to a different numbering scheme (Figure 1). We preserved the genome annotation tags in the names of the ALS genes (e.g., CoALS800 came from CORT_0B00800). This scheme was carried forward to the gene names for the relatively unannotated C. metapsilosis genome assembly. Because the C. orthopsilosis and C. metapsilosis species were more closely related to each other than either was to C. parapsilosis (Tavanti et al., 2005), C. metapsilosis ALS gene names were matched to the C. orthopsilosis names. The name of the new gene, CmALS2265, reflected its location in the C. metapsilosis genome, while paying homage to the C. orthopsilosis assembly (i.e., it occupied the orthologous physical location between CORT_0F02260 and CORT_0F02270).
Als Protein Structure Encoded by the C. parapsilosis Species Complex ALS Genes
ALS gene sequences were translated to amino acids, then used to draw a schematic of Als protein features (Figure 2). Color coding indicated similarities between the proteins. Visualization of the protein sequences provided additional insight into the gene arrangement displayed in Figure 1. For example, CpALS4790 and CpALS4800 each encoded proteins with a central tandem repeat domain, while proteins predicted by CpALS4770 and CpALS4780 were very similar to each other. These observations suggested that CpALS4780 and CpALS4800 arose by duplication of genes CpALS4770 and CpALS4790, respectively. CoALS4210 and CmALS4210 were orthologs of CpALS4770 while a similar relationship was evident for CoALS4220, CmALS4220, and CpALS4790. Proteins predicted from genes CpALS0660, CoALS800, and CmALS800 were all very similar, consistent with the conserved genomic context for these ORFs.
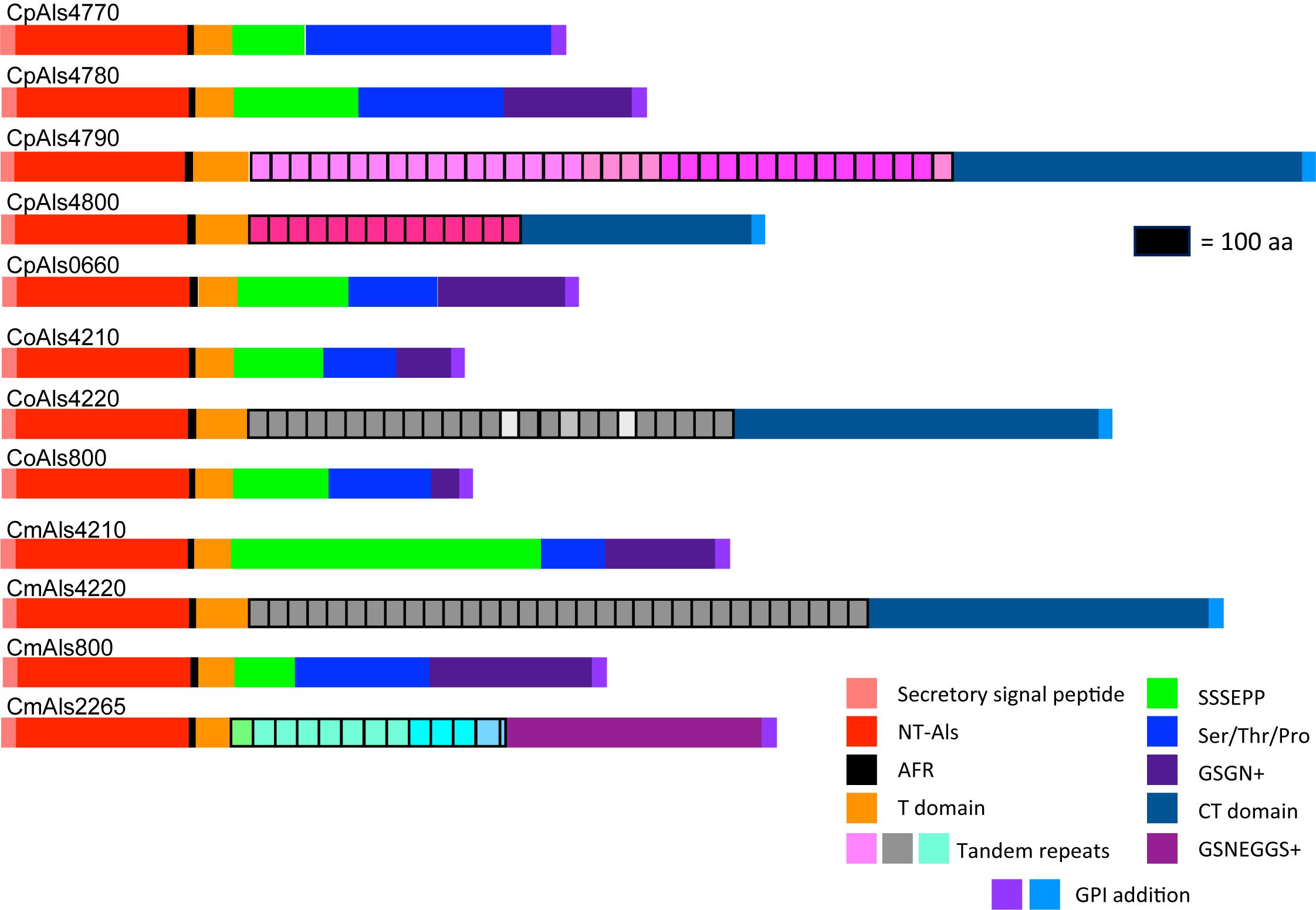
Figure 2. Schematic of proteins predicted from the ALS genes of the C. parapsilosis species complex. Each predicted protein had a secretory signal peptide, a classical NT-Als domain with eight conserved Cys residues to direct folding, an amyloid-forming region (AFR; Garcia et al., 2011), a Thr-rich sequence (T domain), a C-terminal domain rich in Ser/Thr, and a signal for addition of a GPI anchor that directs the mature protein to a final localization linked to β-1,6-glucan in the fungal cell wall (Lu et al., 1994). Only 5 of the 12 proteins included a central domain of tandemly repeated sequences like those found in C. albicans Als proteins (Hoyer et al., 2008). Different colors of the repeated units indicated differences in consensus sequence; shading within the same protein indicated repeat units that varied in the number of amino acids. The other proteins had regions of short, imperfect repeated sequences such as Ser-Ser-Ser-Glu-Pro-Pro (SSSEPP) and/or Gly-Ser-Gly-Asn (GSGN). CmAls2265 had the NT-Als domain attached to tandemly repeated sequences from the Iff/Hyr family (Bates et al., 2007; Boisramé et al., 2011) and a C-terminal region more-characteristic of Iff/Hyr proteins than C. albicans Als proteins.
Each predicted protein from the C. parapsilosis species complex (Figure 2) fulfilled various criteria for inclusion in the Als family. The proteins had a secretory signal peptide and a site for GPI anchor addition, consistent with a final localization in the fungal cell wall, linked to β-1,6-glucan (Lu et al., 1994). The high percentage of Ser and Thr in each protein, particularly in repeated sequences and toward the C-terminal end, also was consistent with composition of C. albicans Als proteins (Hoyer et al., 2008). Most importantly, each protein had an N-terminal domain that was similar to C. albicans NT-Als3 where adhesive function resides (Lin et al., 2014). NT-Als folding is driven by eight conserved Cys residues (Salgado et al., 2011), that were found in each predicted protein from the C. parapsilosis species complex.
NT-Als adhesive function is due to a PBC that can accommodate up to six C-terminal amino acids from peptide ligands in an extended conformation (reviewed in Hoyer and Cota, 2016). Als proteins recognize diverse peptide ligands with different Als proteins demonstrating higher-affinity interactions for specific sequences. Nucleotide sequences encoding NT-Als from the newly characterized genes were included in a phylogenetic analysis with the corresponding regions from the C. albicans ALS genes (Figure 3). C. albicans sequences grouped together in a manner that reflected known protein function (reviewed in Hoyer and Cota, 2016).
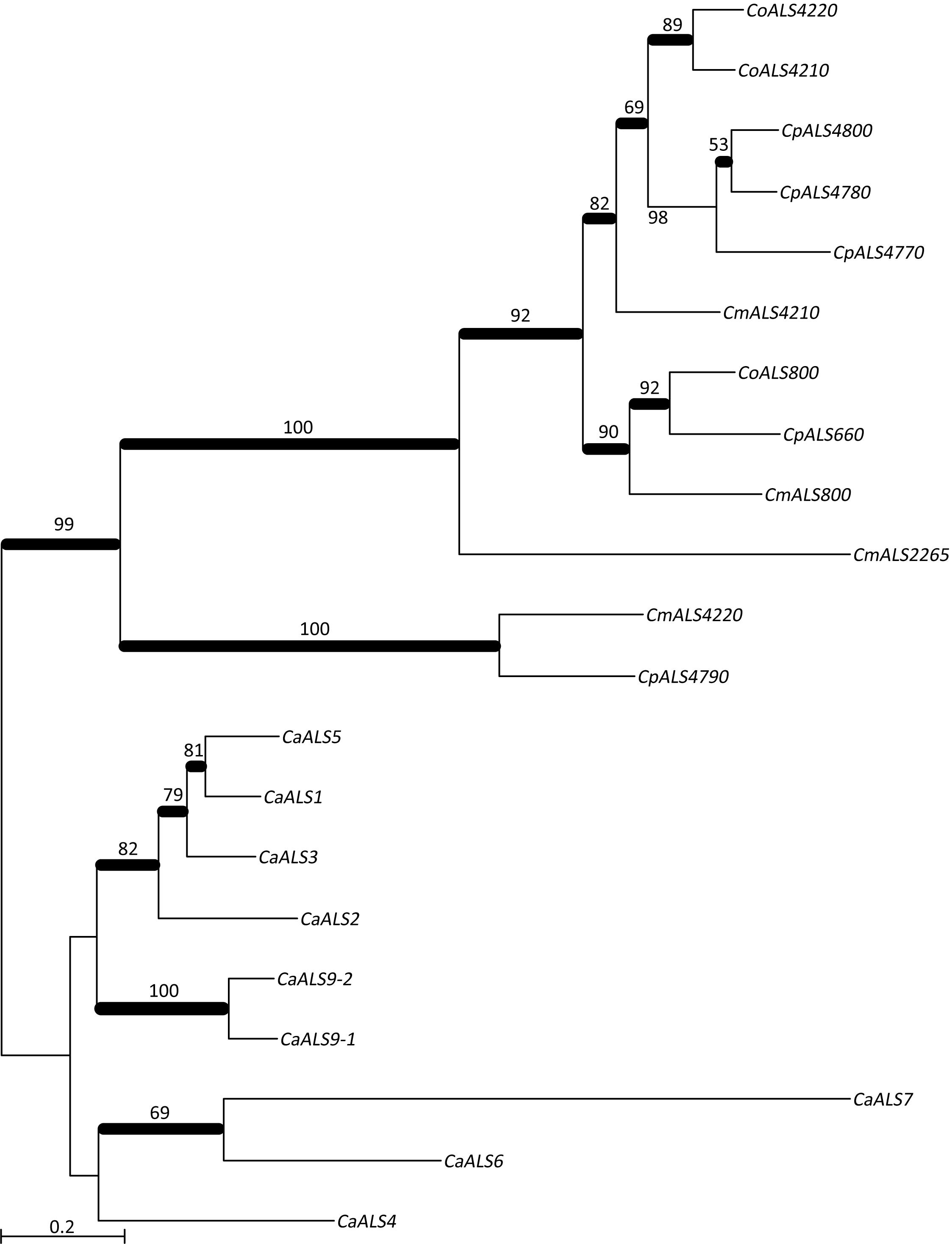
Figure 3. Phylogenetic tree of ALS sequences from C. albicans and the C. parapsilosis species complex. The tree was drawn using a ML analysis after removing ambiguous regions using Gblocks. Thickened branches indicated Bayesian posterior probabilities >95% while the numbers above the branches were bootstrap values >50%. The tree showed strong support for many branches.
Based on positions of the genes in the phylogenetic tree, a few examples were selected for further examination of variation within the predicted PBC. Models of CpAls4790, CoAls4210, and CmAls2265 were created based on the structure of C. albicans Als9-2 (CaAls9-2) bound to the C-terminal residues of human fibrinogen gamma peptide (Protein Data Bank ID 2y7n; Salgado et al., 2011). For CaAls9-2, peptide binding is anchored by a salt bridge with an invariant Lys59 and further stabilized by main chain hydrogen bonding interactions residues S170, T293, and W295. The peptide has an extended conformation with side chains oriented alternatively on opposite sides of the peptide backbone similar to residues in a β strand. The amino acid side chains are inserted into conserved pockets of the protein. Amino acids lining the cavity are variable among known Als protein sequences, presumably modulating binding specificities among proteins in the Als family (Lin et al., 2014).
Peptide-binding cavities for the new Als proteins were analyzed by sequence and structural comparisons (Figure 4A). Differences in size and electrostatic potential were assessed to compare peptide-binding capacities among the NT-Als proteins. The PBC was considered as multiple pockets (called C1 through C5) to facilitate this analysis. The sixth amino acid was not included in the analysis since the position was highly solvent-exposed in the CaAls9-2 structure.
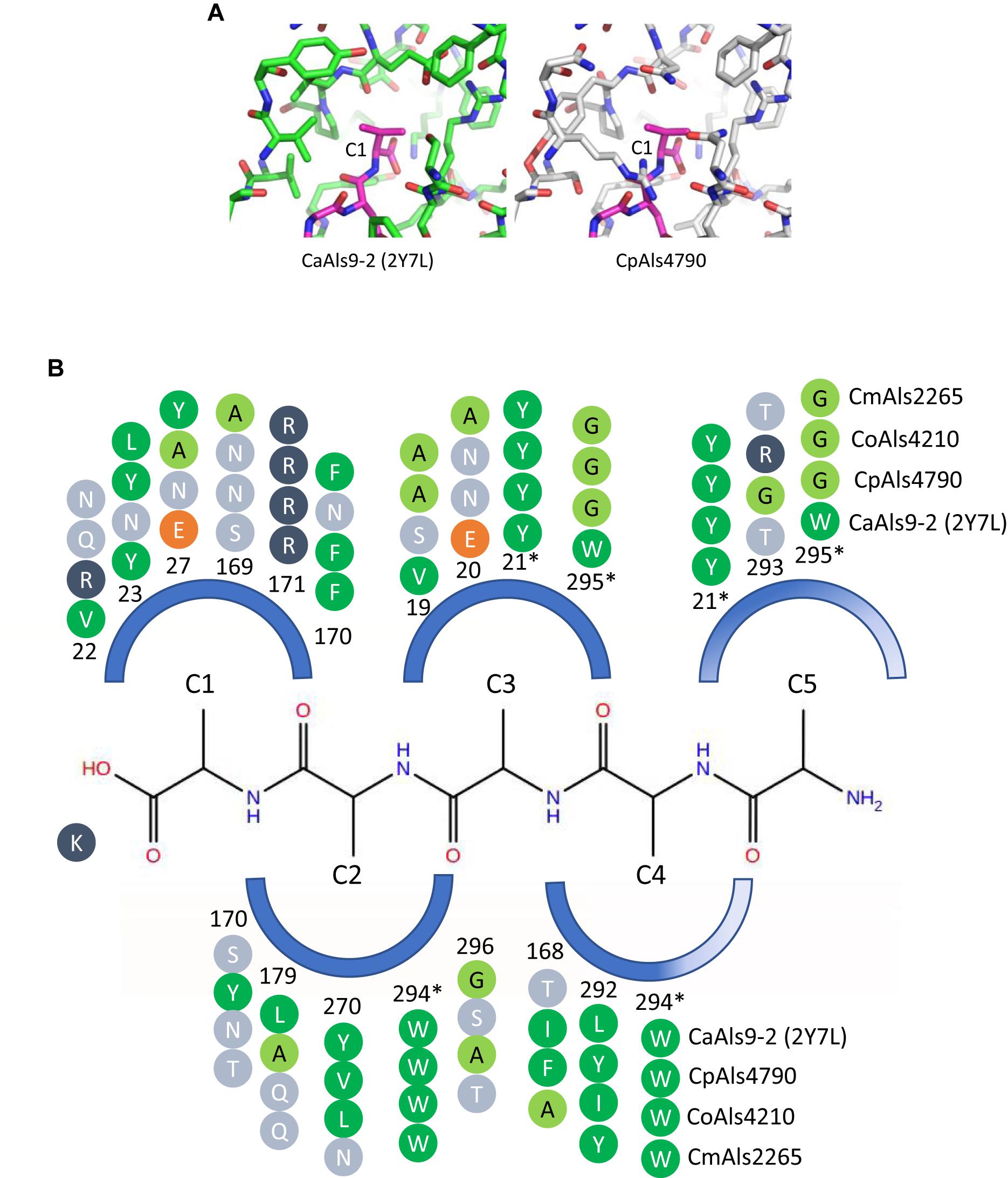
Figure 4. Structural comparison among peptide-binding cavities (PBCs) of a model Als protein and selected proteins from the C. parapsilosis species complex. (A) Side-by-side comparison of the C1 binding pocket of CaAls9-2 (left, green carbons; Protein Data Bank ID 2Y7N; Salgado et al., 2011) and a structural model of CpAls4790 (right, gray carbons) bound to fibrinogen gamma peptide (magenta carbons). The pocket was more constrained in CpAls4790 by the Val22Arg substitution, suggesting a more-selective binding activity than present in CaAls9-2. (B) Schematic of the NT-Als PBC showing the pockets surrounding amino acids of the bound peptide ligand (C1 through C5, with C1 proximal to the invariant Lys at the bottom of the PBC). Only residues which form the binding pocket were shown. Example proteins (CpAls4790, CoAls4210, CmAls2265) were selected from diverse parts of the phylogenetic tree in Figure 3. Side chain residues were indicated as circles and color-coded green (medium to large hydrophobic), light green (small hydrophobic), dark blue (positively charged), orange (negatively charged), or light blue (hydrophilic), and numbered to indicate their position within each protein. Residues with an asterisk were part of two pockets and the sequence was repeated for each pocket. The schematic highlights conserved positions (e.g., Y21, R171, W294) and illustrates variability found throughout the PBC.
There were large differences in PBC shape and charge among the proteins consistent with accommodation of peptides of various sequences. For example, the pocket surrounding the first C-terminal residue (proximal to the invariant Lys) in CaAls9-2 was formed by hydrophobic and hydrophilic residues suggesting the likelihood to accommodate a variety of amino acids. However, in CpAls4790, the pocket was much more constrained by substitution of CaAls9-2 Val22 with Arg in CpAls4790. The Arg residue filled a large part of the pocket and presented a positive charge directly adjacent to the C1 amino acid of the bound peptide. The substitution of Val22Arg in CpAls4790 suggested a more-selective ability to accommodate amino acids in the C1 position of the peptide sequence compared to CaAls9-2.
Further analysis of the first five C-terminal amino acids of the bound peptide identified the nearest residues within 8 Å of the β carbon of each amino acid in the peptide (or hypothetical β carbon in the case of glycine residues) which form the surface of the amino acid binding pocket. The sequence patterns of the C1 through C5 pockets showed variations in ability to accommodate peptide residues by both size and electrostatics (Figure 4B). Pocket C3 was highly constrained by close packing of neighboring residues allowing for accommodation of only very small or small residues like Gly, Ala, or Ser. Pockets C4 and C5 were partially solvent-exposed suggesting the likelihood of higher size promiscuity in accepting hydrophilic residues in these positions. Overall, the analysis was consistent with the conclusion that the C. parapsilosis species complex Als proteins possessed peptide-ligand binding activity similar to the highly characterized examples in C. albicans and, like the CaAls proteins, the newly identified proteins will demonstrate differences in selectivity for peptide binding.
Another common feature of C. albicans Als proteins is a central domain of multiple tandem copies of a 36-amino acid Ser/Thr-rich sequence. Previous thought had considered the C. albicans tandem repeats as grouping into three consensus sequences, represented by CaAls3, CaAls5, and CaAls9-2 (Hoyer et al., 2008). Although the absolute sequences are notably variable among different C. albicans Als proteins, a consensus sequence can be derived (Figure 5A). This consensus sequence was also found among proteins in the C. parapsilosis species complex tandemly repeated sequence units in the center of the protein (Figure 2). However, the repeated unit in CmAls2265 was clearly different in length and sequence from the other proteins. A BLAST search using the CmAls2265 repeat sequence as a query revealed the sequence in the C. albicans Iff family (Figure 5B) (Bates et al., 2007; Boisramé et al., 2011). This protein family was initially defined during annotation of the C. albicans genome sequence with “Iff” denoting Ipf family F, a group of proteins with similar sequence. The C. albicans Iff family also includes Hyr1 (Bailey et al., 1996). Subsequent investigations demonstrated various roles for these proteins, with a common feature being exposure on the cell surface (Boisramé et al., 2011). Supplementary Table S4 lists the currently recognized Iff proteins in C. albicans, as well as those revealed by BLAST searches of the C. parapsilosis, C. orthopsilosis, and C. metapsilosis genomes.

Figure 5. Conservation of tandemly repeated sequence units and short, imperfect repeats in various C. albicans and C. parapsilosis species complex proteins. (A) Consensus tandem repeat sequences were derived for C. albicans Als3 (CaAls3; GenBank accession number AY223552), CaAls5 (AY227440), and CaAls9-1 (AY269423). In C. albicans, each tandemly repeated unit was a perfect 36-aa length. Consensus sequences required that 80% or more of the amino acids were identical at each position. A similar approach was taken to aligning the consensus tandem repeat units from proteins from C. parapsilosis, C. orthopsilosis, and C. metapsilosis. GenBank accession numbers were shown in Table 3. In these proteins, the 36-amino acid repeat unit length was not necessarily conserved in each copy as color coded in Figure 2. The consensus repeat unit from CmAls2265 was 42 amino acids and different from the other Als proteins. (B) Alignment of consensus tandemly repeated sequence units from CmAls2265 and proteins from the Iff family. Repeat unit length was not necessarily conserved among the proteins, but a consensus sequence emerged from comparison among multiple proteins. GenBank accession numbers for the proteins were shown in Supplementary Table S4. (C) Top panel: C. parapsilosis species complex proteins that encode the short, imperfect SSSEPP (blue)/SSEPP (green)/SSEP (gray) repeated motif. Lower panel: Proteins that encode the GSGN (yellow) repeated motif. Protein identifiers were noted at the left of each sequence. The sequence labeled CmContig4 (GenBank accession number MK215077) most closely matched CORT_0E05950, which was annotated as Cdc24 GDP-GTP exchange factor, an unlikely identification of its function. Other proteins were labeled with identifiers from GenBank sequences or from the proteins listed in Supplementary Table S4. Names for proteins that had both SSEP/SSSEPP and GSGN short, imperfect repeats were listed in bold type in the top and lower panels.
Analysis of the Iff family revealed its own consensus repeat sequence (Figure 5B) although the repeat was not found in each protein, and protein length varied considerably (Supplementary Table S4). A thorough description of the Iff family in C. parapsilosis and C. orthopsilosis, and comparison to the C. albicans Iff family was presented by Riccombeni et al. (2012) so will not be expanded upon here. Of highest importance to the current discussion was the idea that CmAls2265 was a hybrid protein, comprised of an NT-Als adhesive domain placed upon the “foundation” of an Iff protein. CmAls2265 was evidence of a recombination event between genes from two different cell wall-protein-encoding families as defined in C. albicans. The recombination event appeared specific to C. metapsilosis, as a similar gene was not found in either C. parapsilosis or C. orthopsilosis, highlighting another difference between the closely related species.
Some of the predicted Als proteins (Figure 2) had short, imperfect repeated sequences instead of the longer tandemly repeated units. Predominant sequence motifs in these proteins included SSSEPP, which tended to immediately follow the classical Als T domain, and GSGN, which was located more-proximal to the C-terminal end of the protein. These sequences were present in other cell-wall proteins from C. albicans and the C. parapsilosis species complex (Figure 5C). The GSGN motif was common among the Iff proteins including CaIff2, CaIff6, CPAR2_600430, CPAR2_600440, CPAR2_806400, and CmIff104. The SSSEPP motif was found in other proteins, such as CPAR2_303790, a hypothetical protein that also included the GSGN motif. The SSSEPP sequence sometimes was truncated (SSEPP or SSEP) and found in proteins that also included the GSGN sequences. One example was CORT_0C02745 (an Iff protein) and another was CPAR2_403520, which was annotated as a hypothetical protein with similarity to C. albicans Hwp1 (Staab et al., 1999). Sequence alignments between CPAR2_403520 and C. albicans Hwp1 did not produce convincing evidence of sequence conservation that suggested CPAR2_403520 had similar function to CaHwp1 (data not shown). The final protein that had both GSGN and SSEP was located on contig 4 of the new C. metapsilosis assembly and deposited in GenBank under accession number MK215077. The best match to this sequence was CORT_0E05950, currently annotated as Cdc24 GDP-GTP exchange factor. The presence of several sequences with the potential for such diverse function suggested more widespread domain swapping among proteins than indicated for CmALS2265. These observations also suggested the need for more-careful annotation of the sequences of the pathogenic fungi with respect to genes that encode cell-wall proteins with potential to interact with the host, other microbes, and the external environment.
Allelic Variability Among the C. parapsilosis Complex ALS Genes
Allelic variability can be considered at multiple levels: the number of ALS genes in a specific fungal isolate is perhaps the most general. PCR was used to monitor ALS gene presence in additional strains of each species (Table 1). Primers were detailed in Supplementary Table S1. C. parapsilosis strains 949, 950, and 1125 each had the five known ALS genes, suggesting at least five ALS genes in each strain. Similar results were obtained for C. orthopsilosis and reported by Lombardi et al. (2019) where four different clinical isolates each encoded the three known C. orthopsilosis ALS genes. Analysis of C. metapsilosis isolates 61, 88, 397, and 482 indicated that, like the genome isolate ATCC 96143, each encoded four ALS genes.
C. parapsilosis, C. orthopsilosis, and C. metapsilosis are diploid, suggesting the potential for sequence and gene-length polymorphisms within each isolate. Current genome sequences for C. parapsilosis (CDC317; GenBank accession ASM18276v2) and C. orthopsilosis (90-125; ASM31587v1) are presented as haploid assemblies. These sequences were used as the basis for design of PCR primers to assess sequence and length polymorphisms among the ALS genes. Sequence polymorphisms were detected by Sanger sequencing of the PCR product. Length polymorphisms were visualized on agarose gels. This approach was effective at general validation of the published genome sequences although limitations must be recognized. For example, Sanger sequence reads were approximately 800-nt long, indicating the largest DNA fragment that could be assembled accurately was around 1.5 kb. PCR products larger than this size, or those that provided diploid allelic fragments of considerably different size were not amenable to this analysis. Accurate assignment of sequence polymorphisms to a specific allele would require construction of a heterozygous deletion mutant for each gene.
Overall, however, these methods validated the high degree of accuracy of the CDC317 genome assembly. Because PCR products represented both alleles of the diploid species, sequence ambiguities were present in the Sanger data where there was variation in the allelic sequences. In general, sequence variation was greater toward the 3′ end of the gene, perhaps due to conservation of the sequence that encodes the NT-Als binding domain. Polymorphic nucleotides were less than 1% of the total sequence reads among the fragments meeting the criteria described above. In one instance, the CDC317 sequence diverged from our Sanger validation (Figure 6A), but only by 12 nucleotides in a region of short, imperfect repeats. Repeated DNA was the largest source of length variation between alleles in the same strain and between different strains. Allelic variation in the “classic” ALS repeat units was the most notable, with the potential to vary by as much as a few kb between alleles. Figure 6B shows an example of length variation within a region of short, imperfect repeats of CpALS4770 in five different C. parapsilosis isolates. Length polymorphisms between alleles in the same and different C. orthopsilosis isolates were documented clearly by Lombardi et al. (2019).

Figure 6. Examples of allelic sequence variation among ALS genes in the C. parapsilosis species complex. (A) The representative genome sequence for C. parapsilosis CDC317 (GenBank accession ASM18276v2) presented ALS genes in a highly accurate manner. Sanger sequencing in the current study revealed many sequence polymorphisms in the diploid C. parapsilosis ALS genes that were not reflected in the haploid GenBank assembly. However, there was only one notable example where Sanger sequence varied from the GenBank genome: CpALS4780 had 12 fewer nucleotides in the region of the gene that encodes the GSGN short, imperfect repeat. (B) Regions of repeated sequences were often variable in length as shown in this example of CpALS4770 alleles from various C. parapsilosis isolates (949, CDC317, 1125, 950). Manual alignment of the amino acid sequences underscored the different length of CpALS4770 alleles in this region, leading to differing numbers of copies of the SSSEPP short, imperfect repeat. (C) The new C. metapsilosis genome assembly resolved into alleles and revealed previously unrecognized allelic variation that affected data interpretation. Primers to amplify C. metapsilosis ALS genes initially were designed from the representative genome sequence in GenBank (CBZN0200000000). The original forward primer to amplify CmALS4210 (highlighted in green) amplified only allele 1 due to mismatches and extra nucleotides in the sequence for allele 2. Re-design of the primer (highlighted in blue) led to successful amplification of the CmALS4210-2 allele, distinguishing it from CmALS4210-1. The CmALS4210 start codon was highlighted in yellow. (D) Alignment between amino acid sequences predicted from the CmALS4220 alleles. Considerable sequence variation between the alleles was observed including an expanded repeated sequence (Gly-Ala-Thr; GAT) in CmAls4220-1. These differences were predicted by the new C. metapsilosis genome sequence and verified by Sanger sequencing. (E) Allelic variation between strains of C. metapsilosis that affected interpretation of TaqMan assay data (see below). TaqMan assays were designed from the sequence of C. metapsilosis strain ATCC 96143. Nucleotide polymorphisms at the 3′ end of CmALS2265 in strain 61 resulted in reduced detection by the TaqMan assay. The stop codon for CmALS2265 was highlighted in yellow. The mismatches between the sequences for the forward primer (green), reverse primer (blue), and probe (gray) were sufficient to severely under-estimate relative gene expression for CmALS2265 in strain 61, suggesting that the gene was barely transcribed.
Advances in genome assembly software provide the tempting possibility to resolve allelic variation without the tedious and self-limiting validation steps described above. The newly derived C. metapsilosis ATCC 96143 genome assembly separated into allelic sequences. CmALS800 was a complete, accurate sequence in both alleles, without any differences between the allelic nucleotide sequences. CmALS800 was deposited in GenBank as a single allele (Table 3). In contrast, CmALS4210 and CmALS4220 each produced distinct allelic sequences in the new genome assembly that were deposited separately into GenBank (Table 3). Sequence polymorphisms were present throughout the coding region. In fact, sequence polymorphisms prevented CmALS4210-2 from amplification with PCR primers designed against the published genome sequence (Figure 6C). Sequence differences within the region encoding NT-Als produced conservative changes unlikely to affect protein function. Length polymorphisms were most evident in regions of repeated sequences, with size estimates confirmed by agarose gel analysis of PCR products. The 3′ end of CmALS4220-1 encoded expansion of a novel repeated sequence (Figure 6D).
CmALS2265 was essentially identical in the genome assembly allelic sequences, although neither produced a complete open reading frame that encoded a full-length protein. The CmALS2265 sequence had to be corrected by the PCR amplification and Sanger sequencing steps described above. To the extent possible using PCR amplification and Sanger data, the allelic sequences were validated as accurate in length and sequence. In one instance, nucleotide differences between C. metapsilosis strains affected use of a TaqMan assay designed to measure relative gene expression (Figure 6E; see below). Overall, these data provided evidence that the combination of short- and long-read sequence data and use of emerging genome assembly methodologies promise to more-accurately assemble genes that contain repeated sequences and resolve allelic variation in diploid species.
Relative Expression Levels of ALS Genes in the C. parapsilosis Species Complex
One major goal of this work was to design and validate real-time PCR assays that can be used for quantification of relative ALS expression. Assay design was complicated by the high degree of similarity between ALS loci in the same species. TaqMan assays were used to exploit their ability to specifically differentiate between highly similar sequences. Initial assay design attempted to standardize the location of the assay in each gene by using the region approximately 800 to 1,000 nt after the start codon. Primer and probe sequences for each assay were shown in Supplementary Table S2. Initial work focused on C. orthopsilosis since it encoded only three ALS genes.
CoALS4210, CoALS4220, and CoALS800 were >75% identical in the targeted region (Figure 7A), providing the opportunity to demonstrate the ability of TaqMan assays to generate highly specific results. Dilutions of cloned target sequences were tested with the various assays and showed specific amplification only for matches between a given TaqMan assay and its intended target. Mismatched assay and targets were generally undetectable, even when adding large quantities of cloned plasmid to the reaction (Figure 7B).
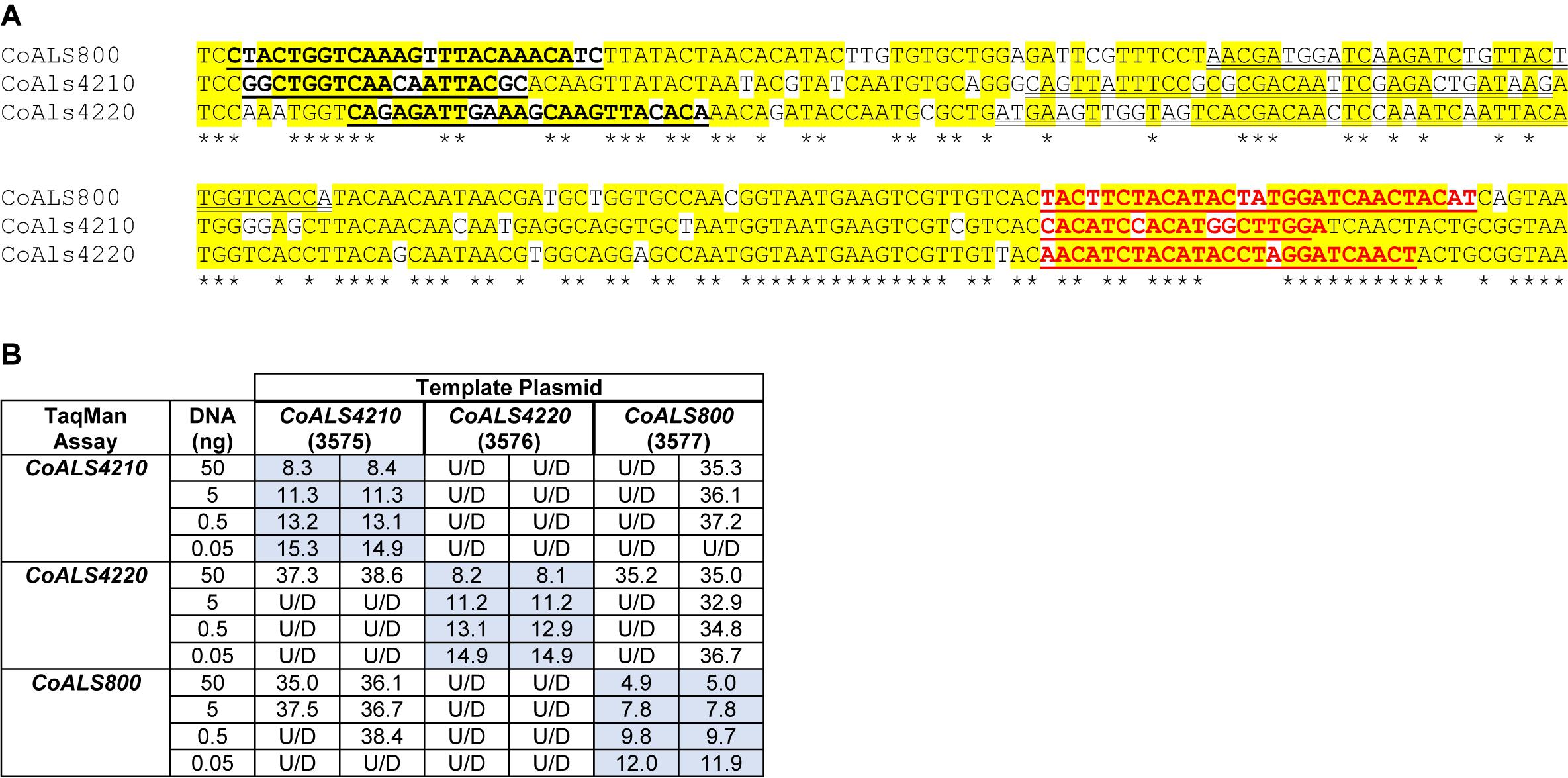
Figure 7. Demonstration of TaqMan assay specificity. TaqMan assays were selected for quantification of relative gene expression because of their exquisite specificity, which was needed to distinguish between highly similar loci in the same species. The most-extreme example was from C. orthopsilosis where sequences in the 5′ end of the gene were >75% identical. (A) Nucleotide sequences that were the most dissimilar in the 5′ end of CoALS4210, CoALS4220, and CoALS800. Yellow highlighting marked positions where at least two of the three sequences were identical. Asterisks marked the positions that were identical in all three sequences. In each sequence, the forward TaqMan primer sequence was underlined and shown in bold type. The TaqMan probe sequence was double underlined; each was in the sense orientation. The TaqMan reverse primer was underlined in bold, red type. (B) Ct values from TaqMan assays using cloned plasmid DNA as the reaction template. Dilutions of plasmid DNA were added to replicate TaqMan assays where the assay and template matched or were mismatched to gauge assay specificity. Ct values were recorded in the table and demonstrated recognition of the plasmid only when matched with the correct assay. For example, 5 ng of the cloned 5′ end of CoALS4210 gave a Ct value of 11.3 when assayed with the CoALS4210 TaqMan primers and probe. Undetectable signal (U/D) or a signal that was nearly undetectable (cycle 36.1) were recorded when the same amount of plasmid DNA was tested with the CoALS4220 or CoALS800 TaqMan assays. The data clearly demonstrated specificity of the TaqMan assays for their intended targets, even among genes as similar as the ALS genes of C. orthopsilosis.
Validated CoALS TaqMan results were compared to results from a previously reported SYBR Green-based assay (Lombardi et al., 2019). C. orthopsilosis strain 90-125 was grown in YPD Medium for 1 h and gene expression assessed using the SYBR Green and TaqMan assays. For SYBR Green, CoALS4220 showed the highest relative expression, followed by CoALS4210 (Figure 8A). The expression level for CoALS800 was considerably lower than the other two genes. Results from the TaqMan assays showed the highest expression for CoALS4220, with CoALS4210 and CoALS800 at a similarly low expression level (Figure 8A).
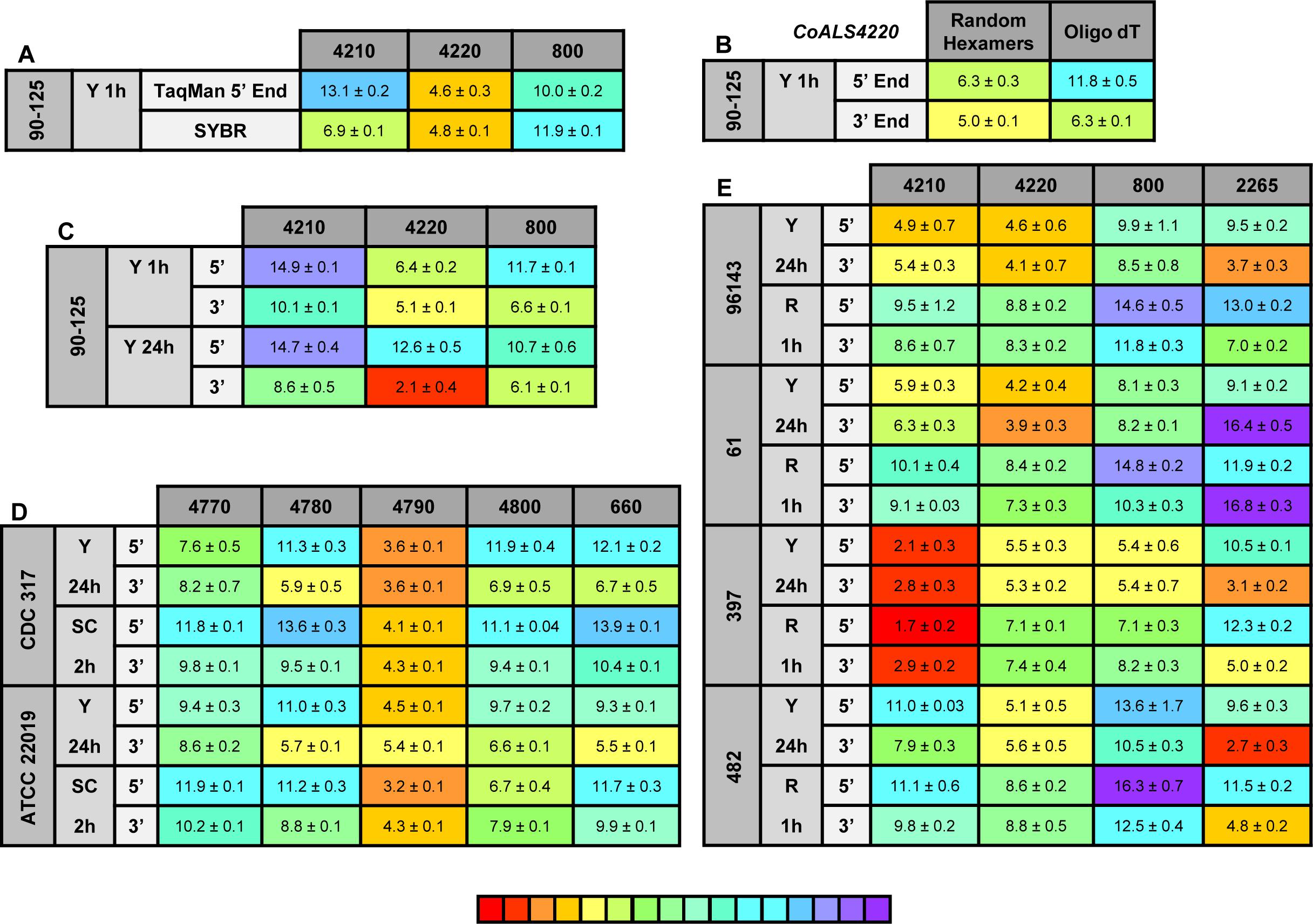
Figure 8. Relative gene expression measured by TaqMan assays. (A) C. orthopsilosis strain 90-125 was grown in YPD for 1 h. RNA preparations from these cells were assayed by TaqMan or the SYBR Green method of Lombardi et al. (2019). Mean (± standard error of the mean) Ct values were reported. Results were shaded to indicate the intensity of relative gene expression. The color key at the bottom of the diagram was shaded from red (high relative gene expression) to purple (low relative gene expression) to facilitate comparison between results. CoALS4220 was expressed more highly than either of the other genes in the growth conditions tested. The SYBR Green method suggested a higher relative expression level for CoALS4210 than did the TaqMan assay. (B) CoALS4220 was used as a model gene to test the effect of cDNA synthesis priming method and TaqMan assay location on estimates of relative gene expression. Placement of the TaqMan assay at the 3′ end of the gene produced higher expression estimates than placement at the 5′ end of the gene. Priming with random hexamers produced a higher gene expression estimate than oligo dT priming. (C) Themes illustrated in (A,B) carried forward to a more-extensive analysis of C. orthopsilosis ALS gene expression. Strain 90-125 was grown for 1 h and 24 h in YPD medium. CoALS4220 was more-highly expressed than the other genes. Estimates of relative gene expression were higher for TaqMan assays at the 3′ end of the gene than at the 5′ end of the gene. (D) Relative expression of C. parapsilosis ALS genes in two strains (CDC317, ATCC 22019) grown in two different conditions (YPD for 24 h; SC with fetal bovine serum for 2 h). CpALS4790 was more-highly expressed than the other genes, even CpALS4800 which was proposed to arise from duplication of CpALS4790. (E) Relative expression of C. metapsilosis ALS genes in four strains (ATCC 96143, 61, 397, 482) in two different growth conditions (YPD for 24 h; RPMI 1640 for 1 h). Considerable strain variation was observed with genes that were relatively quiet in one isolate (e.g., CmALS4210 in strain 482) but extremely highly expressed in another (strain 397). In many instances, gene expression estimates from the 5′ end and 3′ end TaqMan assays provided similar results, although several notable exceptions were present. The apparent lack of CmALS2265 expression in strain 61 (as measured with the 3′ end assay) prompted examination of the gene sequence at the site of the TaqMan assay (Figure 6E). Sequence variation between the TaqMan assay and gene sequence in this region explained the falsely low estimate.
One set of the SYBR Green assay primers (CoALS800) was located within the region to which the TaqMan assays were targeted while the others were located at the 3′ end of each gene (Lombardi et al., 2019). We used the gene with the highest expression level, CoALS4220, to test the effect of TaqMan assay location (5′ end vs. 3′ end of the gene), and method for priming cDNA synthesis (random hexamers vs. oligo dT). Since CoALS4220 was 6078 bp long, we expected a low estimate of gene expression for the combination of the 5′ end assay and oligo dT priming of cDNA synthesis (Figure 8B). The relative gene expression from the combination of random hexamer priming and the 3′ end TaqMan assay was significantly higher than the other combinations (P < 0.0001). For each cDNA priming method, the 3′ end TaqMan assay gave a significantly higher gene expression estimate than the 5′ end assay (P = 0.002 for random hexamers; P < 0.0001 for oligo dT). Subsequent assays used random hexamer priming to make cDNA and both the 5′ and 3′ assays. The trend of lower gene expression (higher Ct) estimates from the 5′ end assay continued in cells grown for 24 h in YPD medium (Figure 8C). CoALS4220 was expressed more highly than the other genes in the two growth conditions studied (P < 0.0001).
Because of these results, both 5′ and 3′ end assays were developed for each of the 5 ALS genes in C. parapsilosis (Figure 8D). Two different strains were studied (genome strain CDC317, and ATCC 22019, the C. parapsilosis type strain). Cells were grown under two different conditions to dissect potential effects of strain, growth stage, and growth medium. One growth condition was suggested by the work of Lackey et al. (2013) who indicated that it promoted C. parapsilosis morphological change. CpALS4790 was the most highly expressed gene in each growth condition, with similar estimates produced by the 5′ and 3′ end TaqMan assays. In many instances, the gene expression estimates from the 5′ end and 3′ end TaqMan assays were similar. Color coding in the image highlighted exceptions where the 3′ end assay provided as much as five cycles higher gene expression estimate than for the 5′ end assay. CpALS4780 and CpALS660 were more highly expressed at 24 h compared to 2 h, which could indicate a growth-stage or growth-medium effect on gene expression.
The same approach was used to assess relative ALS gene expression levels in 4 isolates of C. metapsilosis (Figure 8E) where gene expression differences between strains were more apparent than for the smaller sample of C. parapsilosis isolates. For example, CmALS4210 expression was much higher in strain 397 than the others. The trend of higher gene expression estimates for the 3′ end assay was generally observed. One notable exception was CmALS2265 in strain 61, for which the 3′ end assay produced considerably lower gene expression estimates than the 5′ end assay. PCR amplification of the assay site indicated that the 3′ end assay reverse primer sequence was not present in strain 61 due to sequence polymorphisms in that portion of the coding region (Figure 6E). Overall, these results demonstrated the complications with assay design to estimate relative gene expression and highlighted the need for sequence verification prior to studying gene expression in a variety of isolates.
Discussion
The increasingly common availability of draft genome sequences for microbial pathogens is a tremendous benefit to the scientific community. Insight into genome composition promotes development of testable hypotheses regarding host-pathogen interaction. Genes in the ALS family have proven difficult to assemble because each fungal species encodes multiple, highly similar loci, and the loci tend to have extensive tracts of repeated DNA sequences (Hoyer et al., 2008); these repeated sequence units are similar in size to the read lengths from second-generation DNA sequencers posing a difficult assembly problem. Therefore, it is not surprising that genome assemblies frequently break within ALS coding regions or genes are misassembled with the 5′ end from one locus computationally attached to the 3′ end of a gene from a different physical location. The goal of this work was to develop an accurate description of the ALS family in each species within the C. parapsilosis species complex.
ALS gene sequences were well-assembled in the C. parapsilosis representative genome (CDC317) that was generated from whole-genome shotgun sequencing and available in GenBank. PCR amplification and Sanger DNA sequencing of C. parapsilosis ALS genes revealed numerous instances of nucleotide polymorphisms that are not specified in the public, haploid genome assembly, but only identified one instance where the genome assembly sequence diverged from the Sanger verification (Figure 6). In contrast, ALS genes were not completely assembled in the C. orthopsilosis representative genome (Co90-125), with gaps located in the repeated sequences. This observation prompted generation of a new Co90-125 sequence derived from a combination of long- and short-read sequence data (Lombardi et al., 2019). The effort aided completion of the three ALS gene sequences in this C. orthopsilosis isolate. The C. metapsilosis representative genome sequence was assembled from primarily short-read data from more than one strain (Pryszcz et al., 2015). Despite the published claim of one ALS gene in this species, four ALS genes were detected in both the representative genome and a new sequence generated from long- and short-read data from strain ATCC 96143. Overall, availability of long-read sequence data facilitated accurate ALS gene assembly although Sanger sequencing of PCR products was still needed in some instances to correct premature stop codons and produce a complete open reading frame.
C. albicans ALS genes were named by numbering them in the order in which they were characterized (ALS1 to ALS7, ALS9; Hoyer et al., 2008). This traditional approach to naming genes was initiated before C. albicans genome data were available. These C. albicans names set a precedent and provide ample potential for confusion if applied to ALS genes in other fungal species. Orthology could be used as a criterion for naming genes among the fungal species considered here (Maguire et al., 2013); genes sharing a common physical location in different species could be given the same name with the expectation of similar function. These ideas become complicated for the ALS family because most species have a different number of ALS genes, gene order is not conserved well among the fungal species of interest, and there is a tendency toward tandem duplications of ALS loci (Figure 1). Als proteins are adhesins, yet function promiscuously, so looking toward shared function as the means for assigning names is also not an effective method. Some authors have referred to ALS genes using the label assigned in the annotation of the representative genome assembly (Bertini et al., 2016; Neale et al., 2018; Zoppo et al., 2018). This appealing approach provides a unique name for each gene and, like a street address, communicates physical location information. For example, it is obvious that CPAR2_404770, CPAR2_404780, CPAR2_404790, and CPAR2_404800 are contiguous in the C. parapsilosis genome. The final gene names proposed here (CpALS4770, CpALS4780, CpALS4790, and CpALS4800) included physical location information from the genome annotation labels, an abbreviation to indicate species of origin, and “ALS” to signal inclusion in the gene family. A similar method was used to name the genes in C. orthopsilosis and C. metapsilosis.
Hoyer and Cota (2016) used “NT/T/TR/CT” to describe the standard composition of Als proteins in C. albicans where NT is the NT-Als domain that contains the PBC, T is the Thr-rich region, TR denotes the tandem repeats of the consensus 36-amino-acid sequence (Figure 2), and CT is the Ser/Thr-rich C-terminal domain. C. albicans ALS genes were used as BLAST queries to pull sequences from the C. parapsilosis species complex genomes. However, only four of the resulting Als protein sequences fit the ideal C. albicans definition. Seven of the predicted proteins lacked the TR domain and in the last protein, the TR domain was from a different family as defined in C. albicans. Other predicted proteins that lacked the TR domain instead encoded regions with short, imperfect repeats. Finding these short, imperfect repeats in other cell-surface proteins suggests additional recombination between ALS sequences and others. To incorporate the new C. parapsilosis species complex proteins into the Als family, the definition must shift away from NT/T/TR/CT. Instead, the Als family appears to include proteins that have an Als adhesive domain presented on a structure that promotes cell-surface display. This alternative Als family definition encompasses all proteins identified in this study and designates CmAls2265 as an Als protein, rather than an Iff/Hyr protein. Indeed, several of the genes discussed here have been tested for their contributions to cell adhesion. Bertini et al. (2016) showed that deletion of CpALS4800 resulted in a C. parapsilosis strain with reduced adhesion to human buccal epithelial cells. Neale et al. (2018) documented a role for CpAls4800 in adhesion of C. parapsilosis to extracellular matrix proteins in a microfluidics assay using physiological fluid shear conditions. Zoppo et al. (2018) deleted CoALS4210 and showed decreased adhesion to human buccal epithelial cells for the resulting C. orthopsilosis mutant strain. Overall, data presented here suggest that definitions of gene families established in C. albicans will need to be modified to accommodate data from other fungi.
Extensive analyses of C. albicans ALS gene expression revealed differential expression regulated by morphological form, growth medium, and stage of culture growth (reviewed in Hoyer et al., 2008). This information provided the foundation to assess Als protein function in vitro. TaqMan assays specific for individual ALS genes were developed to collect similar data for ALS genes in the C. parapsilosis species complex. Since ALS genes are large, assays were placed at a similar position within each coding region to avoid effects of assay location on gene expression estimates. Initial assays were placed within the region that encodes the NT-Als domain; a second set of assays was designed at the 3′ end of each gene. Surprisingly, results from the 5′ end and 3′ end assays frequently varied by several Ct units with the tendency toward higher expression estimates from the 3′ end assays. Since the TaqMan assays quantify cDNA as a reflection of RNA abundance, results showed that cDNA from the 3′ end of the genes is more abundant than cDNA from the 5′ region of the gene. Several factors can be invoked to explain this imbalance between cDNA abundance including the 3′ end of the transcript being more accessible to reverse transcription than the 5′ end, selective degradation of the 5′ end of the transcript, or the presence of another promoter that overlaps with the 3′ end of the transcript and boosts its relative abundance. Examination of the genome sequence did not provide obvious evidence for the presence of an overlapping ORF that could result in greater abundance of 3′ end transcripts. Use of the TaqMan assays to compare gene expression across several isolates of the same species revealed sequence heterogeneity that interfered with assay function in certain strains. Lack of target recognition could also partially explain the relative gene expression estimate disparities discussed above. For example, sequence variation between alleles within the same strain could reduce relative estimates of gene expression if the assay recognizes only one of the alleles in the diploid species. Regardless of the issues, TaqMan data pointed to certain genes as being the most-highly expressed and indicated considerable strain variation in gene expression. The TaqMan assays are useful for gene expression analyses but must be validated with the specific strains to be tested. Using more than one assay per locus is also recommended to avoid potential misinterpretations as described above.
The literature contains a few reports regarding relative expression of ALS genes in the C. parapsilosis species complex. Comparisons of C. orthopsilosis TaqMan data to gene expression studies conducted in the laboratory of Dr. Arianna Tavanti (University of Pisa) were presented in the “Results” section. Results were not directly comparable, leading to questions of whether gene expression is influenced by local factors. One potential factor that is noted in the Candida literature is water quality, which despite modern purification systems, may vary widely and influence biological function of fungal cells (Nickerson et al., 2012). Neale et al. (2018) measured expression of the five C. parapsilosis ALS genes using a SYBR Green-based method. Differential expression of CpALS4800 and CpALS4780 was noted when cells were transferred from saturated YPD cultures to tissue culture medium for 3 h. Like the TaqMan data presented here, expression of CpALS4790 was highest in the saturated YPD culture, with CpALS4780 next-most abundant and others at a lower level. Collectively, the data highlight and prioritize research questions to pursue. CpALS4790 may be at the top of this list because of its high relative expression that predicts an abundance of CpAls4790 on the cell surface. However, structural predictions suggest that ligand binding may be more selective than for some of the other Als proteins (Figure 4). Perhaps CpAls4790 serves a more-focused adhesive role than deduced for the C. albicans Als proteins for which function was studied in detail (reviewed in Hoyer and Cota, 2016).
Future studies should also pursue greater insights into ALS gene expression patterns within the C. parapsilosis species complex. The current study was intended to assess whether ALS gene expression displayed the same differential patterns as observed in C. albicans and to identify genes that may produce proteins most abundantly on the fungal cell surface. In C. albicans, ALS genes that always showed a low expression level produced protein at a level undetectable by immunolabeling with specific monoclonal antibodies (Zhao et al., 2011). Genes expressed at a high level produced Als protein that could be long-lived on the cell surface (Coleman et al., 2010). It is also important to consider that gene expression and protein production can vary between in vitro and in vivo environments, suggesting the need for in vivo work to best understand the contributions of Als protein function in the host (Coleman et al., 2012).
Data presented here also set up intriguing experiments to pursue in C. metapsilosis. For example, the previous report that claimed only one ALS gene in the species was perhaps accepted without question because C. metapsilosis does not readily display phenotypes in vitro that are associated with pathogenic species. C. metapsilosis is less adherent to human buccal epithelial cells than either C. parapsilosis or C. orthopsilosis and shows a lower fungal burden at early stages of a murine vaginal infection model (Bertini et al., 2013). However, C. metapsilosis has four ALS genes, some are expressed at high levels, and the predicted proteins have an NT-Als domain like those in C. albicans. This new information must be reconciled with the relative lack of an adhesive phenotype for C. metapsilosis when assayed in vitro. Overall, data presented here highlight key questions that remain to be answered regarding the Als proteins and demonstrate the hypothesis-generating power of accurate genome assemblies.
Author Contributions
CF, AH, and LH designed the experiments. S-HO, BSm, BSt, AM, CF, AH, and LH conducted the experiments. All authors analyzed the data, and wrote and approved the manuscript.
Funding
This work was funded by grant number R15 DE026401 from the National Institute of Dental and Craniofacial Research, National Institutes of Health.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Jessie Kirk, Erica Forbes, Allyson Isenhower, Quinn Nguyen, Anton Bershanskiy, Mariah McNamer, and Kaia Ball for assistance with DNA sequence analysis. Drs. Travis Wilcoxen and Laura Zimmerman, Millikin University Department of Biology, coordinated student participation and supervised research credit for students in the Undergraduate Program in Fungal Genomics.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00781/full#supplementary-material
Footnotes
- ^ https://blast.ncbi.nlm.nih.gov/Blast.cgi
- ^ http://www.cbs.dtu.dk/services/SignalP
- ^ http://mendel.imp.ac.at/sat/gpi/gpi_server.html
- ^ https://www.ebi.ac.uk/services
- ^ https://www.ebi.ac.uk/Tools/msa/clustalo
- ^ https://sites.duke.edu/vilgalyslab/rdna_primers_for_fungi
- ^ www.ncbi.nlm.nih.gov
- ^ www.candidagenome.org
References
Alfaro, M. E., Zoller, S., and Lutzoni, F. (2003). Bayes or bootstrap? A simulation study comparing the performance of Bayesian Markov chain Monte Carlo sampling and bootstrapping in assessing phylogenetic confidence. Mol. Biol. Evol. 20, 255–266. doi: 10.1093/molbev/msg028
Applied Biosystems Inc. (2008). Guide to Performing Relative Quantitation of Gene Expression using Real-Time Quantitative PCR. Available at: https://www.thermofisher.com/document-connect/document-connect.html?url=https://assets.thermofisher.com/TFS-Assets/LSG/manuals/cms-042380.pdf&title=Guide%20to%20Performing%20Relative%20Quantitation%20of%20Gene%20Expression%20Using%20Real-Time%20Quantitative%20PCR (accessed December 20, 2017).
Bailey, D. A., Feldmann, P. J. F., Bovey, M., Gow, N. A. R., and Brown, A. J. P. (1996). The Candida albicans HYR1 gene, which is activated in response to hyphal development, belongs to a gene family encoding yeast cell wall proteins. J. Bacteriol. 178, 5353–5360.
Bates, S., de la Rosa, J. M., MacCallum, D. M., Brown, A. J. P., Gow, N. A. R., and Odds, F. C. (2007). Candida albicans Iff1, a secreted protein required for cell wall structure and virulence. Infect. Immun. 75, 2922–2928. doi: 10.1128/IAI.00102-07
Bertini, A., De Bernardis, F., Hensgens, L. A. M., Sandini, S., Senesi, S., and Tavanti, A. (2013). Comparison of Candida parapsilosis, Candida orthopsilosis, and Candida metapsilosis adhesive properties and pathogenicity. Int. J. Med. Microbiol. 303, 98–103. doi: 10.1016/j.ijmm.2012.12.006
Bertini, A., Zoppo, M., Lombardi, L., Rizzato, C., De Carolis, E., Vella, A., et al. (2016). Targeted gene disruption in Candida parapsilosis demonstrates a role for CPAR2_404800 in adhesion to a biotic surface and in a murine model of ascending urinary tract infection. Virulence 7, 85–97. doi: 10.1080/21505594.2015.1112491
Bliss, J. M. (2015). Candida parapsilosis: an emerging pathogen developing its own identity. Virulence 6, 109–111. doi: 10.1080/21505594.2015.1008897
Boisramé, A., Cornu, A., Da Costa, G., and Richard, M. L. (2011). Unexpected role for a serine/threonine-rich domain in the Candida albicans Iff protein family. Eukaryot. Cell 10, 1317–1330. doi: 10.1128/EC.05044-11
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Butler, G., Rasmussen, M. D., Lin, M. F., Santos, M. A., Sakthikumar, S., Munro, C. A., et al. (2009). Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature 459, 657–662. doi: 10.1038/nature08064
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552.
Chu, W. S., Magee, B. B., and Magee, P. T. (1993). Construction of an SfiI macrorestriction map of the Candida albicans genome. J. Bacteriol. 175, 6637–6651. doi: 10.1128/jb.175.20.6637-6651.1993
Coleman, D. A., Oh, S.-H., Manfra-Maretta, S. L., and Hoyer, L. L. (2012). A monoclonal antibody specific for Candida albicans Als4 demonstrates overlapping localization of Als family proteins on the fungal cell surface and highlights differences between Als localization in vitro and in vivo. FEMS Immunol. Med. Micobiol. 64, 321–333. doi: 10.1111/j.1574-695X.2011.00914.x
Coleman, D. A., Oh, S.-H., Zhao, X., and Hoyer, L. L. (2010). Heterogeneous distribution of Candida albicans cell-surface antigens demonstrated with an Als1-specific monoclonal antibody. Microbiology 156, 3645–3659. doi: 10.1099/mic.0.043851-0
Collart, M. A., and Oliviero, S. (1993). “Preparation of yeast RNA,” in Current Protocols in Molecular Biology, Vol. 2, ed. F. M. Ausubel (New York: Wiley). doi: 10.1099/mic.0.043851-0
Cook, C. E., Bergman, M. T., Cochrane, G., Apweiler, R., and Birney, E. (2017). The European Bioinformatics Institute in 2017: data coordination and integration. Nucleic Acids Res. 46, D21–D29. doi: 10.1093/nar/gkx1154
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Eisenhaber, B., Bork, P., and Eisenhaber, F. (1999). Prediction of potential GPI-modification sites in proprotein sequences. Mol. Cell Biol. 292, 741–758. doi: 10.1006/jmbi.1999.3069
Emsley, P., Lohkamp, B., Scott, W. G., and Cowtan, K. (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501. doi: 10.1107/S0907444910007493
Garcia, M. C., Lee, J. T., Ramsook, C. B., Alsteens, D., Dufrene, Y., and Lipke, P. N. (2011). A role for amyloid in cell aggregation and biofilm formation. PLoS One 6:e17632. doi: 10.1371/journal.pone.0017632
Gouy, M., Guindon, S., and Gascuel, O. (2010). SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224. doi: 10.1093/molbev/msp259
Green, C. B., Cheng, G., Chandra, J., Mukherjee, P., Ghannoum, M. A., and Hoyer, L. L. (2004). RT-PCR detection of Candida albicans ALS gene expression in the reconstituted human epithelium (RHE) model of oral candidiasis and in model biofilms. Microbiology 150, 267–275. doi: 10.1099/mic.0.26699-0
Guindon, S., and Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. doi: 10.1080/10635150390235520
Hoyer, L. L., and Cota, E. (2016). Candida albicans agglutinin-like sequence (Als) family vignettes: a review of Als protein structure and function. Front. Microbiol. 7:280. doi: 10.3389/fmicb.2016.00280
Hoyer, L. L., Green, C. B., Oh, S.-H., and Zhao, X. (2008). Discovering the secrets of the Candida albicans agglutinin-like sequence (ALS) gene family—a sticky pursuit. Med. Mycol. 46, 1–15. doi: 10.1080/13693780701435317
Huang, S., Kang, M., and Xu, A. (2017). HaploMerger2: rebuilding both haploid sub-assemblies from high-heterozygosity diploid genome assembly. Bioinformatics 33, 2577–2579. doi: 10.1093/bioinformatics/btx220
Kelly, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. E. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi: 10.1038/nprot.2015.053
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Lackey, E., Vipulananadan, G., Childers, D. S., and Kadosh, D. (2013). Comparative evolution of morphological regulatory functions in Candida species. Eukaryot. Cell 12, 1356–1368. doi: 10.1128/EC.00164-13
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Lin, J., Oh, S.-H., Jones, R., Garnett, J. A., Salgado, P. S., Rushnakova, S., et al. (2014). The peptide-binding cavity is essential for Als3-mediated adhesion of Candida albicans to human cells. J. Biol. Chem. 289, 18401–18412. doi: 10.1074/jbc.M114.547877
Lombardi, L., Zoppo, M., Rizzato, C., Bottai, D., Hernandez, A., Hoyer, L. L., et al. (2019). Characterization of the Candida orthopsilosis agglutinin-like sequence (ALS) genes. PLoS One (in press).
Lu, C. F., Kurjan, J., and Lipke, P. N. (1994). A pathway for cell wall anchorage of Saccharomyces cerevisiae alpha-agglutinin. Mol. Cell. Biol. 14, 4825–4833. doi: 10.1128/MCB.14.7.4825
Madoui, M. A., Engelen, S., Cruaud, C., Belser, C., Bertrand, L., Alberti, A., et al. (2015). Genome assembly using nanopore-guided long and error-free DNA reads. BMC Genom. 16:327. doi: 10.1186/s12864-015-1519-z
Maguire, S. L., OhEigeartaigh, S. S., Byrne, K. P., Schroder, M. S., O’Gaora, P., Wolfe, K. H., et al. (2013). Comparative genome analysis and gene finding in Candida species using CGOB. Mol. Biol. Evol. 30, 1281–1291. doi: 10.1093/molbev/mst042
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES science gateway for inference of large phylogenetic trees,” in Proceedings of the Gateway Computing Environments Workshop (GCE), (New Orleans, LA). doi: 10.1109/GCE.2010.5676129
Neale, M. N., Glass, K. A., Longley, S. J., Kim, D. J., Laforce-Nesbitt, S. S., Wortzel, J. D., et al. (2018). Role of the inducible adhesin, CpAls7, in binding of Candida parapsilosis to extracellular matrix under fluid shear. Infect. Immun. 86, e00892–e00917. doi: 10.1128/IAI.00892-17
Nickerson, K. W., Atkin, A. L., Hargarten, J. C., Pathirana, R. U., and Hasim, S. (2012). “Thoughts on quorum sensing and fungal dimorphism,” in Biocommunication of Fungi, ed. G. Witzany (Dordrecht: Springer),189–204.
Nielsen, H. (2017). “Predicting secretory proteins with SignalP,” in Protein Function Prediction, Methods in Molecular Biology, ed. D. Kihara 59–73. doi: 10.1007/978-1-4939-7015-5_6
Posada, D. (2008). jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256. doi: 10.1093/molbev/msn083
Pryszcz, L. P., Nemeth, T., Gacser, A., and Gabaldon, T. (2013). Unexpected genomic variability in clinical and environmental strains of the pathogenic yeast Candida parapsilosis. Genome Biol. Evol. 5, 2382–2392. doi: 10.1093/gbe/evt185
Pryszcz, L. P., Nemeth, T., Saus, E., Ksiezopolska, E., Hegedusova, E., Nosek, J., et al. (2015). The genomic aftermath of hybridization in the opportunistic pathogen Candida metapsilosis. PLoS Genet. 11:e1005626. doi: 10.1371/journal.pgen.1005626
Riccombeni, A., Vidanes, G., Proux-Wera, E., Wolfe, K. H., and Butler, G. (2012). Sequence and analysis of the genome of the pathogenic yeast Candida orthopsilosis. PLoS One 7:e35750. doi: 10.1371/journal.pone.0035750
Salgado, P. S., Yan, R., Taylor, J. D., Burcehll, L., Jones, R., Hoyer, L. L., et al. (2011). Structural basis for the broad specificity to host-cell ligands by the pathogenic fungus Candida albicans. Proc. Natl. Acad. Sci. U.S.A. 108, 15775–15779. doi: 10.1073/pnas.1103496108
Senol Cali, D., Kim, J. S., Ghose, S., Alkan, C., and Mutlu, O. (2018). Nanopore sequencing technology and tools for genome assembly: computational analysis of the current state, bottlenecks and future directions. Brief. Bioinform. doi: 10.1093/bib/bby017 [Epub ahead of print].
Sherman, F., Fink, G. R., and Hicks, J. B. (1986). Laboratory Course Manual for Methods in Yeast Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Press.
Staab, J. F., Bradway, S. D., Fidel, P. L., and Sundstrom, P. (1999). Adhesive and mammalian transglutaminase substrate properties of Candida albicans Hwp1. Science 283, 1535–1538.
Swofford, D. L. (2002). PAUP∗. Phylogenetic Analysis Using Parsimony (∗and Other Methods). Version 4. Sunderland, MA: Sinauer Associates.
Tavanti, A., Davidson, A. D., Gow, N. A., Maiden, M. C., and Odds, F. C. (2005). Candida orthopsilosis and Candida metapsilosis spp. nov. replace Candida parapsilosis groups II and III. J. Clin. Microbiol. 43, 284–292. doi: 10.1128/JCM.43.1.284-292.2005
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
White, T. J., Bruns, T., Lee, S., and Taylor, J. W. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in PCR Protocols: A Guide to Methods and Applications, eds M. A. D. Innis, D. H. Gelfand, J. J. Sninsky, and T. J. White (New York: Academic Press, Inc.),315–322.
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017a). Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genom. 3:e000132. doi: 10.1099/mgen.0.000132
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017b). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Zhao, X., Oh, S.-H., Coleman, D. A., and Hoyer, L. L. (2011). ALS51, a newly discovered gene in the Candida albicans ALS family, created by intergenic recombination: analysis of the gene and protein, and implications for evolution of microbial gene families. FEMS Immunol. Med. Microbiol. 61, 245–257. doi: 10.1111/j.1574-695X.2010.00769.x
Zhao, X., Pujol, C., Soll, D. R., and Hoyer, L. L. (2003). Allelic variation in the contiguous loci encoding Candida albicans ALS5, ALS1 and ALS9. Microbiology 149, 2947–2960. doi: 10.1099/mic.0.26495-0
Keywords: agglutinin-like sequence genes, ALS family, Candida species, adhesion, cell-wall proteins, fungi
Citation: Oh S-H, Smith B, Miller AN, Staker B, Fields C, Hernandez A and Hoyer LL (2019) Agglutinin-Like Sequence (ALS) Genes in the Candida parapsilosis Species Complex: Blurring the Boundaries Between Gene Families That Encode Cell-Wall Proteins. Front. Microbiol. 10:781. doi: 10.3389/fmicb.2019.00781
Received: 07 February 2019; Accepted: 27 March 2019;
Published: 26 April 2019.
Edited by:
Joshua D. Nosanchuk, Albert Einstein College of Medicine, United StatesReviewed by:
Maria Rapala-Kozik, Jagiellonian University, PolandRenata Toth, University of Szeged, Hungary
Copyright © 2019 Oh, Smith, Miller, Staker, Fields, Hernandez and Hoyer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lois L. Hoyer, bGhveWVyQGlsbGlub2lzLmVkdQ==