 Shankar Prasad Sha
Shankar Prasad Sha Mangesh Vasant Suryavanshi
Mangesh Vasant Suryavanshi Jyoti Prakash Tamang
Jyoti Prakash Tamang- DAICENTRE (DBT-AIST International Centre for Translational and Environmental Research) and Bioinformatics Centre, Department of Microbiology, School of Life Sciences, Sikkim University, Gangtok, India
Chowan, dawdim, humao, hamei, khekhrii, and phut are sun-dried starters used for preparation of alcoholic beverages in North East regions of India. We attempted to profile the mycobiome community in these starters by high-throughput sequencing (HTS) method. All fungal populations were found to be restricted to Ascomycota (67–99%), Zygomycota (0.7–29%), Basidiomycota (0.03–7%), and Chytridiomycota (0.0003%). We found 45 core operational taxonomic units (OTUs) which were universally present and were further weighed to 41 genera level and 22 species level taxonomy. A total number of 594 fungal species were detected by HTS including common species (224), unique species (133) and rare-species (237) in samples of starters. Unique species were recorded in phut (40 species), khekhrii (28), hamei (23), dawdim (21), chowan (13), and humao (8), respectively. Most of the fungal families were found to correlate to a type of nutritional mode and growth morphologies of the community, where saprotrophic mode of mold species were more dominant, whereas morphotypes were more dominant in yeast species.
Introduction
Traditionally prepared sun-dried cereal-based amylolytic/alcoholic starters, in the form of round/oval/flattened balls of varied sizes for production of mild-alcoholic beverages, are common in South East Asia (Hesseltine, 1983; Steinkraus, 1996; Nout and Aidoo, 2002; Tamang, 2010a). Usually three types of mixed cultures are traditionally used as starters to convert cereal starch to sugar and then to alcohol and organic acids (Hesseltine et al., 1988; Tamang and Fleet, 2009; Tamang, 2010a,b). These are (1): dried starter consisting of consortia of amylase/alcohol producing-yeasts, filamentous molds and bacteria, which are locally called marcha in India, Nepal and Bhutan (Tsuyoshi et al., 2005), chiu/chu/daque in China (Chen et al., 2014; Xu et al., 2017), nuruk in Korea (Jung et al., 2012), ragi in Indonesia (Surono, 2016), loog-pang in Thailand (Limtong et al., 2002), benh men in Vietnam (Dung et al., 2007) and dombea in Cambodia (Ly et al., 2018); (2): mixed culture of molds Aspergillus oryzae and A. sojae in the form of a starter called koji in Japan for making saké, distilled liquor, and several fermented soybean products such as miso and shoyu (Kitamura et al., 2016), and (3): large compact cakes made up of whole-wheat flour with yeasts and filamentous molds to ferment starchy substrates for production of alcohol, mostly in China (Tamang, 2010a). Microbiota associated with traditionally prepared Asian dried starters are starch-degrading genera of molds Actinomucor, Amylomyces, Aspergillus, Mucor, Neurospora, Penicillium, Rhizopus (Hesseltine et al., 1988; Tamang et al., 1988; Nikkuni et al., 1996; Nout and Aidoo, 2002; Chen et al., 2014; Tamang et al., 2016a); amylolytic and alcohol-producing yeasts genera mostly Candida, Debaryomyces, Dekkera, Galactomyces, Geotrichum, Hansenula, Hanseniaspora, Issatchenkia, Kazachstania, Kluyveromyces, Pichia, Saccharomyces, Saccharomycodes, Saccharomycopsis, Schizosaccharomyces, Torulaspora, Torulopsis, Wickerhamomyces, and Zygosaccharomyces (Hesseltine and Kurtzman, 1990; Tamang and Sarkar, 1995; Tsuyoshi et al., 2005; Jeyaram et al., 2008; Lv et al., 2012, 2013; Chakrabarty et al., 2014; Sha et al., 2016, 2017, 2018) and few genera of bacteria, mostly Pediococcus, Lactobacillus (Hesseltine and Ray, 1988; Tamang and Sarkar, 1995; Sujaya et al., 2001; Tamang et al., 2007; Chakrabarty et al., 2014).
Since the culture-dependent method can only isolate the culturable microorganisms from samples using media, the culture-independent method may profile all microbial communities, including both those that are culturable and unculturable in food samples, by extracting the whole genomic DNA directly from small amount of samples (Roh et al., 2010; Jung et al., 2012; Puerari et al., 2015; Sha et al., 2018). Culture-independent methods, including pyrosequencing and high-throughput amplicon sequencing, are commonly applied for profiling microbiome of natural food fermentation within a short time and with more accuracy (Alegría et al., 2011; Cocolin et al., 2013; Chen et al., 2014; Mayo et al., 2014; Puerari et al., 2015; Tamang et al., 2016b; Shangpliang et al., 2018). Application of the amplicon-based high-throughput sequencing has been demonstrated for the monitoring of microbial populations between different strains within a species (Ercolini et al., 2012), and inter- and intra-species diversity within a particular genus or among genera (Yan et al., 2013).
Drinking of traditional alcoholic beverages and drinks is the distinct dietary culture and practices of ethnic people of North East India1 with strong ritualistic and ethnical importance (Tamang, 2010a; Tamang et al., 2016a). Traditionally prepared sun-dried starters such as dawdim, hamei, humao, khekhrii, chowan, phut, etc., in North East states of India (Anupma et al., 2018) are commonly used by diverse groups of ethnic people to prepare mild-alcoholic (4–5%) beverages with sweet taste, providing a high source of calories and minerals (Thapa and Tamang, 2004, 2006; Tamang and Thapa, 2006; Tamang et al., 2012). In this study we selected six different starters, such as chowan of Tripura, dawdim of Mizoram, hamei of Manipur, humao of Assam, khekhrii of Nagaland and phut of Arunachal Pradesh, from North East states of India (Figure 1). All these amylolytic/alcoholic starters are dry, hard, with different shapes of round to flattened solid ball like structure, sizes ranging from 1.2 to 11.2 cm in diameter, and all creamy to dusty white in color. Except for khekhrii, all other starters are traditionally prepared from soaked rice/wheat, mixed with some locally available wild plants, added with previously prepared powdered starters (1–2%), and kneaded into round to flattened cakes by adding water. The mixtures are covered with fern fronds/paddy straws/jute sags, fermented for 1–3 days at room temperature; and finally sun dried (2–3 days) to get dry starters, which can be kept for a year or more (Tamang et al., 2016a; Anupma et al., 2018). Khekhrii is the only amylolytic/alcoholic starter in North East India, which is prepared by fermenting germinated sprouted-rice grains and then sun-dried to use as dry starters to prepare the local alcoholic drink (Anupma et al., 2018). Sha et al. (2018) studied the fungal diversity in chowan, dawdim, hamei, humao, khekhrii, and phut, based on the culture-dependent method using ITS-PCR and a culture-independent approach by PCR-DGGE analysis. In this paper, we attempted to understand the “ethno-microbiology” of mycobiome diversity in chowan, dawdim, hamei, humao, khekhrii, and phut by using the high-throughput sequencing method supported by bioinformatics interpretation.
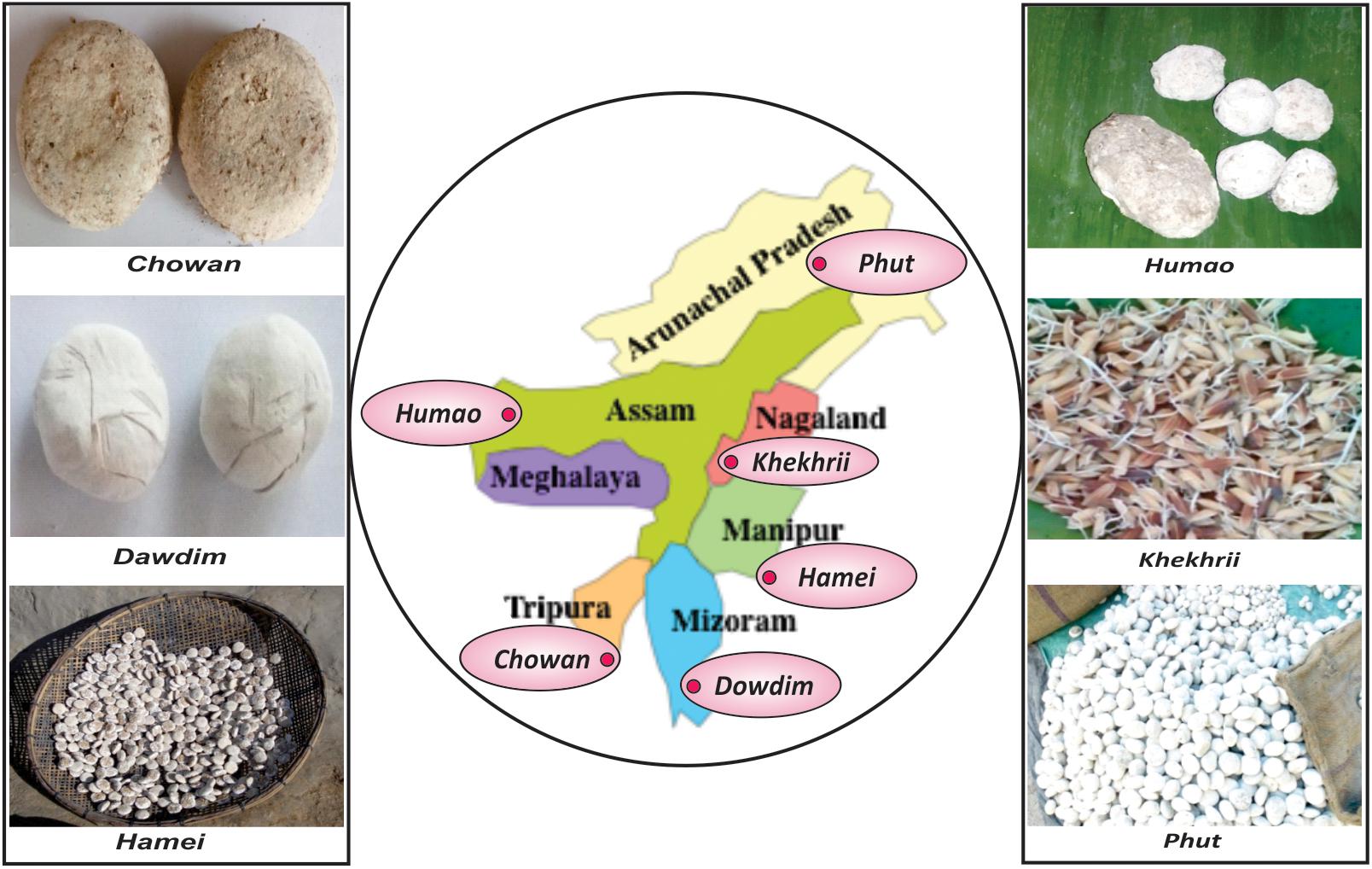
Figure 1. Origin and types of sun-dried alcoholic starter cultures in eight North East states of India.
Materials and Methods
Sample Collection
Six samples of starter: chowan of Tripura, dawdim of Mizoram, hamei of Manipur, humao of Assam, khekhrii of Nagaland and phut of Arunachal Pradesh (Figure 1) were collected immediately after the preparation (fermentation and sun-drying) from different places of North East India. The average pH of these starter samples was 4.9 ± 0.2. Samples were kept in sterile containers, leveled, transported to the laboratory and stored at room temperature in a desiccator; dried starter can retain its potency in situ for more than a year in the moist-free condition (Tamang et al., 1988).
Community DNA Extraction
Firstly, dried starter samples were powdered with the help of sterile mortar and pestle and 1g of powdered sample were taken and homogenized in 9 ml of 0.85% physiological saline and subsequently filtered through 4 layers of sterile cheese-cloth. The resulting filtered solutions were centrifuged at 14000 g for 10 min at 4°C (Lv et al., 2013; Sha et al., 2018) and then the pellets were subjected to total community DNA extraction using the ProMega DNA extraction kit (ProMega, United States) according to the manufacturer’s instructions. Subsequently, the RNA was eliminated from the cellular lysate by administering the RNase solution after incubation at 35°C for 15 min. The residual proteins were removed by adding protein precipitation solution and then centrifuging at maximum speed. Finally, the DNA was precipitated by adding isopropanol, and purified with two washes of 70% ethanol. Quality of DNA was checked on 0.8% agarose gel by measuring the concentration using Nano-Drop spectrophotometer (AG-6135, Eppendorf, Germany). The DNA was kept at -20°C until further processing.
Sequencing of Fungal ITS2 Gene Region and Pre-processing
Internal Transcribed Spacer (ITS) 2 region of fungi was targeted for taxonomic profiling by amplification using ITS2F (GCATCGATGAAGAACGCAGC) and ITS2R (TCCTCCGCTTATTGATATGC) primers (Blaalid et al., 2013) due to its high interspecific variability which also conserved primer sites with multiple copies (Schoch et al., 2012). A composite sample for sequencing was made by combining equimolar ratios of amplicons from the samples, followed by gel purification with a QiagenMinElute gel extraction kit to remove potential contaminants and PCR artifacts. Amplicon libraries were purified by 1X AMpureXP beads, which were checked on an Agilent DNA 1000 chip on Bioanalyzer 2100, and finally quantified by Qubit Fluorometer 2.0 using Qubit dsDNA HS Assay kit (Life Technologies). MiSeqIllumina platform using 2 × 250 bp chemistry sequencing was performed. Pre-processing of downstream analysis for raw read was completed as described by Comeau et al. (2017), as follows: firstly, raw read quality from sequencer was checked for the average and range of the Phred quality scores along the reads (1 to 300 bp), for both forward and reverse reads independently, to pass it to the next steps using FastQC2; secondly, removal of adapter sequences through cut adapt tool (Martin, 2011); thirdly, adapter cleaned paired-end reads files merged using the PEAR (v0.9.10) program (Zhang et al., 2014) with default settings; fourthly, FASTQ stitched files were converted to FASTA and removed any sequences that had an “N” in them with run_fastq_to_fasta.pl command lin; and lastly, chimeric sequences were removed with VSEARCH tool (Rognes et al., 2016) using UNITE_uchime_ITS2only_01.01.2016.fasta reference dataset.
Downstream Analysis of ITS Gene Region Reads
The downstream analysis of chimera free FASTA files was done for detecting the taxonomic classification and their functional guided activity. For taxonomic classification of each sequence, we performed the diversity analysis in the QIIME 1.9 environment (Caporaso et al., 2010). Sequence reads were combined in a single FASTA file with guided metadata files and further steps were done accordingly as described by Comeau et al. (2017). Fungal operational taxonomic units (OTUs) were analyzed by an open reference-based OTU picking approach using UNITE reference database as UNITE_sh_refs_qiime_ver7_dynamic_20.11.2016.fasta. The OTU picking was carried out using the sortmerna_sumaclust method with a similarity threshold of 97%. Taxonomic assignments were performed using mother classifier (Schloss et al., 2009).
We performed the analysis with PIPITS (Gweon et al., 2015) and FUNGuild environments (Nguyen et al., 2016) for functional guided activity determination. The ITS2 region was extracted with ITSx, clustered into OTUs with VSEARCH3 at 97% sequence similarity, and chimera removal was performed using the UNITE UCHIME reference data set. Representative sequences were assigned taxonomic classification with the RDP classifier against the UNITE fungal ITS reference data set at a confidence threshold of 0.85. We generated otu_table_funguild file by using pipits_funguild.py command line. This OTU table was used to run the online Guilds application to assign functional information to OTUs in high-throughput sequencing datasets4.
Other Data Analysis
Alpha diversity analyses of the mycobiome were tested using QIIME platform and with the alpha_diversity.py script. For the continuous variables, non-parametric t-test was used, and for categorical variables between groups, either the Pearson chi-square or Fisher’s exact test was used depending on assumption validity. Data analyses were performed by statistical environment R5. Phylum level abundance plots, bubble plots and heatmap were derived by ggplot2 package (Wickham, 2016), core microbiome heatmap were derived by microbiome package (Lahti and Shetty, 2017) and correlation plot by corplot package (Wei and Simko, 2017). The filtered OTUs based (less than 1% abundance value) rare-phylotypes heatmap were derived by ggplot2 package (Wickham, 2016). UPGMA based dendrogram was created using the Pearson similarity coefficient. Alpha diversity indices like Chao, Shannon, and Simpson were calculated after rarefying all samples to the same sequencing depth (Cox et al., 2017). Non-metric multidimensional scaling plots (NMDS) based on Bray-Curtis distance matrix was constructed to carry out the beta-diversity analysis.
Data Availability
The sequences obtained from high-throughput sequencing effort were submitted to NCBI, which are available under SRA accession: SRP150043 and BioProject ID:PRJNA474271.
Results and Discussion
The present study reveals the mycobiome diversity in the same samples of chowan, dawdim, hamei, humao, khekhrii, and phut by culture-independent method using high-throughput sequencing approach, which permits the analysis of hundreds of nucleotide sequences (Roh et al., 2010). We generated 5213436 paired end sequences and were clustered into operational taxonomic units (OTUs) by single linkage clustering with 97% sequence similarity. About 2488812 high quality sequences (sequence lengths: 374 ± 31 nucleotides) and normalization were done on 292996 per sample for the study, which were assembled into 6097 global and species-level OTUs. All OTUs with <2 reads in total and those not representing fungi were omitted. OTU-table was generated for further taxonomy-based analysis. Samples diversity surveillance for the fungal population was analyzed by intra-sample variations through the alpha diversity measures (Table 1). The diversity indices provide an idea about the expected diversity values, like goods coverage index within 0.990 to 0.998. Observed OTUs values were found to be wide and within the range of 702 to 3037. Among the six starters, hamei had the highest OTUs. Phylum level abundance varied in each sample and was mostly limited to taxa Ascomycota, Basidiomycota, Chytridiomycota and Zygomycota (Figure 2A). The samples were discrete on the NMDS plots using OTUs level variations (Figure 2B). All fungal populations were found to be restricted to only Ascomycota (67–99% of abundance), Zygomycota (0.7–29%) and Basidiomycota (0.03–7%), however, Chytridiomycota (0.0003%) was also enlisted in khekhrii sample. High prevalence of Ascomycota phylum was also reported in similar types of dry starters of Asia such as nuruk of Korea (Jung et al., 2012; Bal et al., 2016), and daqua of China (Li et al., 2011; Chen et al., 2014; Xu et al., 2017). In the present study quantitative differences were observed for the presence of fungal taxa in all six starters, which could be the consequence of differences in the traditional methods of production of starters, use of wrapping materials and varied fermentation time (Jeyaram et al., 2011; Bora et al., 2016; Anupma et al., 2018). The Alpha diversity estimation of all starters using species richness and non-parametric Shannon index showed dominance of phylum Ascomycota over the Zygomycota. A similar observation was also reported in similar types of dry starters of India: thiat of Meghalaya state (Sha et al., 2017), and in marcha of Sikkim state (Tamang et al., 1988).
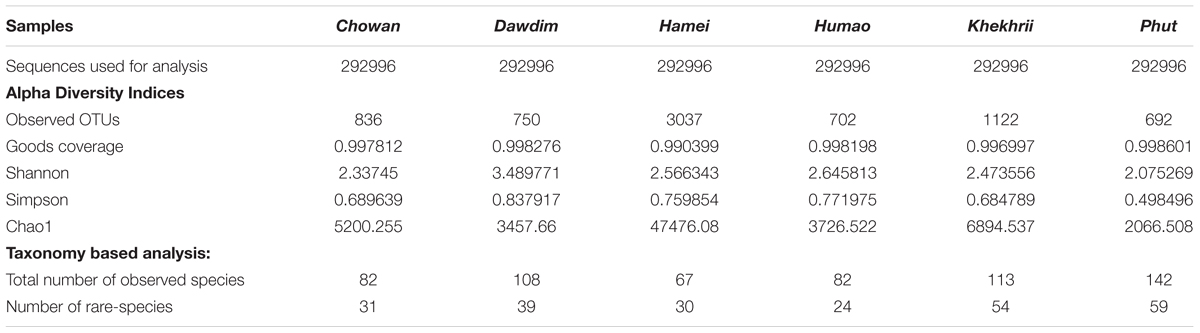
Table 1. Sequence statistics, alpha diversity matrix and species level taxonomy number observed for starter culture samples.
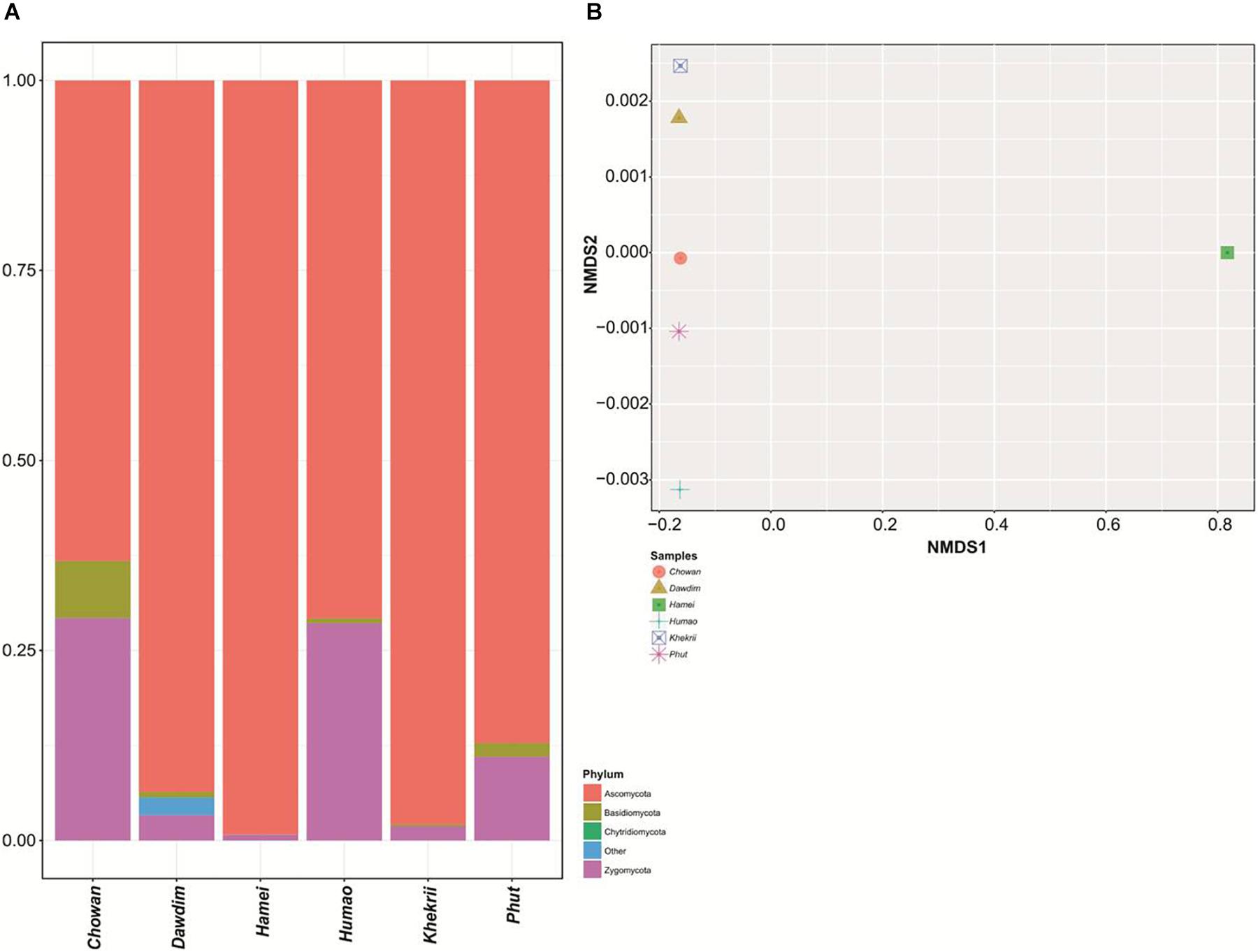
Figure 2. (A) Barplot showing the phylum level diversity and their abundance distribution found in starters. (B) NMDS plot showing the beta diversity clustering pattern in tested subjects.
We found 45 core OTUs which were universally present in all starter samples tested and were further weighed to 41 genera level (Figure 3A) and 22 species level taxonomy (Figure 3B). A wide diversity of fungal species, as well as various unique species in samples has been observed in this study. A total number of 594 fungal species were detected by HTS including noble or unique species (133), common species (224) and rare-species (237), in samples of chowan, dawdim, hamei, humao, khekhrii, and phut (Supplementary Table 1). A total of 133 fungal species were found to be noble or unique species with reference to diversity compared to the common species, out of which 40 species were sample-specific in phu,t followed by khekhrii (28), hamei (23), dawdim (21), chowan (13), and humao (8), respectively (Supplementary Table 1). Dominant unique species based on abundance were Tetracladium setigerum in khekhrii, Saccharomyces eubayanus in chowan, Solicoccozyma terrea in hamei, Penicillium sumatraense in phut, Acremonium implicatum in humao, and Thermomyces lanuginosus in hamei. Species with less than 1% abundances are known as rare-phylotypes (Li et al., 2018). We found 237 species within the rare-phylotypes category [those with less than 1% abundances (Supplementary Table 1)] including 19 different class level taxa (Supplementary Figure 1). Interestingly, samples of phut had the highest number of 59 rare-species, followed by khekhrii (54), dawdim (39), chowan (31), hamai (30), and humao (24), respectively (Table 1). A phylotype, often referred to as OTUs, is an environmental DNA fragment or group of sequences sharing greater than 97–98% similarity of a particular gene marker (Bhadury et al., 2011; Rivett and Bell, 2018). Importantly, in such lesser-known traditionally prepared dry starters, the presence of sizable number of rare-phylotypes may have some functional or biochemical properties, and sometimes these rare-species may have human health perspectives (Bhute et al., 2017).
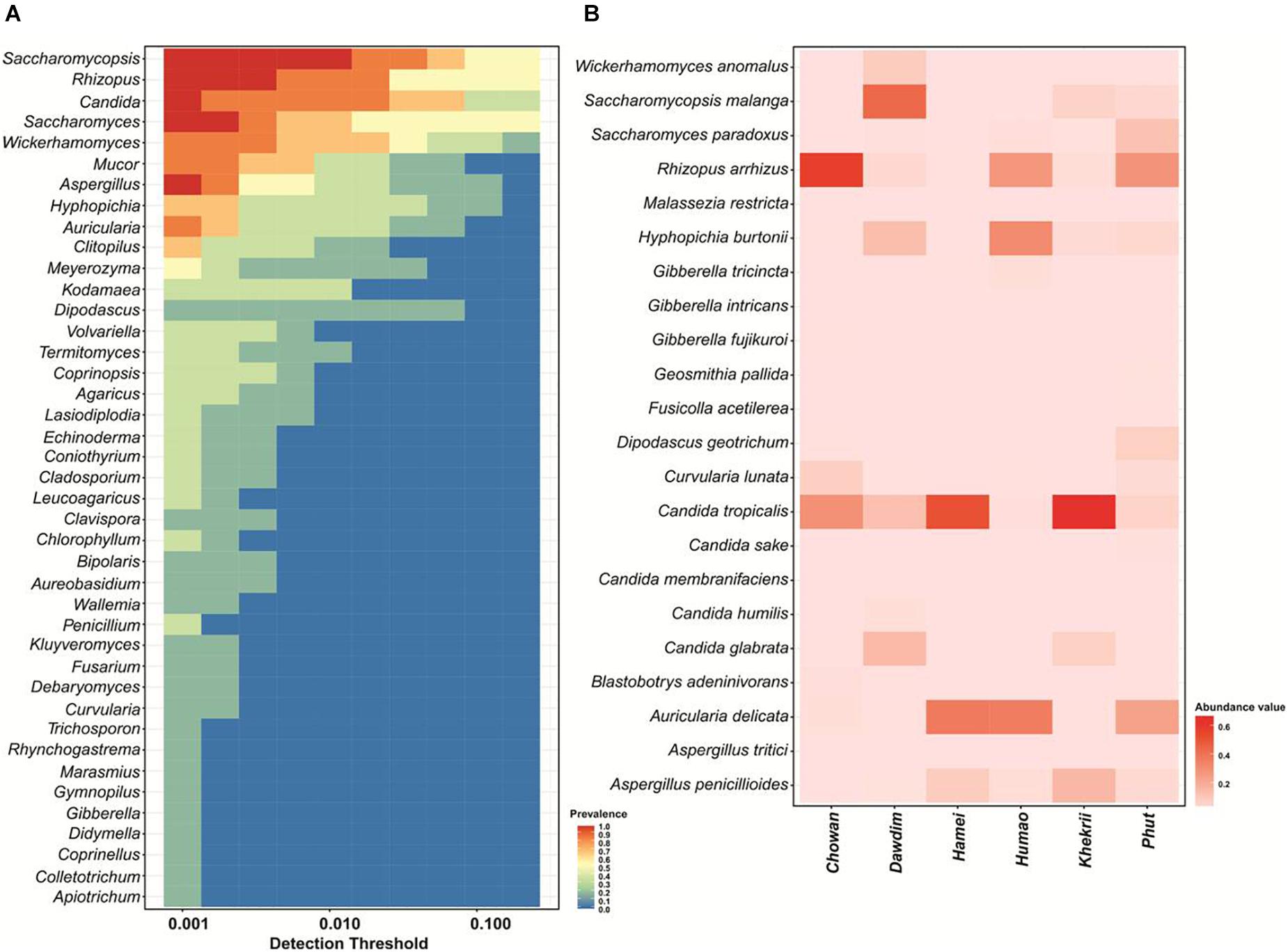
Figure 3. Heatmap of OTUs showing (A) Core genus distribution pattern and (B) Core species and their relative abundance present in all samples.
The unique mold species recorded in dry starters of North East India are Aspergillus penicillioides, Rhizopus arrhizus, Rhizopus microsporus and the unique yeast species are Kluyveromyce smarxianus, Trichomona scusciferrii, Candida humilis, Candida metapsilosis, Saccharomyces paradoxus, Saccharomycopsis malanga, and Wickerhamomyces sydowiorum. Earlier reports demonstrated the presence of common yeasts in most of the Asian dried starters, which were similar to starters of North East India, including Candida glabrata, Cryptococcus heveanensis, Cry. albidus, Pichia fabianii, P. guilliermondii, Rhodosporidium toruloides, Rhodotorula mucilaginosa, Saccharomyces cerevisiae, Saccharomycopsis fibuligera, Saccharomycopsis malanga, Sporobolomyces nylandii, and Wickerhamomyces anomalus (Tsuyoshi et al., 2005; Xie et al., 2007; Jeyaram et al., 2008; Thanh et al., 2008; Lv et al., 2013; Bora et al., 2016; Sha et al., 2016, 2017, 2018). We assume that the higher yeast diversity in our study could have resulted from a larger sampling population. The yeast Saccharomycopsis fibuligera, possessing amylase and ethanol producing capacity, is one of the most common yeasts present in dried starters of Asia (Hesseltine and Kurtzman, 1990; Limtong et al., 2002; Tsuyoshi et al., 2005; Thanh et al., 2008; Sha et al., 2018).
We correlated mycobiome diversity which was earlier detected in six samples of dry starters of India viz. (chowan, dawdim, hamei, humao, khekhrii, and phut) by the culture-dependent method (ITS-PCR) (6 species) and culture-independent method using PCR-DGGE analysis (24 species) (Sha et al., 2018), with that of 594 fungal species detected by high-throughput sequencing method (Supplementary Figure 2). Based on OTUs, the HTS method could detect 594 fungal species showing a diverse profile of mycobiome communities in the six different types of starters in this study, which were not earlier detected by ITS-PCR and PCR-DGGE methods (Sha et al., 2018). The read length required by HTS platforms for DNA metabarcoding is preferably 200–400 bp (Banchi et al., 2018), which is used for ITS2 gene amplification that can generate the amplicons up to 400bp in size necessary for library preparation on Illumina platform (Blaalid et al., 2013). The shorter sequences for HTS platform using ITS2 primers favor the identification of a wide range of fungi, which is a major advantage of the ITS2 primer (Bellemain et al., 2013). Whereas in ITS-PCR, the read length of ITS gene sequence amplified by primers, ITS1 and ITS4, is ranging from 600 to 750 bp (White et al., 1990), which may not be used for the library preparation in Illumina sequencing platform for HTS (Banchi et al., 2018). Amplicon-based high-throughput sequencing reveals comprehensive microbial communities with superior sequence coverage and inter- and intra-species diversity within a particular genus or among genera (Bokulich and Mills, 2013; Yan et al., 2013; Połka et al., 2015), comparable to other molecular tools. This is because HTS can generate far more reads than traditional culture-independent methods such as PCR-DGGE and facilitates the discovery of more microbiota diversity (Ercolini, 2013). However, a combined (culture-dependent and culture-independent) approach can be an appropriate strategy to investigate entire microbial communities of any food sample.
We assume that the geographical environment (including altitudes and climate) play important roles, over the production methods of dried starter cultures, when influencing the composition of microbiota (Jeyaram et al., 2011; Nam et al., 2012; Lv et al., 2012, 2013). Besides these, other factors that may affect the composition of mycobiome communities in dried starters include the level of hygienic conditions, quality of the glutinous rice or other cereal substrates, quality and source of water, as well as the back-slopping techniques used by the ethnic people (Capozzi and Spano, 2011; Gonelimali et al., 2018; Sha et al., 2018). There may also be the possibility of air-borne resources of fungal diversity in these tested samples (Cuadros-Orellana et al., 2013; Aguayo et al., 2018), probably during traditional preparation of starter.
The percentage distribution of total yeast and mold species found in different starters with their respective morphology and mode nutrition is shown in Supplementary Table 2. Saprotrophic mode of mold species was encountered in starters with a dominance range of 64 to 99% over other modes. In other hands, yeast morphotypes were more dominants in all samples (Figure 4A). Several families were enlisted for the diversity players inside the starters; most of them were saprophytes irrespective of the taxonomy. The Saccharomycopsidaceae family showing the saprotrophic mode of nutrition were found to be abundant, and the pathogenic Pleosporaceae family (Ariyawansa et al., 2015) had a lower abundance in Ascomycota phylum (Figure 4B). Most of the families were associated with the functional attributes to the KEGG Orthologous for the eubacterial diversity. Some important correlations have been observed between families and functional guilds (Figure 5). Interestingly, Pichaceae was negatively correlated to the micro-fungus morphotypes, and such correlations have been suggesting the extrusion of the diversity simulation (Schoch et al., 2012).
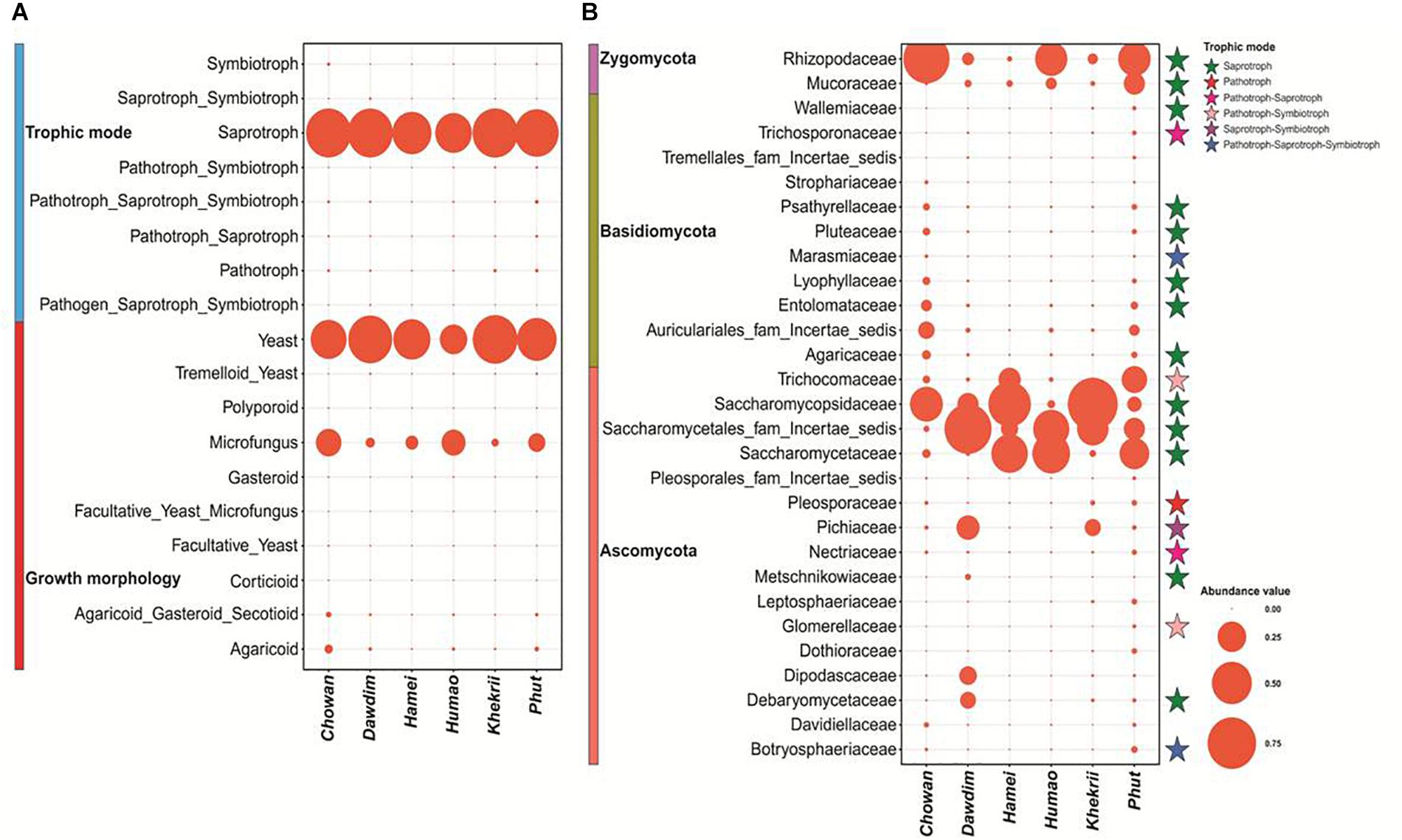
Figure 4. (A) Bubble plot describing the features of mycobiome with relative abundance distribution present in starters. (B) Bubble plot describing the relative abundance distribution of family level diversity and their trophic mode of nutrition found in dried starters. Family level diversity having >1.0% abundance in each sample was taken for this plot.
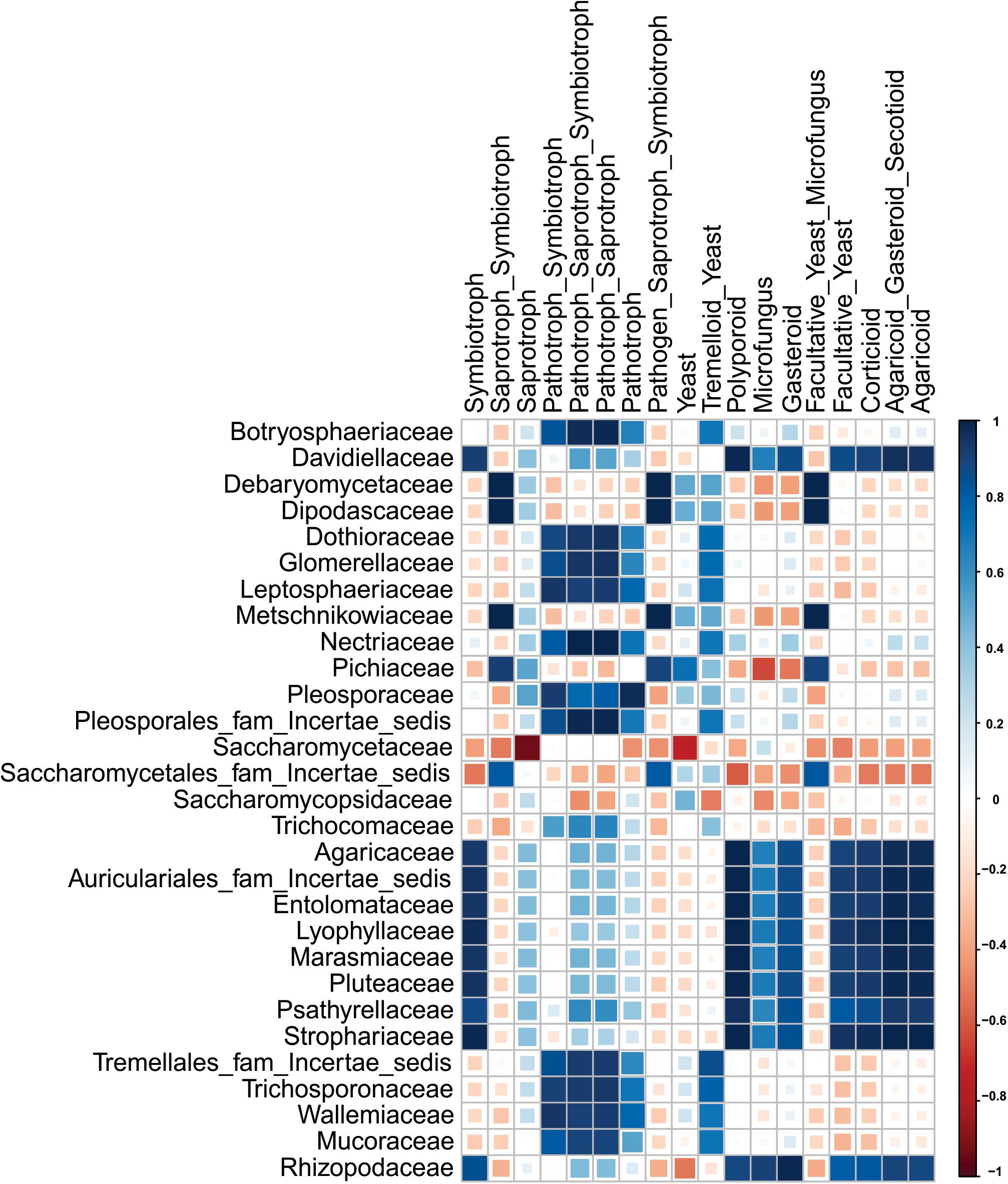
Figure 5. Plot showing the spearman correlation values for family level diversity having >1.0% abundance in each sample and different features of mycobiome.
Functional attributes of the fungal diversity were formulated with bioinformatics tools, based on methods described by Gweon et al. (2015) and Nguyen et al. (2016). Since the ITS region has been recognized as the universal barcode for identification of fungi (Schoch et al., 2012), we used this region for fungal bar code with reference to database UNITE for OTU assignment. We applied the PIPITS pipeline since it extracts the ITS sub-region from raw reads and assigns taxonomy with a trained RDP Classifier. Total 662461 sequences were identified out of 689459 sequences, as containing an ITS2 sub-region. After quality filtering and removal of contaminants, we obtained results in 2402833 quality sequences. We set 59612 sequences per sample for further analysis to form 354 OTUs, which yielded 190 phylotypes. Several functions of the mycobiome were observed after the funguild function analysis (Supplementary Table 3). However, comparing with culture-independent method, the culturable diversity is more relevant for development of a potent starter in beverage industries (Sha et al., 2018).
Conclusion
Our study has shown a wide diversity of yeast and mold species (594 fungal species) in dry starters of North East India, based on nucleotides sequences clustered into OTUs, following the amplicon sequencing using a high-through sequence platform as well as bioinformatics tools. Taxonomical identifications of some sample-specific species of mycobiome in these starters are a remarkable observation in lesser-known, traditionally prepared dry starters for alcohol production in India. The present study demonstrated the baseline data for mycobiome diversity in traditionally prepared dry starters of India.
Author Contributions
SS conducted the major experiments. MS has assisted with the bioinformatics. JT supervised the experiments and finalized the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are gratefully to Department of Biotechnology (DBT), Government of India for financial support. SS was grateful to DBT for award of Senior Research Fellowship. MS for the Research Associateship from DBT-Bioinformatics Centre sanctioned to JT.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00348/full#supplementary-material
FIGURE S1 | Distribution of rare-phylotypes category with 19 different class level taxa in six starters of North East India.
FIGURE S2 | Correlation among mycobiome diversity of six starters (chowan, dawdim, hamei, humao, khekhrii, and phut) A: 6 species of yeasts by culture-dependent method (ITS-PCR) (Sha et al., 2018), B: 24 species (yeasts = 6 and molds = 24) by PCR-DGGE analysis (Sha et al., 2018) and C: Total 594 species (yeasts = 83 and molds = 511) by high-throughput sequencing technique. Number and identity of each species are presented in Supplementary Table 1.
TABLE S1 | Table showing total number of 594 fungal species detected by HTS including common species (224), unique species (133) and rare-species (237), and their taxonomic affiliations in samples of chowan, dawdim, hamei, humao, khekhrii, and phut. Table also shows number of yeast species (6) detected by ITS-PCR, and yeast (18) and mold species (6) by PCR-DGGE in the same samples as reported earlier by Sha et al. (2018).
TABLE S2 | Table showing percent distribution of total yeast and mold species in different starters with their respective morphology and mode nutrition.
TABLE S3 | Different operational taxonomic units (OTUs) with taxonomy and their respective functional attributes observed in different dried starters. Generation of OTUs and functional attributes were derived through PIPITS (Gweon et al., 2015) and FUNGuild (Nguyen et al., 2016) environments.
Footnotes
- ^www.northeasttourism.gov.in
- ^http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- ^https://github.com/torognes/vsearch
- ^http://www.stbates.org/guilds/app.php
- ^https://www.r-project.org/
References
Aguayo, J., Fourrier-Jeandel, C., Husson, C., and Ioos, R. (2018). Assessment of passive traps combined with high-throughput sequencing to study airborne fungal communities. Appl. Environ. Microbiol. 84:e2637-17. doi: 10.1128/AEM.02637-17
Alegría,Á, González, R., Díaz, M., and Mayo, B. (2011). Assessment of microbial populations dynamics in a blue cheese by culturing and denaturing gradient gel electrophoresis. Curr. Microbiol. 62, 888–893. doi: 10.1007/s00284-010-9799-7
Anupma, A., Pradhan, P., Sha, S. P., and Tamang, J. P. (2018). Traditional skill of ethnic people of the Eastern Himalayas and North East India in preserving microbiota as dry amylolytic starters. Indian J. Trad. Knowl. 17, 184–190.
Ariyawansa, H. A., Thambugala, K. M., Manamgoda, D. S., Jayawardena, R., Camporesi, E., Boonmee, S., et al. (2015). Towards a natural classification and backbone tree for Pleosporaceae. Fungal Divers. 71, 85–139. doi: 10.1007/s13225-015-0323-z
Bal, J., Yun, S. H., Chun, J., Kim, B. T., and Kim, D. H. (2016). Taxonomic characterization, evaluation of toxigenicity, and saccharification capability of Aspergillus section Flavi isolates from Korean traditional wheat-based fermentation starter nuruk. Mycobiology 3, 155–161. doi: 10.5941/MYCO.2016.44.3.155
Banchi, E., Stankovic, D., Fernández-Mendoza, F., Gionechetti, F., Pallavicini, A., and Muggia, L. (2018). ITS2 metabarcoding analysis complements lichen mycobiome diversity data. Mycolog. Prog. 9, 1049–1066. doi: 10.1007/s11557-018-1415-4
Bellemain, E., Davey, M. L., and Kauserud, H. (2013). Fungal palaeodiversity revealed using high-throughput metabarcoding of ancient DNA from arctic permafrost. Environ. Microbiol. 15, 1176–1189. doi: 10.1111/1462-2920.12020
Bhadury, P., Bik, H., Lambshead, J. D., Austen, M. C., Smerdon, G. R., and Rogers, A. D. (2011). Molecular diversity of fungal phylotypes co-amplified alongside nematodes from coastal and deep-sea marine environments. PLoS One 6:e26445. doi: 10.1371/journal.pone.0026445
Bhute, S. S., Ghaskadbi, S. S., and Shouche, Y. S. (2017). “Rare biosphere in human gut: a less explored component of human gut microbiota and its association with human health,” in The Mining of Microbial Wealth and MetaGenomics, ed. V. C. Kalia (Singapore: Springer), 133–142.
Blaalid, R., Kumar, S., Nilsson, R. H., Abarenkov, K., Kirk, P. M., and Kauserud, H. (2013). ITS1 versus ITS2 as DNA metabarcodes for fungi. Mol. Eco. Res. 13, 218–224. doi: 10.1111/1755-0998.12065
Bokulich, N. A., and Mills, D. A. (2013). Improved selection of internal transcribed spacer-specific primers enables quantitative, ultra-high-throughput profiling of fungal communities. Appl. Environ. Microbiol. 79, 2519–2526. doi: 10.1128/AEM.03870-12
Bora, S. S., Keot, J., Das, S., Sarma, K., and Barooah, M. (2016). Metagenomics analysis of microbial communities associated with a traditional rice wine starter culture (Xaj-pitha) of Assam, India. 3 Biotech. 6:153. doi: 10.1007/s13205-016-0471-1
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Capozzi, V., and Spano, G. (2011). Food microbial biodiversity and “microbes of protected origin”. Front. Microbiol. 2:237. doi: 10.3389/fmicb.2011.00237
Chakrabarty, J., Sharma, G. D., and Tamang, J. P. (2014). Traditional technology and product characterization of some lesser-known ethnic fermented foods and beverages of North Cachar Hills district of Assam. Indian J. Trad. Knowl. 13, 706–715.
Chen, B., Wu, Q., and Xu, Y. (2014). Filamentous fungal diversity and community structure associated with the solid state fermentation of Chinese Maotai-flavor liquor. Int. J. Food Microbiol. 179, 80–84. doi: 10.1016/j.ijfoodmicro.2014.03.011
Cocolin, L., Alessandria, V., Dolci, P., Gorra, R., and Rantsiou, R. (2013). Culture-independent methods to assess the diversity and dynamics of microbiota during food fermentation. Int. J. Food Microbiol. 167, 29–43. doi: 10.1016/j.ijfoodmicro.2013.05.008
Comeau, A. M., Douglas, G. M., and Langille, M. G. I. (2017). Microbiome helper: a custom and streamlined workflow for microbiome research. mSystems 2:e127-16. doi: 10.1128/mSystems.00127-16
Cox, K. D., Black, M. J., Filip, N., Miller, M. R., Mohn, K., Mortimor, J., et al. (2017). Community assessment techniques and the implications for rarefaction and extrapolation with Hill numbers. Ecol. Evol. 7:11213. doi: 10.1002/ece3.3580
Cuadros-Orellana, S., Leite, L. R., Smith, A., Medeiros, J. D., Badotti, F., Fonseca, P. L. C., et al. (2013). Assessment of fungal diversity in the environment using metagenomics: a decade in review. Fungal Genom. Biol. 3:2. doi: 10.4172/2165-8056.1000110
Dung, N. T. P., Rombouts, F. M., and Nout, M. J. R. (2007). Characteristics of some traditional Vietnamese starch-based rice wine fermentation starters (men). LWT Food Sci. Technol. 40, 130–135. doi: 10.1016/J.LWT.2005.08.004
Ercolini, D. (2013). High-throughput sequencing and metagenomics: moving forward in the culture-independent analysis of food microbial ecology. Appl. Environ. Microbiol. 79, 3148–3155. doi: 10.1128/AEM.00256-213
Ercolini, D., Filippis, F. D., Storia, A. L., and Iacono, M. (2012). A “remake” of the microbiota involved in the production of water buffalo mozzarella cheese by high throughput sequencing. Appl. Environ. Microbiol. 78, 8142–8145. doi: 10.1128/AEM.02218-12
Gonelimali, F. D., Lin, J., Miao, W., Xuan, J., Charles, F., Chen, M., et al. (2018). Antimicrobial properties and mechanism of action of some plant extracts against food pathogens and spoilage microorganisms. Front. Microbiol. 9:1639. doi: 10.3389/fmicb.2018.01639
Gweon, H. S., Oliver, A., Taylor, J., Booth, T., Gibbs, M., and Read, D. S. (2015). PIPITS: an automated pipeline for analyses of fungal internal transcribed spacer sequences from the Illumina sequencing platform. Methods Ecol. Evol. 6, 973–980. doi: 10.1111/2041-210X.12399
Hesseltine, C. W. (1983). Microbiology of oriental fermented foods. Ann. Rev. Microbiol. 37, 575–601. doi: 10.1146/annurev.mi.37.100183.003043
Hesseltine, C. W., and Kurtzman, C. P. (1990). Yeasts in amylolytic food starters. Anal. Inst. Biol. Univ. Nac. Autón. México Ser. Bot. 60, 1–7. doi: 10.3389/fmicb.2018.00894
Hesseltine, C. W., and Ray, M. L. (1988). Lactic acid bacteria in murcha and ragi. J. Appl. Microbiol. 64, 395–401. doi: 10.1111/j.1365-2672.1988.tb05096.x
Hesseltine, C. W., Rogers, R., and Winarno, F. G. (1988). Microbiological studies on amylolytic Oriental fermentation starters. Mycopathology 101, 141–155. doi: 10.1002/jsfa.2740440410
Jeyaram, K., Singh, W., Capece, A., and Romano, P. (2008). Molecular identification of yeast species associated with “Hamei” — A traditional starter used for rice wine production in Manipur, India. Int. J. Food Microbiol. 124, 115–125. doi: 10.1016/j.ijfoodmicro.2008.02.029
Jeyaram, K., Tamang, J. P., Capece, A., and Romano, P. (2011). Geographical markers for Saccharomyces cerevisiae strains with similar technological origins domesticated for rice-based ethnic fermented beverages production in North East India. Antonie Van Leeuwenhoek 100, 569–578. doi: 10.1007/s10482-011-9612-z
Jung, M. J., Nam, Y. D., Roh, S. W., and Bae, J. W. (2012). Unexpected convergence of fungal and bacterial communities during fermentation of traditional Korean alcoholic beverages inoculated with various natural starters. Food Microbiol. 30, 112–123. doi: 10.1016/j.fm.2011.09.008
Kitamura, Y., Kusumoto, K. I., Oguma, T., Nagai, T., Furukawa, S., Suzuki, C., et al. (2016). “Ethnic fermented foods and alcoholic beverages of Japan,” in Ethnic Fermented Foods and Alcoholic Beverages of Asia, ed. J. P. Tamang (New Delhi: Springer), 193–236. doi: 10.1007/978-81-322-2800-4_9
Lahti, L., and Shetty, S. (2017). Tools for Microbiome Analysis in R. Version 1.1.10013. Available at: http://microbiome.github.com/microbiome
Li, P., Xue, Y., Shi, J., Pan, A., Tang, X., and Ming, F. (2018). The response of dominant and rare taxa for fungal diversity within different root environments to the cultivation of Bt and conventional cotton varieties. Microbiome 6:184. doi: 10.1186/s40168-018-0570-9
Li, X. R., Ma, E. B., Yan, L. Z., Meng, H., Du, X. W., Zhang, S. W., et al. (2011). Bacterial and fungal diversity in the traditional Chinese liquor fermentation process. Int. J. Food Microbiol. 146, 31–37. doi: 10.1016/j.ijfoodmicro.2011.01.030
Limtong, S., Sintara, S., and Suwannarit, P. (2002). Yeast diversity in Thai traditional alcoholic starter. Kasetsart J. Nat. Sci. 36, 149–158.
Lv, X., Weng, X., and Huang, R. (2012). Research on biodiversity of yeasts associated with Hongqu glutinous rice wine starters and the traditional brewing process. J. Chinese Inst. Food Sci. Technol. 12, 182–190.
Lv, X. C., Huang, X. L., Zhang, W., Rao, P. F., and Ni, L. (2013). Yeast diversity of traditional alcohol fermentation starters for Hong Qu glutinous rice wine brewing, revealed by culture-dependent and culture-independent methods. Food Control 34, 183–190. doi: 10.1016/J.FOODCONT.2013.04.020
Ly, S., Mith, H., Tarayre, C., Taminiau, B., Daube, G., Fauconnier, M. L., et al. (2018). Impact of microbial composition of Cambodian traditional dried starters (Dombea) on flavor compounds of rice wine: combining amplicon sequencing with HP-SPME-GCMS. Front. Microbiol. 9:894. doi: 10.3389/fmicb.2018.00894
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12. doi: 10.14806/ej.17.1.200
Mayo, B., Rachid, C. T., Alegría,Á, Leite, M. O., Peixoto, R. S., and Delgado, S. (2014). Impact of next generation sequencing techniques in food microbiology. Currt. Genomics 15, 293–309. doi: 10.2174/1389202915666140616233211
Nam, Y. D., Lee, S. Y., and Lim, S. I. (2012). Microbial community analysis of Korean soybean pastes by next-generation sequencing. Int. J. Food Microbiol. 155, 36–42. doi: 10.1016/j.ijfoodmicro.2012.01.013
Nguyen, N. H., Song, Z., Bates, S. T., Branco, S., Tedersoo, L., and Menke, J. (2016). FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 20, 241–248. doi: 10.1016/j.funeco.2015.06.006
Nikkuni, S., Karki, T. B., Terao, T., and Suzuki, C. (1996). Microflora of mana, a Nepalese rice koji. J. Ferment. Bioengin. 81, 168–170. doi: 10.1016/0922-338X(96)87597-0
Nout, M. J. R., and Aidoo, K. E. (2002). “Asian fungal fermented foods,” in Mycota: A Comprehensive Treatise on Fungi as Experimental Systems and Applied Research, Industrial Applications, ed. H. D. Osiewacz (Berlin: Springer-Verlag), 23–47.
Połka, J., Rebecchi, A., Pisacane, V., Morelli, L., and Puglisi, E. (2015). Bacterial diversity in typical Italian salami at different ripening stages as revealed by high-throughput sequencing of 16S rRNA amplicons. Food Microbiol. 46, 342–356. doi: 10.1016/j.fm.2014.08.023
Puerari, C., Magalhães-Guedes, K. T., and Schwan, R. F. (2015). Physicochemical and microbiological characterization of chicha, a rice-based fermented beverage produced by Umutina Brazilian Amerindians. Food Microbiol. 46, 210–217. doi: 10.1016/j.fm.2014.08.009
Rivett, D. W., and Bell, T. (2018). Abundance determines the functional role of bacterial phylotypes in complex communities. Nat. Microbiol. 3, 767–772. doi: 10.1038/s41564-018-0180-0
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ. 4:e2584. doi: 10.7717/peerj.2584
Roh, S. W., Abell, G. C., Kim, K. H., Nam, Y. D., and Bae, J. W. (2010). Comparing microarrays and next-generation sequencing technologies for microbial ecology research. Trends Biotechnol. 28, 291–299. doi: 10.1016/j.tibtech.2010.03.001
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schoch, C. L., Seifert, K. A., Huhndorf, S., Robert, V., Spouge, J. L., Levesque, C. A., et al. (2012). Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for fungi. Proc. Natl. Acad. Sci. 109, 6241–6246. doi: 10.1073/pnas.1117018109
Sha, S. P., Anupama, A., Pradhan, P., Prasad, G. S., and Tamang, J. P. (2016). Identification of yeasts by polymerase-chain-reaction-mediated denaturing gradient gel electrophoresis in marcha, an ethnic amylolytic starter of India. J. Ethnic Foods 3, 292–296. doi: 10.1016/J.JEF.2016.11.009
Sha, S. P., Jani, K., Sharma, A., Anupma, A., Pradhan, P., Shouche, Y., et al. (2017). Analysis of bacterial and fungal communities in Marcha and Thiat, traditionally prepared amylolytic starters of India. Sci. Rep. 7:10967. doi: 10.1038/s41598-017-11609-y
Sha, S. P., Suryavanshi, M. V., Jani, K., Sharma, A., Shouche, Y. S., and Tamang, J. P. (2018). Diversity of yeasts and molds by culture-dependent and culture-independent methods for mycobiome surveillance of traditionally prepared dried starters for the production of Indian alcoholic beverages. Front. Microbiol. 9:2237. doi: 10.3389/fmicb.2018.02237
Shangpliang, H. N. J., Rai, R., Keisam, S., Jeyaram, K., and Tamang, J. P. (2018). Bacterial community in naturally fermented milk products of Arunachal Pradesh and Sikkim of India analysed by high-throughput amplicon sequencing. Sci. Rep. 8:1532. doi: 10.1038/s41598-018-19524-6
Steinkraus, K. H. (1996). Handbook of Indigenous Fermented Food, 2nd Edn. New York, NY: Marcel Dekker, Inc.
Sujaya, I. N., Amachi, S., Yokota, A., Asano, K., and Tomita, F. (2001). Identification and characterization of lactic acid bacteria in ragi tape. World J. Microbiol. Biotech. 17, 349–357. doi: 10.1023/A:1016642315022
Surono, I. S. (2016). “Ethnic fermented foods and beverages of Indonesia,” in Ethnic Fermented Foods and Alcoholic Beverages of Asia, ed. J. P. Tamang (New Delhi: Springer), 341–382. doi: 10.1007/978-81-322-2800-4_14
Tamang, J. P. (2010a). “Diversity of fermented beverages,” in Fermented Foods and Beverages of the World, eds J. P. Tamang and K. Kailasapathy (New York, NY: CRC Press), 85–125. doi: 10.1201/EBK1420094954-c3
Tamang, J. P. (2010b). Himalayan Fermented Foods: Microbiology, Nutrition, and Ethnic Values. New York, NY: CRC Press.
Tamang, J. P., Dewan, S., Tamang, B., Rai, A., Schillinger, U., and Holzapfel, W. H. (2007). Lactic acid bacteria in hamei and marcha of North East India. Indian J. Microbiol. 47, 119–125. doi: 10.1007/s12088-007-0024-8
Tamang, J. P., and Fleet, G. H. (2009). “Yeasts diversity in fermented foods and beverages,” in Yeasts Biotechnology: Diversity and Applications, eds T. Satyanarayana and G. Kunze (New York, NY: Springer), 169–198. doi: 10.1007/978-1-4020-8292-4_9
Tamang, J. P., and Sarkar, P. K. (1995). Microflora of murcha: an amylolytic fermentation starter. Microbios 81, 115–122.
Tamang, J. P., Sarkar, P. K., and Hesseltine, C. W. (1988). Traditional fermented foods and beverages of Darjeeling and Sikkim–a review. J. Sci. Food Agric. 44, 375–385. doi: 10.1002/jsfa.2740440410
Tamang, J. P., Tamang, N., Thapa, S., Dewan, S., Tamang, B. M., Yonzan, H., et al. (2012). Microorganisms and nutritional value of ethnic fermented foods and alcoholic beverages of North East India. Indian J. Trad. Knowl. 11, 7–25.
Tamang, J. P., Thapa, N., Savitri, and Bhalla, T. C. (2016a). “Ethnic fermented foods and beverages of India,” in Ethnic Fermented Foods and Alcoholic Beverages of Asia, ed. J. P. Tamang (New Delhi: Springer), 17–72.
Tamang, J. P., Watanabe, K., and Holzapfel, W. H. (2016b). Review: diversity of microorganisms in global fermented foods and beverages. Front. Microbiol. 7:377. doi: 10.3389/fmicb.2016.00377
Tamang, J. P., and Thapa, S. (2006). Fermentation dynamics during production of bhaatijaanr, a traditional fermented rice beverage of the Eastern Himalayas. Food Biotechnol. 20, 251–261. doi: 10.1080/08905430600904476
Thanh, V. N., Mai, L. T., and Tuan, D. A. (2008). Microbial diversity of traditional Vietnamese alcohol fermentation starters (banh men) as determined by PCR-mediated DGGE. Int. J. Food Microbiol. 128, 268–273. doi: 10.1016/j.ijfoodmicro.2008.08.020
Thapa, S., and Tamang, J. P. (2004). Product characterization of kodo ko jaanr: fermented finger millet beverage of the Himalayas. Food Microbiol. 21, 617–622. doi: 10.1016/J.FM.2004.01.004
Thapa, S., and Tamang, J. P. (2006). Microbiological and physico-chemical changes during fermentation of kodokojaanr, a traditional alcoholic beverage of the Darjeeling hills and Sikkim. Indian J. Microbiol. 46, 333–341.
Tsuyoshi, N., Fudou, R., Yamanaka, S., Kozaki, M., Tamang, N., Thapa, S., et al. (2005). Identification of yeast strains isolated from marcha in Sikkim, a microbial starter for amylolytic fermentation. Int. J. Food Microbiol. 99, 135–146. doi: 10.1016/j.ijfoodmicro.2004.08.011
Wei, T., and Simko, V. (2017). R Package “Corrplot”: Visualization of a Correlation Matrix (Version 0.84). Available at: https://github.com/taiyun/corrplot.
White, T., Bruns, T., Lee, S., and Taylor, J. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in PCR Protocols: a Guide to Methods and Applications, eds M. Innis, D. Gelfand, J. Sninsky, and T. White (San Diego, CA: Academic Press), 315–322.
Wickham, H. (2016). GGplot2: Elegant Graphics for Data Analysis. Berlin: Springer. doi: 10.1007/978-3-319-24277-4
Xie, G., Li, W., Lu, J., Cao, Y., Fang, H., Zou, H., et al. (2007). Isolation and Identification of representative fungi from Shaoxing rice wine wheat Qu using a polyphasic approach of culture-based and molecular-based methods. J. Inst. Brew. 113, 272–279. doi: 10.1002/j.2050-0416.2007.tb00287.x
Xu, Y., Zhi, Y., Wu, Q., Du, R., and Xu, Y. (2017). Zygosaccharomyces bailii is a potential producer of various flavor compounds in Chinese Maotai-flavor liquor fermentation. Front. Microbiol. 8:2609. doi: 10.3389/fmicb.2017.02609
Yan, Y., Qian, Y., Ji, F., Chen, J., and Han, B. (2013). Microbial composition during Chinese soy sauce koji-making based on culture dependent and independent methods. Food Microbiol. 34, 189–195. doi: 10.1016/j.fm.2012.12.009
Keywords: dry starter, mycobiome, yeasts, molds, high-throughput sequencing
Citation: Sha SP, Suryavanshi MV and Tamang JP (2019) Mycobiome Diversity in Traditionally Prepared Starters for Alcoholic Beverages in India by High-Throughput Sequencing Method. Front. Microbiol. 10:348. doi: 10.3389/fmicb.2019.00348
Received: 13 October 2018; Accepted: 11 February 2019;
Published: 05 March 2019.
Edited by:
Teresa Zotta, National Research Council (CNR), ItalyReviewed by:
Angela Capece, University of Basilicata, ItalyKeshab Chandra Mondal, Vidyasagar University, India
Copyright © 2019 Sha, Suryavanshi and Tamang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jyoti Prakash Tamang, anlvdGlfdGFtYW5nQGhvdG1haWwuY29t