 Sabine Matallana-Surget1*
Sabine Matallana-Surget1* Johannes Werner2
Johannes Werner2 Ruddy Wattiez3
Ruddy Wattiez3 Karine Lebaron1
Karine Lebaron1 Laurent Intertaglia4,5Callum Regan1James Morris1Hanno Teeling6
Laurent Intertaglia4,5Callum Regan1James Morris1Hanno Teeling6 Manuel Ferrer7
Manuel Ferrer7 Peter N. Golyshin8Dimitrios Gerogiorgis9Simon I. Reilly8Philippe Lebaron4,5
Peter N. Golyshin8Dimitrios Gerogiorgis9Simon I. Reilly8Philippe Lebaron4,5- 1Division of Biological and Environmental Sciences, Faculty of Natural Sciences, University of Stirling, Stirling, United Kingdom
- 2Department of Biological Oceanography, Leibniz Institute of Baltic Sea Research, Rostock, Germany
- 3Department of Proteomics and Microbiology, Interdisciplinary Mass Spectrometry Center (CISMa), University of Mons, Mons, Belgium
- 4Sorbonne Universites, UPMC Univ Paris 06, CNRS, Laboratoire de Biodiversité et Biotechnologies Microbiennes (LBBM), Observatoire Océanologique, Banyuls/Mer, France
- 5Sorbonne Universites, UPMC Univ Paris 06, CNRS, Observatoire Océanologique de Banyuls (OOB), Banyuls/Mer, France
- 6Department of Molecular Ecology, Max Planck Institute for Marine Microbiology, Bremen, Germany
- 7Department of Applied Biocatalysis, Institute of Catalysis, CSIC, Madrid, Spain
- 8School of Natural Sciences, University of Bangor, Bangor, United Kingdom
- 9Institute for Materials and Processes, School of Engineering, University of Edinburgh, The King's Buildings, Edinburgh, United Kingdom
Epibacterium mobile BBCC367 is a marine bacterium that is common in coastal areas. It belongs to the Roseobacter clade, a widespread group in pelagic marine ecosystems. Species of the Roseobacter clade are regularly used as models to understand the evolution and physiological adaptability of generalist bacteria. E. mobile BBCC367 comprises two chromosomes and two plasmids. We used gel-free shotgun proteomics to assess its protein expression under 16 different conditions, including stress factors such as elevated temperature, nutrient limitation, high metal concentration, and UVB exposure. Comparison of the different conditions allowed us not only to retrieve almost 70% of the predicted proteins, but also to define three main protein assemblages: 584 essential core proteins, 2,144 facultative accessory proteins and 355 specific unique proteins. While the core proteome mainly exhibited proteins involved in essential functions to sustain life such as DNA, amino acids, carbohydrates, cofactors, vitamins and lipids metabolisms, the accessory and unique proteomes revealed a more specific adaptation with the expression of stress-related proteins, such as DNA repair proteins (accessory proteome), transcription regulators and a significant predominance of transporters (unique proteome). Our study provides insights into how E. mobile BBCC367 adapts to environmental changes and copes with diverse stresses.
Introduction
With oceans covering ~70% of the planet's surface they represent the largest habitat on Earth. Living biomass in the oceans is dominated by microorganisms (Church, 2009; DeVries et al., 2017). In contrast to blue water open oceans, coastal marine environments represent more heterogeneous habitats that provide a wider spectrum of accessible dissolved organic matter. These conditions favor copiotrophic generalist bacteria over specialists (Lauro et al., 2009). These generalists are characterized by large pools of catabolic and transporters as well as stress response functions, which enable them to profit from the ample nutrient supply and to cope with changing environmental conditions in their coastal ecosystems (Lauro et al., 2009; Christie-Oleza et al., 2012).
Epibacterium mobile BBCC367, formerly known as Ruegeria mobilis BBCC367 (Wirth and Whitman, 2018), a member of the Roseobacter clade (Lee et al., 2012), is a key component of marine bacterioplankton, as 15% of bacterial cells in the open ocean and 20% in coastal waters are members of this group from tropical to polar regions (Moran et al., 2007; Gram et al., 2010). Members of the Roseobacter clade feature diverse metabolic capabilities that foster their widespread abundance, particularly in temperate and deep pelagic oceans (Luo and Moran, 2014).
E. mobile BBCC367 is a dark-brown pigmented, facultative aerobic bacterium (Lee et al., 2007) that was isolated from the South Pacific Ocean, off the coast of Chile in 2004 (Claustre et al., 2008; Matallana-Surget et al., 2012b). E. mobile BBCC367 is closely related to Ruegeria pomeroyi (formerly Silicibacter pomeroyi, Figure 1) and both species share similar geographical distributions (Sonnenschein et al., 2017). R. pomeroyi was the first member of the Roseobacter clade to have its genome completely sequenced and annotated, revealing a large pool of genes with roles in adaptations to changing environmental conditions (Moran et al., 2007). Nevertheless, little is known about the fraction of expressed proteins that is continuously expressed for maintaining housekeeping functions and the tightly regulated fraction that is expressed only in response to environmental changes (Christie-Oleza et al., 2012).
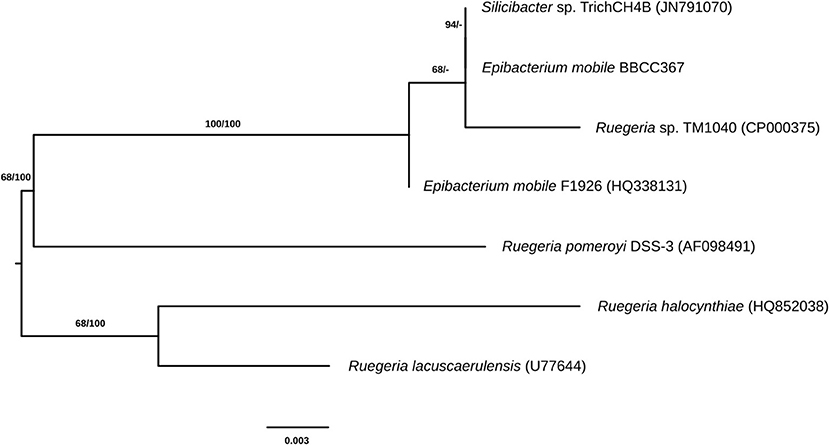
Figure 1. Phylogenetic tree of closely related species to E. mobile (the identifiers in brackets are IDs from the ribosomal database of full-length 16S sequences).
Marine bacteria near the ocean surface are particularly impacted by solar radiation. E. mobile BBCC367 showed a high resistance to UVB exposure (Matallana-Surget et al., 2012b) and thus, was selected as a model organism to study UV resistance. Most bacterial species when exposed to elevated levels of solar UV radiation (UVR) respond with decreases in abundance as well as their amino acid uptake, exo-enzymatic activities, oxygen consumption, protein, and DNA synthesis (Alonso-Saez et al., 2006; Matallana-Surget and Wattiez, 2013). Nonetheless, Farías et al. (2009) showed Roseobacter abundances to increase with solar exposure, even at very high altitudes observed in the Laguna Vilama, a hypersaline Andean lake in Chile (4,650 m) exposed to high doses of UVR. Within the two dominant subgroups of Alphaproteobacteria in the Mediterranean sea, bacteria belonging to the Roseobacter clade showed higher resistance than members of the SAR11 cluster (Alonso-Saez et al., 2006). UVR resistance is often attributed to a low GC content, which limits cytosine-containing photoproduct formation (Agogué et al., 2005; Matallana-Surget et al., 2009). However, E. mobile BBCC367 has a DNA G+C content of 58 mol%, which makes the high UV tolerance in E. mobile BBCC367 even more remarkable.
Proteogenomics is the integrated study of proteomics and genomics with the aim to obtain the complete resolution of a species proteome (Armengaud, 2012). In our study, we sequenced and annotated the genome of E. mobile BBCC367 and used high-throughput proteomics to assess its proteome. We cultivated E. mobile BBCC367 under 16 different conditions to access as many non-redundant proteins as possible and to increase coverage of the theoretical proteome. E. mobile BBCC367 was cultivated under different temperatures (4°C, 40°C), oxic vs. anoxic conditions, in presence of different metals (copper, nickel, zinc, cobalt), under different times of UVB exposure, and harvested at different physiological states (exponential phase, late stationary phase, very late stationary phase). The overall aim of this study was the investigation of E. mobile BBCC367's proteome with a particular focus on its resistance to diverse forms of stress, in particular UVB radiation.
Materials and Methods
Bacterial Strains and Culture Conditions
E. mobile BBCC367 was isolated during the 2004 BIOSOPE cruise from the coastal waters off Chile (Claustre et al., 2008). E. mobile BBCC367 is maintained in the Banyuls Bacterial Culture Collection or BBCC (https://collection.obs-banyuls.fr/catalogue.php). Pre-cultures were grown aerobically on a rotary shaker (120 rpm) at 25°C in either marine broth (MB) for the proteogenomic study or in artificial sea water (250 mL) with 3 mM D-glucose (ASW-G), vitamins and trace elements (Eguchi et al., 1996) for the quantitative proteomics study. E. mobile BBCC367 was cultured under 16 conditions to obtain the highest proteome coverage. For proteogenomics (conditions 1 to 16, Table 1), cells were cultivated in MB. In this first study, no biological replicate was performed for each condition. For the 16 conditions, cells were pelleted by centrifugation at 8,000 g for 15 min and the pellets were washed using phosphate buffer saline to remove proteins derived from the MB medium. Label-free quantitative proteomics allowed assessment of the impact of UVB. Experiments were carried out for UVB treatments and dark controls in triplicates. For quantitative proteomics cells were pelleted by centrifugation at 8,000 g for 15 min and were stored at −80°C until further use.
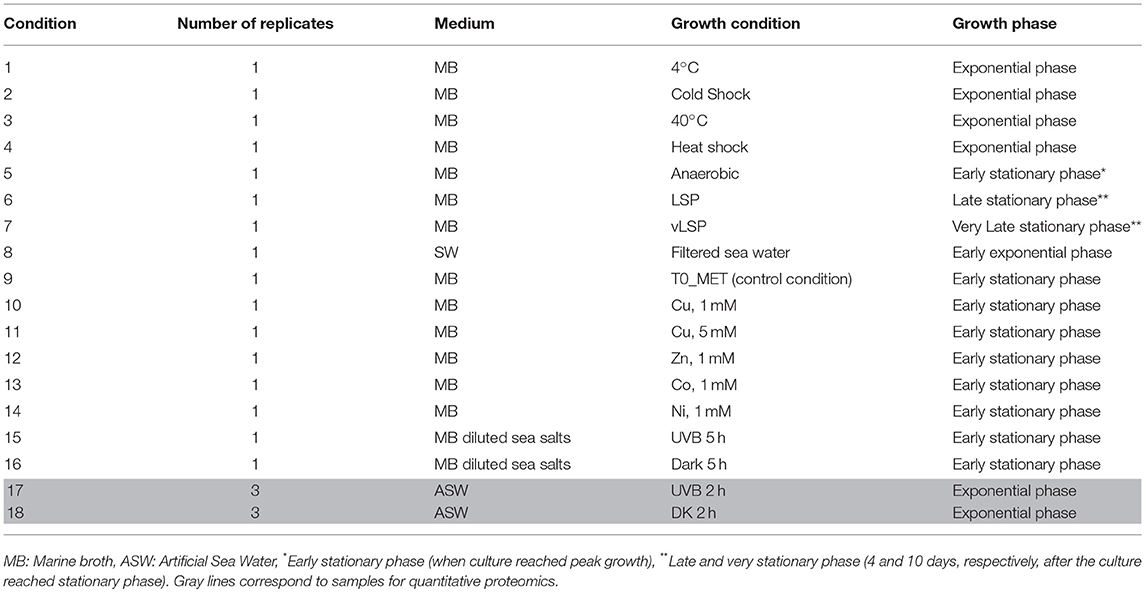
Table 1. Growth conditions used for proteogenomics and quantitative proteomics.
DNA Extraction and Sequencing
For DNA extraction, the pellet of a fresh culture (50 ml, 25°C, 2 days at 100 RPM) of the strain E. mobile BBCC367 was suspended in 9.5 ml TE (10 mM Tris, 1 mM EDTA) buffer. 0.5 ml of 10% SDS, 5 μl RNase A 10 mg/ml and 50 μl of 20 mg/ml of proteinase K were added, mixed thoroughly and incubated 1 h at 37°C. Then, 1.8 ml of 5 M NaCl was added and mixed thoroughly. 1.5 ml CTAB/NaCl solution was added, mixed thoroughly and incubated 20 min at 65°C. An equal volume of chloroform/isoamyl alcohol 24:1 solution was added to the previous solution and after 10 min at 6,000 g at room temperature, the supernatant was transferred into a 0.6 vol of isopropanol until the DNA appeared. The DNA string was hooked with the end of a Pasteur pipette and transfer into a 70% ethanol solution until further analysis.
De novo sequencing data production for E. mobile BBCC367 was conducted at the Liverpool University Genome Centre on a 454 FLX Ti (454 Life Sciences, Branford, CT, USA) using a standard library (ca. 16x coverage). In parallel, a library sequencing using Illumina HiSeq1500 was done at Fidelity Systems Ltd. (Gaithersburg, MD, USA) with short paired-end 400 bp, average read length of 100 bp and 256x coverage. Genome assembly and gap closure were performed by Fidelity Systems using Phred/Phrap and Consed for the final sequence assembly (Ewing and Green, 1998b; Ewing et al., 1998a; Gordon, 2003). DupFinisher (Han and Chain, 2006) was used for the correction of repeat mis-assemblies. For the full closure, a number of direct sequencing was conducted (Malykh et al., 2004).
Genome Annotation
Prediction of genes and functional annotation were carried out through the Rapid Annotation using Subsystem Technology (RAST) server (Aziz et al., 2008). Resulting annotations were subsequently imported into a local installation of the GenDB v.2.2 annotation system (Meyer et al., 2003) for data mining, using similarity searches against the NCBI non-redundant protein (Pruitt et al., 2012), InterPro (Hunter et al., 2012), PFAM (Sonnhammer et al., 1997), KEGG (Ogata et al., 1999), COG (Tatusov et al., 2003) databases, as well as predictions of signal peptides with SignalP v3.0 (Nielsen et al., 1997) and transmembrane regions with TMHMM v2.0c (Krogh et al., 2001). Annotations of selected genes were manually curated using JCOAST (Richter et al., 2008). The annotated genome sequence of E. mobile BBCC367 was submitted to ENA (LR027553-LR027556).
Protein Extraction and Quantification
For protein extraction, the thawed cell pellet was re-suspended in one pellet volume of lysis buffer (6 M guanidine chloride), and cells were mechanically broken by sonication on ice (5 cycles of 1 min with tubes on ice, amplitude 30%, 0.5 pulse rate). Broken cells were centrifuged at 16,000 g at 4°C for 15 min. Protein samples were reduced with 25 mM dithiothreitol (DTT) at 56°C for 30 min and alkylated with 50 mM iodoacetamide at room temperature for 30 min. Proteins were precipitated with cold acetone overnight at −80°C, with an acetone/aqueous protein solution ratio of 4:1. The protein pellet was dissolved in 100 mM phosphate buffer (pH 8) containing 2 M urea. Total protein concentration was determined by a Bradford assay. For LC-MS/MS analysis, a trypsic digestion (sequencing grade modified trypsin, Promega) was performed overnight at 37°C, with an enzyme/substrate ratio of 1:25.
Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS)
Protein identification and quantification were conducted following a label-free strategy on an ultra-high-performance liquid chromatography–high-resolution tandem mass spectroscopy (UHPLC-HRMS/MS) platform (Eksigent 2D Ultra and AB Sciex TripleTOF 5600 system). Peptides were separated on a 25 cm C18 column (Acclaim pepmap 100, 3 μm, Dionex/LC Packings, Amsterdam, the Netherlands) by a linear acetonitrile gradient [5–35% (v/v), in 15 or 120 min] in water containing 0.1% (v/v) formic acid at a flow rate of 300 nL min−1. The instrument was operated in DDA data-dependent acquisition (DDA) mode and MS/MS were acquired across 100–1,800 m/z. A long run procedure was used to acquire quantitative data, and a duty cycle of 3 s per cycle was used to ensure that high quality extracted ion chromatograms (XIC) could be obtained. Protein searches were performed against E. mobile BBCC367 genome using ProteinPilot v4.1. Search parameters included differential amino acid mass shifts for oxidized methionine (+15.9949 Da). The identification of the overall set of proteins was validated by manual inspection of the MS/MS ion spectra, ensuring that a series of consecutives sequence-specific b- and y-type ions was observed. For quantification, the quant application of PeakView was used to calculate XIC for all peptides identified with a confidence >0.99 using ProteinPilot.
Quantified proteins were kept with a p < 0.05. The false discovery rate (FDR) was calculated at the peptide level for all experimental runs using the decoy option in Mascot; this rate was estimated to be lower than 1% using the identity threshold as the scoring threshold system. The cut-off for a significantly differential regulation was used for a protein showing average increase in abundance above 2 or below 0.5 fold change in the UVB radiation samples relative to their controls.
Phylogenetic Analysis
Pairwise sequence similarities and phylogenies of the complete 16S rRNA genes of E. mobile BBCC367 and six further genomes among the genera Epibacterium, Ruegeria, and Silicibacter were calculated by the GGDC web server (http://ggdc.dsmz.de/; Meier-Kolthoff et al., 2013). A multiple sequence alignment was created with MUSCLE (Edgar, 2004), maximum likelihood (ML) and maximum parsimony (MP) trees were inferred from the alignment with RAxML (Stamatakis, 2014) and TNT (Goloboff et al., 2008), respectively. For ML, rapid bootstrapping in conjunction with the autoMRE bootstopping criterion (Pattengale et al., 2010) and subsequent search for the best tree was used; for MP, 1000 bootstrapping replicates were used in conjunction with tree-bisection-and-reconnection branch swapping and ten random sequence addition replicates. The sequences were checked for a compositional bias using the X2 test as implemented in PAUP (Swofford, 2002).
Protein Sequences Cluster Analysis
Proteins that behaved similarly across a set of experiments, or with similar abundance profiles were grouped together. Dendrogram construction was performed by means of complete linkage hierarchical clustering, using the “farthest neighbor” distance metric (Murtagh and Contreras, 2011; Khachumov, 2012). The oncoprint was generated using R v 3.5.1 and the ComplexHeatmap package v. 1.17.1 (Gu et al., 2016).
Results and Discussion
Theoretical vs. Expressed Proteome
The genome sequence of E. mobile BBCC367 consists of 4,712,067 bp, including 4,557 coding sequences (CDS) and 75 RNAs (15 rRNAs and 60 tRNAs). E. mobile BBCC367 comprises two chromosomes (3,073 and 1,167 CDS) and two plasmids (125 and 192 CDS) (Table 2). It is noteworthy that second chromosomes are not common among members of the Rhodobacteraceae family, and were observed in only 6 out of 74 completely sequenced genomes namely: Paracoccus denitrificans PD1222, three strains of Rhodobacter sphaeroides (2.4.1, ATCC_17029 and KD131), Yangia pacifica YSBP01 and Yangia sp. CBB-11M3. The theoretical proteome comprises the complete set of predicted proteins (Matallana-Surget et al., 2009). The expressed proteome represents the complete set of proteins identified by mass spectrometry in the 16 conditions presented in Table 1. One of the main limitations in proteogenomics often relates to the low theoretical proteome coverage. This issue can be overcome by investigating at multiple growth conditions together with several separation and/or fractionation techniques prior to MS/MS analysis (Kucharova and Wiker, 2014). While there are many different techniques to fractionate the proteome all producing different results (Castellana and Bafna, 2010), our experimental design using 16 different growth conditions enabled us to successfully obtain a coverage of 67.6% using only biological fractionation (Table 2). This represents a high coverage compared to other studies, e.g., that of R. pomeroyi where 46% of its theoretical proteome was found to be expressed (Christie-Oleza et al., 2012). Chromosomal and plasmid coding genes comprised 94% and 6% of the expressed E. mobile BBCC367 proteome, respectively.
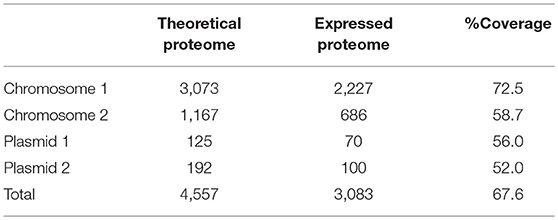
Table 2. Comparison of the theoretical (coding sequences) and expressed proteome.
The most abundant proteins identified in the theoretical proteome were found in COG S (unknown function) and COG R (general function) with 26 and 11%, respectively, with a small portion of COG S characterized in the expressed proteome (Figure 2). The two largest contributors of the expressed proteome were also found to belong to COGs S and R. The most abundant COGs with a related known function and representing about 1/4 of the genome were found to be involved in proteins metabolism including translation (COG J), protein turnover (proteases, chaperones) (COG O) and amino-acid synthesis (COG E) (Figures 2, 3, green parts). COG J (translation, ribosomal structure and biogenesis) presented a 100% coverage value where all 200 proteins were identified from the theoretical proteome. COG J is ubiquitous and expressed predominantly as these proteins form the universal core of life (Tatusov et al., 1997). In a proteomic analysis of R. pomeroyi it was also found that proteins related to COG J (translation, ribosomal structure and biogenesis) (16%) and COG E (amino acid transport and metabolism) (14%) were among the most abundant proteins detected (Christie-Oleza et al., 2012). Moreover, COG E (amino acid transport) represents the largest COG category in 7 different Epibacterium, Ruegeria, and Silicibacter species (Figure 3), formerly all belonging to Ruegeria (Figure 1, Wirth and Whitman, 2018). The second main COGs gathered proteins involved in DNA metabolism including transcription (COG K), DNA repair (COG L), nucleotide metabolism (COG F), and cell division (COG D) (Figures 2, 3, red parts). Finally the third group of proteins was involved in energy production (Figure 2: COGs C and G and Figure 3, blue part).
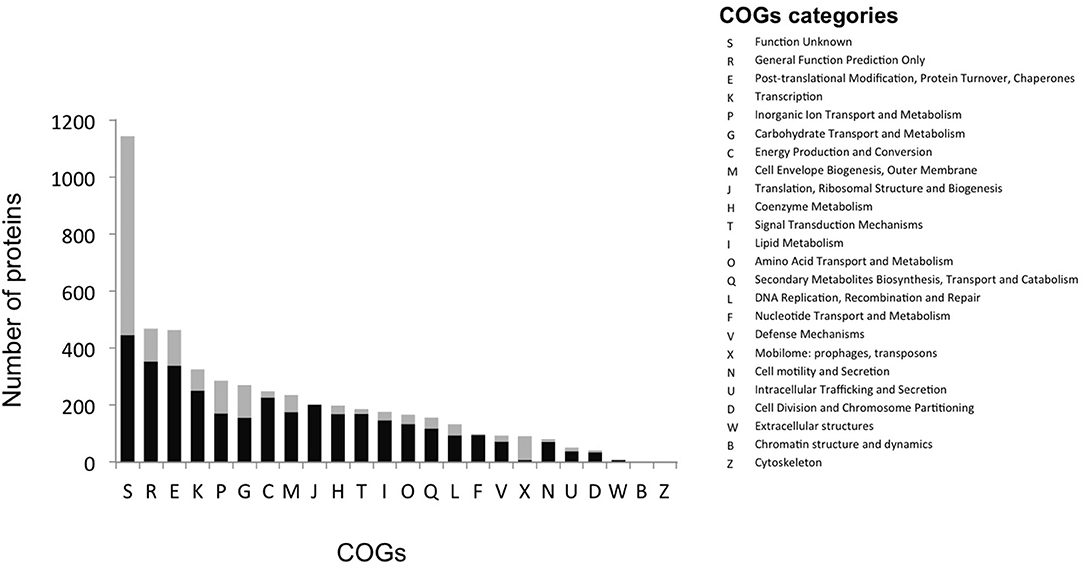
Figure 2. Distribution of proteins into COG categories of the theoretical (gray) and the expressed proteome (black).
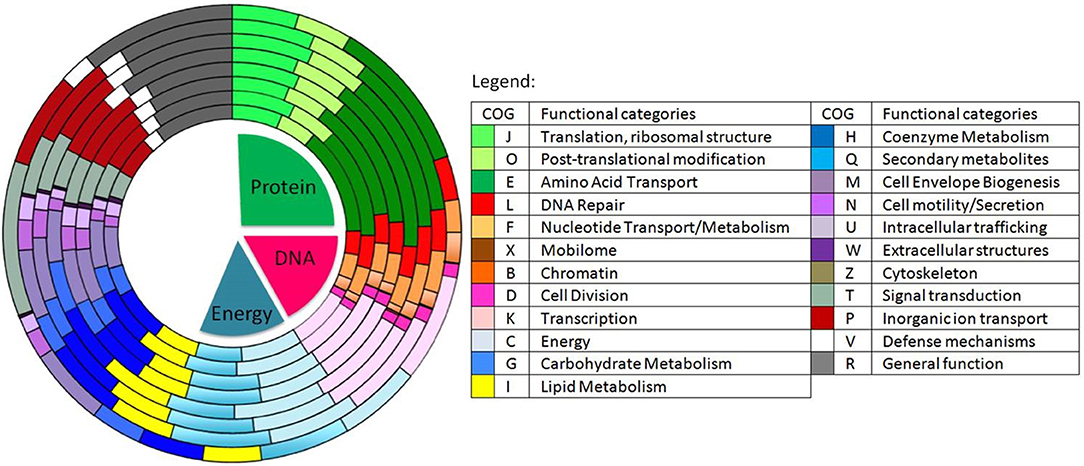
Figure 3. Distribution of the predicted genes into COGs in 8 different species of Ruegeria and Epibacterium. From the innermost to the outermost circle: R. halocynthiae, R. lacuscaerulensis, E. mobile F1926, R. pomeroyi DSS-3, R. TM1040, R. trichCH4B and our strain E. mobile BBCC367. Shades of green represent predicted genes involved in protein functions (e.g., amino acid transport), shades of red to purple represent predicted genes involved in DNA processes (e.g., DNA repair) and shades of light blue to yellow represent predicted genes involved in energy metabolism processes.
Genomic comparison of 7 different strains belonging to Epibacterium, Ruegeria, and Silicibacter (Figure 1) revealed that the most distinct strain was E. mobile BBCC367 (external circle, Figure 3). Predicted genes involved in protein and DNA metabolisms and energy presented a comparable distribution between all strains. Both COG R (general function only) and COG T (signal transduction mechanism) were found to be overrepresented in E. mobile BBCC367 (Figure 3). Signal transduction occurs when an extracellular signaling molecule activates a specific receptor; generating responses such as changes in enzyme activity or gene expression. Thus, E. mobile BBCC367 might be more adapted to respond to environmental variations.
Theoretical Proteome—Diversity of Metabolic Pathways
Diversity of Transporters
Four main categories of transporters were characterized, i.e., ABC (ATP-binding cassette) transporters, TRAP (tripartite ATP-independent periplasmic) transporters, TAT secretion VI (twin-arginine translocation pathway) and Ton-Tol system transporters (Figure 4). ABC transporters were found to be mainly involved in a wide diversity of substrates uptake: amino acid, lipids, sterols or drugs transport (265 proteins) (Wilkens, 2015). On the contrary to specialist bacteria, generalists have evolved a wide diversity of broad-specificity and energy intensive transporters. In this way, E. mobile BBCC367 could rapidly and tightly regulate its metabolism and use energetically expensive transporters for nutrient acquisition. TRAP transporters (55 proteins) are widely used by marine bacteria that live in Na+ rich environments. TRAP transporters co-transport two Na+ ions with one substrate molecule; the energetic cost is thus lower than ABC transporters, while the use of the substrate-binding protein allows the transporter to function with an affinity similar to an ABC transporter (Mulligan et al., 2011). Both ABC and TRAP transporters were found to be abundant in Roseobacter clade bacteria compared to other bacterial groups, thus making them more adapted for dilute and heterogeneous growth substrates (Tang et al., 2012). Other transporters such as TAT secretion pathway serve to export proteins across the cytoplasmic membrane (Posey et al., 2006). ABC transporters play a crucial role in metal resistance and detoxification of metals via metal efflux transport (Bruins et al., 2000). Ton-Tol transporters (26 proteins) allow bacteria to take up scarce resources when nutrients are limited in the environments (Tang et al., 2012). Interestingly, it was previously shown, that generalist bacteria had a higher number of secreted proteins compared to specialist bacteria, due to the higher ectoenzymatic activity observed in particle-attached bacteria (mainly generalist) vs. free-living (mainly specialist) bacteria (Lauro et al., 2009).
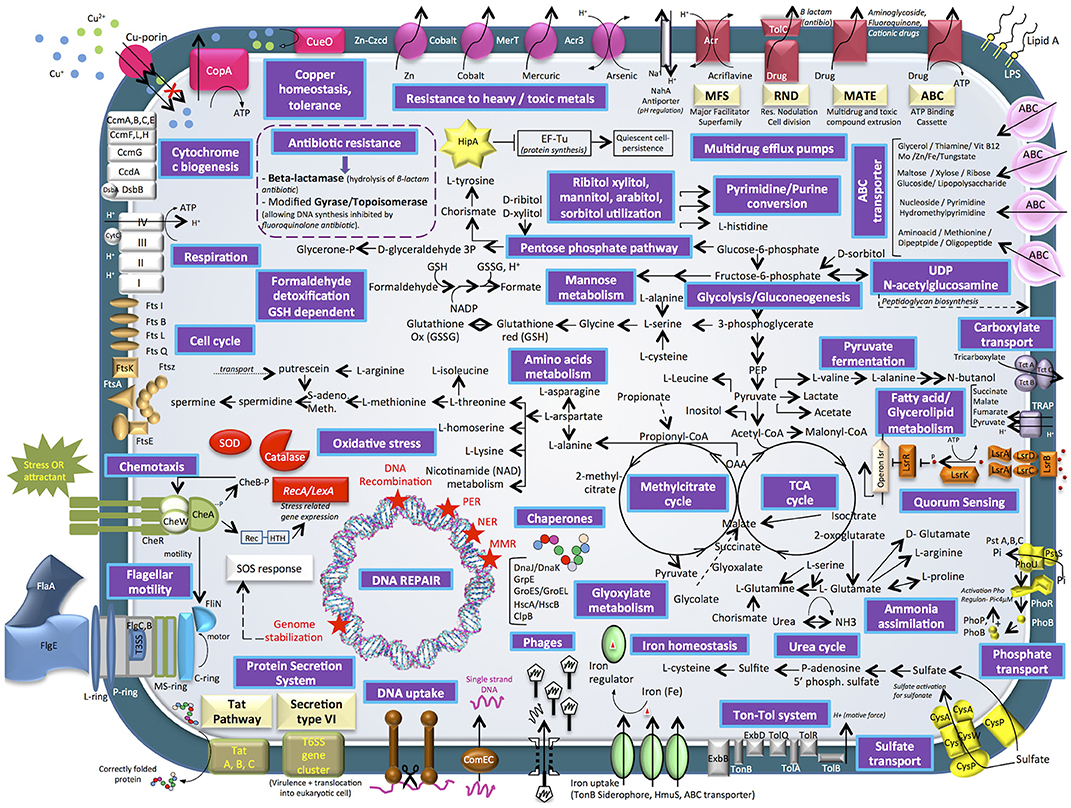
Figure 4. Cell diagram showing the main metabolism pathways of E. mobile BBCC367 obtained from the theoretical proteome.
Heavy metal tolerance to zinc, cobalt, mercury, arsenic, and copper is facilitated by the following proteins in E. mobile BBCC367: Zn- Czcd, Cobalt transporter, MerT, Acr3, CueO, and CopA pumps (Anton et al., 1999; Bondarczuk and Piotrowska-Seget, 2013; Hobman and Crossman, 2015; Ladomersky and Petris, 2015). E. mobile BBCC367's theoretical proteome harbors 12 proteins involved in copper resistance including a copper translocating protein (CopA), a multi-copper oxidase (CueO) that confers copper tolerance (Cooksey, 1993). Another type of bacterial resistance to toxic compounds includes multidrug efflux pumps including MFS, RND, MATE, and ABC transporters. Toxic compounds removed through this system include acrilavine, beta-lactam, aminoglycoside, fluoroquinone, and cationic drugs (Figure 4).
Life in Nutrient-Limited Environments
E. mobile BBCC367 has many pathways involved in ammonia, phosphate, and sulfate assimilation (Figure 4) thus making E. mobile BBCC367 efficient at utilizing the available nutrients. E. mobile BBCC367's theoretical proteome harbors hipA gene involves in the dormancy or quiescence to allow survival in adverse conditions, commonly triggered by a lack of nutrients (Rittershaus et al., 2013). HipA is a kinase, which phosphorylates and inactivates the translation factor EF-Tu (Figure 4, yellow star), facilitating persistence or quiescence (Dawson et al., 2011).
Quorum Sensing and Chemotaxis
Generalist bacteria are enriched in genes involved in motility (N), defense mechanisms (V), and signal transduction (T) (Lauro et al., 2009). Quorum sensing (QS) is a density-dependent bacterial communication mechanism that functions by secretion and detection of small signaling molecules called auto-inducers (AIs). In E. mobile BBCC367, lipolysis-stimulated lipoprotein receptor (LSR) family proteins facilitate QS (depicted in orange, Figure 4). LsrA, LsrB, LsrC and LsrD (mid right of Figure 4) are all protein compounds, which allow extracellular detection of autoinducers (Hooshangi and Bentley, 2011) whereby the lsrACDB genes encode the ABC transporters allowing the import of autoinducers (Taga et al., 2001). In many Gram-negative bacteria QS is regulated by the LuxI/LuxR type system where LuxR senses the autoinducer (Taga et al., 2003). Different types of autoinducers were identified as being produced and sensed in bacteria (LaSarre and Federle, 2013). Within the Roseobacter clade some Ruegeria species including R. atlantica were shown to produce and detect N-acyl homoserine lactone (AHL) signaling molecules (Mohamed et al., 2008). In E. mobile BBCC367, autoinducer 2 (AI-2), another type of signaling molecule, was characterized. AI-2 (Figure 4) facilitates interspecies cell-cell signaling, motility and chemotaxis (Marques et al., 2011). Chemotaxis is a way of bacteria to respond to external stimuli that may either be attractants or repellents. The stimulus is detected via chemoreceptors, which are transmembrane proteins. In E. mobile BBCC367 the complex of histidine protein kinase CheA and linker proteins CheW allows chemotactic modulation of flagellar activity depending on environmental stimuli (Wang et al., 2010).
DNA Repair Mechanisms
A total of 45 genes were found to be involved in oxidative stress defense and five DNA repair mechanisms: nucleotide excision repair (NER), mismatch repair proteins (MMR), photoenzymatic repair (PER), DNA recombination and genome stabilization (Friedberg, 2003) (Figure 4, red stars indicate DNA repair mechanisms). This suggests that E. mobile BBCC367 can cope with a variety of different stresses. Interestingly, this strain was found to be capable of single strand DNA uptake to undergo genetic transformation. DNA uptake can be useful to enhance the genetic diversity (e.g. acquisition of metabolic functions, virulence traits or antibiotic resistance), for DNA repair or as source of carbon, nitrogen, and phosphorus (Chen and Dubnau, 2004).
Phage-Related Genes
The E. mobile BBCC367's theoretical proteome harbors 25 predicted phage genes including five lysozyme prophages. Viruses represent the largest source of genetic material on the planet, and are likely the major vehicle for gene transfer in the oceans (Duhaime et al., 2011). According to Paul (2008), half of the cultivable marine bacteria contain phage-like particles by prophage induction, allowing the repression host growth in times of resource partitioning. The phage shock protein system (Psp) is responsible for regulating proton motor protein force in conditions exerting stress on the inner membrane of bacteria (Jovanovic et al., 2010). PspA was found to be relevant in response to different conditions (8/16, Table S1) including heavy metal stress.
Proteomic Investigation of E. Mobile BBCC367
A total of 3,083 proteins were classified into three different proteomes according to their protein expression patterns under the 16 different conditions: (i) core proteome (584 proteins) (ii) accessory proteome (2,144 proteins) and (iii) unique proteome (355 proteins). The core proteome is the set of proteins, which were expressed in all 16 conditions and is the minimal set of proteins required to sustain life (Yang et al., 2015). The accessory proteome refers to the situational or adaptive set of proteins, which were expressed under at least 2 conditions. Finally, the unique proteome refers to those proteins, which were expressed in a single condition and are highly situational and specifically adaptive. Most proteins identified in E. mobile BBCC367 (the accessory and unique proteomes, 81% of expressed proteome) were subject to change in expression, whereas relatively few proteins were constitutively expressed (core proteome, 19% of the expressed proteome).
Core Proteome (584 Proteins)
Major differences between the core and accessory proteomes include the greater prominence of COGs E (amino acid transport and metabolism) and J (Translation, ribosomal structure, and biogenesis) and an under-representation of hypothetical proteins (COG S—function unknown and COG R—general function predicted only) in the core proteome (Figure 5). This indicates the importance of translation/ribosomal proteins and ribonucleases that control the rates of RNA maturation/decay, which influences the rate of encoded protein synthesis (Nicholson, 1999). Furthermore, COG C (energy production and conversion) and COG O (post translational modifications, protein turnover and chaperones) were also more prominent in the core proteome than the accessory proteome (Figure 5). The prominence of COG C facilitates utilization of diverse carbohydrate sources in E. mobile BBCC367. Protein chaperones function to prevent protein aggregation and efficient protein folding (Hartl et al., 2011) and play an important role in protecting cells from different stress conditions (Skorko-Glonek et al., 2008). Seven protein chaperones were identified in the core proteome of E. mobile BBCC367 (Table S1) including HtrA (EPIB1_1000), chaperone protein DnaK (EPIB1_180), chaperone protein DnaJ (EPIB1_179), heat shock protein 60 family co-chaperone GroES (EPIB1_2287), and heat shock protein 60 family chaperone GroEL (EPIB1_2286). Survival protein SurA precursor (EPIB1_1904) was identified in E. mobile BBCC367's core proteome and was found to be involved in correctly folding outer membrane proteins (Lazar and Kolter, 1996).
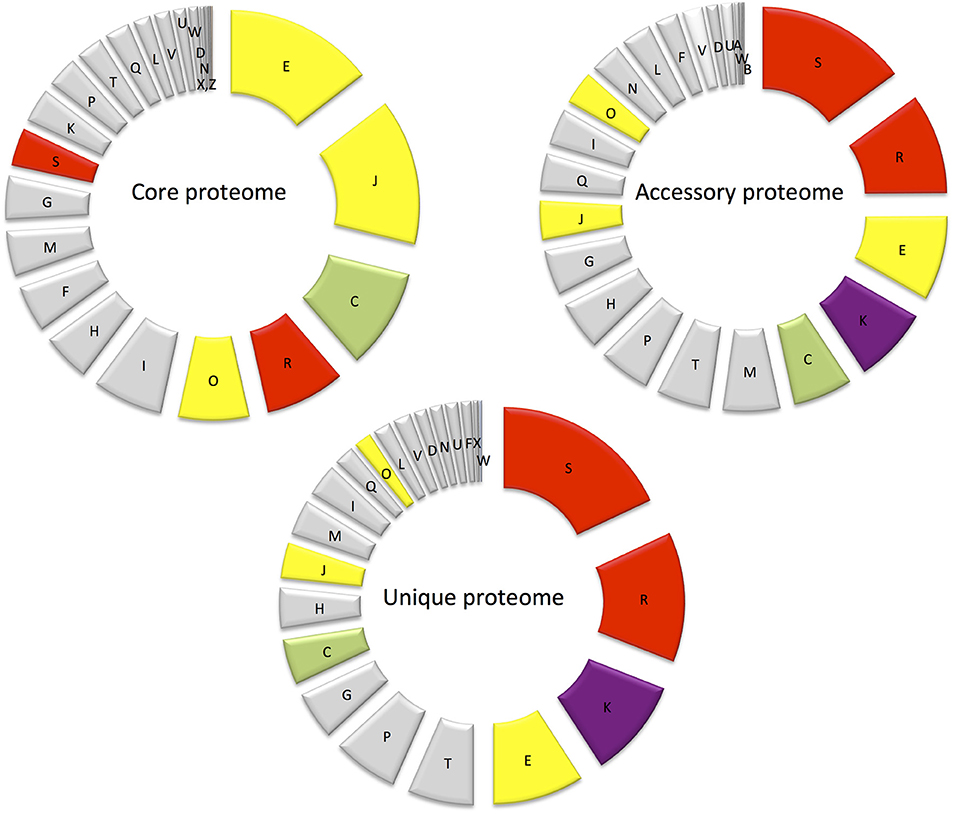
Figure 5. Diagram showing the abundance of proteins in each COG in the core proteome, randomly expressed and unique proteomes.
In E. mobile BBCC367's core proteome 54 ATP-dependent transporters (Table S1) were identified, indicating the significance of ATP hydrolysis for transportation of substrates. Eight TRAP transporters were found in the core proteome (Table S1) suggesting they play a crucial role in transporting essential substrates for cell functionality like ABC transporters. Universal stress protein UspA (EPIB1_2652) was also found in the core proteome (Table S1). UspA can be expressed under a large variety of stress conditions such as stationary phase, exposure to heat or metals (Nyström and Neidhardt, 1994; Kvint et al., 2003). This suggests that UspA can be involved in multiple stresses in E. mobile BBCC367 (Table S1).
COGs W (extracellular structures), Q (Secondary metabolites biosynthesis, transport, and catabolism) and I (Lipid metabolism) were not represented in the core proteome, but were only found in the accessory and unique proteomes and are thus non-essential, yet adaptive.
Accessory Proteome (2,144 Proteins)
This is the largest of the three identified proteomes in E. mobile BBCC367 and comprised proteins whose expression was facultative but could not be linked to dedicated conditions. Still, some non-random patterns could be identified across the 16 tested conditions (Figures 6, 7). We observed two main groups of conditions, showing similarities in terms of protein expression. The four following conditions namely 40°C, anoxic, 5 mM copper and filtered sea water showed significant differences in the pattern of the expressed proteome compared to other conditions (Figure 6). Overall E. mobile BBCC367 would repress the expression of many genes under those four conditions that we could qualify as the most stressful conditions. Indeed high temperature, high concentration of copper, lack of oxygen, or low nutrient concentration (filtered seawater) would lead to gene expression being repressed and/or protein degradation. We identified significantly fewer proteins in those four conditions (1,383, 1,438, 1,365, and 786 proteins for the conditions 3, 5, 9, and 11, respectively) in comparison to the other bacterial growth conditions (about 2,000 proteins) (Figure 6 and Table S2).
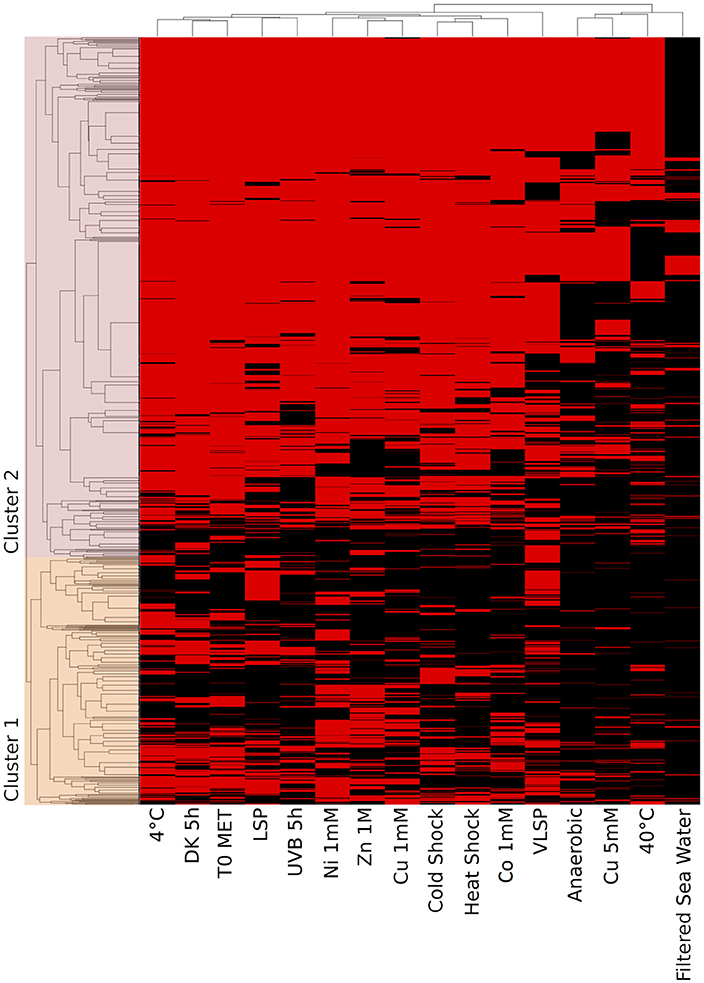
Figure 6. Hierarchical cluster analysis (HCA) of the accessory proteome. The heat map is linked by a dendrogram representing clustering of the different experimental treatments (top), and protein expression profiles (side). Color code: Black: protein not identified/Red: protein identified by mass spectrometry.
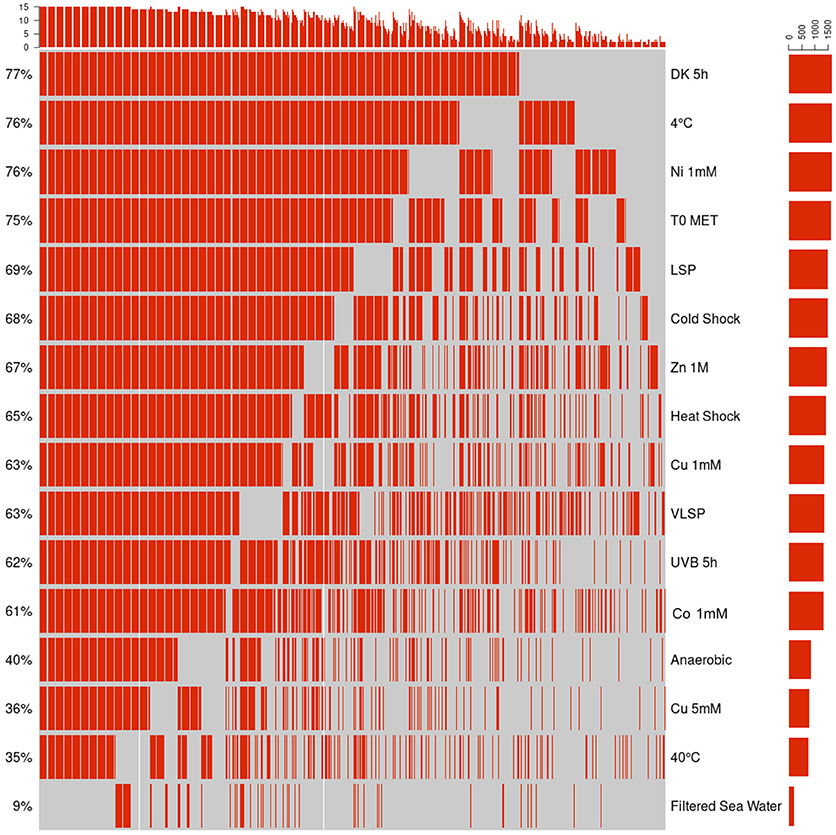
Figure 7. Qualitative visualization (oncoprint) of the accessory proteome. The x-axis represents the identified proteins (red, non-identified proteins in gray), the y-axis the different conditions.
Regarding the grouping according to the protein function, we observed two main branches that divided two groups of proteins (Clusters 1 & 2, Figure 6). The expression of the proteins from Cluster 1 was found to be more variable from one condition to another and this group of proteins was over-represented by energy related proteins (COG C) and amino acid transporters (COG E). Proteins from Cluster 2 were found to be more constitutively expressed across all conditions. This group was dominated by proteins involved in transcription (COG K) and hypothetical proteins (COG S).
A total of 11 proteins involved in DNA repair were found in the accessory proteome including DNA mismatch repair protein MutS (EPIB1_13), MutL (EPIB1_285), DNA repair protein RecN, (EPIB1_2179) and LexA (EPIB1_1683) with RecA expressed in E. mobile BBCC367's core proteome (Table S1). MutS, MutL, RadA and RecN were all expressed in at least 10 conditions with MutS, LexA, and RecN expressed in 15/16 conditions (Table S1). The expression of these proteins in E. mobile BBCC367 demonstrate that DNA repair strategies and the SOS response can provide resistance under different stresses, thus maintaining the integrity of the genome in E. mobile BBCC367 as early as possible in the DNA damage process.
Unique Proteome (355 Proteins)
This is the smallest of the three proteomes defined in E. mobile BBCC367. Proteomic investigation of the unique proteome can infer information on particular protein functions associated with specific environmental conditions. COGs S (unknown function) and R (general function) are the largest contributors of the unique proteome followed by COG K (transcription) as shown in Figure 5. Regulation at the level of transcription is less energetically costly than regulation at the level of translation as transcriptional regulation prevents the production of unnecessary proteins. For clarity purposes, the unique proteome obtained for each individual condition will be discussed according to the three main stressor groups: temperature (90 proteins), nutritional limitation (103 proteins), and metals toxicity (162 proteins) (Table S1).
Proteins Expressed Under Temperature Related Conditions
A total of 31 proteins involved in transcriptional regulation were identified in the unique proteome of E. mobile BBCC367 (S1) (9.2% of the unique proteome). The LysR-type transcriptional regulator is the largest family of transcription regulators in prokaryotes (Santiago et al., 2015), having a regulatory role over genes whose products can be involved in metabolism, cell division, QS, secretion and oxidative stress response (Maddocks and Oysten, 2008). Ten LysR-type proteins were identified in E. mobile BBCC367's unique proteome (S1), including: transcriptional regulator, LysR family (EPIB2_971), three of which were expressed in condition 3 (40°C). These transcriptional regulators most likely play a role in tolerating higher temperatures (40°C), by controlling protein expression. Interestingly, several proteins of the flagellum were found exclusively in the temperature-related conditions. Temperature impacts the motility by changing the polarity of the membrane (Lewus and Ford, 1999). In the unique proteome six proteins involved in motility/QS/chemotaxis were identified: flagellar M-ring protein FliF (EPIB1_88), flagellar hook-associated protein FlgK (EPIB1_82) and autoinducer-binding transcriptional regulator LuxR (EPIB2_776). LuxR is an autoinducer-dependent activator of transcription of the lux operon to activate the signal transduction pathway for QS (Qin et al., 2007). The expression of FliF and FlgK in the unique proteome indicates that they play an important role in flagellar motility/function (which may coincide with chemotaxis strategies) under different temperature conditions.
Proteins Expressed Under Nutrient Limitation
During starvation (LSP/vLSP condition, Table 1) bacterial cells strive to uptake nutrients from the limited environment and thus, proteins actively involved in the transport of nutirents are key for cell survival (Helloin et al., 2003). We identified a significant predominance of ABC transporters specialized in the transport of oligopeptides, sugar, polyamine, tricarboxylate as well as several TRAP transporters that are less energetically costly. Eleven ABC transporters and three TRAP transporters were identified in E. mobile BBCC367's unique proteome as being expressed under conditions 6 and 7 (LSP and vLSP). A range of ABC transporters were expressed including a sugar ABC transporter (EPIB1_641), a histidine ABC transporter (EPIB2_734) and an oligopeptide ABC transporter, the periplasmic oligopeptide-binding protein OppA (PEPIB1_22). This suggests that a wide range of molecules could be transported in conditions 6 and 7 (LSP and vLSP). Other transporters including such as the oligopeptide transport system permease protein OppC and the phosphate homeostasis/uptake protein PhoU (EPIB2_537 and EPIB1_1570, respectively), as well as a few membrane/transmembrane proteins denoted to COG R (general function) were also expressed in conditions 6 and 7. The abundance of transporters and especially ABC transporters was also identified in R. pomeroyi in nutrient-poor conditions (Christie-Oleza and Armengaud, 2010). It is therefore suggested that E. mobile BBCC367 can withstand nutrient limiting conditions through the expression of different membrane transporter proteins and other general membrane proteins.
Proteins Expressed Under Metal Related Conditions
The minimum inhibitory concentration (MIC) in E. mobile BBCC367 for the 4 tested metals was 1 mM for Zinc, 2 mM for Cobalt, 4 mM for Nickel and 5 mM for Copper (Figure 8). In line with those results, the largest proportions of the unique proteomes related to metal stresses were dedicated to nickel, zinc and copper tolerance implying that resistance to nickel, zinc and copper at low concentration (1 mM) demands the greatest physiological change (Table S1). Active transport or efflux systems represent the largest category of metal resistance systems (Nies, 1999; Bruins et al., 2000). In E. mobile BBCC367, fourteen ABC transporters were characterized when the cells were grown in presence of metals as well as a mercuric transport protein MerT (PEPIB2_188), twin-arginine translocation protein TatC (EPIB1_1226) and cobalt-zinc-cadmium resistance protein CzcD (PEPIB2_31). CzcD was only expressed in condition 12 (Zn 1mM), which indicates that CzcD has an important role in providing resistance and tolerance to zinc in E. mobile BBCC367. Two copper translocating P-type ATPases were found to be expressed in Copper, Zinc, and Nickel (1 mM) conditions. ATPases including P-type ATPases have crucial roles in heavy metal resistance and constitute the basic defense against heavy metal cations (Nies, 2003). Those proteins would play a key role in detoxifying metals via efflux transport. In E. mobile BBCC367, the blue copper oxidase CueO precursor (PEPIB2_173) was identified in condition 10 (Cu 1 mM), however not in condition 11 (Cu 5 mM) (Table S1). Higher concentrations of copper cause lipid peroxidation and protein damage (Dupont et al., 2011) and might have altered CueO at 5 mM in E. mobile BBCC367. Indeed as presented in Figure 8, bacterial growth was significantly impacted by the highest concentration of copper of 5 mM. Two other multicopper oxidases were expressed in both conditions 10 and 11 (Cu 1 mM and Cu 5 mM) (Table S1) allowing resistance of E. mobile BBCC367 to copper. Both copper resistance protein B's and copper homeostasis protein CutE were expressed in both conditions 10 and 11 (Cu 1 mM and Cu 5 mM) (Table S1). Copper chaperone CopZ, a metal chaperone involved in zinc homeostasis was identified in the copper and nickel conditions, respectively and would allow an effective storage of copper ions. Colony forming unit (CFU) on agar plate containing copper showed a change of color from orange to dark brown due to the storage of the copper inside the cells (Figure 9).
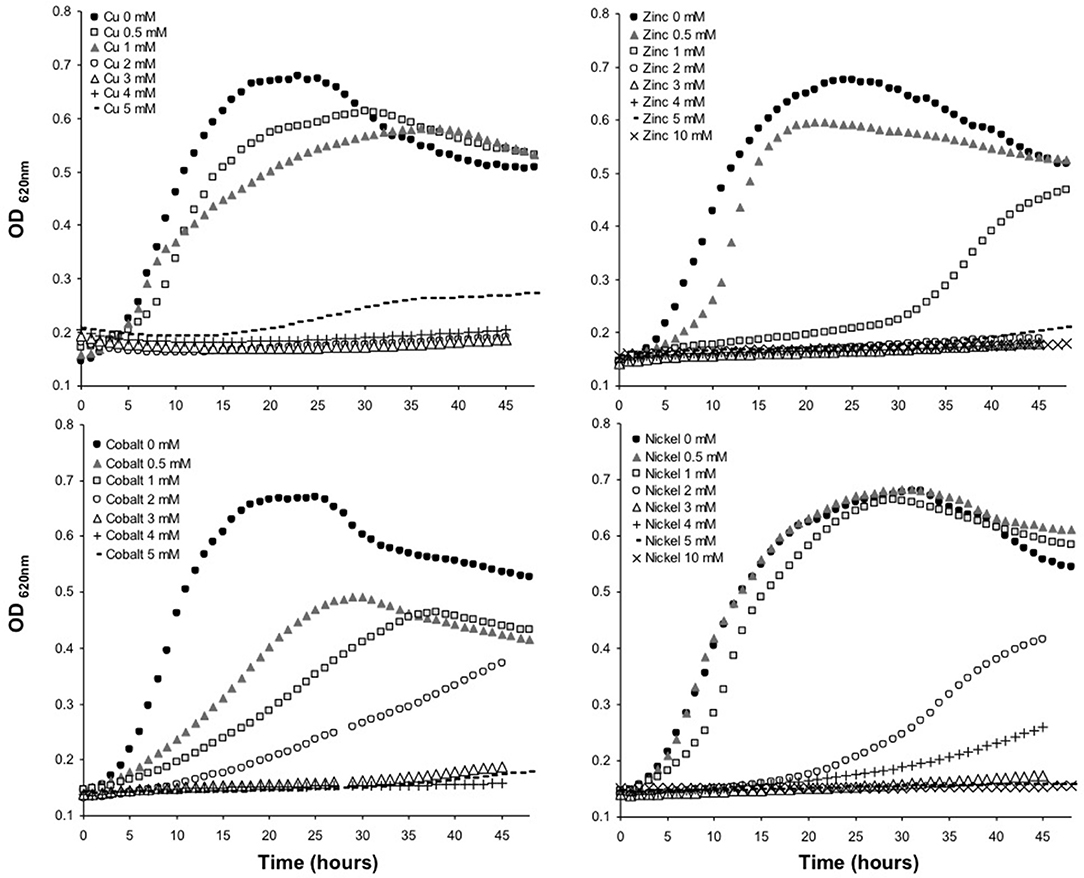
Figure 8. Growth curves of E. mobile BBCC367 in presence of different concentration of copper, zinc, nickel and cobalt.
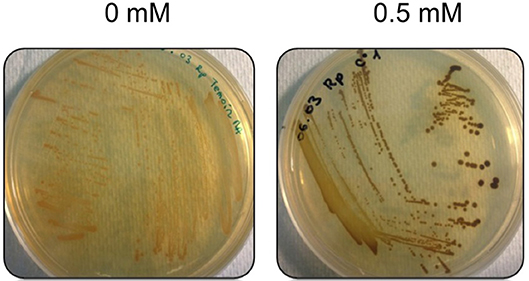
Figure 9. Morphotypes of E. mobile BBCC367 in presence of copper.
Finally, the unique proteome of E. mobile BBCC367 showed that transcriptional regulators are key actors in the resistance to metals. 14 transcriptional regulators including LysR-family transcriptional regulators were identified as being expressed under metal stress conditions (Table S1). LysR-family transcriptional regulators were previously identified to be important in cellular survival upon metal treatments (Latorre et al., 2014; Santiago et al., 2015).
Impact of UVB Radiation on E. mobile BBCC367 Assessed by Quantitative Proteomics
By quantitative proteomics, a total of 65 proteins were quantified, out of which 22 were up-regulated (gray), 11 were down-regulated (white) and 32 showed no significant differential regulation (not shown) following UV exposure compared to the dark control (Table 3).
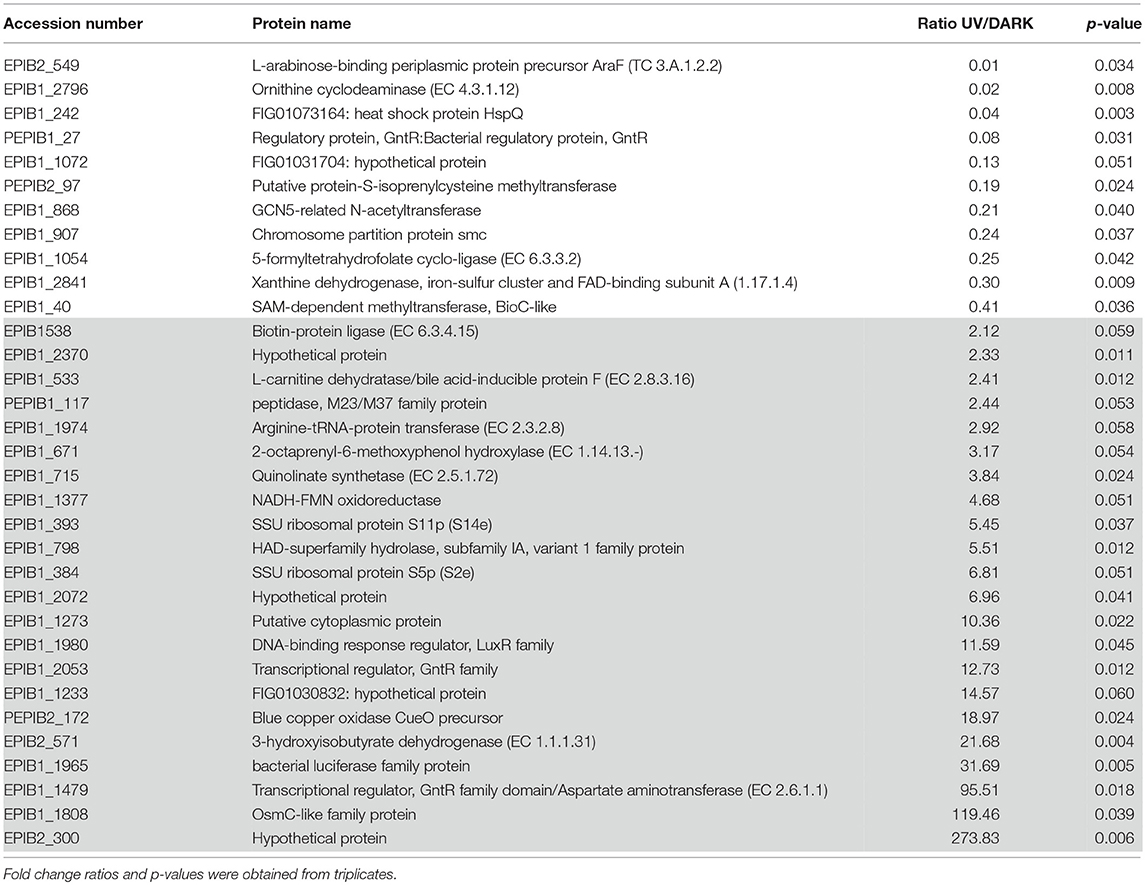
Table 3. List of the quantified proteins differentially regulated between the UV stress and Dark control conditions (white: down-regulated proteins/gray: up-regulated proteins).
Transcription and Translation
Transcription factors belonging to the GntR family were identified as being involved in extreme resistance to gamma radiation, UV radiation and ROS response in Deinococcus radiodurans and Rhodobacter sp. (Dulermo et al., 2015; Pérez et al., 2017). Three GntR family-associated proteins were quantified in E. mobile BBCC367: GntR: bacterial regulatory protein (PEPIB1_27; ratio value 0.08), transcriptional regulator, GntR family (EPIB1_2053; ratio value 12.73) and transcriptional regulator, GntR family domain/aspartate aminotransferase (EPIB1_1479; ratio value 95.51). Although GNAT proteins transferring acetyl groups to a wide variety of substrates (Favrot et al., 2016) are known to be involved in stress response (Xie et al., 2014), the GCN5-related N-acetyltransferase (GNAT) (EPIB1_868) was found to be down-regulated (ratio value 0.21) under UVB radiation (Table 3). Methyltransferases including the putative protein-S-isoprenylcysteine methyltransferase and the BioC-like SAM-dependent methyltransferase were both down-regulated under UVB radiation (Table 3). Methyltransferases are associated with DNA methylation, heterochromatin formation, and repression of DNA synthesis. In the absence of UVR, these methyltransferases were more abundant, potentially being involved in heterochromatin formation of UV-related genes. Finally, ribosomal proteins (EPIB1_393 and EPIB1_384; ratio values 5.45 and 6.81, respectively) were found to be up-regulated upon UVB radiation such as in Photobacterium angustum S14 (Matallana-Surget et al., 2012a) and in Sphingopyxis alaskensis (Matallana-Surget et al., 2009). Ribosomal proteins play a role in UV resistance in E. mobile BBCC367 by maintaining translational/ribosomal stability.
Metabolism and Amino Acid Transport
The L-arabinose-binding periplasmic protein precursor AraF (EPIB2_549; ratio value 0.01), and ornithine cyclodeaminase (EPIB1_2796; ratio value 0.02) both involved in amino acid transport pathways, showed the lowest ratio values in E. mobile BBCC367 and significantly low ratio values compared to previous UV quantitative proteomic studies (Matallana-Surget and Wattiez, 2013). Proteins involved in amino acid synthesis in P. angustum S14 were also down-regulated under UVB treatment (Matallana-Surget et al., 2012a).
The arginine-tRNA-protein transferase exhibited a 2.92 fold change in protein abundance in the UV stress condition compared with the dark control. This protein contributes to protein degradation pathways and thus, may be up-regulated to degrade UV-damaged proteins.
Two proteins involved in folate metabolism (5-formyltetrahydrofolate cyclo-ligase; EPIB1_1054; ratio value 0.25 and xanthine dehydrogenase, iron-sulfur cluster and FAD-binding subunit A; EPIB1_2841; ratio value 0.30) were down-regulated in response to UVB radiation (Table 3), confirming former studies showing UV stressed cells would preferentially use energy for particular UV resistance mechanisms rather than general metabolic processes (Matallana-Surget et al., 2012a; Matallana-Surget and Wattiez, 2013). The oxydoreductase, 3-hydroxyisobutyrate dehydrogenase (EPIB2_571), which carries out valine, leucine and isoleucine degradation and is involved in the production of acetyl-CoA was found to be up-regulated in the dark condition (0.41 UV: dark ratio). Similarly in Chlorella variabilis, the 3-hydroxyisobutyrate dehydrogenase was down-regulated in response to UVR stress (Poong et al., 2018). The relatively higher expression of 3-hydroxyisobutyrate dehydrogenase in the dark condition causes a reduction in valine, leucine, and isoleucine, this is known to stimulate protein degradation and inhibit protein synthesis (Freund and Hanani, 2002). Biotin-protein ligase (EPIB1_538; ratio value 2.12) was found to be up-regulated under UVB radiation in E. mobile BBCC367 (Table 3). Biotin is an essential co-factor aiding the replenishment of the tricarboxylic acid cycle and amino acid metabolism (Salaemae et al., 2016). Thus, the up-regulated biotin-protein ligase is likely to maintain the functionality of these metabolic processes under UV stress. Quinolinate synthetase (EPIB1_715) also known as NadA in E. coli (Ollagnier-de Choudens et al., 2005) was up-regulated (ratio value 3.84) in response to UVB radiation (Table 3). This protein is involved in the NAD biosynthesis pathway and could lead to an increase amount of reduced NADH in E. mobile BBCC367, which allows for the pumping of sodium ions and an increase in ATP synthesis (Matallana-Surget et al., 2012a).
Proteins Involved in Oxidative Stress Response
The OsmC-like family protein (EPIB1_1808) was found to be up-regulated (ratio value 119.46) in E. mobile BBCC367 upon exposure to UVB radiation compared to the dark control (Table 3). The OsmC protein was shown to have a key function in oxidative stress defense (Dubbs and Mongkolsuk, 2007).
Interestingly the blue copper oxidase CueO precursor (PEPIB2_172), involved in periplasmic detoxification of copper, was up-regulated (ratio value 18.97) under UVB radiation compared to the dark control (Table 3). The periplasmic multicopper oxidase CueO was reported to be involved in copper homeostasis and protection against oxidative stress in E. coli (Grass et al., 2004). Copper oxidizes catechols, which leads to the production of hydrogen peroxide and hydroxyl radicals, and catechols are able to reduce Cu2+ to Cu+ (Grass et al., 2004). Overall, these reactions lead to increased levels of ROS and increased intake of toxic Cu+. CueO can reduce the amount of Cu+ and thus, the accumulation of ROS (Grass et al., 2004). Blue copper oxidase CueO precursor is proposed to play an important role in reducing the amount of oxidative stress by reducing the amount of UV induced ROS in E. mobile BBCC367.
Conclusion
Our proteogenomic analysis allowed us to identify almost 70% of the E. mobile BBCC367 theoretical proteome, and revealed that 81% of the proteins were subject to changes in expression depending on growth conditions, whereas 19% were constitutively expressed. E. mobile BBCC367 tolerates and resists environmental stress including temperature, heavy metal exposure, nutrient deprivation and UVB radiation. Cell envelope biogenesis proteins were the only specific class of proteins expressed in response to all tested metals, indicating holistic importance of cell envelope biogenesis in metal tolerance. Findings from the late stationary phase revealed amino acid metabolism and transport proteins to be highly important in the limited environment associated with late stationary phase growth. Proteins involved in transcription/translation regulation and amino acid transport were found to be essential to favor the resistance of E. mobile BBCC367 under UVR. The diversity of stress responses analyzed in this study provides a valuable platform for future more targeted in-depth studies.
Author Contributions
SM-S conceived and designed the experiments. SM-S and LI performed the experiments. PNG and MF genome sequencing. SM-S, JW, KL, CR, JM, DG and SIR analyzed the data. SM-S, HT, PL, and RW contributed reagents, materials, and analysis tools. SM-S, CR, JM, and KL wrote the paper. SM-S, JW, HT, and LI proofreading.
Funding
This work was supported by the European community project MAMBA (FP7-KBBE-2008-226977). PNG thanks ERA Net IB2 Project MetaCat through UK Biotechnology and Biological Sciences Research Council (BBSRC) Grant BB/M029085/1 and the support of the Centre of Environmental Biotechnology Project part-funded by the European Regional Development Fund (ERDF) through the Welsh Government. SM-S and DG also gratefully acknowledge a Scottish Crucible Award (MicMAc project).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.03125/full#supplementary-material
References
Agogué, H., Joux, F., Obernosterer, I., and Lebaron, P. (2005). Resistance of marine bacterioneuston to solar radiation. Appl. Environ. Microbiol. 71, 5282–5289. doi: 10.1128/AEM.71.9.5282-5289.2005
Alonso-Saez, L., Gasol, J. M., Lefort, T., Hofer, J., and Sommaruga, R. (2006). Effect of natural sunlight on bacterial activity and differential sensitivity of natural bacterioplankton groups in northwestern Mediterranean coastal waters. Appl. Environ. Microbiol. 72, 5806–5813. doi: 10.1128/AEM.00597-06
Anton, A., Grosse, C., Reissmann, J., Pribyl, T., and Nies, D. H. (1999). CzcD Is a heavy metal ion transporter involved in regulation of heavy metal resistance in Ralstonia sp. Strain CH34. J. Bacteriol. 181, 6876–6881.
Armengaud, J. (2012). Microbiology and proteomics, getting the best of both worlds!. Environ. Microbiol. 15, 12–23. doi: 10.1111/j.1462-2920.2012.02811.x
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Bondarczuk, K., and Piotrowska-Seget, Z. (2013). Molecular basis of active copper resistance mechanisms in Gram-negative bacteria. Cell Biol. Toxicol. 29, 397–405. doi: 10.1007/s10565-013-9262-1
Bruins, M. R., Kapil, S., and Oehme, F. W. (2000). Microbial resistance to metals in the environment. Ecotoxicol. Environ. Saf. 45, 198–207. doi: 10.1006/eesa.1999.1860
Castellana, N., and Bafna, V. (2010). Proteogenomics to discover the full coding content of genomes: a computational perspective. J. Proteomics 73, 2124–2135. doi: 10.1016/j.jprot.2010.06.007
Chen, I., and Dubnau, D. (2004). DNA uptake during bacterial transformation. Nat. Rev. Microbiol. 2, 241–249. doi: 10.1038/nrmicro844
Christie-Oleza, J. A., and Armengaud, J. (2010). In-depth analysis of exoproteomes from marine bacteria by shotgun liquid chromatography-tandem mass spectrometry: the Ruegeria pomeroyi DSS-3 case-study. Mar. Drugs 8, 2223–2239. doi: 10.3390/md8082223
Christie-Oleza, J. A., Fernandez, B., Nogales, B., Bosch, R., and Armengaud, J. (2012). Proteomic insights into the lifestyle of an environmentally relevant marine bacterium. ISME J. 6, 124–135. doi: 10.1038/ismej.2011.86
Church, M. J. (2009). The trophic tapestry of the sea. Proc. Natl. Acad. Sci. U.S.A. 106, 15519–15520. doi: 10.1073/pnas.0908881106
Claustre, H., Sciandra, A., and Vaulot, D. (2008). Introduction to the special section bio-optical and biogeochemical conditions in the South East Pacific in late 2004: The BIOSOPE program. Biogeosciences 5, 679–691. doi: 10.5194/bg-5-679-2008
Cooksey, D. A. (1993). Copper uptake and resistance in bacteria. Mol. Microbiol. 7, 1–5. doi: 10.1111/j.1365-2958.1993.tb01091.x
Dawson, C. C., Intapa, C., and Jabra-Rizk, M. A. (2011). “Persisters”: survival at the cellular level. PLoS Pathog. 7:e1002121. doi: 10.1371/journal.ppat.1002121
DeVries, T., Holzer, M., and Primeau, F. (2017). Recent increase in oceanic carbon uptake driven by weaker upper-ocean overturning. Nature 542, 215–218. doi: 10.1038/nature21068
Dubbs, J. M., and Mongkolsuk, S. (2007). Peroxiredoxins in bacterial antioxidant defense. Subcell. Biochem. 44, 143–193. doi: 10.1007/978-1-4020-6051-9_7
Duhaime, M. B., Wichels, A., Waldmann, J., Teeling, H., and Glöckner, F. O. (2011). Ecogenomics and genome landscapes of marine Pseudoalteromonas phage H105/1. ISME J. 5, 107–121. doi: 10.1038/ismej.2010.94
Dulermo, R., Onodera, T., Coste, G., Passot, F., Dutertre, M., Porteron, M., et al. (2015). Identification of new genes contributing to the extreme radioresistance of Deinococcus radiodurans using a Tn5-based transposon mutant library. PLoS ONE 10:e0124358. doi: 10.1371/journal.pone.0124358
Dupont, C. L., Grass, G., and Rensing, C. (2011). Copper toxicity and the origin of bacterial resistance–new insights and applications. Metallomics 3, 1109–1118. doi: 10.1039/c1mt00107h
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Eguchi, M., Nishikawa, T., Macdonald, K., Cavicchioli, R., Gottschal, J. C., and Kjelleberg, S. (1996). Responses to stress and nutrient availability by the marine Ultramicrobacterium Sphingomonas sp. Strain RB2256. Appl. Environ. Microbiol. 62, 1287–1294.
Ewing, B., and Green, P. (1998b). Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8, 186–194. doi: 10.1101/gr.8.3.186
Ewing, B., Hiller, L., Wendt, M. C., and Green, P. (1998a). Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8, 175–185. doi: 10.1101/gr.8.3.175
Farías, Farías, M. E., Fernández-Zenoff, V., Flores, R., Ordóñez, O., and Estévez, C. (2009). Impact of solar radiation on bacterioplankton in Laguna Vilama, a hypersaline Andean lake (4650 m). J. Geophys. Res. Biogeosci. 114:G00D04. doi: 10.1029/2008JG000784
Favrot, L., Blanchard, J. S., and Vergnolle, O. (2016). Bacterial GCN5-Related N-Acetyltransferases: from resistance to regulation. Biochemistry 55, 898–1002. doi: 10.1021/acs.biochem.5b01269
Freund, H. R., and Hanani, M. (2002). The metabolic role of branched-chain amino acids. Nutrition 18, 287–288. doi: 10.1016/S0899-9007(01)00740-7
Goloboff, P. A., Farris, J. S., and Nixon, K. C. (2008). TNT, a free program for phylogenetic analysis. Cladistics 24, 774–786. doi: 10.1111/j.1096-0031.2008.00217.x
Gordon, D. (2003). Viewing and editing assembled sequences using Consed. Curr. Protoc. Bioinformatics. 11, Unit 11.12. doi: 10.1002/0471250953.bi1102s02
Gram, L., Melchiorsen, J., and Bruhn, J. B. (2010). Antibacterial activity of marine culturable bacteria collected from a global sampling of ocean surface waters and surface swabs of marine organisms. Mar. Biotechnol. 12, 439–451. doi: 10.1007/s10126-009-9233-y
Grass, G., Thakali, K., Klebba, P. E., Thieme, D., Müller, A., Wildner, G. F., et al. (2004). Linkage between catecholate siderophores and the multicopper oxidase CueO in Escherichia coli. J. Bacteriol. 186, 5826–5833. doi: 10.1128/JB.186.17.5826-5833.2004
Gu, Z., Eils, R., and Schlesner, M. (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. doi: 10.1093/bioinformatics/btw313
Han, C. S., and Chain, P. (2006). “Finishing repetitive regions automatically with Dupfinisher,” in Proceedings of the 2006 International Conference on Bioinformatics and Computational Biology, eds H. R. Arabnia and H. Valafar (Las Vegas, NV: CSREA Press), 142–147.
Hartl, F. U., Bracher, A., and Hayer-Hartl, M. (2011). Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332. doi: 10.1038/nature10317
Helloin, E., Jänsch, L., and Phan-Thanh, L. (2003). Carbon starvation survival of Listeria monocytogenes in planktonic state and in biofilm: a proteomic study. Proteomics 3, 2052–2064. doi: 10.1002/pmic.200300538
Hobman, J. L., and Crossman, L. C. (2015). Bacterial antimicrobial metal ion resistance. J. Med. Microbiol. 64(Pt 5), 471–497. doi: 10.1099/jmm.0.023036-0
Hooshangi, S., and Bentley, W. (2011). LsrR Quorum Sensing “Switch” is revealed by a bottom-up approach. PLoS Comp Biol. 7:e1002172. doi: 10.1371/journal.pcbi.1002172
Hunter, S., Jones, P., Mitchell, A., Apweiler, R., Attwood, T. K., Bateman, A., et al. (2012). InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 40, D306–D312. doi: 10.1093/nar/gkr948
Jovanovic, G., Engl, C., Mayhew, A. J., Burrows, P. C., and Buck, M. (2010). Properties of the phage-shock-protein (Psp) regulatory complex that govern signal transduction and induction of the Psp response in Escherichia coli. Microbiology. 156(Pt 10), 2920–2932. doi: 10.1099/mic.0.040055-0
Khachumov, M. V. (2012). Distances, metrics and cluster analysis. Sci. Tech. Information Process. 39, 310–316. doi: 10.3103/S0147688212060020
Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Kucharova, V., and Wiker, H. G. (2014). Proteogenomics in microbiology: taking the right turn at the junction of genomics and proteomics. Proteomics 14, 2360–2375. doi: 10.1002/pmic.201400168
Kvint, K., Nachin, L., Diez, A., and Nyström, T. (2003). The bacterial universal stress protein: function and regulation. Curr. Opin. Microbiol. 6, 140–145. doi: 10.1016/S1369-5274(03)00025-0
Ladomersky, E., and Petris, M. J. (2015). Copper tolerance and virulence in bacteria. Metallomics 7, 957–964. doi: 10.1039/c4mt00327f
LaSarre, B., and Federle, M. J. (2013). Exploiting quorum sensing to confuse bacterial pathogens. Microbiol. Mol. Biol. Rev. 77, 73–111. doi: 10.1128/MMBR.00046-12
Latorre, M., Galloway-Peña, J., Roh, J. H., Budinich, M., Reyes-Jara, A., and Murray, B. E. (2014). Enterococcus faecalis reconfigures its transcriptional regulatory network activation at different copper levels. Metallomics 6, 572–581. doi: 10.1039/c3mt00288h
Lauro, F. M., McDougald, D., Thomas, T., Williams, T. J., Egan, S., Rice, S., et al. (2009). The genomic basis of trophic strategy in marine bacteria. Proc. Natl. Acad. Sci. U.S.A. 106, 15527–15533. doi: 10.1073/pnas.0903507106
Lazar, S. W., and Kolter, R. (1996). SurA assists the folding of Escherichia coli outer membrane proteins. J. Bacteriol. 178, 1770–1773.
Lee, J., Whon, T. W., Shin, N. R., Roh, S. W., Kim, J., and Park, S. K. (2012). Ruegeria conchae sp. nov., isolated from the ark clam Scapharca broughtonii. Int J Syst. Evol. Microbiol. 62(Pt 12), 2851–2857. doi: 10.1099/ijs.0.037283-0
Lee, K., Choo, Y. J., Giovannoni, S. J., and Cho, J. C. (2007). Ruegeria mobilis BBCC367 sp. nov., isolated from the Sargasso Sea, Atlantic Ocean. Int. J. Syst. Evol. Microbiol. 57(Pt 8), 1815–1818. doi: 10.1099/ijs.0.65032-0
Lewus, P., and Ford, R. M. (1999). Temperature-sensitive motility of Sulfolobus acidocaldarius influences population distribution in extreme environments. J. Bacteriol. 181, 4020–4025.
Luo, H., and Moran, M. A. (2014). Evolutionary ecology of the marine Roseobacter clade. Microbiol. Mol. Biol. Rev. 78, 573–587. doi: 10.1128/MMBR.00020-14
Maddocks, S. E., and Oysten, P. C. (2008). Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology 154(Pt 12), 3609–3623. doi: 10.1099/mic.0.2008/022772-0
Malykh, A., Malykh, O., Polushin, N., Kozyavkin, S., and Slesarev, A. (2004). Finishing ‘Working Draft’ BAC projects by directed sequencing with thermofidelase and fimers. Method Mol. Biol. 255, 295–308. doi: 10.1385/1-59259-752-1:295
Marques, J. C., Lamosa, P., Russell, C., Ventura, R., Maycock, C., Semmelhack, M. F., et al. (2011). Processing the interspecies quorum-sensing signal autoinducer-2 (AI-2): characterization of phospho-(S)-4,5-dihydroxy-2,3-pentanedione isomerization by LsrG protein. J. Biol. Chem. 286, 18331–18343. doi: 10.1074/jbc.M111.230227
Matallana-Surget, S., Joux, F., Raftery, M. J., and Cavicchioli, R. (2009). The response of the marine bacterium Sphingopyxis alaskensis to solar radiation assessed by quantitative proteomics. Environ. Microbiol. 11, 2660–2675. doi: 10.1111/j.1462-2920.2009.01992.x
Matallana-Surget, S., Joux, F., Wattiez, R., and Lebaron, P. (2012a). Proteome analysis of the UVB-resistant marine bacterium Photobacterium angustum S14. PLoS ONE 7:E42299. doi: 10.1371/journal.pone.0042299
Matallana-Surget, S., Villette, C., Intertaglia, L., Joux, F., Bourrain, M., and Lebaron, P. (2012b). Response to UVB radiation and oxidative stress of marine bacteria isolated from South Pacific Ocean and Mediterranean Sea. J. Photochem. Photobiol. B Biol. 117, 254–261. doi: 10.1016/j.jphotobiol.2012.09.011
Matallana-Surget, S., and Wattiez, R. (2013). Impact of solar radiation on gene expression in bacteria. Proteomes 1, 70–86. doi: 10.3390/proteomes1020070
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60
Meyer, F., Goesmann, A., McHardy, A. C., Bartels, D., Bekel, T., Clausen, J., et al. (2003). GenDB–an open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 31, 2187–2195. doi: 10.1093/nar/gkg312
Mohamed, N. M., Cicirelli, E. M., Kan, J., Chen, F., Fuqua, C., and Hill, R. T. (2008). Diversity and quorum-sensing signal production of Proteobacteria associated with marine sponges. Environ. Microbiol. 10, 75–86. doi: 10.1111/j.1462-2920.2007.01431.x
Moran, M. A., Belas, R., Schell, M. A., González, J. M., Sun, F., Sun, S., et al. (2007). Ecological genomics of marine Roseobacters. Appl. Environ. Microbiol. 73, 4559–4569. doi: 10.1128/AEM.02580-06
Mulligan, C., Fischer, M., and Thomas, G. H. (2011). Tripartite ATP-independent periplasmic (TRAP) transporters in bacteria and archaea. FEMS Microbiol. Rev. 35, 68–86. doi: 10.1111/j.1574-6976.2010.00236.x
Murtagh, F., and Contreras, P. (2011). Methods of hierarchical clustering. arXiv:1105.0121 38, 1–21. doi: 10.1007/978-3-642-04898-2_288
Nicholson, A. W. (1999). Function, mechanism and regulation of bacterial ribonucleases. FEMS Microbiol. Rev. 23, 371–390. doi: 10.1111/j.1574-6976.1999.tb00405.x
Nielsen, H., Engelbrecht, J., Brunak, S., and von Heijne, G. (1997). Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 10, 1–6.
Nies, D. H. (2003). Efflux-mediated heavy metal resistance in prokaryotes. FEMS Microbiol. Rev. 27, 313–339.
Nyström, T., and Neidhardt, F.-C. (1994). Expression and role of the universal stress protein, UspA, of Escherichia coli during growth arrest. Mol. Microbiol. 11, 537–544. doi: 10.1111/j.1365-2958.1994.tb00334.x
Ogata, H., Goto, S., Sato, K., Fujibuchi, W., Bono, H., and Kanehisa, M. (1999). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 27, 29–34.
Ollagnier-de Choudens, S., Loiseau, L., Sanakis, Y., Barras, F., and Fontecave, M. (2005). Quinolinate synthetase, an iron–sulfur enzyme in NAD biosynthesis. FEBS Lett. 579, 3737–3743. doi: 10.1016/j.febslet.2005.05.065
Pattengale, N. D., Alipour, M., Bininda-Emonds, O. R., Moret, B. M., and Stamatakis, A. (2010). How many bootstrap replicates are necessary? J. Comput. Biol. 17, 337–354. doi: 10.1089/cmb.2009.0179
Paul, J. H. (2008). Prophages in marine bacteria: dangerous molecular time bombs or the key to survival in the seas? ISME J. 2, 579–589. doi: 10.1038/ismej.2008.35
Pérez, V., Hengst, M., Kurte, L., Dorador, C., Jeffrey, W. H., Wattiez, R., et al. (2017). Bacterial Survival under Extreme UV radiation: a comparative proteomics study of Rhodobacter sp., isolated from high altitude wetlands in Chile. Front. Microbiol. 8:1173. doi: 10.3389/fmicb.2017.01173
Poong, S., Lim, P., Phang, S., Wong, C., Pai, T., Chen, C., et al. (2018). Transcriptome sequencing of an Antarctic microalga, Chlorella sp. (Trebouxiophyceae, Chlorophyta) subjected to short-term ultraviolet radiation stress. J. Appl. Phycol. 30, 87–99. doi: 10.1007/s10811-017-1124-4
Posey, J. E., Shinnick, T. M., and Quinn, F. D. (2006). Characterization of the twin-arginine translocase secretion system of Mycobacterium smegmatis. J. Bacteriol.. 188, 1332–1340. doi: 10.1128/JB.188.4.1332-1340.2006
Pruitt, K. D., Tatusova, T., Brown, G. R., and Maglott, D. R. (2012). NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 40(Database issue): D130-D135. doi: 10.1093/nar/gkr1079
Qin, N., Callahan, S. M., Dunlap, P. V., and Stevens, A. M. (2007). Analysis of LuxR regulon gene expression during quorum sensing in Vibrio fischeri. J. Bacteriol. 189, 4127–4134. doi: 10.1128/JB.01779-06
Richter, M., Lombardot, T., Kostadinov, I., Kottmann, R., Duhaime, M. B., Peplies, J., et al. (2008). JCoast - a biologist-centric software tool for data mining and comparison of prokaryotic (meta)genomes. BMC Bioinformatics 9:177. doi: 10.1186/1471-2105-9-177
Rittershaus, E. S., Baek, S. H., and Sassetti, C. M. (2013). The normalcy of dormancy: common themes in microbial quiescence. Cell Host Microbe, 13, 643–651. doi: 10.1016/j.chom.2013.05.012
Salaemae, W., Booker, G. W., and Polyak, S. W. (2016). The role of biotin in bacterial physiology and virulence: a novel antibiotic target for Mycobacterium tuberculosis. Microbiol. Spectr. 4:2015. doi: 10.1128/microbiolspec.VMBF-0008-2015
Santiago, A. S., Santos, C. A., Mendes, J. S., Toledo, M. A., Beloti, L. L., Souza, A. A., et al. (2015). Characterization of the LysR-type transcriptional regulator YcjZ-like from Xylella fastidiosa overexpressed in Escherichia coli. Protein Expr. Purif. 113, 72–78. doi: 10.1016/j.pep.2015.05.003
Skorko-Glonek, J., Sobiecka-Szkatula, A., Narkiewicz, J., and Lipinska, B. (2008). The proteolytic activity of the HtrA (DegP) protein from Escherichia coli at low temperatures. Microbiology 154(Pt 12), 3649–3658. doi: 10.1099/mic.0.2008/020487-0
Sonnenschein, E. C., Nielsen, K. F., D'Alvise, P., Porsby, C. H., Melchiorsen, J., Heilmann, J., et al. (2017). Global occurrence and heterogeneity of the Roseobacter-clade species Ruegeria mobilis. ISME J. 11, 569–583. doi: 10.1038/ismej.2016.111
Sonnhammer, E. L., Eddy, S. R., and Durbin, R. (1997). Pfam: a comprehensive database of protein domain families based on seed alignments. Proteins 38, 405–420. doi: 10.1002/(SICI)1097-0134(199707)28:3<405::AID-PROT10>3.0.CO;2-L
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Swofford, D. L. (2002). PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods), Version 4.0 b10. Sinauer Associates, Sunderland.
Taga, M., Miller, S. T., and Bassler, B. L. (2003). Lsr-mediated transport and processing of AI-2 in Salmonella typhimurium. Mol. Microbiol. 50, 1411–1427. doi: 10.1046/j.1365-2958.2003.03781.x
Taga, M. E., Semmelhack, J. L., and Bassler, B. L. (2001). The LuxS-dependent autoinducer AI-2 controls the expression of an ABC transporter that functions in AI-2 uptake in Salmonella typhimurium. Mol. Microbiol. 42, 777–793. doi: 10.1046/j.1365-2958.2001.02669.x
Tang, K., Jiao, N., Liu, K., Zhang, Y., and Li, S. (2012). Distribution and functions of TonB-dependent transporters in marine bacteria and environments: implications for dissolved organic matter utilization. PLoS ONE 7:e41204. doi: 10.1371/journal.pone.0041204
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Kiryutin, B., Koonin, E. V., et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41: doi: 10.1186/1471-2105-4-41
Tatusov, R. L., Koonin, E. V., and Lipman, D. J. (1997). A genomic perspective on protein families. Science 278, 631–637. doi: 10.1126/science.278.5338.631
Wang, Q., Zhang, Y., Yang, C., Xiong, H., Lin, Y., Yao, J., et al. (2010). Acetylation of Metabolic Enzymes Coordinates Carbon Source Utilization and Metabolic Flux. Science 327, 1004–1007. doi: 10.1126/science.1179687
Wilkens, S. (2015). Structure and mechanism of ABC transporters. F1000 Prime Rep. 7:14. doi: 10.12703/P7-14
Wirth, J. S., and Whitman, W. B. (2018). Phylogenomic analyses of a clade within the roseobacter group suggest taxonomic reassignments of species of the genera Aestuariivita, Citreicella, Loktanella, Nautella, Pelagibaca, Ruegeria, Thalassobius, Thiobacimonas, and Tropicibacter, and the proposal of six novel genera. Int. J. Syst. Evol. Microbiol. 68, 2393–2411. doi: 10.1099/ijsem.0.002833
Xie, L., Zeng, J., Luo, H., Pan, W., and Xie, J. (2014). The roles of bacterial GCN5-related N-acetyltransferases. Crit. Rev. Eukaryot. Gene Expr. 24, 77–87. doi: 10.1615/CritRevEukaryotGeneExpr.2014007988
Keywords: Epibacterium mobile, proteogenomic, roseobacter, stress response and adaptation, quantitative proteomics
Citation: Matallana-Surget S, Werner J, Wattiez R, Lebaron K, Intertaglia L, Regan C, Morris J, Teeling H, Ferrer M, Golyshin PN, Gerogiorgis D, Reilly SI and Lebaron P (2018) Proteogenomic Analysis of Epibacterium Mobile BBCC367, a Relevant Marine Bacterium Isolated From the South Pacific Ocean. Front. Microbiol. 9:3125. doi: 10.3389/fmicb.2018.03125
Received: 16 September 2018; Accepted: 03 December 2018;
Published: 21 December 2018.
Edited by:
David A. Walsh, Concordia University, CanadaReviewed by:
Brook Leanne Nunn, University of Washington, United StatesKai Tang, Xiamen University, China
Copyright © 2018 Matallana-Surget, Werner, Wattiez, Lebaron, Intertaglia, Regan, Morris, Teeling, Ferrer, Golyshin, Gerogiorgis, Reilly and Lebaron. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sabine Matallana-Surget, c2FiaW5lLm1hdGFsbGFuYXN1cmdldEBzdGlyLmFjLnVr