 Ritesh Mewalal1†‡
Ritesh Mewalal1†‡ Hengfu Yin2†
Hengfu Yin2† Rongbin Hu1
Rongbin Hu1 Sara Jawdy1
Sara Jawdy1 Patrice Vion3Gerald A. Tuskan1
Patrice Vion3Gerald A. Tuskan1 François Le Tacon3
François Le Tacon3 Jessy L. Labbé1*
Jessy L. Labbé1* Xiaohan Yang1*
Xiaohan Yang1*- 1Biosciences Division, Oak Ridge National Laboratory, Oak Ridge, TN, United States
- 2State Key Laboratory of Tree Genetics and Breeding, Research Institute of Subtropical Forestry, Chinese Academy of Forestry, Fuyang, China
- 3INRA, UMR 1136 INRA-University of Lorraine, Interactions Arbres/Microorganismes, Laboratory of Excellence ARBRE, INRA-Nancy, VandIJuvre-lès-Nancy, France
Ecto- and endo-mycorrhizal colonization of Populus roots have a positive impact on the overall tree health and growth. A complete molecular understanding of these interactions will have important implications for increasing agricultural or forestry sustainability using plant:microbe-based strategies. These beneficial associations entail extensive morphological changes orchestrated by the genetic reprogramming in both organisms. In this study, we performed a comparative analysis of two Populus species (Populus deltoides and P. trichocarpa) that were colonized by either an arbuscular mycorrhizal fungus (AmF), Rhizophagus irregularis or an ectomycorrhizal fungus (EmF), Laccaria bicolor, to describe the small RNA (sRNA) landscape including small open reading frames (sORFs) and micro RNAs (miRNAs) involved in these mutualistic interactions. We identified differential expression of sRNAs that were, to a large extent, (1) within the genomic regions lacking annotated genes in the Populus genome and (2) distinct for each fungal interaction. These sRNAs may be a source of novel sORFs within a genome, and in this regard, we identified potential sORFs encoded by the sRNAs. We predicted a higher number of differentially-expressed miRNAs in P. trichocarpa (4 times more) than in P. deltoides (conserved and novel). In addition, 44 miRNAs were common in P. trichocarpa between the EmF and AmF treatments, and only 4 miRNAs were common in P. deltoides between the treatments. Root colonization by either fungus was more effective in P. trichocarpa than in P. deltoides, thus the relatively few differentially-expressed miRNAs predicted in P. deltoides might reflect the extent of the symbiosis. Finally, we predicted several genes targets for the plant miRNAs identified here, including potential fungal gene targets. Our findings shed light on additional molecular tiers with a role in Populus-fungal mutualistic associations and provides a set of potential molecular targets for future enhancement.
Introduction
Mycorrhizal fungi are a diverse group of beneficial organisms that colonize the roots of more than 90% of higher plant species. Typically, two main types of fungi are described: (1) arbuscular mycorrhizal fungi (AmF), developing inside the root cell walls, e.g., Rhizophagus irregularis, and (2) ectomycorrhizal fungi (EmF), colonizing the intercellular spaces of the roots, e.g., Laccaria bicolor (Bonfante and Genre, 2010). These fungi play an important role in the maintenance of the plant health and growth by promoting water cycling, nutrient exchange and enhanced tolerance/resistance to biotic and abiotic stresses, while in exchange, the fungi receive plant-fixed carbon (Smith and Read, 2008; Bonfante and Genre, 2010). Several studies have shown that field application of mycorrhizal fungi improves the overall productivity of a number of crops including cereals, legumes, fruits and trees (Abbott and Robson, 1977; Brundrett et al., 1996; Al-Karaki et al., 2004; Powell, 2018). To address the challenge to food and energy security caused by increases in the global population, and decreases in agricultural and forest land, it is important to gain a deeper understanding of the molecular mechanism underlying beneficial symbiosis between plant and fungi to effectively design and develop plant:microbe-based strategies to enhance forestry and agriculture health and sustainability (Martin et al., 2017).
Much progress has been made in understanding the establishment and maintenance of these mutualistic associations (Bonfante and Genre, 2010; Plett and Martin, 2011). Many studies support the hypothesis that fungi-derived protein signals or effectors facilitate and/or maintain the symbiotic interactions (Daguerre et al., 2017). For example, the genome of L. bicolor encodes a large number of mycorrhizal-induced small secreted proteins (MiSSPs), many of which are expressed and accumulated in the fungal hyphae during colonization (Martin et al., 2008). Plett et al. (2011) reported that the effector protein of L. bicolor, MiSSP7 could enter the nucleus of Populus roots cells to affect transcription and promote symbiosis. MiSSP7 protects the Populus jasmonate zim-domain protein 6 (PtJAZ6), which is a negative regulator of jasmonic acid (JA)-induced gene regulation in Populus, and promotes symbiosis by blocking the action of jasmonic acid (Plett et al., 2014). Similarly, a secreted protein of R. irregularis, SP7, interacts with ERF19, a pathogenesis-related transcription factor that counteracts the plant immune response and facilitates root colonization by this AmF (Kloppholz et al., 2011).
On the plant side, colonization by fungi requires that the plant distinguish between beneficial and pathogenic fungi (el Zahar Haichar et al., 2014). Plant root exudates have been shown to (1) mediate chemotaxis signaling that facilitate the colonization of roots by flagellated bacteria (Scharf et al., 2016) and (2) contain secondary metabolites, strigolactones, which promote hyphal branching and stimulates fungal spore germination (Akiyama et al., 2005). Moreover, transcriptome analysis of Populus trichocarpa roots colonized by L. bicolor has revealed 417 putative plant-encoded small secreted proteins (SSPs) with 39% of them appearing to be specific to Populus, where several of these SSPs were able to enter the hyphae and affect the growth of L. bicolor (Plett et al., 2017). These studies suggest that the genetic contributions from a plant in mutualistic association may be more complex than our current understanding and may involve several levels of regulation. It is unclear if this molecular toolbox for symbiosis, i.e., set of molecular determinants (e.g., protein-encoding genes, non-coding RNAs) are shared across different plant species when colonized by the same fungi or alternatively, the same plant species colonized by different types of symbiotic fungi.
In recent years, the role of small non-coding RNAs (sRNAs), broadly defined as regulatory RNA molecules ranging in size from 20 to 300 nucleotides (Großhans and Filipowicz, 2008), have become apparent in biotic stresses and regulation of plant development and physiology (Mallory and Vaucheret, 2006; Großhans and Filipowicz, 2008; Ruiz-Ferrer and Voinnet, 2009; Chen, 2012; Zhang and Chen, 2013). These regulatory RNA molecules include small interfering RNAs (siRNAs), microRNAs (miRNAs), piRNAs (Piwi-associated RNAs), and long non-coding RNAs (lncRNAs), which may originate from intergenic, intronic, or antisense transcripts. Several detailed reviews of molecular mechanism of these different population of non-coding RNAs (ncRNAs) were published recently (Ruiz-Ferrer and Voinnet, 2009; Chekanova, 2015; Mohanta and Bae, 2015; Huang et al., 2016). miRNAs, typically between 20 and 25 nucleotides, are processed from single-stranded RNA to form imperfect base-paired hairpin secondary structures, and generally negatively regulate their targets including mRNAs (Chen, 2008; Lanet et al., 2009) and ncRNAs such as TAS RNAs (Vaucheret, 2006). Many lines of evidence now confirm that miRNAs are necessary for plant association with AmF (Branscheid et al., 2010; Devers et al., 2011; Lauressergues et al., 2012; Etemadi et al., 2014). For example, colonization of Medicago truncatula roots by R. irregularis was reduced when miR171h was overexpressed (Lauressergues et al., 2012; Hofferek et al., 2014). In plants, sRNAs have also been shown to move between cells via the plasmodesmata, and transverse long distance via the phloem (Brosnan and Voinnet, 2011). Furthermore, sRNA can transverse into interacting fungi through the haustorium, to trigger gene silencing (Molnar et al., 2011; Huang et al., 2016). Consequently, sRNAs are now viewed as an important part of the genetic response network for evolutionary adaption (Amaral et al., 2013).
Studies have reported that sRNAs can also have protein-coding potential and encode small open reading frames (sORFs) which typically contain between 30 and 100 amino acids (Ulveling et al., 2011; Andrews and Rothnagel, 2014; Li et al., 2014; Ruiz-Orera et al., 2014; Kumari and Sampath, 2015). The portfolio of organisms with functional sORFs include bacteria, yeast, Drosophila, plants and human (Oyama et al., 2004; Kastenmayer et al., 2006; Hanada et al., 2007; Ladoukakis et al., 2011; Yang et al., 2011; Aspden et al., 2014; Ruiz-Orera et al., 2014; Shell et al., 2015; Ma et al., 2016). sORFs are partially classified based on their genomic position, e.g., upstream or downstream annotated genes, within annotated genes but out-of-frame, antisense to annotated genes, within novel transcripts and within ncRNAs. The biological significance for these types of sORFs varies. For example, the tarsel-less (tal) gene from Drosophila was classified as ncRNA but the transcript does encode four sORFs (11–32 AAs) that affect Drosophila gene expression and development (Galindo et al., 2007). The intergenic region of Arabidopsis has been predicted to encode approximately 33,809 sORFs (Hanada et al., 2007). Additionally, Castellana et al. (2008) identified ∼5,000 small peptides in Arabidopsis in a proteomic study, several of which were novel and/or identified in the aforementioned study by Hanada et al. (2007). Despite an unclear molecular mechanism, Hanada et al. (2013) showed varying morphological changes caused by overexpression of sORFs in Arabidopsis. Thus, it is plausible that sORFs derived from ncRNAs may provide another level of developmental regulation in mutualistic symbiosis.
In this study, we (1) identify and compare the Populus-derived sRNAs response to two different mutualistic symbionts (i.e., R. irregularis and L. bicolor) and (2) predict and compare the encoded sORFs, as well as their miRNAs and putative gene targets potentially involved in formation and maintenance of the beneficial associations. To achieve this goal, we profiled the sRNA response of two Populus species (P. deltoides and P. trichocarpa) that were each colonized by either of the two different fungi (Tagu et al., 2001; Labbé et al., 2011). Our study provides insight into the common and differential regulation that may be involved in both the endo- and ecto-mycorrhizal associations among members of the genus Populus.
Materials and Methods
Plant Material and Inoculation
Internode cutting of P. trichocarpa (genotype 101–74) and P. deltoides (genotype 73028-62) of the 54B F1 pedigree (Jorge et al., 2005) parental lines were rooted and individually inoculated with a 1/9 (v/v) mixture of L. bicolor inoculum and calcined clay (Oil Dri US Special, Damolin, Denmark). Eight replicates per genotype and treatment were carried out. L. bicolor S238N inoculum was produced by growing mycelium on an autoclaved peat-vermiculite nutrient mixture in 1-L glass jars for 2 months in the dark at 25°C and kept at 4°C before use (Duponnois and Garbaye, 1991). R. irregularis inoculum consisted of spores (200 spores per litter of calcinate clay). Inoculated cuttings were grown for 3 months post-inoculation in a greenhouse at 15–28°C, 12 h photoperiod at INRA-Nancy, France during summer and then harvested. One portion of the sampled root (about 25%) was used to assess successful root colonization, which was confirmed under a stereomicroscope by counting ectomycorrhizas for L. bicolor and for R. irregularis by counting arbuscules and measuring how much of the root length is colonized after Trypan blue staining procedure as described by Koske and Gemma (1989). The remaining lateral root samples (about 75%) were immediately frozen in liquid nitrogen at -80°C for RNA isolation.
RNA Isolation and Sequencing
Total RNA was extracted from the root system of three of the eight replicates per genotype per treatment by combining a CTAB extraction method and the SpectrumTM Total Plant RNA extraction kit (Sigma) as reported in Bryan et al. (2016). Prior to synthesizing sequencing libraries, RNA templates were quantified using a NanoDrop Spectrophotometer (Thermo Scientific) and RNA quality was determined using BioRad ExperionTM. Samples with a 260/280 reading between 2.0 and 2.2 and a RIN > 7 were determined to be of high enough quality and the high-quality RNA samples were used for sRNA-sequencing library preparation with an Illumina TruSeq small RNA sample prep kit (Illumina, CA, United States). As specified in the Illumina protocol, small RNA fragments were selected after PCR amplification by gel purification to capture RNA approximately 20–170 bp in length. The Illumina TruSeq small RNA library prep workflow is considered to be stranded because adapters are ligated directionally, facilitating maintenance of strand information during analysis. A total of 18 sRNA-sequencing libraries (three biological replicates of the control P. deltoides and control P. trichocarpa, and from the treatments, EmF-P. deltoides, EmF- P. trichocarpa, and AmF-P. deltoides and AmF-P. trichocarpa) were constructed. The libraries were sequenced on an Illumina MiSeq system (Illumina, CA, United States) to generate singe-end reads (1 × 150 bp).
sRNA-Seq Data Analysis
In this study, small RNAs are defined as the RNA molecules with size ranging from 20 to 300 nucleotides, consistent with the definition by Großhans and Filipowicz (2008). The raw RNA-Seq data in Fastq format were quality checked and trimmed for low-quality regions and adaptor sequences using Trimmomatic v0.35 (Bolger et al., 2014), parameters of “2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:20.” The trimmed sequencing reads from P. trichocarpa and P. deltoides were mapped to the P. trichocarpa genome v3.0 and P. deltoides WV94 v2.11, respectively, using TopHat2 (Kim et al., 2013). Novel genes were assembled and transcript abundance in RPKM (Reads Per Kilobase of transcript per Million mapped reads) was estimated using Cufflinks (Trapnell et al., 2012). Analysis of differential gene expression was performed using Cuffdiff (Trapnell et al., 2012). The fold-changes of differential expression was calculated by log2(RPKM ratio), with a pseudo-count of 1 added to each RPKM value. To identify the homologs to the significantly differentially expressed transcripts, the sequences were used as query for BLASTN search against the publicly available genomes in the Phytozome2 using an E-value cutoff of 1.0E-3.
sORF Annotation
ORFfinder (Wheeler et al., 2003) was used to annotate putative ORFs within the differentially expressed sRNAs. We searched for 6 translational frames with the standard genetic code between 30 and 300 nucleotides for each prediction and selected the longest full-length sORF (including start and stop codon). The subcellular localization was predicted for each sORF using Loctree3 (Goldberg et al., 2014).
miRNA Identification
The small RNA sequencing reads were filtered to remove adaptors and low-quality bases by FastQC (Andrews, 2010). The sequencing reads were processed to remove adaptors and cleaned by Q30 value. Reads with over 20% bases less than Q30, and N base more than 10% were filtered. The clean reads were further selected by length between 18 and 30 bp for miRNA identification process. The miRDP1.3 package (Yang and Li, 2011) was used for miRNA precursor prediction with size set at 250 nt. To validate the miRNAs, the secondary structure and reads distribution were evaluated to generate the final set of miRNAs (Yin et al., 2016). The precursors and mature sequences were aligned with the searching toolbox in PNRD database for annotation of miRNAs (Yi et al., 2015). To define the conservation of miRNAs, a 17 bp match in the mature sequences were used to identify conserved miRNA families, allowing two mismatches. To quantify the abundance of miRNA, transcripts per million (TPM) value was defined as “counts of read mapped to miRNA∗1,000,000”/”reads mapped to reference genome” (Fahlgren et al., 2007). To identify differentially expressed miRNA DESeq2 software was used, and expression folder change greater than 2 and p-value (Benjamini-Hochberg FDR corrected) less than 0.01 were selected (Love et al., 2014). The miRNA expression data were normalized by row z-score method. Hierarchical clustering of gene expression was performed by clustergram function in MATLAB Bioinformatics toolbox with minor changes.
Target Prediction and Degradome Analysis
The miRNA targets were annotated by standard settings of psRNATarget (Dai and Zhao, 2011) with maximum expectation value 2.0. The transcripts of P. deltoides (WV94_445 v2.1) and P. trichocarpa (Nisqually-1 v3.0) were downloaded from Phytozome 123 while the transcripts for L. bicolor (20110203) and R. irregularis (Gloin1_GeneCatalog_transcripts_20120510.nt) was downloaded from MycoCosm4. To validate the targets, the degradome datasets in P. trichocarpa were downloaded from NCBI Short Read Archive (SRR4010497, SRR4010498). The clean reads from degradome library were obtained for target site identification. The CleaveLand4.0 pipeline (Addo-Quaye et al., 2009) was used for target scanning. The reads were mapped to transcript datasets and the alignment scores and p-value were calculated according the signatures (abundances of potential slicing end based on reads distributions). The t-plots were generated to visualize the miRNA directed slicing to targets and p-value of less than 0.05 was used to identify highly confident targets. Gene ontology enrichment for biological processes and molecular functions were performed online at PopGenIE5 and visualized with REVIGO using default settings (Sjödin et al., 2009; Supek et al., 2011).
Results
Differential Expression of sRNAs During Populus-AmF/EmF Colonization
The AmF, R. irregularis, and the EmF, L. bicolor, were able to colonize both species of Populus, i.e., P. deltoides and P. trichocarpa, though colonization rates were higher for P. trichocarpa (Table 1). To identify differentially expressed sRNAs in response to the AmF and EmF treatments, we performed RNA-sequencing (RNA-Seq) of RNA fragments less than 300 nt isolated from roots of P. deltoides colonized with the AmF R. irregularis or the EmF L. bicolor (PDA and PDE, respectively) and from roots of P. trichocarpa colonized with R. irregularis or L. bicolor (PTA and PTE, respectively, Figure 1A). Roots without fungal inoculation from P. deltoides and P. trichocarpa were used as controls (PTC and PDC, respectively). Thus, a total of 18 sRNA-sequencing libraries (three biological replicates per treatment PDA, PDE, PTA, PTE, PDC, and PTC) were constructed and sequenced. Following trimming, the RNA-Seq reads were mapped to the respective P. trichocarpa v3.0 and P. deltoides WV94 v2.1 genome sequences (Table 2). Based on a corrected p ≤ 0.05 and > 2-fold change in expression compared to the controls, we found 81 and 59 differentially expressed transcripts in P. deltoides and P. trichocarpa, respectively, relative to the control (Supplementary Tables S1, S2 and Supplementary Files S1, S2). Among the 81 fungal-responsive transcripts in P. deltoides, 12 and 69 transcripts were differentially expressed in the PDA and PDE interactions, respectively. Furthermore, among the 59 fungal-responsive transcripts in P. trichocarpa, 32 and 27 transcripts were differentially expressed in the PTA and PTE interactions, respectively. Interestingly, the differential transcripts were mostly specific to each plant-fungal combination, except for 7 transcripts that were shared between the PDA and PDE (all down-regulated except Podel.CUFF.90 which was up-regulated) and 3 transcripts shared between PTA and PTE (two down-regulated while Potri.013G036500 showed an opposite trend in each treatment; Figure 1B). The expression fold-change trend is shown in Figure 1C and Supplementary Tables S1, S2. Across species comparison showed that 5 homologous transcripts could be detected between species, of which three homologous transcript pairs showed the opposite expression trend between PDE and PTA and the other two homologous transcripts pairs were up-regulated in PDE and PTE (Figure 1D). These results suggest that, to a large extent, the expression of the sRNAs were distinct for each plant species within corresponding fungal treatments despite both fungi having a mutualistic relationship with these plants.
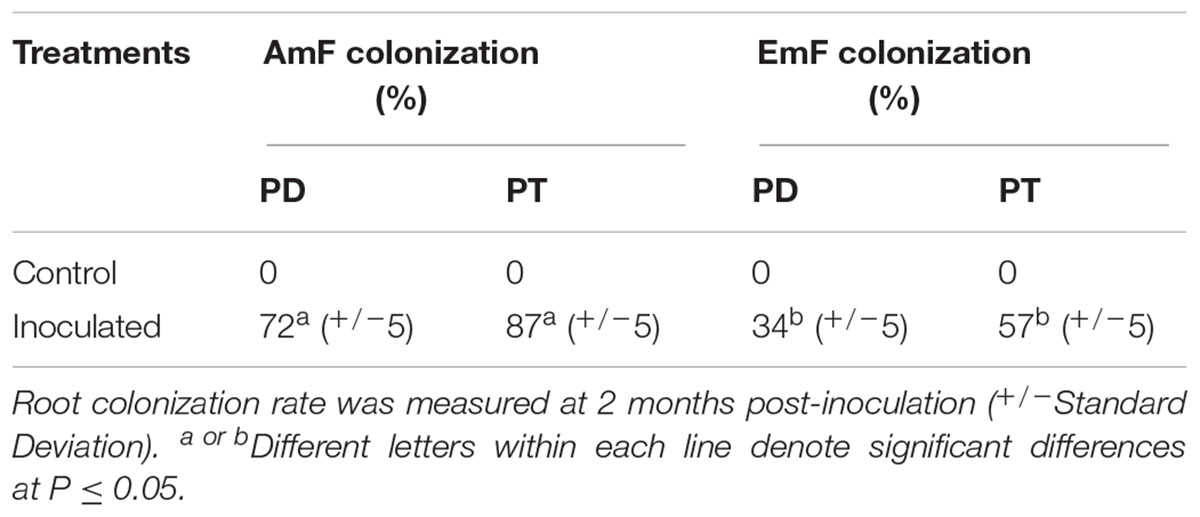
Table 1. Frequency of root colonization by arbuscular (AmF) and ectomycorrhizal (EmF) mycorrhizal fungus in Populus deltoides (PD) and Populus trichocarpa (PT).
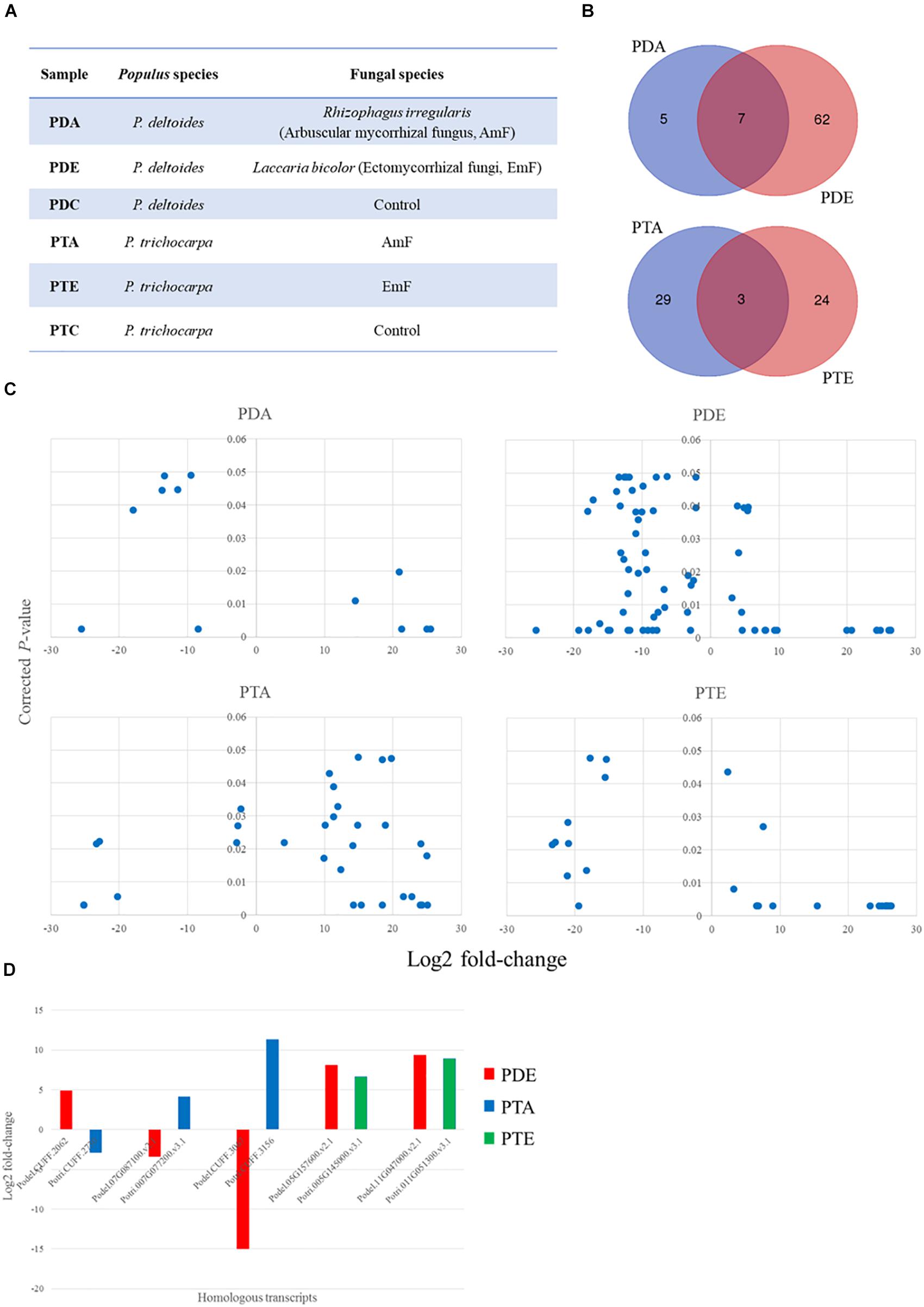
Figure 1. sRNA landscape of Populus species in response to the arbuscular mycorrhizal fungus (AmF), Rhizophagus irregularis and the ectomycorrhizal fungus (EmF), Laccaria bicolor. (A) Populus species and corresponding treatments used in the current study; (B) Venn diagram showing the transcript overlap from the different treatments in P. deltoides and P. trichocarpa; (C) Significant differentially expressed transcripts from the controls (p ≤ 0.05) and greater/less than two-fold change in the expression level; and (D) comparison of fold-change and trend for conserved transcripts across species. Transcript IDs and fold change can be found in Supplementary Tables S1, S2 and transcript sequences can be found in Supplementary Files S1, S2.
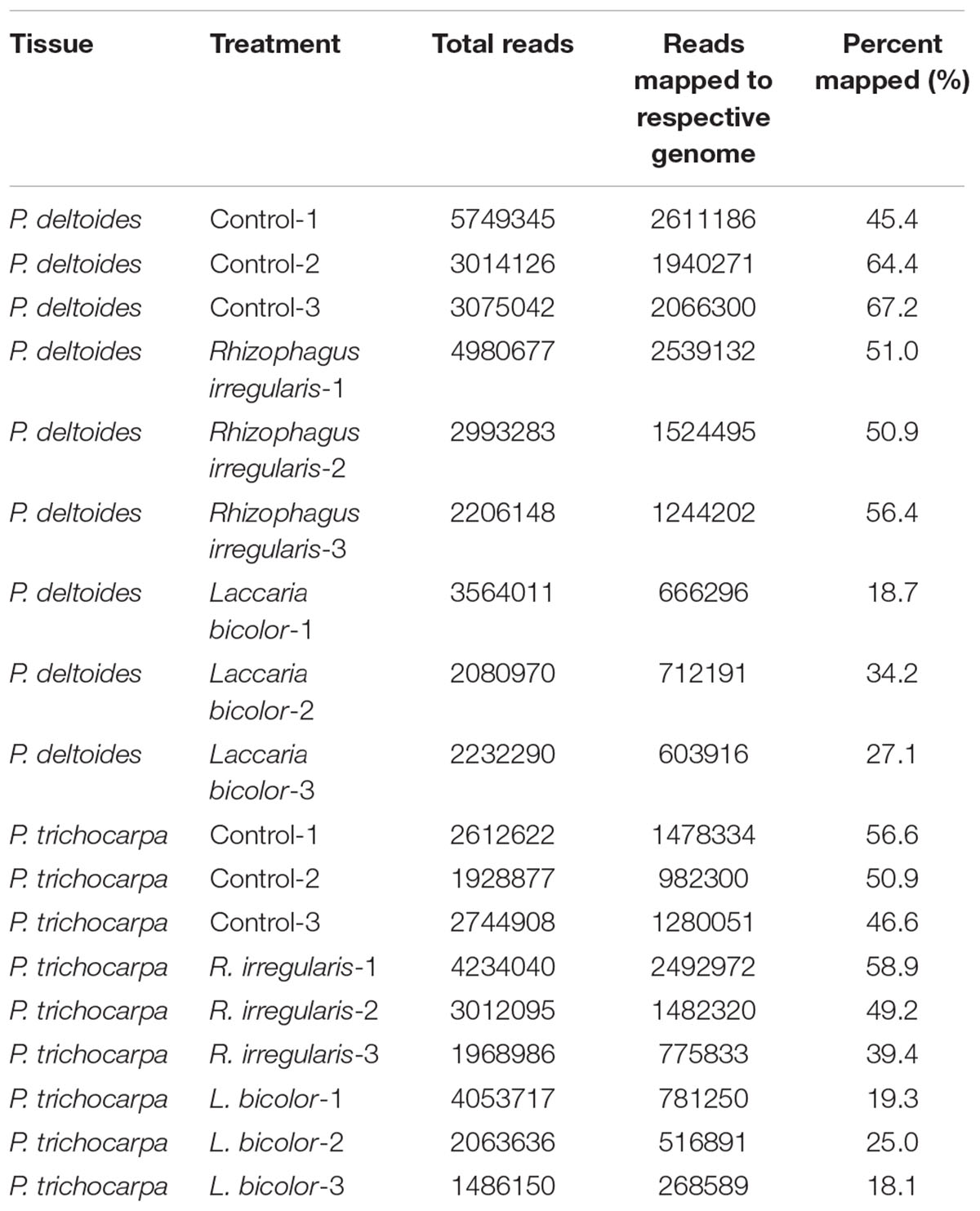
Table 2. Total number of RNA-Seq reads (in the range of three biological replicates per genotype/treatment), following trimming, that were mapped to Populus deltoides WV94 v2.1 and Populus trichocarpa genome v3.0 available on Phytozome 12.
Apart from the 17 transcripts that were previously annotated within the respective genomes (Supplementary Table S3), the remaining significantly differentially expressed fungus-responsive plant sRNAs (with name starting as “CUFF”) are located in the genomic regions without previously annotated genes. From the 17 previously annotated genes, 6 were proteins of known function (e.g., MYB-LIKE DNA-BINDING PROTEIN and AMMONIUM TRANSPORTER 1 MEMBER), while the remaining 11 genes were annotated as proteins of unknown function (Supplementary Table S3). We assessed the conservation of the differentially expressed sRNA across 63 plant genomes available on Phytozome 12.0 and found homology to 47 P. deltoides and 20 P. trichocarpa sRNAs in at least one genome analyzed (E-value ≤ 03; Supplementary Table S4).
Novel sORFs in Populus in Response to Mycorrhizal Fungi
Since sRNAs can potentially encode sORFs (Li et al., 2014; Ruiz-Orera et al., 2014), we used ORFfinder (Wheeler et al., 2003) to scan the differentially expressed sRNAs for potential sORF translations. In total, we could predict 22 and 6 full-length sORFs from the treatments in P. deltoides and P. trichocarpa, respectively (Supplementary Tables S5, S6). The shortest predicted sORF was 39 bp (12 AA) while the longest was 273 bp (90 AA). The transcript, coding, and protein sequences of these sORFs can be found in Supplementary Files S1, S2, S5, S6. Prediction of subcellular localization showed that 26 sORFs are secreted extracellularly while a single sORF is targeted to the nucleus and another to the chloroplast (Supplementary Table S7).
Identification of miRNAs in Populus in Response to Mycorrhizal Fungi
To gain insight into the role of miRNAs in mutualistic interactions, we analyzed the sRNA datasets to identify miRNAs that were responsive to the different mycorrhizal treatments. Using the miRDeep-P pipeline (Yang and Li, 2011), we identified 287 putative miRNAs in P. deltoides and 357 putative miRNAs in P. trichocarpa, including precursor and mature sequences (Supplementary Files S3, S4 and Supplementary Tables S8, S9). In P. deltoides and P. trichocarpa, 174 and 130 miRNAs were classified into “Plant conserved” miRNA families, respectively, and 113 and 227 miRNAs were classified as “Plant novel” miRNAs, respectively (Figure 2A and Supplementary Tables S8, S9). Note that the corresponding known families for the miRNAs identified in this study can found in Supplementary Tables S8, S9 while the miRNA abundance expressed as TPM for each treatment can be found in Supplementary Tables S10, S11. To refine the list of miRNAs, we focused on those that were significantly differentially expressed (negative binomial test, FDR corrected p ≤ 0.05; fold-change > 2; Soneson and Delorenzi, 2013) between the treatment and control. In this regard, we only considered miRNAs with greater than 50 counts in total and found 34 and 130 unique miRNAs with significant differential expression in P. deltoides and P. trichocarpa, respectively, including Plant Novel miRNAs (Supplementary Tables S12, S13). Hierarchical clustering of differentially expressed miRNAs showed distinct differences in expression pattern in response to EmF and AmF in both P. deltoides and P. trichocarpa (Figure 2B).
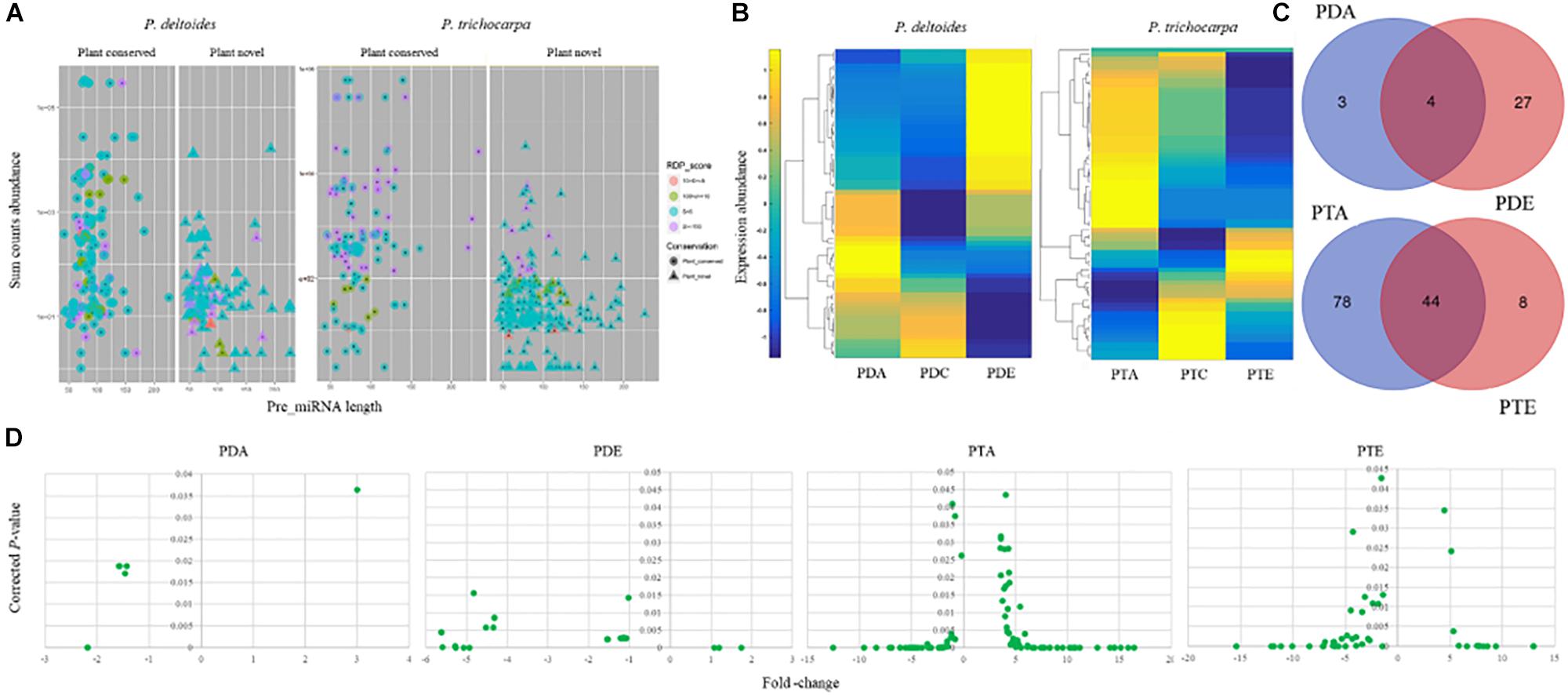
Figure 2. miRNA landscape Populus deltoides and P. trichocarpa roots colonized with Rhizophagus irregularis (PDA and PTA, respectively) and Laccaria bicolor (PDE and PTE, respectively). (A) miRNAs identified were classified as “Plant conserved” or “Plant novel”; (B) Hierarchical clustering of differential expression in transcripts per million of identified miRNAs; (C) Venn showing significant differentially expressed miRNAs that were common and unique miRNAs to each treatment; and (D) Fold-change and the significance (p ≤ 0.05) of differentially expressed miRNAs.
We next investigated whether each species shared a common set of miRNAs during the different mutualistic symbiosis. We found that in P. deltoides 4 miRNAs were common between PDA and PDE and in P. trichocarpa 44 miRNAs were common between PTA and PTE (Figure 2C and Supplementary Tables S12, S13). In P. deltoides, all 4 common miRNAs were down-regulated relative to the control while in P. trichocarpa, 16 shared miRNAs were up-regulated, 23 were down-regulated and 5 showed the opposite fold-change trend (Table 3). Further, this analysis also revealed that, within species, 3 and 27 miRNAs were unique to PDA and PDE, respectively, while 78 and 8 miRNAs were unique to PTA and PTE, respectively (Figure 2C and Supplementary Tables S12, S13). We further compared the Best-Hit of the significantly differentially expressed miRNAs across species, i.e., PDA vs. PTA and PDE vs. PTE (Supplementary Tables S12, S13). We did not find any miRNAs in common between different species colonized by a common fungus (data not shown). Also, we compared the differential transcripts listed in DATA 1 and 2 with the miRNA precursor sequences using the CAP3 program implemented in http://biosrv.cab.unina.it/webcap3/ and identified that five differential transcripts were miRNA precursors (Supplementary Table S14). The miRNA expression fold-change trend can be seen in Figure 2D and Supplementary Tables S12, S13. In P. trichocarpa, we found that 23 and 7 miRNAs were only detected in the PTA and PTE treatments, respectively, and not in the controls.
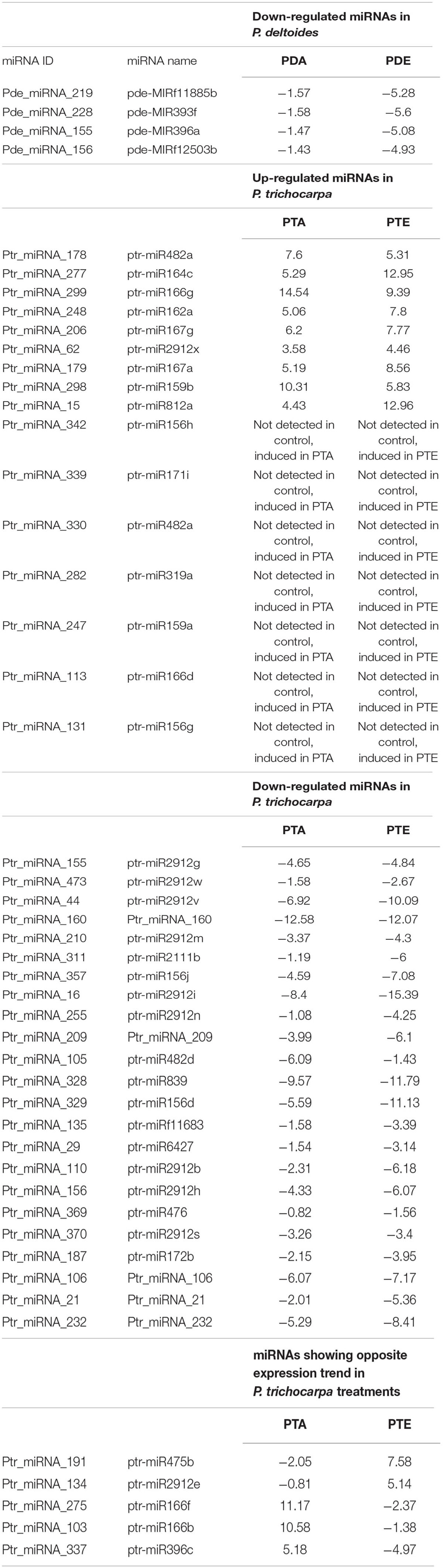
Table 3. Expression fold-change trend of miRNAs in Populus deltoides and P. trichocarpa in response to Rhizophagus irregularis (PDA and PTA, respectively) and Laccaria bicolor (PTA and PTE, respectively).
Identification of Target Genes of miRNAs in Populus in Response to Mycorrhizal Fungi
To determine the biological function of all identified miRNAs, we searched the “degradome” libraries of public databases for potential gene targets in Populus. While we identified 61 gene targets to 41 unique miRNAs in P. deltoides and 117 gene targets to 46 unique miRNAs in P. trichocarpa (p ≤ 0.05; Supplementary Tables S15, S16), there were only 12 and 59 unique gene targets (including alleles) to 7 and 25 unique differentially expressed P. deltoides and P. trichocarpa miRNAs, respectively (Supplementary Table S17). We also identified putative targets to the “Plant novel” miRNAs category, i.e., 10 miRNA-gene targets in P. deltoides and 18 miRNA-gene targets in P. trichocarpa (Table 4). In both P. deltoides and P. trichocarpa, gene ontology enrichment analysis of the miRNA targets (Supplementary Tables S15, S16) for biological processes included terms associated with regulation of metabolism, response to hormones and endogenous stimulus and regulation of transcription (Figures 3A,B) while molecular functions in P. trichocarpa were enriched in transcription factor activity, nucleic acid binding and DNA binding (Figure 3C). There were no enrichments for molecular functions in P. deltoides.
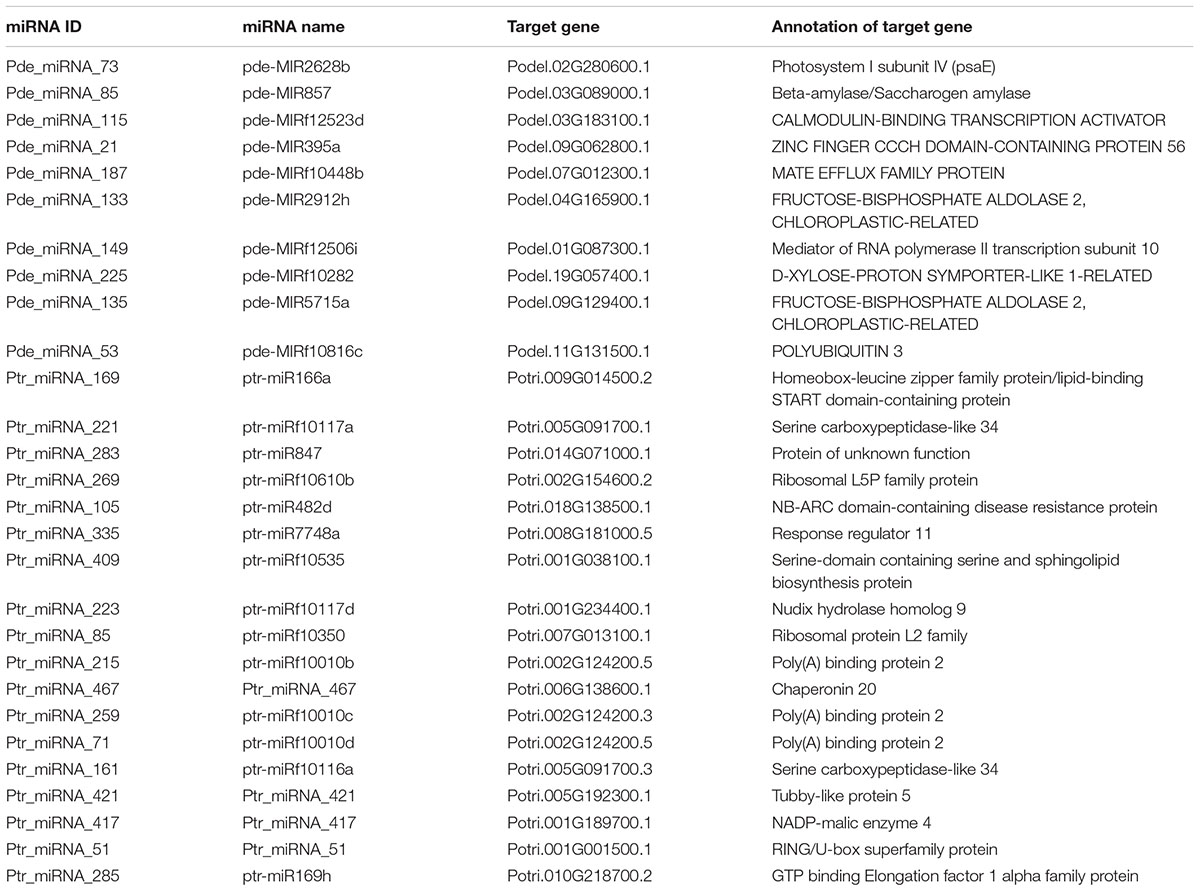
Table 4. Gene targets predicted for the “plant novel” Populus miRNAs in response to Rhizophagus irregularis and Laccaria bicolor.
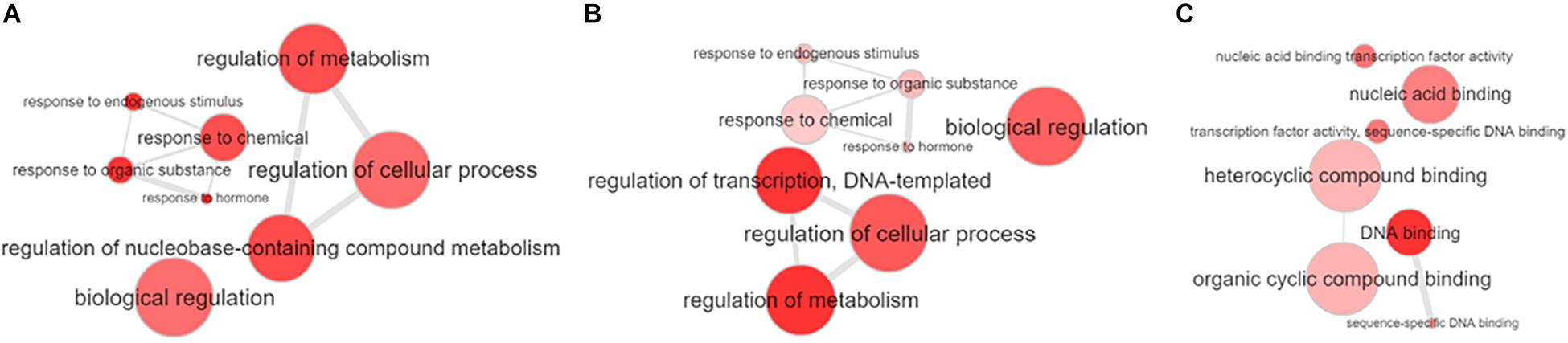
Figure 3. Gene ontology enrichment of the target genes of the miRNAs identified in Populus deltoides and P. trichocarpa roots colonized with the arbuscular mycorrhizal fungus (AmF), Rhizophagus irregularis, and the ectomycorrhizal fungus (EmF), Laccaria bicolor. (A) Gene ontology biological processes for P. deltoides with AmF and EmF; (B) Gene ontology biological processes for P. trichocarpa with AmF and EmF; and (C) Gene ontology molecular processes for P. trichocarpa with AmF and EmF. Enrichment was performed online at PopGenIE (http://popgenie.org/) and visualized with REVIGO (Sjödin et al., 2009; Supek et al., 2011).
Identification of Putative Target Genes for Populus-Derived miRNAs in R. irregularis and L. bicolor
We predicted potential miRNA-gene targets in the respective fungi since small RNA may transverse between symbionts via extracellular vesicles to mediate RNA interference and effect physiological changes across species (Lefebvre and Lécuyer, 2017). We found 5 gene targets in L. bicolor for 14 significantly differentially expressed P. deltoides miRNAs, 4 gene targets in L. bicolor for 6 significantly differentially expressed P. trichocarpa miRNAs and 5 gene targets in R. irregularis for 5 significantly differentially expressed P. trichocarpa miRNAs (Supplementary Table S18). We did not find any genes targets in R. irregularis to the significantly differentially expressed P. deltoides miRNAs. The majority of the predicted gene targets within the respective fungi were hypothetical protein/predicted proteins while a transport protein particle complex subunit (TRAPP 20 K subunit) was a common target for several P. deltoides and P. trichocarpa miRNAs. In addition, despite an annotation as a hypothetical protein, the target genes of two miRNAs had a KOG description as transcription factors.
Discussion
In the current study, we applied high-throughput RNA sequencing to describe the sRNA landscape of the root of two Populus species (P. deltoides and P. trichocarpa) colonized by an EmF, L. bicolor, or an AmF, R. irregularis, with specific focus on the sORF and miRNA response. A striking observation was that these beneficial plant-fungal interactions triggered the expression of otherwise intergenic regions of the genome. Furthermore, despite being mutualistic symbiosis, only a small number of these transcripts were conserved within the species between the two fungi (7 in P. deltoides and 3 in P. trichocarpa; Figure 1B). This may be a reflection of the different classes of fungi, i.e., arbuscular mycorrhizal vs. ectomycorrhizal, suggesting that at least on the plant side and for the sRNAs, mutualistic associations do not necessarily have a common response/regulatory mechanism. Additionally, only 5 transcripts were shared between P. deltoides and P. trichocarpa (Figure 1D), leading to a hypothesis that the molecular differences (i.e., the low number of sRNAs shared between two Populus species under two different fungal treatments) in the expression of plant small RNAs in response to fungi might contribute to the phenotypic difference in colonization efficiency (see Table 1; Tagu et al., 2001). This hypothesis needs to be tested using experimental approaches such as gain-of-function via gene overexpression and loss-of-function via gene knockout in the future.
While we do not know for certain what the end products of each sRNA is, several lines of evidence exist for the functionality of sORFs translated from sRNAs (Andrews and Rothnagel, 2014; Ruiz-Orera et al., 2014). Given that non-coding RNA may be an important source of new peptides, our predicted coding potential of the sRNAs advocates for a functional role for several sORFs in the symbiotic interactions. This is further supported by the predicted subcellular targeting of these sORFs (Supplementary Table S7). Recently, Plett et al. (2017) showed that several Populus-encoded SSPs (≤ 250 AA), including a novel SSP that was not annotated in the P. trichocarpa genome, could enter the fungal hyphae, localize to the nucleus and affect growth of L. bicolor.
The differential expression of a large number of miRNAs identified in response to the fungal treatments (34 in P. deltoides and 130 P. trichocarpa) suggest that the plant miRNAs are an important regulator of this mutualistic symbiosis. In this respect, our differentially expressed miRNA list (Supplementary Tables S12, S13) contained previously identified miRNAs that are previously reported to be involved in mutualistic interactions, albeit in different plant species such as Medicago, tomato and rice, including miR156, miR160, miR170, miR167, miR393, and miR396 (Bazin et al., 2013; Etemadi et al., 2014; Wu et al., 2016). Unlike the sRNA landscape, several significantly differentially expressed miRNAs were shared between PTA and PTE treatments (Figure 2C), indicating that the miRNA landscape and hence, the molecular mechanism imparted by this level of regulation may be conserved to a certain extent within species. The latter is further substantiated by the same expression trend seen for 39 of the 44 miRNAs in common between PTA and PTE and all 4 miRNAs in common between PDA and PDE, where both miRNAs were either up- or down-regulated in the respective treatments (highlighted in blue in Supplementary Tables S12, S3). We however, also noted that the expression pattern of miRNAs belonging to the same family did also vary within a treatment. For example, miR156 family targeting members of the squamosa promoter binding and binding-like transcription factors were up- and down-regulated in PTA (Ptr_miRNA_245 [ptr-miR156b], Ptr_miRNA_329 [ptr-miR156d], Ptr_miRNA_127 [ptr-miR156e], Ptr_miRNA_131 [ptr-miR156g], and Ptr_miRNA_433 [ptr-miR156k]; Supplementary Table S13). This suggests a very intricate role for members of the same miRNA family in these plant-microbe interactions, and further, indicates the specificity to which the miRNAs functions in these tested relationships.
Given that the number of miRNAs identified in P. trichocarpa was approximately 4 times higher than in P. deltoides, and that a large number of miRNAs were in common between PTA and PTE, many with the same expression trend, we hypothesize that species-specific responsive miRNAs may play an important role in the preferential colonization of P. trichocarpa over P. deltoides, as previously reported (Tagu et al., 2001; Labbé et al., 2011). For example, consistent with previous findings in Solanum, Medicago, and Oryza, miR393, which negatively regulates arbuscule formation via an obstruction to the auxin perception, was also down-regulated in PTA in our study (Ptr_miRNA_289 [ptr-miR393b]) but was absent in PDA. The predicted target for Ptr_miRNA_289 [ptr-miR393b] was Potri.002G207800.1, annotated as Transport Inhibitor Response I (TIR/F-box/RNI-like superfamily protein; Supplementary Table S17). When auxin is perceived, TIR of the E3 ligase receptor complex targets the Aux/IAA protein for degradation via ubiquitination which releases the Auxin Response Factors (ARFs) to allow auxin induced gene expression of their targets (Sukumar et al., 2013; Etemadi et al., 2014). Gene components of this auxin signaling are associated with root development where auxin typically stimulates lateral root development and inhibits root elongation (Casimiro et al., 2003; Sukumar et al., 2013). Interestingly, in our study, the two ARFs targets identified, Potri.002G055000.2 and Potri.011G091900.4, should have increased expression since their corresponding miRNA, Ptr_miRNA_407 (ptr-miR167b) was down-regulated as well; thereby supporting the auxin- perception and signaling previously described (Sukumar et al., 2013).
It is also possible that AmF colonization of the P. deltoides roots, which still occurs albeit at a low frequency, proceeds via alternative means. Lauressergues et al. (2012) showed that there was an up-regulation of miR171h during the colonization of Medicago truncatula roots by R. irregularis. The gene target of miR171h is a GRAS transcription factor, NSP2, that is believed to be involved in the biosynthesis of a hormone, strigolactone which promotes germination and growth of AmF. Interestingly, the overexpression of miR171h is associated with a decrease in the target gene expression and an overall decrease in the fungal colonization; while the overexpression of a miR171h-resistant NSP2 gene showed over-colonization. These results suggest that that miR171h is a negative regulator of NSP2 thereby preventing over-colonization. Another study reported increased NSP2 gene levels in mycorrhizal roots despite the presence of miR171h; therefore, suggesting the presence of an addition component regulating miR171h-NSP2 (Hofferek et al., 2014). In our study, we noted an up-regulation of miR171 in PTA and PTE (Ptr_miRNA_339 [ptr-miR171i], common between the treatments; Supplementary Table S13) and down-regulation of miR171 in PDA and PDE (Pde_miRNA_228 [pde-MIR393f], also common between the treatments; Supplementary Table S12). According to Lauressergues et al. (2012), if miR171 is down-regulated, as in the case of PDA, then NSP expression will not be down-regulated thereby increasing mycorrhizal colonization. The alternative proposed by Hofferek et al. (2014) is also true where the NSP2 transcript was elevated during the progression of mycorrhizal colonization independent of miR171h levels, and in the case of PDA will once again promote increased colonization. This hypothesized route may assist in the symbiotic interaction in P. deltoides which has a lower propensity for colonization. A similar trend was noted with pathogenic fungi where a specific miRNAs were either repressed or induced in different plant species, suggesting functional differences in these alternant plants to common pathogens (Gupta et al., 2014). Moreover, as AmF symbiosis progresses miR171 levels should increase (Lauressergues et al., 2012; Hofferek et al., 2014). Ptr_miRNA_339 (ptr-miR171i) was upregulated in the PTA interactions. If miR171 is a negative regulator of NSP2 thereby preventing over-colonization (Lauressergues et al., 2012), the increase in expression in miR171 in PDA could serve as a checkpoint to prevent an imbalance in the mutualism between P. trichocarpa, with its high propensity to be colonized, and the fungus. Future experimental characterization and profiling of these responsive miRNAs and targets should shed some light on this method of colonization and control.
We were also able to identify putative gene targets to the significantly differentially expressed conserved and novel Populus miRNAs (Supplementary Table S17). Our gene ontology enrichment analysis showed consistency with a previous genome-wide study investigating the miRNAs responsive to arbuscular mycorrhiza in tomato, including transcription regulation, biological regulation and response to stimulus (Figure 3; Wu et al., 2016). It was interesting to note that the molecular functions of P. trichocarpa in mycorrhizal associations included transcription factor activity, nucleic acid binding and DNA binding. Plett et al. (2017) showed that SSPs could enter the hyphal nucleus of L. bicolor to affect the fungal physiology. Closer inspection of the miRNAs and their corresponding gene target revealed a large number of transcription factors associated with root formation including growth-regulating factors (GRFs), homeodomain-leucine zipper transcription factor, AP2 transcription factor and MYB transcription factors (Supplementary Table S17). We also found phytohormone related genes including ethylene-responsive transcription factor and auxin response factor, as noted in other studies.
In plants, sRNAs move to adjacent cells via the plasmodesmata and systemically via the vasculature system (Baulcombe, 2004). Recently, it was shown that cross-kingdom/organism RNA interference occurs via exosome-like extracellular vesicles that transfer the host sRNAs to the interacting partner at the infection site, which then consequently causes gene silencing (Wang et al., 2017; Cai et al., 2018; Shahid et al., 2018). Therefore, it is highly probable that plant-encoded sRNAs, during mycorrhizal symbiosis, extends beyond the self-regulation. In support of this hypothesis, we used the plant-encoded miRNAs to predict fungal gene targets in the respective genomes. We identified several fungal gene targets to the Populus-derived miRNAs, including transport proteins, transcription factors and several genes encoding proteins of unknown function in the two fungi analyzed (Supplementary Table S18).
Finally, our study adds to the growing number of reports suggesting the active role of plants in mutualistic associations with microbes (Akiyama et al., 2005; Scharf et al., 2016; Plett et al., 2017). While a number of studies have focused on the arbuscular mycorrhizal-responsive miRNAs (Devers et al., 2011; Wu et al., 2016), genome-wide studies on the endomycorrhizal-responsive miRNAs is in its infancy. Given the role of miRNAs in fungal interactions (Bazin et al., 2013; Etemadi et al., 2014) and the expanding evidence for sORFs in plant physiology and mutualistic symbiosis (Hanada et al., 2007, 2013; Andrews and Rothnagel, 2014; Plett et al., 2017), the current study provides a useful resource of potential regulators that are involved in arbuscular/endomycorrhizal symbiosis with Populus.
Data Availability
The Department of Energy (DOE) will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan). The RNA-Seq reads, along with the processed data (including the sequence and abundance measurements of differentially-expressed transcripts) are deposited in NCBI GEO (https://www.ncbi.nlm.nih.gov/geo/) with the accession code GSE117158.
Author Contributions
XY and JL conceived and supervised the project. RM and HY carried out bioinformatic analysis, interpreted the data, and wrote the manuscript. SJ, RH, and PV carried out the experiments. GT and FL supervised the study and interpreted the data. All authors read and commented on the manuscript.
Funding
This research was sponsored by the Genomic Science Program, U.S. Department of Energy, Office of Science, Biological and Environmental Research as part of the Plant-Microbe Interfaces Scientific Focus Area (http://pmi.ornl.gov). Oak Ridge National Laboratory is managed by UT-Battelle, LLC, for the US Department of Energy under contract DE-AC05-00OR22725. Also, this work was supported by a visiting grant to JL and FL from the Laboratory of Excellence ARBRE (ANR-11-LABX-0002-01) and the Region Lorraine Research Council.
This manuscript has been authored by UT-Battelle, LLC under Contract No. DE-AC05-00OR22725 with the U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00515/full#supplementary-material
Footnotes
- ^https://phytozome.jgi.doe.gov
- ^www.phytozome.com
- ^https://phytozome.jgi.doe.gov/pz/portal.html
- ^https://genome.jgi.doe.gov/Lacbi2/Lacbi2.home.html
- ^http://popgenie.org/
References
Abbott, L. K., and Robson, A. (1977). Growth stimulation of subterranean clover with vesicular arbuscular mycorrhizas. Aust. J. Agric. Res. 28, 639–649. doi: 10.1071/AR9770639
Addo-Quaye, C., Miller, W., and Axtell, M. J. (2009). CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 25, 130–131. doi: 10.1093/bioinformatics/btn604
Akiyama, K., Matsuzaki, K., and Hayashi, H. (2005). Plant sesquiterpenes induce hyphal branching in arbuscular mycorrhizal fungi. Nature 435, 824–827. doi: 10.1038/nature03608
Al-Karaki, G., McMichael, B., and Zak, J. (2004). Field response of wheat to arbuscular mycorrhizal fungi and drought stress. Mycorrhiza 14, 263–269. doi: 10.1007/s00572-003-0265-2
Amaral, P. P., Dinger, M. E., and Mattick, J. S. (2013). Non-coding RNAs in homeostasis, disease and stress responses: an evolutionary perspective. Brief. Funct. Genomics 12, 254–278. doi: 10.1093/bfgp/elt016
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Andrews, S. J., and Rothnagel, J. A. (2014). Emerging evidence for functional peptides encoded by short open reading frames. Nat. Rev. Genet. 15, 193–204. doi: 10.1038/nrg3520
Aspden, J. L., Eyre-Walker, Y. C., Phillips, R. J., Amin, U., Mumtaz, M. A. S., Brocard, M., et al. (2014). Extensive translation of small open reading frames revealed by Poly-Ribo-Seq. eLife 3:e03528. doi: 10.7554/eLife.03528
Bazin, J., Khan, G. A., Combier, J. P., Bustos-Sanmamed, P., Debernardi, J. M., Rodriguez, R., et al. (2013). miR396 affects mycorrhization and root meristem activity in the legume Medicago truncatula. Plant J. 74, 920–934. doi: 10.1111/tpj.12178
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bonfante, P., and Genre, A. (2010). Mechanisms underlying beneficial plant–fungus interactions in mycorrhizal symbiosis. Nat. Commun. 1:48. doi: 10.1038/ncomms1046
Branscheid, A., Sieh, D., Pant, B. D., May, P., Devers, E. A., Elkrog, A., et al. (2010). Expression pattern suggests a role of MiR399 in the regulation of the cellular response to local Pi increase during arbuscular mycorrhizal symbiosis. Mol. Plant Microbe Interact. 23, 915–926. doi: 10.1094/MPMI-23-7-0915
Brosnan, C. A., and Voinnet, O. (2011). Cell-to-cell and long-distance siRNA movement in plants: mechanisms and biological implications. Curr. Opin. Plant Biol. 14, 580–587. doi: 10.1016/j.pbi.2011.07.011
Brundrett, M., Bougher, N., Dell, B., and Grove, T. (1996). Working Ylith Mycorrhizas in Forestry and Agriculture. Canberra: Australian Centre for International Agricultural Research.
Bryan, A. C., Jawdy, S., Gunter, L., Gjersing, E., Sykes, R., Hinchee, M. A., et al. (2016). Knockdown of a laccase in Populus deltoides confers altered cell wall chemistry and increased sugar release. Plant Biotechnol. J. 14, 2010–2020. doi: 10.1111/pbi.12560
Cai, Q., Qiao, L., Wang, M., He, B., Lin, F.-M., Palmquist, J., et al. (2018). Plants send small RNAs in extracellular vesicles to fungal pathogen to silence virulence genes. Science 360, 1126–1129. doi: 10.1126/science.aar4142
Casimiro, I., Beeckman, T., Graham, N., Bhalerao, R., Zhang, H., Casero, P., et al. (2003). Dissecting Arabidopsis lateral root development. Trends Plant Sci. 8, 165–171. doi: 10.1016/S1360-1385(03)00051-7
Castellana, N. E., Payne, S. H., Shen, Z., Stanke, M., Bafna, V., and Briggs, S. P. (2008). Discovery and revision of Arabidopsis genes by proteogenomics. Proc. Natl. Acad. Sci. U.S.A. 105, 21034–21038. doi: 10.1073/pnas.0811066106
Chekanova, J. A. (2015). Long non-coding RNAs and their functions in plants. Curr. Opin. Plant Biol. 27, 207–216. doi: 10.1016/j.pbi.2015.08.003
Chen, X. (2008). MicroRNA metabolism in plants. Curr. Top. Microbiol. Immunol. 320, 117–136. doi: 10.1007/978-3-540-75157-1_6
Chen, X. (2012). Small RNAs in development–insights from plants. Curr. Opin. Genet. Dev. 22, 361–367. doi: 10.1016/j.gde.2012.04.004
Daguerre, Y., Plett, J. M., and Veneault-Fourrey, C. (2017). “Signaling pathways driving the development of ectomycorrhizal symbiosis,” in Molecular Mycorrhizal Symbiosis, ed. F. Martin (Hoboken, NJ: John Wiley & Sons, Inc), 141–157.
Dai, X., and Zhao, P. X. (2011). psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 39, W155–W159. doi: 10.1093/nar/gkr319
Devers, E. A., Branscheid, A., May, P., and Krajinski, F. (2011). Stars and symbiosis: microRNA-and microRNA∗-mediated transcript cleavage involved in arbuscular mycorrhizal symbiosis. Plant Physiol. 156, 1990–2010. doi: 10.1104/pp.111.172627
Duponnois, R., and Garbaye, J. (1991). Mycorrhization helper bacteria associated with the Douglas fir-Laccaria laccata symbiosis: effects in aseptic and in glasshouse conditions. Ann. Sci. For. 48, 239–251. doi: 10.1051/forest:19910301
el Zahar Haichar, F., Santaella, C., Heulin, T., and Achouak, W. (2014). Root exudates mediated interactions belowground. Soil Biol. Biochem. 77, 69–80. doi: 10.1016/j.soilbio.2014.06.017
Etemadi, M., Gutjahr, C., Couzigou, J.-M., Zouine, M., Lauressergues, D., Timmers, A., et al. (2014). Auxin perception is required for arbuscule development in arbuscular mycorrhizal symbiosis. Plant Physiol. 166, 281–292. doi: 10.1104/pp.114.246595
Fahlgren, N., Howell, M. D., Kasschau, K. D., Chapman, E. J., Sullivan, C. M., Cumbie, J. S., et al. (2007). High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS One 2:e219. doi: 10.1371/journal.pone.0000219
Galindo, M. I., Pueyo, J. I., Fouix, S., Bishop, S. A., and Couso, J. P. (2007). Peptides encoded by short ORFs control development and define a new eukaryotic gene family. PLoS Biol. 5:e106. doi: 10.1371/journal.pbio.0050106
Goldberg, T., Hecht, M., Hamp, T., Karl, T., Yachdav, G., Ahmed, N., et al. (2014). LocTree3 prediction of localization. Nucleic Acids Res. 42, W350–W355. doi: 10.1093/nar/gku396
Großhans, H., and Filipowicz, W. (2008). Molecular biology: the expanding world of small RNAs. Nature 451, 414–416. doi: 10.1038/451414a
Gupta, O. P., Sharma, P., Gupta, R. K., and Sharma, I. (2014). Current status on role of miRNAs during plant–fungus interaction. Physiol. Mol. Plant Pathol. 85, 1–7. doi: 10.1016/j.pmpp.2013.10.002
Hanada, K., Higuchi-Takeuchi, M., Okamoto, M., Yoshizumi, T., Shimizu, M., Nakaminami, K., et al. (2013). Small open reading frames associated with morphogenesis are hidden in plant genomes. Proc. Natl. Acad. Sci. U.S.A. 110, 2395–2400. doi: 10.1073/pnas.1213958110
Hanada, K., Zhang, X., Borevitz, J. O., Li, W.-H., and Shiu, S.-H. (2007). A large number of novel coding small open reading frames in the intergenic regions of the Arabidopsis thaliana genome are transcribed and/or under purifying selection. Genome Res. 17, 632–640. doi: 10.1101/gr.5836207
Hofferek, V., Mendrinna, A., Gaude, N., Krajinski, F., and Devers, E. A. (2014). MiR171h restricts root symbioses and shows like its target NSP2 a complex transcriptional regulation in Medicago truncatula. BMC Plant Biol. 14:199. doi: 10.1186/s12870-014-0199-1
Huang, J., Yang, M., and Zhang, X. (2016). The function of small RNAs in plant biotic stress response. J. Integr. Plant Biol. 58, 312–327. doi: 10.1111/jipb.12463
Jorge, V., Dowkiw, A., Faivre-Rampant, P., and Bastien, C. (2005). Genetic architecture of qualitative and quantitative Melampsora larici-populina leaf rust resistance in hybrid poplar: genetic mapping and QTL detection. New Phytol. 167, 113–127. doi: 10.1111/j.1469-8137.2005.01424.x
Kastenmayer, J. P., Ni, L., Chu, A., Kitchen, L. E., Au, W.-C., Yang, H., et al. (2006). Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Res. 16, 365–373. doi: 10.1101/gr.4355406
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, S. L. (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14:R36. doi: 10.1186/gb-2013-14-4-r36
Kloppholz, S., Kuhn, H., and Requena, N. (2011). A secreted fungal effector of Glomus intraradices promotes symbiotic biotrophy. Curr. Biol. 21, 1204–1209. doi: 10.1016/j.cub.2011.06.044
Koske, R., and Gemma, J. (1989). A modified procedure for staining roots to detect VA mycorrhizas. Mycol. Res. 92, 486–488. doi: 10.1016/S0953-7562(89)80195-9
Kumari, P., and Sampath, K. (2015). cncRNAs: Bi-functional RNAs with protein coding and non-coding functions. Semin. Cell Dev. Biol. 47–48, 40–51. doi: 10.1016/j.semcdb.2015.10.024
Labbé, J., Jorge, V., Kohler, A., Vion, P., Marçais, B., Bastien, C., et al. (2011). Identification of quantitative trait loci affecting ectomycorrhizal symbiosis in an interspecific F1 poplar cross and differential expression of genes in ectomycorrhizas of the two parents: Populus deltoides and Populus trichocarpa. Tree Genet. Genomes 7, 617–627. doi: 10.1007/s11295-010-0361-3
Ladoukakis, E., Pereira, V., Magny, E. G., Eyre-Walker, A., and Couso, J. P. (2011). Hundreds of putatively functional small open reading frames in Drosophila. Genome Biol. 12:R118. doi: 10.1186/gb-2011-12-11-r118
Lanet, E., Delannoy, E., Sormani, R., Floris, M., Brodersen, P., Crété, P., et al. (2009). Biochemical evidence for translational repression by Arabidopsis microRNAs. Plant Cell 21, 1762–1768. doi: 10.1105/tpc.108.063412
Lauressergues, D., Delaux, P. M., Formey, D., Lelandais-Brière, C., Fort, S., Cottaz, S., et al. (2012). The microRNA miR171h modulates arbuscular mycorrhizal colonization of Medicago truncatula by targeting NSP2. Plant J. 72, 512–522. doi: 10.1111/j.1365-313X.2012.05099.x
Lefebvre, F. A., and Lécuyer, E. (2017). Small luggage for a long journey: transfer of vesicle-enclosed small RNA in interspecies communication. Front. Microbiol. 8:377. doi: 10.3389/fmicb.2017.00377
Li, H.-M., Hu, C.-S., and Bai, L. (2014). Genome-wide identification of coding small open reading frames: the unknown transcriptome. J. Shanghai Jiaotong Univers. 19, 663–668. doi: 10.1371/journal.pone.0152363
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Ma, J., Diedrich, J. K., Jungreis, I., Donaldson, C., Vaughan, J., Kellis, M., et al. (2016). Improved identification and analysis of small open reading frame encoded polypeptides. Anal. Chem. 88, 3967–3975. doi: 10.1021/acs.analchem.6b00191
Mallory, A. C., and Vaucheret, H. (2006). Functions of microRNAs and related small RNAs in plants. Nat. Genet. 38, S31–S36.
Martin, F., Aerts, A., Ahrén, D., Brun, A., Danchin, E., Duchaussoy, F., et al. (2008). The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature 452, 88–92. doi: 10.1038/nature06556
Martin, F. M., Uroz, S., and Barker, D. G. (2017). Ancestral alliances: plant mutualistic symbioses with fungi and bacteria. Science 356:eaad4501. doi: 10.1126/science.aad4501
Mohanta, T. K., and Bae, H. (2015). Functional genomics and signaling events in mycorrhizal symbiosis. J. Plant Interact. 10, 21–40. doi: 10.1080/17429145.2015.1005180
Molnar, A., Melnyk, C., and Baulcombe, D. C. (2011). Silencing signals in plants: a long journey for small RNAs. Genome Biol. 12:215. doi: 10.1186/gb-2010-11-12-219
Oyama, M., Itagaki, C., Hata, H., Suzuki, Y., Izumi, T., Natsume, T., et al. (2004). Analysis of small human proteins reveals the translation of upstream open reading frames of mRNAs. Genome Res. 14, 2048–2052. doi: 10.1101/gr.2384604
Plett, J. M., Daguerre, Y., Wittulsky, S., Vayssières, A., Deveau, A., Melton, S. J., et al. (2014). Effector MiSSP7 of the mutualistic fungus Laccaria bicolor stabilizes the Populus JAZ6 protein and represses jasmonic acid (JA) responsive genes. Proc. Natl. Acad. Sci. U.S.A. 111, 8299–8304. doi: 10.1073/pnas.1322671111
Plett, J. M., Kemppainen, M., Kale, S. D., Kohler, A., Legué, V., Brun, A., et al. (2011). A secreted effector protein of Laccaria bicolor is required for symbiosis development. Curr. Biol. 21, 1197–1203. doi: 10.1016/j.cub.2011.05.033
Plett, J. M., and Martin, F. (2011). Blurred boundaries: lifestyle lessons from ectomycorrhizal fungal genomes. Trends Genet. 27, 14–22. doi: 10.1016/j.tig.2010.10.005
Plett, J. M., Yin, H., Mewalal, R., Hu, R., Li, T., Ranjan, P., et al. (2017). Populus trichocarpa encodes small, effector-like secreted proteins that are highly induced during mutualistic symbiosis. Sci. Rep. 7:382. doi: 10.1038/s41598-017-00400-8
Powell, C. L. (2018). “Field inoculation with VA mycorrhizal fungi,” in VA Mycorrhiza, eds C. L. Powell and D. J. Bagyaraj (Boca Raton, FL: CRC Press), 205–222.
Ruiz-Ferrer, V., and Voinnet, O. (2009). Roles of plant small RNAs in biotic stress responses. Annu. Rev. Plant Biol. 60, 485–510. doi: 10.1146/annurev.arplant.043008.092111
Ruiz-Orera, J., Messeguer, X., Subirana, J. A., and Alba, M. M. (2014). Long non-coding RNAs as a source of new peptides. eLife 3:e03523. doi: 10.7554/eLife.03523
Scharf, B. E., Hynes, M. F., and Alexandre, G. M. (2016). Chemotaxis signaling systems in model beneficial plant–bacteria associations. Plant Mol. Biol. 90, 549–559. doi: 10.3389/fmicb.2016.01982
Shahid, S., Kim, G., Johnson, N. R., Wafula, E., Wang, F., Coruh, C., et al. (2018). MicroRNAs from the parasitic plant Cuscuta campestris target host messenger RNAs. Nature 553, 82–85. doi: 10.1038/nature25027
Shell, S. S., Wang, J., Lapierre, P., Mir, M., Chase, M. R., Pyle, M. M., et al. (2015). Leaderless transcripts and small proteins are common features of the mycobacterial translational landscape. PLoS Genet. 11:e1005641. doi: 10.1371/journal.pgen.1005641
Sjödin, A., Street, N. R., Sandberg, G., Gustafsson, P., and Jansson, S. (2009). The Populus Genome Integrative Explorer (PopGenIE): a new resource for exploring the Populus genome. New Phytol. 182, 1013–1025. doi: 10.1111/j.1469-8137.2009.02807.x
Soneson, C., and Delorenzi, M. (2013). A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinformatics 14:91. doi: 10.1186/1471-2105-14-91
Sukumar, P., Legue, V., Vayssieres, A., Martin, F., Tuskan, G. A., and Kalluri, U. C. (2013). Involvement of auxin pathways in modulating root architecture during beneficial plant–microorganism interactions. Plant Cell Environ. 36, 909–919. doi: 10.1111/pce.12036
Supek, F., Bošnjak, M., Škunca, N., and Šmuc, T. (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6:e21800. doi: 10.1371/journal.pone.0021800
Tagu, D., Rampant, P. F., Lapeyrie, F., Frey-Klett, P., Vion, P., and Villar, M. (2001). Variation in the ability to form ectomycorrhizas in the F1 progeny of an interspecific poplar (Populus spp.) cross. Mycorrhiza 10, 237–240. doi: 10.1007/PL00009997
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Ulveling, D., Francastel, C., and Hubé, F. (2011). When one is better than two: RNA with dual functions. Biochimie 93, 633–644. doi: 10.1016/j.biochi.2010.11.004
Vaucheret, H. (2006). Post-transcriptional small RNA pathways in plants: mechanisms and regulations. Genes Dev. 20, 759–771. doi: 10.1101/gad.1410506
Wang, M., Thomas, N., and Jin, H. (2017). Cross-kingdom RNA trafficking and environmental RNAi for powerful innovative pre-and post-harvest plant protection. Curr. Opin. Plant Biol. 38, 133–141. doi: 10.1016/j.pbi.2017.05.003
Wheeler, D. L., Church, D. M., Federhen, S., Lash, A. E., Madden, T. L., Pontius, J. U., et al. (2003). Database resources of the national center for biotechnology. Nucleic Acids Res. 31, 28–33. doi: 10.1093/nar/gkg033
Wu, P., Wu, Y., Liu, C.-C., Liu, L.-W., Ma, F.-F., Wu, X.-Y., et al. (2016). Identification of arbuscular mycorrhiza (AM)-responsive microRNAs in tomato. Front. Plant Sci. 7:429. doi: 10.3389/fpls.2016.00429
Yang, X., and Li, L. (2011). miRDeep-P: a computational tool for analyzing the microRNA transcriptome in plants. Bioinformatics 27, 2614–2615. doi: 10.1093/bioinformatics/btr430
Yang, X., Tschaplinski, T. J., Hurst, G. B., Jawdy, S., Abraham, P. E., Lankford, P. K., et al. (2011). Discovery and annotation of small proteins using genomics, proteomics, and computational approaches. Genome Res. 21, 634–641. doi: 10.1101/gr.109280.110
Yi, X., Zhang, Z., Ling, Y., Xu, W., and Su, Z. (2015). PNRD: a plant non-coding RNA database. Nucleic Acids Res. 43, D982–D989. doi: 10.1093/nar/gku1162
Yin, H., Fan, Z., Li, X., Wang, J., Liu, W., Wu, B., et al. (2016). Phylogenetic tree-informed microRNAome analysis uncovers conserved and lineage-specific miRNAs in Camellia during floral organ development. J. Exp. Bot. 67, 2641–2653. doi: 10.1093/jxb/erw095
Keywords: arbuscular mycorrhizal fungus, ectomycorrhizal fungus, Laccaria, microRNA, mutualistic symbiosis, Populus, Rhizophagus, small RNA
Citation: Mewalal R, Yin H, Hu R, Jawdy S, Vion P, Tuskan GA, Le Tacon F, Labbé JL and Yang X (2019) Identification of Populus Small RNAs Responsive to Mutualistic Interactions With Mycorrhizal Fungi, Laccaria bicolor and Rhizophagus irregularis. Front. Microbiol. 10:515. doi: 10.3389/fmicb.2019.00515
Received: 08 April 2018; Accepted: 28 February 2019;
Published: 18 March 2019.
Edited by:
Pierre-Emmanuel Courty, Institut National de la Recherche Agronomique (INRA), FranceReviewed by:
Deqiang Zhang, Beijing Forestry University, ChinaWeiYi Lin, National Taiwan University, Taiwan
Copyright © 2019 Mewalal, Yin, Hu, Jawdy, Vion, Tuskan, Le Tacon, Labbé and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jessy L. Labbé, bGFiYmVqakBvcm5sLmdvdg== Xiaohan Yang, eWFuZ3hAb3JubC5nb3Y=
†These authors have contributed equally to this work
‡Present address: Ritesh Mewalal, Environmental Genomics and Systems Biology Division, Lawrence Berkeley National Laboratory, Walnut Creek, CA, United States