
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 06 March 2019
Sec. Microbial Symbioses
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.00435
This article is part of the Research Topic Advances In The Understanding of The Commensal Eukaryota And Viruses Of The Herbivore Gut View all 10 articles
 Yuanfei Li1
Yuanfei Li1 Yuqi Li1
Yuqi Li1 Wei Jin1,2
Wei Jin1,2 Thomas J. Sharpton3
Thomas J. Sharpton3 Roderick I. Mackie4
Roderick I. Mackie4 Isaac Cann4,5,6,7
Isaac Cann4,5,6,7 Yanfen Cheng1,2*
Yanfen Cheng1,2* Weiyun Zhu1,2
Weiyun Zhu1,2In this study, the effects of a syntrophic methanogen on the growth of Pecoramyces sp. F1 was investigated by characterizing fermentation profiles, as well as functional genomic, transcriptomic, and proteomic analysis. The estimated genome size, GC content, and protein coding regions of strain F1 are 106.83 Mb, 16.07%, and 23.54%, respectively. Comparison of the fungal monoculture with the methanogen co-culture demonstrated that during the fermentation of glucose, the co-culture initially expressed and then down-regulated a large number of genes encoding both enzymes involved in intermediate metabolism and plant cell wall degradation. However, the number of up-regulated proteins doubled at the late-growth stage in the co-culture. In addition, we provide a mechanistic understanding of the metabolism of this fungus in co-culture with a syntrophic methanogen. Further experiments are needed to explore this interaction during degradation of more complex plant cell wall substrates.
In the rumen, microorganisms, which are mainly composed of anaerobic fungi, bacteria, archaea, and protozoa, have coevolved for millions of years, making the rumen one of the most effective and highly evolved systems regarding degradation of recalcitrant lignocellulosic plant material in nature (Russell and Rychlik, 2001; Weimer et al., 2009). Within this system, the diverse microbial communities cooperate efficiently in the digestion and conversion of plant biomass in feeds to various compounds crucial for body maintenance and performance (Kim et al., 2011; Mao et al., 2016). Anaerobic fungi, bacteria, and protozoa degrade and ferment ingested plant biomass and release hydrogen during this process (Akin et al., 1988). However, the accumulation of hydrogen is energetically unfavorable and can inhibit the fermentation of ingested feed (Ungerfeld, 2015). Ruminal methanogens are effective hydrogen utilizers and can use the hydrogen generated to reduce carbon dioxide (which is also a product of primary fermentation) to methane, thereby keeping the steady-state hydrogen concentration low and the rumen operating more efficiently (Janssen and Kirs, 2008). Thus, trophic interactions exist between the methanogenic archaea and the hydrogen-producing microorganisms that includes both anaerobic fungi and bacteria.
Anaerobic fungi assigned to the phylum Neocallimastigomycota play key roles in the decomposition of recalcitrant plant lignocellulosic materials in the rumen. Since the identification of anaerobic fungi by Orpin (1975), 11 genera assigned to the phylum Neocallimastigomycota have been described: Neocallimastix (Heath et al., 1983), Caecomyces (Gold et al., 1988), Piromyces (Gold et al., 1988), Orpinomyces (Barr et al., 1989), Anaeromyces (Breton et al., 1990), Cyllamyces (Ozkose et al., 2001), Buwchfawromyces (Callaghan et al., 2015), Oontomyces (Dagar et al., 2015), Pecoramyces (Hanafy et al., 2017), Feramyces (Hanafy et al., 2018), and Liebetanzomyces (Joshi et al., 2018). Despite their potent capacities for lignocellulose degradation, anaerobic fungi and their enzymes are yet to be exploited in biotechnological processes. This is largely due to their obligately anaerobic lifestyle and a poor understanding of their growth requirements and metabolic characteristics. Anaerobic fungi can ferment a wide range of fermentable sugars, such as glucose, fructose, xylose, and cellobiose as energy sources. These are utilized to produce H2, CO2, formate, acetate, lactate, and ethanol as the major fermentation end products (Lowe et al., 1987; Teunissen et al., 1993). In their natural habitat in the rumen and hind-gut of large mammalian herbivores, anaerobic fungi grow together in communities with other microbes. Anaerobic fungi and closely associated methanogens can be isolated from mixed microbial communities and can be cultured in stable co-culture in media that do not contain appreciable amounts of compounds that methanogens need to grow (Cheng et al., 2009). Anaerobic fungal-methanogen co-cultures have been shown to be stable with robust growth evident over long periods of time (Bauchop and Mountfort, 1981; Cheng et al., 2009). Additionally, in co-cultures, as a consequence of inter-species hydrogen transfer, the metabolite profile of the anaerobic fungus alters, shifting away from more reduced products, such as lactate and ethanol, toward acetate and formate. The formate and hydrogen, end products of fungal fermentation, are used by the methanogens to produce methane (Cheng et al., 2009; Jin et al., 2011; Li et al., 2016). Meanwhile, the fiber-degrading ability of the anaerobic fungus in co-cultures was improved (Jin et al., 2011). Thus, the metabolic profile of anaerobic fungi in the co-culture is comparable to that of their counterparts in the rumen, where hydrogen and formate are known to be transient and low (Hungate, 1967; Hungate et al., 1970), and the fiber-degrading ability is known to be high (Krause et al., 2003). Thus, investigating the interaction between anaerobic fungi and co-cultured methanogen might provide insights into the complex microbial interactions in the rumen.
In recent years, omics-based techniques have been used to study the diversity, ecology, and biology of anaerobic fungi. Five genomes of anaerobic fungal strains have been reported, including Piromyces sp. E2, Pecoramyces ruminantium C1A, Anaeromyces robustus, Neocallimastix californiae, and Piromyces finnis (Youssef et al., 2013; Haitjema et al., 2017). The transcriptomes of Pecoramyces ruminantium C1A, Piromyces finnis, Neocallimastix californiae, Caecomyces churrovis, Anaeromyces mucronatus, Neocallimastix frontalis, Orpinomyces joyonii, Piromyces rhizinflata, and Anaeromyces robustus have been described (Couger et al., 2015; Solomon et al., 2016; Henske et al., 2017; Gruninger et al., 2018). To our knowledge, there are no studies that apply functional genomic, transcriptomic, and proteomic approaches to interrogate the effect of co-culturing a methanogen on the metabolism, including expression of fiber-degrading enzymes, of an anaerobic fungus.
In the present study, we used genomic, transcriptomic, and metabolomic data of the anaerobic fungal monoculture to draw a metabolic pathway of the fungus. The mRNA expression profile of the anaerobic fungus Pecoramyces sp. F1 in the presence and absence of its syntrophic methanogen, Methanobrevibacter thaueri, was also investigated. By combining the foregoing analysis with the anaerobic fungal proteome dynamics and analysis of the metabolites induced by growth with the methanogen, we reveal the effects of the archaeon on the metabolism of the anaerobic fungus.
The anaerobic fungus Pecoramyces sp. F1, formerly described as Piromyces sp. F1, and its symbiotic methanogen, Methanobrevibacter thaueri, were isolated and identified from goat rumen by Jin et al. (2011). The culture was maintained in rumen fluid media (Davies et al., 1993) with 1% (w/v) rice straw as substrate and transferred every 3 days. The media was prepared according to Cheng et al. (2009) and 90 ml media was dispensed into 160 ml serum bottle with 1 g rice straw as substrate. At each transfer, 10 ml of 3-day-old culture was inoculated into 90 ml of fresh media and incubated at 39°C for 3 days. The fungal monoculture was obtained by adding chloramphenicol (50 mg l-1 final concentration) to inhibit the growth of the associated methanogen (Cheng et al., 2009). The relative abundance of methane in the head-space gas of the monoculture was analyzed by GC-TCD (Agilent 7890B, Agilent, Santa Clara, CA, United States) to ensure that no methane was being produced by the culture to confirm that the methanogen was no longer present in fungal pure culture studies.
In the current study, the medium used for experiments was a modified medium M2 (Barichevich and Calza, 1990) with 2.16 g l-1 (12 mM) glucose as substrate. The medium was prepared and dispensed under anaerobic conditions into serum bottles (90 ml/bottle), with pH adjusted to 6.8 (Li et al., 2016). For anaerobic fungal genome sequencing, 40 bottles of Pecoramyces sp. F1 monoculture were incubated at 39°C for 72 h without shaking. The fungal cells were then harvested by centrifugation at 10,000 × g for 15 min.
To investigate the effects of co-culturing with M. thaueri on the metabolism of Pecoramyces sp. F1, the anaerobic fungus was grown alone (monoculture) and also in co-culture with the methanogen at 39°C without shaking. A total of 72 bottles were used for the experiment; details of the protocol information are shown in Supplementary Figure 1. Samples were collected from each replicate for transcriptomic, proteomic, and metabolite analysis. The total volume of gas accumulated in each culture over the incubation period was also measured using the pressure transducer technique (Theodorou et al., 1994). After each reading, the head-space was vented to return the pressure to ambient conditions. Furthermore, the gas drawn was analyzed for CH4 and H2 content. Samples from the cultures were collected at approximately 50% and 100% of maximum gas production (i.e., mid- and late-growth stages) as determined from previously generated gas accumulation curves. The pH was measured at each time point immediately upon removing crimp-seals and stoppers from the serum vials. Aliquots of 5 ml supernatant were then collected and stored at -20°C for subsequent analysis of water-soluble metabolites. The rest of the culture was then centrifuged at 8,000 × g for 15 min, and 1 ml of supernatant was used for the analysis of residual glucose with a commercial glucose kit (Nanjing Jiancheng Biotechnology Institute, Nanjing, China). The cells from the remaining six bottles, representing each replicate, were then mixed and split into two parts for RNAseq and iTRAQ analysis. Two bottles of each replicate were used for the analysis of gas, glucose, pH, and water-soluble metabolites.
Genomic DNA was extracted from a 3-day-old anaerobic fungal monoculture with the CTAB method (Cheng et al., 2017). Briefly, the culture was centrifuged and ground in liquid nitrogen. CTAB buffer was added to dissolve the powder and phenol/chloroform/isoamyl alcohol (25:24:1) was then used to purify the DNA. Three libraries with insert sizes of 170 bp, 350 bp, and 6,000 bp were prepared at BGI (Beijing Genomics Institute, Shenzhen, China) according to the manufacturer’s instructions (Illumina). Paired-end sequencing was conducted on an Illumina Hiseq 2000 platform (BGI, Shenzhen, China). A total of 28.67 Gb in 159,302,966 quality-filtered paired-end reads were used for assembly (Supplementary Table 1). The quality-filtered reads were assembled with SOAPdenovo V1.05 (Li et al., 2008, 2010) using a kmer value of 43. The assembly was then optimized by the paired-end and overlap relationship of reads through mapping reads to assembled contigs. Gene calling was then conducted using a combination of Augustus V2.6.1 and Genemarkes V2.3e (Ter-Hovhannisyan et al., 2008; Keller et al., 2011). Transposable elements (TEs) were identified by RepeatMasker (Repbase) and RepeatProteinMasker1. Tandem repeats were identified by Tandem Repeat Finder (TRF) (Benson, 1999). The number of simple sequence repeats (SSRs) were calculated using the results of TRF according to Youssef et al. (2013). The rRNAs and tRNAs were identified using RNAmmer 1.2 (Lagesen et al., 2007) and tRNAscan-SE 1.23 (Lowe and Eddy, 1997), respectively. BLAST was used for the annotation of gene models against KEGG, GO, CAZy, Uniprot_Swissprot and non-redundant (NR) databases (Bard and Winter, 2000; Kanehisa et al., 2004; Cantarel et al., 2009; The UniProt Consortium, 2015). The genome assembly and gene calling and annotation were conducted by BGI (Shenzhen, China). The raw data was submitted to SRA under the accession number: PRJNA517297.
The RNA for RNAseq analysis were isolated from the mid- and late-growth stages of the anaerobic fungal monoculture and co-culture. The RNAseq libraries, which included only mRNA, were generated according to the Illumina TruSeq RNA sample protocol. The mRNA was enriched using oligo-dT (Rio et al., 2010). Paired-end sequencing was conducted on an Illumina Hiseq 2000 platform (BGI, Shenzhen, China). All quality-filtered reads were mapped to the genome and genes by BWA (Li and Durbin, 2009) and Bowtie (Langmead et al., 2009), respectively. The number of reads produced per sample and the mapping results are provided in Supplementary Table 2. The quantification of gene expression was calculated in fragments per kilobase of transcript per million mapped reads (FPKM) with the RSEM package (Li and Dewey, 2011). To assess variability between biological replicates, the coefficient of determination R2 was calculated between biological replicate pairs using RSEM-generated FPKM values (Supplementary Table 3). The raw data was submitted to SRA under the accession number: PRJNA517315. Differentially expressed genes were screened with the NOISeq package (Tarazona et al., 2015) according to the following criteria: fold change > ±2 and divergence probability >0.8.
Proteins for iTRAQ analysis were collected from the mid- and late-growth stages of the anaerobic fungal monoculture and co-culture. The cells were digested and labeled according to Yan et al. (2016). One biological replicate from each sample (four samples in total) was then mixed as one iTRAQ set resulting in three iTRAQ sets that were analyzed. The mixed fractions were then separated by liquid chromatography (LC) and analyzed by two-step mass spectrometry (MS) (Yan et al., 2016). All procedures were conducted at BGI (Shenzhen, China). The MGF files, converted from the raw data using a 5600 msconverter, were used for protein identification with the Mascot search engine (Matrix Science, London, United Kingdom; version 2.3.02) against the fungal transcriptome containing 17,639 sequences (Yan et al., 2016). The identification of proteins in the three sets is shown in Supplementary Table 4. The proteomic dataset was deposited in the iPROX database under the accession number IPX0001499000. The criteria for differential expression of proteins was a P-value < 0.05 and fold change > ±1.2 in at least two iTRAQ sets.
The genomic DNA from the anaerobic fungal monoculture was used to amplify the 28S rRNA gene using the primer pair AF-LSU (5′-GCTCAAAYTTGAAATCTTMAAG-3′) and AF-LSU (5′-CTTGTTAAMYRAAAAGTGCATT-3′) (Dollhofer et al., 2016). To amplify the ITS sequence, primers ITS1 (5′-TCCGTAGGTGAACCTGCGG-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) were used (White et al., 1990). The PCR reaction (20 μl) consisted of 0.5 μl of each primer, 1 μl of the template DNA and 10 μl of PCR Master Mix. For 28S rRNA gene amplification, after initial denaturation at 94°C for 3 min, 36 cycles of amplification were performed, with 94°C for 20 s denaturation, 61°C for 45 s annealing, 72°C for 45 s extension, and a final extension of 72°C for 10 min. For the amplification of ITS sequence, we performed an initial denaturation at 95°C for 3 min, followed by 39 cycles of amplification with 95°C for 30 s denaturation, 52°C for 1 min annealing, 72°C for 1 min extension, and a final extension of 72°C for 5 min. The sequences were deposited at the GenBank under accession numbers MG250475 and MG250482 for 28S rRNA gene and ITS sequences, respectively. 28S rRNA gene and ITS sequences from representatives of the anaerobic fungal genera were retrieved and used to construct phylogenetic trees with MEGA 6 (Tamura et al., 2013).
The head-space gas of the culture was collected and analyzed for relative abundances of H2 and CH4 using GC-TCD (Agilent 7890B, Agilent, Santa Clara, CA, United States) according to Li et al. (2016). The volumes of H2 and CH4 were then calculated according to the total gas production. The concentration of ethanol was measured by GC-TCD using a method described by Li et al. (2016). The concentrations of formate, acetate, lactate, malate, citrate, and succinate were analyzed by HPLC (1220 Infinity LC system, Agilent, Santa Clara, CA, United States) with a reversed phase column ZorbaxSB-Aq (Agilent, Santa Clara, CA, United States) according to Li et al. (2016). The statistical analysis of glucose, gas, pH, and fermentation end products was conducted in RStudio2 and a significant effect was declared at P < 0.05.
The anaerobic fungus in the present study was isolated in co-culture with the methanogen, M. thaueri and the results were published by Jin et al. (2011). Based on the fungal morphology, particularly the monocentric fungal thallus and presence of monoflagellated zoospores, the fungal component of the co-culture was assigned to the genus Piromyces (Jin et al., 2011). Subsequently it became apparent that the newly discovered genus, Pecoramyces was morphological similar to some Piromyces isolates (Hanafy et al., 2017). To obtain a more accurate identification of our fungal isolate, we applied molecular techniques based on the amplification and sequencing of the gene encoding the 28S rRNA and its ITS sequences. Using these sequences information, two phylogenetic trees were constructed based on the 28S rRNA gene sequence and the ITS sequence, respectively (Supplementary Figure 2). Both phylogenetic trees confirmed that the fungus isolated in co-culture with M. thaueri (Jin et al., 2011) is a member of the newly described anaerobic fungal genus, Pecoramyces (Hanafy et al., 2017), and is subsequently referred to as Pecoramyces sp. F1.
The genome of Pecoramyces sp. F1 was sequenced using paired-end Illumina technology with approximately 268× coverage. Results estimated the genome size of this fungus to be 106.83 Mb (Table 1). As observed in previously reported anaerobic fungal genomes (Youssef et al., 2013; Haitjema et al., 2017), Pecoramyces sp. F1 exhibited low GC content (16.07%) with a very low proportion of the genome used in coding for proteins (23.54%). From the data, it was estimated that the genome encoded 17,740 genes with an average length of 1,918 bp. A comparison of the Pecoramyces sp. F1 genome with five published anaerobic fungal genomes is shown in Table 2. The implications relating to genome structure are discussed later. The putative pathway for metabolism of glucose by Pecoramyces sp. F1 was demonstrated in Supplementary Figure 3 based on genomic and transcriptomic data. Comparison of the genomes of anaerobic fungi and aerobic fungi was demonstrated in Supplementary Figure 4.
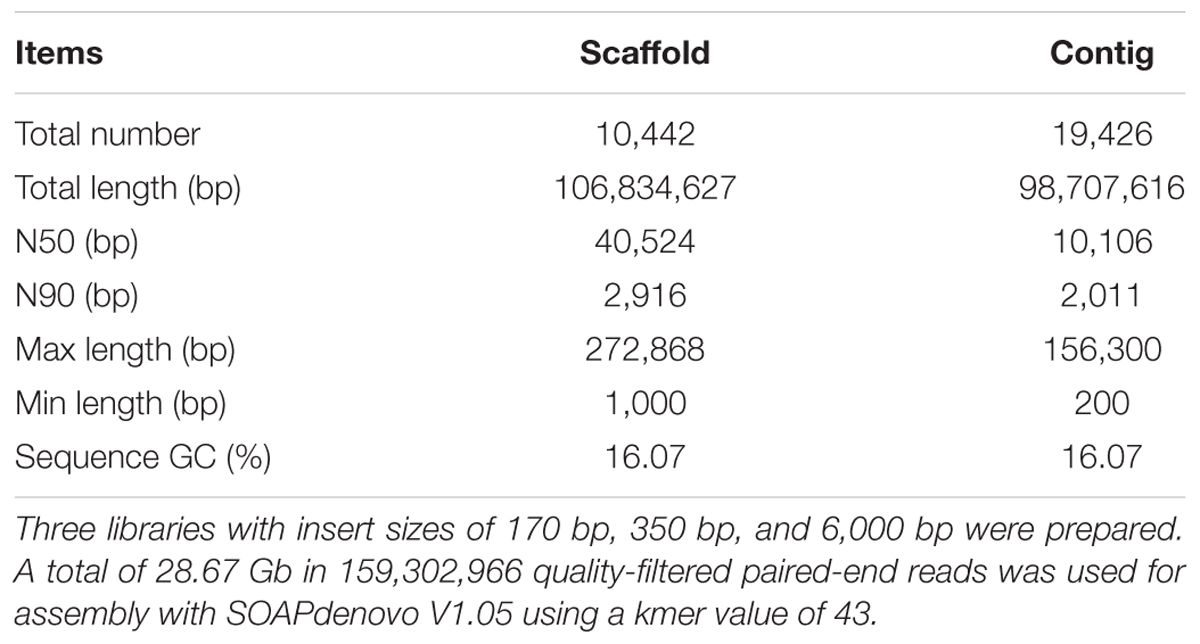
Table 1. The assembly of the genome of Pecoramyces sp. F1.
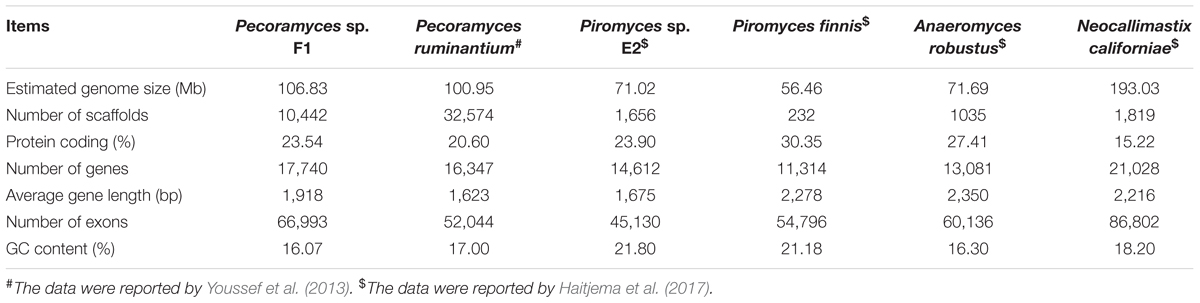
Table 2. A comparison of genomes of Pecoramyces sp. F1 and other anaerobic fungi.
The gas production curves of the anaerobic fungal monoculture and co-culture with the methanogen are shown in Figure 1. The co-culture grew more rapidly and produced more gas, reaching mid- and late-growth stages sooner than the corresponding axenic cultures (Figure 1A). A total gas volume of 107 ml in the anaerobic fungus/methanogen co-culture was measured after 66 h of cultivation compared with 90 ml after a longer incubation time of 80 h of the monoculture. Large amounts of H2 accumulated in the monoculture whereas it was undetectable in the co-culture. As expected, CH4 accumulated in the anaerobic fungus/methanogen co-culture (Figure 1B). For further molecular analysis, samples were taken at mid- and late-growth stages.
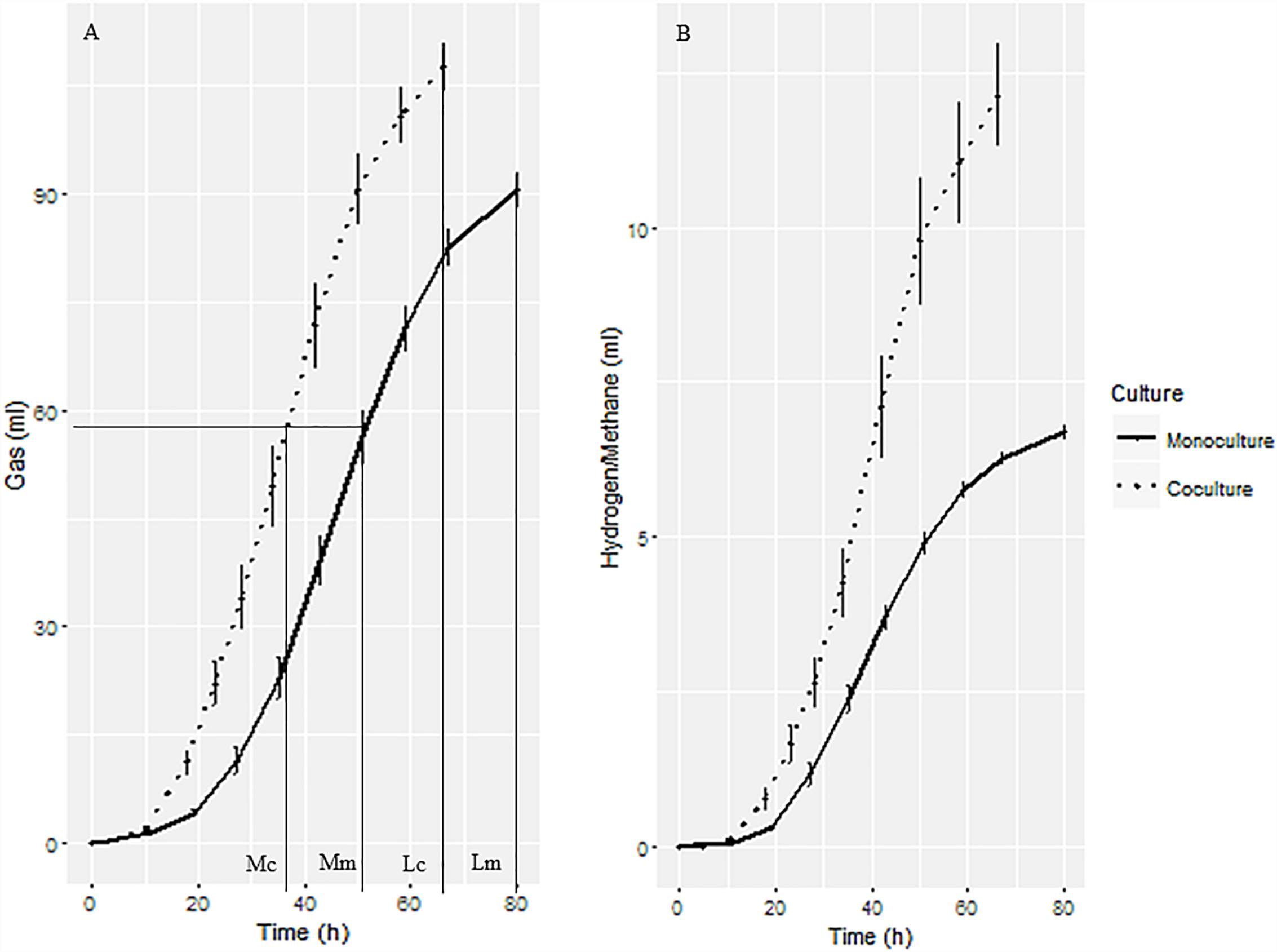
Figure 1. Gas production of anaerobic fungal monoculture and fungal-methanogen co-culture. (A) Cumulative gas production curve showing the sampling time for transcriptomic, proteomic, and fermentation end products analysis. Mc, mid-growth stage of co-culture; Mm, mid-growth stage of monoculture; Lc, late-growth stage of co-culture; Lm, late-growth stage of monoculture. (B) H2 and CH4 production from fungal monoculture and co-culture, respectively.
Based on transcriptional analysis (mRNA data), at the mid-growth stage 12,262 ± 171 and 12,176 ± 311 genes were expressed in the monoculture and co-culture, respectively (P > 0.05). In comparison to the monoculture, it was observed that 62 and 121 genes were up-regulated and down-regulated, respectively, in the co-culture (Supplementary Table 5). The top 10 up-regulated and down-regulated genes and their functional annotations are shown in Table 3. Half of the top 10 up-regulated genes were annotated as fiber-degrading enzymes. The number of genes undergoing alternative splicing were examined and 8,281 ± 878 and 7,727 ± 169 alternatively spliced genes were detected in the monoculture and co-culture, respectively (P > 0.05).
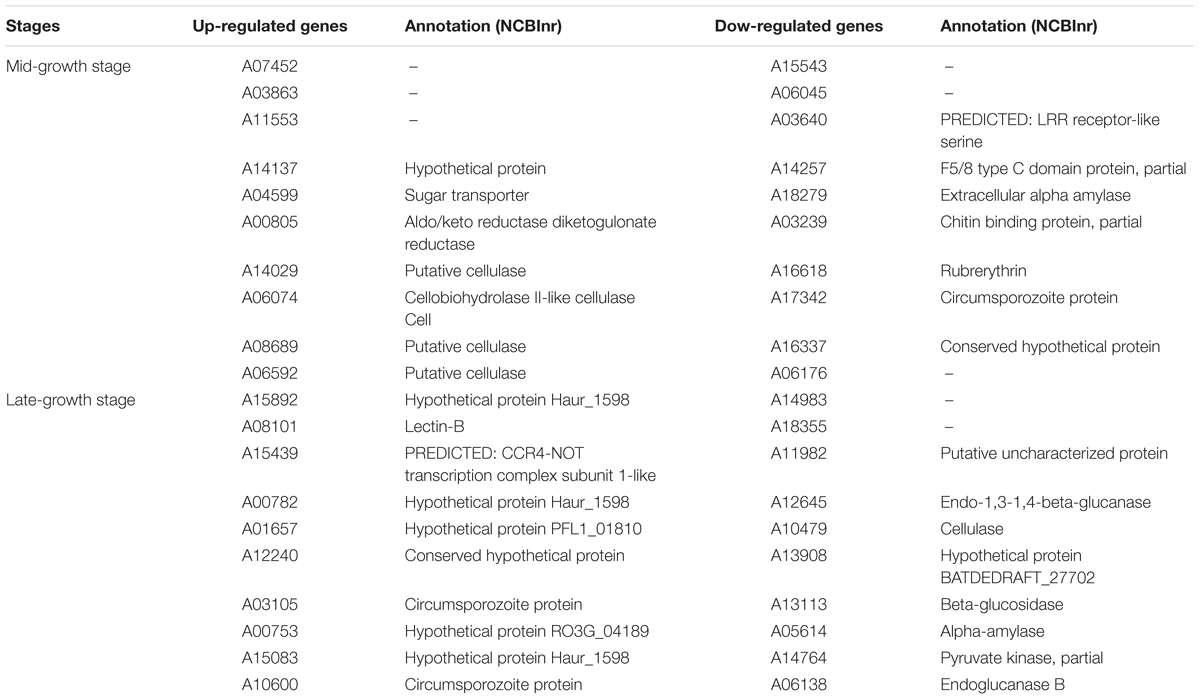
Table 3. The top 10 up-/down-regulated genes of anaerobic fungus Pecoramyces sp. F1 at mid- and late-growth stages.
In addition to the transcriptional analysis, proteomic analysis was carried out on total proteins at mid- and late-growth stages using the iTRAQ approach. A total of 2,149 proteins were identified (MASCOT) and quantified in all three replicates in both cultures at the mid- and late-growth stages. In comparison with the monoculture, it was observed that 117 and 162 proteins were up-regulated and down-regulated, respectively, in the co-culture at the mid-growth stage. The top 10 up-regulated and down-regulated proteins and their functions are shown in Table 4. It is significant that many of the transcripts and proteins that were highly up-regulated or down-regulated had no matches in the databases included in this study (Tables 3, 4), however, a large number of proteins associated with cellular-binding and transmembrane activities were moderately up-regulated (>2 and <100 folds) (Supplementary Table 6).
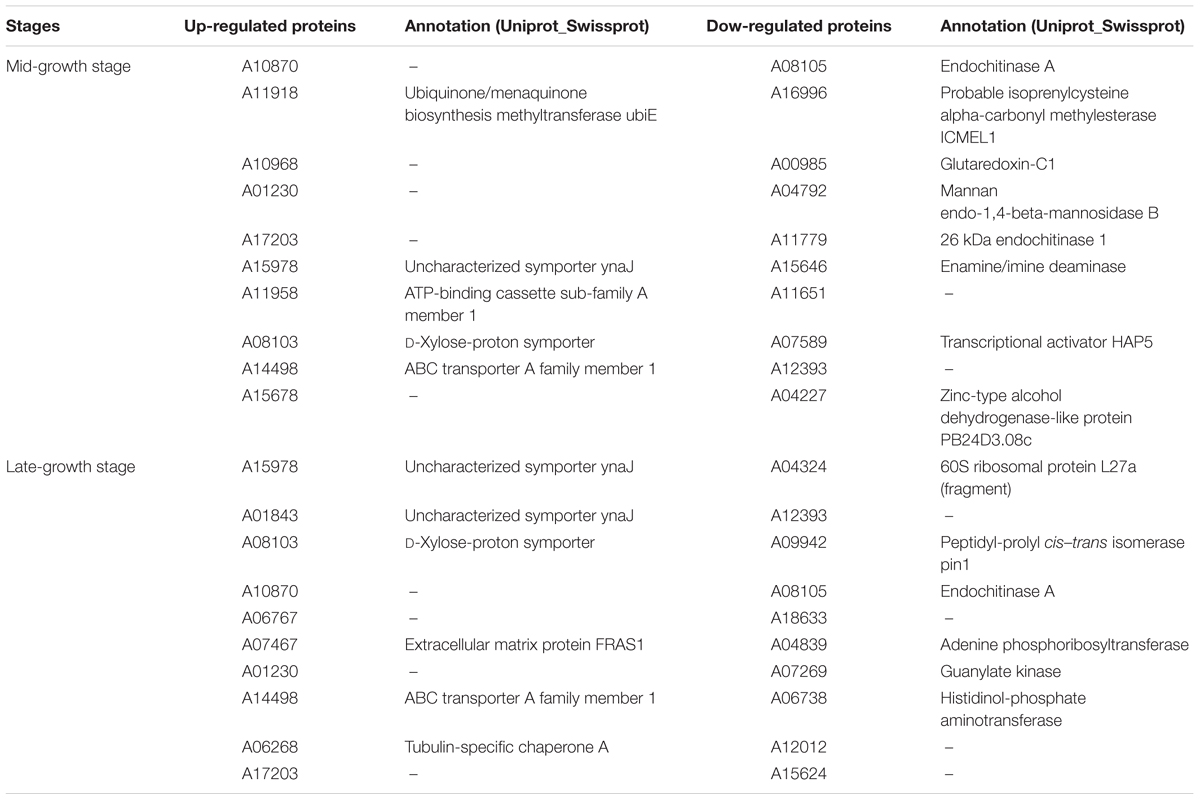
Table 4. The top 10 up-/down-regulated proteins of anaerobic fungus Pecoramyces sp. F1 at mid- and late-growth stages.
The pH value of the co-culture, although not very different, was significantly higher (6.5 ± 0.03) than that of the monoculture (6.4 ± 0.03) (P < 0.05) at the mid-growth stage. Metabolites, including formate, lactate, acetate, ethanol, succinate, malate, and citrate were detected in the supernatant of the monoculture; formate, lactate, succinate, malate, and citrate concentrations were significantly decreased when measured in the anaerobic fungus/methanogen co-culture when compared with the fungal monoculture (Figure 2).
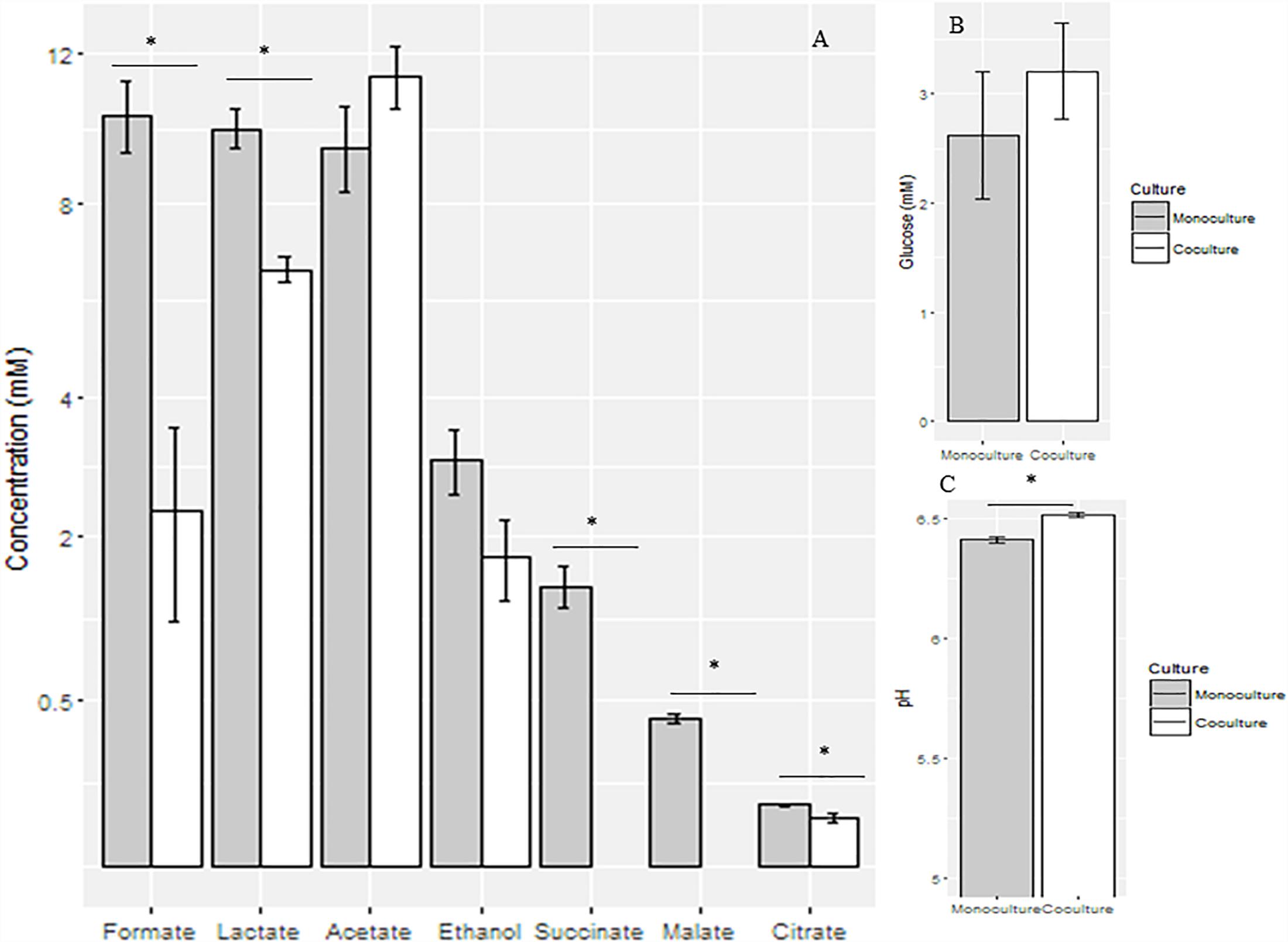
Figure 2. Concentrations of fermentation end products (A) and glucose (B) and pH values (C) in the supernatant of anaerobic fungal monoculture and co-culture at the mid-growth stage. ∗Means significant difference between two groups.
The effects of the methanogen on the metabolism of Pecoramyces sp. F1 in co-culture at the mid-growth stage is presented in Supplementary Figure 5. The expression levels of aconitase and NADH dehydrogenase genes were down-regulated in the co-culture, while no significant differences were observed at the protein level. The expression levels of lactate dehydrogenase and pyruvate formate lyase (PFL) genes were not affected by co-culturing the fungus with the methanogen, although they were up-regulated at the protein level. In the case of aldehyde/alcohol dehydrogenase, it was found to be down-regulated at both the transcription and protein levels.
Measurements made at the late-growth stage showed that 11,978 ± 237 and 10,010 ± 348 genes were expressed in the monoculture and co-culture, respectively (P < 0.05). Relative to the monoculture, 42 and 852 of the expressed genes were up-regulated and down-regulated, respectively, in the co-culture. It was observed that most of the highly up-regulated genes at the transcriptional level in the co-culture (RNA fold change > ±100) were related to binding activities in the cell (Supplementary Table 7). The top 10 up-regulated and down-regulated genes and their functional annotations are shown in Table 3. In comparison to the mid-growth stage, at the late-growth stage, fewer genes were alternatively spliced. Thus, we observed 5,908 ± 603 and 2,061 ± 226 genes were alternatively spliced in the monoculture and the co-culture, respectively (P < 0.05).
In the late-growth stage, the number of proteins up-regulated was double that at the mid-growth stage cultures (276 versus 117). In the case of the down-regulated proteins, however, there was no difference in the numbers observed for the mid- and late-growth stage cultures (168 versus 162). Most of the highly up-regulated proteins (protein ratio > ±2) were related to sporulation, transmembrane, and cellular-binding activities (Supplementary Table 8). The top 10 up-regulated and down-regulated proteins and their functions are shown in Table 4.
As observed in the mid-growth stage, the pH value of the co-culture (6.53 ± 0.002) was significantly higher than that of the monoculture (6.24 ± 0.01) (P < 0.05) at the late-growth stage. Relative to the monoculture, the concentrations of formate, lactate, malate, and citrate were significantly decreased in the co-culture (P < 0.05), while the concentrations of acetate and succinate were significantly increased in the co-culture (P < 0.05). In contrast, the concentration of ethanol did not vary between the monoculture and the co-culture (Figure 3).
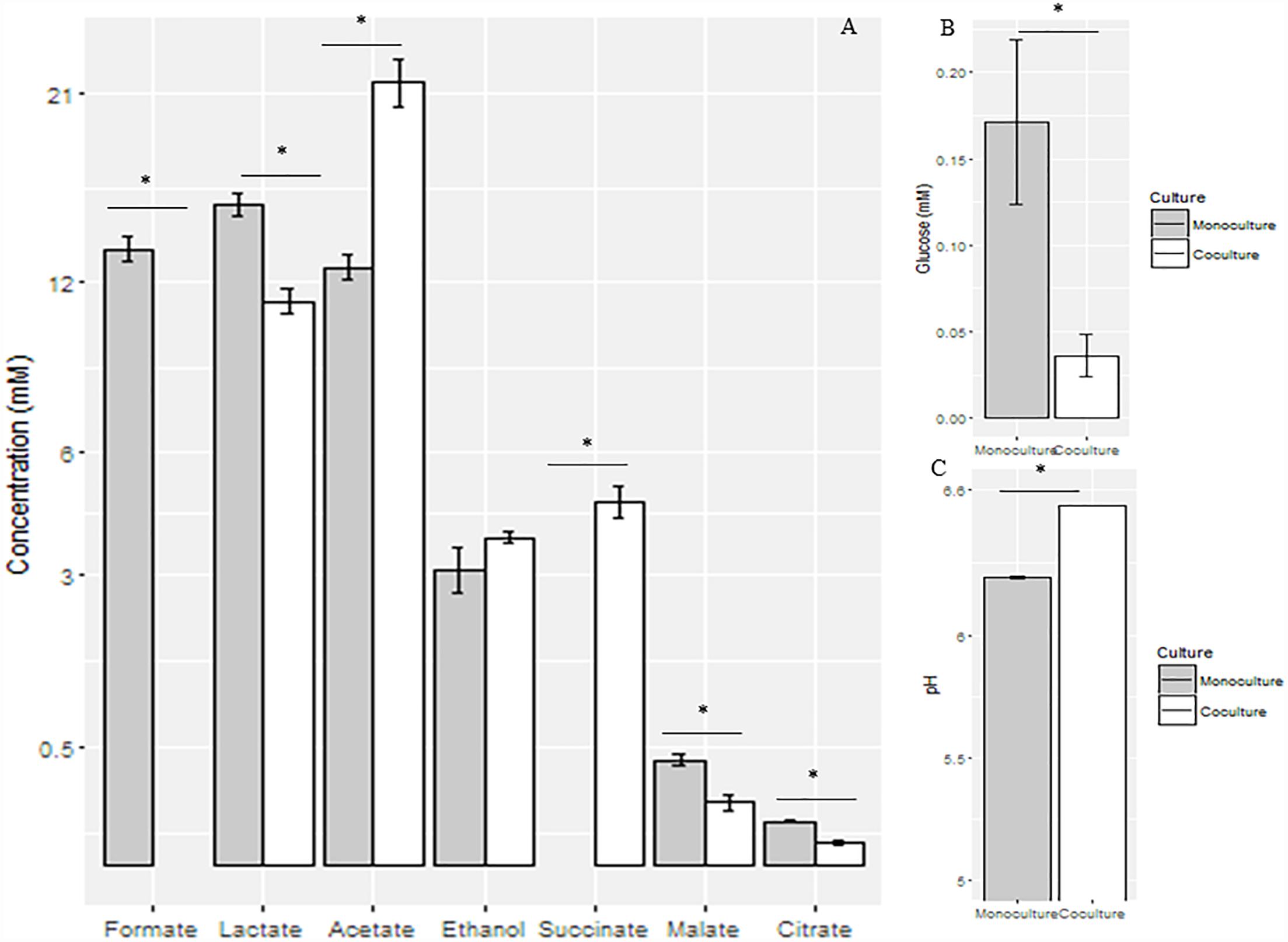
Figure 3. Concentrations of fermentation end products (A) and glucose (B) and pH values (C) in the supernatant of anaerobic fungal monoculture and co-culture at the late-growth stage. ∗Means significant difference between two groups.
The effects of co-culturing the methanogen with Pecoramyces sp. F1 on metabolism at the late-growth stage are shown in Supplementary Figure 6. At the gene expression level, all of the enzymes, except for fumarase, involved in the metabolism of glucose in the anaerobic fungus were down-regulated when cultured with the methanogen, while only glyceraldehyde-3-phosphate dehydrogenase (GAPDH), phosphoglycerate mutase (PGM), PFL, and aldehyde/alcohol dehydrogenase (ADH) were also down-regulated at the protein level.
The top 20 differentially expressed fiber-degrading enzymes at the mid- and late-growth stages were examined (Table 5). Importantly, all of these genes were down-regulated at the late-growth stage. Although it was not anticipated that polysaccharide-degrading enzymes are required for the metabolism of glucose, our search for the top 20 fiber-degrading enzymes showed that several genes coding for such enzymes were up-regulated in the mid-growth stage. The up-regulated genes included the afore-mentioned putative cellulase and others with encoded polypeptides annotated as cellulases, endoxylanases, alpha-amylases and a feruloyl esterase. Thus, a broad range of polysaccharide-degrading enzymes were released during the early stages of glucose metabolism in co-culturing of Pecoramyces sp. F1 with its syntrophic methanogen. However, these polysaccharide targeting enzymes were down-regulated during the late-growth stage.
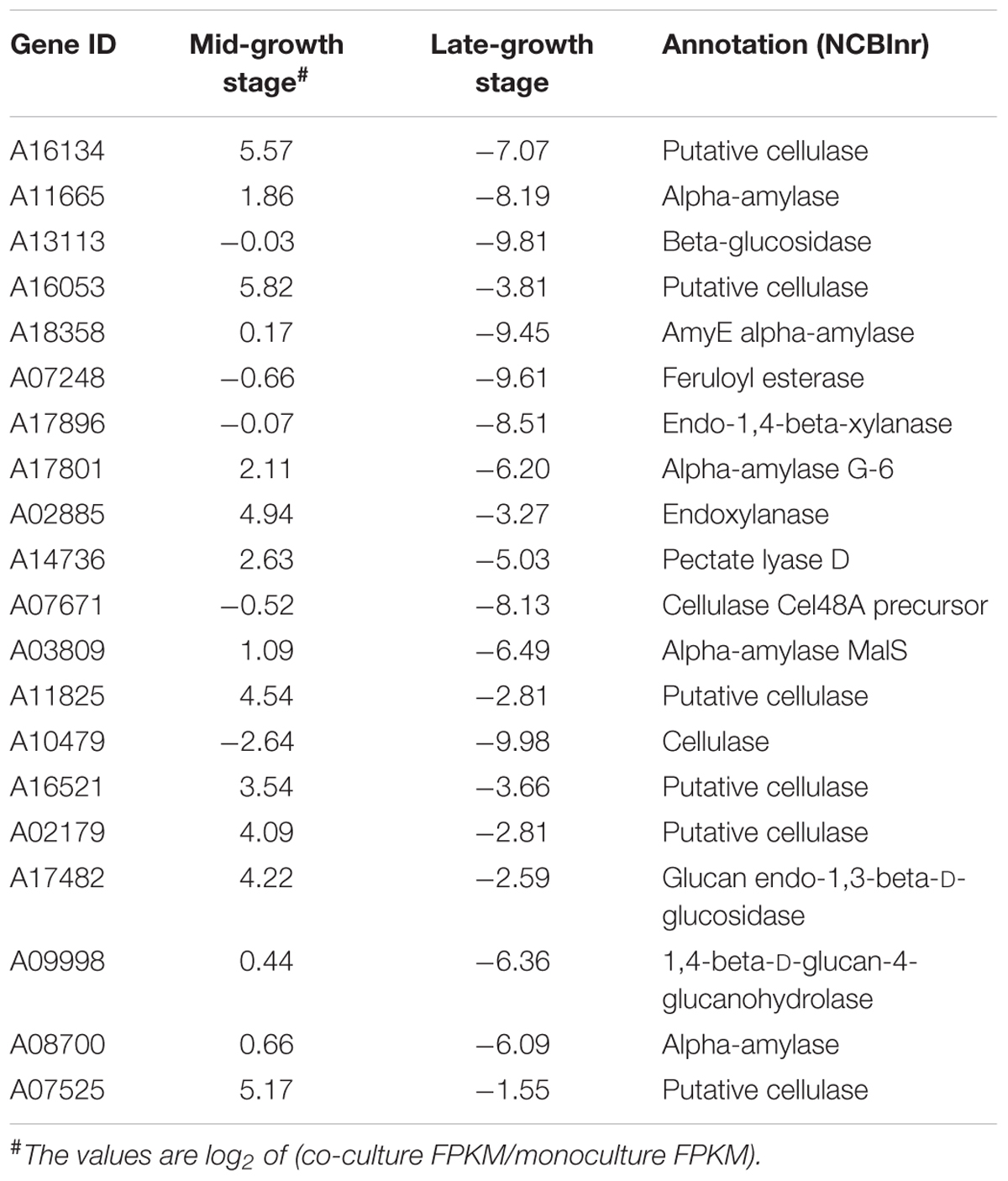
Table 5. Top 20 differentially expressed fiber degrading enzymes at mid- and late-growth stages.
Isolation and maintenance of anaerobic fungi requires a relatively complex, strictly anaerobic culture methodology limiting their study to relatively few research groups world-wide. Consequently, we do not have a good understanding of the diversity and taxonomy of these unique fungi. According to classical taxonomy, zoospore ultrastructure and to a lesser extent, fungal morphology were used to assign generic and specific names to isolates (Theodorou et al., 1996; Ozkose et al., 2001). More recently, molecular techniques based on the amplification and sequencing of genes encoding the 28S rRNA gene and ITS sequences have been used to aid classification. In the current work, molecular techniques were used to reassign the isolate Piromyces sp. F1 to Pecoramyces sp. F1. To date, only one species of Pecoramyces (P. ruminantium) has been described (Youssef et al., 2013; Hanafy et al., 2017). To date, a limited number of publications have studied fungal/methanogen interactions (Mountfort et al., 1982; Nakashimada et al., 2000; Jin et al., 2011; Li et al., 2016). In much of the original work, fungi and methanogens were isolated separately from different ruminal environments (Mountfort et al., 1982; Nakashimada et al., 2000). In working with the new isolate of Pecoramyces and its syntrophically associated methanogen, M. thaueri, we studied the metabolism of this isolate on glucose to obtain primary information about a Pecoramyces strain grown in monoculture and in co-culture with a syntrophic methanogen.
The estimated genome size of Pecoramyces sp. F1 matched that of the previously reported estimate for Pecoramyces ruminantium, as shown in Table 2 (Youssef et al., 2013). This observation shows that Pecoramyces has a larger genome size compared with the Piromyces and Anaeromyces genera, although the estimated genome size of a Neocallimastix is double the size of Pecoramyces. In contrast, the data further demonstrates that the anaerobic fungal genomes are consistently AT-rich (GC% content range from 16 to 22; Table 2). The genera Piromyces and the Anaeromyces appear to have fewer genes (∼13,000) compared to the genus Pecoramyces (∼17,000), while the gene number reported for Neocallimastix is almost twice that for Piromyces and Anaeromyces. While Piromyces finnis codes for approximately 11,000 genes, the number of genes coded by Piromyces sp. E2 is not very different from that of Pecoramyces ruminantium (Youssef et al., 2013). The number of genes coded by Neocallimastix californiae (Haitjema et al., 2017) shows that this fungal species uses about twice the genome of the Pecoramyces strains to encode a number of genes only slightly higher than that of the Pecoramyces strains. Therefore, the protein coding percentage of the genome of the reported Neocallimastix strain is very low in comparison with the other genera discussed in this manuscript.
The results from this study show that alternative splicing occurs in Pecoramyces sp. F1, as reported in the aerobic fungi (Grutzmann et al., 2014). Meanwhile, the average alternative splicing rates of Pecoramyces sp. F1 (∼45% and 22% at the mid- and late-growth stages, respectively) seem higher than the aerobic fungi, which was 6.4% on average (Grutzmann et al., 2014). Furthermore, it was observed that alternative splicing in the co-culture of the fungus with the methanogen was significantly lower than the fungal monoculture at the late-growth stage. The decreased splicing might be due to the limitation of the substrate in the culture or a slower growth rate associated with substrate depletion; Birch et al. (1995) reported that differential splicing in Phanerochaete chrysosporium might regulate the specificities of substrate of this fungus.
As observed in previous reports (Cheng et al., 2009; Jin et al., 2011; Li et al., 2016) during co-culturing of anaerobic fungi with methanogens, total gas production exceeded that of the gut fungal culture alone and the rate of gas production was faster. This observation confirms the increased efficiency with which the anaerobic fungi ferment substrates in the presence of the hydrogen-utilizing methanogen. Sampling at the mid-growth stage showed that total mRNA expression was not different between the monoculture and co-culture and the number of up-regulated genes was half the number of the down-regulated genes. However, by the late-growth phase, mRNA expression of the co-culture was significantly lower than that of the monoculture and the ratio of the up-regulated and down-regulated genes were dramatically decreased. The mRNA expression profiles suggest that on encountering a glucose energy source, Pecoramyces sp. F1 secretes a large number of polysaccharide degrading enzymes including endoglucanases, chitinases, amylases, and licheninases. In the case of the monoculture, this enzyme secretion appears to continue throughout growth, perhaps due to comparatively inefficient substrate utilization. On coupling the fermentation of the anaerobic fungus with the methanogen, the efficiency of the fermentation increased, leading to a down-regulation of the expression of the polysaccharide degrading enzymes. As shown in Table 5, this is particularly so for the putative enzymes involved in cellulose metabolism, including about six putative cellulases, likely reflecting the hydrolysis of the cellulose backbone. The efficiency of the fermentation in anaerobic fungus/methanogen co-culture increases is likely due to the removal of H2 through interspecies transfer to the syntrophic methanogen to produce CH4. In the fungal cell, the oxidization of NADH into NAD+ and H+ is associated with the production of acetic acid. This pathway is likely to be favorable for obtaining higher amounts of ATP, compared with the more reduced electron sinks end-products (e.g., lactate, ethanol) used by anaerobic fungi to regenerate NAD+ for glycolysis (Bauchop and Mountfort, 1981; Marvin-Sikkema et al., 1990).
The results in the present study are in agreement with the observation that during syntrophic interactions between several ruminal organisms with hydrogen-removing methanogens, a shift in the metabolism occurs leading to extra ATP gain by the organism co-cultured with the methanogen (Bauchop and Mountfort, 1981; Marvin-Sikkema et al., 1990). Unlike the transcriptomic data, major shifts in the proteomic data were not observed in the present study. This may be due to the fact that the proteins in the cell have a much longer lifetime than that of the mRNAs. A genome-wide study showed that the lifetime of mRNAs in Escherichia coli were between 3 and 8 min (Bernstein et al., 2002). However, the rate of intracellular protein degradation in E. coli was 4 h (Koch and Levy, 1955).
The changes in the metabolites observed in the present study are similar to the results observed in our previous studies (Li et al., 2016, 2017). In brief, the pH value increased significantly at the late-growth stage as formate was utilized by the co-cultured methanogen and the lactate decreased due to a reduced demand for electron sink products for regeneration of reducing equivalents. Finally, acetate increased significantly because metabolism in the hydrogenosome became more efficient.
Combining the data reported in the present study and previous reports on anaerobic fungi and methanogens co-culture (Cheng et al., 2009; Li et al., 2016, 2017), we found that in the early growth stage of the co-culture, the metabolism in the fungal cell improved and large amounts of end products were produced. At this growth stage, the substrate was adequate and only H2 was used by co-cultured methanogens to reduce the gas pressure, which could inhibit the microbial growth (Li et al., 2016). At the late-growth stage, the substrate was inadequate for anaerobic fungi to produce enough H2 and methanogens would use formate to produce methane, which increased the pH value of the culture. The metabolic interaction between the two organisms would help both of them to be competitive in the rumen. For the anaerobic fungus, the fiber-degrading ability was improved and feedback inhibition (both gas pressure and water-soluble metabolites) was eliminated. For the methanogen, it could obtain H+ as soon as it was produced.
In summary, in the present report we have used modern molecular approaches to assign phylogenetic placement to a new anaerobic fungal isolate and concomitantly provided a mechanistic understanding of its intermediary metabolism in co-culture with a syntrophic methanogen. We look forward to future experiments that explore interactions during degradation of more complex substrates.
YC and WZ conceived and designed the experiments. WJ, YfL, and YqL performed the experiments. YfL, YqL, WJ, YC, TS, RM, and IC generated and analyzed the data. YC, TS, RM, IC, and WZ wrote and revised the paper. All authors read and approved the final manuscript.
This work was supported by Natural Science Foundation of China (31772627) and the Fundamental Research Funds for the Central Universities (KYDK201701).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a past co-authorship with one of the authors YC.
We thank Professor Michael K. Theodorou for his contribution to critical discussion on the manuscript. We are thankful to BGI, China for sequencing services.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00435/full#supplementary-material
Akin, D. E., Borneman, W. S., and Windham, W. R. (1988). Rumen fungi: morphological types from Georgia cattle and the attack on forage cell walls. Biosystems 21, 385–391. doi: 10.1016/0303-2647(88)90037-8
Bard, J., and Winter, R. (2000). Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Barichevich, E. M., and Calza, R. E. (1990). Supernatant protein and cellulase activities of the anaerobic ruminal fungus Neocallimastix frontalis EB188. Appl. Environ. Microbiol. 56, 43–48.
Barr, D. J. S., Kudo, H., Jakober, K. D., and Cheng, K. J. (1989). Morphology and development of rumen fungi: Neocallimastix sp., Piromyces communis, and Orpinomyces bovis gen. nov., sp. nov. Can. J. Bot. 67, 2815–2824. doi: 10.1139/b89-361
Bauchop, T., and Mountfort, D. O. (1981). Cellulose fermentation by a rumen anaerobic fungus in both the absence and the presence of rumen methanogens. Appl. Environ. Microbiol. 42, 1103–1110.
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bernstein, J. A., Khodursky, A. B., Lin, P. H., Lin-Chao, S., and Cohen, S. N. (2002). Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proc. Natl. Acad. Sci. U.S.A. 99, 9697–9702. doi: 10.1073/pnas.112318199
Birch, P. R. J., Sims, P. F. G., and Broda, P. (1995). Substrate-dependent differential splicing of introns in the regions encoding the cellulose binding domains of two exocellobiohydrolase I-like genes in Phanerochaete chrysosporium. Appl. Environ. Microbiol. 61, 3741–3744.
Breton, A., Bernalier, A., Dusser, M., Fonty, G., Gaillard-Martinie, B., and Guillot, J. (1990). Anaeromyces mucronatus nov. gen., nov. sp. A new strictly anaerobic rumen fungus with polycentric thallus. FEMS Microbiol. Lett. 70, 177–182. doi: 10.1111/j.1574-6968.1990.tb13974.x
Callaghan, T. M., Podmirseg, S. M., Hohlweck, D., Edwards, J. E., Puniya, A. K., Dagar, S. S., et al. (2015). Buwchfawromyces eastonii gen. nov., sp. nov.: a new anaerobic fungus (Neocallimastigomycota) isolated from buffalo faeces. MycoKeys 9, 11–28. doi: 10.3897/mycokeys.9.9032
Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V., and Henrissat, B. (2009). The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238. doi: 10.1093/nar/gkn663
Cheng, Y., Wang, Y., Li, Y., Zhang, Y., Liu, T., Wang, Y., et al. (2017). Progressive colonization of bacteria and degradation of rice straw in the rumen by Illumina sequencing. Front. Microbiol. 8:2165. doi: 10.3389/fmicb.2017.02165
Cheng, Y. F., Edwards, J. E., Allison, G. G., Zhu, W. Y., and Theodorou, M. K. (2009). Diversity and activity of enriched ruminal cultures of anaerobic fungi and methanogens grown together on lignocellulose in consecutive batch culture. Bioresour. Technol. 100, 4821–4828. doi: 10.1016/j.biortech.2009.04.031
Couger, M. B., Youssef, N. H., Struchtemeyer, C. G., Liggenstoffer, A. S., and Elshahed, M. S. (2015). Transcriptomic analysis of lignocellulosic biomass degradation by the anaerobic fungus isolate Orpinomyces sp. strain C1A. Biotechnol. Biofuels 8:208. doi: 10.1186/s13068-015-0390-0
Dagar, S. S., Kumar, S., Griffith, G. W., Edwards, J. E., Callaghan, T. M., Singh, R., et al. (2015). A new anaerobic fungus (Oontomyces anksri gen. nov., sp. nov.) from the digestive tract of the Indian camel (Camelus dromedarius). Fungal Biol. 119, 731–737. doi: 10.1016/j.funbio.2015.04.005
Davies, D. R., Theodorou, M. K., Lawrence, M. I., and Trinci, A. P. (1993). Distribution of anaerobic fungi in the digestive tract of cattle and their survival in faeces. J. Gen. Microbiol. 139, 1395–1400. doi: 10.1099/00221287-139-6-1395
Dollhofer, V., Callaghan, T. M., Dorn-In, S., Bauer, J., and Lebuhn, M. (2016). Development of three specific PCR-based tools to determine quantity, cellulolytic transcriptional activity and phylogeny of anaerobic fungi. J. Microbiol. Methods 127, 28–40. doi: 10.1016/j.mimet.2016.05.017
Gold, J. J., Heath, I. B., and Bauchop, T. (1988). Ultrastructural description of a new chytrid genus of caecum anaerobe, Caecomyces equi gen. nov., sp. nov., assigned to the Neocallimasticaceae. Biosystems 21, 403–415. doi: 10.1016/0303-2647(88)90039-1
Gruninger, R. J., Nguyen, T. T. M., Reid, I. D., Yanke, J. L., Wang, P., Abbott, D. W., et al. (2018). Application of transcriptomics to compare the carbohydrate active enzymes that are expressed by diverse genera of anaerobic fungi to degrade plant cell wall carbohydrates. Front. Microbiol. 9:1581. doi: 10.3389/fmicb.2018.01581
Grutzmann, K., Szafranski, K., Pohl, M., Voigt, K., Petzold, A., and Schuster, S. (2014). Fungal alternative splicing is associated with multicellular complexity and virulence: a genome-wide multi-species study. DNA Res. 21, 27–39. doi: 10.1093/dnares/dst038
Haitjema, C. H., Gilmore, S. P., Henske, J. K., Solomon, K. V., Groot, R., Kuo, A., et al. (2017). A parts list for fungal cellulosomes revealed by comparative genomics. Nat. Microbiol. 2:17087. doi: 10.1038/nmicrobiol.2017.87
Hanafy, R. A., Elshahed, M. S., Liggenstoffer, A. S., Griffith, G. W., and Youssef, N. H. (2017). Pecoramyces ruminantium, gen. nov., sp. nov., an anaerobic gut fungus from the feces of cattle and sheep. Mycologia 109, 231–243. doi: 10.1080/00275514.2017.1317190
Hanafy, R. A., Elshahed, M. S., and Youssef, N. H. (2018). Feramyces austinii, gen. nov., sp. nov., an anaerobic gut fungus from rumen and fecal samples of wild Barbary sheep and fallow deer. Mycologia 110, 513–525. doi: 10.1080/00275514.2018.1466610
Heath, I. B., Bauchop, T., and Skipp, R. A. (1983). Assignment of the rumen anaerobe Neocallimastix frontalis to the Spizellomycetales (Chytridiomycetes) on the basis of its polyflagellate zoospore ultrastructure. Can. J. Bot. 61, 295–307. doi: 10.1139/b83-033
Henske, J. K., Gilmore, S. P., Knop, D., Cunningham, F. J., Sexton, J. A., Smallwood, C. R., et al. (2017). Transcriptomic characterization of Caecomyces churrovis: a novel, non-rhizoid-forming lignocellulolytic anaerobic fungus. Biotechnol. Biofuels 10:305. doi: 10.1186/s13068-017-0997-4
Hungate, R. E. (1967). Hydrogen as an intermediate in the rumen fermentation. Arch. Microbiol. 59, 158–164. doi: 10.1007/BF00406327
Hungate, R. E., Smith, W., Bauchop, T., Yu, I., and Rabinowitz, J. C. (1970). Formate as an intermediate in the bovine rumen fermentation. J. Bacteriol. 102, 389–397.
Janssen, P. H., and Kirs, M. (2008). Structure of the archaeal community of the rumen. Appl. Environ. Microbiol. 74, 3619–3625. doi: 10.1128/AEM.02812-07
Jin, W., Cheng, Y. F., Mao, S. Y., and Zhu, W. Y. (2011). Isolation of natural cultures of anaerobic fungi and indigenously associated methanogens from herbivores and their bioconversion of lignocellulosic materials to methane. Bioresour. Technol. 102, 7925–7931. doi: 10.1016/j.biortech.2011.06.026
Joshi, A., Lanjekar, V. B., Dhakephalkar, P. K., Callaghan, T. M., Griffith, G. W., and Dagar, S. S. (2018). Liebetanzomyces polymorphus gen. et sp. nov., a new anaerobic fungus (Neocallimastigomycota) isolated from the rumen of a goat. MycoKeys 40, 89–110. doi: 10.3897/mycokeys.40.28337
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y., and Hattori, M. (2004). The KEGG resource for deciphering the genome. Nucleic Acids Res. 32, D277–D280. doi: 10.1093/nar/gkh063
Keller, O., Kollmar, M., Stanke, M., and Waack, S. (2011). A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 27, 757–763. doi: 10.1093/bioinformatics/btr010
Kim, H., Lee, I., Kwon, Y., Kim, B. C., Ha, S., Lee, J. H., et al. (2011). Immobilization of glucose oxidase into polyaniline nanofiber matrix for biofuel cell applications. Biosens. Bioelectron. 26, 3908–3913. doi: 10.1016/j.bios.2011.03.008
Koch, A. L., and Levy, H. R. (1955). Protein turnover in growing cultures of Escherichia coli. J. Biol. Chem. 217, 947–957.
Krause, D. O., Denman, S. E., Mackie, R. I., Morrison, M., Rae, A. L., Attwood, G. T., et al. (2003). Opportunities to improve fiber degradation in the rumen: microbiology, ecology, and genomics. FEMS Microbiol. Rev. 27, 663–693. doi: 10.1016/S0168-6445(03)00072-X
Lagesen, K., Hallin, P., Rodland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. doi: 10.1186/gb-2009-10-3-r25
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, R., Li, Y., Kristiansen, K., and Wang, J. (2008). SOAP: short oligonucleotide alignment program. Bioinformatics 24, 713–714. doi: 10.1093/bioinformatics/btn025
Li, R., Zhu, H., Ruan, J., Qian, W., Fang, X., Shi, Z., et al. (2010). De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272. doi: 10.1101/gr.097261.109
Li, Y., Jin, W., Cheng, Y., and Zhu, W. (2016). Effect of the associated methanogen Methanobrevibacter thaueri on the dynamic profile of end and intermediate metabolites of anaerobic fungus Piromyces sp. F1. Curr. Microbiol. 73, 434–441. doi: 10.1007/s00284-016-1078-9
Li, Y., Jin, W., Mu, C., Cheng, Y., and Zhu, W. (2017). Indigenously associated methanogens intensified the metabolism in hydrogenosomes of anaerobic fungi with xylose as substrate. J. Basic Microbiol. 57, 933–940. doi: 10.1002/jobm.201700132
Lowe, S. E., Theodorou, M. K., and Trinci, A. P. (1987). Growth and fermentation of an anaerobic rumen fungus on various carbon sources and effect of temperature on development. Appl. Environ. Microbiol. 53, 1210–1215.
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Mao, S. Y., Huo, W. J., and Zhu, W. Y. (2016). Microbiome–metabolome analysis reveals unhealthy alterations in the composition and metabolism of ruminal microbiota with increasing dietary grain in a goat model. Environ. Microbiol. 18, 525–541. doi: 10.1111/1462-2920.12724
Marvin-Sikkema, F. D., Richardson, A. J., Stewart, C. S., Gottschal, J. C., and Prins, R. A. (1990). Influence of hydrogen-consuming bacteria on cellulose degradation by anaerobic fungi. Appl. Environ. Microbiol. 56, 3793–3797.
Mountfort, D. O., Asher, R. A., and Bauchop, T. (1982). Fermentation of cellulose to methane and carbon dioxide by a rumen anaerobic fungus in a triculture with Methanobrevibacter sp. strain RA1 and Methanosarcina barkeri. Appl. Environ. Microbiol. 44, 128–134.
Nakashimada, Y., Srinivasan, K., Murakami, M., and Nishio, N. (2000). Direct conversion of cellulose to methane by anaerobic fungus Neocallimastix frontalis and defined methanogens. Biotechnol. Lett. 22, 223–227. doi: 10.1023/A:1005666428494
Orpin, C. G. (1975). Studies on the rumen flagellate Neocallimastix frontalis. J. Gen. Microbiol. 91, 249–262. doi: 10.1099/00221287-91-2-249
Ozkose, E., Thomas, B. J., Davies, D. R., Griffith, G. W., and Theodorou, M. K. (2001). Cyllamyces aberensis gen. nov. sp. nov., a new anaerobic gut fungus with branched sporangiophores isolated from cattle. Can. J. Bot. 79, 666–673. doi: 10.1139/b01-047
Rio, D. C., Ares, M. Jr., Hannon, G. J., and Nilsen, T. W. (2010). Enrichment of poly(A)+ mRNA using immobilized oligo(dT). Cold Spring Harb. Protoc. 7:db.rot5454. doi: 10.1101/pdb.prot5454
Russell, J. B., and Rychlik, J. L. (2001). Factors that alter rumen microbial ecology. Science 292, 1119–1122. doi: 10.1126/science.1058830
Solomon, K. V., Haithema, C. H., Henske, J. K., Gilmore, S. P., Borges-Rivera, D., Lipzen, A., et al. (2016). Early-branching gut fungi possess a large, comprehensive array of biomass-degrading enzymes. Science 351, 1192–1195. doi: 10.1126/science.aad1431
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tarazona, S., Furio-Tari, P., Turra, D., Pietro, A. D., Nueda, N. J., Ferrer, A., et al. (2015). Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 43:e140. doi: 10.1093/nar/gkv711
Ter-Hovhannisyan, V., Lomsadze, A., Chernoff, Y. O., and Borodovsky, M. (2008). Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 18, 1979–1990. doi: 10.1101/gr.081612.108
Teunissen, M. J., de Kort, G. V., Op den Camp, H. J., and Vogels, G. D. (1993). Production of cellulolytic and xylanolytic enzymes during growth of anaerobic fungi from ruminant and nonruminant herbivores on different substrates. Appl. Biochem. Biotechnol. 39–40, 177–189. doi: 10.1007/BF02918988
The UniProt Consortium (2015). UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–D212. doi: 10.1093/nar/gku989
Theodorou, M. K., Williams, B. A., Dhanoa, M. S., Mcallan, A. B., and France, J. (1994). A simple gas production method using a pressure transducer to determine the fermentation kinetics of ruminant feeds. Anim. Feed Sci. Technol. 48, 185–197. doi: 10.1016/0377-8401(94)90171-6
Theodorou, M. K., Zhu, W. Y., Rickers, A., Nielsen, B. B., Gull, K., and Trinci, A. P. J. (1996). “Biochemistry and ecology of anaerobic fungi,” in The Mycota: VI Human and Animal Relationship, eds D. H. Howard and J. D. Miller (New York, NY: Springer-Verlag), 265–295.
Ungerfeld, E. M. (2015). Limits to dihydrogen incorporation into electron sinks alternative to methanogenesis in ruminal fermentation. Front. Microbiol. 6:1272. doi: 10.3389/fmicb.2015.01272
Weimer, P. J., Russell, J. B., and Muck, R. E. (2009). Lessons from the cow: what the ruminant animal can teach us about consolidated bioprocessing of cellulosic biomass. Bioresour. Technol. 100, 5323–5331. doi: 10.1016/j.biortech.2009.04.075
White, T. J., Bruns, T., Lee, S., and Taylor, J. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in PCR Protocols: A Guide to Methods and Applications, eds M. A. Innis, D. H. Gelfand, J. J. Sninsky, and T. J. White (San Diego, CA: Academic Press), 315–322.
Yan, G., Lestari, R., Long, B., Fan, Q., Wang, Z., Guo, X., et al. (2016). Comparative proteomics analysis reveals L-arginine activates ethanol degradation pathways in HepG2 cells. Sci. Rep. 6:23340. doi: 10.1038/srep23340
Youssef, N. H., Couger, M. B., Struchtemeyer, C. G., Liggenstoffer, A. S., Prade, R. A., Najar, F. Z., et al. (2013). The genome of the anaerobic fungus Orpinomyces sp. strain C1A reveals the unique evolutionary history of a remarkable plant biomass degrader. Appl. Environ. Microbiol. 79, 4620–4634. doi: 10.1128/AEM.00821-13
Keywords: anaerobic fungus, methanogen, metabolism, genome, RNAseq, iTRAQ
Citation: Li Y, Li Y, Jin W, Sharpton TJ, Mackie RI, Cann I, Cheng Y and Zhu W (2019) Combined Genomic, Transcriptomic, Proteomic, and Physiological Characterization of the Growth of Pecoramyces sp. F1 in Monoculture and Co-culture With a Syntrophic Methanogen. Front. Microbiol. 10:435. doi: 10.3389/fmicb.2019.00435
Received: 13 December 2018; Accepted: 19 February 2019;
Published: 06 March 2019.
Edited by:
Sumit Singh Dagar, Agharkar Research Institute, IndiaReviewed by:
Robert J. Gruninger, Agriculture and Agri-Food Canada, CanadaCopyright © 2019 Li, Li, Jin, Sharpton, Mackie, Cann, Cheng and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanfen Cheng, eWFuZmVuY2hlbmdAbmphdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.