 Miho Watanabe
Miho Watanabe Hisaya Kojima
Hisaya Kojima Kazuhiro Umezawa
Kazuhiro Umezawa Manabu Fukui
Manabu Fukui- 1Institute of Low Temperature Science, Hokkaido University, Sapporo, Japan
- 2Japan Society for the Promotion of Science, Tokyo, Japan
Desulfonema ishimotonii strain Tokyo 01T is a filamentous sulfate-reducing bacterium isolated from a marine sediment. In this study, the genome of this strain was sequenced and analyzed with a focus on gene transfer from phylogenetically distant organisms. While the strain belongs to the class Deltaproteobacteria, hundreds of proteins encoded in the genome showed the highest sequence similarities to those of organisms outside of the class Deltaproteobacteria, suggesting that more than 20% of the genome is putatively of foreign origins. Many of these proteins had the highest sequence identities with proteins encoded in the genomes of filamentous bacteria, including giant sulfur oxidizers of the orders Thiotrichales, cyanobacteria of various genera, and uncultured bacteria of the candidate phylum KSB3. As mobile genetic elements transferred from phylogenetically distant organisms, putative inteins were identified in the GyrB and DnaE proteins encoded in the genome of strain Tokyo 01T. Genes involved in DNA recombination and repair were enriched in comparison to the closest relatives in the same family. Some of these genes were also related to those of organisms outside of the class Deltaproteobacteria, suggesting that they were acquired by horizontal gene transfer from diverse bacteria. The genomic data suggested significant genetic transfer among filamentous gliding bacteria in phylogenetically dispersed lineages including filamentous sulfate reducers. This study provides insights into the genomic evolution of filamentous bacteria belonging to diverse lineages, characterized by various physiological functions and different ecological roles.
Introduction
The genus Desulfonema belongs to the family Desulfobacteraceae in the class Deltaproteobacteria (Kuever, 2014). This genus encompasses mesophilic filamentous sulfate-reducing bacteria which have gliding motility (Kuever et al., 2015). At present, the genus consists of three marine species: D. limicola (Widdel et al., 1983), D. magnum (Widdel et al., 1983), and D. ishimotonii (Fukui et al., 1999). Previous studies have reported that Desulfonema species attach to filamentous giant sulfur oxidizers which form conspicuous mats on the sea floor (Fukui et al., 1999; Teske et al., 2009). In a hypersaline lake, Desulfonema organisms were abundant in the oxic layer of cyanobacterial mats (Teske et al., 1998; Minz et al., 1999). These findings of physical association imply that Desulfonema species have close relationships with filamentous sulfur oxidizers and cyanobacteria, which may drive cycles of sulfur and carbon.
The type strain of D. ishimotonii, strain Tokyo 01T, was isolated from sulfidic marine sediment of Tokyo Bay, Japan. The 16S rRNA gene sequence analysis demonstrated that strain Tokyo 01T showed 89–90% of sequence similarity to the type strains of D. limicola and D. magnum. Utilization of alcohols is the distinctive characteristic of D. ishimotonii, which has not been found in other Desulfonema species (Widdel et al., 1983; Fukui et al., 1999). Here, we describe the complete genome sequence of Desulfonema ishimotonii strain Tokyo 01T. The sequenced genome was analyzed with a focus on gene transfer among phylogenetically distant lineages.
Materials and Methods
DNA Preparation, Sequencing, and Assembly
Desulfonema ishimotonii strain Tokyo 01T (=DSM 9680T) was grown in artificial seawater medium containing aluminum salt, as described previously (Fukui et al., 1999). Genomic DNA was extracted from the collected cells using a Wizard® Genomic DNA Purification kit (Promega).
Sequencing and library construction were carried out using the PacBio RS II platform. In total, 84,039 polymerase reads (659,435,372 bp) were generated and filtered. The resulting sequences (655,821,304 bp) were assembled into nine linear contigs using FALCON version 0.4.0 (Chin et al., 2016). For the longest contig (6,610,565 bp), a specific PCR primer set was designed by Primer-BLAST (Ye et al., 2012) to bridge both ends of the linear contig. With the primer pair, a PCR product with a length of 1,799 bp was obtained and sequenced. The resulting sequence completely matched that of the contig at both ends, and a circular contig representing a chromosome was constructed by aligning them. The other eight short contigs (max length = 6,714) were directly subjected to further analysis.
Genome Annotation and Functional Analysis
In the obtained genome, genes were predicted by the MiGAP pipeline (Sugawara et al., 2009). In this pipeline, open-reading frames likely to encode proteins were identified as coding sequences (CDSs) by MetaGene Annotator (Noguchi et al., 2008). In addition, RNAmmer (Lagesen et al., 2007) and tRNAscan-SE (Lowe et al., 1997) were used to identify genes for rRNA and tRNA, respectively. Based on the results of automatic gene prediction by the pipeline, further manual annotation was performed using IMC-GE software (In Silico Biology, Yokohama, Japan), as described below. Putative CDSs possessing BLASTP matches with more than 60% coverage, 30% identity, and E-values less than e-8 were considered as functional genes. The CDSs were annotated as hypothetical proteins when these standard values were not satisfied or the function of the hit was unidentified. Transcription start sites were corrected based on multiple sequence alignments. The eight linear contigs were excluded from further genomic analysis because there was no solid evidence to indicate that they are constituents of the genome of the strain Tokyo 01T. CRISPR loci were distinguished using the CRISPR Recognition Tool (Bland et al., 2007). The chromosome was also analyzed with CheckM version 1.0.8 (Parks et al., 2015), to assess its genome completeness and possible contamination in it.
To evaluate the phylogenetic affiliation of each protein-coding gene, all putative protein sequences were searched against the NCBI-nr database (released in 18 February 2018). The top hits with E-values less than e-5 were regarded as significant hits by BLASTP analysis. The protein-coding genes in the genome sequence were also subjected to analysis on the WebMGA (Wu et al., 2011) for COG annotations. In addition, sequence analysis was carried out using REBASE (Roberts et al., 2015) to identify the DNA restriction and modification system in the genome. Functional domains within proteins were identified by using Pfam protein families database (El-Gebali et al., 2019). For a comparative analysis, COGs annotations of four strains from the genera Desulfosarcina and Desulfococcus were retrieved from the IMG/M database (Markowitz et al., 2013). These strains were selected as the closest relatives of strain Tokyo 01T, on the basis of the 16S rRNA gene sequence similarities higher than 89% (of 1,390 total nucleotide positions).
Phylogenetic Tree Construction
Phylogenetic analyses were performed for several proteins and inteins encoded in the genome based on their amino acid sequences. They were searched against the public databases (GenBank/EMBL/DDBJ) and aligned with reference sequences using the program CLUSTAL X version 2.1 (Larkin et al., 2007). Selection of the best nucleotide substitution models and construction of the phylogenetic trees were performed using the program MEGA version 7.0.20 (Kumar et al., 2016). All positions with gaps in the alignments were excluded from the calculation.
Results and Discussion
General Genomic Features
The obtained genome was comprised of a single 6,610,564 bp circular chromosome (Supplementary Figure S1) and 8 short contigs (total length = 28,713 bp) (Table 1). The complete genome sequence of D. ishimotonii Tokyo 01T (=DSM 9680) has been deposited at DDBJ/EMBL/GenBank under the accession number of BEXT01000000.

Table 1. General genomic features of Desulfonema ishimotonii strain Tokyo 01T.
Overall, 78.8% of the genome sequence was DNA-coding, with a G + C content of 53.5%. In total, 76 tRNA genes and 12 rRNA genes (four copies each of the 16S, 23S, and 5S rRNA genes) were predicted in the genome. The four copies of the 16S rRNA gene had almost identical sequences, which showed the highest sequence similarities to Desulfosarcina and Desulfococcus species, rather than the other species in the genus Desulfonema. The highest similarity was observed with Desulfococcus multivorans DSM 2059T (89.8% in 1,440 nucleotide positions), followed by Desulfosarcina variabilis DSM 7267T (89.2% in 1,440 positions). The sequence similarity to the type strains of D. magnum and D. limicola was 89.0 and 88.6%, respectively (1,394 positions). Approximately 70.3% of proteins encoded in the genome of strain Tokyo 01T were assigned to 20 COG functional categories, and their relative abundances are shown in Figure 1. The completeness of the chromosome as estimated by CheckM was 99.68%, and no contamination was detected using the same tool. The GC skew was unstable throughout the chromosome (Supplementary Figure S1), while that of the D. multivorans genome showed the typical pattern, with clear separation at the putative replication origin (Dörries et al., 2016). The atypical pattern of GC skewing was also reported in the genomes of filamentous sulfur oxidizers, Thioploca ingrica (Kojima et al., 2015) and Beggiatoa leptomitoformis (Orlova et al., 2018).
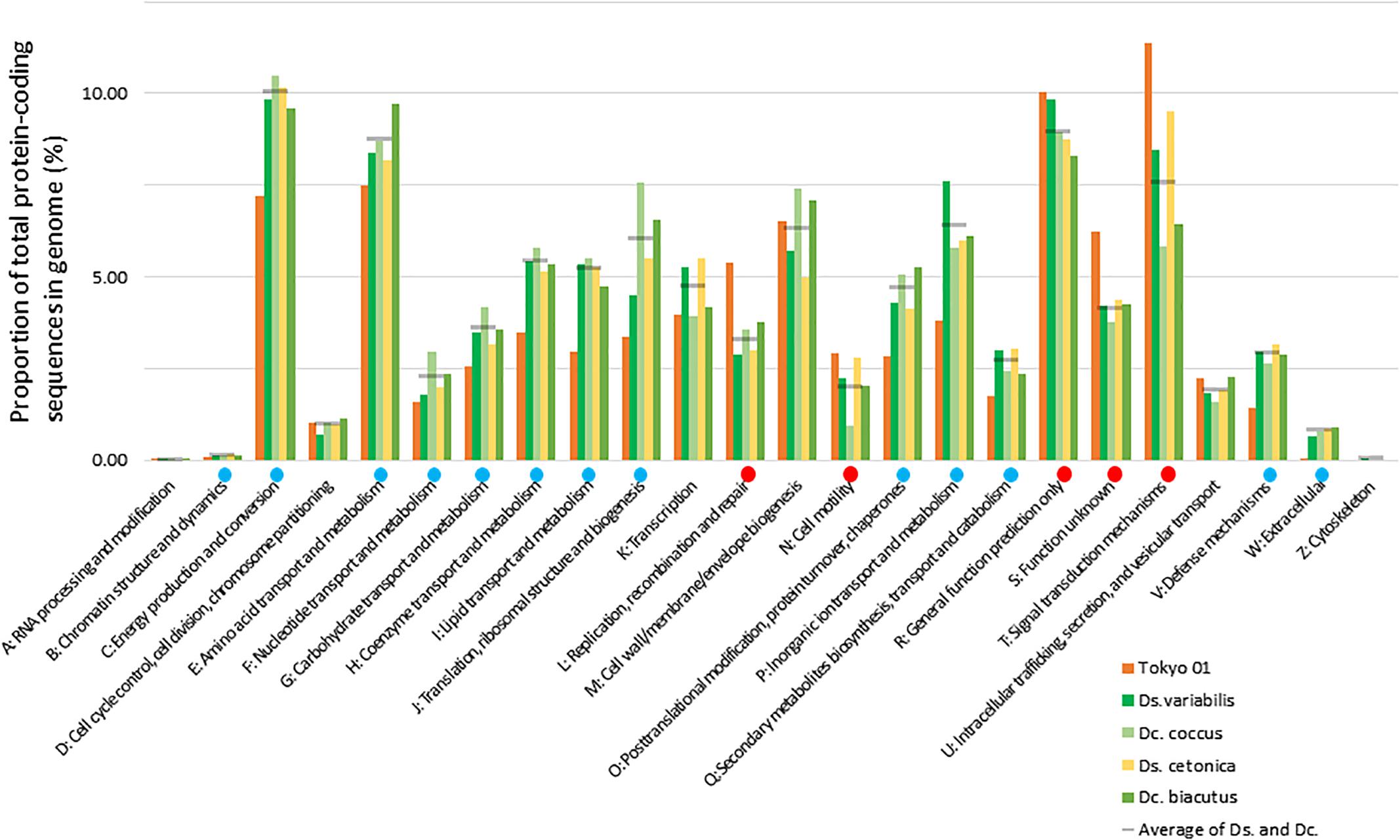
Figure 1. Relative abundances of COG categories in the genomes of strain Tokyo 01T and its closest relatives in the genera Desulfosarcina (Ds.) and Desulfococcus (Dc.). Gray lines indicate average of the Desulfosarcina and Desulfococcus species included in the analysis. Red or blue circles indicate that the marked category is significantly enriched or depleted in Tokyo 01’s genome compared to the other genomes, as calculated by the method previously described (Sekiguchi et al., 2015).
Genetic Basis for Carbon Metabolism and Oxygen Tolerance
A previous study reported that the strain oxidizes organic substrates to CO2 via the anaerobic C1 pathway (Fukui et al., 1999). The strain Tokyo 01T possesses the genes encoding CO dehydrogenase/CO-methylating acetyl-CoA synthase complex (locus tags: DENIS_4589–4592), which is the key enzyme in this pathway. For alcohol oxidation, the genome of the strain harbors five genes encoding alcohol dehydrogenases (DENIS_0326, 0442, 0444, 1430, and 2670). The protein predicted for one of these ORFs (locus tag DENIS_1430) was most similar in amino acid sequence to predicted alcohol dehydrogenases from filamentous or chain-forming sulfur oxidizers, Beggiatoa sp. 4572_84 (68%), “Candidatus (Ca.) Thiomargarita nelsonii” (66%) and T. ingrica (65%) (Kojima et al., 2015; Flood et al., 2016; Dombrowski et al., 2017). The other alcohol dehydrogenases predicted from the genome are phylogenetically related to those of the family Desulfobacteraceae, consistent with the 16S rRNA gene phylogeny.
The ability to tolerate and/or respire oxygen has been detected in various sulfate-reducing bacteria, including members of the family Desulfobacteraceae (Hewitt and Morris, 1975; Hatchikian et al., 1977; Dilling and Cypionka, 1990; Lemos et al., 2001; Kjeldsen et al., 2005). The closest relative of strain Tokyo 01T, D. multivorans DSM 2059T, also possesses genes for oxygen detoxifying enzymes (Dörries et al., 2016). Although strain Tokyo 01T is a strict anaerobe isolated from a sulfidic marine sediment, putative genes for proteins required for oxygen tolerance systems were identified in its genome, including the genes encoding cytochrome c oxidase (DENIS_0725, 0727, 0728), cytochrome ubiquinol oxidase (DENIS_3441, 3442, 3677, 3678), superoxide dismutase (DENIS_3256, 5148), and catalase (DENIS_4322). These defense systems against oxidative stress may also be used by Desulfonema organisms inhabiting oxygenic environments (Teske et al., 1998; Minz et al., 1999). Despite their close phylogenetic relatedness, the predicted catalase encoded in the genome of strain Tokyo 01T is distinct from that of D. multivorans DSM 2069T, with an amino acid sequence similarity of 58%. The putative catalase showed the highest sequence similarity (80%) to that of “Ca. Kuenenia stuttgartiensis,” a bacterium belonging to the phylum Planctomycetes (Parks et al., 2017; Frank et al., 2018). These results suggest that the catalase gene was acquired from a foreign lineage by horizontal gene transfer.
Phylogenetic Affiliation of Protein-Coding Genes
In addition to the cases of putative alcohol dehydrogenase and catalase mentioned above, many proteins encoded in the genome of strain Tokyo 01T showed the highest sequence similarities to organisms outside the class Deltaproteobacteria (Table 2). These results suggest that more than 20% of genes in this genome are putatively of foreign origin.
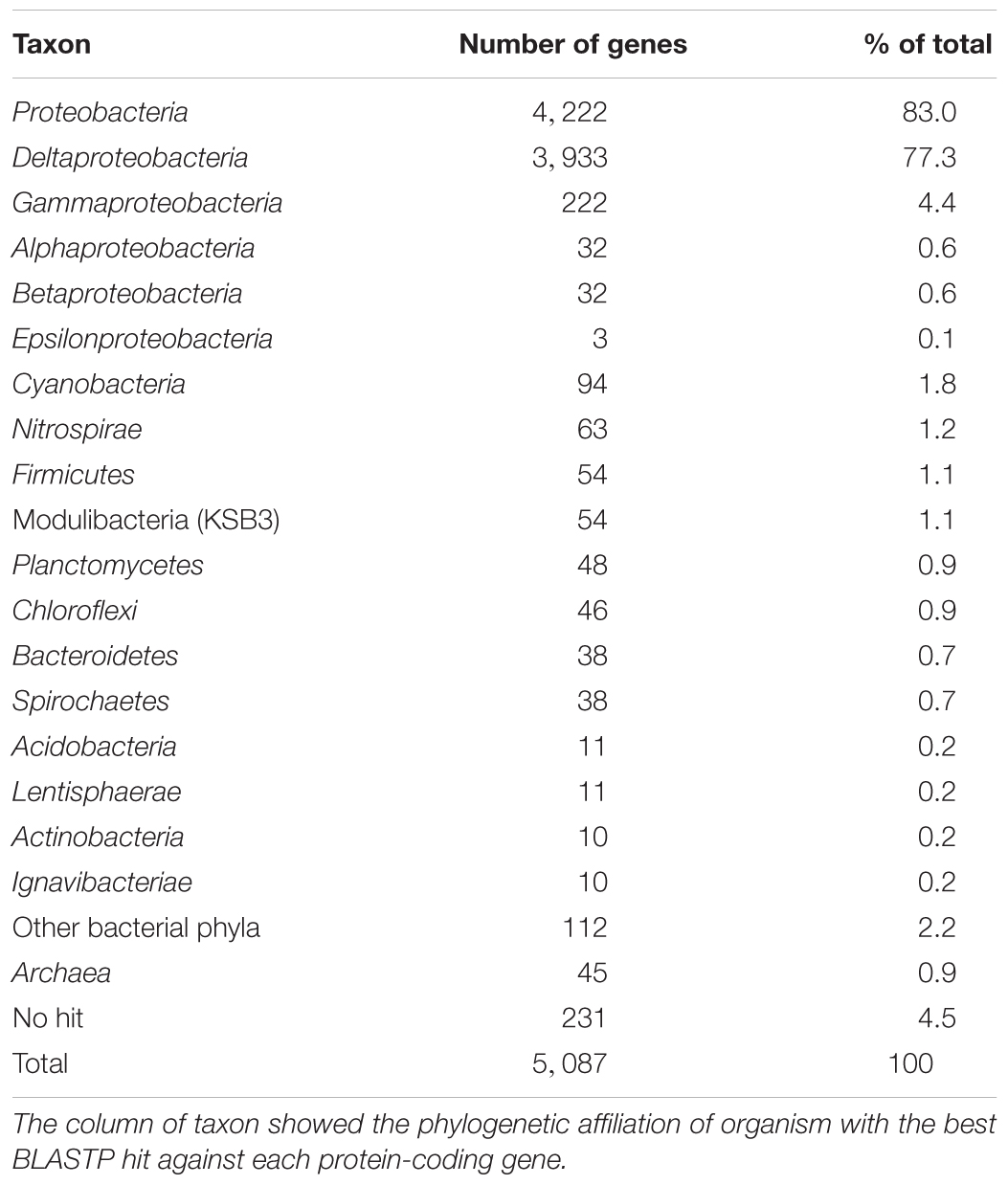
Table 2. Distribution of phylogenetic affiliations of protein-coding genes in circular chromosome of strain Tokyo 01T.
In total, 222 proteins (approx. 4.4% of total CDSs) encoded in the genome of strain Tokyo 01T had the best BLASTP hits to gammaproteobacterial proteins, including 55 and 29 proteins from the orders Thiotrichales and Chromatiales, respectively (Table 3 and Supplementary Table S1). The majority of proteins of these orders are encoded in the genomes of sulfur-oxidizing bacteria (Supplementary Table S1). In particular, those of Thiotrichales were mainly identified in the genomes of filamentous and/or giant sulfur oxidizers, including the genera Beggiatoa, Thioploca, Thiomargarita, Thiothrix, and “Ca. Marithrix.” Most of these proteins with the highest similarities to sulfur-oxidizing bacteria were annotated as hypothetical proteins, and few were identified as functional proteins such as chemotaxis protein CheB, alcohol dehydrogenase, and DNA modification methylase (Supplementary Table S2).
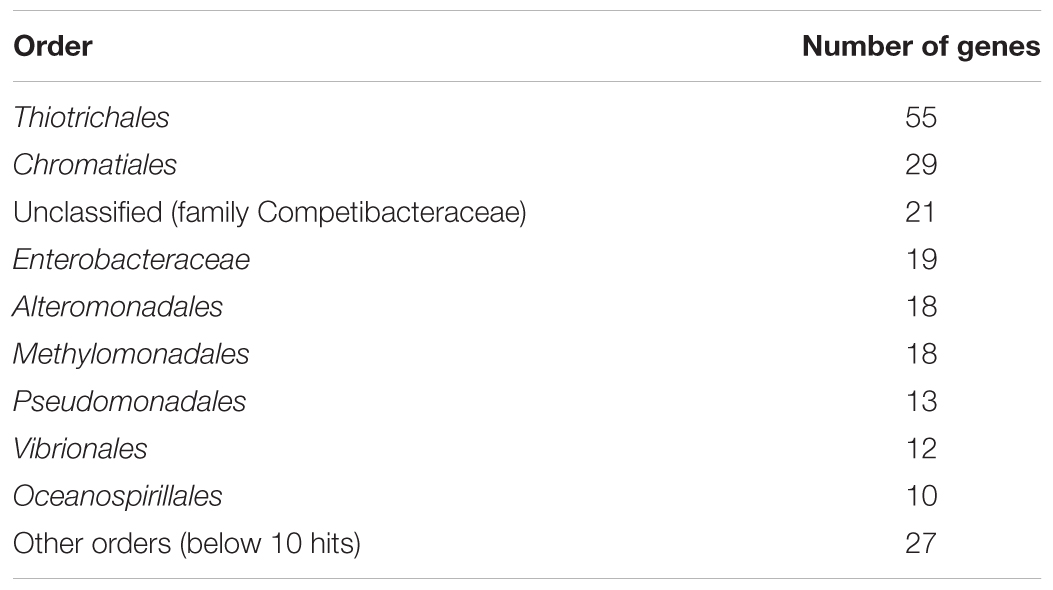
Table 3. Order-level classification of phylogenetic distributions of protein-coding genes with the greatest identity to the class Gammaproteobacteria.
As shown in Table 2, genes potentially transferred from cyanobacteria comprise 1.8% (94 genes) of the protein-coding genes in the genome of Tokyo 01T. Genus-level classification indicated that Leptolyngbya may be a major genetic donor in this lineage. Members of the genus Leptolyngbya are known as gliding filamentous bacteria, which form clusters or mats (Anagnostidis and Komárek, 1988; Komárek and Anagnostidis, 2005). In addition to this genus, many genera identified in this analysis are known to have filamentous morphology (Table 4). The total count of filamentous genera accounted for half of the best hits against cyanobacteria. The functions of these genes with cyanobacterial origins are still unknown, but some were identified as genes encoding transposases (Supplementary Table S3).
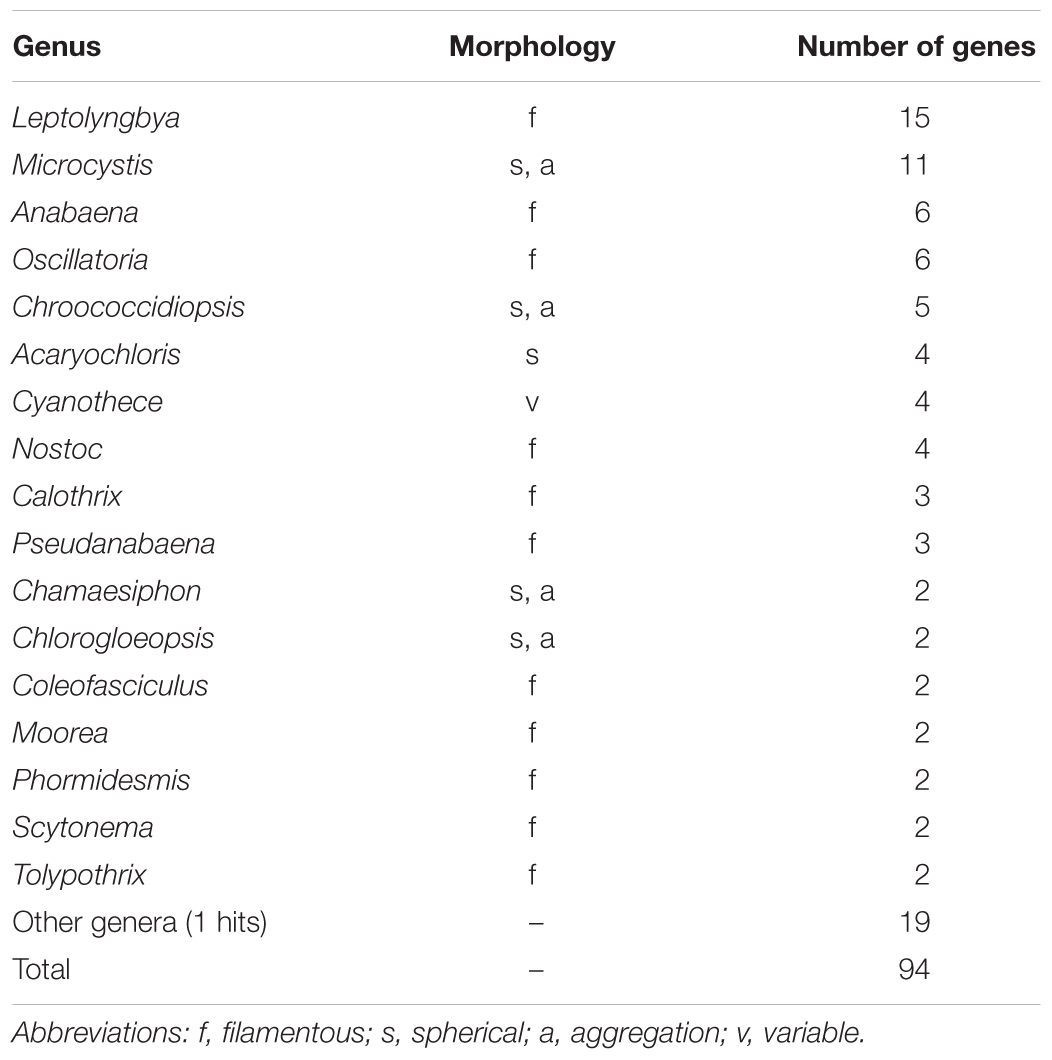
Table 4. Genus-level classification of phylogenetic distributions of protein-coding genes with the greatest identity to cyanobacteria.
The cases of the order Thiotrichales and cyanobacteria indicate that the genome of Tokyo 01T harbors many genes that may have been transferred from filamentous bacteria of phylogenetically distant lineages. In addition, putative genetic donors with filamentous morphology were also found in another lineage without cultivated representatives, known as the candidate phylum KSB3. Among the proteins encoded in the genome of strain Tokyo 01T, 100 proteins (2.0% of total) showed the best BLASTP hits to proteins of uncultured bacteria belonging to candidate phyla. Over half of them were potentially transferred from the candidate phylum KSB3 also known as the phylum “Modulibacteria” (Table 2). Among the 54 best hits against KSB3, 29 belonged to “Ca. Vecturithrix glanuli,” and 14 showed the greatest identity to “Ca. Moduliflexus floccans” (Sekiguchi et al., 2015). Both of these bacteria are known to exhibit filamentous morphology and gliding motility (Sekiguchi et al., 2015). The bacteria of KSB3 are known as the predominant filamentous bacteria in bulking sludge of anaerobic digestion systems (Tanner et al., 2000; Yamada et al., 2007). KSB3 organisms have also been detected as minor members of microbial mats in natural habitats (Wong et al., 2015), but their ecology in the environments remains unknown. In contrast to the genes putatively transferred from the order Thiotrichales and cyanobacteria, the genes putatively transferred from KSB3 encode proteins with known functions in most cases. The majority of these genes were predicted as functional genes encoding signal transduction, transporter, or transcriptional proteins (Supplementary Table S4).
The phylogenetic positions of the protein-coding genes showed that the genome of strain Tokyo 01T appears to have been affected by genetic transfer from the order Thiotrichales, cyanobacteria, and candidate phylum KSB3. Interestingly, gene sharing between giant sulfur oxidizers and cyanobacteria has repeatedly been reported in previous studies (Mußmann et al., 2007; MacGregor et al., 2013; Flood et al., 2014, 2016; Winkel et al., 2016). As mentioned in the section “Introduction,” organisms from both these groups have been shown to have ecological relationships with filamentous sulfate reducers of the genus Desulfonema in the same habitats (Teske et al., 1998; Fukui et al., 1999; Minz et al., 1999; Teske et al., 2009).
Genetic Elements Relevant to Horizontal Transfer
As described above, many protein-coding genes in the genome of strain Tokyo 01T showed high similarities to those of giant sulfur oxidizers and cyanobacteria. In previous studies, genetic sharing between giant sulfur oxidizers and cyanobacteria was suggested by presence of potential mobile elements and horizontally transferred genes in their genomes (Mußmann et al., 2007; MacGregor et al., 2013; Flood et al., 2014). Further genome analysis of strain Tokyo 01T was performed with a specific focus on these genetic elements investigated in the previous studies as described below.
The DNA gyrase subunit B (GyrB) is a type II topoisomerase indispensable for DNA replication in bacteria (Khodursky et al., 2000). In the genome of strain Tokyo 01T, the predicted gyrB gene (locus tag DENIS_4377) is located apart from the gene encoding the other subunit of the topoisomerase (gyrA gene, locus tag DENIS_5021), as in the genome of D. multivorans DSM 2059T. GyrB possesses a putative mini-intein which consists of a protein splicing domain but lacks the endonucleolytic domain. The intein constitutes 123 amino acid residues of the 1,042 amino acids in the entire GyrB sequence. A phylogenetic tree of the putative GyrB intein showed that it has a phylogenetic relationship with those found in several candidate phyla, Archaea, and Cyanobacteria (Figure 2).
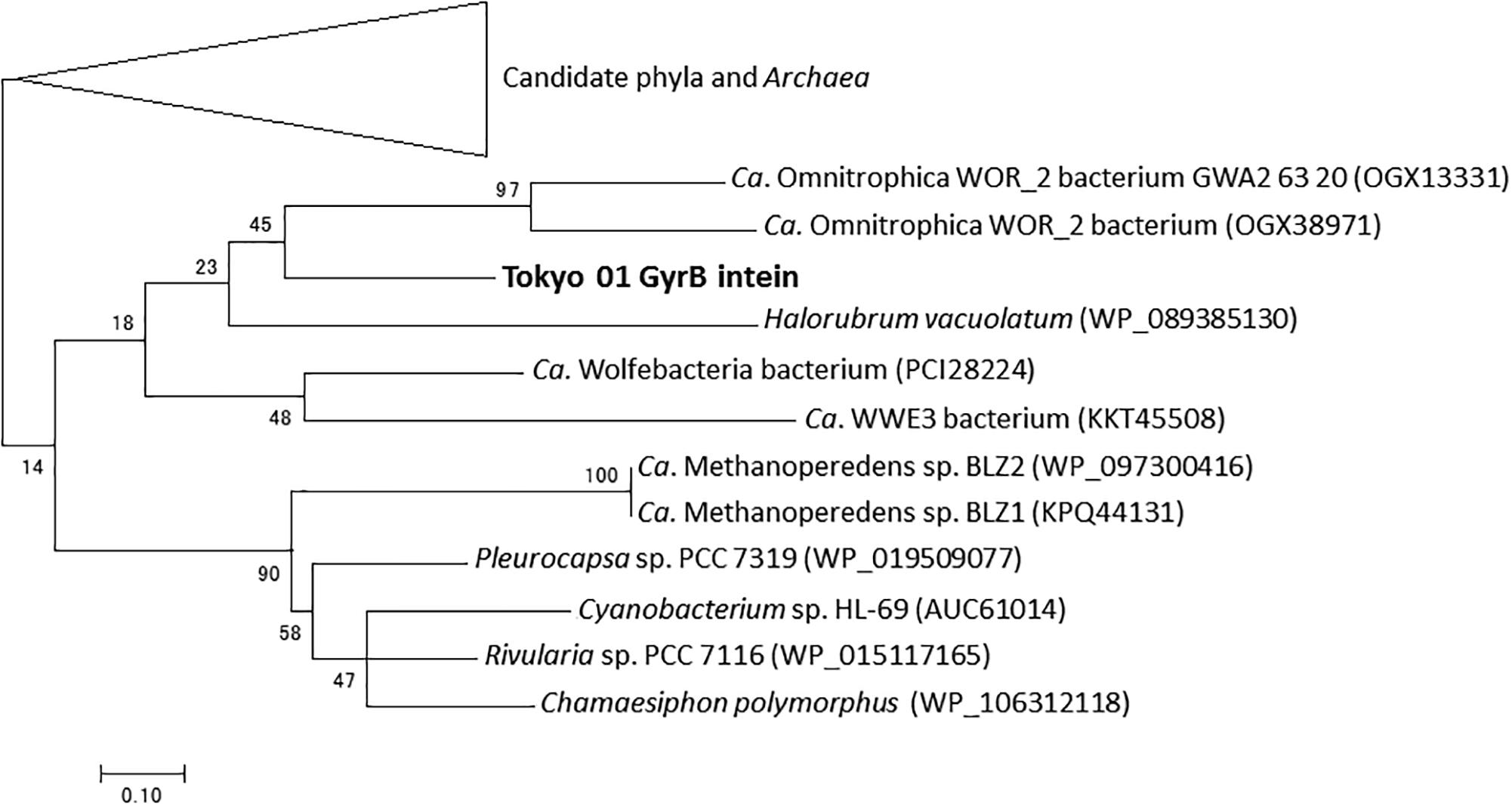
Figure 2. Phylogenetic tree of putative GyrB intein of strain Tokyo 01T and related sequences identified as the top 50 BLAST hits. This tree was constructed by using maximum-likelihood method with LG + G + I model. There were 114 positions in the final dataset. Bootstrap values (percentages of 1,000 replications) are shown at nodes. Neighbor-joining tree are present for comparison in Supplementary Figure S1 in the Supplementary Material.
dnaE is the gene for the catalytic subunit of DNA polymerase III subunit alpha (Welch and McHenry, 1982). The putative DnaE protein is encoded by a split gene, with the locus tags DENIS_0455 and DENIS_0456; these two CDSs are adjacent on the genome but are each in different reading frames. A possible intein with a splicing domain was also found in the DnaE protein in the region encoded by DENIS_0456. This intein sequence comprised 415 amino acid residues of the 1,156 amino acids encoded by DENIS_0456, which represents the major part of the DnaE protein. The phylogenetic tree of the putative DnaE intein demonstrated that the intein is related to that of the candidate phylum KSB3 and Cyanobacteria (Figure 3). A separate DnaE protein divided by the split inteins was reported in the cyanobacterial lineage (Wu et al., 1998; Caspi et al., 2003), but this was not the case for the strain Tokyo 01T.
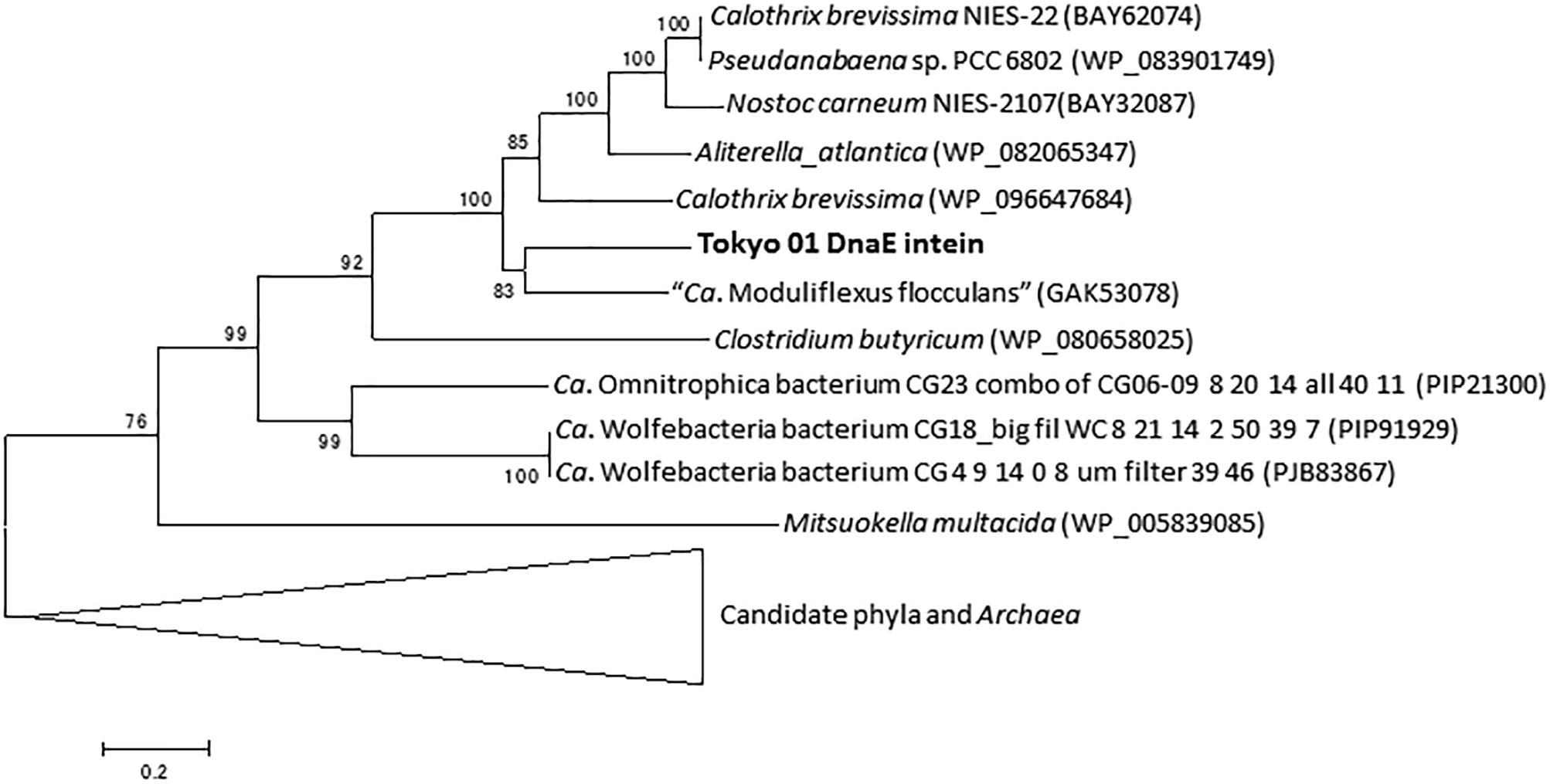
Figure 3. Phylogenetic tree of putative DnaE intein of strain Tokyo 01T and related sequences identified as the top 50 BLAST hits. This tree was constructed by using maximum-likelihood method with LG + G + I model. There were 284 positions in the final dataset. Bootstrap values (percentages of 1,000 replications) are shown at nodes. Neighbor-joining tree are present for comparison in Supplementary Figure S2 in the Supplementary Material.
As shown in Figures 2, 3, the inteins of strain Tokyo 01T in GyrB and DnaE appear to be most similar to possible inteins of several candidate phyla, Archaea, and Cyanobacteria. With the exceptions of the region corresponding to these inteins, the amino acid sequences of GyrB and DnaE proteins of strain Tokyo 01T are closely related to those of the Desulfobacteraceae species. The inteins in GyrB and DnaE have not been found in the closest relatives of strain Tokyo 01T. The GyrB and DnaE inteins were found in the genomes of Beggiatoaceae (MacGregor et al., 2013; Flood et al., 2016) which may be a major donors of foreign genes to strain Tokyo 01T. Nevertheless, their intein sequences were not closely related to that of strain Tokyo 01T.
As another genetic element of foreign origin, introns in the 23S rRNA gene were identified in the genomes of Beggiatoaceae organisms and cyanobacteria (MacGregor et al., 2013). In the 23S rRNA gene of strain Tokyo 01T, however, no intron was identified. XisH and XisI are proteins involved in heterocyst differentiation of some cyanobacteria (Carrasco et al., 2005; Henson et al., 2008), and genes encoding these proteins have been identified in the genomes of giant filamentous sulfur oxidizers (Mußmann et al., 2007; MacGregor et al., 2013). The genes encoding proteins related to XisH or XisI were not found in the genome of strain Tokyo 01T.
DNA Replication, Recombination, and Repair Systems
To reveal the genomic feature of strain Tokyo 01T, comparative analysis against the closest relatives was performed on the basis of the relative abundance of CDSs classified into COG categories (Figure 1). This analysis revealed that the genome of Tokyo 01T harbors a high proportion of genes involved in DNA recombination and repair (L), cell motility (N), and signal transduction (T). Consequently, genes of other categories were generally depleted in comparison to the closest relatives. Among the enriched categories, COG category L was the most highly enriched in the genome of strain Tokyo 01T. The genes in this category may also be related to genetic transfer from giant sulfur oxidizers and cyanobacteria. More detailed properties of the genes for replication, recombination, and repair systems in the genome of strain Tokyo 01T are described below.
The DNA repair systems protect bacterial cells against genetic damage. The nucleotide excision repair (NER) system is one of the most important mechanisms for DNA damage removal and is widely conserved in bacteria. The complete gene set for the NER systems has also been found in some euryarchaeota, whereas archaea of other lineages may have other unknown systems (Grogan, 2000; Costa et al., 2003). In the genome of strain Tokyo 01T, uvrA, uvrB, uvrC, and uvrD were identified as the NER genes (Supplementary Table S5). There were two copies of predicted uvrB and uvrD genes with different sequences. One of the putative uvrD genes (DENIS_2690) encoded a protein more closely related to that of archaea, rather than deltaproteobacteria. Previous studies have demonstrated that the phylogeny of the NER system genes exhibit significant incongruence as a result of horizontal gene transfers (Denamur et al., 2000; Martins-Pinheiro et al., 2004). However, phylogenetic positions of the other NER genes seemed to be generally consistent with the 16S rRNA gene phylogeny.
The RecQ family DNA helicases play an important role in genome maintenance and are ubiquitously conserved in life forms ranging from bacteria to eukaryotes (Nakayama et al., 1984; Kusano et al., 1994; Chu and Hickson, 2009). In general, bacterial genomes encode only one RecQ homolog (Kaiser et al., 2017), although exceptions can be seen in genomes of Staphylococcus aureus subsp. aureus DR10 (accession number: AIDT01000000) and S. aureus subsp. aureus 71193 (CP003045). In the genome of strain Tokyo 01T, two genes encoding RecQ were identified (DENIS_2369 and DENIS_3915). The proteins encoded by these predicted genes are phylogenetically distant from each other. The protein encoded by DENIS_2369 protein had the highest amino acid sequence similarity (78%) with RecQ of Bathymodiolus septemdierum, a gammaproteobacterial thioautotrophic gill symbiont (Ikuta et al., 2016). The closer relationship with gammaproteobacterial proteins was confirmed by constructing a phylogenetic tree (Figure 4). The other putative RecQ, encoded by DENIS_3915, had the highest sequence similarity with the RecQ protein of candidate phylum “Desantisbacteria” bacterium CG2_30_40_21 (Probst et al., 2017), with a similarity of 58%. Phylogenetic analysis showed that this protein is distantly related to the RecQ helicases of some bacteria of the family Desulfobacteraceae (Figure 5). While all known functional domains of the RecQ helicase were identified in the protein encoded by DENIS_3915, the protein encoded by DENIS_2369 seemed to possess only the ATPase domain, which is the most well-conserved element among three RecQ-characteristic domains (Kaiser et al., 2017).
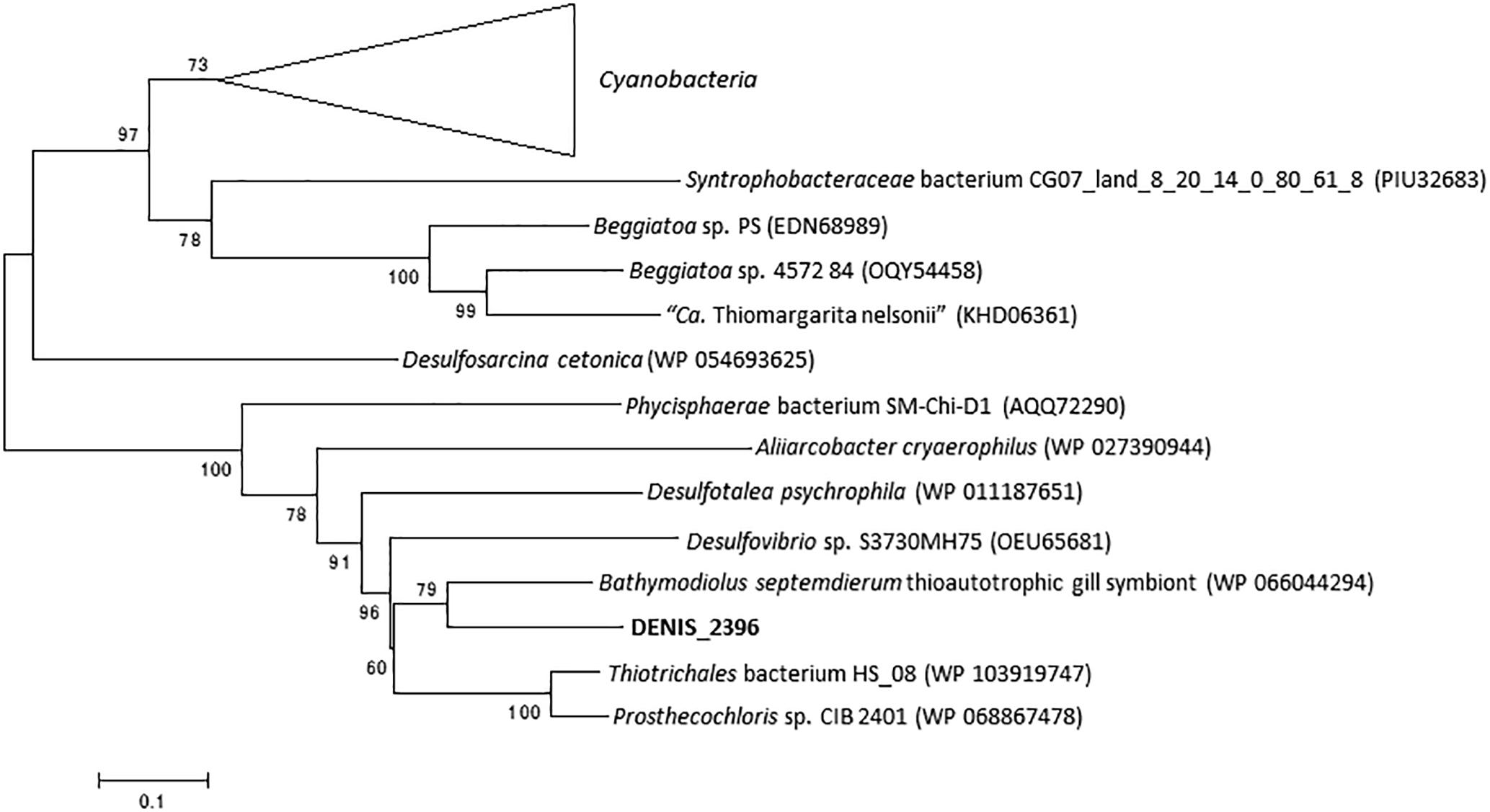
Figure 4. RecQ phylogeny of the strain Tokyo 01T and Desulfosarcina cetonica. The top 55 BLAST hits to DENIS_2396 were used as the dataset to construct the tree. There were 662 positions in the final dataset. This tree was constructed by using maximum-likelihood method with LG + G + I model. Bootstrap values (percentages of 1,000 replications) are shown at nodes.
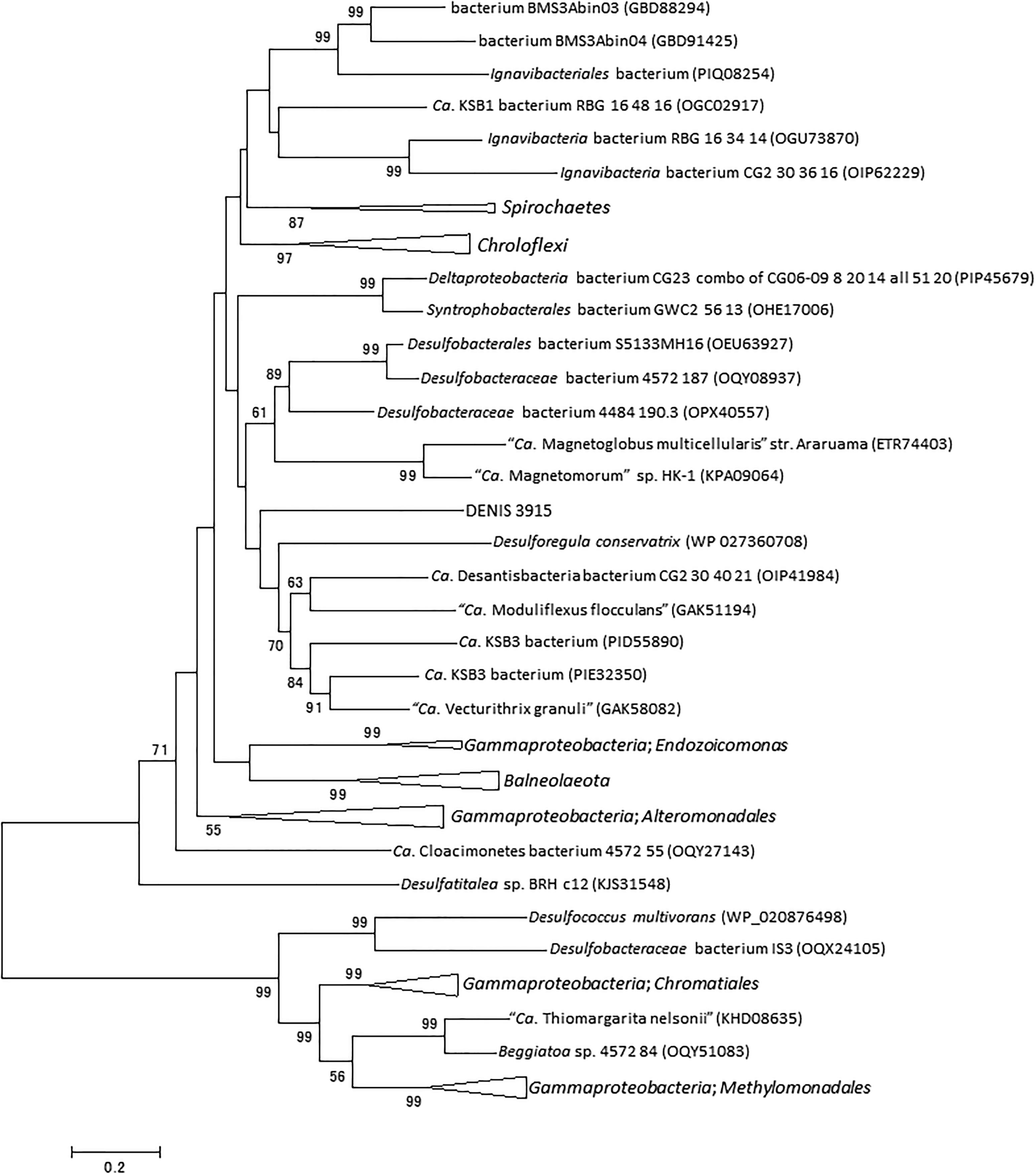
Figure 5. RecQ phylogeny of the strain Tokyo 01T and D. multivorans. The dataset consists the top 50 BLAST hits to DENIS_3915 and 20 hits to the RecQ of D. multivorans. There were 560 positions in the final dataset. This tree was constructed by using maximum-likelihood method with LG + G + I model. Bootstrap values (percentages of 1,000 replications) lower than 50% are shown at nodes.
A search against the REBASE database identified 40 genes for restriction-modification systems in the genome of strain Tokyo 01T (Supplementary Table S6). The proteins encoded by these predicted genes showed the highest sequence similarities to proteins of prokaryotes belonging to diverse taxa, including Gammaproteobacteria (8 out of 40) and Cyanobacteria (5), as major lineages outside of Deltaproteobacteria. Despite their crucial role in defense against foreign DNA, previous studies have demonstrated that the genes for restriction–modification systems have undergone frequent genetic exchange between species (Kita et al., 1999; Kobayashi et al., 1999; Rocha et al., 1999; Nobusato et al., 2000) and quick evolution (Lauster, 1989; Jeltsch and Pingoud, 1996). It was also shown that these genes in “Ca. Maribeggiatoa” are related to a phylogenetically wide range of organisms, suggesting their foreign origins (MacGregor et al., 2013).
Duplications of the genes for DNA repair system possibly improve response to environmental stress (Van Sluys et al., 2002; Martins-Pinheiro et al., 2004). The multiple genes for RecQ helicases and other DNA repair genes in strain Tokyo 01T may function to maintain genomic stability during aggressive horizontal genetic transfer and facilitate response to environmental changes.
Conclusion
In this study, we showed that the genome of strain Tokyo 01T appears to have many protein-coding genes with wide phylogenetic affiliations, implying extensive genetic transfer from diverse organisms. A considerable number of genetic elements were inferred to be transferred from filamentous bacteria, including gammaproteobacterial sulfur oxidizers, cyanobacteria, and organisms of the candidate phylum KSB3. The enhanced DNA repair systems of strain Tokyo 01T might have helped genomic maintenance during horizontal gene transfer. We conclude that the strain Tokyo 01T obtained genetic elements from gliding filamentous bacteria belonging to various lineages. The acquired foreign genes might have facilitated acquisition of new niches (Lawrence, 1999). This study highlights the unexplored aspect of genomic evolutions of filamentous bacteria, which may be intertwined with organisms of different physiological functions and ecological roles.
Author Contributions
MF and HK supervised the study. HK performed the cultivation and genomic DNA preparation for the analysis. KU carried out data analysis to obtain the genome sequence. MW designed the study and carried out bioinformatics analysis based on the obtained genome. MW and HK wrote the manuscript. All authors discussed the data and approved the final manuscript.
Funding
This study was supported by a grant-in-aid for Research Fellow of Japan Society for the Promotion Science to MW and JSPS KAKENHI grant number 22370005 to MF.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Professor Friedrich Widdel for many helpful discussions. Genome analysis was performed on the National Institute of Genetics (NIG) supercomputer at the Research Organization of Information and Systems (ROIS) National Institute of Genetics.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00227/full#supplementary-material
FIGURE S1 | Circular map of the single chromosome of strain Tokyo 01T. From outside to the center; genes on forward strand, genes on reverse strand, G + C content, GC skew. Positive or negative values in G + C content and GC skew were shown as pink or green, respectively.
FIGURE S2 | Neighbor-joining tree of putative GyrB intein of strain Tokyo 01T and other organisms.
FIGURE S3 | Neighbor-joining tree of putative DnaE intein of strain Tokyo 01T and other organisms.
References
Anagnostidis, K., and Komárek, J. (1988). Modern approach to the classification system of cyanophytes. 3-Oscillatoriales. Algol Stud für Hydrobiol. 50-53, 327–472.
Bland, C., Ramsey, T. L., Sabree, F., Lowe, M., Brown, K., Kyrpides, N. C., et al. (2007). CRISPR Recognition Tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 8:209. doi: 10.1186/1471-2105-8-209
Carrasco, C. D., Holliday, S. D., Hansel, A., Lindblad, P., and Golden, J. W. (2005). Heterocyst-specific excision of the Anabaena sp. strain PCC 7120 hupL element requires xisC. J. Bacteriol. 187, 6031–6038. doi: 10.1128/JB.187.17.6031-6038.2005
Caspi, J., Amitai, G., Belenkiy, O., and Pietrokovski, S. (2003). Distribution of split DnaE inteins in cyanobacteria. Mol. Microbiol. 50, 1569–1577. doi: 10.1046/j.1365-2958.2003.03825.x
Chin, C.-S., Peluso, P., Sedlazeck, F. J., Nattestad, M., Concepcion, G. T., Clum, A., et al. (2016). Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054. doi: 10.1038/nmeth.4035
Chu, W. K., and Hickson, I. D. (2009). RecQ helicases: multifunctional genome caretakers. Nat. Rev. Cancer 9, 644–654. doi: 10.1038/nrc2682
Costa, R. M. A., Chiganças, V., da Silva Galhardo, R., Carvalho, H., and Menck, C. F. M. (2003). The eukaryotic nucleotide excision repair pathway. Biochimie 85, 1083–1099. doi: 10.1016/j.biochi.2003.10.017
Denamur, E., Lecointre, G., Darlu, P., Tenaillon, O., Acquaviva, C., Sayada, C., et al. (2000). Evolutionary implications of the frequent horizontal transfer of mismatch repair genes. Cell 103, 711–721. doi: 10.1016/S0092-8674(00)00175-6
Dilling, W., and Cypionka, H. (1990). Aerobic respiration in sulfate-reducing bacteria. FEMS Microbiol. Lett. 71, 123–127. doi: 10.1111/j.1574-6968.1990.tb03809.x
Dombrowski, N., Seitz, K. W., Teske, A. P., and Baker, B. J. (2017). Genomic insights into potential interdependencies in microbial hydrocarbon and nutrient cycling in hydrothermal sediments. Microbiome 5:106. doi: 10.1186/s40168-017-0322-2
Dörries, M., Wöhlbrand, L., Kube, M., Reinhardt, R., and Rabus, R. (2016). Genome and catabolic subproteomes of the marine, nutritionally versatile, sulfate-reducing bacterium Desulfococcus multivorans DSM 2059. BMC Genomics 17:918. doi: 10.1186/s12864-016-3236-7
El-Gebali, S., Mistry, J., Bateman, A., Eddy, S. R., Luciani, A., Potter, S. C., et al. (2019). The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432. doi: 10.1093/nar/gky995
Flood, B. E., Bailey, J. V., and Biddle, J. F. (2014). Horizontal gene transfer and the rock record: comparative genomics of phylogenetically distant bacteria that induce wrinkle structure formation in modern sediments. Geobiology 12, 119–132. doi: 10.1111/gbi.12072
Flood, B. E., Fliss, P., Jones, D. S., Dick, G. J., Jain, S., Kaster, A.-K., et al. (2016). Single-cell (meta-) genomics of a dimorphic Candidatus Thiomargarita nelsonii reveals genomic plasticity. Front. Microbiol. 7:603. doi: 10.3389/fmicb.2016.00603
Frank, J., Lücker, S., Vossen, R. H. A. M., Jetten, M. S. M., Hall, R. J., Op den Camp, H. J. M., et al. (2018). Resolving the complete genome of Kuenenia stuttgartiensis from a membrane bioreactor enrichment using Single-Molecule Real-Time sequencing. Sci. Rep. 8:4580. doi: 10.1038/s41598-018-23053-7
Fukui, M., Teske, A., Aßmus, B., Muyzer, G., and Widdel, F. (1999). Physiology, phylogenetic relationships, and ecology of filamentous sulfate-reducing bacteria (genus Desulfonema). Arch. Microbiol. 172, 193–203. doi: 10.1007/s002030050760
Grogan, D. W. (2000). The question of DNA repair in hyperthermophilic archaea. Trends Microbiol. 8, 180–185. doi: 10.1016/S0966-842X(00)01729-7
Hatchikian, C. E., LeGall, J., and Bell, G. R. (1977). “Significance of superoxide dismutase and catalase activities in the strict anaerobes, sulfate reducing bacteria,” in Superoxide and superoxide dismutase, eds A. M. Michael, J. M. McCord, and I. Fridovich (New York, NY: Academic Press), 159–172.
Henson, B. J., Pennington, L. E., Watson, L. E., and Barnum, S. R. (2008). Excision of the nifD element in the heterocystous cyanobacteria. Arch. Microbiol. 189, 357–366. doi: 10.1007/s00203-007-0326-6
Hewitt, J., and Morris, J. G. (1975). Superoxide dismutase in some obligately anaerobic bacteria. FEBS Lett. 50, 315–318. doi: 10.1016/0014-5793(75)90058-7
Ikuta, T., Takaki, Y., Nagai, Y., Shimamura, S., Tsuda, M., Kawagucci, S., et al. (2016). Heterogeneous composition of key metabolic gene clusters in a vent mussel symbiont population. ISME J. 10, 990–1001. doi: 10.1038/ismej.2015.176
Jeltsch, A., and Pingoud, A. (1996). Horizontal gene transfer contributes to the wide distribution and evolution of type II restriction-modification systems. J. Mol. Evol. 42, 91–96. doi: 10.1007/BF02198833
Kaiser, S., Sauer, F., and Kisker, C. (2017). The structural and functional characterization of human RecQ4 reveals insights into its helicase mechanism. Nat. Commun. 8:15907. doi: 10.1038/ncomms15907
Khodursky, A. B., Peter, B. J., Schmid, M. B., DeRisi, J., Botstein, D., Brown, P. O., et al. (2000). Analysis of topoisomerase function in bacterial replication fork movement: use of DNA microarrays. Proc. Natl. Acad. Sci. 97, 9419–9424. doi: 10.1073/pnas.97.17.9419
Kita, K., Tsuda, J., Kato, T., Okamoto, K., Yanase, H., and Tanaka, M. (1999). Evidence of horizontal transfer of the ecoO109I restriction-modification gene to Escherichia coli chromosomal DNA. J. Bacteriol. 181, 6822–6827.
Kjeldsen, K. U., Joulian, C., and Ingvorsen, K. (2005). Effects of oxygen exposure on respiratory activities of Desulfovibrio desulfuricans strain DvO1 isolated from activated sludge. FEMS Microbiol. Ecol. 53, 275–284. doi: 10.1016/j.femsec.2004.12.010
Kobayashi, I., Nobusato, A., Kobayashi-Takahashi, N., and Uchiyama, I. (1999). Shaping the genome–restriction–modification systems as mobile genetic elements. Curr. Opin. Genet. Dev. 9, 649–656. doi: 10.1016/S0959-437X(99)00026-X
Kojima, H., Ogura, Y., Yamamoto, N., Togashi, T., Mori, H., Watanabe, T., et al. (2015). Ecophysiology of Thioploca ingrica as revealed by the complete genome sequence supplemented with proteomic evidence. ISME J. 9, 1166–1176. doi: 10.1038/ismej.2014.209
Komárek, J., and Anagnostidis, K. (2005). “Cyanoprokaryota. 2. teil. part: oscillatoriales,” in Süsswasserflora von Mitteleuropa, eds B. Büdel, G. Gärtner, L. Krienitz, and M. Schagerl (München: Elsevier GmbH), 1–759.
Kuever, J. (2014). “The family Desulfobacteraceae,” in The Prokaryotes, eds E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin: Springer), 45–73. doi: 10.1007/978-3-642-39044-9-266
Kuever, J., Rainey, F. A., and Widdel, F. (2015). “Desulfonema,” in Bergey’s Manual of Systematic Bacteriology, (Hoboken, NJ: John Wiley & Sons, Inc).
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kusano, K., Sunohara, Y., Takahashi, N., Yoshikura, H., and Kobayashi, I. (1994). DNA double-strand break repair: genetic determinants of flanking crossing-over. Proc. Natl. Acad. Sci. U.S.A. 91, 1173–1177. doi: 10.1073/PNAS.91.3.1173
Lagesen, K., Hallin, P., Rødland, E. A., Stærfeldt, H.-H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Lauster, R. (1989). Evolution of type II DNA methyltransferases: a gene duplication model. J. Mol. Biol. 206, 313–321. doi: 10.1016/0022-2836(89)90481-6
Lawrence, J. G. (1999). Gene transfer, speciation, and the evolution of bacterial genomes. Curr. Opin. Microbiol. 2, 519–523. doi: 10.1016/S1369-5274(99)00010-7
Lemos, R. S., Gomes, C. M., Santana, M., LeGall, J., Xavier, A. V., and Teixeira, M. (2001). The ‘strict’ anaerobe Desulfovibrio gigas contains a membrane-bound oxygen-reducing respiratory chain. FEBS Lett. 496, 40–43. doi: 10.1016/S0014-5793(01)02399-7
Lowe, T. M., Eddy, S. R., and Meyuhas, O. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.0955
MacGregor, B. J., Biddle, J. F., and Teske, A. (2013). Mobile elements in a single-filament orange guaymas basin Beggiatoa (”Candidatus Maribeggiatoa”) sp. draft genome: evidence for genetic exchange with cyanobacteria. Appl. Environ. Microbiol. 79, 3974–3985. doi: 10.1128/AEM.03821-12
Markowitz, V. M., Chen, I. M. A., Chu, K., Szeto, E., Palaniappan, K., Pillay, M., et al. (2013). IMG/M4 version of the integrated metagenome comparative analysis system. Nucleic Acids Res. 42, D568–D573. doi: 10.1093/nar/gkt919
Martins-Pinheiro, M., Galhardo, R. S., Lage, C., Lima-Bessa, K. M., Aires, K. A., and Menck, C. F. (2004). Different patterns of evolution for duplicated DNA repair genes in bacteria of the Xanthomonadales group. BMC Evol. Biol. 4:29. doi: 10.1186/1471-2148-4-29
Minz, D., Fishbain, S., Green, S. J., Muyzer, G., Cohen, Y., Rittmann, B. E., et al. (1999). Unexpected population distribution in a microbial mat community: sulfate-reducing bacteria localized to the highly oxic chemocline in contrast to a eukaryotic preference for anoxia. Appl. Environ. Microbiol. 65, 4659–4665.
Mußmann, M., Hu, F. Z., Richter, M., de Beer, D., Preisler, A., Jørgensen, B. B., et al. (2007). Insights into the genome of large sulfur bacteria revealed by analysis of single filaments. PLoS Biol. 5:e230. doi: 10.1371/journal.pbio.0050230
Nakayama, H., Nakayama, K., Nakayama, R., Irino, N., Nakayama, Y., and Hanawalt, P. C. (1984). Isolation and genetic characterization of a thymineless death-resistant mutant of Escherichia coli K12: identification of a new mutation (recQ1) that blocks the RecF recombination pathway. MGG Mol. Gen. Genet. 195, 474–480. doi: 10.1007/BF00341449
Nobusato, A., Uchiyama, I., and Kobayashi, I. (2000). Diversity of restriction–modification gene homologues in Helicobacter pylori. Gene 259, 89–98. doi: 10.1016/S0378-1119(00)00455-8
Noguchi, H., Taniguchi, T., and Itoh, T. (2008). MetaGeneAnnotator: detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res. 15, 387–396. doi: 10.1093/dnares/dsn027
Orlova, M. V., Tarlachkov, S. V., Kulinchenko, E. I., Dubinina, G. A., Tutukina, M. N., and Grabovich, M. Y. (2018). Genomics and biochemistry of metabolic pathways for the C1 compounds utilization in colorless sulfur bacterium Beggiatoa leptomitoformis D-402. Indian J. Microbiol. 58, 415–422. doi: 10.1007/s12088-018-0737-x
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Parks, D. H., Rinke, C., Chuvochina, M., Chaumeil, P. A., Woodcroft, B. J., Evans, P. N., et al. (2017). Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2, 1533–1542. doi: 10.1038/s41564-017-0012-7
Probst, A. J., Castelle, C. J., Singh, A., Brown, C. T., Anantharaman, K., Sharon, I., et al. (2017). Genomic resolution of a cold subsurface aquifer community provides metabolic insights for novel microbes adapted to high CO2 concentrations. Environ. Microbiol. 19, 459–474. doi: 10.1111/1462-2920.13362
Roberts, R. J., Vincze, T., Posfai, J., and Macelis, D. (2015). REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 43, D298–D299. doi: 10.1093/nar/gku1046
Rocha, E. P., Danchin, A., and Viari, A. (1999). Analysis of long repeats in bacterial genomes reveals alternative evolutionary mechanisms in Bacillus subtilis and other competent prokaryotes. Mol. Biol. Evol. 16, 1219–1230. doi: 10.1093/oxfordjournals.molbev.a026212
Sekiguchi, Y., Ohashi, A., Parks, D. H., Yamauchi, T., Tyson, G. W., and Hugenholtz, P. (2015). First genomic insights into members of a candidate bacterial phylum responsible for wastewater bulking. PeerJ 3:e740. doi: 10.7717/peerj.740
Sugawara, H., Ohyama, A., Mori, H., and Kurokawa, K. (2009). “Microbial Genome Annotation Pipeline (MiGAP) for diverse users. in Software Demonstrations S001-1-2L,” in Proceedings of the 20th International Conference Genome Information (GIW2009) Poster Software Demonstrations, (Tokyo: Yokohama).
Tanner, M. A., Everett, C. L., Coleman, W. J., Yang, M. M., and Youvan, D. C. (2000). Complex microbial communities inhabiting sulfide-rich black mud from marine coastal environments. Biotechnol. Alia 8, 1–16.
Teske, A., Jørgensen, B. B., and Gallardo, V. A. (2009). Filamentous bacteria inhabiting the sheaths of marine Thioploca spp. on the Chilean continental shelf. FEMS Microbiol. Ecol. 68, 164–172. doi: 10.1111/j.1574-6941.2009.00659.x
Teske, A., Ramsing, N. B., Habicht, K., Fukui, M., Küver, J., Jørgensen, B. B., et al. (1998). Sulfate-reducing bacteria and their activities in cyanobacterial mats of Solar Lake (Sinai, Egypt). Appl. Environ. Microbiol. 64, 2943–2951.
Van Sluys, M. A., Monteiro-Vitorello, C. B., Camargo, L. E. A., Menck, C. F. M., da Silva, A. C. R., Ferro, J. A., et al. (2002). Comparative genomic analysis of plant-associated bacteria. Annu. Rev. Phytopathol. 40, 169–189. doi: 10.1146/annurev.phyto.40.030402.090559
Welch, M. M., and McHenry, C. S. (1982). Cloning and identification of the product of the dnaE gene of Escherichia coli. J. Bacteriol. 152, 351–356.
Widdel, F., Kohring, G. W., and Mayer, F. (1983). Studies on dissimilatory sulfate-reducing bacteria that decompose fatty-acids. 3. Characterization of the filamentous gliding Desulfonema limicola gen. nov. sp. nov., and Desulfonema magnum sp. nov. Arch. Microbiol. 134, 286–294. doi: 10.1007/BF00407804
Winkel, M., Salman-Carvalho, V., Woyke, T., Richter, M., Schulz-Vogt, H. N., Flood, B. E., et al. (2016). Single-cell sequencing of Thiomargarita reveals genomic flexibility for adaptation to dynamic redox conditions. Front. Microbiol. 7:964. doi: 10.3389/fmicb.2016.00964
Wong, H. L., Smith, D.-L., Visscher, P. T., and Burns, B. P. (2015). Niche differentiation of bacterial communities at a millimeter scale in Shark Bay microbial mats. Sci. Rep. 5:15607. doi: 10.1038/srep15607
Wu, H., Hu, Z., and Liu, X. Q. (1998). Protein trans-splicing by a split intein encoded in a split DnaE gene of Synechocystis sp. PCC6803. Proc. Natl. Acad. Sci. U.S.A. 95, 9226–9231. doi: 10.1073/PNAS.95.16.9226
Wu, S., Zhu, Z., Fu, L., Niu, B., and Li, W. (2011). WebMGA: a customizable web server for fast metagenomic sequence analysis. BMC Genomics 12:444. doi: 10.1186/1471-2164-12-444
Yamada, T., Yamauchi, T., Shiraishi, K., Hugenholtz, P., Ohashi, A., Harada, H., et al. (2007). Characterization of filamentous bacteria, belonging to candidate phylum KSB3, that are associated with bulking in methanogenic granular sludges. ISME J. 1, 246–255. doi: 10.1038/ismej.2007.28
Keywords: complete genome sequence, genetic transfer, filamentous sulfate-reducing bacterium, Desulfonema, genome analysis
Citation: Watanabe M, Kojima H, Umezawa K and Fukui M (2019) Genomic Characteristics of Desulfonema ishimotonii Tokyo 01T Implying Horizontal Gene Transfer Among Phylogenetically Dispersed Filamentous Gliding Bacteria. Front. Microbiol. 10:227. doi: 10.3389/fmicb.2019.00227
Received: 28 November 2018; Accepted: 28 January 2019;
Published: 19 February 2019.
Edited by:
Marc Mussmann, University of Vienna, AustriaReviewed by:
Jan Kuever, Bremen Institute for Materials Testing, GermanyBarbara J. MacGregor, University of Minnesota, United States
Copyright © 2019 Watanabe, Kojima, Umezawa and Fukui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miho Watanabe, bS53YXRhbmFiZUBwb3AubG93dGVtLmhva3VkYWkuYWMuanA=