 Xiaoli Zhang
Xiaoli Zhang Beibei Sun
Beibei Sun Qiaoran Tang
Qiaoran Tang Rui Chen
Rui Chen Su Han
Su Han- 1Department of Parasitology, Harbin Medical University, Harbin, China
- 2Department of Orthopaedics, The Fourth Affiliated Hospital of Harbin Medical University, Harbin, China
Studying the genetic diversity of parasite is important for understanding their biogeography and molecular epidemiology, as well as for establishing disease prevention and control strategies. Clonorchis sinensis is an important foodborne parasite worldwide. However, despite its epidemiological significance, the genetic diversity of C. sinensis has not been well studied from human in northeastern China. In this study, a total of 342 fecal specimens were collected from residents living in five villages in Heilongjiang Province and analyzed for the presence of C. sinensis by PCR amplification and sequencing of the internal transcribed spacer 1 (ITS1) and ITS2 regions of nuclear ribosomal DNA. 21.64% (74/342) of fecal samples were found to be positive for C. sinensis by PCR. The sequences of the ITS1 region in 34 of the 74 samples (45.95%) matched that of MK179278, Genetic polymorphisms were observed at six nucleotide sites. The ITS2 gene sequence of 37 of the 74 samples (50%) matched that of MK179281. In conclusion, a low degree of genetic diversity between C. sinensis isolates from China and different geographical regions was found at ITS loci. Despite this conservation, sequencing of the rDNA region has provided important data that will be useful for future studies addressing the molecular evolution, biology, medical implications and ecology of C. sinensis.
Introduction
Clonorchis sinensis (C. sinensis) is an important foodborne zoonotic pathogen. Hosts become infected with C. sinensis by ingesting raw or undercooked freshwater fish containing metacercariae. Adults then parasitize the peripheral intrahepatic bile ducts. Typically, C. sinensis infections cause no obvious clinical symptoms or only mild symptoms (Zhang et al., 2008). However, high intensity or long-term C. sinensis infections can potentially lead to liver damage such as cholelithiases, cholecystitis and hepatic fibrosis (Choi et al., 2004). Furthermore, C. sinensis is considered a group I carcinogen-metazoan parasite that can potentially induce cholangiocarcinoma (Bouvard et al., 2009).
Clonorchis sinensis is endemic in Asia, particularly in China, Japan, Korea and Vietnam (Tang et al., 2016). In China, approximately 15 million people are estimated to be infected, mainly in southeast and northeast areas such as Guangdong, Guangxi and Heilongjiang Provinces (Lun et al., 2005). National sampling surveys showed that the prevalence of clonorchiasis in China increased by 75% from 1990 to 2003 and that Heilongjiang Province was an endemic focus (Rim, 2005; Tang et al., 2016). A study conducted in Heilongjiang Province between 2009 and 2012 found that the mean prevalence of C. sinensis was 25.93% (Han et al., 2013). However, despite its epidemiological significance, the genetic diversity of C. sinensis has not been sufficiently studied. Knowledge of genetic variation of C. sinensis is important for understanding its epidemiology and for disease control.
Molecular biology methods not only contribute to the understanding of parasite epidemiology but also allow for exploration of parasite characteristics, including host specificity, transmission patterns and genetic diversity (Choi et al., 2010; Huang et al., 2012). Researchers have investigated the genetic variation in C. sinensis isolates from Russia, Vietnam, China and Korea using molecular biology methods (Chelomina et al., 2014). Although C. sinensis is represented by a single species with a low divergence, karyotypic variation between C. sinensis isolates from China, Korea and eastern Russia suggests that this taxon might contain sibling species (Wang et al., 2017). To date, there have been no comprehensive studies exploring population genetic variation among large numbers of C. sinensis isolates from disparate geographic locations using complete or partial mitochondrial and/or nuclear genomic data sets.
Nuclear ribosomal DNA (rDNA) is widely used for molecular investigations and genetic analyses (Park, 2007). Ribosomal genes, including 18S, 5.8S and 28S rRNA, are typically organized into tandem repeats separated by two internal transcribed spacers (ITS1 and ITS2) (Prasad et al., 2008; Tatonova et al., 2017). Due to their high interspecific and low intraspecific variability, the ITS1 and ITS2 genes have been used extensively for ecological genetic studies and phylogenetic and evolutionary analyses at various taxonomic levels for different organisms (Dai et al., 2012; Buathong et al., 2017). Several studies have also been conducted on the genetic variability of trematode species such as Schistosoma japonicum, Fasciola hepatica, and Opisthorchis felineus (Katokhin et al., 2008; Zhao et al., 2010; Dar et al., 2012). However, limited information is available on the genetic diversity and molecular epidemiological surveys based on the ITS gene of C. sinensis from human in Heilongjiang Province.
In the present study, C. sinensis isolates from human feces in Heilongjiang Province were identified and genotyped by PCR amplification of ITS genes. The ITS1 and ITS2 gene sequences obtained here were compared with sequences previously published in GenBank. The sequence analysis were assessed by comparing the ITS1 and ITS2 gene sequences obtained here with those previously published in GenBank. These genetic data will be crucial for understanding the prevalence and genetic structure of C. sinensis and for developing disease treatment and control strategies.
Materials and Methods
Ethics Statement
This research study was approved by the Medical Ethics Review Committee of Harbin Medical University. The objectives, procedures and potential risks were explained to all participants. Written informed consent was obtained directly from all adult participants. If the participants were children, written informed consent were obtained from the next of kin, carers, or guardians on the behalf of the minors/children participants.
Specimen Collection
Between June 2014 and March 2015, a total of 342 fecal samples were collected from residents living five villages along Songhua river in Heilongjiang Province. Information about five villages is given in Figure 1. Approximately, 15–20 g of fecal specimens were randomly collected from each of participants. All fecal specimens were transported to the laboratory in a cooler with ice packs within 24 h and stored at 4°C until they were extracted.
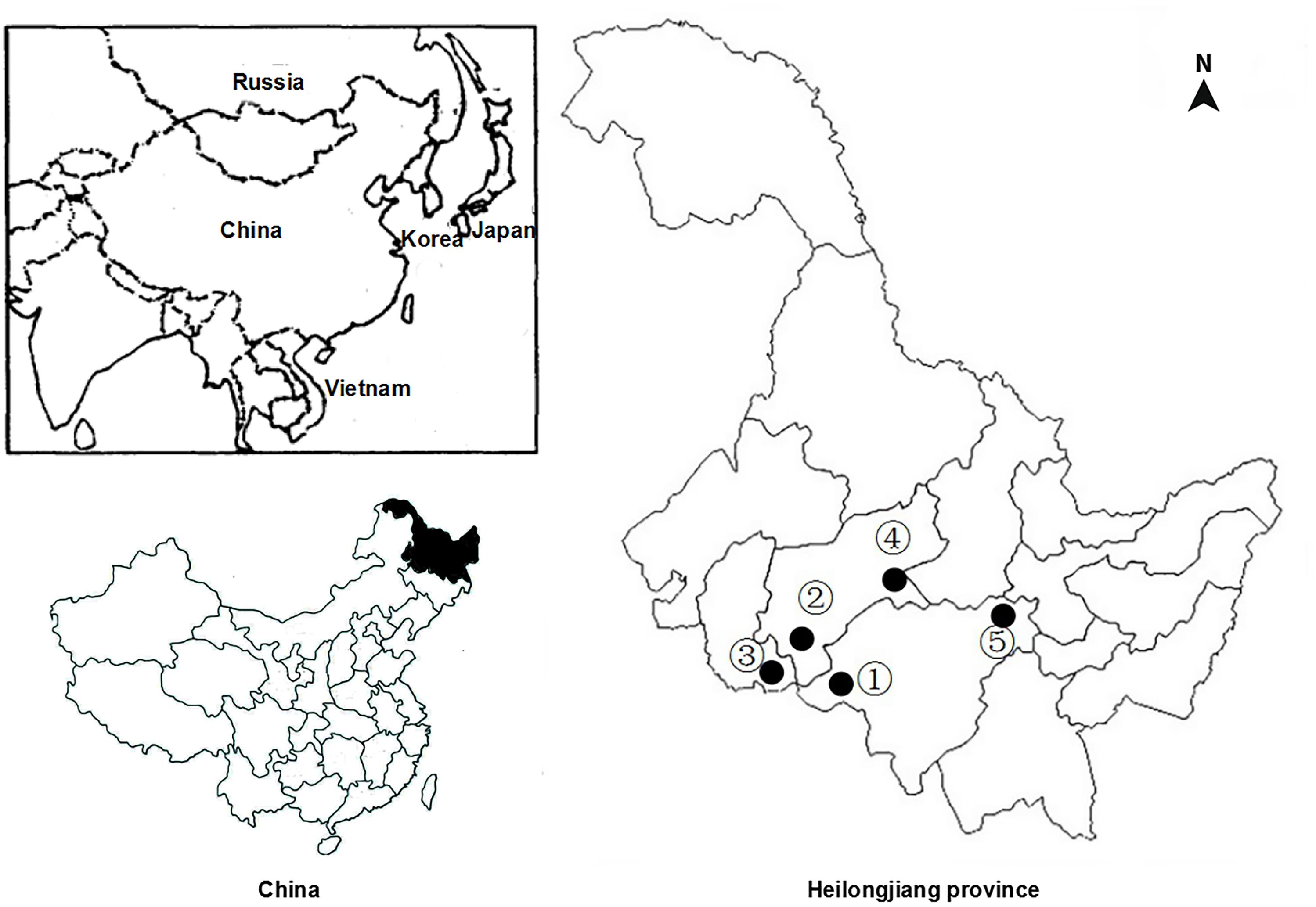
Figure 1. Sampling locations for Clonorchis sinensis in heilongjiang province, China. Numbers correspond with those in Table 1.
DNA Extraction
The fecal specimens were sieved and centrifuged at 1500 g for 10 minutes at room temperature, and then washed with distilled water for three times. Genomic DNA was extracted from 200 mg fecal specimen by a QIAamp DNA Stool Mini Kit (QIAgen, Hilden, Germany). The procedures and utilized reagents were utilized according to the manufacturer’s protocol. The eluted DNA was finally stored at -20°C in freezers until PCR analysis.
PCR Amplification of the ITS1 and ITS2 Genes
Each of the DNA specimens was detected for the presence of C. sinensis by amplifying 572 bp of the ITS1 gene and 249 bp of the ITS2 gene, respectively. The primes for ITS1 are CsITS1F (5′-CGATTCTAGTTCCGTCATCT-3′) and CsITS1R (5′-CCGCTCAGAGTACTCAT-3′) (Liu et al., 2007). The primers of the ITS2 gene are CsITS2F (5′-TATAAACTATCACGACGCCC-3′) and CsITS2R (5′-TACTGAAGCCTCAACCAAAG-3′) (Yang et al., 2014).
Amplification of the ITS1 gene was used the following cycling conditions: a 4 min initial denaturation step at 95°C; 35 cycles of 30 s at 94°C, 1 min at 62°C, 1 min at 72°C; and a 10 min extension at 72°C. The cycling parameters of amplification of the ITS2 gene were as follows: a 3 min initial denaturation step at 95°C; 35 cycles of 30 s at 95°C, 30 s at 55°C, 60 s at 72°C; and a 3 min extension at 72°C. Negative and positive controls with both primers were included, respectively. TaKaRa Taq DNA polymerase (TaKaRa Bio Inc., Tokyo, Japan) was used for all the PCR amplifications. Then the PCR products were separated in 1.5% agarose gel electrophoresis and visualized under UV light after staining with ethidium bromide.
Nucleotide Sequencing
All the PCR products of expected size were directly sequenced on an ABI PRISM 3730 XL DNA Analyzer by Sinogeno-max Biotechnology Co., Ltd. (Beijing, China), using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, United States).
DNA Sequence Analysis
The positive results in electrophoresis were selected for sequencing. All the nucleotide sequences were aligned with each other. According to the identity percentage and query coverage parameter, the reference sequences downloaded from GenBank database using the Basic Local Alignment Search Tool (BLAST)1 and Clustal X 1.832 to determine isolates of C. sinensis.
Nucleotide Sequence Accession Numbers
If the isolates obtained in this study with the cutoff values of above 95% in sequence similarity were identical to those published in GenBank, they were identified to be known isolates and given the first published name. If not, they were considered to be novel isolates. Representative nucleotide sequences obtained in the present study were deposited in the GenBank database under accession numbers MK179278 to MK179280 (ITS1), MK179281 to MK179283 (ITS2).
Date Analysis
To better present the diversity of all the isolates of C. sinensis obtained in this study and to assess the genetic relationship of the novel ones here to the known ones, intraspecific phylogenies were reconstructed with the neighbor-joining (NJ) and maximum likelihood (ML) methods in MEGA7.0 program. NJ and ML trees were constructed using 1000 and 100 bootstrap replicates, respectively. In addition, Bayesian analysis was also used and proceed as follow: the posterior probabilities are determined by Markov chain Monte Carlo sampling (MCMC) in MrBayes v3.2 (Ronquist et al., 2012) based on the models from MrModeltest (Posada and Crandall, 1998; Posada, 2008). There were 275000 generation for ITS1 and 170000 generation for ITS2 run in MrBayes program. An average standard deviation of <0.01 for split frequencies is used to suggest a convergence between parallel runs. Twenty-five percent of the total trees are discarded as burn-in. Additional, the network was also generated with SplitsTree (SplitsTree 4.0 program) using the Neighbor Network method to display the samples (Morrison, 2005; Huson and Bryant, 2006).
The ITS1 sequences of Metorchis bilis (KY356536), M.xanthosomus (KY356540), M.orientalis (KX832894), M.orientalis (KX857496), Opisthorchis felineus (EU038139), O. felineus (KR995729), and O. felineus (KT020830) were used as the outgroup. The ITS2 sequences of M.bilis (KT740982), M.orientalis (KX857496), M.xanthosomus (KT740977), O. noverca (KJ767634), O. lobatus (HQ328545) and O. parageminus (KX258657) were chosen as the outgroup to root the trees. The appropriate nucleotide substitution model for each gene is determined using MrModeltest MrModeltest v2.3 (Nylander, 2008). The Kimura-2-parameter “K2” model is the best model for both ITS1 and ITS2 sequence data.
SPSS (version 10.0 software for windows; Chicago, IL, United States) was used for analyzing the date. The chi-square test was used to evaluate the assessment between qualitative variable to check for statistical differences. P < 0.05 was regarded as statistically significant.
Results
Prevalence of C. sinensis
In the present study, a total of 342 human fecal specimens were examined for C. sinensis by PCR amplification of the ITS1 and ITS2 genes. C. sinensis was found in all five villages examined, with infection rates ranging from 12.31 to 33.33% (Table 1 and Figure 1). Specimens that were found to be positive for C. sinensis by PCR were confirmed by sequencing, and an overall infection rate of 21.64% was found (74/342).
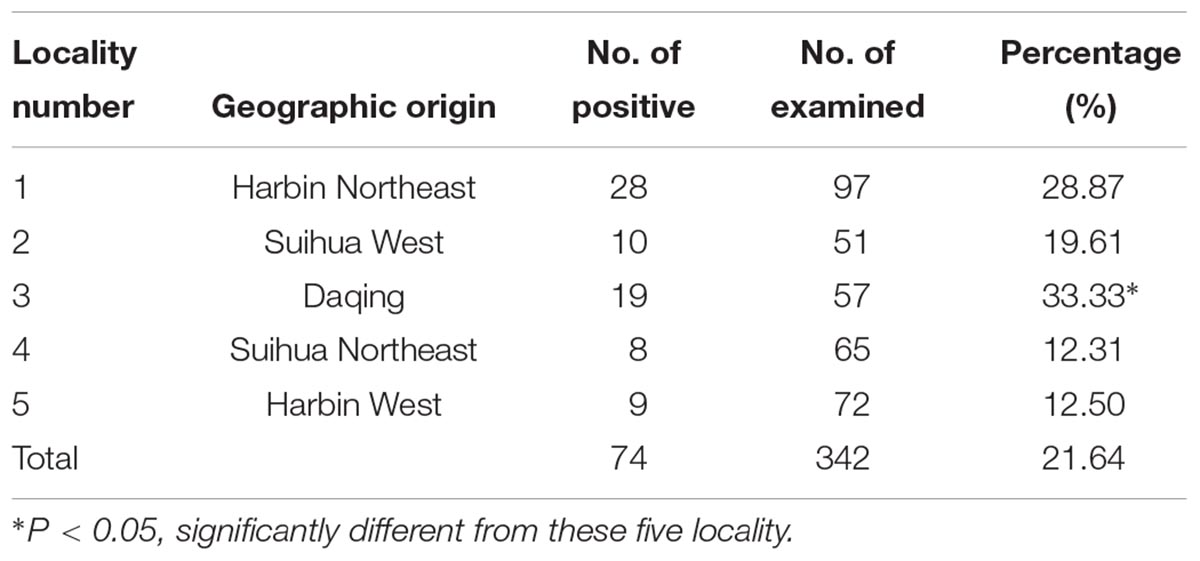
Table 1. Regional distribution of C. sinensis infection in five localities from Heilongjiang Province based on ITS gene.
Genetic Characterization of the C. sinensis ITS1 and ITS2 Gene
The ITS1 gene sequence of 34 of the 74 samples (45.95%) matched that of MK179278, followed by MK179280 (25.68%, 19/74), MK179279 (24.32%, 18/74), and MF319641, HQ874526, JQ048598 (one each, 1.35%, 1/74) (Table 2). The genetic polymorphisms of one to two base variations were observed at six nucleotide sites (Table 3).
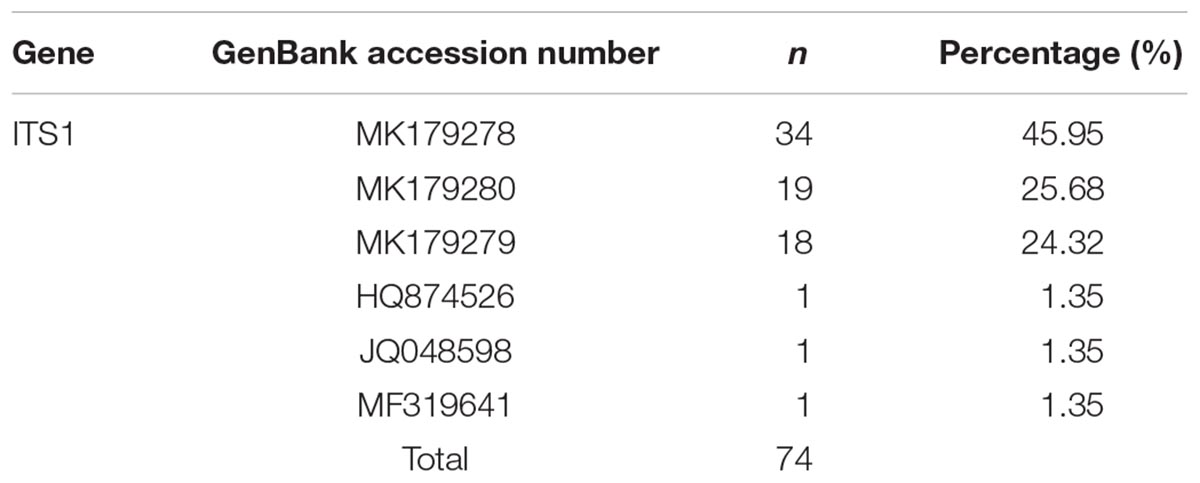
Table 2. ITS1 sequences of C. sinensis from Heilongjiang Province in this study.
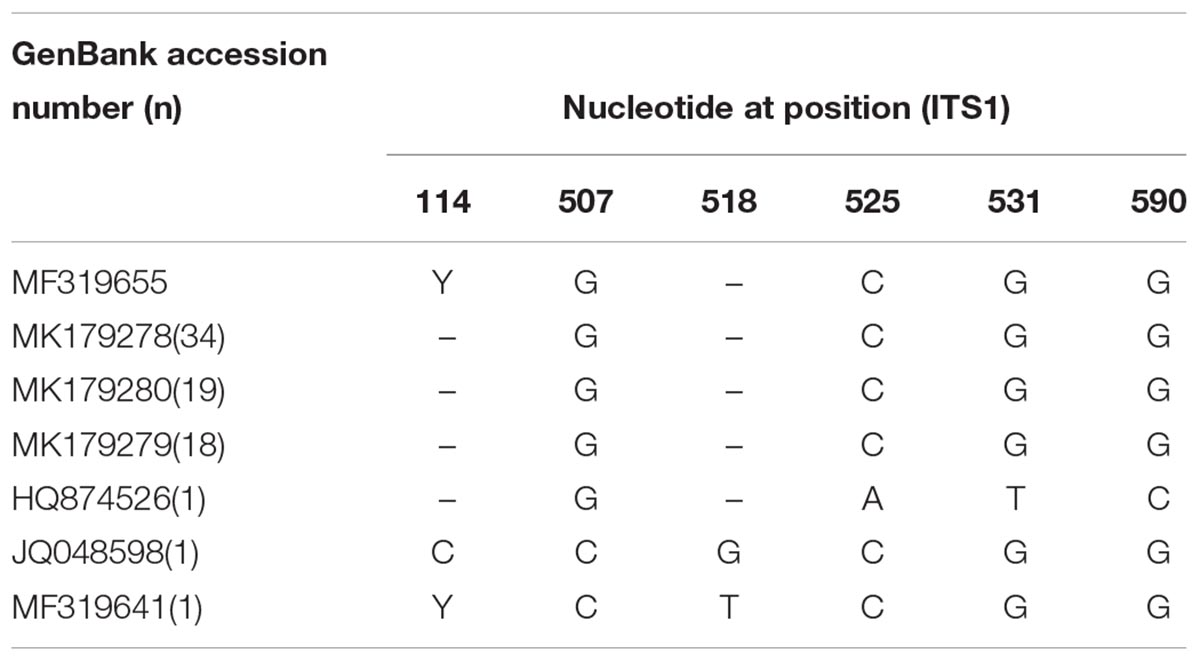
Table 3. Nucleotide variation at six polymorphic sites in ITS1 gene region of C. sinensis isolates from human in this study.
The ITS2 gene sequence of 37 of the 74 samples (50%) matched that of MK179281, followed by MK179283 (25.68%, 19/74) and MK179282 (24.32%, 18/74), respectively. There were no genetic polymorphisms observed.
Phylogenetic Relationships of C. sinensis
The phylogenetic analysis was carried out based on the ITS gene sequences of the isolates of C. sinensis and some isolates published previously, inferred by neighbor-joining (NJ), maximum likelihood (ML) and Bayesian (Bayes) analyses, with M. bilis, M. xanthosomus, M. orientalis, O. lobatus, O. felineus, O. noverca, and O. parageminus as outgroup.
The topologies of NJ, ML and Bayesian (Figures 2, 3) trees were very similar and only small differences in bootstrap support values were obtained among those. These trees demonstrated unresolved topology with low bootstrap support for most branches. None of those corresponded to the geographical localities based on ITS1 gene; nevertheless, only a few clusters were statistically supported according to the geographical distance based on ITS2 gene. Additional, the Neighbor Network was also used to display the samples (Supplementary Figures S1, S2). Instead of conventional single phylogenetic tree, Splits Tree makes a phylogenetic network with reticulations. It also showed that C. sinensis isolates clustered together with the exclusion of other liver fluke representatives.

Figure 2. Phylogenetic relationships between the C. sinensis isolates based on the partial ITS1 rDNA sequences. Nodal support of >50% is shown for maximum likelihood/neighbor-joining/Bayesian Inference analyses (as indicated). Opisthorchis felineus and Metorchis bilis et al. were used for comparison. Each sequence is identified by its accession number and host origin. Genotypes with black triangle are genotypes identified in this study, respectively.

Figure 3. Phylogenetic relationships between the C. sinensis isolates based on the partial ITS2 rDNA sequences. Nodal support of >50% is shown for maximum likelihood/neighbor-joining/Bayesian Inference analyses (as indicated). Opisthorchis noverca and Opisthorchis lobatus et al. were used for comparison. Each sequence is identified by its accession number and host origin. Genotypes with black triangle are genotypes identified in this study, respectively.
Discussion
Genetic data are crucial for understanding the biological history of parasitic diseases and developing treatment and control strategies (Gasser and Newton, 2000). In addition, genetic variation is common in parasite populations and is a valuable resource for studying the population biology, epidemiology, and genetic structure of parasites (Liu et al., 2012). Although intraspecific and interspecific variations between C. sinensis from different geographic regions have recently been studied using nuclear rDNA and mitochondrial DNA sequences (Park, 2007; Sun et al., 2013), there is a paucity of information on the genetic variation among C. sinensis populations from disparate geographic locations, where C. sinensis infection remains a significant health problem (Han et al., 2016).
In the present study, ITS genes were targeted to detect genetic variations in C. sinensis isolates from human fecal samples collected in five villages in Heilongjiang Province, China. We found that the C. sinensis infection rate was 21.64%. In contrast, the prevalence of C. sinensis infection in Korea was from 1.6 to 9.0% during 1993 – 2006 (Kim et al., 2010). Infection rates are related to many factors, including the sensitivity and specificity of the detection method, infection intensity, overall sample size, climate and experimental design (Tang et al., 2016). The high infection rate in the present study may have been due to the age group, sex, or other demographic factors of the individuals assessed (Han et al., 2013; Lai et al., 2017). For example, males prefer to eat more fish and have more frequent social activities and eating opportunities at restaurants compared with females (Lun et al., 2005). This results in a higher infection rate among males than females. Prevalence rates also increased with age, reaching a plateau among adolescents and young adults and decreasing in elderly people (Han et al., 2013). As an C.sinensis endemic area, Heilongjiang Province has numerous rivers, including the Songhua River, which is an important site for carp, grass carp, and catfish and for the freshwater fish industry (Han et al., 2013). The consumption of raw or undercooked freshwater fish and/or shrimp has been prevalent in this area for a long time, and it is difficult to change eating habits in the short time (Qian et al., 2016). Thus, further prevention and control strategies should be strengthened in these villages, especially the promotion of public health among local residents (Guoqing et al., 2001).
In the present study, 74 C. sinensis samples were distributed among six genotypes based on their ribosomal ITS1 region sequences. Additionally, six variations in the ITS1 gene region were observed in this study. Nucleotide diversity within ITS1 region was unequally distributed and mainly located at the ends of the sequence. Similarly, another study of genetic variation within C. sinensis in Korea and China also demonstrated a high degree of similarity in the ITS rDNA sequences (Lee and Huh, 2004). In trematodes, including C. sinensis, intraspecific variation in the ITS sequences is minimal (Liu et al., 2007; Xiao et al., 2013). This low genetic diversity may be due to high levels of gene flow between parasite populations, which counteracts local adaptation (Sun et al., 2013). However, the small number of polymorphisms in the ITS sequence (one locus) does not necessarily mean that there was no genetic differentiation. Analysis of the rDNA region has provided new data that will be useful for future studies addressing the molecular evolution, biology, and ecology of C. sinensis (Tatonova et al., 2017). Indeed, due to selective pressures from the human host, many parasites, such as Giardia species, have low levels of genetic variation. The features of the ITS region might reflect an adaptation strategy of C. sinensis to environmental conditions and different hosts. No variations in length or nucleotide composition were detected in the ITS2 region of three genotypes in this study. The lack of intraspecific variability in the ITS2 region was also confirmed by a genetic diversity study conducted in eastern Russian (Tatonova et al., 2012). Therefore, ITS2 might be a suitable and sensitive marker for species-level analysis (Tatonova et al., 2017).
Phylogenetic analysis was used to assess the genetic relationships of C. sinensis. The phylogenetic tree of intraspecific and interspecific relationships based on the ITS1 and ITS2 sequences showed low divergence among all tested isolates. Phylogenetic trees constructed using the NJ, ML and Bayesian analyze and Neighbor Network showed that all C. sinensis isolates clustered together with the exclusion of other liver fluke representatives. Nonetheless, the clustering within C. sinensis was not well supported in the analyses. Such information can be useful to improve our understanding of the molecular mechanisms of species adaptation and evolution and parasite infection strategies (Chelomina et al., 2014). ITS rDNA spacers usually diverge among species but are homogeneous within species due to concerted evolution (Bower et al., 2008). Nevertheless, we believe that these genetic data could have important epidemiological, evolutionary and medical implications.
Based on the analysis of ITS region form GenBank sequences, this was the first genetic analysis based on ITS1 and ITS2 sequences of C. sinensis from human in different regions of Heilongjiang Province (Supporting Information Supplementary Tables S1–S4). The results could provide an important reference for future studies on C. sinensis, including species identification studies and assessments of molecular variations between disparate geographical locations. Most importantly, our research will be valuable for the further classification and identification of C. sinensis for the purposes of developing suitable disease prevention and control strategies.
There were some limitations to this study. Firstly, the number of fecal samples collected for this study was relatively small, and our findings were potentially related to the limited number of specimens. Although collection of fecal samples from humans can be difficult, more extensive investigations with a larger number of animal and human specimens are required in the future. Secondly, future studies analyzing other molecular markers are necessary to provide additional information on the population genetic structure of this parasite.
Conclusion
In conclusion, this study provided molecular evidence of C. sinensis in northeast China. A low degree of genetic diversity between C. sinensis isolates from China and different geographical regions was found at ITS loci. The rDNA region data has provided important information that will be useful for future studies addressing the molecular evolution, biology, medical implications, and ecology of C. sinensis. In the future, analysis of additional samples and molecular markers is necessary to provide further information on the population genetic structure of C. sinensis, enhancing our ability to prevention and control strategies of C. sinensis.
Author Contributions
SH and XZ conceived and designed the experiments. SH, BS, QT, and XZ performed the experiments. SH and XZ analyzed the data. SH, BS, QT, and RC contributed to reagents, materials, and analysis tools. SH, BS, and XZ wrote the paper. All authors edited the manuscript, read, and approved the final version of the manuscript.
Funding
This work was supported by National Natural Science Foundation of China (81601785), Heilongjiang province Post-doctoral Science Foundation (LBH-Q17114), Heilongjiang province College students innovative entrepreneurial training program (201710226044, 201810226003, and 201810226027). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00026/full#supplementary-material
FIGURE S1 | The phylogenetic split network of the Clonorchis sinensis isolates based on the partial ITS1 rDNA sequences. Genotypes with black triangle are genotypes identified in this study, respectively.
FIGURE S2 | The phylogenetic split network of the Clonorchis sinensis isolates based on the partial ITS2 rDNA sequences. Genotypes with black triangle are genotypes identified in this study, respectively.
TABLE S1 | ITS1 sequences of C. sinensis worldwide.
TABLE S2 | ITS1 sequences of C. sinensis by geographical location in China.
TABLE S3 | ITS2 sequences of C. sinensis worldwide.
TABLE S4 | ITS2 sequences of C. sinensis by geographical location in China.
Footnotes
References
Bouvard, V., Baan, R., Straif, K., Grosse, Y., Secretan, B., Ei Ghissassi, F., et al. (2009). A review of human carcinogens–Part B: biological agents. Lancet Oncol. 10, 321–322. doi: 10.1016/S1470-2045(09)70096-8
Bower, J. E., Dowton, M., Cooper, R. D., and Beebe, N. W. (2008). Intraspecific concerted evolution of the rDNA ITS1 in Anopheles farauti sensu stricto (Diptera: Culicidae) reveals recent patterns of population structure. J. Mol. Evol. 67, 397–411. doi: 10.1007/s00239-008-9161-x
Buathong, S., Leelayoova, S., Mungthin, M., Ruang-Areerate, T., Naaglor, T., Suwannahitatorn, P., et al. (2017). Molecular discrimination of Opisthorchis-like eggs from residents in a rural community of central Thailand. PLoS Negl. Trop. Dis. 11:e0006030. doi: 10.1371/journal.pntd.0006030
Chelomina, G. N., Tatonova, Y. V., Hung, N. M., and Ngo, H. D. (2014). Genetic diversity of the Chinese liver fluke Clonorchis sinensis from Russia and Vietnam. Int. J. Parasitol. 44, 795–810. doi: 10.1016/j.ijpara.2014.06.009
Choi, B. I., Han, J. K., Hong, S. T., and Lee, K. H. (2004). Clonorchiasis and cholangiocarcinoma: etiologic relationship and imaging diagnosis. Clin. Microbiol. Rev. 17, 540–552. doi: 10.1128/CMR.17.3.540-552.2004
Choi, M. H., Park, S. K., Li, Z., Ji, Z., Yu, G., Feng, Z., et al. (2010). Effect of control strategies on prevalence, incidence and re-infection of clonorchiasis in endemic areas of China. PLoS Negl. Trop. Dis. 4:e601. doi: 10.1371/journal.pntd.0000601
Dai, R. S., Liu, G. H., Song, H. Q., Lin, R. Q., Yuan, Z. G., Li, M. W., et al. (2012). Sequence variability in two mitochondrial DNA regions and internal transcribed spacer among three cestodes infecting animals and humans from China. J. Helminthol. 86, 245–251. doi: 10.1017/S0022149X11000319
Dar, Y., Amer, S., Mercier, A., Courtioux, B., and Dreyfuss, G. (2012). Molecular identification of Fasciola spp. (Digenea: Fasciolidae) in Egypt. Parasite 19, 177–182. doi: 10.1051/parasite/2012192177
Gasser, R. B., and Newton, S. E. (2000). Genomic and genetic research on bursate nematodes: significance, implications and prospects. Int. J. Parasitol. 30, 509–534. doi: 10.1016/S0020-7519(00)00021-7
Guoqing, L., Xiaozhu, H., and Kanu, S. (2001). Epidemiology and control of Clonorchiasis sinensis in China. Southeast Asian J. Trop. Med. Public Health 2, 8–11.
Han, S., Tang, Q., Lu, X., Chen, R., Li, Y., Shu, J., et al. (2016). Dysregulation of hepatic microRNA expression profiles with Clonorchis sinensis infection. BMC Infect. Dis. 16:724. doi: 10.1186/s12879-016-2058-1
Han, S., Zhang, X., Chen, R., Wen, J., Li, Y., Shu, J., et al. (2013). Trends in prevalence of clonorchiasis among patients in Heilongjiang province, Northeast China (2009-2012): implications for monitoring and control. PLoS One 8:e80173. doi: 10.1371/journal.pone.0080173
Huang, S. Y., Zhao, G. H., Fu, B. Q., Xu, M. J., Wang, C. R., Wu, S. M., et al. (2012). Genomics and molecular genetics of Clonorchis sinensis: current status and perspectives. Parasitol. Int. 61, 71–76. doi: 10.1016/j.parint.2011.06.008
Huson, D. H., and Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
Kang, S., Sultana, T., Loktev, V. B., Wongratanacheewin, S., Sohn, W. M., Eom, K. S., et al. (2008). Molecular identification and phylogenetic analysis of nuclear rDNA sequences among three opisthorchid liver fluke species (Opisthorchiidae: Trematoda). Parasitol. Int. 57, 191–197. doi: 10.1016/j.parint.2007.12.007
Katokhin, A. V., Shekhovtsov, S. V., Konkow, S., Yurlova, N. I., Serbina, E. A., Vodianitskai, S. N., et al. (2008). Assessment of the genetic distinctions of Opisthorchis felineus from O. viverrini and Clonorchis sinensis by ITS2 and CO1 sequences. Dokl. Biochem. Biophys. 421, 214–217. doi: 10.1134/S1607672908040133
Kim, Y. J., Lee, S. M., Choi, G. E., Hwang, S. H., Kim, H. H., Lee, E. Y., et al. (2010). Performance of an enzyme-linked immunosorbent assay for detection of Clonorchis sinensis infestation in high- and low-risk groups. J. Clin. Microbiol. 48, 2365–2367. doi: 10.1128/JCM.02506-09
Lai, Y. S., Zhou, X. N., Pan, Z. H., Utzinger, J., and Vounatsou, P. (2017). Risk mapping of clonorchiasis in the People’s Republic of China: a systematic review and Bayesian geostatistical analysis. PLoS Negl. Trop. Dis. 11:e0005239. doi: 10.1371/journal.pntd.0005239
Lee, S. U., and Huh, S. (2004). Variation of nuclear and mitochondrial DNAs in Korean and Chinese isolates of Clonorchis sinensis. Korean J. Parasitol. 42, 145–148. doi: 10.3347/kjp.2004.42.3.145
Liu, G. H., Li, B., Li, J. Y., Song, H. Q., Lin, R. Q., Cai, X. Q., et al. (2012). Genetic variation among Clonorchis sinensis isolates from different geographic regions in China revealed by sequence analyses of four mitochondrial genes. J. Helminthol. 86, 479–484. doi: 10.1017/S0022149X11000757
Liu, W. Q., Liu, J., Zhang, J. H., Long, X. C., Lei, J. H., and Li, Y. L. (2007). Comparison of ancient and modern Clonorchis sinensis based on ITS1 and ITS2 sequences. Acta Trop. 101, 91–94. doi: 10.1016/j.actatropica.2006.08.010
Lun, Z. R., Gasser, R. B., Lai, D. H., Li, A. X., Zhu, X. Q., Yu, X. B., et al. (2005). Clonorchiasis: a key foodborne zoonosis in China. Lancet Infect. Dis. 5, 31–41. doi: 10.1016/S1473-3099(04)01252-6
Morrison, D. A. (2005). Networks in phylogenetic analysis: new tools for population biology. Int. J. Parasitol. 35, 567–582. doi: 10.1016/j.ijpara.2005.02.007
Nylander, J. A. A. (2008). MrModeltest v2. Program Distributed by the Author. Uppsala: Evolutionary Biology Centre, Uppsala University.
Park, G. M. (2007). Genetic comparison of liver flukes, Clonorchis sinensis and Opisthorchis viverrini, based on rDNA and mtDNA gene sequences. Parasitol. Res. 100, 351–357. doi: 10.1007/s00436-006-0269-x
Posada, D. (2008). Model test: phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256. doi: 10.1093/molbev/msn083
Posada, D., and Crandall, K. A. (1998). MODELTEST: testing the model of DNA substitution. Bioinformatics 14, 817–818. doi: 10.1093/bioinformatics/14.9.817
Prasad, P. K., Tandon, V., Biswal, D. K., Goswami, L. M., and Chatterjee, A. (2008). Molecular identification of the Indian liver fluke, Fasciola (Trematoda: Fasciolidae) based on the ribosomal internal transcribed spacer regions. Parasitol. Res. 103, 1247–1255. doi: 10.1007/s00436-008-1121-2
Qian, M. B., Utzinger, J., Keiser, J., and Zhou, X. N. (2016). Clonorchiasis. Lancet 387, 800–810. doi: 10.1016/S0140-6736(15)60313-0
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Sun, J., Huang, Y., Huang, H., Liang, P., Wang, X., Mao, Q., et al. (2013). Low divergence of Clonorchis sinensis in China based on multilocus analysis. PLoS One 8:e67006. doi: 10.1371/journal.pone.0067006
Sun, J., Xu, J., Liang, P., Mao, Q., Huang, Y., Lv, X., et al. (2011). Molecular identification of Clonorchis sinensis and discrimination with other opisthorchid liver fluke species using multiple Ligation-depended Probe Amplification (MLPA). Parasit. Vectors 4:98. doi: 10.1186/1756-3305-4-98
Tang, Z. L., Huang, Y., and Yu, X. B. (2016). Current status and perspectives of Clonorchis sinensis and clonorchiasis: epidemiology, pathogenesis, omics, prevention and control. Infect. Dis. Poverty 5:71. doi: 10.1186/s40249-016-0166-1
Tatonova, Y. V., Chelomina, G. N., and Nguyen, H. M. (2012). Genetic diversity of nuclear ITS1-5.8S-ITS2 rDNA sequence in Clonorchis sinensis Cobbold, 1875 (Trematoda: Opisthorchidae) from the Russian Far East. Parasitol. Int. 61, 664–674. doi: 10.1016/j.parint.2012.07.005
Tatonova, Y. V., Chelomina, G. N., and Nguyen, H. M. (2017). Inter-individual and intragenomic variations in the ITS region of Clonorchis sinensis (Trematoda: Opisthorchiidae) from Russia and Vietnam. Infect. Genet. Evol. 55, 350–357. doi: 10.1016/j.meegid.2017.10.008
Wang, D., Young, N. D., Koehler, A. V., Tan, P., Sohn, W. M., Korhonen, P. K., et al. (2017). Mitochondrial genomic comparison of Clonorchis sinensis from South Korea with other isolates of this species. Infect. Genet. Evol. 51, 160–166. doi: 10.1016/j.meegid.2017.02.015
Xiao, J. Y., Gao, J. F., Cai, L. S., Dai, Y., Yang, C. J., Luo, L., et al. (2013). Genetic variation among Clonorchis sinensis isolates from different hosts and geographical locations revealed by sequence analysis of mitochondrial and ribosomal DNA regions. Mitochondrial DNA 24, 559–564. doi: 10.3109/19401736.2013.770490
Yang, Q. L., Shen, J. Q., Jiang, Z. H., Yang, Y. C., Li, H. M., Chen, Y. D., et al. (2014). Identification of Clonorchis sinensis metacercariae based on PCR targeting ribosomal DNA ITS regions and COX1 gene. Zhong Guo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi 32, 217–220.
Zhang, X., Jin, Z., Da, R., Dong, Y., Song, W., Chen, X., et al. (2008). Fas/FasL-dependent apoptosis of hepatocytes induced in rat and patients with Clonorchis sinensis infection. Parasitol. Res. 103, 393–399. doi: 10.1007/s00436-008-0985-5
Zhang, Y., Chang, Q. C., Zhang, Y., Na, L., Wang, W. T., Xu, W. W., et al. (2014). Prevalence of Clonorchis sinensis infection in freshwater fishes in northeastern China. Vet. Parasitol. 204, 209–213. doi: 10.1016/j.vetpar.2014.05.007
Keywords: Clonorchis sinensis, molecular identification, phylogenetic analysis, ITS1, ITS2, Heilongjiang Province
Citation: Zhang X, Sun B, Tang Q, Chen R and Han S (2019) Molecular Identification and Phylogenetic Analysis of Nuclear rDNA Sequences of Clonorchis sinensis Isolates From Human Fecal Samples in Heilongjiang Province, China. Front. Microbiol. 10:26. doi: 10.3389/fmicb.2019.00026
Received: 23 May 2018; Accepted: 09 January 2019;
Published: 28 January 2019.
Edited by:
Shu Hui Chen, National Institutes of Health (NIH), United StatesReviewed by:
Kin-Ming (Clement) Tsui, Weill Cornell Medicine-Qatar, QatarGuo-Hua Liu, Hunan Agricultural University, China
Copyright © 2019 Zhang, Sun, Tang, Chen and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Su Han, aHNoc2hzMTk4MkAxMjYuY29t
†These authors have contributed equally to this work