 Jingli Liu1,2†
Jingli Liu1,2† Ji Liu1,2†Sheng-Hui Zhang3Jinchang Liang1
Ji Liu1,2†Sheng-Hui Zhang3Jinchang Liang1 Heyu Lin1Delei Song1Gui-Peng Yang3,4Jonathan D. Todd2*
Heyu Lin1Delei Song1Gui-Peng Yang3,4Jonathan D. Todd2* Xiao-Hua Zhang1,4*
Xiao-Hua Zhang1,4*- 1College of Marine Life Sciences, Ocean University of China, Qingdao, China
- 2School of Biological Sciences, University of East Anglia, Norwich, United Kingdom
- 3College of Chemistry and Chemical Engineering, Ocean University of China, Qingdao, China
- 4Laboratory for Marine Ecology and Environmental Science, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China
The compatible solute dimethylsulfoniopropionate (DMSP), made by many marine organisms, is one of Earth's most abundant organosulfur molecules. Many marine bacteria import DMSP and can degrade it as a source of carbon and/or sulfur via DMSP cleavage or DMSP demethylation pathways, which can generate the climate active gases dimethyl sulfide (DMS) or methanthiol (MeSH), respectively. Here we used culture-dependent and -independent methods to study bacteria catabolizing DMSP in the East China Sea (ECS). Of bacterial isolates, 42.11% showed DMSP-dependent DMS (Ddd+) activity, and 12.28% produced detectable levels of MeSH. Interestingly, although most Ddd+ isolates were Alphaproteobacteria (mainly Roseobacters), many gram-positive Actinobacteria were also shown to cleave DMSP producing DMS. The mechanism by which these Actinobacteria cleave DMSP is unknown, since no known functional ddd genes have been identified in genome sequences of Ddd+ Microbacterium and Agrococcus isolates or in any other sequenced Actinobacteria genomes. Gene probes to the DMSP demethylation gene dmdA and the DMSP lyase gene dddP demonstrated that these DMSP-degrading genes are abundant and widely distributed in ECS seawaters. dmdA was present in relatively high proportions in both surface (19.53% ± 6.70%) and bottom seawater bacteria (16.00% ± 8.73%). In contrast, dddP abundance positively correlated with chlorophyll a, and gradually decreased with the distance from land, which implies that the bacterial DMSP lyase gene dddP might be from bacterial groups that closely associate with phytoplankton. Bacterial community analysis showed positive correlations between Rhodobacteraceae abundance and concentrations of DMS and DMSP, further confirming the link between this abundant bacterial class and the environmental DMSP cycling.
Introduction
The tertiary sulfonium compound dimethylsulfoniopropionate (DMSP) is made in prodigious amounts (several petagrams, worldwide annually) in marine environments (Ksionzek et al., 2016). DMSP is synthesized by many marine microalgae, e.g., coccolithophores, dinoflagellates, and diatoms (Curson et al., 2018; Kageyama et al., 2018), macroalgae (Reed, 1983), a few angiosperms (Otte et al., 2004), and some corals (Raina et al., 2013). Recently, heterotrophic bacteria have also been reported to synthesize DMSP (Curson et al., 2017). In these organisms DMSP may function in e.g., storage of excess sulfur and carbon (Stefels, 2000), cryoprotection, oxidative damage protection (Sunda et al., 2002), signaling pathways acting as chemoattractant (Seymour et al., 2010) and enhancing the production of quorum-sensing molecules (Johnson et al., 2016). The major ecological significance of DMSP lies in it being an important nutrient for marine microorganisms (Curson et al., 2011b) providing carbon, sulfur and/or energy to microbes catabolizing it. Microbial DMSP catabolism can generate environmentally important catabolites including the climate active gases dimethylsulfide (DMS) and methanethiol (MeSH). DMS, largely derived from DMSP catabolism, is the most significant biogenic sulfur compound transferred from oceans to the atmosphere [~3 × 108 tons, worldwide annually; (Andreae, 1990)], where its oxidative products act as cloud condensation nuclei (CCN) affecting cloud cover and thus the radiation reaching Earth's surface (Vallina and Simó, 2007).
Although many marine phytoplankton can catabolize DMSP (Alcolombri et al., 2015; Johnston, 2015), it is believed that marine bacteria are significant contributors to global DMSP catabolism once this molecule is released into the dissolved pool of DMSP in seawater (Curson et al., 2011b). Indeed, DMSP supports 1–13% of the bacterial carbon demand in surface seawater (Kiene and Linn, 2000) and it is especially important as a reduced organic sulfur source, e.g., for the dominant heterotrophic bacteria SAR11, which require exogenous sources of reduced sulfur for growth (Tripp et al., 2008). A wide variety of marine microorganism import (Vila et al., 2004; Howard et al., 2008) and catabolize DMSP via two enzymatic pathways: demethylation and cleavage (Curson et al., 2011b; Moran et al., 2012). Demethylation is believed to be the predominant DMSP catabolic pathway, converting ~75% dissolved DMSP into 3-methylmercaptopropionate (MMPA), further into MeSH, and then into microbial biomass (Kiene and Linn, 2000). This pathway does not liberate DMS. The maker gene for DMSP demethylation “dmdA” is only found in bacteria and is prevalent in the SAR11 lineage and another abundant marine Alphaproteobacteria lineage known as the Roseobacters (Howard et al., 2006; Reisch et al., 2011). The dmdA genes can be grouped into five clades and fourteen subclades based on their nucleotide and amino acid sequences (Howard et al., 2006, 2008; Varaljay et al., 2010).
By comparison, there is far more biodiversity in the DMSP cleavage pathway where DMSP lyase enzymes generate DMS from DMSP in bacteria, some fungi and phytoplankton (Curson et al., 2011b; Alcolombri et al., 2015; Sun et al., 2016). To date, eight different DMSP lyase genes, dddD, dddL, dddP, dddQ, dddW, dddY, dddK, and Alma1, encoding distinct polypeptides in various protein families, have been identified in a wide range of microbes, demonstrating a high level of biochemical and genetic diversity in DMSP lyase enzymes (Curson et al., 2011b; Alcolombri et al., 2015; Johnston, 2015; Johnston et al., 2016; Sun et al., 2016). With the exception of dddY, the bacterial ddd genes are common in Roseobacters (Curson et al., 2011b), which can account for up to 30% of bacterioplankton cells (Gonzalez and Moran, 1997) in eutrophic coastal regions where DMS emission is intense and likely exerts influence on climate, e.g., the East China Sea (ECS). Of the identified bacterial DMSP lyases, the DMSP lyase genes dddP and dddQ are by far the most prevalent in ocean microbial reference gene catalog (OM-RGC) metagenomic and Tara Oceans metatranscriptomic datasets apportioned mainly to marine bacteria (Curson et al., 2018).
The ECS is the largest marginal sea of the western Pacific (Figure 1). It is influenced by the Yangtze River effluent and the Kuroshio water current, and acts as a transition zone where terrigenous and anthropogenic materials are discharged from the mainland to the ocean. A previous study of the ECS in the summer of 2011 found the surface waters to contain moderate concentrations of DMSP, DMS and chlorophyll a (28.25 nM, 5.64 nM and 0.84 mg L−1, respectively) (Yang et al., 2011). To date, our understanding of microbial DMSP metabolism in marine environments comes mainly from studies conducted in open sea, salt marsh and estuarine environments (e.g., Ansede et al., 2001) or with phytoplankton-attached bacteria (e.g., Hatton et al., 2012). Studies on the spatial and temporal distribution of bacterial DMSP-degrading genes have been carried out in the Sargasso Sea (Levine et al., 2012), the Pacific Ocean (Varaljay et al., 2012; Cui et al., 2015), and the Arctic Kongsfjorden (Zeng et al., 2016). However, studies of the abundance and diversity of bacterial DMSP catabolism in marginal sea environments are limited. In this study, we collected and characterized seawater samples from seven ECS sites for their oceanographic parameters and DMSP compositions. Culture-dependent methods were used to study DMSP catabolizing bacteria in samples from two of these sites and led to the identification of novel DMSP catabolizing bacterial taxa. Culture-independent methods were used to explore the spatial distribution and diversity of key DMSP catabolic genes in a transect of five ECS stations from inshore to the offshore waters, and the results further demonstrate the importance of bacterial DMSP-catabolism in the ECS.
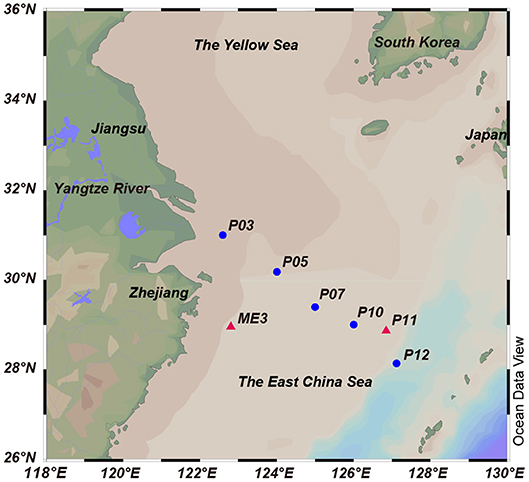
Figure 1. Locations of the sampling sites in the ECS. The red triangles indicate sites from 2013 cruise, and the blue circles indicate sites from 2015 cruise. Stations plotted in Ocean Data View (Schlitzer, 2002).
Materials and Methods
Sampling and Environmental Parameters
Surface seawater (SW) and bottom seawater (BW) samples were collected onboard the R/V “Dong Fang Hong 2” in the ECS during two cruises from 14 July to 1 August 2013 (two sites, ME3 located near the continent and P11 further from the land) and 19 October to 2 November 2015 (five sites, P03, P05, P07, P10, and P12; along a transect from coast to the ocean), respectively (Figure 1, Table 1). Seawater was collected by Niskin bottles equipped on a standard conductivity-temperature-depth rosette (CTD). One liter of each sample was immediately filtered through 0.22 μm pore size polycarbonate membranes filters (Millipore Corporation, Billerica, MA, USA). Filters from 2013 cruise were soaked in sterile 0.85% (w/v) saline supplemented with 15% (v/v) glycerol before stored at −80°C until in-lab bacterial isolation. Filters from 2015 cruise were stored in liquid nitrogen onboard and at −80°C in lab for nucleic acid extraction. Salinity, temperature and dissolved oxygen were recorded with a Seabird 911 conductivity-temperature-depth (CTD).
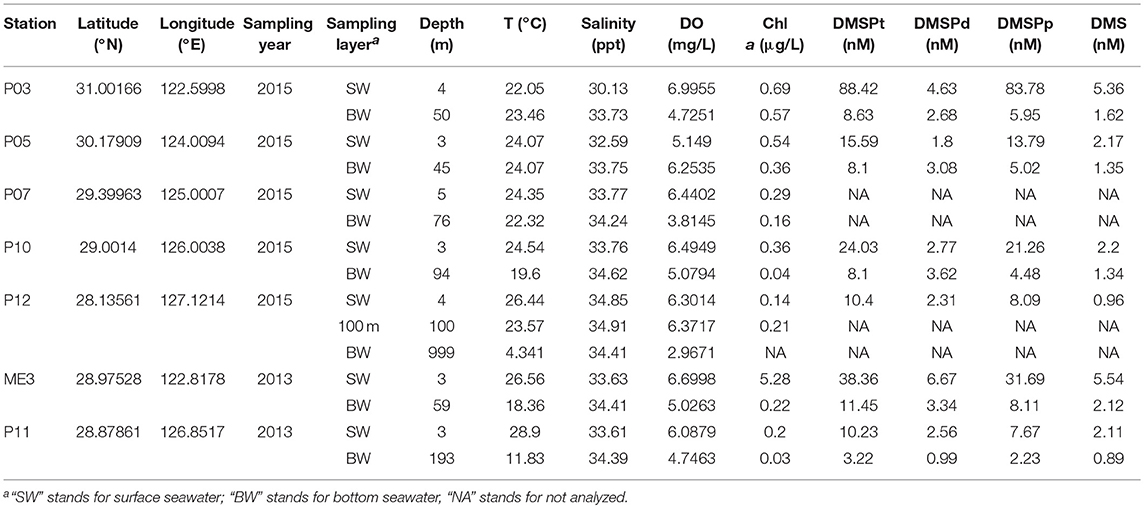
Table 1. Seawater sampling stations and environmental parameters.
DMS and DMSP concentrations in the seawater samples were measured as described by Zhang et al. (2014). DMS samples were measured onboard immediately after sampling using a modified purge and trap method. Briefly, a sample of 2 mL was collected into a glass bubbling chamber through a GF/F filter. Sulfur gases were sparged from the seawater with nitrogen and trapped in a loop of Teflon tubing immersed in liquid nitrogen. The trapped gases were desorbed with hot water (90°C) and analyzed on a Shimadzu GC-2014 gas chromatograph equipped with a flame photometric detector. A 3 m × 3 mm glass column packed with 10% DEGS on Chromosorb W-AW-DMCS was used to separate sulfur gases at 70°C.
Gravity filtering of samples for dissolved DMSP (DMSPd) was conducted as described by Kiene and Slezak (2006) with the following modifications. Total DMSP (DMSPt) and DMSPd samples were fixed with 50% sulfuric acid and stored on ship at room temperature for 2 days. For DMSPd samples, the first few drops of filtrate was discarded, and a 4 mL sample was transferred to a glass vial containing 40 μL of concentrated sulfuric acid and sealed. For DMSPt samples, 100 μL of 50% sulfuric acid was directly added to 10 mL of unfiltered seawater samples and then sealed. When analyzed, 300 μL of 10 M KOH was injected into 2 mL of the preserved DMSP sample and incubated in the dark at 4°C for at least 24 h, allowing for complete conversion of DMSP into DMS and acrylate. The liberated DMS was measured using the method described above.
For chlorophyll a (Chl a) analysis, the seawater samples were filtered through 47 mm Whatman GF/F filters. The filters were soaked in 10 ml of 90% acetone and then stored in the dark at 4°C. After 24 h, the concentration of Chl a was measured using a F4500 (Hitachi) fluorometer (Parsons et al., 1984).
Bacteria Isolation and Phylogenetic Analysis
Filters from the 2013 cruise were rinsed with sterile 0.85% (w/v) saline supplemented with 15% (v/v) glycerol. The cells were resuspended by vortexing, spread on Marine Agar (MA) plates and incubated at 28°C for 1 week. Single colonies were picked randomly and purified three times on fresh plates prior to further studies. Genomic DNA of the isolates was extracted by phenol/chloroform extraction, and the 16S rRNA genes were amplified using the 27F/1492R primer set (Lane, 1991) and sequenced to determine their taxonomy. Calculation of pairwise similarity values for the 16S rRNA gene of the cultivated strains and the most closely related type strains were achieved from the Ezbiocloud server (http://www.ezbiocloud.net/identify). The 16S rRNA gene sequences of representative cultivated bacterial strains and the most closely related type strains were aligned using the CLUSTAL_X program (Thompson et al., 1997). Phylogenetic trees based on the neighbor-joining (NJ) algorithms were constructed by MEGA version 5.0 (Tamura et al., 2011) with K2+G model. The tree topology was checked by 1000 bootstrap replicates.
Sole Carbon Source Test
Fifty-seven representative bacterial isolates were selected and tested for their growth on MBM minimal medium (Baumann and Baumann, 1981) with DMSP (2 mM; TCI, Japan) as sole carbon source. The same medium lacking a carbon source (negative control) or supplied with glucose (2 mM), succinate (2 mM), sucrose (2 mM), pyruvic acid sodium salt (2 mM), and glycerol (2 mM) as the carbon source was used. Briefly, cells were grown in Marine Broth (MB; Becton Dickinson) and harvested after incubation at 28°C for 2 days, and then washed three times with 3% (w/v) NaCl saline. Washed cells were diluted to OD600nm = 0.4–0.6, then 1% (w/v) were inoculated in triplicate into the media supplied with DMSP or mixed carbon source. Cells added to MBM medium without any carbon source were set up as non-carbon control. Growth was measured spectrophotometrically (OD600nm) after 1 week. Significance was determined using a Student's t-test (P < 0.05).
DMSP Catabolism Assay
To measure DMSP catabolism of cultivated bacteria, bacterial strains were grown overnight in MB at 28°C. Cells were washed twice in MAMS media (Table S1) and diluted to OD600 = 0.3 prior to 1 in 10 dilution into vials (CNW, China) containing MAMS media supplied with 0.45% glycerol (v/v) and 0.05% glucose (w/v) as the mixed carbon source and 1 μM, 0.5 mM or 5 mM DMSP as the catabolism substrate to get a final volume of 2 mL. After gas-tight sealing and incubation at 28°C for ~36 h, the 2 mL cultures were directly assayed for DMS and MeSH production as described above. Ratified DMSP degrading strains, Roseovarius nubinhibens ISM and Ruegeria pomeroyi DSS-3, which can produce both DMS and MeSH from DMSP (González et al., 1999, 2003), were used as positive controls. Abiotic media controls of MAMS media supplied with the same concentration of DMSP were set up and incubated under the same conditions to monitor the background chemical lysis of DMSP to DMS. Bacterial Ddd production was calculated by subtracting the abiotically generated DMS from the total detected DMSP-dependent DMS in the bacterial cultures.
An eight-point (2.06–103 nM) calibration curve was made with a gradient DMS concentrations to calculate the DMSP dependent DMS production rate of tested bacterial strains. The detection limit for the purge and trap GC analysis of DMS was 0.8 pmol DMS, and the square root of the DMS peak area was linear to the DMS concentration. Total protein concentration in the cells was estimated using Bradford assays (Bio-Rad). The rate of DMS production was expressed in nmol DMS per mg protein per hour.
A similar eight-point (10–0.1 mM) calibration curve was constructed for MeSH as was done for DMS, see above. However, unlike DMS, the detected peak area of MeSH was not linear with MeSH concentration. The detection limit for the purge and trap GC analysis of MeSH was 0.1 μmol. Thus, MeSH assays carried out here, as described above, are considered as only qualitative and not quantitative for DMSP dependent MeSH production.
Strains that could use DMSP as sole carbon source were tested for their DMSP dependent DMS and MeSH production with and without the addition of mixed carbon source to MBM media (as above) to test whether the addition of extra carbon source significantly affected MeSH and/or DMS production from DMSP.
BLASTp Analysis of Ddd and DmdA Homologs
The bacterial genome sequences of the strains that are of the same genus as our 57 representative isolates were retrieved from the NCBI database as reference genomes (https://www.ncbi.nlm.nih.gov/assembly, Table S2). Ratified DMSP lyases, i.e., DddD, DddL, DddP, DddQ, DddY and DddW, and demethylase DmdA (Table S3) were used to interrogate the putative proteins in all reference genome sequences, using the BLASTp program (E-value ≤ 1e-5), and then the produced sequences were manually curated with the thresholds of identity ≥ 40%, coverage ≥ 70%, and length difference ≤ 20%.
Degenerate dddP Primer Design, PCR Amplification and Sequencing of ddd and dmdA genes of DMSP-Degrading Isolates
DMSP lyase coding genes dddD, dddL and dmdA were PCR amplified using the published primer pairs dddDf/dddDr, dddLf/dddLr (Raina et al., 2009) and dmdA primers for subclade A/1, A/2, B/3, and E/2 which contain culturable bacterial dmdA sequences (Varaljay et al., 2010), while the degenerate primer set DddPUf (ATGTTCGACCCGATGAACathmgntaygc) and DddPUr (CCGCACTCCTGGAACcanggrttngt) (Table S4) for dddP were acquired by the j-CODEHOPE designer (Rose et al., 2003; Boyce et al., 2009) based on the ratified DddP sequences in Table S3 and the validity of their dddP-targeting property was verified on Ruegeria pomeroyi DSS-3 and Roseovarius nubinhibens ISM as positive controls and sequenced marine strains without dddP in their genomes as negative controls. The PCR system included 250 μM of each deoxyribonucleotide triphosphate and 5 μL of 10 × rTaq buffer containing MgCl2, 0.4 μM of each primer, 1 U of rTaq DNA polymerase of TaKaRa (5 U/μL), ~50 ng bacterial genomic DNA and nuclease-free water to adjust to a final volume of 50 μL. The reaction conditions for dddD and dddL were the same as used by Raina et al. (2009) except that two rounds of PCR (using the product of the first round as template for the second round) were performed to enhance the intensity of the product. For dddP, two rounds of PCR were also performed and the reaction condition for each round was as follows: 95°C for 5 min; followed by 30 cycles of 95°C for 1 min, 58°C for 1 min, and 72°C for 40 s; and then a final extension of 72°C for 10 min. PCR products were visualized by electrophoresis on 1% agarose gel, purified by TIANgel Mini Purification Kit (TIANGEN Biotech, Beijing), then cloned into the pUCm-T (TaKaRa) and sequenced by M13F primer (Table S4) using an automated DNA sequencer (model ABI3730; Applied BioSystems) at BGI, Qingdao, China. All PCR amplicons were subjected to BLASTx analysis against the RefSeq database and hits were counted as encoding functional Ddd or DmdA sequence if they were most similar, ≥68% amino acid identity, to ratified Ddd or DmdA enzymes.
Genomic Sequencing of Two Representative Actinobacteria
Genomic DNA of Microbacterium sp. ZYF042 and Agrococcus sp. LZB059 were extracted using E.Z.N.A. Bacterial DNA kit (Omega). Genome sequencing was performed by Shanghai Majorbio Bio-Pharm Technology Co. (China) using the Illumina HiSeq 2000 sequencer system with a 500 bp pair-end library. The reads were assembled using SOAPdenovo v2.04. The genome coverages for strains ZYF042 and LZB059 were 247 × and 450 ×, respectively. Putative genes were identified using Glimmer 3.02. Annotation was performed with BLAST+2.2.24, searching against the National Center for Biotechnology Information (NCBI) Non-Redundant Proteins (NR), Clusters of Orthologous Groups of Proteins (COG), the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) databases, respectively. Ddd and DmdA homolog searching was performed by BLASTP analysis with verified Ddd sequences as query sequences, as above (Table S3).
Cloning and Expression of Predicted ddd-Like Genes
The predicted dddL gene of Ahrensia LZD062 and dddD-like gene of Microbacterium ZYFD042 were amplified from their genomic DNA and ligated into the pET24a (+) vector (Novagen), transformed into E.coli BL21 (DE3) and incubated at 37°C in Luria-Bertani (LB) complete medium (Sambrook et al., 1989) supplemented with 100 μg mL−1 kanamycin. At the mid-exponential growth phase, isopropyl β-D-1-thiogalactopyranoside (IPTG) was added at a final concentration of 0.1 mM. Cultivation was continued at 16°C and 150 rpm until the cell density reached an OD600 of 1.2. To measure Ddd+ of recombinant E. coli cells, IPTG-induced culture was washed twice by M9 medium (Sambrook et al., 1989), then resuspended in M9 medium containing a final concentration of 500 nM DMSP and mixed carbon source as above, following by incubation with shaking in vials at 37°C. After 2 h, DMS was quantified by GC as described above.
Environmental DNA Extraction, Pyrosequencing and Data Analysis
Total DNA of seawater samples were extracted using the method described by Yin et al. (2013) with a modified step to maximize the output, in which a Fast Prep-24 Homogenization System (MP Biomedicals, Irvine, CA, USA) was used to intensify cell lysis at maximum speed for 1 min. The V4-V5 regions of bacterial 16S rRNA gene were amplified with primer sets 515F/907R (Chen et al., 2016) in triplicate and pooled. Pyrosequencing was performed on Illumina MiSeq PE300 platform at Majorbio Bio-Pharm Technology Co., Ltd., Shanghai, China. Chimeras were excluded during assigning OTUs based on 97% similarity level. Taxonomic assignment was against the SILVA database (Release 123) with 80% similarity threshold. Archaeal 16S rRNA gene sequences were removed. After rarefaction to the minimum sequence number for each sample, estimators such as Chao 1′, Shannon indexes, and Good's coverage were calculated (Table S5). All the above analyzes were performed via Qiime pipelining (Caporaso et al., 2010).
Quantification of dddP, dmdA and 16S rRNA Genes
qPCR was performed on StepOne ABI (Applied Biosystems, Foster City, CA, USA). The abundance of dddP was quantified with primer sets dddP_874F/dddP_971R (Table S4) targeting Roseobacter clade (Levine et al., 2012). Seven primer sets designed to target different dmdA subclades (A/1, A/2, B/3, D/1, D/3, C/2, E/2) were used for dmdA quantification (Varaljay et al., 2010). Environmental sample SW of P03 were subjected to Sanger sequencing to confirm dmdA and dddP gene specificity. The abundance of 16S rRNA gene was quantified using the primer set Eub338F/518R (Yin et al., 2013) (Table S4). All PCR reactions were performed in triplicates in 20 μL system using 10 μL 2 × SYBR Premix Ex Taq II (Takara Bio Inc.), 0.4 μL 50 × ROX reference dye, 0.2–0.4 μM each primer, 2 μL 1/10 diluted template DNA. The PCR reaction conditions referred to the primer designers. Amplification efficiencies for each gene ranged from 0.70 to 0.93, with all R2 values higher than 0.99. The relative abundance of bacterial DMSP-degrading genes was acquired by normalizing their copy numbers to the copy number of bacterial 16S rRNA gene. Pearson's correlation analysis was conducted among environmental parameters, DMSP-degrading gene relative abundance and 16S rRNA gene abundance in pyrosequencing data using R's Hmisc package. Statistical significance of the differences of functional gene relative abundance and 16S rRNA gene abundance between SW and BW samples were tested by Student t-test.
Construction and Analyzes of dddP Clone Libraries
To study the diversity of dddP, dddP amplicons from different samples with primers designed by Peng et al. (2012) (Table S4) were used to construct clone libraries and the insertions in the vector were sequenced. The procedures were essentially as what described by Yin et al. (2013). The OTUs of dddP was determined with nucleotide similarity of 80% by Mothur. Estimators like Shannon, Simpson indexes and Good's coverage were calculated (Table S6). Representative sequences of each OTU were translated into protein sequences and used for phylogenetic tree construction as described above with the Poisson model.
Data Availability
Bacterial 16S rRNA gene sequences and accompanying metadata produced from pyrosequencing were deposited in the NCBI Short Read Archive database under accession number SRP138803. Partial 16S rRNA genes of cultivated bacterial isolates were under the GenBank accession numbers KP639130 to KP639182; partial sequences of dddP gene from clone libraries were under accession numbers MH193618 to MH193931; amplified partial dddL genes from cultivated strains were under accession numbers MH193936 to MH193939, amplified dddP genes from cultivated strains were under accession numbers MH193932 to MH193935, amplified dmdA genes from cultivated isolates were under accession numbers MH193940 to MH193947. The Whole Genome Shotgun project of Microbacterium ZYFD042, Agrococcus sp. LZB059 have been deposited at DDBJ/ENA/GenBank under the accession RBZY00000000 and RBZZ00000000 respectively.
Results
Environmental Characteristics of the ECS Seawater
The oceanographic parameters, environmental factors and DMSP/DMS concentrations of the seawater samples were recorded (Table 1). As expected, SW samples had higher Chl a, DMSP, and DMS concentrations than BW samples. Coincidently, bacterial numbers, estimated by 16S rRNA gene quantitative PCR (qPCR), were also much higher in SW than in BW samples (3-fold higher in average) (Table S7, Figure S1). Chl a concentration ranged from 0.03 μg/L to 5.28 μg/L (average 0.65 μg/L) and showed a negative correlation with longitude/distance from shore (r = −0.78, P < 0.01) and salinity (r = −0.84, P < 0.05) (Figure S2). The average total DMSP (DMSPt) concentration in SW samples was 31.17 nM (ranging from 10.23 nM to 88.42 nM), which was ca. four times higher than that in BW samples (average 7.90 nM). Particulate DMSP (DMSPp) accounted for 55.31 to 94.75% (75.76% on average) of the DMSPt. DMS concentrations ranged from 0.89 to 5.54 nM (3.06 nM in SW and 1.46 nM in BW on average). These results are consistent with the higher light levels in SW over BW favoring photosynthetic algae which are thought to be the major producers of DMSP. This would in turn result in the higher levels of DMSP substrate observed in SW and ultimately higher microbial DMSP dependent DMS production.
Isolation of Marine Bacteria From the ECS Seawater
Heterotrophic marine bacteria from sites ME3 and P11 seawater were isolated on MA plates. There was no additional selective pressure applied during the incubation and isolation processes, enabling us to later assess the proportion of cultivable marine bacteria that could catabolize DMSP. In total 211 bacterial strains were isolated and identified from their 16S rRNA gene sequences. These isolates belonged to the phyla Proteobacteria, Actinobacteria, Bacteroidetes, or Firmicutes, comprising 37 genera and 54 species (Figure 2). Alphaproteobacteria comprised the largest percentage of bacterial isolates, accounting for 60.66% of the total cultivated bacterial community (73.60% in ME3 and 41.86% in P11). Not surprisingly, Roseobacters were well represented within the isolates, accounting for 29.86% (35.20% in ME3 and 22.09% in P11), while Gammaproteobacteria accounted for 5.2% of the total bacterial isolates (6.40% in ME3 and 3.49% in P11). In addition, Actinobacteria (22.33% in total, 12.80% in ME3 and 36.05% in P11), Bacteroidetes (9.95% in total, 4.00% in ME3 and 18.60% in P11), and Firmicutes (1.90% in total, 3.20% in ME3 and none in P11) were also represented. Very little is known about DMSP catabolism in any Bacteroidetes, Actinobacteria or Firmicutes and, to our knowledge, none of these bacteria are known to contain functional dmdA or any DMSP lyase genes.
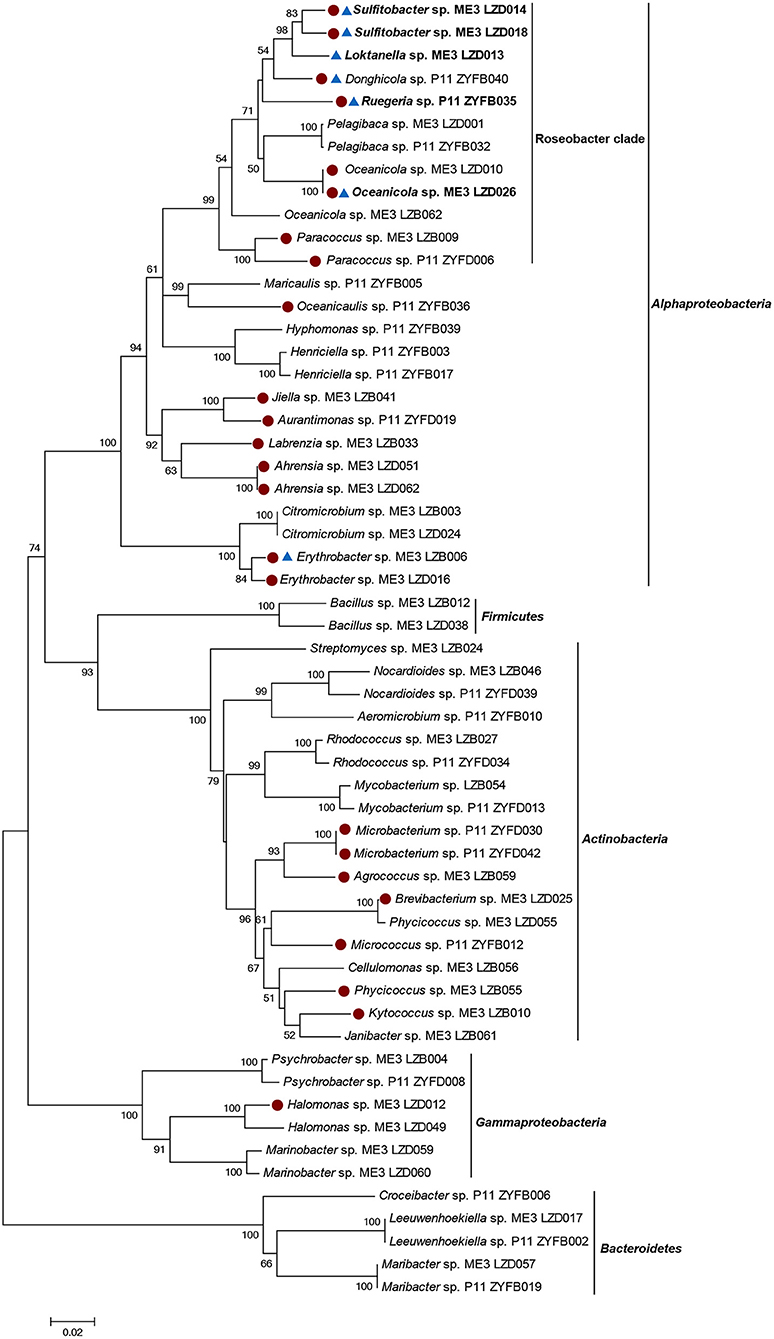
Figure 2. Neighbor-joining tree of 16S rRNA gene sequences of 57 representative cultivated strains. Solid circle represents Ddd+ strains. Triangle represents Ddm+ strains. The genera shown in bold represents bacteria which only showed Ddm+ activity when 0.5 and 5 mM DMSP was added. Bootstrap coefficients below 50% were not shown. Scale bar 0.02 substitutions per nucleotide position.
DMSP Catabolism by Cultivated Bacterial Strains and Their ddd and dmdA Genes
Fifty-seven representative isolates from the different genera were screened for their ability to utilize DMSP as sole carbon source (Figure 2; Table S2). Of these bacterial isolates, only alphaproteobacterial strains of the genera Sulfitobacter and Ahrensia, and gammaproteobacterial Halomonas could use DMSP as sole carbon source (Table S8). Some Halomonas isolates, e.g., HTNK1 are known to use DMSP as sole carbon source, cleave DMSP and contain the CoA transferase family DMSP lyase DddD (Todd et al., 2010). We also tested the ability of these strains to degrade DMSP with and without a mixed carbon source. The strains of genera Ahrensia and Halomonas, which only produced DMS from DMSP, had much higher DMS production when only supplied with DMSP as sole carbon source compared to the presence of the mixed carbon sources (Figure S3). The same was true for Sulfitobacter sp. LZD018, which produced comparatively higher levels of MeSH when DMSP was used as sole carbon source. Note the MeSH could not be accurately quantified by the GC method used here, likely due to the highly reactive properties of MeSH. These observations implied that the ability of these strains to catabolize DMSP is likely underestimated if catabolism is assayed in the presence of other carbon sources.
It is well known that many bacteria containing functional ddd and/or dmdA genes are not able to utilize DMSP as a sole carbon source under lab conditions (Curson et al., 2011b). For this reason, all the representative isolates were tested for their DMSP-dependent DMS (Ddd) and MeSH production (Ddm) when grown in the presence of mixed “regular” carbon sources (0.45% [v/v] glycerol and 0.05% [w/v] glucose). Under this condition, 24 (42.11%) of the tested isolates showed Ddd+ activity (Figure 2 Table 2; Table S2).
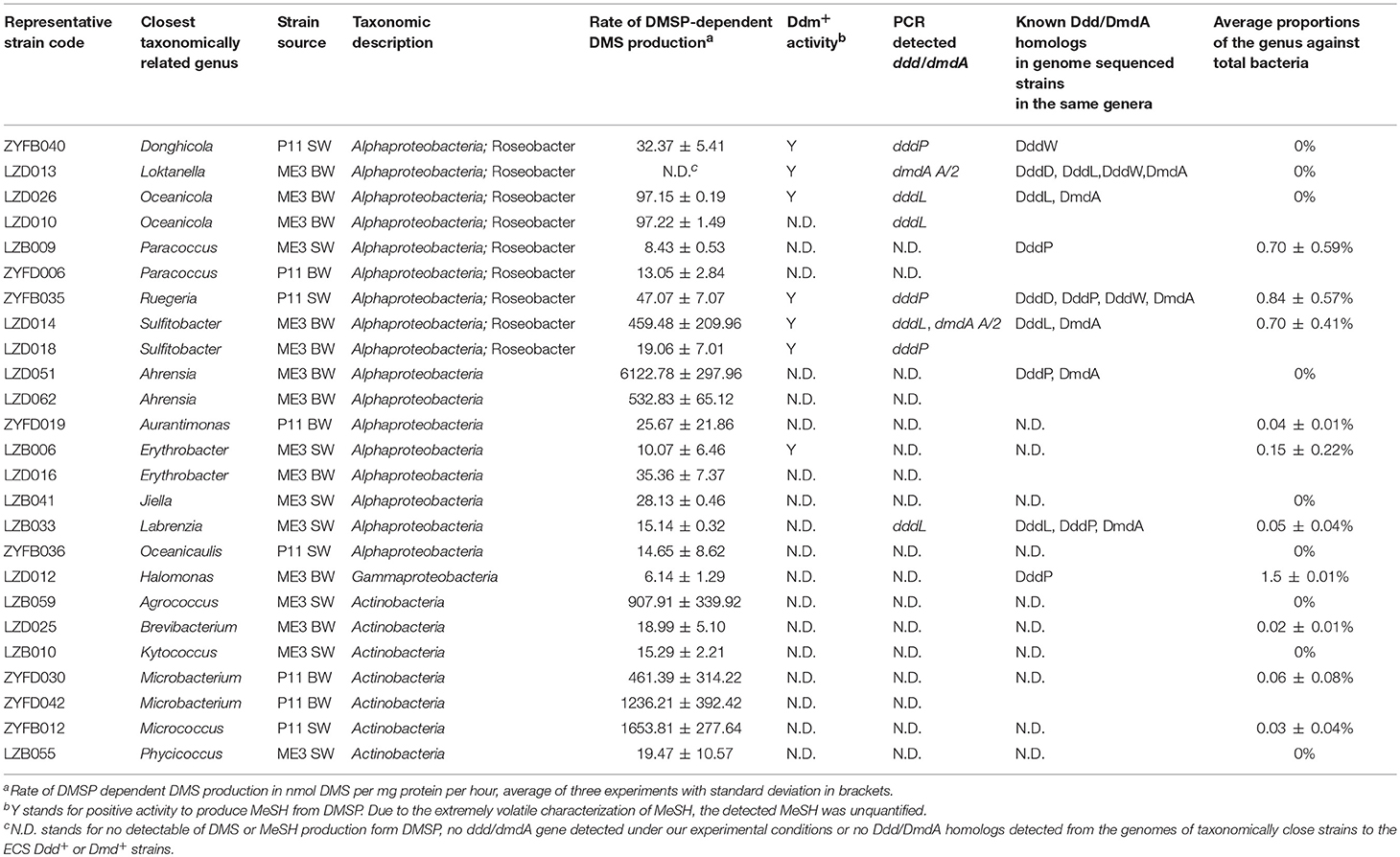
Table 2. Characteristics of DMSP-degrading bacterial isolates from the ECS sites ME3 and P11 and their proportions in the total bacteria community.
As expected, the majority of Ddd+ isolates were Proteobacteria of which Alphaproteobacteria dominated (66.67%), including many Roseobacters (accounting for 33.33% of all tested Ddd+ isolates), i.e., Donghicola, Ruegeria, Oceanicola, Paracoccus, and Sulfitobacter isolates; some non-Roseobacter strains, like strains of known Ddd+ genera Ahrensia and Labrenzia; and of many other unreported Ddd+ alphaproteobacterial genera Aurantimonas, Erythrobacter, Jiella, and Oceanicaulis. Surprisingly, only one gammaproteobacterial strain of Halomonas was shown to be Ddd+, while another Halomonas isolate was not.
Interestingly, the second largest group (12.28%) of cultivated Ddd+ isolates was Actinobacteria. These included Agrococcus, Brevibacterium, Kytococcus, Microbacterium, Micrococcus, and Phycicoccus genera which together accounted for 3.79% of total bacterial isolates. Until now, only members of Rhodococcus and Clostridium genera of gram-positive bacteria isolated from fresh water had been reported to be Ddd+ (Yoch et al., 2001). Furthermore, no functional ddd or dmdA genes have been identified in any gram-positive bacteria.
Under our conditions, strains of Ahrensia, Oceanicola, Ruegeria, Sulfitobacter, Agrococcus, Micrococcus, and Microbacterium showed similar or higher Ddd production rates (> 40 nmol DMS per mg protein per hour) when compared to the positive control strains, i.e., Roseovarius nubinhibens ISM, which contains functional dddQ and dddP genes (Todd et al., 2011), and Oceanimonas doudoroffii J495, which contains functional dddP and dddD genes (Curson et al., 2012). The other Ddd+ isolates showed relatively low Ddd production levels (Table 2).
In contrast to the high proportion isolates shown to be Ddd+, only two alphaproteobacterial strains, Donghicola sp. ZYFB040 (a Roseobacter) and Erythrobacter sp. LZB006, showed detectable Ddm production activity when tested with 1 μM DMSP concentration (Table 2). To exclude the possibility that DMSP added to the ECS isolates was insufficient to produce detectable MeSH levels, incubations with higher DMSP concentrations were conducted (0.5 and 5 mM). When exposed to higher DMSP levels, most strains likely to contain dmdA (i.e., dmdA was amplified from their genomic DNA or closely related strains have dmdA homologs in their genomes, see below) showed detectable MeSH production, including Rhodobacteraceae strains Loktanella sp. LZD013, Oceanicola LZD026, Ruegeria ZYFB035 and Sulfitobacter LZD018 and LZD014 (Table 2; Table S2). These strains were considered as DMSP demethylating bacteria. However, alphaproteobacterial strains of Aurantimonas ZYFD019 and Henriciella ZYFB017, and the gram-positive strains of Mycobacterium LZB054 and ZYFD013 (which were shown to contain dmdA by PCR) had no Ddm+ activity even with the increased DMSP concentrations. We tried to quantify the disappearance of DMSP of these strains in addition to the production of MeSH, but only R. nubinhibens ISM, the positive control, showed a statistically significant reduction in DMSP levels (P < 0.05) (Table S9). It is possible that these cells were degrading very low levels of DMSP via demethylation, but the MeSH produced from DMSP was quickly incorporated into biomass during the incubation, leaving a MeSH concentration below the MeSH detection limit used here. Thus, the absence of detectable DMSP dependent MeSH production does not necessarily indicate that a strain cannot demethylate DMSP. A similar situation could exist for Ddd in bacteria where DMS produced from DMSP is rapidly transformed, e.g., oxidized to dimethylsulfoxide. It would be interesting to see if the dmdA and/or ddd genes are functional and/or transcribed in strains that contain them but which show no detectable Ddd or Ddm phenotype.
To further investigate the potential molecular mechanisms of the Ddd and Ddm production in our isolates, we used published dddD, dddL, dmdA primers (Raina et al., 2009; Varaljay et al., 2010) and our newly designed and ratified dddP degenerate primers (Table S4) to screen for the presence of these functional DMSP lyase and demethylase genes in all representative strains (Table 2; Table S2). dddL homologs were amplified from genomic DNA of Ddd+ Oceanicola sp. LZD010 and LZD026, Sulfitobacter sp. LZD014 and Labrenzia sp. LZB033, displaying 52.90 to 100% amino acid identity to the cupin-containing DddL from Sulfitobacter EE36 (Curson et al., 2008). As expected, dddP homologs were amplified from Ddd+ Sulfitobacter sp. LZD018, Ruegeria sp. ZYFB035 and Donghicola sp. ZYFB040 with 79.68 to 88.24% amino acid identity to R. nubinhibens ISM DMSP lyase DddP (Todd et al., 2009). Labrenzia sp. LZB033 possessed DddP homologs, with 35.16% identity to O. doudoroffii J495 DddP2 (Curson et al., 2012). For the other isolates, no ddd gene could be amplified by the primers we used.
When probing the ECS isolates for DMSP demethylase gene dmdA using the published universal dmdA primer sets dmdAU (Varaljay et al., 2010), only Loktanella sp. LZD013, a strain that showed no detectable Ddm+ activity, gave the correct PCR product (Table S2). Given far more of the isolates were expected to contain dmdA, primers specifically targeting dmdA subclades (A/1, A/2, B/3, E/2) were also used (Varaljay et al., 2010). Using these primers, 12.28% of the representative isolates were shown to contain dmdA and thus the genetic potential to demethylate DMSP (Table S2). dmdA A/2 amplicons likely encoding functional enzymes were detected in Oceanicola, Loktanella, Sulfitobacter, and Henriciella isolates, which are genera of Rhodobacteraceae, and surprisingly, dmdA A/2 was also present in one actinobacterium of the genus Mycobacterium. Indeed, a recent metagenomic study suggested that some Actinobacteria can catabolize DMSP via the demethylation pathway and contain dmdA (Mizuno et al., 2015). Gammaproteobacterial E/2 subclade-targeting primers gave no PCR products with any gammaproteobacterial isolate but did from one alphaproteobacterial Aurantimonas strain and another actinobacterium of the genus Mycobacterium.
Using BLASTp, we interrogated the available genomes of closely related strains that are in the same genera as our 57 representative strains with ratified Ddd lyase sequences (Table S3). DddD, DddL, DddP, DddQ, and DddW homologs were found in many of the alphaproteobacterial strains (Table S2). However, no known Ddd homologs were identified in the genomes of bacteria most closely related to the Ddd+ actinobacterial isolates or alphaproteobacterial Erythrobacter, Aurantimonas, Jiella, and Oceanicaulis isolates. DmdA homologs were present in sequenced genomes of Ahrensia, Labrenzia, Oceanicola, Ruegeria, and Sulftobacter strains, but none of our isolates in these genera showed detectable levels of Ddm+ activity.
Bacterial Community Structure
Microbial community analysis was carried out by analyzing the diversity of 16S rRNA genes in SW and BW samples from five ECS sites across a transect from inshore to offshore waters. Based on 97% nucleotide identity level at the 16S rRNA gene level, a total of 3089 operational taxonomic units (OTUs) were obtained from 11 seawater samples. In all seawater samples, Alphaproteobacteria (34.31 ± 6.46%) and Gammaproteobacteria (17.70 ± 8.19%) were the most abundant classes (Figure 3).
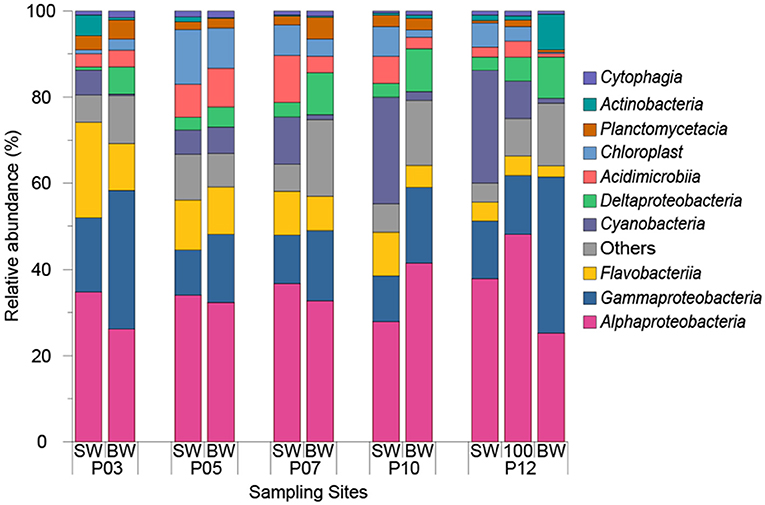
Figure 3. Relative abundance of top 10 dominant classes in seawater samples from 2015 cruise. “SW” stands for surface seawater, “BW” stands for bottom seawater.
The ECS Alphaproteobacteria largely comprised SAR11 clade and Rhodobacterales bacteria which together constituted 79.09 ± 5.25% of total Alphaproteobacteria. SAR11 dominated in almost all sampling sites (22.40 ± 6.88% of the total bacteria) except P03 where Rhodobacterales constituted 26.36% and 12.92% of total bacteria in SW and BW respectively. The relative abundance of Rhodobacterales showed positive correlations with DMS, DMSPt, and DMSPp concentrations (r = 0.89, P < 0.01), and Chl a concentration (r = 0.70, P < 0.05). Of all annotated Rhodobacterales, the genera Ruegeria, Sulfitobacter, Paracoccus, and Labrenzia, which we find to be Ddd+, represented on average 0.84 ± 0.57%, 0.70 ± 0.41%, 0.70 ± 0.59%, and 0.05 ± 0.04% of the total bacteria (Table 2). The genus Roseovarius, a member of which is also known to catabolize DMSP (González et al., 2003), was also identified and represented 0.63 ± 0.43% of the total bacteria. Consistent with the cultivation results, the above Ddd+ genera represented a large proportion (19.2% to 54.1%) of total Rhodobacteraceae. The Ddd+ genus Aurantimonas was found in most samples (represented 0.04 ± 0.01% of total bacteria) except P05 and BW of P03. Erythrobacter, which had Ddd+ representatives, made up 0.01 to 0.80% of total bacteria (Table 2). Although not isolated in this study, the most abundant genus of SAR11 was Candidatus Pelagibacter, representing 59.17 ± 17.89% of SAR11 clade (10.94 ± 5.65% of total bacteria). SAR11 bacteria contain dmdA and demethylate DMSP (Howard et al., 2006) and many SAR11 bacteria also contain dddK and cleave DMSP (Sun et al., 2016).
Of ECS Gammaproteobacteria, Oceanospirillales (7.37 ± 3.78% of total bacteria) and Alteromonadales (4.70 ± 3.67% of total bacteria) were the dominant orders representing 67.76 ± 15.73% of total Gammaproteobacteria. Correspondingly, Halomonas from Oceanospirillales and Alteromonas from Alteromonadales were the most abundant genera representing 0.08% to 1.33% (1.11% on average) and 0.42% to 3.44% (2.03% on average) of total bacteria, respectively. Halomonas representatives from both our ECS bacterial isolates and an isolate from the macroalgae Ulva lactuca are known to have Ddd+ activity (Todd et al., 2010). However, to our knowledge no Alteromonas isolates have been shown to catabolize DMSP or contain know ddd or dmdA genes.
Cyanobacteria which are known to import DMSP (Vila-Costa et al., 2006), were abundant in SW samples (5.56% to 26.16%) and were positively correlated with longitude (r = 0.94, P < 0.05). As expected, their abundance decreased in BW samples (ranged from 0.37% to 6.20%) with decreased light levels. At the genus level, Synechococcus made up 9.35 ± 7.58% of total bacteria in SW samples and was the second most abundant genera. To our knowledge, no Cyanobacteria has been shown to catabolize DMSP. Only one sequenced cyanobacterial strain, Synechococcus sp. KORDI-100, and one metagenome assembled Synechococcus genome (Synechococcus sp. TMED20) (Lei et al., 2017) have a putative DMSP lyase gene, this being dddY-like gene. Thus, Cyanobacteria are not believed to be significant DMSP catabolisers. In contrast to Cyanobacteria, Deltaproteobacteria were always more abundant in BW (8.01 ± 2.19%) compared to SW samples (2.67 ± 0.95%). Although no Ddd+ or Ddm+ Deltaproteobacteria were isolated in this study likely due to the isolation conditions, Desulfovibrio acrylicus is known to contain a DMSP lyase, likely DddY, and cleave DMSP (Der Maarel et al., 1996; Curson et al., 2011a).
The Abundance and Diversity of dmdA and dddP in the ECS Samples
The dmdA and dddP genes are the most abundant environmental indicators of DMSP demethylation and cleavage (Curson et al., 2018) and qPCR primers targeting these genes have been designed (Varaljay et al., 2010). These primer sets were used to investigate the abundance of dddP and dmdA in ECS samples. It should be noted that the dmdA primer sets only target about half of known dmdA sequences.
The relative abundance of DMSP-degrading genes (normalizing ddd and dmdA gene copy numbers to that of the bacterial 16S rRNA gene copy numbers) are shown in Figure 4. As expected, dmdA was abundant in the ECS seawaters, with the total relative abundances of all the dmdA subclades ranging from 2.89 to 29.88% (Figure 4A). Apart from BW sample of P12 (999 m in depth), the total relative abundance of dmdA subclades was 19.57% on average, and had no significant difference (P > 0.05) between the SW and BW samples, but was positively correlated with temperature (r = 0.72, P < 0.05) (Figure S2). Within samples excluding BW sample of P12, dmdA SAR11 subclades D/1 (2.51 × 106 to 6.56 × 108 copies L−1) and D/3 (1.69 × 106 to 4.68 × 108 copies L−1) were the most abundant; SAR11 subclade C/2 (6.89 × 105 to 4.30 × 107 copies L−1) was comparatively abundant in the offshore site P12, particularly in the SW and 100 m depth samples (with relative abundance of 5.56% and 8.12%, respectively). On average, the relative abundances of Roseobacter subclades A/1 and A/2 were 0.98% and 0.82%, respectively in SW samples, and 0.41% and 1.35%, respectively in BW samples. The relative abundance of Gammaproteobacteria-derived subclade E/2 showed a decreasing trend along the transection from inshore (3.04%) to offshore (0.36%), and negatively correlated with longitude (r = −0.75, P < 0.01) (Figure S2). The dmdA subclade B/3 that is represented by the SAR116 group member “Candidatus Puniceispirillum marinum” (Oh et al., 2010) was more abundant in BW samples (relative abundance of 2.61% on average) than in SW samples (relative abundance of 0.94% on average), with P < 0.05.
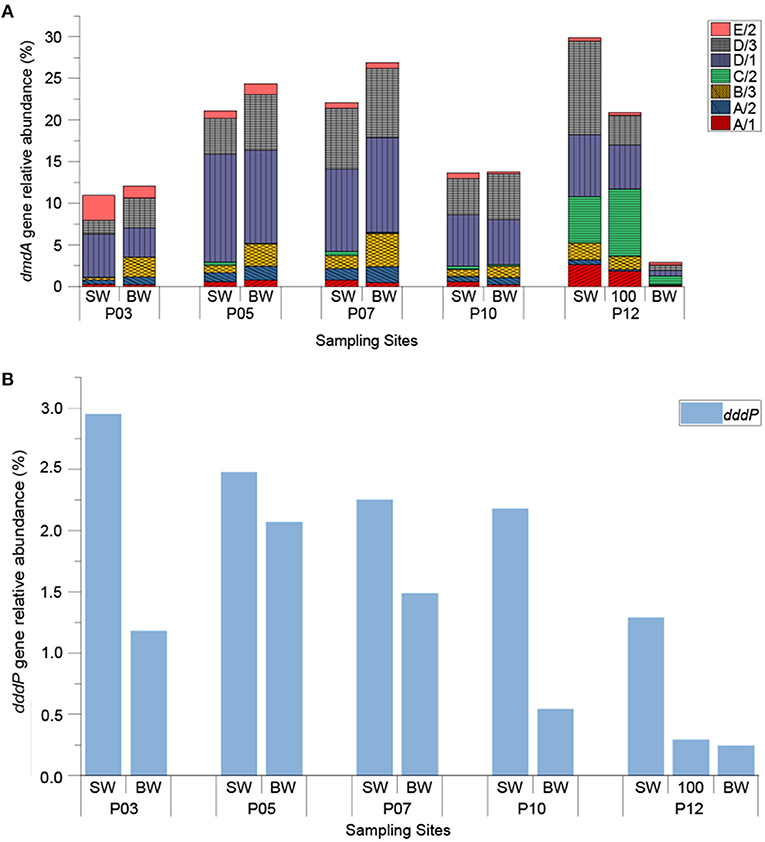
Figure 4. The relative abundance of bacterial DMSP-degrading genes in ECS seawater samples. The dddP and dmdA genes were normalized against bacterial 16S rRNA copy numbers. (A) Seven different subclades of dmdA; (B) dddP. “SW” stands for surface seawater; “BW” stands for bottom seawater; “100” stands for seawater of 100 m depth.
It was clear that the genetic potential to cleave DMSP via the DddP DMSP lyase is far less prominent in the ECS samples than that for DMSP demethylation (Figure 4B). This is expected and fits with previous metagenomic predictions (Moran et al., 2012). The abundance of dddP ranged from 1.91 × 104 to 2.34 × 108 copies L−1, with an average of 6.03 × 107 copies L−1. Unlike dmdA, the dddP gene was significantly more abundant in all SW samples than in BW samples (P < 0.05). This fits with DMSP cleaving bacteria being most abundant in SW where DMSP concentrations are higher. The relative abundance of dddP was highest in the SW of P03 site (2.95%) and steadily decreased as the sites moved away from land (to 0.244% in BW of P12), showing a negative correlation with longitude (r = −0.7, P < 0.05), and a positive correlation with Chl a concentration (r = 0.68, P < 0.05) (Figure S2). This data implies that dddP may be more prominent in bacteria that closely associate with phytoplankton, like Rhodobacteraceae. DMS concentration in ECS samples positively correlated with the proportion of the dddP gene in the total DMSP-degrading genes (dddP/total dmdA + dddP) (r = 0.94, P < 0.01) (Figure S2). Although this value does not include other prominent DMSP lyase genes, correlation still implies the competing relationship between DMSP demethylation and cleavage.
Clone libraries of dddP were constructed from samples of P11, ME3, P03, P05, P10, and P12 (except BW samples of ME3 and P12, since no PCR amplicon could be achieved). In total 314 clones were sequenced and classified into 13 OTUs (Table S6), among which OTU1, OTU2, and OTU3 were dominant. Most representative OTU sequences were clustered with functional DddP sequences of Roseobacter clade bacteria (Figure 5), except those affiliated in cluster 4 in which representative OTUs were clustered with DddP homologs from uncultured bacteria, including OTU2 (represented 31.53% of total sequences). This implies that uncultured bacteria, likely of the Rhodobacteraceae, make a significant contribution to bacterial DMSP cleavage in the ECS, highlighting the need to combine culture-dependent and -independent techniques.
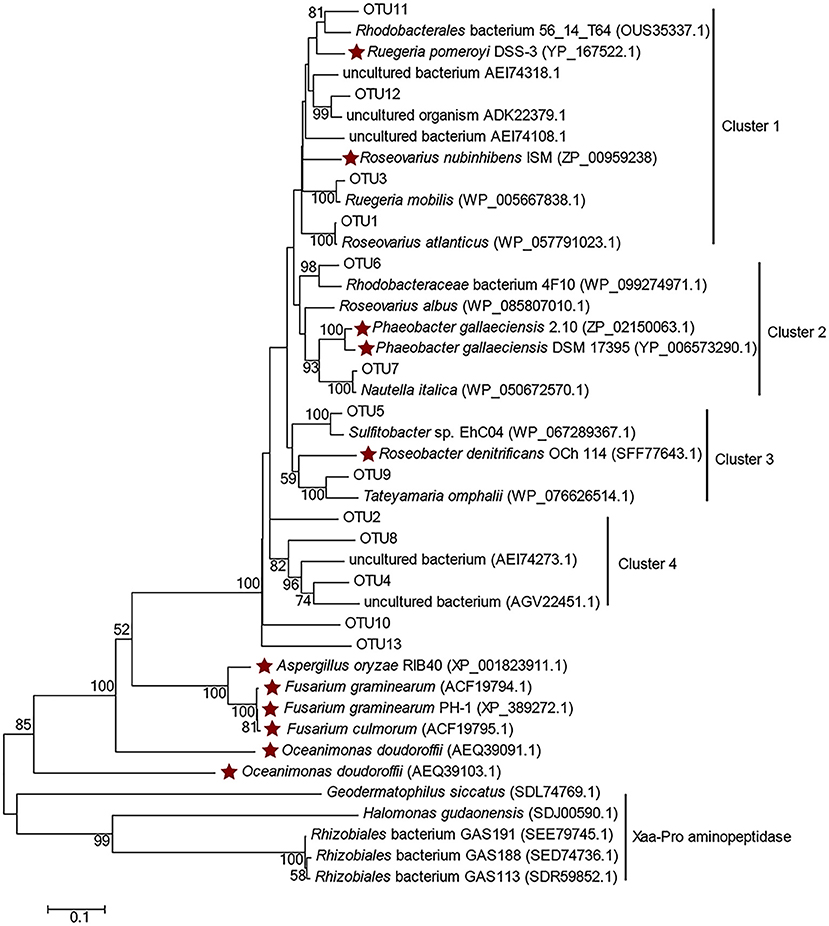
Figure 5. Amino acid tree of representative DddP OTU sequences and their top-hit sequences in GenBank. Xaa-Pro aminopeptidase sequences were used as the outgroup. Stars represent the DddPs which have been experimentally ratified to be functional. The neighbor-joining tree was made with the Passion model.
Discussion
Bacterial demethylation and lysis of DMSP are important and well-established components of the sulfur cycle in marine environment. This study found that a relatively high proportion (42.11%) of the tested cultivated heterotrophic bacteria had the capacity to cleave DMSP generating DMS. Of course, this is not necessarily reflecting the total bacterial community since the composition of the tested cultivable bacteria is not wholly reflecting the composition of the total cultivable isolates, and moreover, we cannot reliably predict the proportion of uncultivated Ddd+ bacteria via the methods used here. Curson et al. (2018) predicted that ~20% of total bacteria in the OM-RGC metagenomic dataset, mainly apportioned surface ocean bacteria, contain known ddd homologs and thus the genetic potential to cleave DMSP. Although this value is relatively lower than the culture-dependent work presented here, both methods confirmed that Ddd+ bacteria are abundant in the natural marine environment.
Many of the ECS Ddd+ isolates are of genera whose members have been demonstrated to cleave DMSP and contain known DMSP lyases, including the alphaproteobacterial Ruegeria, Labrenzia, Donghicola, Sulfitobacter, Oceanicola, and gammaproteobacterial Halomonas (Curson et al., 2008, 2011b). Every Ddd+ roseobacterial isolate and the Labrenzia isolate were found to contain either DddL and/or DddP DMSP lyases that were all very closely related to functionally ratified DMSP lyases (Curson et al., 2008; Todd et al., 2009). The Ahrensia DddL proteins predicted from the genome sequence of LZD062 (Liu et al., 2016) was only 46.98% to that of Sulfitobacter EE-36 (Curson et al., 2008), but was cloned and shown to encode a functional DMSP lyase by heterogenous expression in Escherichia coli BL21 (DE3) (140.01 ± 14.63 nmol DMS mg protein−1 h−1). These data extend our knowledge as to what constitutes a functional DddL DMSP lyase protein.
The Ddd+ alphaproteobacterial isolates in the genera of Jiella and Aurantimonas are in the same Aurantimonadaceae family as Ddd+ Fulvimarina pelagi that contains DddL (Curson et al., 2008). However, both Jiella and Aurantimonas isolates gave no products when probed for dddL, dddP, and dddD primers, thus we do not know the identity of the DMSP lyase in these isolates. The Ddd+ Halomonas isolate which could use DMSP as sole carbon source did not give a product with dddD or any other ddd primer sets used. Since Halomonas HTNK1 has dddD, uses DMSP as sole carbon source (Todd et al., 2010), and other sequenced bacteria of this genera contain DddP homologs, it is most likely that the dddD and/or dddP genes in the ECS Halomonas were not captured by the primer sets used. However, it cannot be ruled out that the ECS Halomonas isolate contains other known ddd genes, like dddL, dddQ, dddY or dddW, or even novel ddd gene(s).
Many Ddd+ isolates from ECS were of genera not reported or even suspected to catabolize DMSP, including Erythrobacter isolates in order Sphingomonadales and many gram-positive Actinobacteria isolates of the Agrococcus, Brevibacterium, Kytococcus, Microbacterium, Micrococcus, and Phycicoccus genera. This is the first report of any gram-positive marine actinobacteria making DMS from DMSP. None of the Ddd+ actinobacterial isolates gave any products with the dddL, dddP or dddD primer sets, nor were there any Ddd homologs in the sequenced genomes of closely related bacteria. We sequenced the genomes of two Ddd+ Actinobacteria (Microbacterium ZYFD042 and Agrococcus sp. LZB059, which grew well in MAMS media and also showed relatively high Ddd+ levels), finding only dddD-like gene in the genome of ZYFD042, with 35.64% amino acid similarity to DddD of Marinomonas sp. MWYL1 (Todd et al., 2007). When cloned and expressed it in E. coli, this DddD-like enzyme did not cleave DMSP. Thus, it is most likely these bacteria contain novel DMSP lyase enzymes. Future molecular work is required to identify the novel DMSP lyase gene(s).
In comparison to e.g., SAR11 and Roseobacter bacteria, Ddd+ genera of Actinobacteria were not abundant in the ECS samples and were not detected in all samples (Table 2). Brevibacterium was only seen in SW of P05 (0.01%) and BW of P12 (0.02%). Microbacterium was found in most samples (representing 0.06 ± 0.08% of total bacteria) except in the BW of P03, P05, and P07. Micrococcus was only found in BW and comprised less than 0.01% of total bacteria on average. It is unlikely these Ddd+ Actinobacteria constitute major contributors to DMS production in these environments. Further molecular work on these bacteria is required to test these hypotheses.
Quantification of dmdA subclade genes (normalized to 16S rRNA gene copy numbers) indicated their abundant distribution in both SW (19.53 ± 6.70%) and BW (16.00 ± 8.73%) ECS samples (Figure 4A), although at lower levels than previously reported (~60% of bacteria) in the GOS metagenomic data (Howard et al., 2008). This discrepancy may partly be due to the limitation of primer sets that do not cover all the dmdA subclades and/or to the selection of 16S rRNA gene as the normalizing gene, since some bacterial genomes have multiple copies of 16S rRNA gene (Cui et al., 2015). Nevertheless, this work confirms that the genetic potential for the bacterial DMSP demethylation is prevalent in the ECS. Culture-dependent work showed that in the majority of cases where dmdA was found in a bacterium, the said bacterium had detectable MeSH production when grown in the presence of DMSP. However, this was not always the case. Further work involving RNA and/or proteomics on the environmental and pure cultures is required to establish why bacteria, e.g., Aurantimonas ZYFD019, Henriciella ZYFB017, and the gram-positive strains of Mycobacterium LZB054 and ZYFD013, that contain dmdA homologs but do not generate MeSH from DMSP.
In the transect of ECS studied here, we found that the concentrations of Chl a, DMS, and DMSP (in the surface seawater) gradually decreased with the distance to the land (longitude), while the salinity and temperature showed increasing trends (Table 1; Figure S2), which provided us natural environmental gradients to study correlations between bacterial DMSP degradation and environmental factors. The predicted major DMSP-degrading bacterial groups, Rhodobacteraceae and SAR11, showed contradictory changes in relative abundances based on the 16S rRNA gene amplicon sequencing data (Figure S2H) and had different correlations with Chl a, DMSPt, and DMS concentrations (Figure S2). Rhodobacteraceae are normally abundant in coastal environments and associated with oceanic algal blooms (Gonzalez and Moran, 1997; Simo et al., 2000; Moran et al., 2004). Their positive correlations with DMS and DMSPt concentrations implied that Rhodobacteraceae might be the major DMS producer from DMSP in the ECS study sites. However, since some phytoplankton directly cleave DMSP to DMS, the correlations might also be due to the association of Rhodobacteraceae and phytoplankton (Figures S2I–K). SAR11 bacteria are widely distributed in marine seawaters and particularly abundant in the open ocean where relatively low DMSP concentrations exist (Giovannoni, 2005, 2017). SAR11 bacteria lack a sulfur reductase and require reduced organosulfur compounds, like DMSP, in surrounding seawater (Tripp et al., 2008). The negative correlation between SAR11 and DMS, DMSPt concentrations implies that SAR11 are not significant contributions to DMS production from DMSP in the studied areas of the ECS (Figures S2L–N). Although, it should be noted that standing stock concentrations of DMSP and DMS are not always good indicators of activity. The positive correlation between DMS and DMSP concentrations with the total relative abundance of dmdA SAR11 subclade D1 and D3 (r = 0.72, P < 0.05) (Figure S2O) implies that SAR11 might degrade DMSP mainly through the demethylation pathway.
Overall, using culture-dependent methods, we identify DMSP catabolizing bacterial taxa in the ECS, many of which are novel gram-positive DMSP-cleaving Actinobacteria. This further extends the phylogenetic territory of marine microorganisms that can carry out this globally influential process. The work provides necessary model organisms to discover novel pathway(s) and gene(s) for DMSP degradation. Also, through culture-independent methods, we find a high proportion of ECS bacteria have the genetic potential to catabolize DMSP, and highlight heterotrophic Alphaproteobacteria, particularly SAR11 and Roseobacter bacteria, as key degraders. Further work studying the activity and regulation of key DMSP lyase and demethylase genes in this environment will shed light on the relative contribution of these two competing pathways.
Author Contributions
X-HZ and JT designed the experiments and wrote the manuscript. JingL and JL collected samples, performed experiments and wrote the manuscript. S-HZ, G-PY performed experiments (DMS measurement on GC, Chl a concentration measurement). JincL helped to perform experiments (qPCR and clone library construction). HL conducted BLASTp analysis of DmdA and Ddd homologs in genomes of taxonomically related strains to Ddd+ bacterial isolates. DS performed the PCR amplicon of ddd and dmdA from genomic DNA of all the tested bacterial isolates.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all the scientists and crew members on the R/V Dong Fang Hong 2 during the expeditions for their great efforts and help in sample collection. This work was financially supported by the National Natural Science Foundation of China (91751202, 41521064, and 41730530), the National Key Research and Development Program of China (2016YFA0601303) in X-HZ's lab, and the NE/N002385/1 and NE/P012671/1 NERC-funded grants in JT's lab.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.03206/full#supplementary-material
References
Alcolombri, U., Ben-Dor, S., Feldmesser, E., Levin, Y., Tawfik, D. S., and Vardi, A. (2015). Identification of the algal dimethyl sulfide-releasing enzyme: a missing link in the marine sulfur cycle. Science 348, 1466–1469. doi: 10.1126/science.aab1586
Andreae, M. O. (1990). Ocean-atmosphere interactions in the global biogeochemical sulfur cycle. Mar. Chem. 30, 1–29. doi: 10.1016/0304-4203(90)90059-L
Ansede, J. H., Friedman, R., and Yoch, D. C. (2001). Phylogenetic analysis of culturable dimethyl sulfide-producing bacteria from a spartina-dominated salt marsh and estuarine water. Appl. Environ. Microbiol. 67, 1210–1217. doi: 10.1128/AEM.67.3.1210-1217.2001
Baumann, P., and Baumann, L. (1981). The Prokaryotes: A Handbook on Habitats, Isolation and Identification of Bacteria. 1st edn, eds M. P. Starr, H. Stolp, H. G. Trüper, A. Balows, H. G. Schlegel (Berlin: Springer), 1302–1331.
Boyce, R., Chilana, P., and Rose, T. M. (2009). iCODEHOP: a new interactive program for designing COnsensus-DEgenerate Hybrid Oligonucleotide Primers from multiply aligned protein sequences. Nucleic Acids Res. 37, W222–W228. doi: 10.1093/nar/gkp379
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chen, H., Zhang, H., Xiong, J., Wang, K., Zhu, J., Zhu, X., et al. (2016). Successional trajectories of bacterioplankton community over the complete cycle of a sudden phytoplankton bloom in the Xiangshan Bay, East China Sea. Environ. Pollut. 219, 750–759. doi: 10.1016/j.envpol.2016.07.035
Cui, Y., Suzuki, S., Omori, Y., Wong, S.-K., Ijichi, M., Kaneko, R., et al. (2015). Abundance and distribution of dimethylsulfoniopropionate degradation genes and the corresponding bacterial community structure at dimethyl sulfide hot spots in the tropical and subtropical pacific ocean. Appl. Environ. Microbiol. 81, 4184–4194. doi: 10.1128/AEM.03873-14
Curson, A. R. J., Fowler, E. K., Dickens, S., Johnston, A. W. B., and Todd, J. D. (2012). Multiple DMSP lyases in the γ-proteobacterium Oceanimonas doudoroffii. Biogeochemistry 110, 109–119. doi: 10.1007/s10533-011-9663-2
Curson, A. R. J., Liu, J., Bermejo Martínez, A., Green, R. T., Chan, Y., Carrión, O., et al. (2017). Dimethylsulfoniopropionate biosynthesis in marine bacteria and identification of the key gene in this process. Nat. Microbiol. 2:17009. doi: 10.1038/nmicrobiol.2017.9
Curson, A. R. J., Rogers, R., Todd, J. D., Brearley, C. A., and Johnston, A. W. B. (2008). Molecular genetic analysis of a dimethylsulfoniopropionate lyase that liberates the climate-changing gas dimethylsulfide in several marine α-proteobacteria and Rhodobacter sphaeroides. Environ. Microbiol. 10, 757–767. doi: 10.1111/j.1462-2920.2007.01499.x
Curson, A. R. J., Sullivan, M. J., Todd, J. D., and Johnston, A. W. B. (2011a). DddY, a periplasmic dimethylsulfoniopropionate lyase found in taxonomically diverse species of Proteobacteria. ISME J. 5, 1191–1200. doi: 10.1038/ismej.2010.203
Curson, A. R. J., Todd, J. D., Sullivan, M. J., and Johnston, A. W. B. (2011b). Catabolism of dimethylsulphoniopropionate: microorganisms, enzymes and genes. Nat. Rev. Microbiol. 9, 849–859. doi: 10.1038/nrmicro2653
Curson, A. R. J., Williams, B. T., Pinchbeck, B. J., Sims, L. P., Martínez, A. B., Rivera, P. P. L., et al. (2018). DSYB catalyses the key step of dimethylsulfoniopropionate biosynthesis in many phytoplankton. Nat. Microbiol. 3, 430–439. doi: 10.1038/s41564-018-0119-5
Der Maarel, M. J. E. C., Aukema, W., and Hansen, T. A. (1996). Purification and characterization of a dimethylsulfoniopropionate cleaving enzyme from Desulfovibrio acrylicus. FEMS Microbiol. Lett. 143, 241–245. doi: 10.1111/j.1574-6968.1996.tb08487.x
Giovannoni, S. J. (2005). Genome streamlining in a cosmopolitan oceanic bacterium. Science 309, 1242–1245. doi: 10.1126/science.1114057
Giovannoni, S. J. (2017). SAR11 bacteria: the most abundant plankton in the oceans. Ann. Rev. Mar. Sci. 9, 231–255. doi: 10.1146/annurev-marine-010814-015934
González, J. M., Covert, J. S., Whitman, W. B., Henriksen, J. R., Mayer, F., Scharf, B., et al. (2003). Silicibacter pomeroyi sp. nov. and Roseovarius nubinhibens sp. nov., dimethylsulfoniopropionate-demethylating bacteria from marine environments. Int. J. Syst. Evol. Microbiol. 53, 1261–1269. doi: 10.1099/ijs.0.02491-0
González, J. M., Kiene, R. P., and Moran, M. A. (1999). Transformation of sulfur compounds by an abundant lineage of marine bacteria in the α-subclass of the classproteobacteria. Appl. Environ. Microbiol. 65, 3810–3819.
Gonzalez, J. M., and Moran, M. A. (1997). Numerical dominance of a group of marine bacteria in the alpha-subclass of the clas Proteobacteria in coastal seawater. Appl. Environ. Microbiol. 63, 4237–4242.
Hatton, A. D., Shenoy, D. M., Hart, M. C., Mogg, A., and Green, D. H. (2012). Metabolism of DMSP, DMS and DMSO by the cultivable bacterial community associated with the DMSP-producing dinoflagellate Scrippsiella trochoidea. Biogeochemistry 110, 131–146. doi: 10.1007/s10533-012-9702-7
Howard, E. C., Henriksen, J. R., Buchan, A., Reisch, C. R., Bürgmann, H., Welsh, R., et al. (2006). Bacterial taxa that limit sulfur flux from the ocean. Science 314, 649–652. doi: 10.1126/science.1130657
Howard, E. C., Sun, S., Biers, E. J., and Moran, M. A. (2008). Abundant and diverse bacteria involved in DMSP degradation in marine surface waters. Environ. Microbiol. 10, 2397–2410. doi: 10.1111/j.1462-2920.2008.01665.x
Johnson, W. M., Kido Soule, M. C., and Kujawinski, E. B. (2016). Evidence for quorum sensing and differential metabolite production by a marine bacterium in response to DMSP. ISME J. 10, 2304–2316. doi: 10.1038/ismej.2016.6
Johnston, A. W., Green, R. T., and Todd, J. D. (2016). Enzymatic breakage of dimethylsulfoniopropionate — a signature molecule for life at sea. Curr. Opin. Chem. Biol. 31, 58–65. doi: 10.1016/j.cbpa.2016.01.011
Johnston, A. W. B. (2015). Who can cleave DMSP? Science 348, 1430–1431. doi: 10.1126/science.aac5661
Kageyama, H., Tanaka, Y., Shibata, A., Waditee-Sirisattha, R., and Takabe, T. (2018). Dimethylsulfoniopropionate biosynthesis in a diatom Thalassiosira pseudonana: identification of a gene encoding MTHB-methyltransferase. Arch. Biochem. Biophys. 645, 100–106. doi: 10.1016/j.abb.2018.03.019
Kiene, R. P., and Linn, L. J. (2000). The fate of dissolved dimethylsulfoniopropionate (DMSP) in seawater: tracer studies using 35S-DMSP. Geochim. Cosmochim. Acta 64, 2797–2810. doi: 10.1016/S0016-7037(00)00399-9
Kiene, R. P., and Slezak, D. (2006). Low dissolved DMSP concentrations in seawater revealed by small-volume gravity filtration and dialysis sampling. Limnol. Oceanogr. Methods 4, 80–95. doi: 10.4319/lom.2006.4.80
Ksionzek, K. B., Lechtenfeld, O. J., McCallister, S. L., Schmitt-Kopplin, P., Geuer, J. K., Geibert, W., et al. (2016). Dissolved organic sulfur in the ocean: biogeochemistry of a petagram inventory. Science 354, 456–459. doi: 10.1126/science.aaf7796
Lane, D. J. (1991). 16S/23S rRNA sequencing. Nucleic Acid Techniques in Bacterial Systematics. Available online at: https://ci.nii.ac.jp/naid/10005795102/ (Accessed April 15, 2018).
Lei, L., Alcolombri, U., and Tawfik, D. S. (2017). DddY is a Bacterial Dimethylsulfoniopropionate Lyase Representing a New Cupin Enzyme Superfamily With Unknown Primary Function. bioRxiv 161257. doi: 10.1101/161257
Levine, N. M., Varaljay, V. A., Toole, D. A., Dacey, J. W. H., Doney, S. C., and Moran, M. A. (2012). Environmental, biochemical and genetic drivers of DMSP degradation and DMS production in the Sargasso Sea. Environ. Microbiol. 14, 1210–1223. doi: 10.1111/j.1462-2920.2012.02700.x
Liu, J., Wang, Y., Liu, Y., and Zhang, X.-H. (2016). Ahrensia marina sp. nov., a dimethylsulfoniopropionate-cleaving bacterium isolated from seawater, and emended descriptions of the genus Ahrensia and Ahrensia kielensis. Int. J. Syst. Evol. Microbiol. 66, 874–880. doi: 10.1099/ijsem.0.000805
Mizuno, C. M., Rodriguez-Valera, F., and Ghai, R. (2015). Genomes of planktonic Acidimicrobiales: widening horizons for marine Actinobacteria by metagenomics. MBio 6, e02083–e02014. doi: 10.1128/mBio.02083-14
Moran, M. A., Buchan, A., González, J. M., Heidelberg, J. F., Whitman, W. B., Kiene, R. P., et al. (2004). Genome sequence of Silicibacter pomeroyi reveals adaptations to the marine environment. Nature 432, 910–913. doi: 10.1038/nature03170
Moran, M. A., Reisch, C. R., Kiene, R. P., and Whitman, W. B. (2012). Genomic insights into bacterial DMSP transformations. Annu. Rev. Mar. Sci. 4, 523–542. doi: 10.1146/annurev-marine-120710-100827
Oh, H.-M., Kwon, K. K., Kang, I., Kang, S. G., Lee, J.-H., Kim, S.-J., et al. (2010). Complete genome sequence of “Candidatus Puniceispirillum marinum” IMCC1322, a representative of the SAR116 clade in the Alphaproteobacteria. J. Bacteriol. 192, 3240–3241. doi: 10.1128/JB.00347-10
Otte, M. L., Wilson, G., Morris, J. T., and Moran, B. M. (2004). Dimethylsulphoniopropionate (DMSP) and related compounds in higher plants. J. Exp. Bot. 55, 1919–1925. doi: 10.1093/jxb/erh178
Parsons, T., Maita, Y., and Lalli, C. (1984). Manual of Chemical and Biological Methods for Seawater Analysis. Oxford: Pergamon Press.
Peng, M., Xie, Q., Hu, H., Hong, K., Todd, J. D., Johnston, A. W. B., et al. (2012). Phylogenetic diversity of the dddP gene for dimethylsulfoniopropionate-dependent dimethyl sulfide synthesis in mangrove soils. Can. J. Microbiol. 58, 523–30. doi: 10.1139/w2012-019
Raina, J.-B., Tapiolas, D., Willis, B. L., and Bourne, D. G. (2009). Coral-associated bacteria and their role in the biogeochemical cycling of sulfur. Appl. Environ. Microbiol. 75, 3492–3501. doi: 10.1128/AEM.02567-08
Raina, J.-B., Tapiolas, D. M., Forêt, S., Lutz, A., Abrego, D., Ceh, J., et al. (2013). DMSP biosynthesis by an animal and its role in coral thermal stress response. Nature 502, 677–680. doi: 10.1038/nature12677
Reed, R. H. (1983). Measurement of osmotic significance of beta-dimethylsulphoniopropionate in marine macroalgae. Mar. Biol. Lett. 4, 173–181.
Reisch, C. R., Moran, M. A., and Whitman, W. B. (2011). Bacterial catabolism of Dimethylsulfoniopropionate (DMSP). Front. Microbiol. 2:172. doi: 10.3389/fmicb.2011.00172
Rose, T., Henikoff, J. G., and Henikoff, S. (2003). CODEHOP (COnsensus-DEgenerate Hybrid Oligonucleotide Primer) PCR primer design. Nucleic Acids Res. 31, 3763–3766. doi: 10.1093/nar/gkg524
Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). Molecular Cloning, A Laboratory Manual, 2nd Edn, Vol. 3. Cold Spring Harbor Laboratory Press.
Schlitzer, R. (2002). Interactive analysis and visualization of geoscience data with Ocean data view. Comput. Geosci. 28, 1211–1218. doi: 10.1016/S0098-3004(02)00040-7
Seymour, J. R., Simó, R., Ahmed, T., and Stocker, R. (2010). Chemoattraction to dimethylsulfoniopropionate throughout the marine microbial food web. Science 329, 342–345. doi: 10.1126/science.1188418
Simo, R., Casamayor, E. O., Pedro, C., and Moran, M. N. N. (2000). Bacterial community structure associated with a North Atlantic algal bloom. Appl. Environ. Microbiol. 66, 4237–4246. doi: 10.1128/AEM.66.10.4237-4246.2000
Stefels, J. (2000). Physiological aspects of the production and conversion of DMSP in marine algae and higher plants. J. Sea Res. 43, 183–197. doi: 10.1016/S1385-1101(00)00030-7
Sun, J., Todd, J. D., Thrash, J. C., Qian, Y., Qian, M. C., Temperton, B., et al. (2016). The abundant marine bacterium Pelagibacter simultaneously catabolizes dimethylsulfoniopropionate to the gases dimethyl sulfide and methanethiol. Nat. Microbiol. 1:16065. doi: 10.1038/nmicrobiol.2016.65
Sunda, W., Kieber, D. J., Kiene, R. P., and Huntsman, S. (2002). An antioxidant function for DMSP and DMS in marine algae. Nature 418, 317–320. doi: 10.1038/nature00851
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Thompson, J., Gibson, T. J., Plewniak, F., Jeanmougin, F., and Higgins, D. G. (1997). The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882. doi: 10.1093/nar/25.24.4876
Todd, J. D., Curson, A. R. J., Dupont, C. L., Nicholson, P., and Johnston, A. W. B. (2009). The dddP gene, encoding a novel enzyme that converts dimethylsulfoniopropionate into dimethyl sulfide, is widespread in ocean metagenomes and marine bacteria and also occurs in some Ascomycete fungi. Environ. Microbiol. 11, 1376–1385. doi: 10.1111/j.1462-2920.2009.01864.x
Todd, J. D., Curson, A. R. J., Kirkwood, M., Sullivan, M. J., Green, R. T., and Johnston, A. W. B. (2011). DddQ, a novel, cupin-containing, dimethylsulfoniopropionate lyase in marine roseobacters and in uncultured marine bacteria. Environ. Microbiol. 13, 427–438. doi: 10.1111/j.1462-2920.2010.02348.x
Todd, J. D., Curson, A. R. J., Nikolaidou-Katsaraidou, N., Brearley, C. A., Watmough, N. J., Chan, Y., et al. (2010). Molecular dissection of bacterial acrylate catabolism - unexpected links with dimethylsulfoniopropionate catabolism and dimethyl sulfide production. Environ. Microbiol. 12, 327–343. doi: 10.1111/j.1462-2920.2009.02071.x
Todd, J. D., Rogers, R., Li, Y. G., Wexler, M., Bond, P. L., Sun, L., et al. (2007). Structural and regulatory genes required to make the gas dimethyl sulfide in bacteria. Science 315, 666–669. doi: 10.1126/science.1135370
Tripp, H. J., Kitner, J. B., Schwalbach, M. S., Dacey, J. W. H., Wilhelm, L. J., and Giovannoni, S. J. (2008). SAR11 marine bacteria require exogenous reduced sulphur for growth. Nature 452, 741–744. doi: 10.1038/nature06776
Vallina, S. M., and Simó, R. (2007). Strong relationship between DMS and the solar radiation dose over the global surface ocean. Science 315, 506–508. doi: 10.1126/science.1133680
Varaljay, V. A., Gifford, S. M., Wilson, S. T., Sharma, S., Karl, D. M., and Moran, M. A. (2012). Bacterial dimethylsulfoniopropionate degradation genes in the oligotrophic north pacific subtropical gyre. Appl. Environ. Microbiol. 78, 2775–2782. doi: 10.1128/AEM.07559-11
Varaljay, V. A., Howard, E. C., Sun, S., and Moran, M. A. (2010). Deep sequencing of a dimethylsulfoniopropionate-degrading gene (dmdA) by using PCR primer pairs designed on the basis of marine metagenomic data. Appl. Environ. Microbiol. 76, 609–617. doi: 10.1128/AEM.01258-09
Vila, M., Simó, R., Kiene, R. P., Pinhassi, J., González, J. M., Moran, M. A., et al. (2004). Use of microautoradiography combined with fluorescence in situ hybridization to determine dimethylsulfoniopropionate incorporation by marine bacterioplankton taxa. Appl. Environ. Microbiol. 70, 4648–4657. doi: 10.1128/AEM.70.8.4648-4657.2004
Vila-Costa, M., Simó, R., Harada, H., Gasol, J. M., Slezak, D., and Kiene, R. P. (2006). Dimethylsulfoniopropionate uptake by marine phytoplankton. Science 314, 652–654. doi: 10.1126/science.1131043
Yang, G.-P., Zhang, H.-H., Zhou, L.-M., and Yang, J. (2011). Temporal and spatial variations of dimethylsulfide (DMS) and dimethylsulfoniopropionate (DMSP) in the East China Sea and the Yellow Sea. Cont. Shelf Res. 31, 1325–1335. doi: 10.1016/j.csr.2011.05.001
Yin, Q., Fu, B., Li, B., Shi, X., Inagaki, F., and Zhang, X.-H. (2013). Spatial Variations in microbial community composition in surface seawater from the ultra-oligotrophic center to rim of the South Pacific Gyre. PLoS ONE 8:e55148. doi: 10.1371/journal.pone.0055148
Yoch, D. C., Carraway, R. H., Friedman, R., and Kulkarni, N. (2001). Dimethylsulfide (DMS) production from dimethylsulfoniopropionate by freshwater river sediments: phylogeny of Gram-positive DMS-producing isolates. FEMS Microbiol. Ecol. 37, 31–37. doi: 10.1111/j.1574-6941.2001.tb00850.x
Zeng, Y.-X., Qiao, Z.-Y., Yu, Y., Li, H.-R., and Luo, W. (2016). Diversity of bacterial dimethylsulfoniopropionate degradation genes in surface seawater of Arctic Kongsfjorden. Sci. Rep. 6:33031. doi: 10.1038/srep33031
Keywords: DMSP catabolism, DMS, methanthiol (MeSH), bacterial community, the East China Sea
Citation: Liu J, Liu J, Zhang S-H, Liang J, Lin H, Song D, Yang G-P, Todd JD and Zhang X-H (2018) Novel Insights Into Bacterial Dimethylsulfoniopropionate Catabolism in the East China Sea. Front. Microbiol. 9:3206. doi: 10.3389/fmicb.2018.03206
Received: 13 July 2018; Accepted: 11 December 2018;
Published: 21 December 2018.
Edited by:
Alison Buchan, University of Tennessee, Knoxville, United StatesReviewed by:
Jozef I. Nissimov, Rutgers State University of New Jersey, United StatesNaomi Levine, University of Southern California, United States
Copyright © 2018 Liu, Liu, Zhang, Liang, Lin, Song, Yang, Todd and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan D. Todd, am9uYXRoYW4udG9kZEB1ZWEuYWMudWs=
Xiao-Hua Zhang, eGh6aGFuZ0BvdWMuZWR1LmNu
†These authors have contributed equally to this work