 Matthew J. Dorman
Matthew J. Dorman Charles J. Dorman
Charles J. Dorman- 1Wellcome Sanger Institute, Wellcome Genome Campus, Hinxton, United Kingdom
- 2Department of Microbiology, Moyne Institute of Preventive Medicine, Trinity College Dublin, Dublin, Ireland
Gram-negative enteropathogenic bacteria use a variety of strategies to cause disease in the human host and gene regulation in some form is typically a part of the strategy. This article will compare the toxin-based infection strategy used by the non-invasive pathogen Vibrio cholerae, the etiological agent in human cholera, with the invasive approach used by Shigella flexneri, the cause of bacillary dysentery. Despite the differences in the mechanisms by which the two pathogens cause disease, they use environmentally-responsive regulatory hierarchies to control the expression of genes that have some features, and even some components, in common. The involvement of AraC-like transcription factors, the integration host factor, the Factor for inversion stimulation, small regulatory RNAs, the RNA chaperone Hfq, horizontal gene transfer, variable DNA topology and the need to overcome the pervasive silencing of transcription by H-NS of horizontally acquired genes are all shared features. A comparison of the regulatory hierarchies in these two pathogens illustrates some striking cross-species similarities and differences among mechanisms coordinating virulence gene expression. S. flexneri, with its low infectious dose, appears to use a strategy that is centered on the individual bacterial cell, whereas V. cholerae, with a community-based, quorum-dependent approach and an infectious dose that is several orders of magnitude higher, seems to rely more on the actions of a bacterial collective.
Introduction
Shigella flexneri and Vibrio cholerae are Gram-negative enteric pathogens of humans, causing the diarrheal diseases dysentery and cholera, respectively. Although both cause diarrheal diseases in humans and enter via the oral route, they differ in the site of infection, the mechanisms by which they cause damage to the host and in their infectious doses. Infections by V. cholerae leading to diarrheal disease arise from ingestion of contaminated food or water and affect the small intestine, with the bacterium remaining extracellular (Fu et al., 2018). V. cholerae uses a T6SS to eliminate competing members of the host microbiota at the site of infection(Zhao et al., 2018), colonizes the cleansed zone with the help of the TCP adhesin before producing CT to alter the physiology of adjacent epithelial cells (Figure 1). V. cholerae produces CT, a potent enterotoxin that is produced in response to environmental signals that are characteristic of the lumen of the human small intestine. CT binds to GM1 gangliosides on the epithelial layer (King and van Heyningen, 1973), enters the epithelial cell where it dysregulates human adenylate cyclase by ADP-ribosylation (Cohen and Chang, 2018), causing over-production of the second messenger cyclic adenosine monophosphate (cAMP) (Chen et al., 1971; Sharp and Hynie, 1971) (Figure 1). This in turn causes watery diarrhea characterized by a rapid loss of water and electrolytes that, if unchecked, can prove fatal to the patient (De Haan and Hirst, 2004).
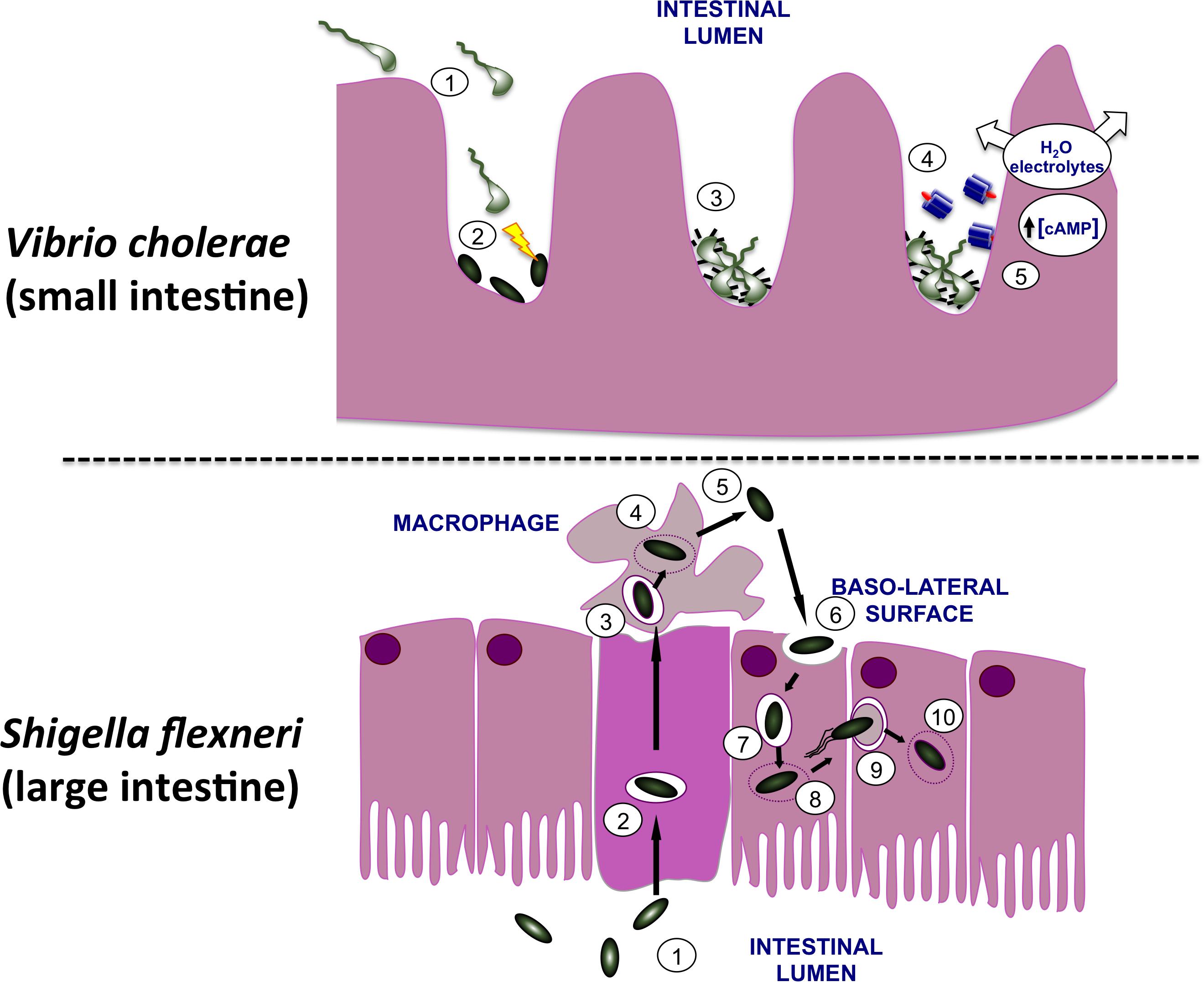
FIGURE 1. Infection of the human gut by Vibrio cholerae and Shigella flexneri. (Top) V. cholerae bacteria are shown moving from the lumen of the small intestine (1) to the surface of microvilli. Here they use their T6SS to eliminate commensal bacteria (2), creating a niche that they may then occupy on the gut wall. In response to signals that are characteristic of this niche, the bacteria produce TCP pili (3) and CT (4). The active moiety of the toxin A subunit enters the host cells and dysregulates adenylate cyclase, causing an increase in the cAMP concentration with a concomitant loss of water and electrolytes. This produces the watery diarrhea that is characteristic of cholera. (Bottom) S. flexneri bacteria (1) use their T3SS to invade the M cells of the lower gut epithelium (2). Following exit from these cells the bacteria encounter macrophage and are engulfed (3). The bacteria escape from the macrophage vacuole (4), triggering inflammatory cell death (5) and then invade gut epithelial cells via the basolateral surface (6). Once in the cell, S. flexneri escapes from the vacuole (7, 8), recruits and polymerizes host actin (9) and moves along the epithelial layer, cell by cell, escaping from each membrane double-envelopment in turn (10). The resulting damage to the epithelial layer produces an inflammatory response that amplifies the damage, causing the bloody diarrhea that characterizes dysentery.
In contrast, S. flexneri invades the M cells in the epithelium of the lower gut (Wassef et al., 1989; Perdomo et al., 1994). Next, S. flexneri undergoes transcytosis to the M cell pocket where it encounters and is engulfed by macrophage (Figure 1). The bacterium escapes from the macrophage vacuole by disrupting it, multiplies in the cytosol before inducing inflammatory cell death (Zychlinsky et al., 1992). The released S. flexneri bacteria invade nearby epithelial cells through their basolateral surfaces, promoting actin cytoskeleton rearrangements that result in engulfment of the bacteria (High et al., 1992). This produces bacteria-containing vacuoles that are soon ruptured (Weiner et al., 2016) with the released bacteria inducing actin polymerization at one pole of each microbial cell to achieve motility in the host cell cytosol (Mauricio et al., 2016). The bacterium is capable of moving from one cell to the next via clathrin-dependent endocytosis (Fukumatsu et al., 2012), escaping from the double-membrane-enclosed vacuole after each new invasion (Welch and Way, 2013). This damages the epithelium, induces an inflammatory response and results in the bloody diarrhea that is characteristic of dysentery (Ashida et al., 2015) (Figure 1).
This article will focus on the genetics of serogroup O1 and O139 V. cholerae biotype El Tor, which are responsible for the seventh cholera pandemic, and form a single phylogenetic lineage, 7PET (Domman et al., 2017). Although there are over 200 serogroups of V. cholerae (Shimada et al., 1994; Chapman et al., 2015), only serogroups O1 and O139 cause epidemic cholera. It should be noted that two biotypes of toxigenic V. cholerae O1, classical and El Tor, have been described, and there are differences in virulence gene expression between the two (Beyhan et al., 2006). Notably, classical strains of V. cholerae express virulence genes under standard laboratory conditions, and produce CT when cultured in rich media. By contrast, El Tor V. cholerae produce CT only when cultured under stringent in vitro growth conditions (Iwanaga and Yamamoto, 1985; Iwanaga et al., 1986). However, laboratory strains of both classical and El Tor V. cholerae have been used to dissect the regulatory systems described in this article.
Shigella flexneri and V. cholerae differ in the sizes of the doses required to initiate a successful infection. In the case of S. flexneri, the infectious dose can be as low as 10 cells whereas V. cholerae requires between 103 and 108 cells (Schmid-Hempel and Frank, 2007). It has been suggested that the presence of an extreme acid resistance (XAR) system in S. flexneri and its absence from V. cholerae may help to explain the difference in infectious dose: fewer V. cholerae bacteria will survive the transit through the low pH environment of the stomach, so a high infectious dose is necessary to establish an infection in the gut (Lund et al., 2014). The difference in infectious dose is also consistent with the important role that is played by cell-to-cell communication in V. cholerae compared with the more individualistic style that characterizes S. flexneri infection. In each pathogen, the genes encoding the principal virulence factors are only produced when the bacterium arrives at the correct location in the host. This indicates that the bacteria use physical and/or chemical information from the host to make a decision at the molecular level to produce or to repress production of virulence and colonization genes. In both examples, many genes have to be regulated collectively and their expression has to be finely controlled in space and time. In both cases, mobile genetic elements carry important virulence genes and their relationships with the bacterial chromosome have a significant influence on the stability of these genes and their expression.
Horizontal Gene Transfer and Virulence in S. flexneri and V. cholerae
Shigella flexneri and V. cholerae both show evidence of an important role for horizontal gene transfer in the their evolution as pathogens. Many of the virulence genes are A + T-rich compared to the A + T content of the core genome, the transcription of these genes is silenced by the H-NS DNA binding protein and important genes are located on genetic elements that are either currently mobile and transmissible, or have been in the past (Buchrieser et al., 2000; Beloin and Dorman, 2003; Ayala et al., 2015a). In the case of S. flexneri, the majority of the virulence genes are carried on a large plasmid that is a genetic mosaic made from precursor plasmids and transposable elements (Buchrieser et al., 2000; Dorman, 2009) while many prominent virulence genes in V. cholerae are found on pathogenicity islands (Karaolis et al., 1998, 1999; Faruque et al., 2003) or in a filamentous bacteriophage (Waldor and Mekalanos, 1996). These features are explored in the following sections.
S. flexneri Has Plasmid-Based Virulence Genes
Shigella flexneri uses a T3SS to inject effector proteins into host cells that produce the alterations to the actin cytoskeleton that are a characteristic of the invasive disease. Other virulence proteins help to create the actin comet tails that generate the movement of the bacteria through the host cytoplasm. Most of the principal T3SS-associated virulence genes are located on a large (213 kbp) plasmid called pINV, with those involved in the production of the T3SS being grouped in a so-called Entry Region (Figure 2) that is actually a pathogenicity island (Buchrieser et al., 2000; Lan et al., 2001; Dorman, 2009). The plasmid is capable of integrating into the chromosome, where expression of the virulence genes is silenced (Zagaglia et al., 1991; Colonna et al., 1995; Pilla et al., 2017). As virulence gene expression on the plasmid has been associated with its structural and genetic instability (Schuch and Maurelli, 1997), integration accompanied by silencing may be a strategy to ensure the vertical transmission of the virulence plasmid with the genes intact. Post-segregational killing by pINV-encoded toxin/anti-toxin systems also favors maintenance of the autonomously replicating plasmid by eliminating any plasmid-free cells that arise at cell division (Pilla et al., 2017; Pilla and Tang, 2018). The S. flexneri virulence plasmid is not self-transmissible (Sansonetti et al., 1982) but it contains genes encoding remnants of a tra transfer apparatus, suggesting that it has evolved from precursor plasmids that were mobile (Buchrieser et al., 2000). Although horizontal gene transfer via a mobile genetic element is not currently a feature of S. flexneri pathogenesis, it is central to that of V. cholerae.
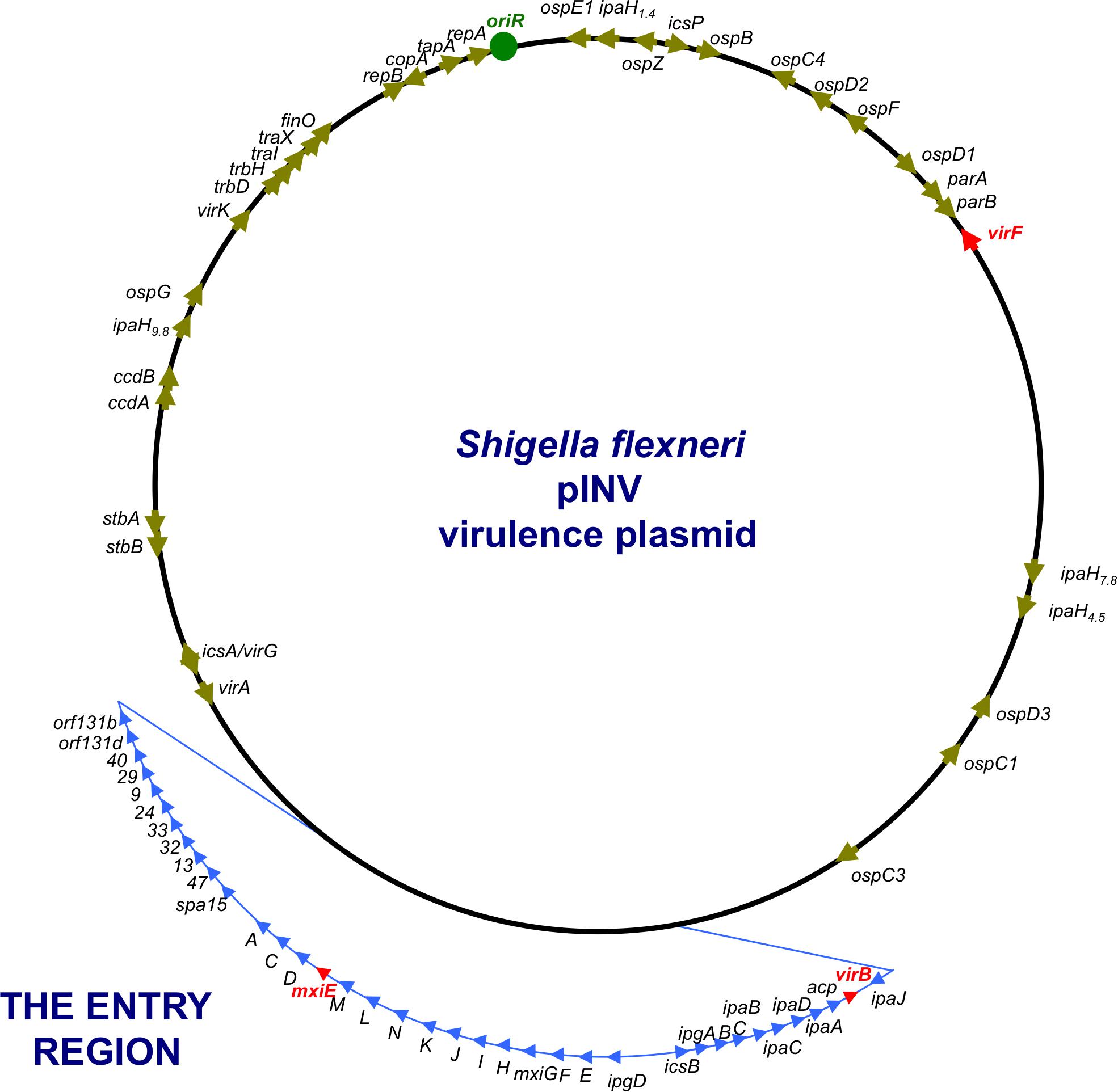
FIGURE 2. A genetic map of the pINV of S. flexneri. The plasmid is approximately 220 kbp in circumference and has a 31-kbp Entry Region (actually a pathogenicity island) where the principal virulence operons are located (light blue). Individual virulence genes can be seen around the plasmid, outside the Entry Region and others are located on the chromosome (not shown). The master regulatory genes virF, virB, and mxiE are shown in red.
The Genes for Cholera Toxin Are on a Bacteriophage
The CT has an A1B5 structure and is encoded by the ctxAB operon (De Haan and Hirst, 2004). This operon is part of the CTXϕ filamentous phage genome (Waldor and Mekalanos, 1996) that usually inserts itself at the dif site on the larger chromosome of V. cholerae using the bacterial XerCD site-specific recombination system to catalyze insertion (Huber and Waldor, 2002; McLeod and Waldor, 2004; Das et al., 2013). Insertion is catalyzed by XerC alone and follows a pathway in which the plus strand of the virus genome is integrated and the complementary strand is generated by DNA replication (Val et al., 2005). V. cholerae has two chromosomes, with the smaller, second chromosome having many of the characteristics of a large plasmid (Val et al., 2016). This is interesting in the context of the comparison with S. flexneri, where the principal virulence genes are found on a large plasmid that replicates independently of the chromosome, almost like a mini-chromosome dedicated to causing disease. This S. flexneri plasmid is actually a mosaic of up to four plasmids and it has two functioning and one vestigial plasmid partition system (Buchrieser et al., 2000; Pilla and Tang, 2018). In an interesting example of the rather ad hoc and modular nature of bacterial gene control circuits, the VirB protein from the vestigial plasmid partitioning system has been integrated into the circuit that governs expression of the Shigella virulence phenotype (Adler et al., 1989; Watanabe et al., 1990; Taniya et al., 2003; Turner and Dorman, 2007).
Transcription of the ctxAB operon is under multifactorial control in response to a range of environmental signals (Figure 3). Among the agents of control is the H-NS nucleoid-associated protein, a DNA binding protein that is a silencer of transcription (Nye et al., 2000; Nye and Taylor, 2003; Stonehouse et al., 2011). H-NS has a preference for binding to A + T-rich DNA sequences and these are frequently a feature of genes that have been acquired by horizontal transfer. Since the ctxAB operon is part of an active bacteriophage, it is de facto a genetic element that is acquired in this way. H-NS has been studied best in the model organisms Escherichia coli and Salmonella enterica (Dorman, 2007). In V. cholerae, H-NS is also known as VicH, and it has approximately 50% amino acid sequence identity with its functional ortholog in E. coli (Tendeng et al., 2000; Cerdan et al., 2003). H-NS is a DNA bridging protein that also has the ability to polymerize along DNA, excluding other proteins (Dame et al., 2006; Dorman, 2007). Relief of H-NS-mediated transcription silencing can be achieved through myriad mechanisms (Stoebel et al., 2008; Dorman and Kane, 2009; Will et al., 2015). Several of these involve the intervention of other DNA binding proteins that antagonize the transcriptionally repressive activity of H-NS. In the case of the ctxAB operon, the ToxR protein binds to DNA using a wHTH and is an important H-NS antagonist. In fact, the primary biological function of ToxR appears to be opposition to the transcriptional silencing activity of H-NS (Kazi et al., 2016). ToxR appears to have relatively relaxed requirements for its DNA binding sites at the level of nucleotide sequence (Higgins and DiRita, 1996), possibly operating primarily via an indirect readout mechanism that relies on DNA shape (Dorman and Dorman, 2017). Among its most important regulatory targets is the toxT gene (Figure 3). The principal features of the ToxT regulatory protein are described in the following section.
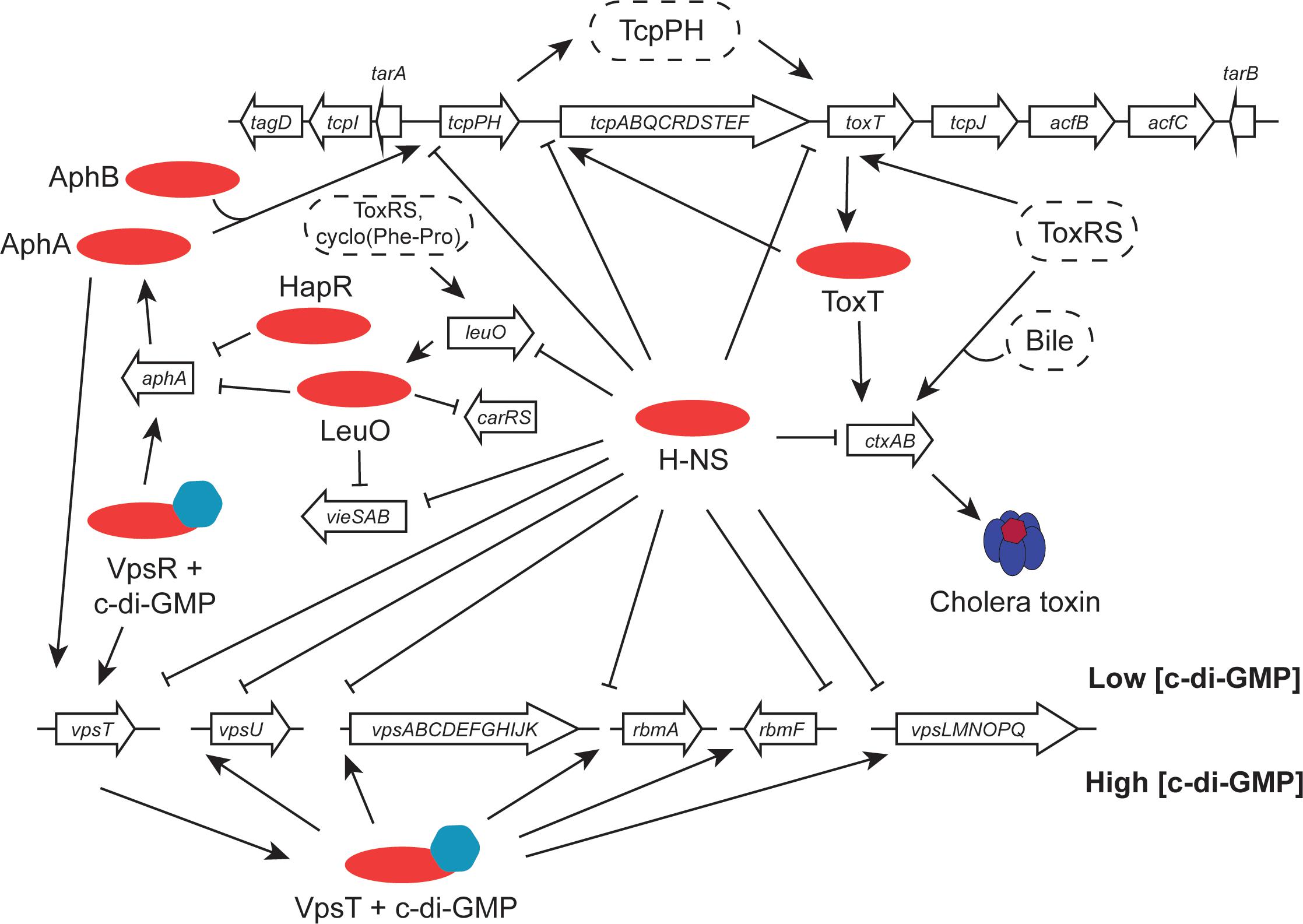
FIGURE 3. Virulence gene clusters in seventh pandemic V. cholerae share regulatory elements. Transcription promoters throughout the VPI-1 pathogenicity island and the Vibrio polysaccharide biosynthesis loci are silenced by the H-NS protein. The cholera toxin operon (ctxAB), located in the CTXϕ prophage at the dif site of chromosome I (classical strains have a second copy integrated at the equivalent site on chromosome II), is activated by the ToxT AraC-like protein and (under certain conditions) by ToxR. ToxR, together with TcpP, activates the toxT gene in VPI-1. It also positively regulates leuO, with LeuO performing important functions elsewhere in the genome, regulating aphA (in association with HapR), carRS and vieSAB. The tcpPH operon is under the positive control of the AphA and AphB proteins. At the Vps cluster, transcription is silenced by H-NS when c-di-GMP concentrations are low. When these rise, the VpsR activator switches on the vpsT regulatory gene and the VpsT protein activates the other chief transcription units. Transcription of vpsT is also stimulated by AphA.
The V. cholerae AraC-Like ToxT Protein
ToxT is an AraC-like protein that binds to DNA using two HTH motifs, making it a member of a large family of bacterial transcription factors (Egan, 2002; Li et al., 2016). The toxT gene is located in VPI-1, a horizontally acquired genetic element that harbors many virulence genes (Karaolis et al., 1998, 1999; Faruque et al., 2003) (Figure 3). Prominent among these are the genes that encode the surface-expressed TCP. Here, the toxin in question is CT, with TCP playing an important role in the acquisition of CTXϕ by acting as the receptor for this filamentous phage (Waldor and Mekalanos, 1996) as well as being an important surface factor for autoaggregation and microcolony formation on the lining of the human gut (Taylor et al., 1987; Lim et al., 2010). Since TCP serves as the CTXϕ receptor, possession of VPI-1 is therefore necessary for V. cholerae to be lysogenised by CTXϕ and to become toxigenic. Given the patterns of CT and TCP production and their linked functions in the pathogenesis of V. cholerae, coordinated regulation of their respective genes is not unexpected. This coordination is achieved, in part, by the ToxT protein (DiRita et al., 1991; Higgins et al., 1992; Champion et al., 1997), which also regulates the transcription of its own gene (Brown and Taylor, 1995; Yu and DiRita, 1999) (Figure 3).
Like many AraC-like proteins, ToxT consists of two functional domains: its DNA binding domain is in the C-terminus of the protein and a ligand-binding domain and the dimerization functions are located in the N-terminus (Lowden et al., 2010). ToxT function is modulated by bile (Schuhmacher and Klose, 1999; Chatterjee et al., 2007) and oleic fatty acids are a component of bile; interestingly, cis-palmitoleic acid has been shown to bind to the N-terminus of ToxT in its crystallized form (Lowden et al., 2010). ToxT plays a dual role in transcription regulation, acting as an anti-repressor that overcomes gene silencing by H-NS as well as serving as a conventional transcription factor by recruiting RNA polymerase to target promoters (Yu and DiRita, 2002). The DNA sequences that are targeted by ToxT are A + T-rich, a feature that is shared by H-NS binding sites, making the two proteins natural antagonists. The ToxT protein binds to the promoter region of the CT operon and activates its transcription; the same operon is silenced by H-NS (Yu and DiRita, 2002; Stonehouse et al., 2011). ToxT is regarded as the primary protein regulator of ctxAB transcription, although ToxR can also play a role independently of ToxT under some growth conditions, e.g., when bile salts are present (Hung and Mekalanos, 2005) (Figure 3). ToxR was recognized early on as a central regulator of virulence gene expression in V. cholerae, transmitting information about the composition of the external environment to its target genes (Miller et al., 1987). It has become clear that ToxR is part of a much more complex regulatory cascade with the toxT gene as one of its key targets (see the following section).
The ToxRS/TcpPH Partnership in V. cholerae
The ToxRS/TcpPH regulatory protein partnership transmits information about growth medium composition, pH, osmolarity, and temperature to the V. cholerae virulence regulon (Taylor et al., 1987; Miller and Mekalanos, 1988; Peterson and Mekalanos, 1988; Thomas et al., 1995; Carroll et al., 1997). ToxR is a cytoplasmic-membrane-associated DNA binding protein that exists in a complex with ToxS, another transmembrane protein (Ottemann and Mekalanos, 1996). The genes that encode these two proteins form an operon that is a component of the core genome of V. cholerae and is expressed constitutively (Miller et al., 1989). The N-terminus of the ToxR protein contains the wHTH DNA binding domain while the C-terminus is located in the periplasm of the bacterium (Miller et al., 1987; DiRita and Mekalanos, 1991). It was noticed early on that ToxR forms either ToxR–ToxR homodimers or ToxR-ToxS heterodimers (Ottemann and Mekalanos, 1996). ToxR undergoes regulated intramembrane proteolytic degradation and is protected from this by ToxS and by an intramolecular disulphide bond between two cysteines in the ToxR periplasmic domain (Fengler et al., 2012; Midgett et al., 2017). Exposure to bile salts or other detergents enhances ToxR–ToxS contact and increases the influence of ToxR on gene expression: the stability of ToxR declines as the bacterium makes its immediate environment more alkaline in stationary phase, disrupting ToxR’s protective interaction with the ToxS protein (Midgett et al., 2017).
Paralogous counterparts to ToxR and ToxS, known as TcpP and TcpH, are encoded by genes on VPI-1 (Figure 3). The AphA and AphB transcription factors, encoded by the ancestral part of the genome, act cooperatively at the tcpPH promoter to activate transcription (Kovacikova and Skorupski, 2001). The cAMP-Crp complex also regulates this promoter (Kovacikova and Skorupski, 2001). TcpP and TcpH form a regulatory complex that is analogous to the ToxRS one, and they also regulate the transcription of the toxT gene (Häse and Mekalanos, 1998; Krukonis et al., 2000). Like ToxR, the TcpP transcriptional activator is subject to regulated intramembrane proteolysis, making it unstable (Teoh et al., 2015). Its TcpH partner protects TcpP from such degradation (Beck et al., 2004). The presence of a disulphide bond within the periplasmic domain of TcpP is important for the protection of both TcpP and TcpH from degradation (Morgan et al., 2015).
ToxR and TcpP cooperate in the positive regulation of toxT (Morgan et al., 2011) and single-molecule-tracking experiments show that ToxR recruits TcpP to the toxT promoter (Haas et al., 2015) (Figure 3). The two proteins make contact through wing-wing interactions in their wHTH DNA binding domains (Krukonis and DiRita, 2003; Goss et al., 2010). Once ToxT is produced, it activates the other genes in its regulon, including the genes for the TCP, the accessory colonization factor (ACF), aldehyde dehydrogenase, and the CT operon itself (DiRita et al., 1991). ToxT also induces the production of the small regulatory RNA TarB, encoded within the tcp gene cluster (Figure 3). When made stable by the Hfq RNA chaperone, TarB down-regulates the VspR transcription repressor resulting in derepression of virulence genes in the Vibrio seventh pandemic pathogenicity island VSP-1 in 7PET V. cholerae (Davies et al., 2012). The expression of these genes produces increased intracellular concentrations of signaling molecules such as cyclic-GMP/cyclic-AMP and possibly c-di-GMP/c-di-AMP, altering the metabolism of the bacterium and its chemotactic behavior (Davies et al., 2012; Dorman, 2015; Severin et al., 2018) (Figure 3). TarB also post-transcriptionally down-regulates TcpF production from VPI-1: here, Hfq does not seem to be required and the production of the sRNA is enhanced during anaerobic growth (Bradley et al., 2011) (Figure 3).
H-NS plays an important role in the control of gene expression in the context of adaptation to environments outside the human host (Ayala et al., 2017) (Figure 3). It is also required for V. cholerae motility (Ghosh et al., 2006; Silva et al., 2008). Apart from the ctxAB operon, most of the genes that are silenced by H-NS are located in VPI-1 on chromosome I and in a 125-kb super-integron located on chromosome II; others include the hlyA hemolysin/cytolysin, the vas major type VI secretion operon and the vps-rbm biofilm and chitin utilization genes (Ayala et al., 2015a,b; Wang et al., 2015; Zamorano-Sánchez et al., 2015; Kazi et al., 2016) (Figures 3–5).
The AphA AphB Regulon
AphA is a wHTH transcription factor (De Silva et al., 2005) that assists the LysR-like protein AphB to bind to the tcpPH promoter (Kovacikova and Skorupski, 2001; Kovacikova et al., 2004) (Figure 3). AphA represses the transcription of the V. cholerae genes that synthesize acetoin and hence helps to manage the pH of the bacterial cytoplasm (Kovacikova et al., 2005). AphA activates the expression of the gene encoding VpsT, a LuxR-like biofilm regulator of biofilm formation (Yang et al., 2010) (Figure 3). The VpsR regulatory protein activates production of VpsT in the presence of high c-di-GMP; VpsR also activates the gene encoding AphA under these conditions (Ayala et al., 2015a,b; Dorman, 2015; Zamorano-Sánchez et al., 2015) (Figure 3). These VpsR/T-AphA connections link the virulence and biofilm gene clusters intimately at the level of cross regulation.
HapR, the chief quorum-sensing regulator of V. cholerae, is known to repress the transcription of the tcpPH gene and hence the virulence regulon (Zhu et al., 2002). HapR binds to, and represses, the aphA promoter when the bacterial culture is at high density, preventing activation of tcpPH by AphA (Kovacikova and Skorupski, 2002; Kovacikova et al., 2003). This link makes the virulence cascade responsive to quorum sensing (Figures 3, 4). The HapR and AphA proteins play reciprocal roles in the response of V. cholerae to cell density: AphA operates at low cell density and HapR takes over the role of master regulator at high cell density. At low cell density, AphA production is upregulated by sRNAs and then AphA and Hfq-dependent sRNAs repress production of HapR (Lenz et al., 2005; Lenz and Bassler, 2007; Rutherford et al., 2011; Basu et al., 2017) (Figure 4). These (redundant) sRNA molecules are four Quorum Regulatory RNAs (Qrr) and they act with Hfq to repress production of HapR. In parallel, the production of the CsrB, CsrC, and CsrD sRNAs (also redundant) is under the control of the V. cholerae two-component system VsrS/VsrA (Lenz et al., 2005) (Figure 4). These Csr sRNAs act on the CsrA global regulatory protein to activate production of the Qrr sRNAs via the LuxO quorum-sensing protein, in an RpoN-dependent manner (Lenz et al., 2005). At high cell density, HapR acts at aphA by antagonizing its activation by the leucine-responsive regulatory protein, Lrp, and the NtrC-like biofilm regulator, VpsR (Lin et al., 2007) (Figure 3). In Shigella, CsrA has a poorly defined impact on the virulence phenotype through effects on expression of the virF and virB regulatory genes, implicating carbon metabolism as an important determinant of Shigella virulence (Gore and Payne, 2010). Quorum sensing seems to play a role in production of the S. flexneri T3SS, affecting in particular expression of the virB regulatory gene. T3SS production is maximal in cells entering the stationary phase of growth, a stage when bacterial cell density is maximal during growth in laboratory culture. However, the details of the signaling cascade remain obscure (Day and Maurelli, 2001).
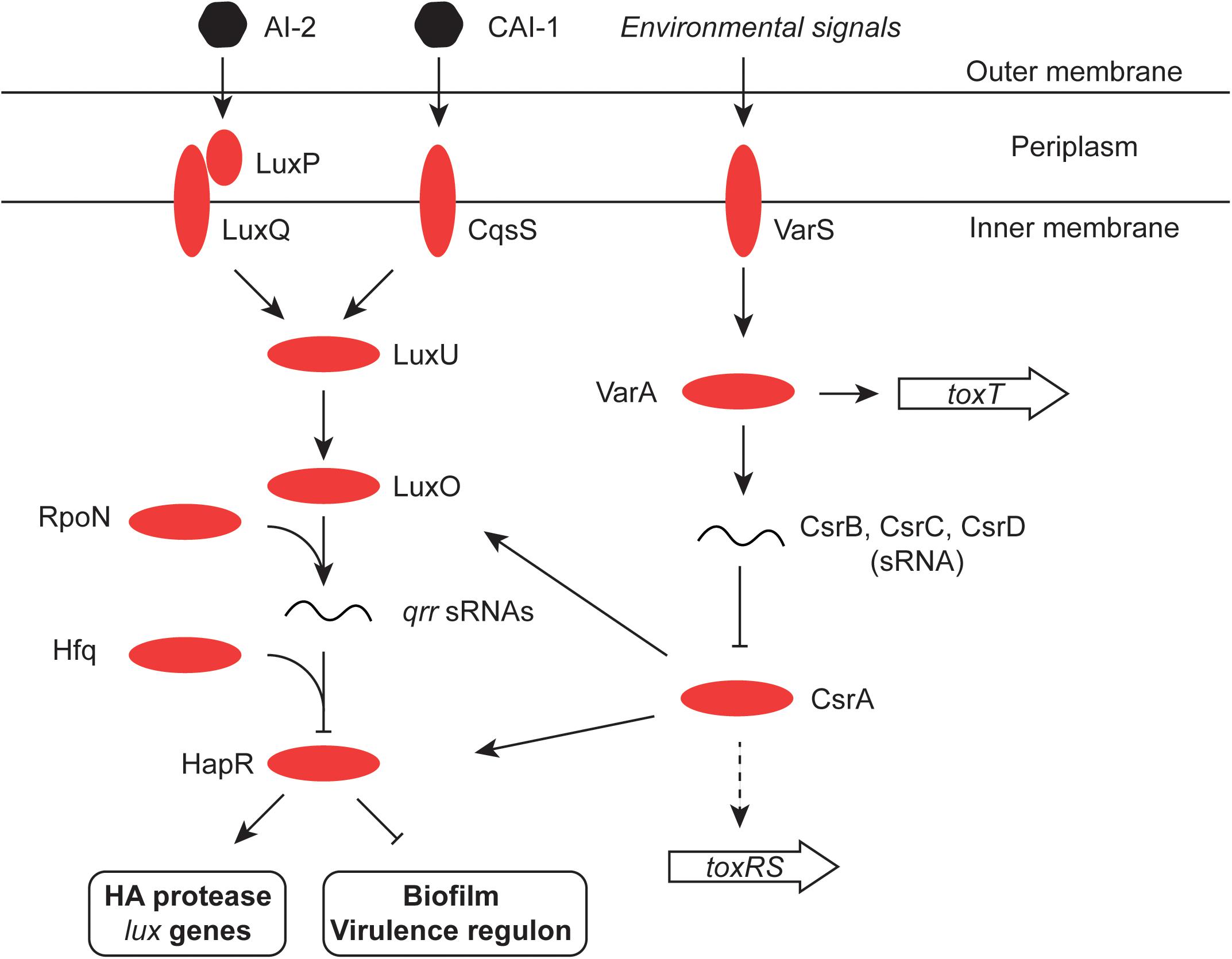
FIGURE 4. Quorum sensing in V. cholerae. The signaling molecules CAI-1 and AI-2 are detected by the cytoplasmic membrane protein CqsS and the periplasmic binding protein LuxP, respectively. LuxP interacts with LuxQ which, like CqsS, interacts in turn with LuxU in the cytoplasm. LuxU activates the LuxO regulatory protein and this promotes expression of the Qrr sRNA from an RpoN (σ54)-dependent promoter. Aided by the RNA chaperone Hfq, Qrr inhibits the production of the HapR regulatory protein (an activator of the HA protease and the lux genes, and an inhibitor of the virulence regulon and the biofilm production). LuxO production is also under the control of the global regulatory protein CsrA, which in turn is inhibited by the redundant sRNA molecules CsrB, C, and D. CsrA also influences production of the HapR transcription factor directly, as well as indirectly through LuxO (Tsou et al., 2011). The two-component VarS–VarA system responds to environmental and quorum-sensing signals and promotes the production of the Csr sRNAs. It has also been reported to modulate the expression of the ToxT protein (Jang et al., 2011). Finally, CsrA may directly promote production of ToxR in response to the amino acids asparagine, arginine, glutamate, and serine (Mey et al., 2015).
AphB plays an important role in V. cholerae pH homeostasis by regulating, among other genes, the expression of cadC (Kovacikova et al., 2010). The CadC protein shares with ToxR and TcpP the properties of being a cytoplasmic-membrane-associated transcription regulator (Merrell and Camilli, 2000). Among its targets is the cadBA operon encoding a lysine decarboxylase (CadA) and a lysine/cadaverine antiporter (CadB). CadA converts lysine to cadaverine and CadB transports cadaverine out of the cell, exchanging it for lysine (Merrell and Camilli, 1999). Interestingly, cadA has been identified as an anti-virulence gene that is deleted in Shigella species: it has been suggested that since cadaverine inhibits Shigella enterotoxin activity, loss of cadA by deletion enhances the virulence of the pathogen (Maurelli et al., 1998).
The cadC gene in El Tor V. cholerae is repressed by the LysR-like wHTH transcription factor LeuO (Ante et al., 2015a), a protein with wide-ranging effects on transcription in Gram-negative bacteria (Shimada et al., 2011; Dillon et al., 2012; Guadarrama et al., 2014; Dorman and Dorman, 2017). ToxR derepresses the H-NS-silenced leuO gene (Bina et al., 2016) and it also mediates the transcription of ompU, a gene encoding an outer membrane porin that confers resistance to bile stress (Ante et al., 2015b) (Figure 3). LeuO represses the carRS operon that controls the remodeling of lipid A for the elaboration of cationic antimicrobial peptide resistance (Bina et al., 2016). In classical strains of V. cholerae, LeuO is a co-repressor, with H-NS of vieSAB transcription, an operon that modulates biofilm formation and cell motility (Ayala et al., 2018). In contrast, El Tor biotype strains produce very little LeuO. The vieSAB operon is almost silent in the El Tor biotype due to repression by the quorum-sensing regulator, HapR, a protein that is reported to be absent from classical biotype strains (Wang et al., 2015). The emerging picture is one in which H-NS and LeuO cooperate to repress vieSAB in classical strains and H-NS and HapR achieve the corresponding effect in El Tor organisms (Ayala et al., 2018). Consistent with this role, leuO mutants exhibit reduced levels of biofilm production (Moorthy and Watnick, 2005). LeuO, whose production is enhanced by ToxR, feeds back negatively onto aphA transcription, leading to downregulation of the ToxR regulon with reductions in CT and TCP production (Figure 3). The signal for the ToxR enhanced transcription of leuO is cyclo(Phe-Pro) (Bina et al., 2013).
Signals Affecting CT Production: Role of the Stringent Response
AphB has also been linked to the anaerobic response in V. cholerae (Kovacikova et al., 2010). Its dimerization and activity in regulating the expression of tcpPH are enhanced by anaerobiosis. A cysteine amino acid has been shown to be essential for the anaerobic enhancement, indicating that AphB uses a thiol-based switch to respond to oxygen limitation (Liu et al., 2011). Anaerobiosis and the stringent response are linked in enhancing the production of CT (Lee et al., 2012). When grown anaerobically using trimethylamine N-oxide (TMAO) as a terminal electron acceptor, V. cholerae exhibits enhanced production of CT. The bacteria also produce the alarmone guanosine tetraphosphate (ppGpp) whereas mutants deficient in ppGpp production are also impaired in CT production. Similarly, mutants lacking DksA, the protein that works synergistically with ppGpp at RNA polymerase, are also impaired in CT production (Oh et al., 2014). The stringent response is a global reaction to nutritional stress that is usually triggered by an accumulation of uncharged tRNAs that indicate a shortage of one or more amino acids. Bacteria experiencing the stringent response accumulate ppGpp and reprogramme RNA polymerase so that genes expressing components of the translational machinery are downregulated. The enzyme RelA produces pppGpp from ATP and GTP and then converts it to ppGpp; SpoT hydrolyses ppGpp (although SpoT has a synthesis role too) (Dalebroux et al., 2010; Dalebroux and Swanson, 2012). In V. cholerae the RelV enzyme synthesizes ppGpp in the absence of RelA and SpoT when the bacterium is experiencing glucose or fatty acid starvation (Das et al., 2009). All three synthases are involved in regulating V. cholerae biofilm production: vpsR transcription requires all three while vpsT requires RelA (He et al., 2012). V. cholerae also has a GTP-binding protein called CgtA that modulates the function of SpoT, repressing the stringent response by keeping ppGpp levels low (Raskin et al., 2007). DksA regulates the production of the hemagglutinin/protease (HAP) that is involved in the shedding stage of the V. cholerae infection (Basu et al., 2017). HAP production is controlled by HapR and RpoS with DksA controlling HapR production positively at the transcriptional and post-transcriptional levels. The post-transcriptional regulation involves Fis-dependent Quorum Regulatory RNAs (Qrr) sRNAs and the levels of these regulatory RNA molecules are strongly reduced in the absence of DksA (Lenz et al., 2005; Lenz and Bassler, 2007). Furthermore, DksA is required for normal levels of RpoS production in stationary phase V. cholerae cells (Basu et al., 2017). The counterpart to CgtA in E. coli (and Shigella) is the evolutionarily highly conserved Obg GTPase (Dewachter et al., 2016). Obg has been described as a cell cycle protein that can arrest chromosome replication, a factor required for stress response and for ribosome assembly. In association with ppGpp it determines persistence through the stochastic induction of a state of dormancy and antibiotic tolerance during periods of nutrient stress (Verstraeten et al., 2015; Gkekas et al., 2017). In S. flexneri, the DksA protein and ppGpp are required for production of the Hfq RNA chaperone (Sharma and Payne, 2006) a protein that controls the level of the central virulence regulator VirB (Mitobe et al., 2008, 2009). Loss of DksA impairs the intercellular spreading of S. flexneri, with dksA mutants having the IcsA protein that is required for actin-dependent motility mislocated on the cell surface (Mogull et al., 2001). The stringent response operates by reprogramming the activity of RNA polymerase in response to amino acid starvation. RNA polymerase is also reprogrammed in response to stress by the replacement of the housekeeping sigma factor RpoD by a stress-specific sigma factor, with RpoS being a prominent example.
General Response to Stress and the Role of the RpoS Sigma Factor
Many commensal and pathogenic bacteria respond to stress and/or the cessation of growth by reprogramming RNA polymerase by replacing the RpoD sigma factor with RpoS (Klauck et al., 2007; Schellhorn, 2014). The role of the RpoS sigma factor in the expression of virulence has been investigated more intensively in V. cholerae than in S. flexneri. In S. flexneri, much of the focus has been on the link between RpoS and resistance to low pH stress. The discovery that Shigella and some pathogenic E. coli strains have a regulator called IraL that stabilizes RpoS in the absence of stress may reveal infection-related RpoS roles in Shigella that have yet to be explored (Hryckowian et al., 2014). In terms of where RpoS sits in the V. cholerae regulatory hierarchy, it is useful to note that transcription silencing by H-NS may be less effective in V. cholerae at RpoS-dependent promoters (Wang et al., 2012), although it is not clear if this is an entirely due to an intrinsic property of the RpoS sigma factor or if some other feature, such as the topology of the promoter DNA during periods of RpoS availability, might play a role. Also, RpoS in V. cholerae enhances the production of IHF, with knock-on effects for IHF-dependent processes (Wang et al., 2012). Early work with V. cholerae showed that loss of RpoS production makes the bacterium sensitive to a variety of environmental stresses in vitro (Yildiz and Schoolnik, 1998) and interferes with its ability to colonize the mouse small intestine (Merrell et al., 2000). The sigma factor is also involved in the latter stages of infection when the bacterium detaches from the epithelial surface. Here, the so-called mucosal escape step requires RpoS with the sigma factor contributing to the expression of genes involved in motility and chemotaxis, functions that are important for the subsequent dispersal of the pathogen (Nielsen et al., 2006; Ringgaard et al., 2015). Motility genes have a complex relationship with the virulence phenotype, with mutants deficient in specific flagellar hierarchy regulators displaying altered production of virulence factors including CT, TCP and the T6SS (Syed et al., 2009). V. cholerae’s re-entry to an aquatic environment may also be RpoS-dependent. The sigma factor is required for the production of the chitinase that renders V. cholerae both competent for the uptake of foreign DNA and which is needed for the growth of the bacterium on otherwise insoluble chitin (Dalia, 2016). This RpoS-competence link is independent of any quorum-sensing link to natural transformation by foreign DNA (Dalia, 2016). HapR and quorum sensing have been reported to enhance the production of RpoS (Joelsson et al., 2007) and RpoS plays a positive role in the expression of the HapR-dependent V. cholerae hapA gene, encoding a hemagglutinin/protease (Silva and Benitez, 2004) (Figure 4). This enzyme plays an important part in the bacterial shedding phase that occurs toward the end of the infection (Basu et al., 2017). Transcription of the rpoS gene is repressed by the VpsT transcription factor, in association with the second messenger cyclic-di-GMP, when V. cholerae is within biofilm (Wang et al., 2014) (Figure 5). While it has been appreciated for some time that RpoS (and quorum sensing) are important influences in determining entry into a biofilm, it is now becoming clear that RpoS and quorum sensing are also important for V. cholerae dispersion from biofilm (Singh et al., 2017).
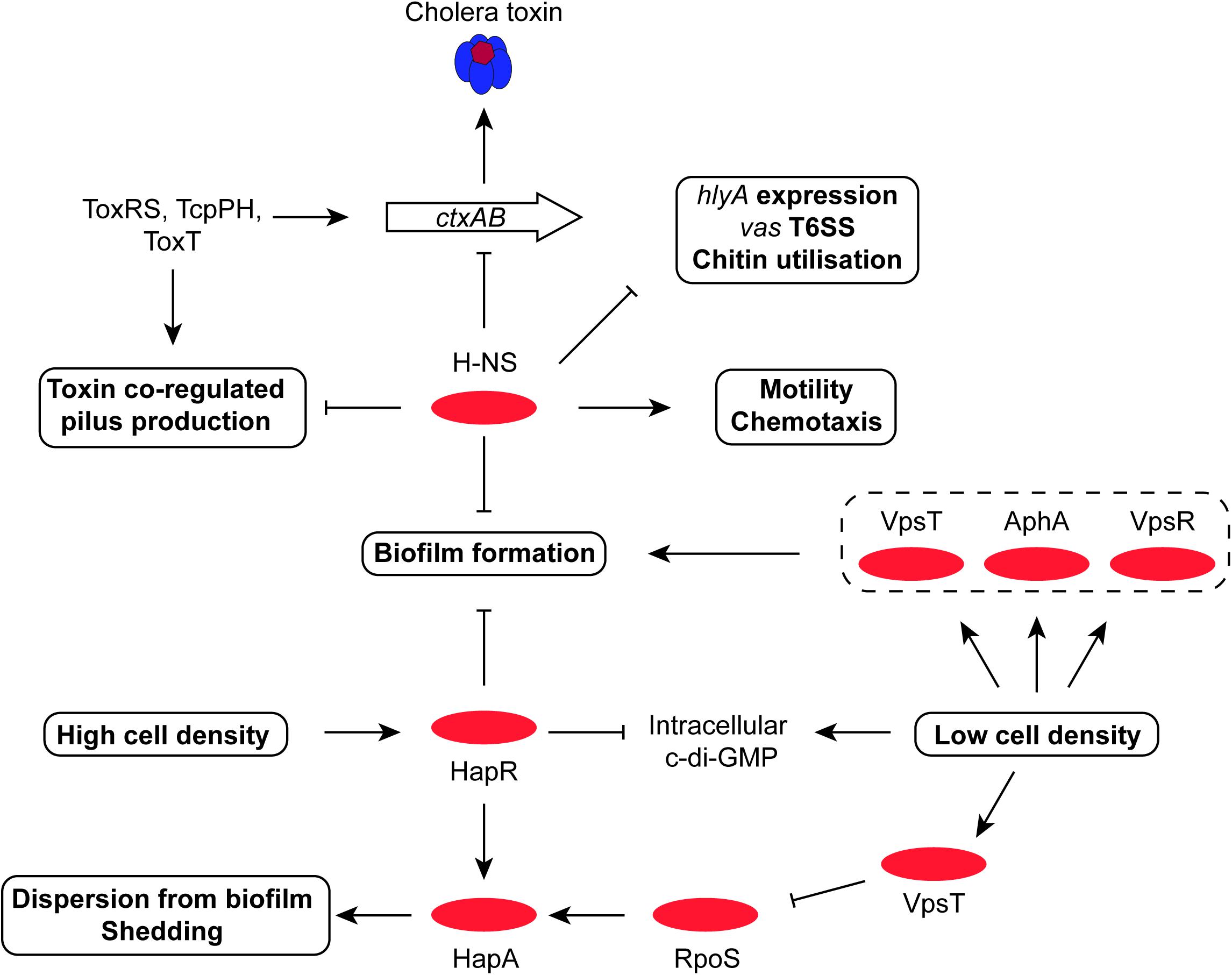
FIGURE 5. The central roles of HapR and H-NS in biofilm formation, motility, T6SS and chitin utilization. The H-NS protein plays a positive role in V. cholerae motility, as it does in other Gram-negative bacteria. It represses expression of genes for production of T6SS, virulence, chitin utilization, and biofilm. At high cell density, the HapR transcription factor also represses biofilm formation while promoting dispersion of bacteria from biofilm and shedding; it also promotes the production of the stress and stationary phase sigma factor RpoS (σ38). RpoS and HapR cooperate to promote the production of the HapA protease, which erodes biofilm. The VpsR-and-c-di-GMP-dependent VpsT regulatory protein down-regulates RpoS, helping to maintain the dominance of RpoD (σ70) in metabolically-active cells.
Chitin and Gene Regulation in V. cholerae
Chitin transport and metabolism depend on the chb operon in V. cholerae. The cell division protein SlmA binds to chb and regulates its expression, possibly in association with an unknown transcription factor that is under the control of the ChiS sensor-kinase (Klancher et al., 2017). Cbp is a periplasmic chitin binding protein that regulates ChiS negatively when chitin is absent; binding chitin by Cbp relieves repression (Li and Roseman, 2004) (Figure 6). TfoS is an independent chitin sensor that activates the transcription of the tfoR gene that encodes an sRNA, TfoR (Dalia et al., 2014; Yamamoto et al., 2014). This Hfq-dependent sRNA positively controls the translation of tfoX mRNA with the TfoX protein, in turn, activating genes involved in foreign DNA uptake in competent cells (Yamamoto et al., 2011) (Figure 6). The tfoX gene is also regulated at the level of transcription by the cAMP-Crp complex (Wu et al., 2015). This is consistent with the roles of cAMP-Crp and chitin in the establishment of competence in Vibrio spp. (Blokesch, 2012). Chitin makes V. cholerae competent for DNA uptake by transformation, allowing it to influence the evolution of the bacterium by horizontal gene transfer (Meibom et al., 2005; Le Roux and Blokesch, 2018). The fate of DNA taken up by V. cholerae that has been made competent by chitin depends on cell density and quorum sensing. At low cell density the HapR regulator is present in low abundance, allowing the Dns nuclease to accumulate and to degrade the incoming DNA extracellularly or in the periplasm (Blokesch and Schoolnik, 2008; Seitz and Blokesch, 2014). At high cell density, HapR is abundant and production of the nuclease is repressed. Under these conditions the TfoX-dependent competence system imports the DNA (Seitz and Blokesch, 2013) (Figure 6). In addition to its roles in controlling competence, the cAMP-Crp complex also controls the virulence regulon at the tcpPH promoter (Kovacikova and Skorupski, 2001) and both the integrase gene promoter and the main promoter for gene cassette transcription in the superintegron found on chromosome II of V. cholerae (Krin et al., 2014). The gene that encodes the outer membrane porin protein OmpT requires cAMP-Crp for expression; ompT is the only member of the ToxR regulon known to be repressed by ToxR and it acts by disrupting the positive influence of the cAMP-Crp complex at the ompT promoter (Caiyi et al., 2002).
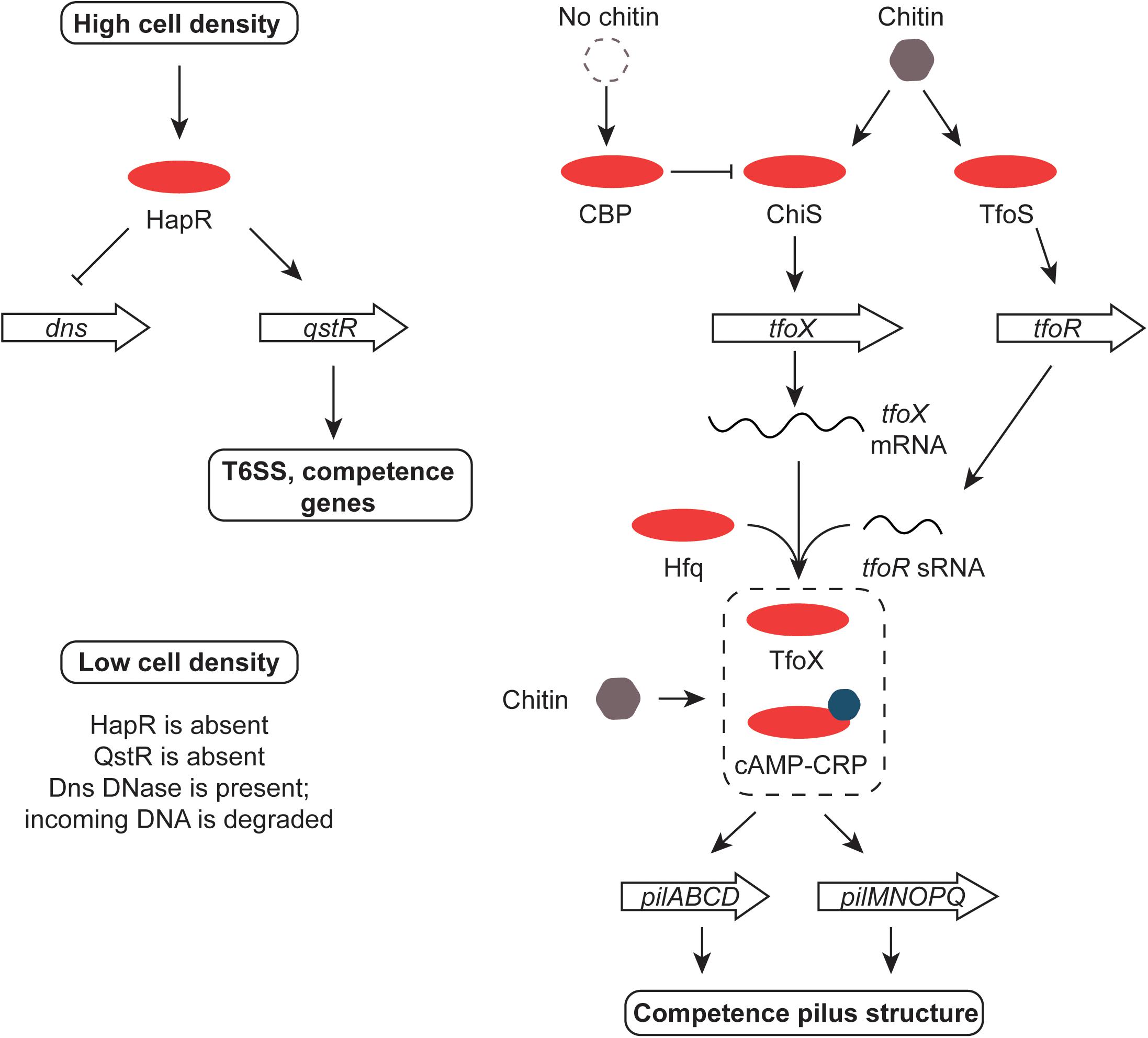
FIGURE 6. Chitin, HapR, QstR, and competence. At high cell density, HapR represses production of the Dns DNase, an enzyme that degrades foreign DNA. It also promotes the production of the QstR regulator, switching on the T6SS and genes for competence. The former kills nearby cells, releasing DNA for uptake by competent V. cholerae. The genes encoding the pilus that forms the DNA uptake system are up-regulated in response to chitin by the cAMP-Crp complex and the TfoX regulatory protein. The tfoX gene is activated by ChiS in response to chitin and tfoX mRNA is translated efficiently in the presence of the Hfq-dependent TfoR sRNA. The switch is reset in the absence of chitin by the CBP protein. Production of the TfoR sRNA is promoted by the TfoS regulator in response to chitin. When cell density is low, HapR and QstR are absent, allowing the Dns enzyme to be produced and foreign DNA to be degraded.
Gene Silencing and Anti-Silencing in S. flexneri
The H-NS protein plays a general role in silencing transcription throughout the virulence regulon of S. flexneri (and enteroinvasive E. coli) (Dorman et al., 2001; Beloin and Dorman, 2003) (Figure 7). Like the V. cholerae system, the S. flexneri regulon uses an AraC-like protein to overcome H-NS-mediated transcription silencing (Dorman, 1992; Porter and Dorman, 2002). The VirF protein performs approximately an equivalent role to ToxT and is encoded by virF, a regulatory gene on the virulence plasmid that lies outside the Entry Region (Adler et al., 1989; Porter and Dorman, 1997b, 2002; Di Martino et al., 2016a). It is closely related to the Rns protein of enterotoxigenic E. coli and the two proteins are at least partly functionally interchangeable (Porter et al., 1998). Transcription activation of virF involves a complicated process in which H-NS-mediated silencing is overcome by an adjustment to the local DNA structure at the virF promoter (Stoebel et al., 2008). This adjustment is triggered by a shift in temperature to 37°C from a lower value. This temperature is one of the environmental signals, along with appropriate values of pH and osmolarity that are characteristic of the human intestine (Di Martino et al., 2016a). It involves a change in the bend angle of a region of A + T-rich DNA that widens the space between the arms of the bend, possibly compromising the integrity of a DNA-H-NS-DNA bridge, an event that is exacerbated by a displacement of the center of the bend that disrupts the parallel placement of the DNA bend arms (Prosseda et al., 2004). This remodeling event does not require the intervention of proteins, although the Fis and IHF nucleoid-associated proteins play a positive role in virF transcription activation and the VirB protein also makes a direct and positive contribution (Porter and Dorman, 1997a; Prosseda et al., 2004; Kane and Dorman, 2012). DNA remodeling to activate virF transcription is consistent with the known role of DNA negative supercoiling in modulating the expression both of virF (Durand et al., 2000) and the wider S. flexneri virulence system (Dorman et al., 1990; Ni Bhriain and Dorman, 1993; Dorman, 1995; McNairn et al., 1995). There is also a positive regulatory role for the CpxA/CpxR two-component system (that transmits information about ambient pH) in the activation of virF expression (Nakayama and Watanabe, 1995, 1998; Marman et al., 2014). CpxA has also been described as influencing positively the stability of invE/virB mRNA (Mitobe et al., 2005).
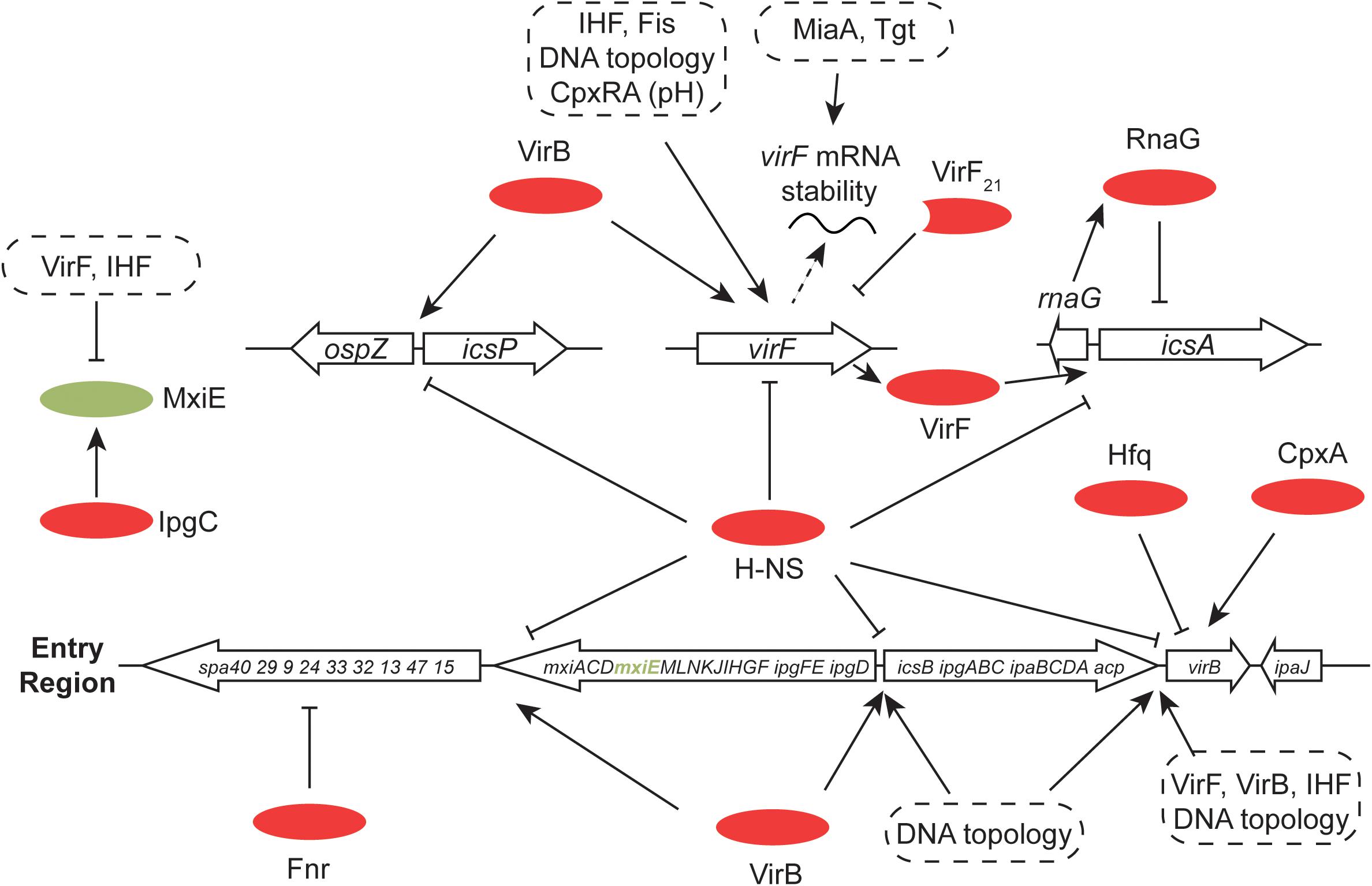
FIGURE 7. Global control of the S. flexneri virulence system. The 31-kbp Entry Region, consisting of three large operons, is indicated. All of the main transcription promoters in this region are repressed by H-NS and this is reversed by the Entry-Region-encoded VirB anti-repressor. Production of VirB depends on the VirF AraC-like protein (encoded by pInv), the correct degree of DNA negative supercoiling and the chromosomally-encoded IHF protein. VirB autoregulates its own gene positively and cross-regulates positively the gene for virF. VirB is also subject to Hfq-dependent post-transcriptional control at the level of mRNA stability. In addition to VirB, production of VirF requires IHF, an appropriate degree of DNA negative supercoiling, the chromosomally encoded Fis protein and the chromosomally encoded pH-responsive CpxRA two-component regulatory system. VirF is controlled post-transcriptionally by the tRNA-modifying MiaA and Tgt factors. The truncated form of VirF, VirF21, acts as an antagonist of full-length VirF production. MxiE is another AraC-like protein with multiple gene targets on pInv (and on the chromosome) whose activity is modulated by the IpgC co-activator and the anti-activators OspD1 and Spa15. The Fnr anaerobic regulator makes negative regulatory inputs at spa32 and spa33. The VirF-dependent icsA gene, repressed transcriptionally by H-NS and RnaG, is shown, together with the divergently-transcribed-and-H-NS-repressed, VirB-activated icsP and ospZ genes.
Control of VirF Production in S. flexneri
The virF transcript is translated into two proteins: a major form known as VirF30 (which has a molecular mass of 30 kDa) and VirF21, lacking the N-terminal segment of the full-length protein (Di Martino et al., 2016b). While VirF30 is responsible for the activation of the virulence regulon, VirF21 is a negative autoregulator of virF expression. The shorter protein is translated from a leaderless mRNA called llmRNA that is transcribed from a separate promoter that is internal to virF: the function of VirF21 may be to restrict production of VirF to environments where S. flexneri will benefit from expression of the full virulence regulon (Di Martino et al., 2016b). Production of the VirF protein is reduced in mutants with deficiencies in tRNA modification: specifically in miaA and tgt mutants (Durand et al., 2000). This reduction in VirF production has a negative impact on S. flexneri virulence and results from poor translation of the virF mRNA. This negative effect can be offset by the presence of methionine and arginine in combination, or by adding putrescine to the growth medium (Durand and Björk, 2003). These observations indicate the extent to which VirF production is keyed into central metabolism in S. flexneri.
A striking feature of the S. flexneri virulence gene regulatory cascade is that VirF does not regulate all of the main structural genes directly. Its direct influence is restricted to the ‘intracellular spread’ gene icsA (also known as virG) a small RNA antagonist of icsA transcription called RnaG, and to an intermediate regulatory gene called virB. The H-NS protein silences each of these genes (Tran et al., 2011) (Figure 7). The icsA gene product is localized to one of the poles of the S. flexneri cell where it promotes the polymerization of actin that, in turn, creates the characteristic comet-tail-like structures that propel the bacterium through the host cell cytoplasm (Suzuki et al., 1996, 1998).
The Unusual VirB Regulatory Protein in S. flexneri
The virB gene, known as invE in S. sonnei (Watanabe et al., 1990), encodes a protein with strong amino acid sequence similarity to plasmid segregation proteins of the ParB family (Adler et al., 1989; Beloin et al., 2002; McKenna et al., 2003; Taniya et al., 2003; Turner and Dorman, 2007). VirB acts between VirF and most of the virulence gene promoters of the S. flexneri virulence regulon, carrying out the role of an intermediate regulator (Figure 7). VirB has a requirement for a specific parS-like DNA sequence at its binding sites and its mode of action consists of remodeling H-NS-DNA complexes to eliminate the transcription silencing activity of the H-NS protein (Taniya et al., 2003; Turner and Dorman, 2007). The icsB gene is first in an operon that encodes components of the S. flexneri T3SS and it is silenced by H-NS (Beloin and Dorman, 2003). The counter-silencing mechanism used at the icsB promoter seems to consist of a combination of DNA wrapping and VirB protein polymerization (Turner and Dorman, 2007). VirB antagonism of H-NS-mediated silencing can act over considerable distances, as at the icsP promoter (Castellanos et al., 2009). The IcsP (SopA) protein is a protease that deactivates IcsA, the cell-pole-located actin-tail-polymerizing protein (Fukuda et al., 1995; d’Hauteville et al., 1996; Shere et al., 1997; Wing et al., 2004). The icsP promoter is silenced by H-NS and VirB acts there as an anti-repressor; contact between the counteracting proteins may involve DNA dependent protein polymerization in vivo (Weatherspoon-Griffin et al., 2018). The ospZ promoter is also controlled by a VirB/H-NS-dependent silencing/anti-silencing mechanism (Basta et al., 2013). The OspZ protein is secreted by the S. flexneri T3SS to manipulate the host inflammatory response (Zurawski et al., 2008; Zhang et al., 2016). The ospZ gene is located immediately upstream of icsP and is transcribed in the opposite direction. The two genes share important cis-acting regulatory sites that are targets for VirB when acting in its capacity as an anti-repressor (Basta et al., 2013). A similar arrangement is found between the icsB and ipgD genes whose promoters are silenced by H-NS and derepressed by VirB (Porter and Dorman, 1997a; Taniya et al., 2003; Turner and Dorman, 2007).
Early work showed that the virB gene is repressed by H-NS and regulated positively by VirF (Tobe et al., 1993) (Figure 7). Although the activation of virB transcription depends on the presence of VirF, the presence of this AraC-like protein is not sufficient: a temperature of 37°C is also required. This thermal requirement can be dispensed with if the topology of the DNA in the vicinity of the virB promoter is manipulated appropriately, an observation that supports a model in which the positive role of VirF is contingent on the conformation of the DNA on which it acts (Tobe et al., 1995). Consistent with this hypothesis is the finding that negative DNA supercoils, generated by a divergently transcribing promoter, activate the virB promoter in the presence of VirF at the normally non-permissive temperature of 30°C (Tobe et al., 1995). The virF and virB genes represent the principal sites for thermal sensing in the S. flexneri virulence gene cascade. Once the VirB protein is produced, the genes under its control will be derepressed as H-NS-mediated transcription silencing is progressively relieved. This can be shown by producing the VirB protein ectopically, e.g., using an arabinose-inducible promoter in bacteria growing at 30°C, a temperature that is normally non-permissive for expression of the virulence regulon. Here, the level of transcription of the virulence genes corresponds to the level of arabinose-induced VirB protein that is present in the cell (Beloin and Dorman, 2003). The virB/invE transcript becomes unstable at 30°C (Mitobe et al., 2008) or if the osmolarity of the growth medium declines (Mitobe et al., 2009). Like temperature, osmotic pressure is an important signal for full activation of the virulence system (Porter and Dorman, 1994). In both cases, virB mRNA turnover is dependent on the RNA chaperone Hfq (Mitobe et al., 2008, 2009). These findings indicate that down-regulation of virB gene expression involves rapid turnover of the mRNA when growth conditions no longer correspond to those found in the human large intestine.
The similarity of the VirB protein to ParB-like plasmid partitioning proteins and the similarity between the binding site in DNA used by VirB and those used by ParB-like proteins (parS) suggests that the VirB protein is a vestige of a mechanism that was concerned with plasmid segregation and that VirB has been co-opted into gene regulation (Adler et al., 1989; Taniya et al., 2003; Turner and Dorman, 2007). The utility of VirB as a general antagonist of transcriptional silencing mediated by H-NS has been shown by using it to overcome silencing by H-NS of the proU operon (Kane and Dorman, 2011). The proU operon encodes a transport system for the osmo-protectants glycine-betaine and proline and is not normally controlled by VirB (Ref for ProU). However, judicial placement at the proU promoter of the parS-like binding site used by VirB at the S. flexneri icsB promoter brings proU under VirB control. This simple experiment demonstrates the modular nature of gene control mechanisms based on H-NS silencing and anti-silencing and illustrates the ease with which novel systems might evolve in nature.
The VirB protein forms a positive feedback loop onto its own gene’s expression and onto that of the virF gene (Kane and Dorman, 2012) (Figure 7). This may facilitate reinforcement of the transition from the ‘off’ to the ‘on’ state once a commitment to expressing the virulence cascade is made. An examination of the fold-induction of the virulence regulon in response to the thermal signal reveals a gearing effect, with virF showing the narrowest range of transcription between its induced and non-induced states, the major virulence operons having the widest range and virB has an induction ratio with an intermediate value (Porter and Dorman, 1997a). The imposition of additional regulatory signals and regulators may help to restrict full production of the T3SS and its effector proteins to circumstances when the bacterium is most likely to benefit from the physiological investment. This is consistent with findings that reveal roles for post-transcriptional control in the system, such as the phenotypes associated with mutants deficient in tgt or miaA (Durand and Björk, 2003). The translation control factors elongation factor P (EF-P) and the enzyme that modifies it (PoxA) are also needed for expression of a full virulence phenotype (Marman et al., 2014). It has been suggested that the stability of virB mRNA is influenced through interaction with the cytoplasmic-membrane-associated RodZ protein, a determinant of bacterial cell morphology (Mitobe et al., 2011). Other examples of additional control include the sRNA known as RnaG (Tran et al., 2011), the MxiE AraC-like transcription factor, the FNR anaerobic regulator (Marteyn et al., 2010; Vergara-Irigaray et al., 2014), the Rho transcription termination factor (Tobe et al., 1994), the Hfq RNA chaperone (Mitobe et al., 2008, 2009) and the osmo-regulatory OmpR/EnvZ two-component regulatory system (Bernardini et al., 1990, 1993).
Additional Virulence Regulators in S. flexneri
The role of the OmpR/EnvZ system in virulence gene expression seems to be restricted to conditions of high osmolarity growth, but precise molecular details are lacking (Bernardini et al., 1990). The gene encoding the OmpC outer membrane porin is known to require OmpR for expression and has been shown to be required for full virulence, but the ompC gene is part of the core genome and is located on the S. flexneri chromosome (Bernardini et al., 1993). The possibility that OmpR is involved in the pH response of the Shigella virulence system does not seem to have been investigated: OmpR plays such a role in controlling the transcription of T3SS genes in the facultative intracellular pathogen Salmonella (Quinn et al., 2014; Chakraborty et al., 2017).
MxiE is encoded by a gene in the Entry Region of the virulence plasmid and plays a role late in the program of virulence gene expression (Kane et al., 2002) (Figures 1, 7). MxiE is an AraC-like transcription factor that operates in partnership with the co-activator IpgC (Pilonieta and Munson, 2008). The IpgC protein is also a chaperone of the IpaB and IpaC effector proteins. MxiE binds to a 17-bp ‘MxiE box’ and regulates several virulence genes on the plasmid and may have targets on the chromosome too (Mavris et al., 2002; Bongrand et al., 2012). Transcriptional slippage by RNA polymerase that introduces an additional base into the MxiE mRNA is required for the production of full-length MxiE protein. The slippage event (and the associated translational frameshift) occurs in about 30% of cases and may represent an additional level of control over mxiE gene expression (Penno et al., 2005). This arrangement also influences the efficiency of expression of the immediately-downstream-and-overlapping mxiD gene (Penno and Parsot, 2006). Control of MxiE activity is complex and involves a number of other proteins: the anti-co-activators IpaB and IpaC oppose the positive action of the co-activator IpgC by competing with MxiE for IpgC. The anti-activator OspD1, in association with Spa15, inhibits MxiE. The secretion of these co- and anti-activators by the T3SS modulates the activity of MxiE and links its activity to different stages in the operation of the virulence system (Parsot et al., 2005).
Production of the S. flexneri T3SS is sensitive to oxygen. The oxygen-sensitive FNR protein, which is related to the cAMP receptor protein (Crp), binds to the large virulence plasmid and represses the transcription of the spa32 and spa33 genes (Figure 7). These genes control protein secretion through the T3SS. When the bacterium approaches the epithelial layer at the gut wall, a low concentration of oxygen emanating from the gut surface relieves the FNR-mediated repression, allowing invasion by the bacterium to proceed (Marteyn et al., 2010). In partnership with Fis, Crp regulates the production of the SigA toxin, a cytotoxic protease, encoded by the SHE pathogenicity island on the S. flexneri chromosome (Al-Hasani et al., 2001; Rossiter et al., 2015).
Concluding Remarks
Shigella flexneri and V. cholerae both cause diarrheal diseases in humans and both employ sophisticated mechanisms for environmental sensing and virulence gene regulation. The sites of the diseases differ in that dysentery is a disease of the lower gut while cholera affects the small intestine and the infection strategies are distinct: host cell invasion (dysentery) and gut lining intoxication (cholera). The infectious dose also differs between the two diseases, being very low for dysentery (about 10 bacterial cells) and much higher for cholera (103 to 108 bacterial cells). These distinctions are consistent with the stronger emphasis on inter-bacterial communication that characterizes V. cholerae and evidence that collectives of cells are required at the site of infection for the disease to proceed. It is also consistent with the absence from V. cholerae of an extreme acid resistance (XAR) system, a system that S. flexneri possesses. The probability that V. cholerae can establish an infection is reduced if large numbers of bacteria are killed in the low pH environment of the stomach, creating the need for a large initial inoculum when the host ingests the organisms (Lund et al., 2014). Both pathogens rely on mobile genetic elements that encode virulence factors: pathogenicity islands, a large mobilizable plasmid (dysentery) and a phage (cholera). They share a dependency on the H-NS protein to achieve transcription silencing of their virulence genes, and in the case of S. flexneri, this seems to be a strategy for stable maintenance of the genes and for transmitting them vertically. AraC-like transcription factors feature prominently in virulence gene regulation in both organisms, although V. cholerae and not S. flexneri has a dependence on regulators that are associated with the cytoplasmic membrane. In S. flexneri, the key positive regulatory genes are located on the pINV plasmid with their chief target genes; V. cholerae uses regulatory genes that are co-imported with the target genes (toxT) and others that are part of the core genome (toxR). In both species, core-genome members (e.g., Crp, Fnr) play important roles in fine-tuning the regulatory outputs, as do non-H-NS NAPs such as Fis and IHF.
The virulence gene regulatory features shared by S. flexneri and V. cholerae are also shared by other Gram-negative pathogens, making many of the points raised in this article generally relevant. Horizontal gene transfer has played an important role in the evolution of pathogenic bacteria and the H-NS protein is used widely to silence those genes (Lucchini et al., 2006; Navarre et al., 2006). The mechanisms by which the silencing is overcome in response to specific environmental signals varies from one example to another, but the underlying principle of silencing and anti-silencing of horizontally acquired genes of high A + T base content is a general feature (Stoebel et al., 2008). AraC-like proteins are widespread among Gram-negative bacteria, with many responding to thermal signals and contributing to transcription control in pathogens of mammals (Egan, 2002; Li et al., 2016). Variable DNA topology that responds to environmental stimuli is also widely reported across pathogenic species (Dorman et al., 2016), as are regulatory inputs from nucleoid-associated proteins, inputs that are not restricted to H-NS but include Fis, IHF, HU, and others (Dillon et al., 2010). The specific aspects of virulence gene control in S. flexneri and V. cholerae are many and reflect the natural histories of those organisms, their lifestyles and infection strategies. This is likely to be true in comparisons of any pathogens, even closely related ones. The general pattern described here, of shared regulatory mechanisms operating as a backdrop for individual, species-specific ones, is likely to hold true across bacteriology.
The research methods used in the field have evolved over the years from those that focus on individual genes and their cis- and trans-acting regulators to those approaches that survey events across the entire genome (Ayala et al., 2015a, 2017) or employ single-molecule methods (Haas et al., 2015). The result has been both a broader and a deeper understanding of the regulatory events in V. cholerae and S. flexneri. Whole genome sequencing is revealing important details of the epidemiology of the diseases that these pathogens cause and their evolution (Domman et al., 2017, 2018). It is anticipated that our understanding of the contributions of gene regulatory circuits to their evolution and geographical spread will become ever clearer in the near future.
Author Contributions
MD and CD researched the topic, wrote the manuscript, and prepared the figures.
Funding
CD was supported by Science Foundation Ireland Principal Investigator Award 13/1A/1875. MD was supported by grant 206194 from Wellcome.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank N. R. Thomson for comments on the manuscript.
Abbreviations
7PET, seventh pandemic El Tor genetic lineage; CT, cholera toxin; Fis, factor for inversion stimulation, HTH, helix-turn-helix; IHF, integration host factor; NAP, nucleoid-associated protein; pINV, plasmid for invasion; T3SS, type III secretion system; T6SS, type VI secretion system; TCP, toxin co-regulated pilus; VPI-1 Vibrio pathogenicity island one; wHTH, winged-helix-turn-helix.
References
Adler, B., Sasakawa, C., Tobe, T., Makino, S., Komatsu, K., and Yoshikawa, M. (1989). A dual transcriptional activation system for the 230 kb plasmid genes coding for virulence-associated antigens of Shigella flexneri. Mol. Microbiol. 3, 627–635. doi: 10.1111/j.1365-2958.1989.tb00210.x
Al-Hasani, K., Rajakumar, K., Bulach, D., Robins-Browne, R., Adler, B., and Sakellaris, H. (2001). Genetic organisation of the she pathogenicity island in Shigella flexneri 2a. Microb. Pathog. 30, 1–8. doi: 10.1006/mpat.2000.0404
Ante, V. M., Bina, X. R., and Bina, J. E. (2015a). The LysR-type regulator LeuO regulates the acid tolerance response in Vibrio cholerae. Microbiology 161, 2434–2443. doi: 10.1099/mic.0.000194
Ante, V. M., Bina, X. R., Howard, M. F., Sayeed, S., Taylor, D. L., and Bina, J. E. (2015b). Vibrio cholerae leuO transcription is positively regulated by ToxR and contributes to bile resistance. J. Bacteriol. 197, 3499–3510. doi: 10.1128/JB.00419-15
Ashida, H., Mimuro, H., and Sasakawa, C. (2015). Shigella manipulates host immune responses by delivering effector proteins with specific roles. Front. Immunol. 6:219. doi: 10.3389/fimmu.2015.00219
Ayala, J. C., Silva, A. J., and Benitez, J. A. (2017). H-NS: an overarching regulator of the Vibrio cholerae life cycle. Res. Microbiol. 168, 16–25. doi: 10.1016/j.resmic.2016.07.007
Ayala, J. C., Wang, H., Benitez, J. A., and Silva, A. J. (2015a). RNA-Seq analysis and whole genome DNA-binding profile of the histone-like nucleoid structuring protein (H-NS). Genomics Data 5, 147–150. doi: 10.1016/j.gdata.2015.05.039
Ayala, J. C., Wang, H., Silva, A. J., and Benitez, J. A. (2015b). Repression by H-NS of genes required for the biosynthesis of the Vibrio cholerae biofilm matrix is modulated by the second messenger cyclic diguanylic acid. Mol. Microbiol. 97, 630–645. doi: 10.1111/mmi.13058
Ayala, J. C., Wang, H., Benitez, J. A., and Silva, A. J. (2018). Molecular basis for the differential expression of the global regulator VieA in Vibrio cholerae biotypes directed by H-NS, LeuO and quorum sensing. Mol. Microbiol. 107, 330–343. doi: 10.1111/mmi.13884
Basta, D. W., Pew, K. L., Immak, J. A., Park, H. S., Picker, M. A., Wigley, A. F., et al. (2013). Characterization of the ospZ promoter in Shigella flexneri and its regulation by VirB and H-NS. J. Bacteriol. 195, 2562–2572. doi: 10.1128/JB.00212-13.
Basu, P., Pal, R. R., Dasgupta, S., and Bhandra, R. K. (2017). DksA-HapR-RpoS axis regulates haemagglutinin protease production in Vibrio cholerae. Microbiology 163, 900–910. doi: 10.1099/mic.0.000469
Beck, N. A., Krukonis, E. S., and DiRita, V. J. (2004). TcpH influences virulence gene expression in Vibrio cholerae by inhibiting degradation of the transcription activator TcpP. J. Bacteriol. 186, 8309–8316. doi: 10.1128/JB.186.24.8309-8316.2004
Beloin, C., and Dorman, C. J. (2003). An extended role for the nucleoid structuring protein H-NS in the virulence gene regulatory cascade of Shigella flexneri. Mol. Microbiol. 47, 825–838. doi: 10.1046/j.1365-2958.2003.03347.x
Beloin, C., McKenna, S., and Dorman, C. J. (2002). Molecular dissection of VirB, a key regulator of the virulence cascade of Shigella flexneri. J. Biol. Chem. 277,15333–15344. doi: 10.1074/jbc.M111429200
Bernardini, M. L., Fontaine, A., and Sansonetti, P. J. (1990). The two-component regulatory system ompR-envZ controls the virulence of Shigella flexneri. J. Bacteriol. 172, 6274–6281. doi: 10.1128/jb.172.11.6274-6281.1990
Bernardini, M. L., Sanna, M. G., Fontaine, A., and Sansonetti, P. J. (1993). OmpC is involved in invasion of epithelial cells by Shigella flexneri. Infect. Immun. 61, 3625–3365.
Beyhan, S., Tischler, A. D., Camilli, A., and Yildiz, F. H. (2006). Differences in gene expression between the classical and El Tor biotypes of Vibrio cholerae O1. Infect. Immun. 74, 3633–3642. doi: 10.1128/IAI.01750-05
Bina, X. R., Howard, M. F., Ante, V. M., and Bina, J. E. (2016). Vibrio cholerae LeuO links the ToxR regulon to expression of lipid A remodeling genes. Infect. Immun. 84, 3161–3171. doi: 10.1128/IAI.00445-16
Bina, X. R., Taylor, D. L., Vikram, A., Ante, V. M., and Bina, J. E. (2013). Vibrio cholerae ToxR downregulates virulence factor production in response to cyclo(Phe-Pro). mBio 4:e00366-13. doi: 10.1128/mBio.00366-13
Blokesch, M. (2012). Chitin colonization, chitin degradation and chitin- induced natural competence of Vibrio cholerae are subject to catabolite repression. Environ. Microbiol. 14, 1898–1912. doi: 10.1111/j.1462-2920.2011.02689.x
Blokesch, M., and Schoolnik, G. K. (2008). The extracellular nuclease Dns and its role in natural transformation of Vibrio cholerae. J. Bacteriol. 190, 7232–7240. doi: 10.1128/JB.00959-08
Bongrand, C., Sansonetti, P. J., and Parsot, C. (2012). Characterization of the promoter, MxiE box and 5’ UTR of genes controlled by the activity of the type III secretion apparatus in Shigella flexneri. PLoS One 7:e32862. doi: 10.1371/journal.pone.0032862
Bradley, E. S., Bodi, K., Ismail, A. M., and Camilli, A. (2011). A genome-wide approach to discovery of small RNAs involved in regulation of virulence in Vibrio cholerae. PLoS Pathog. 7:e10002126 doi: 10.1371/journal.ppat.1002126
Brown, R. C., and Taylor, R. K. (1995). Organization of tcp, acf, and toxT genes within a ToxT-dependent operon. Mol. Microbiol. 16, 425–439. doi: 10.1111/j.1365-2958.1995.tb02408.x
Buchrieser, C., Glaser, P., Rusniok, C., Nedjari, H., D’Hauteville, H., Kunst, F., et al. (2000). The virulence plasmid pWR100 and the repertoire of proteins secreted by the type III secretion apparatus of Shigella flexneri. Mol. Microbiol. 38, 760–771. doi: 10.1046/j.1365-2958.2000.02179.x
Caiyi, C. L., Merrell, D. S., Camilli, A., and Kaper, J. B. (2002). ToxR interferes with CRP-dependent transcriptional activation of ompT in Vibrio cholerae. Mol. Microbiol. 43, 1577–1589. doi: 10.1046/j.1365-2958.2002.02845.x
Carroll, P. A., Tashima, K. T., Rogers, M. B., DiRita, V. J., and Calderwood, S. B. (1997). Phase variation in tcpH modulates expression of the ToxR regulon in Vibrio cholerae. Mol. Microbiol. 25, 1099–1111. doi: 10.1046/j.1365-2958.1997.5371901.x
Castellanos, M. I., Harrison, D. J., Smith, J. M., Labahn, S. K., Levy, K. M., and Wing, H. J. (2009). VirB alleviates H-NS repression of the icsP promoter in Shigella flexneri from sites more than one kilobase upstream of the transcription start site. J. Bacteriol. 191, 4047–4050. doi: 10.1128/JB.00313-09
Cerdan, R., Bloch, V., Yang, Y., Bertin, P., Dumas, C., Rimsky, S., et al. (2003). Crystal structure of the N-terminal dimerisation domain of VicH, the H-NS-like protein of Vibrio cholerae. J. Mol. Biol. 334, 179–185. doi: 10.1016/j.jmb.2003.09.051
Chakraborty, S., Winardhi, R. S., Morgan, L. K., Yan, J., and Kenney, L. J. (2017). Non-canonical activation of OmpR drives acid and osmotic stress responses in single bacterial cells. Nat. Commun. 8:1587. doi: 10.1038/s41467-017-02030-0
Champion, G. A., Neely, M. N., Brennan, M. A., and DiRita, V. J. (1997). A branch in the ToxR regulatory cascade of Vibrio cholerae revealed by characterization of toxT mutant strains. Mol. Microbiol. 23, 323–331. doi: 10.1046/j.1365-2958.1997.2191585.x
Chapman, C., Henry, M., Bishop-Lilly, K. A., Awosika, J., Briska, A., Ptashkin, R. N., et al. (2015). Scanning the landscape of genome architecture of non-O1 and non-O139 Vibrio cholerae by whole genome mapping reveals extensive population genetic diversity. PLoS One 10:e0120311. doi: 10.1371/journal.pone.0120311
Chatterjee, A., Dutta, P. K., and Chowdhury, R. (2007). Effect of fatty acids and cholesterol present in bile on expression of virulence factors and motility of Vibrio cholerae. Infect. Immun. 75, 1946–1953. doi: 10.1128/IAI.01435-06
Chen, L. C., Rohde, J. E., and Sharp, G. W. (1971). Intestinal adenyl-cyclase activity in human cholera. Lancet 1, 939–941. doi: 10.1016/S0140-6736(71)91443-7
Cohen, M. S., and Chang, P. (2018). Insights into the biogenesis, function, and regulation of ADP-ribosylation. Nat. Chem. Biol. 14, 236–243 doi: 10.1038/nchembio.2568
Colonna, B., Casalino, M., Fradiani, P. A., Zagaglia, C., Naitza, S., Leoni, L., et al. (1995). H-NS regulation of virulence gene expression in enteroinvasive Escherichia coli harboring the virulence plasmid integrated into the host chromosome. J. Bacteriol. 177, 4703–4712. doi: 10.1128/jb.177.16.4703-4712.1995
Dalebroux, Z. D., Svensson, S. L., Gaynor, E. C., and Swanson, M. S. (2010). ppGpp conjures bacterial virulence. Microbiol. Mol. Biol. Rev. 74, 171–199. doi: 10.1128/MMBR.00046-09
Dalebroux, Z. D., and Swanson, M. S. (2012). ppGpp: magic beyond RNA polymerase. Nat. Rev. Microbiol. 10, 203–212. doi: 10.1038/nrmicro2720
Dalia, A. B. (2016). RpoS is required for natural transformation of Vibrio cholerae through regulation of chitinases. Environ. Microbiol. 18, 3758–3767. doi: 10.1111/1462-2920.13302
Dalia, A. B., Lazinski, D. W., and Camilli, A. (2014). Identification of a membrane-bound transcriptional regulator that links chitin and natural competence in Vibrio cholerae. mBio 5:e01028-13. doi: 10.1128/mBio.01028-13
Dame, R. T., Noom, M. C., and Wuite, G. J. (2006). Bacterial chromatin organization by H-NS protein unravelled using dual DNA manipulation. Nature 444, 387–390. doi: 10.1038/nature05283
Das, B., Martínez, E., Midonet, C., and Barre, F. X. (2013). Integrative mobile elements exploiting Xer recombination. Trends Microbiol. 21, 23–30. doi: 10.1016/j.tim.2012.10.003
Das, B., Pal, R. R., Bag, S., and Bhadra, R. K. (2009). Stringent response in Vibrio cholerae: genetic analysis of spoT gene function and identification of a novel (p)ppGpp synthetase gene. Mol. Microbiol. 72, 380–398. doi: 10.1111/j.1365-2958.2009.06653.x
Davies, B. W., Bogard, R. W., Young, T. S., and Mekalanos, J. J. (2012). Coordinated regulation of accessory genetic elements produces cyclic di-nucleotides for V. cholerae virulence. Cell 149, 358–370. doi: 10.1016/j.cell.2012.01.053
Day, W. A. Jr., and Maurelli, A. T. (2001). Shigella flexneri LuxS quorum-sensing system modulates virB expression but is not essential for virulence. Infect. Immun. 69, 15–23. doi: 10.1128/IAI.69.1.15-23.2001
De Haan, L., and Hirst, T. R. (2004). Cholera toxin: a paradigm for multi-functional engagement of cellular mechanisms. Mol. Membr. Biol. 21, 77–92. doi: 10.1080/09687680410001663267
De Silva, R. S., Kovacikova, G., Lin, W., Taylor, R. K., and Skorupski, K. (2005). Crystal structure of the virulence gene activator AphA from Vibrio cholerae reveals it is a novel member of the winged helix transcription factor superfamily. J. Bio. Chem. 280, 13779–13783. doi: 10.1074/jbc.M413781200
Dewachter, L., Verstraeten, N., Fauvart, M., and Michiels, J. (2016). The bacterial cell cycle checkpoint protein Obg and its role in programmed cell death. Microb. Cell. 3, 255-256. doi: 10.15698/mic2016.06.507
d’Hauteville, H., Dufourc Qlagelouse, R., Nato, F., and Sansonetti, P. J. (1996). Lack of cleavage of IcsA in Shigella flexneri causes aberrant movement and allows demonstration of a cross-reactive eukaryotic protein. Infect. Immun. 64, 511–517.
Di Martino, M. L., Falconi, M., Micheli, G., Colonna, B., and Prosseda, G. (2016a). The multifaceted activity of the VirF regulatory protein in the Shigella lifestyle. Front. Mol. Biosci. 3:61. doi: 10.3389/fmolb.2016.00061
Di Martino, M. L., Romilly, C., Wagner, E. G., Colonna, B., and Prosseda, G. (2016b). One gene and two proteins: a leaderless mRNA supports the translation of a shorter form of the Shigella VirF regulator. mBio 7, e01860-16. doi: 10.1128/mBio.01860-16.
Dillon, S. C., and Dorman, C. J. (2010). Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nat. Rev. Microbiol. 8, 949–959. doi: 10.1038/nrmicro2261
Dillon, S. C., Espinosa, E., Hokamp, K., Ussery, D. W., Casadesús, J., and Dorman, C. J. (2012). LeuO is a global regulator of gene expression in Salmonella enterica serovar Typhimurium. Mol. Microbiol. 85, 1072–1089. doi: 10.1111/j.1365-2958.2012.08162.x
DiRita, V. J., and Mekalanos, J. J. (1991). Periplasmic interaction between two membrane regulatory proteins, ToxR and ToxS, results in signal transduction and transcriptional activation. Cell 64, 29–37. doi: 10.1016/0092-8674(91)90206-E
DiRita, V. J., Parsot, C., Jander, G., and Mekalanos, J. J. (1991). Regulatory cascade controls virulence in Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A. 88, 5403–5407. doi: 10.1073/pnas.88.12.5403
Domman, D., Chowdhury, F., Khan, A. I., Dorman, M. J., Mutreja, A., Uddin, M. I., et al. (2018). Defining endemic cholera at three levels of spatiotemporal resolution within Bangladesh. Nat. Genet. 50, 951–955. doi: 10.1038/s41588-018-0150-8
Domman, D., Quilici, M. L., Dorman, M. J., Njamkepo, E., Mutreja, A., Mather, A. E., et al. (2017). Integrated view of Vibrio cholerae in the Americas. Science 358, 789–793. doi: 10.1126/science.aao2136
Dorman, C. J. (1992). The VirF protein from Shigella flexneri is a member of the AraC transcription factor superfamily and is highly homologous to Rns, a positive regulator of virulence genes in enterotoxigenic Escherichia coli. Mol. Microbiol. 6:1575. doi: 10.1111/j.1365-2958.1992.tb00879.x
Dorman, C. J. (1995). DNA topology and the global control of bacterial gene expression: implications for the regulation of virulence gene expression. Microbiology 141, 1271–1280. doi: 10.1099/13500872-141-6-1271
Dorman, C. J. (2007). H-NS, the genome sentinel. Nat. Rev. Microbiol. 5, 157–161. doi: 10.1038/nrmicro1598
Dorman, C. J. (2009). The virulence plasmids of Shigella flexneri, in Microbial Megaplasmids, ed. E. Schwartz (Heidelberg: Springer 151–170.
Dorman, C. J. (2015). Integrating small molecule signalling and H-NS antagonism in Vibrio cholerae, a bacterium with two chromosomes. Mol. Microbiol. 97, 612–615. doi: 10.1111/mmi.13063
Dorman, C. J., Colgan, A. M., and Dorman, M. J. (2016). Bacterial pathogen gene regulation: a DNA-structure-centred view of a protein-dominated domain. Clin. Sci. 130, 1165–1177. doi: 10.1042/CS20160024
Dorman, C. J., and Dorman, M. J. (2017). Control of virulence gene transcription by indirect readout in Vibrio cholerae and Salmonella enterica serovar Typhimurium. Environ. Microbiol. 19, 3834–3845. doi: 10.1111/1462-2920
Dorman, C. J., and Kane, K. A. (2009). DNA bridging and antibridging: a role for bacterial nucleoid-associated proteins in regulating the expression of laterally acquired genes. FEMS Microbiol. Rev. 33, 587–592. doi: 10.1111/j.1574-6976.2008.00155.x
Dorman, C. J., Ni Bhriain, N., and Higgins, C. F. (1990). DNA supercoiling and environmental regulation of virulence gene expression in Shigella flexneri. Nature 344, 789–792. doi: 10.1078/1438-4221-00105
Dorman, C.J., McKenna, S., and Beloin, C. (2001). Regulation of virulence gene expression in Shigella flexneri, a facultative intracellular pathogen. Int. J. Med. Microbiol. 291, 89–96. doi: 10.1046/j.1365-2958.2003.03314.x
Durand, J. M., and Björk, G. R. (2003). Putrescine or a combination of methionine and arginine restores virulence gene expression in a tRNA modification-deficient mutant of Shigella flexneri: a possible role in adaptation of virulence. Mol. Microbiol. 47, 519–527. doi: 10.1046/j.1365-2958.2000.01767.x
Durand, J. M., Dagberg, B., Uhlin, B. E., and Björk, G. R. (2000). Transfer RNA modification, temperature and DNA superhelicity have a common target in the regulatory network of the virulence of Shigella flexneri: the expression of the virF gene. Mol. Microbiol. 35, 924–935. doi: 10.1128/JB.184.20.5529-5532.2002
Egan, S. M. (2002). Growing repertoire of AraC/XylS activators. J. Bacteriol. 184, 5529–5532. doi: 10.1128/IAI.71.6.2993-2999.2003
Faruque, S. M., Zhu, J., Asadulghani, Kamruzzaman, M., and Mekalanos, J. J. (2003). Examination of diverse toxin-coregulated pilus-positive Vibrio cholerae strains fails to demonstrate evidence for Vibrio pathogenicity island phage. Infect. Immun. 71, 2993–2999. doi: 10.1371/journal.pone.0047756
Fengler, V. H., Boritsch, E. C., Tutz, S., Seper, A., Ebner, H., Roier, S., et al. (2012). Disulfide bond formation and ToxR activity in Vibrio cholerae. PLoS One 7:e47756. doi: 10.1371/journal.pone.0047756.
Fu, Y., Ho, B. T., and Mekalanos, J. J. (2018). Tracking Vibrio cholerae cell-cell interactions during infection reveals bacterial population dynamics within intestinal microenvironments. Cell Host Microbe 23, 274–281. doi: 10.1016/j.chom.2017.12.006
Fukuda, I., Suzuki, T., Munakata, H., Hayashi, N., Katayama, E., Yoshikawa, M., et al. (1995). Cleavage of Shigella surface protein VirG occurs at a specific site, but the secretion is not essential for intracellular spreading. J. Bacteriol. 177, 1719–1726. doi: 10.1016/j.chom.2012.03.001
Fukumatsu, M., Ogawa, M., Arakawa, S., Suzuki, M., Nakayama, K., Shimizu, S., et al. (2012). Shigella targets epithelial tricellular junctions and uses a noncanonical clathrin-dependent endocytic pathway to spread between cells. Cell Host Microbe 11, 325–336. doi: 10.1128/IAI.74.5.3060-3064.2006
Ghosh, A., Paul, K., and Chowdhury, R. (2006). Role of the histone-like nucleoid structuring protein in colonization, motility, and bile-dependent repression of virulence gene expression in Vibrio cholerae. Infect. Immun. 74, 3060–3064. doi: 10.1074/jbc.M116.761809
Gkekas, S., Singh, R. K., Shkumatov, A. V., Messens, J., Fauvart, M., Verstraeten, N., et al. (2017). Structural and biochemical analysis of Escherichia coli ObgE, a central regulator of bacterial persistence. J. Biol. Chem. 292, 5871–5883. doi: 10.1074/jbc.M116.761809
Gore, A. L., and Payne, S. M. (2010). CsrA and Cra influence Shigella flexneri pathogenesis. Infect. Immun. 78, 4674–4682. doi: 10.1128/IAI.00566-10
Goss, T. J., Seaborn, C. P., Gray, M. D., and Krukonis, E. S. (2010). Identification of the TcpP-binding site in the toxT promoter of Vibrio cholerae and the role of ToxR in TcpP-mediated activation. Infect. Immun. 78, 4122–4133. doi: 10.1128/IAI.00566-10
Guadarrama, C., Medrano-López, A., Oropeza, R., Hernández-Lucas, I., and Calva, E. (2014). The Salmonella enterica serovar Typhi LeuO global regulator forms tetramers: residues involved in oligomerization, DNA binding, and transcriptional regulation. J. Bacteriol. 196, 2143–2154. doi: 10.1128/JB.01484-14
Haas, B. L., Matson, J. S., DiRita, V. J., and Biteen, J. S. (2015). Single-molecule tracking in live Vibrio cholerae reveals that ToxR recruits the membrane-bound virulence regulator TcpP to the toxT promoter. Mol. Microbiol. 96, 4–13. doi: 10.1111/mmi.12834
Häse, C. C., and Mekalanos, J. J. (1998). TcpP protein is a positive regulator of virulence gene expression in Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A. 95, 730–734. doi: 10.1073/pnas.95.2.730
He, H., Cooper, J. N., Mishra, A., and Raskin, D. M. (2012). Stringent response regulation of biofilm formation in Vibrio cholerae. J. Bacteriol. 194, 2962–2972. doi: 10.1128/JB.00014-12
Higgins, D. E., and DiRita, V. J. (1996). Genetic analysis of the interaction between Vibrio cholerae transcription activator ToxR and toxT promoter DNA. J. Bacteriol. 178, 1080–1087. doi: 10.1128/jb.178.4.1080-1087.1996
Higgins, D. E., Nazareno, E., and DiRita, V. J. (1992). The virulence gene activator ToxT from Vibrio cholerae is a member of the AraC family of transcriptional activators. J. Bacteriol. 174, 6974–6980. doi: 10.1128/jb.174.21.6974-6980.1992
High, N., Mounier, J., Preìvost, M. C., and Sansonetti, P. J. (1992). IpaB of Shigella flexneri causes entry into epithelial cells and escape from the phagocytic vacuole. EMBO J. 11, 1991–1999. doi: 10.1002/j.1460-2075.1992.tb05253.x
Hryckowian, A. J., Battesti, A., Lemke, J. J., Meyer, Z. C., and Welch, R. A. (2014). IraL is an RssB anti-adaptor that stabilizes RpoS during logarithmic phase growth in Escherichia coli and Shigella. mBio. 5:e01043-14. doi: 10.1128/mBio.01043-14
Huber, K. E., and Waldor, M. K. (2002). Filamentous phage integration requires the host recombinases XerC and XerD. Nature 417, 656–659. doi: 10.1038/nature00782
Hung, D. T., and Mekalanos, J. J. (2005). Bile acids induce cholera toxin expression in Vibrio cholerae in a ToxT-independent manner. Proc. Natl. Acad. Sci. U.S.A. 102, 3028–3033. doi: 10.1073/pnas.0409559102
Iwanaga, M., and Yamamoto, K. (1985). New medium for the production of cholera toxin by Vibrio cholerae O1 biotype El Tor. J. Clin. Microbiol. 22, 405–408.
Iwanaga, M., Yamamoto, K., Higa, N., Ichinose, Y., Nakasone, N., and Tanabe, M. (1986). Culture conditions for stimulating cholera toxin production by Vibrio cholerae O1 El Tor. Microbiol. Immunol. 30, 1075–1083. doi: 10.1111/j.1348-0421.1986.tb03037.x
Jang, J., Jung, K. -T., Park, J., Yoo, C. -K., and Rhie, G. -E. (2011). The Vibrio cholerae VarS/VarA two-component system controls the expression of virulence proteins through ToxT regulation. Microbiology 157, 1466–1473. doi: 10.1099/mic.0.043737-0
Joelsson, A., Kan, B., and Zhu, J. (2007). Quorum sensing enhances the stress response in Vibrio cholerae. Appl. Environ. Microbiol. 73, 3742–3746. doi: 10.1128/AEM.02804-06
Kane, C. D., Schuch, R., Day, W.A. Jr., and Maurelli, A. T. (2002). MxiE regulates intracellular expression of factors secreted by the Shigella flexneri 2a type III secretion system. J. Bacteriol. 184, 4409–4419. doi: 10.1128/JB.184.16.4409-4419.2002
Kane, K. A., and Dorman, C. J. (2011). Rational design of an artificial genetic switch: Co-option of the H-NS-repressed proU operon by the VirB virulence master regulator. J. Bacteriol. 193, 5950-5960. doi: 10.1128/JB.05557-11
Kane, K. A., and Dorman, C. J. (2012). VirB-mediated positive feedback control of the virulence gene regulatory cascade of Shigella flexneri. J. Bacteriol. 194, 5264–5273. doi: 10.1128/JB.00800-12
Karaolis, D. K., Johnson, J. A., Bailey, C. C., Boedeker, E. C., Kaper, J. B., and Reeves, P. R. (1998). A Vibrio cholerae pathogenicity island associated with epidemic and pandemic strains. Proc. Natl. Acad. Sci. U.S.A. 95, 3134–3139. doi: 10.1073/pnas.95.6.3134
Karaolis, D. K., Somara, S., Maneval, D. R. Jr., Johnson, J. A., and Kaper, J. B. (1999). A bacteriophage encoding a pathogenicity island, a type-IV pilus and a phage receptor in cholera bacteria. Nature 399, 375–379. doi: 10.1038/20715
Kazi, M. I., Conrado, A. R., Mey, A. R., Payne, S. M., and Davies, B. W. (2016). ToxR antagonizes H-NS regulation of horizontally acquired genes to drive host colonization. PLoS Pathog. 12:e1005570. doi: 10.1371/journal.ppat.1005570
King, C. A., and van Heyningen, W. E. (1973). Deactivation of cholera toxin by a sialidase monosialosylganglioside. J. Infect. Dis. 127, 639–647. doi: 10.1093/infdis/127.6.639
Klancher, C. A., Hayes, C. A., and Dalia, A. B. (2017). The nucleoid occlusion protein SlmA is a direct transcriptional activator of chitobiose utilization in Vibrio cholerae. PLoS Genet. 13:e1006877. doi: 10.1371/journal.pgen.1006877
Klauck, E., Typas, A., and Hengge, R. (2007). The sigmaS subunit of RNA polymerase as a signal integrator and master regulator in the general stress response in Escherichia coli. Sci. Prog. 90, 103–127.
Kovacikova, G., Lin, W., and Skorupski, K. (2003). The virulence activator AphA links quorum sensing to pathogenesis and physiology in Vibrio cholerae by repressing the expression of a penicillin amidase gene on the small chromosome. J. Bacteriol. 185, 4825-4836. doi: 10.1128/JB.16.4825-4836.2003
Kovacikova, G., Lin, W., and Skorupski, K. (2004). Vibrio cholerae AphA uses a novel mechanism for virulence gene activation that involves interaction with the LysR-type regulator AphB at the tcpPH promoter. Mol. Microbiol. 53, 129–142. doi: 10.1111/j.1365-2958.2004.04121.x
Kovacikova, G., Lin, W., and Skorupski, K. (2005). Dual regulation of genes involved in acetoin biosynthesis and motility/biofilm formation by the virulence activator AphA and the acetate-responsive LysR-type regulator AlsR in Vibrio cholerae. Mol. Microbiol. 57, 420–433. doi: 10.1111/j.1365-2958.2005.04700.x
Kovacikova, G., Lin, W., and Skorupski, K. (2010). The LysR-type virulence activator AphB regulates the expression of genes in Vibrio cholerae in response to low pH and anaerobiosis. J. Bacteriol. 192, 4181–4191. doi: 10.1128/JB.00193-10
Kovacikova, G., and Skorupski, K. (2001). Overlapping binding sites for the virulence gene regulators AphA, AphB and cAMP-CRP at the Vibrio cholerae tcpPH promoter. Mol. Microbiol. 41, 393–407. doi: 10.1046/j.1365-2958.2001.02518.x
Kovacikova, G., and Skorupski, K. (2002). Regulation of virulence gene expression in Vibrio cholerae by quorum sensing: HapR functions at the aphA promoter. Mol. Microbiol. 46, 1135–1147. doi: 10.1046/j.1365-2958.2002.03229.x
Krin, E., Cambray, G., and Mazel, D. (2014). The superintegron integrase and the cassette promoters are co-regulated in Vibrio cholerae. PLoS One 9:e91194. doi: 10.1371/journal.pone.0091194
Krukonis, E. S., and DiRita, V. J. (2003). DNA binding and ToxR responsiveness by the wing domain of TcpP, an activator of virulence gene expression in Vibrio cholerae. Mol. Cell 12, 157–165. doi: 10.1016/S1097-2765(03)00222-3
Krukonis, E. S., Yu, R. R., and DiRita, V. J. (2000). The Vibrio cholerae ToxR/TcpP/ToxT virulence cascade: distinct roles for two membrane-localized transcriptional activators on a single promoter. Mol. Microbiol. 38, 67–84. doi: 10.1046/j.1365-2958.2000.02111.x
Lan, R., Lumb, B., Ryan, D., and Reeves, P. R. (2001). Molecular evolution of large virulence plasmid in Shigella clones and enteroinvasive Escherichia coli. Infect. Immun. 69, 6303–6309. doi: 10.1128/IAI.69.10.6303-6309.2001
Le Roux, F., and Blokesch, M. (2018). Eco-evolutionary dynamics linked to horizontal gene transfer in vibrios. Annu. Rev. Microbiol. 72, 89–110. doi: 10.1146/annurev-micro-090817-062148
Lee, K.-M., Park, Y., Bari, W., Yoon, M. Y., Go, J., Kim, S. C., et al. (2012). Activation of Cholera toxin production by anaerobic respiration of trimethylamine N-oxide in Vibrio cholerae. J. Biol. Chem. 287, 39742–39752. doi: 10.1074/jbc.M112.394932
Lenz, D.H., and Bassler, B.L. (2007) The small nucleoid protein Fis is involved in Vibrio cholerae quorum sensing. Mol. Microbiol. 63, 859–876. doi: 10.1111/j.1365-2958.2006.05545.x
Lenz, D.H., Miller, M.B., Zhu, J., Kulkarni, R.V., and Bassler, B.L. (2005). CsrA and three redundant small RNAs regulate quorum sensing in Vibrio cholerae. Mol. Microbiol. 58, 1186–1202. doi: 10.1111/j.1365-2958.2005.04902.x
Li, J., Wehmeyer, G., Lovell, S., Battaile, K. P., and Egan, S. M. (2016). 1.65 Å resolution structure of the AraC-family transcriptional activator ToxT from Vibrio cholerae. Acta. Crystallogr. F. Struct. Biol. Commun. 72, 726–731. doi: 10.1107/S2053230X1601298X
Li, X., and Roseman, S. (2004). The chitinolytic cascade in vibrios is regulated by chitin oligosaccharides and a two-component chitin catabolic sensor/kinase. Proc. Natl. Acad. Sci. U.S.A. 101, 627–631. doi: 10.1073/pnas.0307645100
Lim, M. S., Ng, D., Zong, Z., Arvai, A. S., Taylor, R. K., Tainer, J. A., et al. (2010). Vibrio cholerae El Tor TcpA crystal structure and mechanism for pilus-mediated microcolony formation. Mol. Microbiol. 77, 755–770. doi: 10.1111/j.1365-2958.2010.07244.x
Lin, W., Kovacikova, G., and Skorupski, K. (2007). The quorum sensing regulator HapR downregulates the expression of the virulence gene transcription factor AphA in Vibrio cholerae by antagonising Lrp- and VpsR-mediated activation. Mol. Microbiol. 64, 953–967 doi: 10.1111/j.1365-2958.2007.05693.x
Liu, Z., Yang, M., Peterfreund, G. L., Tsou, A. M., Selamoglu, N., Daldal, F., et al. (2011). Vibrio cholerae anaerobic induction of virulence gene expression is controlled by thiol-based switches of virulence regulator AphB. Proc. Natl. Acad. Sci. U.S.A. 108, 810–815. doi: 10.1073/pnas.1014640108
Lowden, M. J., Skorupski, K., Pellegrini, M., Chiorazzo, M. G., Taylor, R. K., and Kull, F. J. (2010). Structure of Vibrio cholerae ToxT reveals a mechanism for fatty acid regulation of virulence genes. Proc. Natl. Acad. Sci. U.S.A. 107, 2860–2865. doi: 10.1073/pnas.0915021107
Lucchini, S., Rowley, G., Goldberg, M. D., Hurd, D., Harrison, M., and Hinton, J. C. (2006). H-NS mediates the silencing of laterally acquired genes in bacteria. PLoS Pathog. 2:e81. doi: 10.1371/journal.ppat.0020081
Lund, P., Tramonti, A., and De Biase, D. (2014). Coping with low pH: molecular strategies in neutralophilic bacteria. FEMS Microbiol. Rev. 38: 1091–1125. doi: 10.1111/1574-6976.12076
Marman, H. E., Mey, A. R., and Payne, S. M. (2014). Elongation factor P and modifying enzyme PoxA are necessary for virulence of Shigella flexneri. Infect. Immun. 82, 3612–3621. doi: 10.1128/IAI.01532-13
Marteyn, B., West, N. P., Browning, D. F., Cole, J. A., Shaw, J. G., Palm, F., et al. (2010). Modulation of Shigella virulence in response to available oxygen in vivo. Nature 465, 355–358. doi: 10.1038/nature08970
Maurelli, A. T., Fernandez, R. E., Bloch, C. A., Rode, C. K., and Fasano, A. (1998). “Black holes” and bacterial pathogenicity: a large genomic deletion that enhances the virulence of Shigella spp. and enteroinvasive Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 95, 3943–3948. doi: 10.1073/pnas.95.7.3943
Mauricio, R. P., Jeffries, C. M., Svergun, D. I., and Deane, J. E. (2016). The Shigella virulence factor IcsA relieves N-WASP autoinhibition by displacing the verprolin homology/cofilin/acidic (VCA) domain. J. Biol. Chem. 292, 134–145. doi: 10.1074/jbc.M116.758003
Mavris, M., Sansonetti, P. J., and Parsot, C. (2002). Identification of the cis-acting site involved in activation of promoters regulated by activity of the type III secretion apparatus in Shigella flexneri. J. Bacteriol. 184, 6751–6759. doi: 10.1128/JB.184.24.6751-6759.2002
McKenna, S., Beloin, C., and Dorman, C.J. (2003). In vitro DNA-binding properties of VirB, the Shigella flexneri virulence regulatory protein. FEBS Lett. 545, 183–187. doi: 10.1016/S0014-5793(03)00524-6
McLeod, S. M., and Waldor, M. K. (2004). Characterization of XerC- and XerD-dependent CTX phage integration in Vibrio cholerae. Mol. Microbiol. 54, 935–947. doi: 10.1111/j.1365-2958.2004.04309.x
McNairn, E., Ni Bhriain, N., and Dorman, C. J. (1995). Overexpression of the Shigella flexneri genes coding for DNA topoisomerase IV compensates for loss of DNA topoisomerase I: effect on virulence gene expression. Mol. Microbiol. 15, 507–517. doi: 10.1111/j.1365-2958.1995.tb02264.x
Meibom, K. L., Blokesch, M., Dolganov, N. A., Wu, C., and Schoolnik, G. K. (2005). Chitin induces natural competence in Vibrio cholerae. Science 310, 1824–1827. doi: 10.1126/science.1120096
Merrell, D. S., and Camilli, A. (1999). The cadA gene of Vibrio cholerae is induced during infection and plays a role in acid tolerance. Mol. Microbiol. 34, 836–849. doi: 10.1046/j.1365-2958.1999.01650.x
Merrell, D. S., and Camilli, A. (2000). Regulation of Vibrio cholerae genes required for acid tolerance by a member of the “ToxR-like” family of transcription regulators. J. Bacteriol. 182, 5342–5350. doi: 10.1128/JB.182.19.5342-5350.2000
Merrell, D. S., Tischler, A. D., Lee, S. H., and Camilli, A. (2000). Vibrio cholerae requires rpoS for efficient intestinal colonization. Infect. Immun. 68, 6691–6696. doi: 10.1128/IAI.68.12.6691-6696.2000
Mey, A. R., Butz, H. A., and Payne, S. M. (2015). Vibrio cholerae CsrA regulates ToxR levels in response to amino acids and is essential for virulence. mBio 6:e01064. doi: 10.1128/mBio.01064-15
Midgett, C. R., Almagro-Moreno, S., Pellegrini, M., Taylor, R. K., Skorupski, K., and Kull, F. J. (2017). Bile salts and alkaline pH reciprocally modulate the interaction between the periplasmic domains of Vibrio cholerae ToxR and ToxS. Mol. Microbiol. 105, 258–272 doi: 10.1111/mmi.13699
Miller, V. L., DiRita, V. J., and Mekalanos, J. J. (1989). Identification of toxS, a regulatory gene whose product enhances toxR-mediated activation of the cholera toxin promoter. J. Bacteriol. 171, 1288–1293. doi: 10.1128/jb.171.3.1288-1293.1989
Miller, V. L., and Mekalanos, J. J. (1988). A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires ToxR. J. Bacteriol. 170, 2575–2583. doi: 10.1128/jb.170.6.2575-2583.1988
Miller, V. L., Taylor, R. K., and Mekalanos, J. J. (1987). Cholera toxin transcriptional activator toxR is a transmembrane DNA binding protein. Cell 48, 271–279. doi: 10.1016/0092-8674(87)90430-2
Mitobe, J., Arakawa, E., and Watanabe, H. (2005). A sensor of the two-component system CpxA affects expression of the type III secretion system through posttranscriptional processing of InvE. J. Bacteriol. 187, 107–113. doi: 10.1128/JB.187.1.107-113.2005
Mitobe, J., Morita-Ishihara, T., Ishihama, A., and Watanabe, H. (2008). Involvement of RNA-binding protein Hfq in the post-transcriptional regulation of invE gene expression in Shigella sonnei. J. Biol. Chem. 283, 5738–5747. doi: 10.1074/jbc.M710108200
Mitobe, J., Morita-Ishihara, T., Ishihama, A., and Watanabe, H. (2009). Involvement of RNA-binding protein Hfq in the osmotic-response regulation of invE gene expression in Shigella sonnei. BMC Microbiol. 9:110. doi: 10.1186/1471-2180-9-110
Mitobe, J., Yanagihara, I., Ohnishi, K., Yamamoto, S., Ohnishi, M., Ishihama, A., et al. (2011). RodZ regulates the post-transcriptional processing of the Shigella sonnei type III secretion system. EMBO Rep. 12, 911–916. doi: 10.1038/embor.2011.132
Mogull, S. A., Runyen-Janecky, L. J., Hong, M., and Payne, S. M. (2001). dksA is required for intercellular spread of Shigella flexneri via an RpoS-independent mechanism. Infect. Immun. 69, 5742–5751. doi: 10.1128/IAI.69.9.5742-5751.2001
Moorthy, S., and Watnick, P. I. (2005). Identification of novel stage-specific genetic requirements through whole genome transcription profiling of Vibrio cholerae biofilm development. Mol. Microbiol. 57, 1623–1635. doi: 10.1111/j.1365-2958.2005.04797.x
Morgan, S. J., Felek, S., Gadwal, S., Koropatkin, N. M., Perry, J. W., Bryson, A. B., et al. (2011). The two faces of ToxR: activator of ompU, co-regulator of toxT in Vibrio cholerae. Mol. Microbiol. 81, 113–128. doi: 10.1111/j.1365-2958.2011.07681
Morgan, S. J., French, E. L., Thomson, J. J., Seaborn, C. P., Shively, C. A., and Krukonis, E. S. (2015). Formation of an intramolecular periplasmic disulfide bond in TcpP protects TcpP and TcpH from degradation in Vibrio cholerae. J. Bacteriol. 198, 498–509. doi: 10.1128/JB.00338-15
Nakayama, S., and Watanabe, H. (1995). Involvement of cpxA, a sensor of a two-component regulatory system, in the pH-dependent regulation of expression of Shigella sonnei virF gene. J. Bacteriol. 177, 5062–5069. doi: 10.1128/jb.177.17.5062-5069.1995
Nakayama, S. I., and Watanabe, H. (1998). Identification of cpxR as a positive regulator essential for expression of the Shigella sonnei virF gene. J. Bacteriol. 180, 3522–3528.
Navarre, W. W., Porwollik, S., Wang, Y., McClelland, M., Rosen, H., Libby, S. J., et al. (2006). Selective silencing of foreign DNA with low GC content by the H-NS protein in Salmonella. Science 313, 236–238 doi: 10.1126/science.1128794
Ni Bhriain, N., and Dorman, C. J. (1993). Isolation and characterization of a topA mutant of Shigella flexneri. Mol. Microbiol. 7, 351–358. doi: 10.1111/j.1365-2958.1993.tb01127.x
Nielsen, A. T., Dolganov, N. A., Otto, G., Miller, M. C., Wu, C. Y., and Schoolnik, G. K. (2006). RpoS controls the Vibrio cholerae mucosal escape response. PLoS Pathog. 2:e109. doi: 10.1371/journal.ppat.0020109
Nye, M. B., Pfau, J. D., Skorupski, K., and Taylor, R. K. (2000). Vibrio cholerae H-NS silences virulence gene expression at multiple steps in the ToxR regulatory cascade. J. Bacteriol. 182, 4295–4303. doi: 10.1128/JB.182.15.4295-4303.2000
Nye, M. B., and Taylor, R. K. (2003). Vibrio cholerae H-NS domain structure and function with respect to transcriptional repression of ToxR regulon genes reveals differences among H-NS family members. Mol. Microbiol. 50, 427–444. doi: 10.1046/j.1365-2958.2003.03701.x
Oh, Y. T., Park, Y., Yoon, M. Y., Bari, W., Go, J., Min, K. B., et al. (2014). Cholera toxin production during anaerobic trimethylamine N-oxide respiration is mediated by stringent response in Vibrio cholerae. J. Biol. Chem. 289, 13232–13242. doi: 10.1074/jbc.M113.540088
Ottemann, K. M., and Mekalanos, J. J. (1996). The ToxR protein of Vibrio cholerae forms homodimers and heterodimers. J. Bacteriol. 178, 156–162. doi: 10.1128/jb.178.1.156-162.1996
Parsot, C., Ageron, E., Penno, C., Mavris, M., Jamoussi, K., d’Hauteville, H., et al. (2005). A secreted anti-activator, OspD1, and its chaperone, Spa15, are involved in the control of transcription by the type III secretion apparatus activity in Shigella flexneri. Mol. Microbiol. 56, 1627–1635. doi: 10.1111/j.1365-2958.2005.04645.x
Penno, C., and Parsot, C. (2006). Transcriptional slippage in mxiE controls transcription and translation of the downstream mxiD gene, which encodes a component of the Shigella flexneri type III secretion apparatus. J. Bacteriol. 188, 1196–1198. doi: 10.1128/JB.188.3.1196-1198.2006
Penno, C., Sansonetti, P., and Parsot, C. (2005). Frameshifting by transcriptional slippage is involved in production of MxiE, the transcription activator regulated by the activity of the type III secretion apparatus in Shigella flexneri. Mol. Microbiol. 56, 204–214. doi: 10.1111/j.1365-2958.2004.04530.x
Perdomo, O. J., Cavaillon, J.M., Huerre, M., Ohayon, H., Gounon, P., and Sansonetti, P.J. (1994). Acute inflammation causes epithelial invasion and mucosal destruction in experimental shigellosis. J. Exp. Med. 180, 1307–1319. doi: 10.1084/jem.180.4.1307
Peterson, K. M., and Mekalanos, J. J. (1988). Characterisation of the Vibrio cholerae ToxR regulon: identification of novel genes involved in intestinal colonization. Infect. Immun. 56, 2822–2829.
Pilla, G., McVicker, G., and Tang, C. M. (2017). Genetic plasticity of the Shigella virulence plasmid is mediated by the intra- and inter-molecular events between insertion sequences. PLoS Genet. 13:e1007014. doi: 10.1371/journal.pgen.1007014
Pilla, G., and Tang, C. M. (2018). Going around in circles: virulence plasmids in enteric pathogens. Nat. Rev. Microbiol. 16, 484–495. doi: 10.1038/s41579-018-0031-2
Pilonieta, M. C., and Munson, G. P. (2008). The chaperone IpgC copurifies with the virulence regulator MxiE. J. Bacteriol. 190, 2249–2251. doi: 10.1128/JB.01824-07
Porter, M. E., and Dorman, C. J. (1994). A role for H-NS in the thermo-osmotic regulation of virulence gene expression in Shigella flexneri. J. Bacteriol. 176, 4187–4191. doi: 10.1128/jb.176.13.4187-4191.1994
Porter, M. E., and Dorman, C. J. (1997a). Differential regulation of the plasmid-encoded genes in the Shigella flexneri virulence regulon. Molec. Gen. Genet. 256, 93–103. doi: 10.1007/s004380050550
Porter, M. E., and Dorman, C. J. (1997b). Positive regulation of Shigella flexneri virulence genes by integration host factor. J. Bacteriol. 179, 6537–6550. doi: 10.1128/jb.179.21.6537-6550.1997
Porter, M. E., Smith, S. G., and Dorman, C. J. (1998). Two highly related regulatory proteins, Shigella flexneri VirF and enterotoxigenic Escherichia coli Rns, have common and distinct regulatory properties. FEMS Microbiol. Lett. 162, 303–309. doi: 10.1111/j.1574-6968.1998.tb13013.x
Porter, M. E., and Dorman, C. J. (2002). In vivo DNA-binding and oligomerization properties of the Shigella flexneri AraC-like transcriptional regulator VirF as identified by random and site-specific mutagenesis. J. Bacteriol. 184, 531–539. doi: 10.1128/JB.184.2.531-539.2002
Prosseda, G., Falconi, M., Giangrossi, M., Gualerzi, C. O., Micheli, G., and Colonna, B. (2004). The virF promoter in Shigella: more than just a curved DNA stretch. Mol. Microbiol. 51, 523–537. doi: 10.1046/j.1365-2958.2003.03848.x
Quinn, H. J., Cameron, A. D., and Dorman, C. J. (2014). Bacterial regulon evolution: distinct responses and roles for the identical OmpR proteins of Salmonella Typhimurium and Escherichia coli in the acid stress response. PLoS Genet. 10:e1004215. doi: 10.1371/journal.pgen.1004215
Raskin, D. M., Judson, N., and Mekalanos, J. J. (2007). Regulation of the stringent response is the essential function of the conserved bacterial G protein CgtA in Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A. 104, 4636–4641. doi: 10.1073/pnas.0611650104
Ringgaard, S., Hubbard, T., Mandlik, A., Davis, B. M., and Waldor, M. K. (2015). RpoS and quorum sensing control expression and polar localization of Vibrio cholerae chemotaxis cluster III proteins in vitro and in vivo. Mol. Microbiol. 97, 660-675. doi: 10.1111/mmi.13053
Rossiter, A. E., Godfrey, R. E., Connolly, J. A., Busby, S. J., Henderson, I. R., and Browning, D. F. (2015). Expression of different bacterial cytotoxins is controlled by two global transcription factors, Crp and Fis, that co-operate in a shared-recruitment mechanism. Biochem. J. 466, 323–335. doi: 10.1042/BJ20141315
Rutherford, S. T., van Kessel, J. C., Shao, Y., and Bassler, B. L. (2011). AphA and LuxR/HapR reciprocally control quorum sensing in vibrios. Genes Dev. 25, 397–408. doi: 10.1101/gad.2015011
Sansonetti, P. J., Kopecko, D. J., and Formal, S. B. (1982). Involvement of a plasmid in the invasive ability of Shigella flexneri. Infect. Immun. 35, 850–860.
Schellhorn, H. E. (2014). Elucidating the function of the RpoS regulon. Future Microbiol. 9, 497–507. doi: 10.2217/fmb.14.9
Schmid-Hempel, P., and Frank, S. A. (2007). Pathogenesis, virulence and infective dose. PLoS Pathog. 3:e147. doi: 10.1371/journal.ppat.0030147
Schuch, R., and Maurelli, A. T. (1997). Virulence plasmid instability in Shigella flexneri 2a is induced by virulence gene expression. Infect. Immun. 65, 3686–3692.
Schuhmacher, D. A., and Klose, K. E. (1999). Environmental signals modulate ToxT-dependent virulence factor expression in Vibrio cholerae. J. Bacteriol. 181,1508–1514.
Seitz, P., and Blokesch, M. (2013). DNA-uptake machinery of naturally competent Vibrio cholerae. Proc. Natl. Acad. Sci. USA 110, 17987–17992. doi: 10.1073/pnas.1315647110
Seitz, P., and Blokesch, M. (2014). DNA transport across the outer and inner membranes of naturally transformable Vibrio cholerae is spatially but not temporally coupled. mBio 5, e01409–e01414. doi: 10.1128/mBio.01409-14
Severin, G. B., Ramliden, M. S., Hawver, L. A., Wang, K., Pell, M. E., Kienunger, A. -K., et al. (2018). Direct activation of a phospholipase by cyclic GMP-AMP in El Tor Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A. 115, E6048–E6055. doi: 10.1073/pnas.1801233115
Sharma, A. K., and Payne, S. M. (2006). Induction of expression hfq by DksA is essential for Shigella flexneri virulence. Mol. Microbiol. 62, 469–479. doi: 10.1111/j.1365-2958.2006.05376.x
Sharp, G. W. G., and Hynie, S. (1971). Stimulation of intestinal adenyl cyclase by cholera toxin. Nature 229, 266–269. doi: 10.1038/229266a0
Shere, K. D., Sallustio, S., Manessis, A., D’Aversa, T. G., and Goldberg, M. B. (1997). Disruption of IcsP, the major Shigella protease that cleaves IcsA, accelerates actin-based motility. Mol. Microbiol. 25, 451–462. doi: 10.1046/j.1365-2958.1997.4681827.x
Shimada, T., Arakawa, E., Itoh, K., Okitsu, T., Matsushima, A., Asai, Y., et al. (1994). Extended serotyping scheme for Vibrio cholerae. Curr. Microbiol. 28, 175–178. doi: 10.1007/BF01571061
Shimada, T., Bridier, A., Briandet, R., and Ishihama, A. (2011). Novel roles of LeuO in transcription regulation of E. coli genome: antagonistic interplay with the universal silencer H-NS. Mol. Microbiol. 82, 378–397. doi: 10.1111/j.1365-2958.2011.07818.x
Silva, A. J., and Benitez, J. A. (2004). Transcriptional regulation of Vibrio cholerae hemagglutinin/protease by the cyclic AMP receptor protein and RpoS. J. Bacteriol. 186, 6374–6382. doi: 10.1128/JB.186.19.6374-6382.2004
Silva, A. J., Sultan, S. Z., Liang, W., and Benitez, J. A. (2008). Role of the histone-like nucleoid structuring protein in the regulation of rpoS and RpoS-dependent genes in Vibrio cholerae. J. Bacteriol. 190, 7335–7345. doi: 10.1128/JB.00360-08
Singh, P. K., Bartalomej, S., Hartmann, R., Jeckel, H., Vidakovic, L., Nadell, C. D., et al. (2017). Vibrio cholerae combines individual and collective sensing to trigger biofilm dispersal. Curr. Biol. 27, 3359–3366. doi: 10.1016/j.cub.2017.09.041
Stoebel, D. M., Free, A., and Dorman, C. J. (2008). Anti-silencing: overcoming H-NS-mediated repression of transcription in Gram-negative enteric bacteria. Microbiology 154, 2533–2545. doi: 10.1099/mic.0.2008/020693-0
Stonehouse, E. A., Hulbert, R. R., Nye, M. B., Skorupski, K., and Taylor, R. K. (2011). H-NS binding and repression of the ctx promoter in Vibrio cholerae. J. Bacteriol. 193, 979–988. doi: 10.1128/JB.01343-09
Suzuki, T., Miki, H., Takenawa, T., and Sasakawa, C. (1998). Neural Wiskott-Aldrich syndrome protein is implicated in the actin-based motility of Shigella flexneri. EMBO J. 17, 2767–2776. doi: 10.1093/emboj/17.10.2767
Suzuki, T., Saga, S., and Sasakawa, C. (1996). Functional analysis of Shigella VirG domains essential for interaction with vinculin and actin-based motility. J. Biol. Chem. 271, 21878–21885. doi: 10.1074/jbc.271.36.21878
Syed, K. A., Beyhan, S., Correa, N., Queen, J., Liu, J., Peng, F., et al. (2009). The Vibrio cholerae flagellar regulatory hierarchy controls expression of virulence factors. J. Bacteriol. 191, 6555–6570. doi: 10.1128/JB.00949-09
Taniya, T., Mitobe, J., Nakayama, S., Mingshan, Q., Okuda, K., and Watanabe, H. (2003). Determination of the InvE binding site required for expression of IpaB of the Shigella sonnei virulence plasmid: involvement of a ParB boxA-like sequence. J. Bacteriol. 185, 5158–5165. doi: 10.1128/JB.185.17.5158-5165.2003
Taylor, R. K., Miller, V. L., Furlong, D. B., and Mekalanos, J. J. (1987). Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc. Natl. Acad. Sci. U.S.A. 84, 2833–2837. doi: 10.1073/pnas.84.9.2833
Tendeng, C., Badaut, C., Krin, E., Gounon, P., Ngo, S., Danchin, A., et al. (2000). Isolation and characterization of vicH, encoding a new pleiotropic regulator in Vibrio cholerae. J. Bacteriol. 182, 2026–2032. doi: 10.1128/JB.182.7.2026-2032.2000
Teoh, W. P., Matson, J. S., and DiRita, V. J. (2015). Regulated intramembrane proteolysis of the virulence activator TcpP in Vibrio cholerae is initiated by the tail-specific protease (Tsp). Mol. Microbiol. 97, 822–831. doi: 10.1111/mmi.13069
Thomas, S., Williams, S. G., and Manning, P. A. (1995). Regulation of tcp genes in classical and El Tor strains of Vibrio cholerae O1. Gene 166, 43–48. doi: 10.1016/0378-1119(95)00610-X
Tobe, T., Yoshikawa, M., Mizuno, T., and Sasakawa, C. (1993). Transcriptional control of the invasion regulatory gene virB of Shigella flexneri: activation by virF and repression by H-NS. J. Bacteriol. 175, 6142–6149. doi: 10.1128/jb.175.19.6142-6149.1993
Tobe, T., Yoshikawa, M., and Sasakawa, C. (1994). Deregulation of temperature-dependent transcription of the invasion regulatory gene, virB, in Shigella by rho mutation. Mol. Microbiol. 12, 267–276. doi: 10.1111/j.1365-2958.1994.tb01015.x
Tobe, T., Yoshikawa, M., and Sasakawa, C. (1995). Thermoregulation of virB transcription in Shigella flexneri by sensing of changes in local DNA superhelicity. J. Bacteriol. 177, 1094–1097. doi: 10.1128/jb.177.4.1094-1097.1995
Tran, C. N., Giangrossi, M., Prosseda, G., Brandi, A., Di Martino, M. L., Colonna, B., et al. (2011). A multifactor regulatory circuit involving H-NS, VirF and an antisense RNA modulates transcription of the virulence gene icsA of Shigella flexneri. Nucleic Acids Res. 39, 8122–8134. doi: 10.1093/nar/gkr521
Tsou, A. M., Liu, Z., Cai, T., and Zhu, J. (2011). The VarS/VarA two-component system modules the activity of the Vibrio cholerae quorum-sensing transcriptional regulator HapR. Microbiol. 157, 1620–1628. doi: 10.1099/mic.0.046235-0
Turner, E. C., and Dorman, C. J. (2007). H-NS antagonism in Shigella flexneri by VirB, a virulence gene transcription regulator that is closely related to plasmid partition factors. J. Bacteriol. 189, 3403–3413. doi: 10.1128/JB.01813-06
Val, M. E., Bouvier, M., Campos, J., Sherratt, D., Cornet, F., Mazel, D., et al. (2005). The single-stranded genome of phage CTX is the form used for integration into the genome of Vibrio cholerae. Mol. Cell 19, 559–566 doi: 10.1016/j.molcel.2005.07.002
Val, M. E., Marbouty, M., de Lemos Martins, F., Kennedy, S. P., Kemble, H., Bland, M. J., et al. (2016). A checkpoint control orchestrates the replication of the two chromosomes of Vibrio cholerae. Sci. Adv. 2:e1501914. doi: 10.1126/sciadv.1501914
Vergara-Irigaray, M., Fookes, M. C., Thomson, N. R., and Tang, C. M. (2014). RNA-seq analysis of the influence of anaerobiosis and FNR on Shigella flexneri. BMC Genomics 15:438. doi: 10.1186/1471-2164-15-438
Verstraeten, N., Knapen, W. J., Kint, C. I., Liebens, V., Van den Bergh, B., Dewachter, L., et al. (2015). Obg and membrane depolarization are part of a microbial bet-hedging strategy that leads to antibiotic tolerance. Mol. Cell 59, 9–21. doi: 10.1016/j.molcel.2015.05.011
Waldor, M. K., and Mekalanos, J. J. (1996). Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 272, 1910–1914. doi: 10.1126/science.272.5270.1910
Wang, H., Ayala, J. C., Benitez, J. A., and Silva, A. J. (2012). Interaction of the histone-like nucleoid structuring protein and the general stress response regulator RpoS at Vibrio cholerae promoters that regulate motility and hemagglutinin/protease expression. J. Bacteriol. 194, 1205–1215. doi: 10.1128/JB.05900-11
Wang, H., Ayala, J. C., Benitez, J. A., and Silva, A. J. (2014). The LuxR-type regulator VpsT negatively controls the transcription of rpoS, encoding the general stress response regulator, in Vibrio cholerae biofilms. J. Bacteriol. 196, 1020–1030. doi: 10.1128/JB.00993-13
Wang, H., Ayala, J. C., Benitez, J. A., and Silva, A. J. (2015). RNA-Seq analysis identifies new genes regulated by the histone-like nucleoid structuring protein (H-NS) affecting Vibrio cholerae virulence, stress response and chemotaxis. PLoS One 10:e0118295. doi: 10.1371/journal.pone.0118295
Wassef, J. S., Keren, D. F., and Mailloux, J. L. (1989). Role of M cells in initial antigen up take and in ulcer formation in the rabbit intestinal loop model of shigellosis. Infect. Immun. 57, 858–863.
Watanabe, H., Arakawa, E., Ito, K., Kato, J., and Nakamura, A. (1990). Genetic analysis of an invasion region by use of a Tn3-lac transposon and identification of a second positive regulator gene, invE, for cell invasion of Shigella sonnei: significant homology of invE with ParB of plasmid P1. J. Bacteriol. 172, 619–629. doi: 10.1128/jb.172.2.619-629.1990
Weatherspoon-Griffin, N., Picker, M. A., Pew, K. L., Park, H. S., Ginete, D. R., Karney, M. M., et al. (2018). Insights into transcriptional silencing and anti-silencing in Shigella flexneri: a detailed molecular analysis of the icsP virulence locus. Mol. Microbiol. 108, 505–518. doi: 10.1111/mmi.13932
Weiner, A., Mellouk, N., Lopez-Montero, N., Chang, Y. -Y., Souque, C., Schmitt, C., et al. (2016). Macropinosomes are key players in early Shigella invasion and vacuolar escape in epithelial cells. PLoS Pathog. 12:e1005602. doi: 10.1371/journal.ppat.1005602
Welch, M. D., and Way, M. (2013). Arp2/3-mediated actin-based motility: a tail of pathogen abuse. Cell Host Microbe 14, 242–255. doi: 10.1016/j.chom.2013.08.011
Will, W. R., Navarre, W. W., and Fang, F. C. (2015). Integrated circuits: how transcriptional silencing and counter-silencing facilitate bacterial evolution. Curr. Opin. Microbiol. 23, 8–13. doi: 10.1016/j.mib.2014.10.005
Wing, H. J., Yan, A. W., Goldman, S. R., and Goldberg, M. B. (2004). Regulation of IcsP, the outer membrane protease of the Shigella actin tail assembly protein IcsA, by virulence plasmid regulators VirF and VirB. J. Bacteriol. 186, 699–705. doi: 10.1128/JB.186.3.699-705.2004
Wu, R., Zhao, M., Li, J., Gao, H., Kan, B., and Liang, W. (2015). Direct regulation of the natural competence regulator gene tfoX by cyclic AMP (cAMP) and cAMP receptor protein (CRP) in Vibrios. Sci. Rep. 5:14921. doi: 10.1038/srep14921
Yamamoto, S., Izumiya, H., Mitobe, J., Morita, M., Arakawa, E., Ohnishi, M., et al. (2011). Identification of a chitin-induced small RNA that regulates translation of the tfoX gene, encoding a positive regulator of natural competence in Vibrio cholerae. J. Bacteriol. 193, 1953–1965. doi: 10.1128/JB.01340-10.
Yamamoto, S., Mitobe, J., Ishikawa, T., Wai, S. N., Ohnishi, M., Watanabe, H., et al. (2014). Regulation of natural competence by the orphan two-component system sensor kinase ChiS involves a non-canonical transmembrane regulator in Vibrio cholerae. Mol. Microbiol. 91, 326–347. doi: 10.1111/mmi.12462
Yang, M., Frey, E. M., Liu, Z., Bishar, R., and Zhu, J. (2010). The virulence transcriptional activator AphA enhances biofilm formation by Vibrio cholerae by activating the expression of the biofilm regulator VpsT. Infect. Immun. 78, 697–703. doi: 10.1128/IAI.00429-09
Yildiz, F. H., and Schoolnik, G. K. (1998). Role of rpoS in stress survival and virulence of Vibrio cholerae. J. Bacteriol. 180, 773–784
Yu, R. R., and DiRita, V. J. (1999). Analysis of an autoregulatory loop controlling ToxT, cholera toxin, and toxin-coregulated pilus production in Vibrio cholerae. J. Bacteriol. 181, 2584–2592.
Yu, R. R., and DiRita, V. J. (2002). Regulation of gene expression in Vibrio cholerae by ToxT involves both antirepression and RNA polymerase stimulation. Mol. Microbiol. 43, 119–134. doi: 10.1046/j.1365-2958.2002.02721.x
Zagaglia, C., Casalino, M., Colonna, B., Conti, C., Calconi, A., and Nicoletti, M. (1991). Virulence plasmids of enteroinvasive Escherichia coli and Shigella flexneri integrate into a specific site on the host chromosome: integration greatly reduces expression of plasmid-carried virulence genes. Infect. Immun. 59, 792–799.
Zamorano-Sánchez, D., Fong, J. C., Kilic, S., Erill, I., and Yildiz, F. H. (2015). Identification and characterization of VpsR and VpsT binding sites in Vibrio cholerae. J. Bacteriol. 197, 1221–1235. doi: 10.1128/JB.02439-14
Zhang, Y., Mühlen, S., Oates, C. V., Pearson, J. S., and Hartland, E. L. (2016). Identification of a distinct substrate-binding domain in the bacterial cysteine methyltransferase effectors NleE and OspZ. J. Biol. Chem. 291, 20149–20162. doi: 10.1074/jbc.M116.734079
Zhao, W., Caro, F., Robins, W., and Mekalanos, J. J. (2018). Antagonism toward the intestinal microbiota and its effect on Vibrio cholerae virulence. Science 359, 210–213. doi: 10.1126/science.aap8775.
Zhu, J., Miller, M. B., Vance, R. E., Dziejman, M., Bassler, B. L., and Mekalanos, J. J. (2002). Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A. 99, 3129–3134. doi: 10.1073/pnas.052694299
Zurawski, D. V., Mumy, K. L., Badea, L., Prentice, J. A., Hartland, E. L., McCormick, B. A., et al. (2008). The NleE/OspZ family of effector proteins is required for polymorphonuclear transepithelial migration, a characteristic shared by enteropathogenic Escherichia coli and Shigella flexneri infections. Infect. Immun. 76, 369–379. doi: 10.1128/IAI.00684-07
Keywords: Vibrio cholerae, Shigella flexneri, cholera toxin, type 3 secretion system, virulence plasmid, chitin, biofilm, H-NS
Citation: Dorman MJ and Dorman CJ (2018) Regulatory Hierarchies Controlling Virulence Gene Expression in Shigella flexneri and Vibrio cholerae. Front. Microbiol. 9:2686. doi: 10.3389/fmicb.2018.02686
Received: 31 July 2018; Accepted: 22 October 2018;
Published: 09 November 2018.
Edited by:
Dongsheng Zhou, Beijing Institute of Microbiology and Epidemiology, ChinaReviewed by:
Yanhong Liu, United States Department of Agriculture, United StatesMiguel A. De la Cruz, Instituto Mexicano del Seguro Social (IMSS), Mexico
Copyright © 2018 Dorman and Dorman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charles J. Dorman, Y2pkb3JtYW5AdGNkLmll