
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol., 12 November 2018
Sec. Virology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.02644
This article is part of the Research TopicInnate Immunity Evasion by Picornaviruses View all 9 articles
 Gisselle N. Medina1,2*
Gisselle N. Medina1,2* Fayna Díaz-San Segundo1,3
Fayna Díaz-San Segundo1,3 Carolina Stenfeldt1,4
Carolina Stenfeldt1,4 Jonathan Arzt1
Jonathan Arzt1 Teresa de los Santos1*
Teresa de los Santos1*Like all pathogens, foot-and-mouth disease virus (FMDV) is recognized by the immune system inducing a heightened immune response mainly mediated by type I and type III IFNs. To overcome the strong antiviral response induced by these cytokines, FMDV has evolved many strategies exploiting each region of its small RNA genome. These include: (a) inhibition of IFN induction at the transcriptional and translational level, (b) inhibition of protein trafficking; (c) blockage of specific post-translational modifications in proteins that regulate innate immune signaling; (d) modulation of autophagy; (e) inhibition of stress granule formation; and (f) in vivo modulation of immune cell function. Here, we summarize and discuss FMDV virulence factors and the host immune footprint that characterize infection in cell culture and in the natural hosts.
Foot-and-mouth disease virus (FMDV) is the prototype member of the Aphthovirus genus within the Picornaviridae family. The virus is the etiologic agent of foot-and-mouth disease (FMD), a disease of cloven-hoofed animals that often causes extensive epizootics in livestock, mostly farmed cattle and swine, although sheep, goats and over 50 wild species can be affected. FMDV exists as seven distinct serotypes: A, Asia-1, C, O and Southern African Territories 1–3 (SAT 1–3), all including numerous subtypes. High morbidity, and broad diversity have made FMD prevention and control challenging.
Similar to other RNA viruses, FMDV bears an error prone polymerase that causes extensive genetic heterogeneity, allowing its existence as viral quasispecies, a phenomenon that permits virus adaptation to rapidly changing environments in the host (Domingo and Perales, 2018). A detailed study of complex virus–host interactions is essential to understand the virus biology and identify potential therapeutic strategies. In this review, we summarize the current knowledge on the strategies FMDV has evolved to evade immune responses in cell culture and in the natural host.
The FMDV genome consists of a positive single stranded RNA of approximately 8,500 nucleotides (nt) covalently linked at the 5′end to a viral encoded protein (3B or VPg). The RNA is organized in a relatively extensive 5′UTR, a single ORF and a short 3′UTR (Figure 1). The 5′UTR of about 1,300 nt, is composed of five specific regions that are critical for virus replication and include the S fragment, poly(C) tract, pseudoknots (PKs), cis acting replication element (cre) and the IRES (Belsham, 2005). Translation of the ORF begins at two alternative AUG codons, producing a 2,300 amino acid polyprotein that is processed by viral encoded enzymes, resulting in precursors and mature protein products (Vakharia et al., 1987; Clarke and Sangar, 1988; Ryan et al., 1991; Medina et al., 1993; Kirchweger et al., 1994). Mature products include four structural [1A (VP4), 1B (VP2), 1C (VP3), 1D (VP1)] and eight non-structural (NS) proteins [Lpro, 2A, 2B, 2C, 3A, 3 distinct copies of 3B (VPg), 3Cpro, and 3Dpol]. The first mature protein in the ORF is Lpro, a cysteine protease that self-cleaves from the growing polypeptide chain. Lpro is also involved in cleaving many cellular proteins, contributing significantly to virus pathogenesis (Strebel and Beck, 1986; Kirchweger et al., 1994; Brown et al., 1996; Belsham et al., 2000). The P1 region codes for the capsid proteins while P2 and P3 encode for NS proteins that are necessary for viral RNA replication (Gao et al., 2016). 3Cpro is a cysteine protease that processes P1, P2, and P3 precursors to generate mature viral products (Vakharia et al., 1987; Bablanian and Grubman, 1993), and 3Dpol is the viral RdRp.
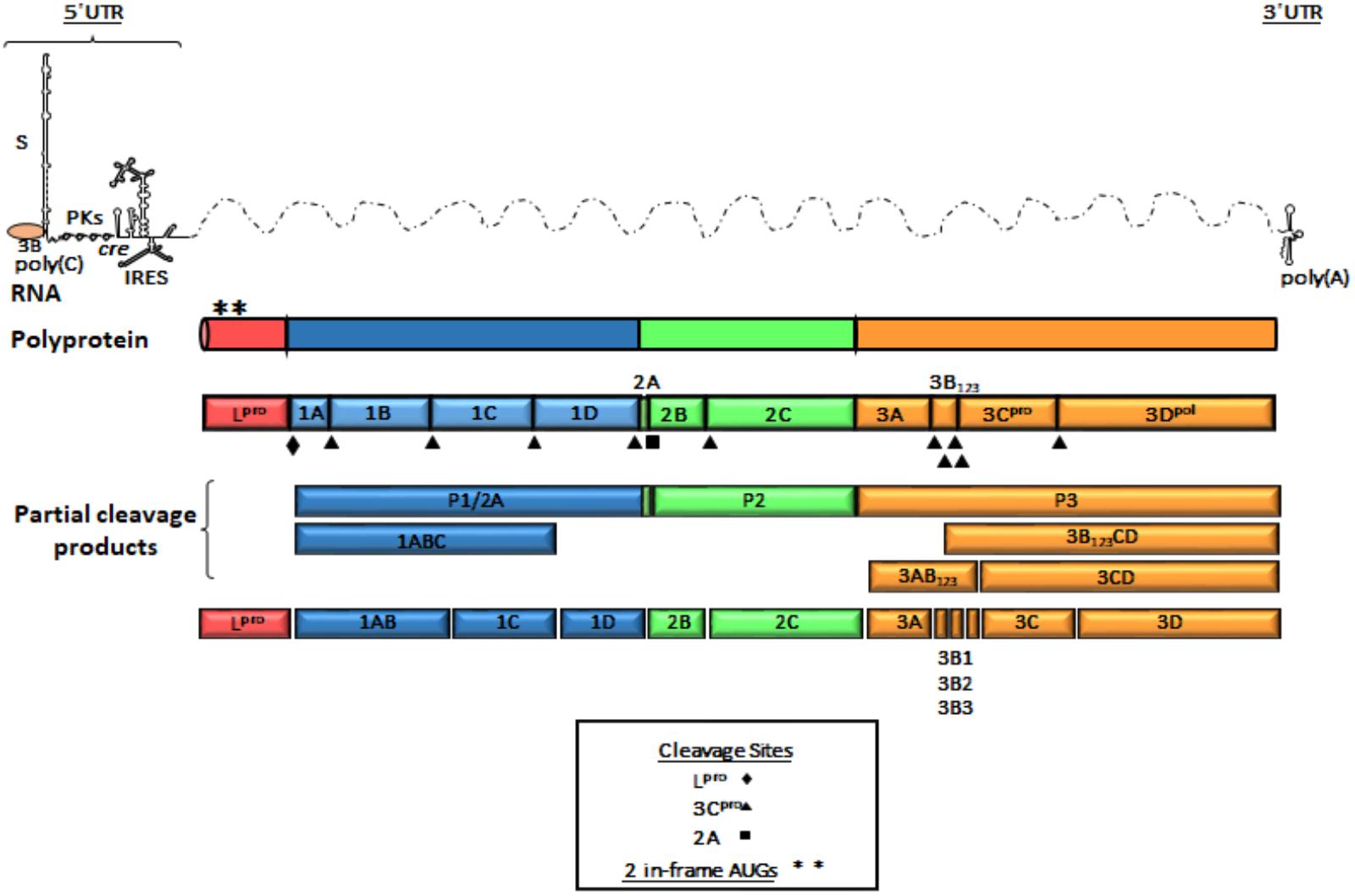
FIGURE 1. Genome organization of FMDV. A schematic representation of linear (+) stranded FMDV RNA is depicted as thin solid lines and dotted lines represent ORF. Viral genomic RNA contains elements shown at the 5′ and 3′ UTR and are represented as thin lines. The 5′UTR consists of S fragment, poly (C) tract, pseudoknots (PKs), cis-acting replicative element (cre) and internal ribosome entry site (IRES). The single ORF encodes a polyprotein and is represented in outlined-open boxes. Filled triangles, squares and diamonds indicate processing sites for 3Cpro, 2A, and Lpro, respectively. Post-translational proteolytic cleavages are shown as partial products. Asterisks describe the two AUG initiation codons. The 3′UTR consist of a short stretch of RNA and a poly (A) sequence. The P1 region encodes the structural polypeptides. The P2 and P3 regions encode the non-structural proteins associated with replication. 3B (VPg) protein is shown as covalently linked to the 5′end of the genomic RNA.
The virus cycle of FMDV can be divided in seven distinct phases: binding, internalization, uncoating, translation, replication, encapsidation and cell lysis. FMDV initiates infection by binding to integrins via the highly conserved (Arg-Gly-Asp) RGD motif displayed on the surface-exposed G-H loop of VP1 (O’Donnell et al., 2009; Monaghan et al., 2005). In cattle, FMDV mainly binds to αVβ6 integrins highly expressed on epithelial cells (Monaghan et al., 2005; Brown et al., 2006; O’Donnell et al., 2009). However, receptors different from integrins can also be used for entry [i.e., Heparan sulfate (Jackson et al., 1996; Baranowski et al., 2000); Jumonji C-domain containing protein 6 (Lawrence et al., 2016)]. Receptor binding induces internalization of FMDV via either CCPs or caveolae–mediated mechanisms (Martín-Acebes et al., 2007; O’Donnell et al., 2008). Following internalization, acidification of the endosomes triggers viral uncoating of the icosahedral capsid and genome release into the cytosol (Vázquez-Calvo et al., 2012). Translation begins at the IRES element (Belsham, 2009) to produce a polyprotein. FMDV efficiently blocks host translation by an Lpro-dependent cleavage of the translation initiation factor eIF4G (Devaney et al., 1988; Kirchweger et al., 1994). The FMDV RNA is replicated by the viral polymerase 3Dpol, which along with other FMDV NS products (i.e., 3A, 2B and 2C) concentrates on membranes of the ER and Golgi leading to RNA synthesis (Polatnick and Wool, 1983; Moffat et al., 2005; Midgley et al., 2013). Culmination of the replication cycle requires encapsidation of the viral genomic RNA and maturation of the capsid. At this stage the intermediate protein VP0 is processed into VP2 and VP4 by an unknown mechanism (Han et al., 2015). Eventually the infected cell is destroyed (cell lysis) causing the egress of newly assembled virus.
During virus infection, conflicting interests can drive the coevolution of hosts and pathogens: while the host needs to detect the pathogen and stop progression of an infection, usually by mounting a timely inflammatory response, viruses subvert the innate immune system, but avoid overreaction to ensure survival in a live host. In parallel, in the host, the innate sensing is anatomically regulated. Viruses that infect epithelial cells trigger a local IFN response resulting in secreted IFN and other cytokines that induce the expression of ISGs in neighboring and distal tissues, thus increasing hematopoiesis and preparing for systemic viral spread (Hermesh et al., 2010). When the local response fails to restrict virus replication, the virus enters the bloodstream causing robust IFN responses, usually produced by circulating pDCs, (Swiecki and Colonna, 2015). Interestingly, the effectiveness of viral clearance relies upon the particular nature of each virus. While some acute viral infections, resolve in 1–2 weeks, others can become persistent and may reappear when conditions are favorable. Thus, a fine-tuning of the virus–host interplay at the site of infection and systemically in the animal, will faithfully determine the outcome of infection.
The primary receptors of innate immunity are composed of a diverse set of PRRs that identify atypical molecules present in viruses, and other microbes, defined as PAMPs, (Kawai and Akira, 2010; Palm et al., 2012; Wu and Chen, 2014) (Figure 2). As PAMPs, viral RNAs are mainly recognized by three types of receptors: endosome associated TLRs, cytosolic RNA helicases known as RIG-I like receptors (RLRs), and NOD-leucine-rich repeat–containing receptors (NLRs). In addition, viral RNA can interact with a family of cellular enzymes such as, dsRNA-dependent PKR, oligoadenylate synthetase proteins (OAS), and others, eliciting a signaling response that limits virus propagation (Kawai and Akira, 2010; Palm et al., 2012; Rathinam et al., 2012; Dempsey and Bowie, 2015; Yoneyama et al., 2015). Of outmost importance in RNA recognition are the TLR and RLR families of proteins (Kawai and Akira, 2010; Wu and Chen, 2014).
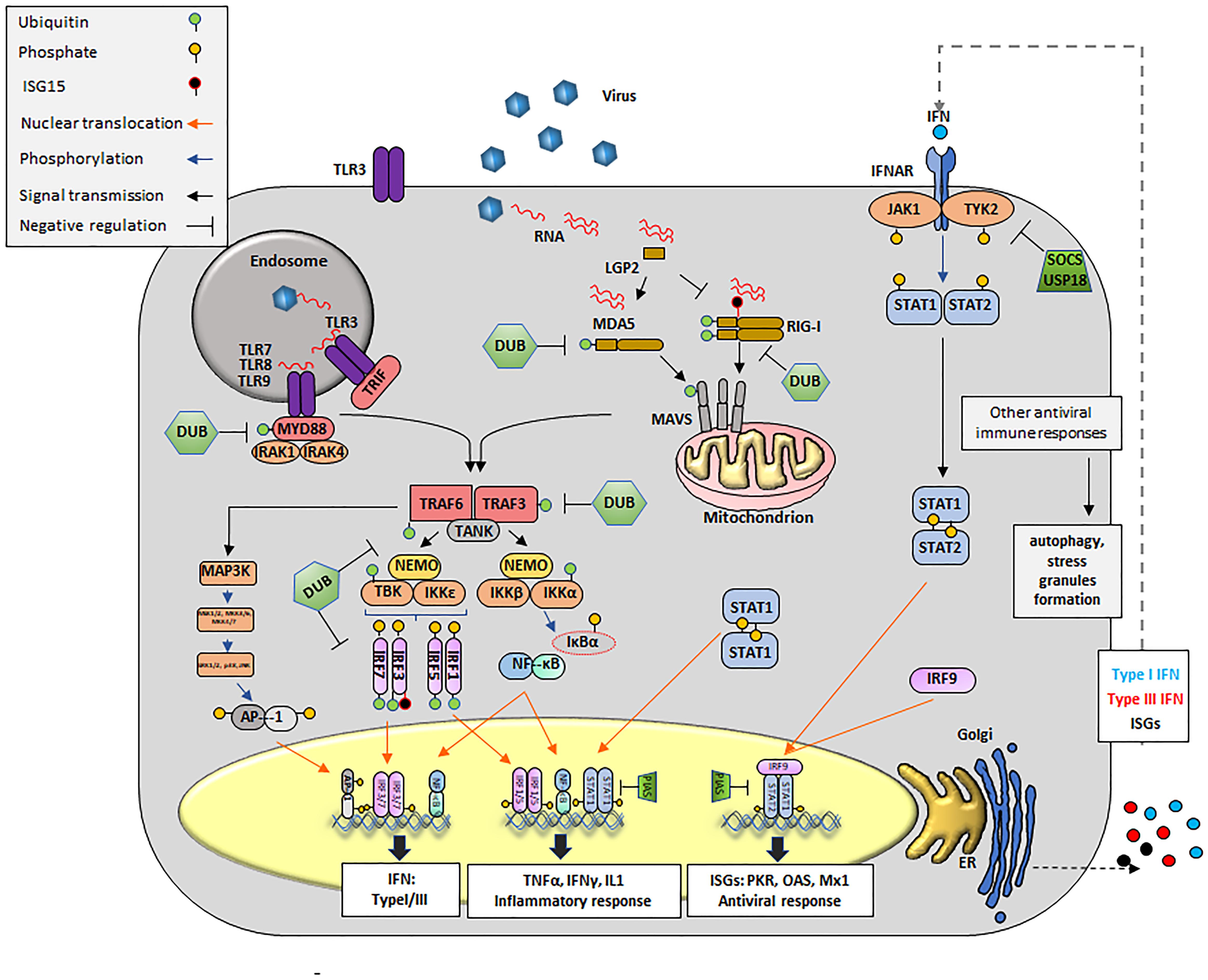
FIGURE 2. Innate antiviral immune responses during viral infection. Upon viral infection, viral RNA is detected by cytosolic RLRs (RIG-I, MDA-5 or LGP2) and/or membrane bound TLRs, triggering a signal transduction cascade that culminates in the transcription of innate molecules including IFN, pro-inflammatory cytokines and ISGs. Cytosolic RNA is detected by RIG-I, MDA5 and presumably LGP2. RIG-I recognizes RNAs containing 5′ppp and panhandle-like secondary structures. MDA5 recognizes dsRNA. LGP2 might regulate the functions of RIG-I and MDA5. Interaction of RIG-I and MDA5 RNA exposes their CARD domains. RIG-I is subsequently ubiquitinated with unanchored K63-linked Ub chains. MDA5 stacks along dsRNA and form helical filaments. Discrete CARD oligomers (with or without Ub) align on CARD domains of mitochondrial MAVS inducing its polymerization. Endosomal RNAs are detected by TLR3 or TLR7/8, which interact with adaptor proteins TRIF and MyD88, respectively. MyD88 uses other adaptors, IRAK1/4 to allow for interaction with TRAF proteins. Polymerization of MAVS or conformational changes on TRIF and MyD88/IRAK1/4 recruit E3 ligases, mainly TRAF3 and TRAF6. These E3 ligases synthesize poly-Ub that are sensed by NEMO to recruit TBK1 or IKK. NEMO/TBK1 complexes lead mainly to phosphorylation of IRF3/7 (although IRF1 or IRF5 might also be phosphorylated). Phosphorylated IRFs dimerize and translocate to the nucleus. The IKK complex phosphorylates IκB causing its degradation and detachment of NF-κB which then translocates to the nucleus. The same IKK complex can phosphorylate IRF7. Nuclear homo and heterodimers of IRF3 and IRF7 bind to the IFNβ promoter to stimulate its transcription. Nuclear NF-κB binds the IFNβ promoter but also promoters of many proinflammatory cytokines such as TNFα, IL-1, etc. Binding of dimers of phosphorylated IRF7 is essential for the expression of IFNα. In parallel, E3 ligases can activate MAPK3 and subsequently other kinases including ERK1/2 and JNK which phosphorylate the components of the AP1 heterodimer followed by translocation to the nucleus to cooperate with the induction of IFNβ transcription. Secreted IFN can act in an autocrine and paracrine manner by binding to its receptor (IFNR). Upon binding JAKs (JAK1 and TYK2) are activated to phosphorylate the IFNAR receptor, also allowing for JAK dependent phosphorylation of STAT1 and STAT2 (although other STATs may also be affected). Heterodimers of STAT1/2 interact with IRF9 to form the ISGF3G complex which translocate to the nucleus and binds DNA to drive the expression of over 300 ISGs, many of which display antiviral activity. Homodimers of STAT1 translocate to the nucleus and mainly enhance transcription of proinflammatory cytokines. Many proteins of the pathway (RIG-I, MDA5, MAVS, TRAF3/6, MyD88, and IRF3/7/1/5) are targets for deUbiquitination and/or deISGylation and serve as negative regulators of the IFN signaling pathway. For instance, USP18 remove ISG15 from ISG15-conjugated substrates by cleaving the isopeptide bond between ISG15 and its targeted substrate. Other negative regulators of IFN signaling include SOCS and PIAS. Formation of SGs and activation of autophagy can be detected during innate immunity signaling responses.
Interaction of a TLR with RNA occurs in the extracellular milieu or inside endosomes and within the TLR family, TLR3 recognizes dsRNA (Alexopoulou et al., 2001), while TLR7 and TLR8 sense ssRNA (Heil et al., 2004). Signals sensed through TLRs are transduced through interactions with adaptor proteins including TIR domain proteins (i.e., TRIF, TRAF, etc.), and with MyD88. Successive TLR-driven signals lead to the nuclear translocation of nuclear factor κB (NF-κB, p65/p50) and/or IRF3/IRF7, key transcription factors for IFN and proinflammatory cytokines (Ikushima et al., 2013). In addition, E3 Ub ligases (i.e., TRAF6) can activate the mitogen activated protein kinase 3 (MAPK3) thus inducing the assembly of the AP1, another transcription factor that facilitates IFN mRNA expression (Shaulian and Karin, 2002) (Figure 2).
Three non-membrane bound RLRs have been well characterized: RIG-I, melanoma differentiation-associated gene 5 (MDA5) and LGP2 (Kang et al., 2002; Yoneyama et al., 2004, 2005). RIG-I recognizes relatively short RNA duplexes that contain a tri- or di-phosphate group at the 5′end (Weber et al., 2015; Devarkar et al., 2016) and MDA5 senses longer dsRNA molecules (Kato et al., 2008; Li et al., 2009; Berke and Modis, 2012). At the N-termini, RIG-I and MDA5 proteins contain tandem CARDs that are normally found in a ‘silent signaling’ conformation. Upon interaction with foreign RNA, the CARD domains of these proteins facilitate binding to the MAVS and subsequent aggregation (Seth et al., 2005). In contrast to RIG-I and MDA5, LGP2 is truncated at the 5′end terminus and lacks the CARD domain (Yoneyama et al., 2005). The role of LGP2 in sensing viral RNA is still not completely understood and opposing functions -LGP2 dependent activation and repression- of the IFN pathway have been proposed (Yoneyama et al., 2005; Venkataraman et al., 2007; Satoh et al., 2010). Subsequent events post-CARD active conformation results in the activation of, TBK1, leading to phosphorylation, dimerization and nuclear translocation of IRF3 and IRF7, which strongly induce IFN transcription (Figure 2).
Downstream from PAMP recognition and ISG induction, IFN autocrine and paracrine loops can perpetuate activation in a calibrated manner. Based on their structural features, receptor usage and biological activities, there are three families of IFNs, types I, II, and III (Donnelly and Kotenko, 2010). As secreted cytokines, IFNs orchestrate a milieu of antiviral responses on infected, neighboring and distal cells including not only virus permissive but also immune cells such as natural killer (NK), APC, and ILCs. IFN signal activation begins extracellularly with the binding to the IFN receptor (i.e., IFNAR1/2), causing the activation of TYK2 and JAK1 which results in the phosphorylation, dimerization and nuclear translocation of the transcription factors signal transducer and activator of transcription 1 (STAT1) and STAT2. STAT1/STAT2 heterodimers can bind to IRF9 to form the complex called the IFN-stimulated gene factor 3 (ISGF3) which in turn binds to DNA to activate the transcription of antiviral genes (i.e., OAS, PKR, MX1, etc.) (Figure 2).
Many steps of the elaborate IFN induction pathway are tightly regulated, and effectively control viral infection while minimizing autoimmune damage. Of significant importance to these processes are PTMs, i.e., Ub and Ub-like incorporation/removal from several components of the pathogen-sensing and transduction pathways (Heaton et al., 2016). For instance, the ubiquitination and/or deubiquitination of TLR3, TLR9, MyD88, TRIF, TRAF3, TRAF6, Inhibitor of κB kinases (IκB), NEMO, TBK1, IRF3, IRF7, RIG-I, MDA5, MAVS and tripartite motif containing protein 25 (TRIM25) play a critical role in the activation of the key components IRFs, NF-κB, and AP1 (Zhou et al., 2017). In addition, the Ub-like protein (UBL) ISG15, has recently emerged as an important tool in the struggle against many viral pathogens (Skaug and Chen, 2010). Unlike Ub, expression of ISG15 is IFN-induced and can be rapidly upregulated in response to viral infection (dos Santos and Mansur, 2017). ISG15 is linked to target proteins by an isopeptide bond in a mechanism known as ISGylation. This modification has been shown to stimulate and/or block several IFN signaling events (Kim et al., 2008), targeting IRF3, PKR, RIG-I, TANK1, and filamin B among others (dos Santos and Mansur, 2017). In parallel, USP18, another ISG, also affects innate immune responses by removing ISG15 from target proteins thus creating a negative feedback loop that has been reported very important in preventing excessive immune stimulation and autoimmune diseases (Honke et al., 2016). Interestingly, direct interaction between USP18 and IFNAR2 have been shown to contribute in the negative regulation of type I IFN independently of USP18-deISGylase/isopeptidase activity (Malakhova et al., 2006). Other unconventional PTM that may affect the induction of the innate response against RNA viruses have been characterized and are reviewed elsewhere (Zhou et al., 2017).
Another line of immunity that has been linked with innate responses, is the utilization of preexisting cellular processes and molecules such as those seen during cellular stress: SGs formation, autophagy and apoptosis.
Stress granules are transient non-membrane bound dynamic organelles that store many untranslated mRNPs. SGs usually accumulate upon stress-induced translation arrest and can interfere with viral replication (McCormick and Khaperskyy, 2017). Since, all viruses require the translation machinery of the host to synthesize their own proteins, blockage of SGs represent an strategy to favor their replication. In fact, it has been demonstrated in poliovirus and other picornavirus that formation of SGs is disrupted during infection due to the cleavage of the SG nucleating protein Ras-GAP SH3 domain binding protein 1 (G3BP1) (White et al., 2007; White and Lloyd, 2011; Yang et al., 2018). Interestingly, recent studies have linked virus-induced SGs with RLRs revealing a new function for SGs in sensing viral RNA. Studies by Onomoto et al. (2012) demonstrated that SGs may serve as a platform for RIG-I dependent recognition of viral RNA, at least for influenza viruses. MDA5 has also been detected in SGs (Langereis et al., 2013) suggesting that RLRs can be recruited to SGs and may play an important role in antiviral innate immunity.
Autophagy is an evolutionarily conserved cell-regulated pathway that degrades and recycles long-lived proteins and cellular components. Specifically, under unfavorable conditions such as nutrient deprivation, autophagy mediates a self-eating process via lysosomes. Autophagy-dependent formation of double-membrane vesicles, termed autophagosomes, engulf long-lived proteins, damaged organelles and transport these cargos to the lysosomes where degradation occurs (He and Klionsky, 2009). Much of the orchestration of autophagy, ranging from its induction to vesicular formation and breakdown, are mediated by proteins encoded by ATG (Kuo et al., 2017). Interestingly, signaling pathways that control innate immunity also regulate autophagy (Deretic et al., 2013). For instance, type I IFN enables autophagy (Schmeisser et al., 2014) and upon TLR activation, MyD88 or TRIF can bind to Beclin-1 (ATG9) promoting the maturation of autophagy (Shi and Kehrl, 2008). Additionally, in pDCs, TLR7-dependent production of IFN is impacted upon depletion of the autophagy protein ATG5 during viral infection (Lee et al., 2007). Unexpectedly, autophagy can be beneficial for viruses. For example, poliovirus and rhinovirus are known to usurp the autophagosomal machinery for viral synthesis (Jackson et al., 2005). Some of the reported hijacking strategies include the utilization of autophagosomes as a home for virus replication due to its protected environment and the use of autophagy-generated metabolites and energy required for replication (Choi et al., 2018).
During viral infection, cellular death can occur as a defensive response to limit the production of viruses (Deretic and Levine, 2009). Apoptosis is controlled by internal or external stimulation, both triggering the activation of caspases which leads to cytoskeletal disintegration, metabolic unbalance and culminates with genomic fragmentation. Interestingly, different studies have shown that viruses can use apoptotic processes to produce sufficient virus progeny or to facilitate virus release (Benedict et al., 2002; Ryan et al., 2016). In the case of poliovirus, apoptosis is induced but active infection inhibits its progression (Belov et al., 2003). Undoubtedly, the ultimate outcome following infection will depend on the balance between the competing host and viral influences on the cell death program.
Foot-and-mouth disease virus is very effective at replicating in the susceptible host. Exposure to the virus results in close to 100% morbidity. Full disease usually develops by 2–5 days post infection in livestock species such as swine and cattle (Stenfeldt et al., 2014, 2015, 2016a,b). In tissue culture, virus end-point titers can be reached by 4–6 h post infection, depending on the serotype and initial multiplicity of infection. This rapid infectivity relies on many factors, including but not exclusive to, the availability of the specific cellular receptor, the intrinsic viral replication fitness, and the effectiveness of distinct viral proteins to counteract the host innate immune response.
Only a limited number of studies have identified cellular sensors for FMDV infection mostly using cultured primary cells such as porcine/bovine/murine epithelial cells (i.e., derived from kidney, pharynx, lung, and thyroid cells), or myeloid cells (i.e., PBMCs, dendritic cells and macrophages). In addition, many studies have been performed in stable cell lines of porcine/bovine origin such as PK15, SK6, IB-RS2, LF-PK, and MDBK cells, in human HEK293, MCF-10A and HeLa cells or in murine BHK-21 cells. It is noteworthy to mention that when using primary cells, most of the IFN sensing and signaling pathways are intact, mimicking the dynamics of infection in the natural host. In contrast, the knowledge gained from studies conducted in established cell lines may be affected by specific intrinsic cellular abnormalities. For example, commonly used BHK-21 cells only produce limited amounts of IFN upon viral infection (MacDonald et al., 2007). HEK293 cells are deficient in some PRRs such as TLRs and STING (Medvedev and Vogel, 2003; Burdette et al., 2011), although they are able to transduce signals in transient expression experiments using RIG-I, MDA5, TBK1, IRF3, etc. (Dou et al., 2017).
Foot-and-mouth disease virus RNA is recognized by MDA5 but not RIG-I or TLR3 in epithelial porcine PK15 cells (Hüsser et al., 2011). Interestingly a new study revealed that overexpression of the RNA helicase LGP2 can inhibit FMDV replication in these cells, presumably due to a decreased in transcripts involved in inflammatory or anti-inflammatory responses such as CCL3LI, TNF-α, IL-6, IL-4, IL-12, TGF-β1, GM-CSF, and IL-10 (Zhu et al., 2017). However, these observations have not been confirmed in the context of an FMDV infection of the animal host. Nevertheless, these results suggest that LGP2 plays a role in FMDV recognition and support a previous hypothesis proposing a synergistic interaction between LGP2 and MDA5 to mediate antiviral signaling (Bruns and Horvath, 2015). On the other hand, although no studies have been published reporting PKR as a molecular sensor of FMDV RNA, it has been shown that depletion of PKR by gene KO or siRNA, significantly increases virus yield in tissue culture (Chinsangaram et al., 2001; de los Santos et al., 2006). Moreover, expression of PKR mRNA is induced to higher levels after infection with FMDV lacking or containing mutations in Lpro in comparison to WT virus (de los Santos et al., 2009). These results suggest that PKR plays a critical role during FMDV infection, presumably not as a RNA sensor, but as an ISG that effectively suppresses host and viral translation upon phosphorylation of eIF2α target (Samuel, 1979).
Besides the limited knowledge on FMDV cellular sensors, it is widely understood that the ability of FMDV to successfully replicate in the host cell, depends on the effective suppression of the induced innate immunity. This activity is contingent upon the expression of Lpro, the FMDV protein that as defined by Agol and Gmyl, has evolved as a ‘security protein’ to warrant the virus counterattack of the host response (Agol and Gmyl, 2010). Hence, almost every region of the FMDV genome is involved in counteracting the immune response ensuring survival in nature as summarized in Table 1.

TABLE 1. Involvement of structural and non-structural FMDV proteins in modulating/counteracting innate immunity signaling pathways.
Leader (Lpro) is a papain-like protease that contains a cysteine (Cys)-Histidine (His)-Aspartic acid (Asp) catalytic triad (Guarne et al., 1998). Alike cardiovirus, Aphthovirus Lpro is encoded at the beginning of the viral ORF, a feature that makes it unique relatively to other picornaviruses. Due to the translation initiation of viral RNA at different AUG codons, two forms of the Lpro are expressed, Lab and Lb, which discern by 28 amino acids. These forms free themselves from the nascent viral polyprotein through intra- and intermolecular self-processing events (Steinberger and Skern, 2014). Notably, it has been reported that the physiologically relevant form during viral infection is Lb (Cao et al., 1995), and its expression is abundantly observed in vitro. Lpro induces cleavage of the translation initiation factor eIF4G, including eIF4GI and eIF4GII (Devaney et al., 1988; Medina et al., 1993; Gradi et al., 2004), resulting in the inhibition of cellular cap-dependent protein synthesis. In addition, phosphorylation of eIF2α, as a response to stress, also contributes to the strong translation arrest induced by the virus infection. However, protein synthesis on the FMDV RNA is maintained due to its dependence on the IRES contained within the 5′-UTR (Belsham, 2009), and the Lpro ability to enhance IRES-driven translation in the presence of phosphorylated eIF2α (Moral-López et al., 2014).
During FMDV infection, Lpro can drive many specific countermeasures to overcome the host innate immune defenses. In addition to its primary function to broadly and efficiently inhibit translation of all host capped mRNAs, including molecules involved in innate and adaptive immunity, FMDV Lpro causes degradation of p65/RelA, a subunit of the transcription factor NF-κB, thus blocking its activity in modulating pro-inflammatory cytokine expression (de los Santos et al., 2007). Degradation of this NF-κB subunit not only requires catalytic activity, but also an intact SAP domain on Lpro (de los Santos et al., 2009). Interestingly, mutations in the SAP domain prevented nuclear retention of Lpro and degradation of NF-κB (de los Santos et al., 2009). Subsequent studies demonstrated that Lpro significantly inhibits NF-κB-dependent gene expression, including IFNβ and many ISGs during infection (Zhu et al., 2010). In overexpression studies in PK15 cells, Lpro can also decrease the IRF-induced IFNα/β expression by reducing IRF3 and IRF7 protein levels independently of its protease catalytic activity. However, the specific Lpro dependent mechanism that regulate IRF3 and IRF7 protein turnover remains undetermined (Wang et al., 2010). In contrast, decrease in the levels of IFNλ1 transcripts in cultured cells overexpressing Lpro requires an intact Lpro catalytic activity (Wang et al., 2011b). These results suggest that the specific determinants of Lpro virulence are not completely understood. Studies directed to examine Lpro cellular protein target affinity and/or the spatial regulation during FMDV infection may provide additional clues.
Many of the approaches evolved by FMDV Lpro, are directed to hinder the connection between transcription factors and IFN promoters. In fact, Lpro can also regulate the host transcriptional machinery by directly binding the transcription factor ADNP (activity-dependent neuroprotective protein). Specifically, during infection, WT FMDV but not LLV (Piccone et al., 1995), induced ADNP binding to IFN-α promoter disrupting the expression of IFN and ISGs (Medina et al., 2017). Furthermore, Lpro-ADNP complex was found in association with chromatin remodeling protein Brg-1 indicating a potential interplay between FMDV and the epigenetics machinery that modulates the antiviral response. Targeting specific epigenetic mechanisms that influence expression of TNFα, NF-κB1a, IFNβ, and IL-12b and IL-6 provides a potential advantage during virus infection (Ramirez-Carrozzi et al., 2009). In fact, in a recent study, the role of such influences during FMDV infection has been examined. Specifically, blocking of the EHMT2 during FMDV infection resulted in the upregulation of IFNβ, ISG15, Mx-1, Mx-2, RIG-I, OAS-1, and PKR transcripts in bovine cells, and led to a significant reduction in virus replication (Singh et al., 2016).
As mentioned above, it has been reported that during infection, FMDV induces transcription of LGP2 mRNA but limits its protein expression (Zhu et al., 2017). Interestingly these authors found that overexpression of NS viral proteins, 2B, Lpro and 3Cpro, were responsible for this function, independently of their intrinsic proteolytic activity. More recently Rodríguez Pulido et al. (2018) demonstrated that Lpro directly cleaves LGP2 resulting in reduced IFNβ mRNA expression. Specific cell type used in these analyses may explain these differences. Notwithstanding, both studies showed that overexpression of LGP2 resulted in a significant reduction in FMDV replication.
Foot-and-mouth disease virus Lpro has also been implicated in removing Ub molecules from several immune signaling molecules including RIG-I, TBK1, TRAF3, and TRAF6 (Wang et al., 2011a), thus inactivating downstream signaling. In elegant experiments, Swatek et al. (2018) have recently shown that FMDV Lpro is able to remove ISG15 from cellular proteins in vitro on synthetic substrates and, on cellular targets during virus infection. Although evidence for Lpro ability to deISGylate innate immune signaling proteins has thus far, not been shown, the peculiar disengagement of ISG15 from substrates upon FMDV Lpro targeting suggests that Lpro could prevent ISG15 recycling and thus affect many host responses.
Foot-and-mouth disease virus 3C (3Cpro) is a protease responsible for the proteolytic cleavage of most of the viral polypeptide into the functional proteins required for virus replication (Vakharia et al., 1987; Clarke and Sangar, 1988). 3Cpro is a chymotrypsin-like cysteine protease (Birtley et al., 2005) that has been associated with inhibition of host cell transcription and translation. While Lpro is responsible for cleavage of eIF4G resulting in the hallmark shutoff of host protein synthesis during FMDV infection, a role for 3Cpro in this process has also been reported. 3Cpro can cleave eIF4G and the cap-binding complex eIF4A although these events take place at later times post infection (Belsham et al., 2000). In the case of cellular transcription interruption, 3Cpro induces the cleavage of histone H3 during FMDV infection (Grigera and Tisminetzky, 1984; Falk et al., 1990; Tesar and Marquardt, 1990).
Targeting of 3Cpro to immune signaling pathways has been recently reported and include the direct cleavage of NEMO, which bridges the activation of NF-κB and IRF signaling pathways (Wang D. et al., 2012). It has been recently reported that 3Cpro also mediates the direct cleavage of Sam68, a component of SGs related to the host stress/antiviral response (Lawrence et al., 2012). Furthermore, Ye et al. have showed that during FMDV infection 3Cpro cleaves G3BP1, another SG marker (Ye et al., 2018). It has been reported that G3BP1 also binds to FMDV IRES hindering viral translation (Galan et al., 2017). Cleavage of G3BP1/2 may favor viral infection not only by preventing SG modulation of host antiviral responses but also by eliminating a negative function on IRES dependent viral translation.
Foot-and-mouth disease virus 3Cpro has been also associated to degradation of other protein host factors, however, no direct targeting has been so far demonstrated. A decrease of endogenous PKR have been correlated to overexpression of FMDV 3Cpro, but protein processing was mediated by lysosomal degradation (Li et al., 2017). In addition, interference of the JAK/STAT signaling pathway has been observed in IFNβ treated HeLa cells overexpressing 3Cpro. Specifically, 3Cpro suppressed the ISRE promoter activities and inhibited the nuclear translocation of STAT1/STAT2 heterodimers due to the degradation of KPNA1 (Du et al., 2014). More recently, a study has reported the targeting of proteins involved in autophagy by FMDV 3Cpro. In the context of FMDV infection, autophagy seems to be beneficial for infection (O’Donnell et al., 2011; Gladue et al., 2012), especially during virus entry (Berryman et al., 2012). However, autophagy-associated antiviral responses can be elicited during virus infection (Xu and Eissa, 2010). To countermeasure these responses, FMDV 3Cpro can stimulate the degradation of autophagy proteins ATG5 and ATG12 negatively regulating the NF-κB pathway, the phosphorylation of TBK1 and the activation of IRF3 (Fan et al., 2017).
The non-structural protein 2B is known to be involved in the rearrangement of host cell membranes and disruption of the cellular secretory pathways (Moffat et al., 2005, 2007), a function that may be further enhanced by the viral 3Cpro which causes Golgi fragmentation (Zhou et al., 2013). FMDV 2B is considered one of the most conserved regions in the entire FMDV genome (Carrillo et al., 2005) and molecular modeling analysis have suggested that it has similar features of a viroporin (Nieva et al., 2012). Specifically, FMDV 2B contains two transmembrane domains and localizes primarily in the ER (Moffat et al., 2007). Nearly alike to known viroporins, 2B oligomerizes to form homomultimers and is involved in membrane rearrangements that are required for efficient virus replication. It is thought that these membrane alterations contribute to the formation of intracellular niches that prevent detection of the virus just like the induction of autophagosomes during infection. In addition, FMDV 2B has been shown to effectively change Ca2+ concentrations in the cytoplasm and consequently stimulate autophagy (Ao et al., 2015). Notably, during FMDV infection, 2B can be found in association with autophagosomes markers (O’Donnell et al., 2011).
Direct connection between FMDV 2B and antiviral response mechanisms have recently become evident. In vitro experiments have identified a direct interaction between FMDV 2B and RIG-I which induced the reduction of RIG-I expression in porcine PK-15 cells (Zhu et al., 2016), a secondary porcine kidney cell line normally used to examined IFN stimulation and IFN-inducible genes due to the presence of an intact IFN signaling pathway (Chinsangaram et al., 2001; de los Santos et al., 2006). Examination of the RIG-I mediated signaling transduction events indicated that PK-15 cells overexpressing FMDV 2B suppressed ISG15 and IFN-induced GBP1, adding a novel mechanism to counteract antiviral responses. Interestingly, FMDV infection sparked an increase in LGP2 transcripts while significantly reducing LGP2 protein abundance in porcine PK-15 cells. Positive and negative regulatory functions on IFN and inflammatory cytokines have been ascribed to LGP2 during a viral infection (Satoh et al., 2010; Si-Tahar et al., 2014). Consistent with these results in the context of FMDV infection, LGP2 can function as a suppressor of expression of TNFα, IL-6 IL-4, and CCL3L1 (Zhu et al., 2017), or as an inducer of IFNβ (Rodríguez Pulido et al., 2018). As mentioned above, similarly to Lpro and 3Cpro, FMDV 2B associates to LGP2 upon co-expression in HEK293T cells (Zhu et al., 2017).
Synergistic functions between FMDV 2B and 2C (Moffat et al., 2007) have been reported and together can interrupt protein secretions that could affect the transport of major histocompatibility class (MHC) molecules by blocking ER-to Golgi traffic (Moffat et al., 2007). Alterations in MHC-I cell surface expression have been previously reported in epithelial cells infected with FMDV (Sanz-Parra et al., 1998) indicating a mechanism that dodges FMDV detection by preventing the formation of MHC-I peptide complexes on the plasma membrane.
The non-structural protein 2C is the largest membrane-binding component of the virus and contains a predicted amphipathic helix domain at its N-terminus (Teterina et al., 2006) which is thought to be required for membrane rearrangements (Monaghan et al., 2004). Given its capacity to enhance membrane alterations, bind to ssRNA and display ATPase activity (Sweeney et al., 2010), 2C acts as an important factor in FMDV viral replication. FMDV 2C has been associated with autophagy as immunofluorescence microscopy experiments indicated the colocalization between FMDV 2C and autophagy markers (O’Donnell et al., 2011). In addition, FMDV 2C can interact with the host protein Beclin-1, a well-known key regulator of autophagy that promotes FMDV replication by inhibiting the fusion of lysosomes with autophagosomes (Gladue et al., 2012). Another aspect of FMDV 2C function involves the interaction with N-myc and STAT (Nmi), a cellular protein known to interact with STATs to augment STAT-mediated transcription in response to cytokines such as IL-2 and IFNγ and can be found in the mitochondria and ER (Wang J. et al., 2012). Interestingly, such interaction resulted in the induction of apoptosis, as defined by the presence of activated caspase-3 and DNA fragmentation markers in BHK-21 cells, albeit not in the context of FMDV infection. Although, induction of apoptosis by FMDV is somewhat controversial and may be cell-dependent, additional examination of this cellular pathway is warranted. Furthermore, co-immunoprecipitation assays and colocalization detected by confocal microscopy, indicated the association of FMDV 2C with the IFN-induced protein 35 (IFP35) (Zheng et al., 2014), a factor with roles in antiviral and cytokine responses. Interestingly, IFP35 have been found to negatively impact the activation of RIG-I favoring virus infection (Das et al., 2014), however, overexpression of FMDV 2C in HEK293T cells resulted in Nmi-induced activation of type I IFN promoters which was found to be dependent on the expression of IFP35 (Zheng et al., 2014) since its depletion obliterated the response.
Among all picornaviral 3A proteins, FMDV 3A is the largest, containing 153 amino acids as compared to only 89 amino acids for poliovirus 3A (Mason et al., 2003). Deletions and point mutations in 3A have been associated to altered host specificity, adaptation, attenuation and virulence. For instance, a deletion in the C-terminus region of 3A was found in a strain of FMDV that caused the 1997 outbreak in Taiwan and exclusively affected swine (Dunn and Donaldson, 1997). Interestingly, FMDV isolates from pigs carrying deletions in 3A grew up well in porcine cells and caused disease in swine but displayed restricted growth in bovine cells in vitro, and only signs of subclinical FMD in bovines in in vivo experiments (Pacheco et al., 2013; Stenfeldt et al., 2018). Furthermore, a single mutation in 3A provided adaptation of FMDV to guinea pigs suggesting this protein participates in determining host range specificity (Nunez et al., 2001). Examination of FMDV 3A membrane topology has revealed a hydrophobic domain that facilitates its interaction with cellular membranes (González-Magaldi et al., 2014). This domain has been detected partially in association with ER and Golgi markers (O’Donnell et al., 2001; Garcia Briones et al., 2006), being the ER a critical organelle for FMDV replication (Midgley et al., 2013). In addition, functionality assays using a DNA-launched luciferase reporter replicon system for FMDV, reported that mutations in FMDV 3A hydrophobic domain resulted in reduced viral replication and nuclear translocation in BHK-21 cells (Lotufo et al., 2018). Recent data points to the involvement of FMDV 3A and the inhibition of RLR-mediated IFNβ induction (Li et al., 2016a). In this study, overexpression of FMDV 3A in HEK293 cells resulted in decreased transcript expression of RIG-I and MDA5, and inhibited SeV-induced activation of IRF3. Interestingly, co-immunoprecipitation experiments indicated that an overall hydrophobic region comprising the first 102 amino acids of 3A are required for association with RIG-I, MDA5, and MAVS. Notably, RIG-I and MAVS associate with mitochondrial-associated membranes (MAM) which can establish innate immune synapses with the ER and can be targeted by viral proteases to ablate RIG-I signaling (Horner et al., 2011). Thus, targeting of these membrane-complexes by FMDV 3A may prevent the effective organization of RLR-signaling pathways.
Reports on the antagonism of structural proteins are limited, since modification of the cellular landscape is mainly accomplished through the actions of the above-mentioned non-structural proteins, i.e., Lpro, 3Cpro, etc. However, recent evidence engaged FMDV structural proteins VP1 and VP3 in the suppression of innate immune signaling responses that are primarily driven by type I IFN. A yeast two-hybrid screen using a swine spleen cDNA library identified the interaction between VP1 and the cellular protein sorcin (soluble resistance-related calcium binding protein), a protein that seems to regulate cell response to viral infections (Li et al., 2013). The functionality of this interaction was examined in HEK293 cells by overexpression of VP1 and indicated a reduction on the TNFα and (SeV)-induced activation of IFNα/β and NF-κB transcription. Interestingly, mouse PMCs treated with recombinant FMDV VP1–VP4 proteins resulted in the downregulation of TNFα and other cytokines, while expression levels of CCL19, IL-15, IL-9, GM-CSF, and Galectin-1 were significantly upregulated (Wang et al., 2016). Furthermore, downregulation of IKK/NF-κB has been observed in human lung cancer cells treated with recombinant FMDV VP1 (Ho et al., 2014) indicating a direct relationship between VP1 and the regulation of innate immunity.
Most recently, a report has highlighted the biological role of FMDV VP2 in the induction of autophagy (Sun et al., 2018). In this study, transfection of porcine cells PK-15 with FMDV VP2 resulted in the activation of eIF2α-ATF4 pathway, which plays a key role in regulating the autophagy gene transcription program in response to stress (B’Chir et al., 2013). In addition, activation of this pathway was dependent on the interaction between FMDV VP2 and the HSPB1. Interestingly, mutations known to affect antigenicity and pathogenicity of FMDV also blocked FMDV VP2 association with HSPB1 leading to the inhibition of autophagy (Xue et al., 2012).
In the case of VP3, direct antiviral activity was reported in overexpression studies using HEK293T cells which resulted in the inhibition of phosphorylation-mediated regulation of STAT, and the blockage of the JAK1/STAT1 complex (Li et al., 2016b). In this study, FMDV VP3 was found in association with JAK1 which affected JAK1 protein levels by promoting degradation through the lysosomal pathway. In addition, FMDV VP3 significantly inhibited SeV-triggered activation of the IFNβ promoter leading to the decrease in transcription of IFNβ, CXCL-10, ISG56, and RANTES. Furthermore, FMDV VP3 has been found to block IRF3 phosphorylation and dimerization and decrease the expression of RIG-I and MDA5 (Li et al., 2016c). Interestingly, co-immunoprecipitation studies showed that FMDV VP3 interacts with MAVS and their association was dependent on the presence of the transmembrane domain in MAVS and the C-terminal domain in VP3.
Overall, the recent discoveries on the involvement of FMDV structural proteins beyond their well-defined role in virus assembly and antigenicity in the animal host, suggest a novel function to modulate innate immunity signaling pathways during internalization and replication. It has been reported that during picornavirus infection molecular chaperones (i.e., Hsp90) alleviate competing constraints determined by protein stability, propensity to aggregation and translation kinetics (Geller et al., 2018). Furthermore, it has been proposed that chaperones interact with PRRs to ensure proper folding and affect innate immune signaling (Binder, 2014). It is possible that during FMDV infection, chaperones assist for the proper assembly of capsid proteins and consequently affect innate immunity signaling. In fact, it was recently demonstrated that Hsp90 is required for FMDV capsid precursor processing and pentamer assembly (Newman et al., 2017). These results further support a role of FMDV structural proteins in modulating the innate immune response.
The 5′ UTR of FMDV is over 1,300 bases in length and exhibits the most intricate organization among picornaviruses, comprising the S region, a poly(C) tract, several pseudoknots, the cis-acting replication element (cre) and the IRES, all involved in many facets of replication (Kühn et al., 1990; Martínez-Salas et al., 2015; Kloc et al., 2018). Recent studies have also implicated the FMDV 5′ UTR in modulation of innate immune signaling pathways. For instance, in vitro transcribed FMDV non-coding regions transfected in SK6 porcine cells or injected into mice triggered immune responses mediated by type I IFN and reduced susceptibility against FMDV (Rodríguez-Pulido et al., 2011). This antiviral cellular state has also been observed in a study targeted to identify the minimal FMDV S fragment sequence required to stimulate IFNβ mediated pathways (Kloc et al., 2017). Specifically, a genetically modified FMDV containing deletions in the S fragment resulted in attenuation in primary bovine kidney cells and in mice. Furthermore, examination of IFN and ISGs mRNA transcripts in cells infected with FMDV S fragment mutant demonstrated an upregulation of Mx-1, IFNβ, IL-6, TNFα, and IRF7 when compared to WT virus infection. Whether or not the establishment of a defined FMDV 5′ UTR fragment-stimulated antiviral response is linked to specific interactions with host proteins, remains to be determined. However, many host cellular proteins have been found to interact with the 5′UTR. Among them, the cellular factors PTBP, IRES transacting factor (ITAF45), PCBP2 and nucleolin, support viral translation or RNA stability (Luz and Beck, 1991; Pilipenko et al., 2000; Andreev et al., 2007). These proteins regulate IRES activity providing cell type specificity and determining virus spread, and some of them are cleaved during infection (Rodríguez Pulido et al., 2007). Other proteins, such as Gemin5, downregulate IRES driven translation, but this effect is neutralized during FMDV infection, since this protein is cleaved by Lpro (Pacheco et al., 2009; Piñeiro et al., 2012). Another host factor that interacts with the 5′ UTR is RHA, a cellular protein that binds the S fragment and the viral NS proteins 2C and 3A. During FMDV infection, RHA, a methylated nuclear protein, is re-localized in a non-methylated form to the cytoplasm of the cell favoring viral replication (Lawrence and Rieder, 2009; Lawrence et al., 2014).
The 3′UTR FMDV comprises a structural sequence of 90 nt folding into two separate stem-loops and a poly (A) tail with variable lengths (Belsham, 2005; Serrano et al., 2006), both involved in viral replication and virulence (García-Nuñez et al., 2014). As displayed by other picornaviruses, FMDV show long-distance interactions between both terminal ends of the genomic RNA to support coordination of viral protein and RNA synthesis (Serrano et al., 2006). Interestingly, construction of a virus containing a deletion in the 3′UTR resulted in a non-viable virus (Sáiz et al., 2001) indicating that this region is critical for FMDV infectivity and replication. Interaction between FMDV 3′UTR and the innate immune response has been reported and is linked to type I IFN. Specifically, examination of IFNβ expression in SK6 porcine cells transfected with FMDV 3′UTR transcripts resulted in an effectual response, which was also confirmed in vivo (Rodríguez-Pulido et al., 2011). Importantly, deletions within the secondary structures of FMDV 3′UTR negatively impacted the stimulation of an antiviral cellular state, presumably because RNA structure conservation in these regions may be important for recognition by PRRs.
The viral and host components of the pathogenesis of FMDV infection have been described in detail in cattle and pigs, delineating commonalities and differences across species. Defined pathogenesis events have guided research into innate immunity on the basis that the host responses are expected to originate at sites of infection and subsequently continue with systemic cascades affecting distant target tissues. In cattle, primary FMDV infection has been localized to distinct regions of lymphoid-associated epithelium of the nasopharyngeal mucosa (Arzt et al., 2010; Stenfeldt et al., 2015). In cattle that are experimentally exposed to aerosolized virus, primary infection of the nasopharynx is followed by a phase of viral amplification in the lungs (Brown et al., 1996; Arzt et al., 2010; Pacheco et al., 2010a). However, this distinct phase of FMDV pathogenesis does not occur in cattle that have been infected through intra-nasopharyngeal inoculation or natural contact exposure (Stenfeldt et al., 2015). The subsequent clinical phase of disease involves systemic generalization and virus amplification in vesicular lesions at peripheral sites, including the oral mucosa and coronary bands of the feet. FMDV pathogenesis in cattle is further complicated by the occurrence of a prolonged subclinical persistent phase of infection, which does not occur in pigs (Stenfeldt et al., 2016b). During this FMDV carrier state, which occurs in approximately 50% of infected cattle, infectious FMDV is similarly restricted to distinct regions of lymphoid-associated epithelium of the nasopharyngeal mucosa, as occurs during primary infection (Zhang and Kitching, 2001; Pacheco et al., 2015; Stenfeldt et al., 2016a).
In pigs, primary FMDV infection has instead been demonstrated to occur within epithelial crypts of the oropharyngeal tonsils (Stenfeldt et al., 2014). This anatomic difference in primary FMDV infection between cattle and pigs is consistent with an apparent difference in susceptibility to infection via inhalation versus oral exposure (Donaldson et al., 1987; Donaldson and Alexandersen, 2001; Alexandersen and Donaldson, 2002). However, despite the different anatomic location, the micro-anatomic and phenotypic characteristics of the distinct regions of lymphoid-associated epithelium that support primary FMDV infection are highly similar in both host species.
There is a strong consensus amongst published works indicating the occurrence of a substantial activation of systemic type I and/or type III IFN activity concurrent with the onset of viremia during the fulminant, acute clinical phase in unvaccinated FMDV-infected cattle (Stenfeldt et al., 2011; Windsor et al., 2011; Perez-Martin et al., 2012; Arzt et al., 2014; Eschbaumer et al., 2016). This innate response has been demonstrated with variable detection of bio-active IFN through reporter assays and/or induction of mRNAs for IFN and ISGs in PBMCs (Perez-Martin et al., 2012). The correlation between the appearance of high quantities of virus in the blood, and upregulated systemic antiviral activation is supported by the finding that vaccinated cattle that are protected from generalization of infection, typically do not have viremia, and similarly lack the systemic IFN activation that occurs in naïve cattle (Eschbaumer et al., 2016). However, thus far it remains unclear, which is the source of systemic IFN in cattle. Two distinct mechanisms have been proposed based on experimental data. Bovine pDCs have been implicated through ex vivo studies, as a potential source of the high levels of IFN in response to FMDV immune complexes (Reid et al., 2011). This finding is further supported by the demonstration of up-regulation of IFNβ and λ3, and ISG [e.g., Mx-1, OAS-1, CXCL10, ISG15, OAS1, and RIG-I] mRNAs in PBMCs concurrent with establishment of viremia (Perez-Martin et al., 2012). Alternatively, several studies have demonstrated significant induction of inflammatory and antiviral factors at sites of lesions with abundant viral amplification (Zhang et al., 2009; Arzt et al., 2014; Stenfeldt et al., 2018). These sites include the characteristic vesicular lesions that develop on the tongue, within and around the oral cavity, as well as in coronary band- and interdigital cleft epithelium during the clinical phase of disease. The finding of extremely high induction of IFNα (>1000-fold) and IFNλ (>2400-fold) mRNAs at lesion sites suggests that innate mediators produced at sites of high levels of viral replication may enter the systemic circulation and thereby induce innate responses at distant sites (Arzt et al., 2014).
There are fewer published records and, in some instances controversial results, characterizing the systemic IFN response to FMDV infection in pigs. Nfon et al measured IFNα protein in porcine serum following infection with FMDV. Interestingly concomitant with the onset of viremia, different IFNα amounts were detected depending on the specific FMDV serotype used for infection (Nfon et al., 2010; Summerfield, 2012). However, only few animals were used for the study, no statistical significance could be determined and the levels of systemic IFN were lower than those detected in cattle upon FMDV infection. A separate study found that there was no detectable induction of systemic IFNα in naïve pigs following challenge with FMDV A24 (Diaz-San Segundo et al., 2010). Further studies are required to determine whether or not, FMDV interferes with the induction of systemic IFN in the swine.
A number of studies have investigated the role of local antiviral response activation at the site of primary infection in the bovine nasopharyngeal mucosa. However, the evidence of an activated innate response in the mucosal tissue is not as consistent as the strong systemic antiviral response detected in the host species (Arzt et al., 2010; Windsor et al., 2011; Stenfeldt et al., 2015). This may be due to the substantially lower level of FMDV replication that occurs at the sites of primary infection as compared to the massive viral amplification that occurs in vesicular lesions in the tongue and the feet. Some investigations have reported a low to moderate upregulation of ISGs, including OAS and Mx-1, in the nasopharyngeal mucosa concurrent with establishment of viremia (Arzt et al., 2014; Stenfeldt et al., 2018). Interestingly, these same publications reported non-significant down-regulation of IFNα and β mRNA in nasopharyngeal tissue samples harvested at 24, 48, and 72 h post aerosol inoculation (hpi), whereas IFNλ mRNA was variable, up- or down- regulated. It is known that IRF7, is the master regulator of IFNα expression and strong upregulation is detected upon viral infection (Honda et al., 2005); however, no consistency in the expression of IRF7 could be detected in bovine tissues isolated from FMDV infected cattle, with up- or down- temporal regulation depending on the study (Stenfeldt et al., 2018). A different study showed upregulation of IFNα, β, γ, and λ mRNA in distinct micro-anatomic compartments of the nasopharyngeal mucosa concurrent with occurrence of viremia in non-vaccinated cattle (Stenfeldt et al., 2015). Interestingly, this upregulation of local antiviral activity was more pronounced, and occurred earlier in cattle that had been vaccinated, and in which FMDV infection was restricted to the nasopharyngeal mucosa (Stenfeldt et al., 2015). Consistent with transcriptomic evidence of activation of inflammatory or antiviral responses at the site of primary infection, investigations by immuno-microscopy have demonstrated recruitment of CD 11c/major histocompatibility complex II (CD11c+/MHCII+) cells (presumptively DCs) to distinct focal regions of FMDV-infected epithelial cells within the nasopharyngeal mucosa at 24–72 hpi (Arzt et al., 2010; Stenfeldt et al., 2015). The identification of these cells at early times after infection suggests mechanisms that are driven by innate immunity mediators. However, similar to regulation of antiviral genes, this distinct influx of APCs was more consistent, and occurred at an earlier stage of infection in cattle that had been vaccinated prior to virus exposure (Stenfeldt et al., 2015), suggesting that priming of adaptive immune processes may enhance the response.
Less work has been published on the characterization of the innate response to FMDV infection in porcine tissues. One investigation found that while the levels of INDO, MIP3α, and MCP-1 mRNAs are induced in skin early post infection, no consistent change could be detected in the pattern of pro-inflammatory cytokines (IL-12, -15, or -18) expression (Diaz-San Segundo et al., 2010).
Overall, these data suggest that cattle generate a robust IFN response during the fulminant phase of FMD in the presence of abundant quantities of virus and viral RNA. However, the mechanisms and magnitudes of innate responses at the primary infection site are less clear, with contrasting evidence for activation and inhibition of innate immune processes. The findings from in vivo investigations in cattle suggest that there is a relationship between the magnitude of FMDV replication, and activation of the innate immune response. Only limited information is available regarding the local and systemic innate response to FMDV infection in pigs. Further research in this field is warranted as new technologies and reagents in immunology become available.
As mentioned above, the FMDV infectious cycle in individual animals is short. The virus infects, replicates, spreads throughout the body and is shed in less than 7 days. Evidently, FMDV accomplishes such a rapid colonization of the host by manipulating the early innate immune response creating a window of opportunity that allows dissemination prior to the establishment of adaptive immunity. Understanding of the host-pathogen interaction and viral escape mechanisms of immunity is particularly important in cattle and swine because they represent one of the most important livestock industries worldwide and in swine, acutely infected pigs shed substantial quantities of virus into the environment. Furthermore, high scale production of swine has gained momentum in recent years with the development and increased demand in many countries of East Asia, particularly China and South Korea.
The interaction of FMDV with the host begins via infection of epithelial cells of the nasopharyngeal mucosa in cattle (Arzt et al., 2010; Stenfeldt et al., 2015) or similar epithelium within crypts of oropharyngeal tonsils in pigs (Stenfeldt et al., 2014) [see previous section for more details]. After the primary infection, the virus may get in contact with NK cells, γδ T cells or APCs, either as a result of lytic infection of epithelial cells and subsequent phagocytosis (Rigden et al., 2002) and/or lytic action on damaged infected tissue (Toka et al., 2009) (Figure 3). Furthermore, immune cells such as Mφ (McCullough et al., 1988; Mason et al., 2003) or DCs can be infected in an antibody-mediated internalization process, thus facilitating cell contact during infection (Díaz-San Segundo et al., 2006; Guzylack-Piriou et al., 2006).
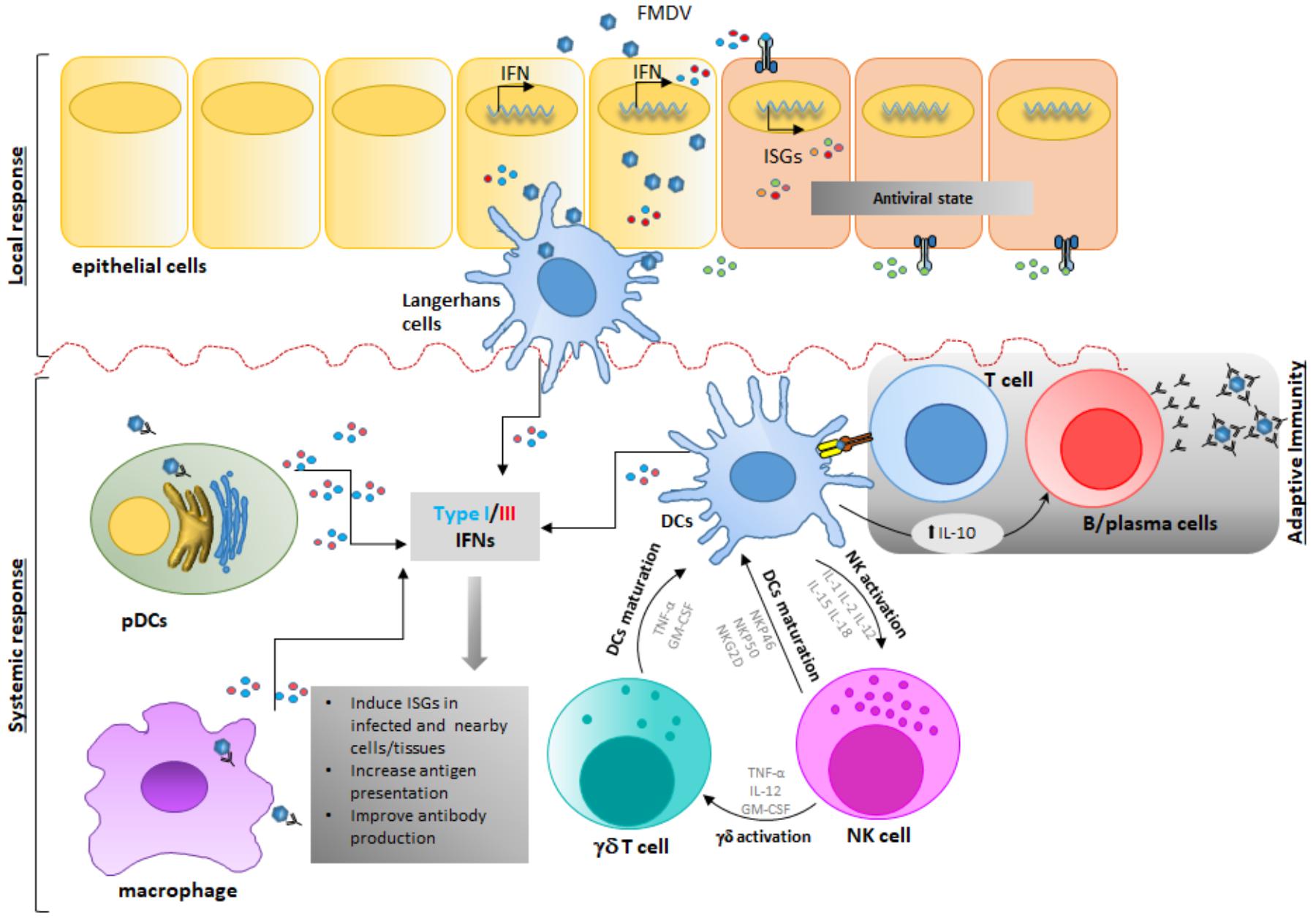
FIGURE 3. Foot-and-mouth disease virus modulates early stages of immune response in vivo. Upon FMDV infection and first round of replication in epithelial cells-the primary infection site-, FMDV gets in contact with cells of the innate immune response inducing functional consequences that affect the host response. FMDV interacts with different players of the immune response either as a result of lytic infection of epithelial cells and subsequent phagocytosis, and/or lytic action on damaged infected tissue or by direct infection of immune cells through a direct or an antibody-dependent internalization process in Mφ, DCs, or NK or γδ T cells. After this stage, DCs, pDCs and Mφ produce IFN and other cytokines that modulate the immune response. Possible interaction of DC, NK and γδ T cells in the innate immune response is depicted, although clear interaction is thus far not completely understood. Ultimately, after FMDV infection and replication, systemic IL-10 produced by cDCs is detected, thus directing the adaptive immune response toward a stronger humoral stimulation rather than a T-cell mediated response.
Similarly to other species, porcine NK cells are identified as CD2+/CD8+/CD3- cells (Denyer et al., 2006). At rest, these cells show minimal cytolytic activity even toward FMDV infected epithelial cells. However, cytotoxicity and expression of IFNγ expression significantly increased upon stimulation with cytokines such as IL-2, IL-12, IL-15, IL-18, or IFNα (Pintarič et al., 2008; Toka et al., 2009), including cytotoxicity against FMDV infected target cells. However, shortly after FMDV infection in swine, the number of circulating NK cells transiently decrease. Moreover, NK cells isolated from FMDV-infected swine are dysfunctional due to the lack of IFNγ secretion and the inability to kill NK-sensitive targets such as K562, a human lymphocytic cell line, or porcine fibroblasts, infected with FMDV (Toka et al., 2009). The same authors have proposed several mechanisms to explain the defective behavior of NKs during FMDV infection in swine. Induction of TLR3 and SOCS3 mRNAs may negatively affect the expression of IFNα, which has shown to be required for NK activation. Additionally, NK unresponsiveness may be caused by a reduction in the levels of cytokines IL-12, IL-15, and IL-18, upon viral infection. Interestingly, it has been shown that the expression of NK surface receptors, NKG2D, NKp80, and cytoplasmic granzyme B, all key molecules involved in cellular activation and inhibition, is minimally affected in infected swine (Toka et al., 2009). Similarly to swine, bovine NK cells appear to have a CD2+/-/CD8+/-/CD3- phenotype, express natural cytotoxicity receptors like CD335 (Boysen et al., 2006b; Bastos et al., 2008) and are capable of lysing infected target cells (Boysen et al., 2006a). However, NK cells originated in FMDV infected cows have an elevated cytotoxic-function against bovine target epithelial cells in vitro (Patch et al., 2014). The ability of cattle NK cells of responding to FMDV infection represents a difference in pathogenesis between cattle and pigs. These data may partially explain the higher morbidity of FMD in outbreak situations in swine as compared to cattle, due to significantly higher levels of virus shedding in this species (Donaldson et al., 1970), however, more studies to understand the importance of NK response during FMDV infection in cattle are needed.
At least 12 different γδ T cell populations have been found in the thymus of pigs, based on the expression of CD1, CD2, CD4, CD8, and CD45RC (Šinkora et al., 2005). Interestingly, in vitro exposure of naïve γδ T cells to high concentrations of inactivated FMDV vaccine antigen can induce the expression of many cytokines and chemokines (Takamatsu et al., 2006). However, further studies are needed to elucidate the role of each γδ T cell swine population in modulating the innate and adaptive immune responses to FMDV infection and vaccination.
On the other hand, the landscape of these cells during FMDV infection in cattle is better characterized. There are two major populations of γδ T cells in cattle whose differentiation is based on the expression of the protein WC1, a cysteine-rich scavenger receptor. Bovine γδ T cells are WC1+/CD3+/CD5+/CD2-/CD6-/CD8- or WC1-/CD3+/CD5+/CD2+/CD6+/CD8+ (Wijngaard et al., 1992; Aruffo et al., 1997). Earlier reports demonstrated that γδ T cells from vaccinated cattle could respond to FMDV antigen (Amadori et al., 1995). More recently, examination of bovine γδ T cells during FMDV infection revealed that WC1+ γδ T cells show a transient activated phenotype characterized by upregulation of CD25, downregulation of CD62L and CD45RO, and increased expression of IFNγ (Toka et al., 2009). Interestingly, it has been shown that WC1+ γδ T cells acquire NK-like capabilities to kill target cells in vitro by increasing the expression of CD335 and perforin. Parallel to those changes, WC1+ γδ T cells also showed upregulation of MHC-II and CD13, suggesting that after exposure to FMDV in vivo they may also act as APCs, as previously suggested (Collins et al., 1998). However, more studies are needed to determine whether the changes observed in γδ T cells are the result of direct virus-cell interactions or a bystander consequence, and how FMDV modulates their activity considering the prevalence of this specific type of T-cells in cattle (Wijngaard et al., 1992).
The DCs can be broadly classified into two lineage populations: pDC, specializing in the production of cytokines, most notably types I and III IFN, and conventional DCs (cDCs), which are potent APCs (Swiecki and Colonna, 2015). cDCs have been defined as sentinel cells that capture, process and present antigen upon migration to lymphoid tissues, resulting in activation and proliferation of rare T cell clones, and linking innate and adaptive immunity (Steinman, 2008). Although it has been shown that the interactions between FMDV and APCs are mostly abortive because no virions are produced (Rigden et al., 2002; Guzylack-Piriou et al., 2006; Díaz-San Segundo et al., 2009), heparan sulfate mediated viral uptake resulted in transient FMDV replication in cDCs (Harwood et al., 2008). It is worth mentioning that DC functionality is affected upon FMDV infection. During acute infection, the virus stimulates swine and cattle cDCs to produce IL-10, a cytokine that directs the immune response toward a stronger humoral rather than a T-cell mediated adaptive response (Díaz-San Segundo et al., 2009; Sei et al., 2016). FMDV also blocks the ability of porcine DCs to differentiate into mature cDCs (Díaz-San Segundo et al., 2009) and impairs the response to stimulation by TLR ligands (Nfon et al., 2008). Similarly, during FDMV infection in cattle, the number of bovine CD11c- cDCs, but not CD11c+ cDCs, is significantly decreased during the peak of viremia, the expression of MHC-II molecules on all bovine cDC populations is dramatically downregulated and the processing of exogenous antigen is impaired (Sei et al., 2016).
Another important set of tissue resident DCs affected during FMDV infection are the LC, a particular subset of DCs that expresses langerin and is found in the epidermis (Clayton et al., 2017). FMDV can attach to, and become internalized by porcine LCs in vitro, although, no viral RNA replication or production of viral proteins upon internalization could be detected (Bautista et al., 2005). Furthermore, after ex vivo stimulation, IFNα production is impaired in LCs derived from FMDV-infected pigs, although the ability to present antigen remains intact (Nfon et al., 2008).
Porcine pDCs also sense FMDV through TLR7 and produce IFNα in response to infection, but the levels of secreted IFN are relatively modest when compared to other viruses such as influenza (Bel et al., 2011; Lannes et al., 2012). Furthermore, in vivo studies in swine have shown that during FMDV infection there is a depletion of pDCs in peripheral blood, and remaining pDCs produce less IFNα upon ex vivo stimulation with TLR ligands or virus (Nfon et al., 2010). In contrast, high levels of type I IFN are produced after ex vivo stimulation of bovine pDCs with TLR9 agonist CpG and FMDV immune complexes (Reid et al., 2011). Furthermore, a robust systemic in vivo type I IFN response is detected in cattle infected with FMDV (Stenfeldt et al., 2011; Perez-Martin et al., 2012) and the number of systemic mature bovine CD4+ MHC-II+ pDCs is increased during FMDV infection, while levels of MHC-II and immature CD4+ MHC-II- pDCs are declined (Sei et al., 2016).
Monocytes/Mφ are part of the innate response to viral infection and are essential for the rapid clearing of pathogens at the sites of infection. Similarly to what happens with pDCs, FMDV also enters Mφ by utilizing the FcγRII receptor in an antibody-dependent internalization manner (McCullough et al., 1988; Baxt and Mason, 1995). Interestingly, it has been reported that, FMDV infectivity endures at least for 10–24 h after viral uptake in the absence of productive infection of porcine Mφ, (Rigden et al., 2002). Implications of these observations favor a model in which Mφ act as transporters and disseminators of viable virions to distant sites of the body where the virus can infect and replicate in other cells. On the other hand, in cattle, CD14+ monocyte frequency increases following inoculation with FMDV (Sei et al., 2016), consistently with previous reports demonstrating that the number of blood monocytes augment following vaccination and challenge with FMDV (Sigal et al., 1992). In this case, blood monocytes may function as cDCs, in which the reduced levels of MHC-II compromise antigen presentation.
Foot-and-mouth disease virus infection also affects the innate immune response at the cytokine level in both, swine and cattle. In vivo cytokine profile analysis during the 1st week of infection shows a systemic decrease of pro-inflammatory cytokines (IL-1β, IL-6, and TNFα), while an increase of the anti-inflammatory cytokine IL-10 and IFNα is detected (Nfon et al., 2010; Diaz-San Segundo et al., 2012, 2013; Perez-Martin et al., 2012; Sei et al., 2016). Most likely, these changes are related with the early T cell unresponsiveness and lymphopenia described in swine and cattle during FMDV infection (Bautista et al., 2005; Díaz-San Segundo et al., 2006; Perez-Martin et al., 2012; Sei et al., 2016). Indeed, it has been reported that, IL-10, inhibits a broad spectrum of cellular responses, and causes immunosuppression and persistence in vivo for other viruses (Brooks et al., 2006). With respect to FMDV-induced lymphopenia, no clear mechanism has thus far been elucidated. Apoptosis mediated cell death has been ruled out in ex vivo infected swine and bovine lymphocytes, despite detection of productive FMDV replication (Díaz-San Segundo et al., 2006; Joshi et al., 2009). These results suggested that lymphopenia could be directly attributed to a cytolytic or alternative mechanism of cell death. Another possibility would be that the observed lymphopenia is the result of a transient egression of lymphocytes from blood to infected tissues, primarily influenced by an increase of systemic IFN. Support for this hypothesis comes from murine studies in which administration of IFNα in vivo resulted in broad lymphopenia which was not observed in IFNα/β receptor KO mice (Kamphuis et al., 2006). Furthermore, these results are consistent with studies in livestock species in which increased levels of type I IFNs were detected in blood of infected swine (Nfon et al., 2010) or cattle (Stenfeldt et al., 2011; Perez-Martin et al., 2012). However, overexpression of IFN using a replication defective human Adenovirus 5 vector (Ad5-IFN) did not induce lymphopenia during the peak of IFN detection in cattle (Perez-Martin et al., 2012). Further studies are needed to understand the mechanisms of FMDV induced lymphopenia and the role IFN might play in this clinical sign.
Despite the differences in response to FMDV between swine and cattle, it is clear that the virus triggers a state of immunosuppression in both species, favoring a Th2 cell/cytokine-like environment that induces a strong FMDV-specific neutralizing antibody response to ultimately clear the virus. In fact, the serological response of naïve pigs and cattle to FMDV infection is characterized by a rapid surge of anti-FMDV immunoglobulin M (IgM) that peaks approximately at 7 days succeeded by a sustained anti-FMDV IgG response which remains at high titers beyond 28 days (Juleff et al., 2009; Pacheco et al., 2010b).
Despite the robust IFN response detected during the peak of viremia in cattle, FMDV is very sensitive to IFN (Ahl and Rump, 1976; Diaz-San Segundo et al., 2013). In fact, it has been demonstrated that when IFN (either type I, type II, or type III IFNs) -mediated antiviral stage is induced prior to virus infection, replication of all seven FMDV serotypes can be dramatically inhibited (Chinsangaram et al., 1999, 2001; Moraes et al., 2007; Díaz-San Segundo et al., 2011; Grubman et al., 2012). Furthermore, animals inoculated with Ad5 vectors expressing type I or type III IFN are efficiently protected against challenge at 1 day post inoculation in swine (Moraes et al., 2003, 2007; Diaz-San Segundo et al., 2010; Dias et al., 2011) and in cattle (Wu et al., 2003; Perez-Martin et al., 2012). Interestingly, treatment of swine with Ad5-IFNα is effective against multiple FMDV serotypes and protection lasts for approximately 3–5 days (Moraes et al., 2003). A synergistic effect against FMD has been detected when types I and II IFNs were co-administered in swine (Moraes et al., 2007; Kim et al., 2014). Studies aimed at elucidating the mechanisms by which IFN protects swine against FMD have demonstrated that protection of swine inoculated with Ad5-IFNα correlates with a local recruitment of skin DCs (Diaz-San Segundo et al., 2010) with partial maturation, increased expression of CD80/86 and decreased phagocytic activity (Diaz-San Segundo et al., 2013).
In essence, combined delivery of FMDV vaccines and select immunomodulatory constructs, such as exogenous sources of IFN, could provide a rapid onset and broad protection to deter primary virus infection prior to the vaccine-induced antibody response. In a proof of concept study in swine, Moraes et al. (2003) treated animals with a combination of Ad5-poIFNα and Ad5-FMD subunit vaccine challenging at 5 dpi with FMDV A24. Treated animals were completely protected against FMD and developed a significant adaptive immune response (Moraes et al., 2003). More recently, similar results were described in cattle treated with a combination of Ad5-boIFN-λ3 and Ad5-O1Manisa (Ad5-O1M) against aerosol challenge with FMDV (Diaz-San Segundo et al., 2016).
In this review, we have summarized a wealth of information that over the years has advanced our understanding of the many components required for the FMDV life cycle and the host restriction mechanisms targeted by the virus to favor its replication. FMDV has evolved an unusually fast replication rate to conquer the host. The virus uses its entire genome to juggle with the cellular machinery involved in induction and modulation of innate immunity accomplishing severe morbidity soon after infection. The most efficient mechanism by which FMDV defeats the host antiviral response is perhaps the early shut-off on host translation, that limits the expression of antiviral molecules during the transition from local to systemic infection. However, the virus also uses many additional strategies to finely modulate and take advantage of the induced antiviral pathways gaining fitness and rapidly proliferating. FMDV successfully achieves these goals by: inhibiting RNA sensing by host PRRs; interfering with PTMs of many factors involved in IFN pathway activation and cellular trafficking; causing degradation of the transcription factors that govern expression of IFN, inflammatory cytokines and ISGs; modifying the chromatin architecture at the IFN/ISG loci; inducing rearrangements of cell membranes to optimize replication and limit protein secretion; disrupting formation of stress organelles such as SGs; modulating metabolic pathways such as autophagy and apoptosis, while simultaneously destabilizing pathways and components involved in stimulating the IFN pathway. Finally, FMDV uses all its proteins, structural and non-structural, to directly interact with most of the proteins that mediate the IFN auto/paracrine loops.
A relatively short replication cycle allows FMDV to quickly establish infection in local tissues and disseminate throughout the body of the animal host in less than a week. Interestingly, despite the local inhibition in the production of IFN in infected cells early post infection, significant levels of IFN protein can be detected systemically following kinetics of viremia. A rapid uptake of virus particles mediated by phagocytosis or Fc dependent internalization by APCs skews the adaptive response to the production of relatively high amounts of antibodies although surface expression of MHC class I and II molecules declines post infection. Furthermore, FMDV infection results in a rapid, but transient lymphopenia, reducing the number of circulating T and B cells and adversely affecting T cell function, although no clear mechanism has thus far been elucidated.
The theme for this successful pathogen appears to be the ability to counteract both, the host innate and adaptive immune response, at many levels. Uncovering additional FMDV host interactions will be instrumental in understanding in detail the molecular mechanisms by which FMDV counteracts the host immune response and target novel improved disease control strategies.
GM and TS planned, coordinated, and wrote the manuscript. FS drafted the sections on analysis of the host immune responses in vivo and contributed scientific comments to the manuscript. CS drafted the section on FMDV pathogenesis and contributed scientific comments to the whole manuscript. JA contributed scientific comments to the manuscript.
This work was funded by USDA ARS-CRIS Project 1940-32000-061-00D. Additional funding came from an interagency agreement with the Science and Technology Directorate of the U.S. Department of Homeland Security (award numbers HSHQPM-13-X-00131 and HSHQPM-13-X-00113).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ADNP, activity dependent neuroprotective protein; AP1, activator protein 1; APC, antigen presenting cell; ATF, activating transcription factor; ATG, autophagy-related gene; BHK, baby hamster kidney; CARD, caspase activation and recruitment domain; CCPs, clathrin-coated pits; CD, cluster of differentiation; cDCs, conventional dendritic cells; cGAS, cyclic GMP-AMP synthase; CXCL10, C-X-C Motif Chemokine Ligand 10; EHMT2, euchromatic histone-lysine N-methyltransferase 2; eIF, eukaryotic translation initiation factor; ER, endoplasmic reticulum; ERK, extracellular signal-regulated kinase; FMDV, foot-and-mouth disease virus; G3BP1/2, GTPase-activating protein binding protein ½; GBP1, guanylate-binding protein 1; HSPB1, heat shock protein B1; IκB, Inhibitor of κB kinases; IFN, interferon; IFNR, IFN receptor; IFP35, IFN induced protein 35; IL, interleukin; ILC, immune lymphoid cell; INDO, Indoleamine-pyrrole 2,3-dioxygenase; IRAK, interleukin-1 receptor-associated kinase; IRES, internal ribosome entry site; IRF, IFN regulatory factor; ISG, IFN stimulated gene; ISGF3G, IFN-stimulated transcription factor 3, gamma; ISRE IFN, stimulated response element; ITAF, IRES transacting factors; JAK, Janus kinase; KO, knockout; KPNA, nuclear localization signal receptor protein; LCs, Langerhans cells; LGP2, laboratory of genetics protein 2; LLV, leaderless virus; MAM, mitochondria associated membranes; MAPK, mitogen activated protein kinase; MAVS, mitochondrial antiviral signaling protein; MCP-1, monocyte chemoattractant protein 1; MDA5, melanoma differentiation associated genes; MHC, major histocompatibility complex; MIP3α, macrophage inflammatory protein-3 alpha; mRNP, messenger ribonucleoprotein; Mx-1, myxovirus resistant 1; MyD88, myeloid differentiation primary response protein 88d; Mφ, macrophage; NEMO, NF-κB essential modulator; NF-κB, nuclear factor-κB; NKC, natural killer cell; NLR, NOD leucine-rich repeat-containing receptors; NOD, nucleotide-binding oligomerization domain; OAS, oligoadenylate synthase proteins; ORF, open reading frame; PAMP, pathogen associated molecular patterns; PBMC, peripheral blood monocyte cell; PCBP2, poly(rC) binding protein 2; pDCs, plasmacytoid dendritic cells; PIAS, protein inhibitor of activated STAT; PK, pseudo knots; PKR, protein kinase R; PMC, peritoneal mast cell; poly-I:C, polyinosinic:polycytidylic acid; poly-Ub, polyubiquitination; PRR, pathogen recognition receptors; PTBP, polypyrimidine tract-binding protein; PTM, post-translational modifications; RANTES, regulated on activation, normal T cell expressed and secreted; RdRp, RNA-dependent RNA polymerase; RHA, RNA helicase A; RIG-I, retinoic acid-inducible gene I; RLR, RIG-I like receptor; SAP, SAF-ACINUS-PIAS; SeV, Sendai virus; SGs, stress granules; SOCS3, suppressor of cytokine signaling 3; STAT, signal transducer and transcription activator; TANK1, TRAF family member associated NF-κB activator 1; TBK, TANK binding kinase; TIR, toll/interleukin-1 receptor; TIRAP, toll/interleukin 1 receptor domain containing adaptor protein; TLR, Toll-like receptor; TNF, tumor necrosis factor; TRAF, TNF receptor associated factor; TRIF, TIR-domain-containing adapter-inducing interferon-β; TRIM, tripartite motif containing protein; TYK2, tyrosine kinase 2; UB, ubiquitin; UBL, ubiquitin-like; USP18, ubiquitin-specific protease 18; UTR, untranslated region; WC1, workshop cluster 1; WT, wild type.
Agol, V. I., and Gmyl, A. P. (2010). Viral security proteins: counteracting host defences. Nat. Rev. Microbiol. 8, 867–878. doi: 10.1038/nrmicro2452
Ahl, R., and Rump, A. (1976). Assay of bovine interferons in cultures of the porcine cell line IB-RS-2. Infect. Immun. 14, 603–606.
Alexandersen, S., and Donaldson, A. I. (2002). Further studies to quantify the dose of natural aerosols of foot-and-mouth disease virus for pigs. Epidemiol. Infect. 128, 313–323. doi: 10.1017/S0950268801006501
Alexopoulou, L., Holt, A. C., Medzhitov, R., and Flavell, R. A. (2001). Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413, 732–738. doi: 10.1038/35099560
Amadori, M., Archetti, I. L., Verardi, R., and Berneri, C. (1995). Role of a distinct population of bovine gamma delta T cells in the immune response to viral agents. Viral Immunol. 8, 81–91. doi: 10.1089/vim.1995.8.81
Andreev, D. E., Fernandez-Miragall, O., Ramajo, J., Dmitriev, S. E., Terenin, I. M., Martinez-Salas, E., et al. (2007). Differential factor requirement to assemble translation initiation complexes at the alternative start codons of foot-and-mouth disease virus RNA. RNA 13, 1366–1374. doi: 10.1261/rna.469707
Ao, D., Guo, H. C., Sun, S. Q., Sun, D. H., Fung, T. S., Wei, Y. Q., et al. (2015). Viroporin activity of the foot-and-mouth disease virus non-structural 2B protein. PLoS One 10:e0125828. doi: 10.1371/journal.pone.0125828
Arias, A., Perales, C., Escarmís, C., and Domingo, E. (2010). Deletion mutants of VPg reveal new cytopathology determinants in a picornavirus. PLoS One 5:e10735. doi: 10.1371/journal.pone.0010735
Aruffo, A., Bowen, M. A., Starling, G. C., Gebe, J. A., Bajorath, J., Patel, D. D., et al. (1997). CD6-ligand interactions: a paradigm for SRCR domain function? Immunol. Today 18, 498–504. doi: 10.1016/S0167-5699(97)01130-4
Arzt, J., Pacheco, J. M., and Rodriguez, L. L. (2010). The early pathogenesis of foot-and-mouth disease in cattle after aerosol inoculation. Identification of the nasopharynx as the primary site of infection. Vet. Pathol. 47, 1048–1063. doi: 10.1177/0300985810372509
Arzt, J., Pacheco, J. M., Smoliga, G. R., Tucker, M. T., Bishop, E., Pauszek, S. J., et al. (2014). Foot-and-mouth disease virus virulence in cattle is co-determined by viral replication dynamics and route of infection. Virology 45, 12–22. doi: 10.1016/j.virol.2014.01.001
Bablanian, G. M., and Grubman, M. J. (1993). Characterization of the foot-and-mouth disease virus 3C protease expressed in Escherichia coli. Virology 197, 320–327. doi: 10.1006/viro.1993.1593
Baranowski, E., Ruiz-Jarabo, C. M., Sevilla, N., Andreu, D., Beck, E., and Domingo, E. (2000). Cell recognition by foot-and-mouth disease virus that lacks the RGD integrin-binding motif: flexibility in aphthovirus receptor usage. J. Virol. 74, 1641–1647. doi: 10.1128/JVI.74.4.1641-1647.2000
Bastos, R. G., Johnson, W. C., Mwangi, W., Brown, W. C., and Goff, W. L. (2008). Bovine NK cells acquire cytotoxic activity and produce IFN-γ after stimulation by Mycobacterium bovis BCG- or Babesia bovis-exposed splenic dendritic cells. Vet. Immunol. Immunopathol. 124, 302–312. doi: 10.1016/j.vetimm.2008.04.004
Bautista, E. M., Ferman, G. S., Gregg, D., Brum, M. C. S., Grubman, M. J., and Golde, W. T. (2005). Constitutive expression of alpha interferon by skin dendritic cells confers resistance to infection by foot-and-mouth disease virus. J. Virol. 79, 4838–4847. doi: 10.1128/JVI.79.8.4838-4847.2005
Baxt, B., and Mason, P. W. (1995). Foot-and-mouth disease virus undergoes restricted replication in macrophage cell cultures following Fc receptor-mediated adsorption. Virology 207, 503–509. doi: 10.1006/viro.1995.1110
B’Chir, W., Maurin, A. C., Carraro, V., Averous, J., Jousse, C., Muranishi, Y., et al. (2013). The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 41, 7683–7699. doi: 10.1093/nar/gkt563
Bel, M., Ocaña-Macchi, M., Liniger, M., McCullough, K. C., Matrosovich, M., and Summerfield, A. (2011). Efficient sensing of avian influenza viruses by porcine plasmacytoid dendritic cells. Viruses 3, 312–330. doi: 10.3390/v3040312
Belov, G. A., Romanova, L. I., Tolskaya, E. A., Kolesnikova, M. S., Lazebnik, Y. A., and Agol, V. I. (2003). The major apoptotic pathway activated and suppressed by poliovirus. J. Virol. 77, 45–56. doi: 10.1128/JVI.77.1.45-56.2003
Belsham, G. J. (2005). Translation and replication of FMDV RNA. Curr. Top. Microbiol. Immunol. 288, 43–70. doi: 10.1007/3-540-27109-0_3
Belsham, G. J. (2009). Divergent picornavirus IRES elements. Virus Res. 139, 183–192. doi: 10.1016/j.virusres.2008.07.001
Belsham, G. J., McInerney, G. M., and Ross-Smith, N. (2000). Foot-and-mouth disease virus 3C protease induces cleavage of translation initiation factors eIF4A and eIF4G within infected cells. J. Virol. 74, 272–280. doi: 10.1128/JVI.74.1.272-280.2000
Benedict, C. A., Norris, P. S., and Ware, C. F. (2002). To kill or be killed: viral evasion of apoptosis. Nat. Immunol. 3, 1013–1018. doi: 10.1038/ni1102-1013
Berke, I. C., and Modis, Y. (2012). MDA5 cooperatively forms dimers and ATP-sensitive filaments upon binding double-stranded RNA. EMBO J. 31, 1714–1726. doi: 10.1038/emboj.2012.19
Berryman, S., Brooks, E., Burman, A., Hawes, P., Roberts, R., Netherton, C., et al. (2012). Foot-and-mouth disease virus induces autophagosomes during cell entry via a class III phosphatidylinositol 3-kinase-independent pathway. J. Virol. 86, 12940–12953. doi: 10.1128/JVI.00846-12
Binder, R. J. (2014). Functions of heat shock proteins in pathways of the innate and adaptive immune system. J. Immunol. 193, 5765–5771. doi: 10.4049/jimmunol.1401417
Birtley, J. R., Knox, S. R., Jaulent, A. M., Brick, P., Leatherbarrow, R. J., and Curry, S. (2005). Crystal structure of foot-and-mouth disease virus 3C protease: new insights into catalytic mechanism and cleavage specificity. J. Biol. Chem. 280, 11520–11527. doi: 10.1074/jbc.M413254200
Boysen, P., Klevar, S., Olsen, I., and Storset, A. K. (2006a). The protozoan Neospora caninum directly triggers bovine NK cells to produce gamma interferon and to kill infected fibroblasts. Infect. Immun. 74, 953–960. doi: 10.1128/IAI.74.2.953-960.2006
Boysen, P., Olsen, I., Berg, I., Kulberg, S., Johansen, G. M., and Storset, A. K. (2006b). Bovine CD2-/NKp46+ cells are fully functional natural killer cells with a high activation status. BMC Immunol. 7:10. doi: 10.1186/1471-2172-7-10
Brooks, D. G., Trifilo, M. J., Edelmann, K. H., Teyton, L., McGavern, D. B., and Oldstone, M. B. A. (2006). Interleukin-10 determines viral clearance or persistence in vivo. Nat. Med. 12, 1301–1309. doi: 10.1038/nm1492
Brown, C. C., Piccone, M. E., Mason, P. W., McKenna, T. S., and Grubman, M. J. (1996). Pathogenesis of wild-type and leaderless foot-and-mouth disease virus in cattle. J. Virol. 70, 5638–5641.
Brown, J. K., McAleese, S. M., Thornton, E. M., Pate, J. A., Schock, A., Macrae, A. I., et al. (2006). Integrin-αvβ6, a putative receptor for foot-and-mouth disease virus, is constitutively expressed in ruminant airways. J. Histochem. Cytochem. 54, 807–816. doi: 10.1369/jhc.5A6854.2006
Bruns, A. M., and Horvath, C. M. (2015). LGP2 synergy with MDA5 in RLR-mediated RNA recognition and antiviral signaling. Cytokine 74, 198–206. doi: 10.1016/j.cyto.2015.02.010
Burdette, D. L., Monroe, K. M., Sotelo-Troha, K., Iwig, J. S., Eckert, B., Hyodo, M., et al. (2011). STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518. doi: 10.1038/nature10429
Cao, X., Bergmann, I. E., Füllkrug, R., and Beck, E. (1995). Functional analysis of the two alternative translation initiation sites of foot-and-mouth disease virus. J. Virol. 69, 560–563.
Carrillo, C., Tulman, E. R., Delhon, G., Lu, Z., Carreno, A., Vagnozzi, A., et al. (2005). Comparative genomics of foot-and-mouth disease virus. J. Virol. 79, 6487–6504. doi: 10.1128/JVI.79.10.6487-6504.2005
Chinsangaram, J., Koster, M., and Grubman, M. J. (2001). Inhibition of L-deleted foot-and-mouth disease virus replication by alpha/beta interferon involves double-stranded RNA-dependent protein kinase. J. Virol. 75, 5498–5503. doi: 10.1128/JVI.75.12.5498-5503.2001
Chinsangaram, J., Piccone, M. E., and Grubman, M. J. (1999). Ability of foot-and-mouth disease virus to form plaques in cell culture is associated with suppression of alpha/beta interferon. J. Virol. 73, 9891–9898.
Choi, Y., Bowman, J. W., and Jung, J. U. (2018). Autophagy during viral infection — a double-edged sword. Nat. Rev. Microbiol. 2018:1. doi: 10.1038/s41579-018-0003-6
Clarke, B. E., and Sangar, D. V. (1988). Processing and assembly of foot-and-mouth disease virus proteins using subgenomic RNA. J. Gen. Virol. 692313–2325. doi: 10.1099/0022-1317-69-9-2313
Clayton, K., Vallejo, A. F., Davies, J., Sirvent, S., and Polak, M. E. (2017). Langerhans cells-programmed by the epidermis. Front. Immunol. 8:1676. doi: 10.3389/fimmu.2017.01676
Collins, R. a, Camon, E. B., Chaplin, P. J., and Howard, C. J. (1998). Influence of IL-12 on interferon-gamma production by bovine leucocyte subsets in response to bovine respiratory syncytial virus. Vet. Immunol. Immunopathol. 63, 69–72. doi: 10.1016/S0165-2427(98)00083-X
Das, A., Dinh, P. X., Panda, D., and Pattnaik, A. K. (2014). Interferon-inducible protein IFI35 negatively regulates RIG-I antiviral signaling and supports vesicular stomatitis virus replication. J. Virol. 88, 3103–3113. doi: 10.1128/JVI.03202-13
de los Santos, T., De Avila Botton, S., Weiblen, R., and Grubman, M. J. (2006). The leader proteinase of foot-and-mouth disease virus inhibits the induction of beta interferon mRNA and blocks the host innate immune response. J. Virol. 80, 1906–1914. doi: 10.1128/JVI.80.4.1906-1914.2006
de los Santos, T., Diaz-San Segundo, F., and Grubman, M. J. (2007). Degradation of nuclear factor kappa B during foot-and-mouth disease virus infection. J. Virol. 81, 12803–12815. doi: 10.1128/JVI.01467-07
de los Santos, T., Segundo, F. D.-S., Zhu, J., Koster, M., Dias, C. C. A, and Grubman, M. J. (2009). A conserved domain in the leader proteinase of foot-and-mouth disease virus is required for proper subcellular localization and function. J. Virol. 83, 1800–1810. doi: 10.1128/JVI.02112
Dempsey, A., and Bowie, A. G. (2015). Innate immune recognition of DNA: a recent history. Virology 47, 146–152. doi: 10.1016/j.virol.2015.03.013
Denyer, M. S., Wileman, T. E., Stirling, C. M. A., Zuber, B., and Takamatsu, H. H. (2006). Perforin expression can define CD8 positive lymphocyte subsets in pigs allowing phenotypic and functional analysis of Natural Killer, Cytotoxic T, Natural Killer T and MHC un-restricted cytotoxic T-cells. Vet. Immunol. Immunopathol. 110, 279–292. doi: 10.1016/j.vetimm.2005.10.005
Deretic, V., and Levine, B. (2009). Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5, 527–549. doi: 10.1016/j.chom.2009.05.016
Deretic, V., Saitoh, T., and Akira, S. (2013). Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 13, 722–737. doi: 10.1038/nri3532
Devaney, M. A., Vakharia, V. N., Lloyd, R., Ehrenfeld, E., and Grubman, M. J. (1988). Leader protein of foot-and-mouth disease virus is required for cleavage of the p220 component of the cap-binding protein complex. J. Virol. 62, 4407–4409.
Devarkar, S. C., Wang, C., Miller, M. T., Ramanathan, A., Jiang, F., Khan, A. G., et al. (2016). Structural basis for m7G recognition and 2’-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc. Natl. Acad. Sci. U.S.A. 113, 596–601. doi: 10.1073/pnas.1515152113
Dias, C. C. A., Moraes, M. P., Diaz-SanSegundo, F., de los Santos, T., and Grubman, M. J. (2011). Porcine type I interferon rapidly protects swine against challenge with multiple serotypes of foot-and-mouth disease virus. J. Interferon Cytokine Res. 31, 227–236. doi: 10.1089/jir.2010.0055
Diaz-San Segundo, F., Montiel, N., de los Santos, T., and Grubman, M. J. (2013). Understanding the mechanisms of interferon-induced protection against foot-and-mouth disease. Rome: Food and Agriculture Organization of the United Nations.
Diaz-San Segundo, F., Montiel, N. A., Sturza, D. F., Perez-Martin, E., Hickman, D., Ramirez-Medina, E., et al. (2016). Combination of Adt-O1Manisa and Ad5-boIFNλ3 induces early protective immunity against foot-and-mouth disease in cattle. Virology 499, 340–349. doi: 10.1016/j.virol.2016.09.027
Diaz-San Segundo, F., Moraes, M. P., de Los Santos, T., Dias, C. C. A., and Grubman, M. J. (2010). Interferon-induced protection against foot-and-mouth disease virus infection correlates with enhanced tissue-specific innate immune cell infiltration and interferon-stimulated gene expression. J. Virol. 84, 2063–2077. doi: 10.1128/JVI.01874-09
Díaz-San Segundo, F., Rodríguez-Calvo, T., de Avila, A., and Sevilla, N. (2009). Immunosuppression during acute infection with foot-and-mouth disease virus in swine is mediated by IL-10. PLoS One 4:e5659. doi: 10.1371/journal.pone.0005659
Díaz-San Segundo, F., Salguero, F. J., de Avila, A., Fernandez, de Marco, M. M., Sanchez-Martin, M. A., et al. (2006). Selective lymphocyte depletion during the early stage of the immune response to foot-and-mouth disease virus infection in swine. J. Virol. 80, 2369–2379. doi: 10.1128/JVI.80.5.2369-2379.2006
Diaz-San Segundo, F., Weiss, M., Perez-Martin, E., Dias, C. C., Grubman, M. J., and de los Santos, T. (2012). Inoculation of swine with foot-and-mouth disease SAP-mutant virus induces early protection against disease. J. Virol. 86, 1316–1327. doi: 10.1128/JVI.05941-11
Díaz-San Segundo, F., Weiss, M., Perez-Martín, E., Koster, M. J., Zhu, J., Grubman, M. J., et al. (2011). Antiviral activity of bovine type III interferon against foot-and-mouth disease virus. Virology 413, 283–292. doi: 10.1016/j.virol.2011.02.023
Domingo, E., and Perales, C. (2018). Quasispecies and virus. Eur. Biophys. J. 47, 443–457. doi: 10.1007/s00249-018-1282-6
Donaldson, A. I., and Alexandersen, S. (2001). Relative resistance of pigs to infection by natural aerosols of FMD virus. Vet. Rec. 148, 600–602. doi: 10.1136/vr.148.19.600
Donaldson, A. I., Gibson, C. F., Oliver, R., Hamblin, C., and Kitching, R. P. (1987). Infection of cattle by airborne foot-and-mouth disease virus: minimal doses with O1 and SAT 2 strains. Res. Vet. Sci. 43, 339–346. doi: 10.1016/S0034-5288(18)30804-X
Donaldson, A. I., Herniman, K. A. J., Parker, J., and Sellers, R. F. (1970). Further investigations on the airborne excretion of foot-and-mouth disease virus. J. Hyg. (Lond). 68, 557–564. doi: 10.1017/S0022172400042480
Donnelly, R., and Kotenko, S. (2010). Interferon-lambda: a new addition to an old family. J Interf. Cytokine Res. 30, 555–564. doi: 10.1089/jir.2010.0078
dos Santos, P. F., and Mansur, D. S. (2017). Beyond ISGlylation: functions of free intracellular and extracellular ISG15. J. Interf. Cytokine Res. 37, 246–253. doi: 10.1089/jir.2016.0103
Dou, Y., Yim, H. C., Kirkwood, C. D., Williams, B. R., and Sadler, A. J. (2017). The innate immune receptor MDA5 limits rotavirus infection but promotes cell death and pancreatic inflammation. EMBO J. 36, 2742–2757. doi: 10.15252/embj.201696273
Du, Y., Bi, J., Liu, J., Liu, X., Wu, X., Jiang, P., et al. (2014). 3Cpro of foot-and-mouth disease virus antagonizes the interferon signaling pathway by blocking STAT1/STAT2 nuclear translocation. J. Virol. 88, 4908–4920. doi: 10.1128/JVI.03668-13
Dunn, C. S., and Donaldson, A. I. (1997). Natural adaption to pigs of a Taiwanese isolate of foot-and-mouth disease virus. Vet. Rec. 141, 147–175. doi: 10.1136/vr.141.7.174
Eschbaumer, M., Stenfeldt, C., Rekant, S. I., Pacheco, J. M., Hartwig, E. J., Smoliga, G. R., et al. (2016). Systemic immune response and virus persistence after foot-and-mouth disease virus infection of naïve cattle and cattle vaccinated with a homologous adenovirus-vectored vaccine. BMC Vet. Res. 12:205. doi: 10.1186/s12917-016-0838-x
Falk, M. M., Grigera, P. R., Bergmann, I. E., Zibert, A., Multhaup, G., and Beck, E. (1990). Foot-and-mouth disease virus protease 3C induces specific proteolytic cleavage of host cell histone H3. J. Virol. 64, 748–756.
Fan, X., Han, S., Yan, D., Gao, Y., Wei, Y., Liu, X., et al. (2017). Foot-and-mouth disease virus infection suppresses autophagy and NF-κB antiviral responses via degradation of ATG5-ATG12 by 3Cpro. Cell Death Dis. 8:e2561. doi: 10.1038/cddis.2016.489
Galan, A., Lozano, G., Piñeiro, D., and Martinez-Salas, E. (2017). G3BP1 interacts directly with the FMDV IRES and negatively regulates translation. FEBS J. 284, 3202–3217. doi: 10.1111/febs.14184
Gao, Y., Sun, S.-Q., and Guo, H.-C. (2016). Biological function of Foot-and-mouth disease virus non-structural proteins and non-coding elements. Virol. J. 13:107. doi: 10.1186/s12985-016-0561-z
Garcia Briones, M., Rosas, M. F., Gonzalez Magaldi, M., Martin Acebes, M. A., Sobrino, F., and Armas Portela, R. (2006). Differential distribution of non-structural proteins of foot-and-mouth disease virus in BHK-21 cells. Virology 349, 409–421. doi: 10.1016/j.virol.2006.02.042
García-Nuñez, S., Gismondi, M. I., König, G., Berinstein, A., Taboga, O., Rieder, E., et al. (2014). Enhanced IRES activity by the 3’UTR element determines the virulence of FMDV isolates. Virology 448, 303–313. doi: 10.1016/j.virol.2013.10.027
Geller, R., Pechmann, S., Acevedo, A., Andino, R., and Frydman, J. (2018). Hsp90 shapes protein and RNA evolution to balance trade-offs between protein stability and aggregation. Nat. Commun. 9:1781. doi: 10.1038/s41467-018-04203-x
Gladue, D. P., O’Donnell, V., Baker-Branstetter, R., Holinka, L. G., Pacheco, J. M., Fernandez-Sainz, I., et al. (2012). Foot-and-mouth disease virus nonstructural protein 2C interacts with beclin1, modulating virus replication. J. Virol. 86, 12080–12090. doi: 10.1128/JVI.01610-12
González-Magaldi, M., Martín-Acebes, M. A., Kremer, L., and Sobrino, F. (2014). Membrane topology and cellular dynamics of foot-and-mouth disease virus 3A protein. PLoS One 9:e106685. doi: 10.1371/journal.pone.0106685
Gradi, A., Foeger, N., Strong, R., Yuri, V., Sonenberg, N., Skern, T., et al. (2004). Cleavage of eukaryotic translation initiation factor 4GII within foot-and-mouth disease virus-infected cells: identification of the l-protease cleavage site in vitro. J. Virol. 78, 3271–3278. doi: 10.1128/JVI.78.7.3271
Grigera, P. R., and Tisminetzky, S. G. (1984). Histone H3 modification in BHK cells infected with foot-and-mouth disease virus. Virology 136, 10–19. doi: 10.1016/0042-6822(84)90243-5
Grubman, M. J., Diaz-San Segundo, F., Dias, C. C., Moraes, M. P., Perez-Martin, E., and de los Santos, T. (2012). Use of replication-defective adenoviruses to develop vaccines and biotherapeutics against foot-and-mouth disease. Future Virol. 7, 767–778. doi: 10.2217/fvl.12.65
Guarne, A., Tormo, J., Kirchweger, R., Pfistermueller, D., Fita, I., and Skern, T. (1998). Structure of the foot and mouth disease virus leader protease: a papain like fold adapted for self processing and eIF4G recognition. Embo J. 1998:74697479. doi: 10.1093/emboj/17.24.7469
Guzylack-Piriou, L., Bergamin, F., Gerber, M., McCullough, K. C., and Summerfield, A. (2006). Plasmacytoid dendritic cell activation by foot-and-mouth disease virus requires immune complexes. Eur. J. Immunol. 36, 1674–1683. doi: 10.1002/eji.200635866
Han, S.-C., Guo, H.-C., and Sun, S.-Q. (2015). Three-dimensional structure of foot-and-mouth disease virus and its biological functions. Arch. Virol. 160, 1–16. doi: 10.1007/s00705-014-2278-x
Harwood, L. J., Gerber, H., Sobrino, F., Summerfield, A., and McCullough, K. C. (2008). Dendritic cell internalization of foot-and-mouth disease virus: influence of heparan sulfate binding on virus uptake and induction of the immune response. J. Virol. 82, 6379–6394. doi: 10.1128/JVI.00021-08
He, C., and Klionsky, D. J. (2009). Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93. doi: 10.1146/annurev-genet-102808-114910
Heaton, S. M., Borg, N. A., and Dixit, V. M. (2016). Ubiquitin in the activation and attenuation of innate antiviral immunity. J. Exp. Med. 213, 1–13. doi: 10.1084/jem.20151531
Heil, F., Hemmi, H., Hochrein, H., Ampenberger, F., Kirschning, C., Akira, S., et al. (2004). Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303, 1526–1529. doi: 10.1126/science.1093620
Hermesh, T., Moltedo, B., Moran, T. M., and López, C. B. (2010). Antiviral instruction of bone marrow leukocytes during respiratory viral infections. Cell Host Microbe 7, 343–353. doi: 10.1016/j.chom.2010.04.006
Ho, M.-Y., Hung, S.-W., Liang, C.-M., and Liang, S.-M. (2014). Recombinant viral capsid protein VP1 suppresses lung cancer metastasis by inhibiting COX-2/PGE2 and MIG-7. Oncotarget 5, 3931–3943. doi: 10.18632/oncotarget.2040
Honda, K., Yanai, H., Negishi, H., Asagiri, M., Sato, M., Mizutani, T., et al. (2005). IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434, 772–777. doi: 10.1038/nature03464
Honke, N., Shaabani, N., Zhang, D. E., Hardt, C., and Lang, K. S. (2016). Multiple functions of USP18. Cell Death Dis. 7:e2444. doi: 10.1038/cddis.2016.326
Horner, S. M., Liu, H. M., Park, H. S., Briley, J., and Gale, M. (2011). Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. U.S.A. 108, 14590–14595. doi: 10.1073/pnas.1110133108
Hüsser, L., Alves, M. P., Ruggli, N., and Summerfield, A. (2011). Identification of the role of RIG-I, MDA-5 and TLR3 in sensing RNA viruses in porcine epithelial cells using lentivirus-driven RNA interference. Virus Res. 159, 9–16. doi: 10.1016/j.virusres.2011.04.005
Ikushima, H., Negishi, H., and Taniguchi, T. (2013). The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb. Symp. Quant. Biol. 78, 105–116. doi: 10.1101/sqb.2013.78.020321
Jackson, T., Ellard, F. M., Ghazaleh, R. A., Brookes, S. M., Blakemore, W. E., Corteyn, A. H., et al. (1996). Efficient infection of cells in culture by type O foot-and-mouth disease virus requires binding to cell surface heparan sulfate. J. Virol. 70, 5282–5287.
Jackson, W. T., Giddings, T. H., Taylor, M. P., Mulinyawe, S., Rabinovitch, M., Kopito, R. R., et al. (2005). Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3:e156. doi: 10.1371/journal.pbio.0030156
Joshi, G., Sharma, R., and Kakker, N. K. (2009). Phenotypic and functional characterization of T-cells and in vitro replication of FMDV serotypes in bovine lymphocytes. Vaccine 27, 6656–6661. doi: 10.1016/j.vaccine.2009.08.107
Juleff, N., Windsor, M., Lefevre, E. A., Gubbins, S., Hamblin, P., Reid, E., et al. (2009). Foot-and-mouth disease virus can induce a specific and rapid CD4+ T-cell-independent neutralizing and isotype class-switched antibody response in naive cattle. J. Virol. 83, 3626–3636. doi: 10.1128/JVI.02613-08
Kamphuis, E., Junt, T., Waibler, Z., Forster, R., and Kalinke, U. (2006). Type I interferons directly regulate lymphocyte recirculation and cause transient blood lymphopenia. Blood 108, 3253–3261. doi: 10.1182/blood-2006-06-027599
Kang, D.-C., Gopalkrishnan, R. V., Wu, Q., Jankowsky, E., Pyle, A. M., and Fisher, P. B. (2002). mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc. Natl. Acad. Sci. U.S.A. 99, 637–642. doi: 10.1073/pnas.022637199
Kato, H., Takeuchi, O., Mikamo-Satoh, E., Hirai, R., Kawai, T., Matsushita, K., et al. (2008). Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid–inducible gene-I and melanoma differentiation–associated gene 5. J. Exp. Med. 205, 1601–1610. doi: 10.1084/jem.20080091
Kawai, T., and Akira, S. (2010). The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat. Immunol. 11, 373–384. doi: 10.1038/ni.1863
Kim, M.-J., Hwang, S.-Y., Imaizumi, T., and Yoo, J.-Y. (2008). Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J. Virol. 82, 1474–1483. doi: 10.1128/JVI.01650-07
Kim, S. M., Kim, S. K., Park, J. H., Lee, K. N., Ko, Y. J., Lee, H. S., et al. (2014). A recombinant adenovirus bicistronically expressing porcine interferon-α and interferon-γ enhances antiviral effects against foot-and-mouth disease virus. Antiv. Res. 104, 52–58. doi: 10.1016/j.antiviral.2014.01.014
Kirchweger, R., Ziegler, E., Lamphear, B. J., Waters, D., Liebig, H. D., Sommergruber, W., et al. (1994). Foot-and-mouth disease virus leader proteinase: purification of the Lb form and determination of its cleavage site on eIF-4 gamma. J. Virol. 68, 5677–5684.
Kloc, A., Diaz-San Segundo, F., Schafer, E. A., Rai, D. K., Kenney, M., de los Santos, T., et al. (2017). Foot-and-mouth disease virus 5′-terminal S fragment is required for replication and modulation of the innate immune response in host cells. Virology 512, 132–143. doi: 10.1016/j.virol.2017.08.036
Kloc, A., Rai, D. K., and Rieder, E. (2018). The roles of picornavirus untranslated regions in infection and innate immunity. Front. Microbiol. 9:485. doi: 10.3389/fmicb.2018.00485
Kühn, R., Luz, N., and Beck, E. (1990). Functional analysis of the internal translation initiation site of foot-and-mouth disease virus. J. Virol. 64, 4625–4631.
Kuo, C. -J., Hansen, M., and Troemel, E. (2017). Autophagy and innate immunity: Insights from invertebrate model organisms. Autophagy 14, 233–242. doi: 10.1080/15548627.2017.1389824
Langereis, M. A., Feng, Q., and van Kuppeveld, F. J. (2013). MDA5 localizes to stress granules, but this localization is not required for the induction of Type I interferon. J. Virol. 87, 6314–6325. doi: 10.1128/JVI.03213-12
Lannes, N., Python, S., and Summerfield, A. (2012). Interplay of foot-and-mouth disease virus, antibodies and plasmacytoid dendritic cells: virus opsonization under non-neutralizing conditions results in enhanced interferon-alpha responses. Vet. Res. 43:64. doi: 10.1186/1297-9716-43-64
Lawrence, P., Conderino, J. S., and Rieder, E. (2014). Redistribution of demethylated RNA helicase A during foot-and-mouth disease virus infection: role of jumonji c-domain containing protein 6 in RHA demethylation. Virology 45, 1–11. doi: 10.1016/j.virol.2013.12.040
Lawrence, P., Rai, D., Conderino, J. S., Uddowla, S., and Rieder, E. (2016). Role of Jumonji C-domain containing protein 6 (JMJD6) in infectivity of foot-and-mouth disease virus. Virology 492, 38–52. doi: 10.1016/j.virol.2016.02.005
Lawrence, P., and Rieder, E. (2009). Identification of RNA helicase A as a new host factor in the replication cycle of foot-and-mouth disease virus. J. Virol. 83, 11356–11366. doi: 10.1128/JVI.02677-08
Lawrence, P., Schafer, E. A., and Rieder, E. (2012). The nuclear protein Sam68 is cleaved by the FMDV 3C protease redistributing Sam68 to the cytoplasm during FMDV infection of host cells. Virology 425, 40–52. doi: 10.1016/j.virol.2011.12.019
Lee, H. K., Lund, J. M., Ramanathan, B., Mizushima, N., and Iwasaki, A. (2007). Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science (80-) 315, 1398–1401. doi: 10.1126/science.1136880
Li, C., Zhu, Z., Du, X., Cao, W., Yang, F., Zhang, X., et al. (2017). Foot-and-mouth disease virus induces lysosomal degradation of host protein kinase PKR by 3C proteinase to facilitate virus replication. Virology 509, 222–231. doi: 10.1016/j.virol.2017.06.023
Li, D., Lei, C., Xu, Z., Yang, F., Liu, H., Zhu, Z., et al. (2016a). Foot-and-mouth disease virus non-structural protein 3A inhibits the interferon-β signaling pathway. Sci. Rep. 6:21888. doi: 10.1038/srep21888
Li, D., Wei, J., Yang, F., Liu, H. -N., Zhu, Z. -X., Cao, W. -J., et al. (2016b). Foot-and-mouth disease virus structural protein VP3 degrades Janus kinase 1 to inhibit IFN-γ signal transduction pathways. Cell Cycle 15, 850–860. doi: 10.1080/15384101.2016.1151584
Li, D., Yang, W., Yang, F., Liu, H., Zhu, Z., Lian, K., et al. (2016c). The VP3 structural protein of foot-and-mouth disease virus inhibits the IFN-β signaling pathway. FASEB J. 30, 1757–1766. doi: 10.1096/fj.15-281410
Li, X., Lu, C., Stewart, M., Xu, H., Strong, R. K., Igumenova, T., et al. (2009). Structural basis of double-stranded RNA recognition by the RIG-I like receptor MDA5. Arch. Biochem. Biophys. 488, 23–33. doi: 10.1016/j.abb.2009.06.008
Li, X., Wang, J., Liu, J., Li, Z., Wang, Y., Xue, Y., et al. (2013). Engagement of soluble resistance-related calcium binding protein (sorcin) with foot-and-mouth disease virus (FMDV) VP1 inhibits type I interferon response in cells. Vet. Microbiol. 166, 35–46. doi: 10.1016/j.vetmic.2013.04.028
Lotufo, C., Wilda, M., Giraldez, A., Grigera, P., and Mattion, N. (2018). Relevance of the N-terminal and major hydrophobic domains of non-structural protein 3A in the replicative process of a DNA-launched foot-and-mouth disease virus replicon. Arch. Virol. 163, 1769–1778. doi: 10.1007/s00705-018-3795-9
Luz, N., and Beck, E. (1991). Interaction of a cellular 57-kilodalton protein with the internal translation initiation site of foot-and-mouth disease virus. J. Virol. 65, 6486–6494.
MacDonald, M. R., Machlin, E. S., Albin, O. R., and Levy, D. E. (2007). The zinc finger antiviral protein acts synergistically with an interferon-induced factor for maximal activity against alphaviruses. J. Virol. 81, 13509–13518. doi: 10.1128/JVI.00402-07
Malakhova, O. A., Kim, K. IL., Luo, J. K., Zou, W., Kumar, K. G. S., Fuchs, S. Y., et al. (2006). UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2358–2367. doi: 10.1038/sj.emboj.7601149
Martín-Acebes, M. A., González-Magaldi, M., Sandvig, K., Sobrino, F., and Armas-Portela, R. (2007). Productive entry of type C foot-and-mouth disease virus into susceptible cultured cells requires clathrin and is dependent on the presence of plasma membrane cholesterol. Virology 369, 105–118. doi: 10.1016/j.virol.2007.07.021
Martínez-Salas, E., Francisco-Velilla, R., Fernandez-Chamorro, J., Lozano, G., and Diaz-Toledano, R. (2015). Picornavirus IRES elements: RNA structure and host protein interactions. Virus Res. 306, 62–73. doi: 10.1016/j.virusres.2015.01.012
Mason, P. W., Grubman, M. J., and Baxt, B. (2003). Molecular basis of pathogenesis of FMDV. Virus Res. 91, 9–32. doi: 10.1016/S0168-1702(02)00257-5
McCormick, C., and Khaperskyy, D. A. (2017). Translation inhibition and stress granules in the antiviral immune response. Nat. Rev. Immunol. 17, 647–660. doi: 10.1038/nri.2017.63
McCullough, K. C., Parkinson, D., and Crowther, J. R. (1988). Opsonization-enhanced phagocytosis of foot-and-mouth disease virus. Immunology 65, 187–191.
Medina, G. N., Knudsen, G. M., Greninger, A. L., Kloc, A., Díaz-San Segundo, F., Rieder, E., et al. (2017). Interaction between FMDV Lpro and transcription factor ADNP is required for optimal viral replication. Virology 505, 12–22. doi: 10.1016/j.virol.2017.02.010
Medina, M., Domingo, E., Brangwyn, J. K., and Belsham, G. J. (1993). The two species of the foot-and-mouth disease virus leader protein, expressed individually, exhibit the same activities. Virology 194, 355–359. doi: 10.1006/viro.1993.1267
Medvedev, A. E., and Vogel, S. N. (2003). Overexpression of CD14, TLR4, and MD-2 in HEK 293T cells does not prevent induction of in vitro endotoxin tolerance. J. Endotoxin Res. 9, 60–64. doi: 10.1179/096805103125001360
Midgley, R., Moffat, K., Berryman, S., Hawes, P., Simpson, J., Fullen, D., et al. (2013). A role for endoplasmic reticulum exit sites in foot-and-mouth disease virus infection. J. Gen. Virol. 94, 2636–2646. doi: 10.1099/vir.0.055442-0
Moffat, K., Howell, G., Knox, C., Belsham, G. J., Monaghan, P., Ryan, M. D., et al. (2005). Effects of foot-and-mouth disease virus nonstructural proteins on the structure and function of the early secretory pathway: 2BC but not 3A blocks endoplasmic reticulum-to-Golgi transport. J. Virol. 79, 4382–4395. doi: 10.1128/JVI.79.7.4382-4395.2005
Moffat, K., Knox, C., Howell, G., Clark, S. J., Yang, H., Belsham, G. J., et al. (2007). Inhibition of the secretory pathway by foot-and-mouth disease virus 2BC protein is reproduced by coexpression of 2B with 2C, and the site of inhibition is determined by the subcellular location of 2C. J. Virol. 81, 1129–1139. doi: 10.1128/JVI.00393-06
Monaghan, P., Cook, H., Jackson, T., Ryan, M., and Wileman, T. (2004). The ultrastructure of the developing replication site in foot-and-mouth disease virus-infected BHK-38 cells. J. Gen. Virol. 85, 933–946. doi: 10.1099/vir.0.19408-0
Monaghan, P., Gold, S., Simpson, J., Zhang, Z., Weinreb, P. H., Violette, S. M., et al. (2005). The alpha(v)beta6 integrin receptor for Foot-and-mouth disease virus is expressed constitutively on the epithelial cells targeted in cattle. J. Gen. Virol. 86, 2769–2780. doi: 10.1099/vir.0.81172-0
Moraes, M. P., Chinsangaram, J., Brum, M. C. S., and Grubman, M. J. (2003). Immediate protection of swine from foot-and-mouth disease: a combination of adenoviruses expressing interferon alpha and a foot-and-mouth disease virus subunit vaccine. Vaccine 22, 268–279. doi: 10.1016/S0264-410X(03)00560-7
Moraes, M. P., de Los Santos, T., Koster, M., Turecek, T., Wang, H., Andreyev, V. G., et al. (2007). Enhanced antiviral activity against foot-and-mouth disease virus by a combination of type I and II porcine interferons. J. Virol. 81, 7124–7135. doi: 10.1128/JVI.02775-06
Moral-López, P., Alvarez, E., Redondo, N., Skern, T., and Carrasco, L. (2014). L protease from foot and mouth disease virus confers eIF2-independent translation for mRNAs bearing picornavirus IRES. FEBS Lett. 588, 4053–4059. doi: 10.1016/j.febslet.2014.09.030
Newman, J., Asfor, A. S., Berryman, S., Jackson, T., Curry, S., and Tuthill, T. J. (2017). The cellular chaperone heat shock protein 90 is required for foot-and-mouth disease virus capsid precursor processing and assembly of capsid pentamers. J. Virol. 92:e01415-17. doi: 10.1128/JVI.01415-17
Nfon, C. K., Ferman, G. S., Toka, F. N., Gregg, D. A., and Golde, W. T. (2008). Interferon-alpha production by swine dendritic cells is inhibited during acute infection with foot-and-mouth disease virus. Viral Immunol. 21, 68–77. doi: 10.1089/vim.2007.0097
Nfon, C. K., Toka, F. N., Kenney, M., Pacheco, J. M., and Golde, W. T. (2010). Loss of plasmacytoid dendritic cell function coincides with lymphopenia and viremia during foot-and-mouth disease virus infection. Viral Immunol. 23, 29–41. doi: 10.1089/vim.2009.0078
Nieva, J. L., Madan, V., and Carrasco, L. (2012). Viroporins: Structure and biological functions. Nat. Rev. Microbiol. 10, 563–574. doi: 10.1038/nrmicro2820
Nunez, J. I., Baranowski, E., Molina, N., Ruiz-Jarabo, C. M., Sanchez, C., Domingo, E., et al. (2001). A single amino acid substitution in nonstructural protein 3A can mediate adaptation of foot-and-mouth disease virus to the guinea pig. J. Virol. 75, 3977–3983. doi: 10.1128/JVI.75.8.3977-3983.2001
O’Donnell, V., LaRocco, M., and Baxt, B. (2008). Heparan sulfate-binding foot-and-mouth disease virus enters cells via caveola-mediated endocytosis. J. Virol. 82, 9075–9085. doi: 10.1128/JVI.00732-08
O’Donnell, V., Pacheco, J. M., Gregg, D., and Baxt, B. (2009). Analysis of foot-and-mouth disease virus integrin receptor expression in tissues from naïve and infected cattle. J. Comp. Pathol. 141, 98–112. doi: 10.1016/j.jcpa.2008.09.008
O’Donnell, V., Pacheco, J. M., LaRocco, M., Burrage, T., Jackson, W., Rodriguez, L. L., et al. (2011). Foot-and-mouth disease virus utilizes an autophagic pathway during viral replication. Virology 410, 142–150. doi: 10.1016/j.virol.2010.10.042
O’Donnell, V. K., Pacheco, J. M., Henry, T. M., and Mason, P. W. (2001). Subcellular distribution of the foot-and-mouth disease virus 3A protein in cells infected with viruses encoding wild-type and bovine-attenuated forms of 3A. Virology 287, 151–162. doi: 10.1006/viro.2001.1035
Onomoto, K., Jogi, M., Yoo, J. S., Narita, R., Morimoto, S., Takemura, A., et al. (2012). Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS One 7:e43031. doi: 10.1371/journal.pone.0043031
Pacheco, A., De Quinto, S. L., Ramajo, J., Fernández, N., and Martínez-Salas, E. (2009). A novel role for Gemin5 in mRNA translation. Nucleic Acids Res. 37, 582–590. doi: 10.1093/nar/gkn979
Pacheco, J. M., Arzt, J., and Rodriguez, L. L. (2010a). Early events in the pathogenesis of foot-and-mouth disease in cattle after controlled aerosol exposure. Vet. J. 183, 46–53. doi: 10.1016/j.tvjl.2008.08.023
Pacheco, J. M., Butler, J. E., Jew, J., Ferman, G. S., Zhu, J., and Golde, W. T. (2010b). IgA antibody response of swine to foot-and-mouth disease virus infection and vaccination. Clin. Vaccine Immunol. 17, 550–558. doi: 10.1128/CVI.00429-09
Pacheco, J. M., Gladue, D. P., Holinka, L. G., Arzt, J., Bishop, E., Smoliga, G., et al. (2013). A partial deletion in non-structural protein 3A can attenuate foot-and-mouth disease virus in cattle. Virology 446, 260–267. doi: 10.1016/j.virol.2013.08.003
Pacheco, J. M., Smoliga, G. R., O’Donnell, V., Brito, B. P., Stenfeldt, C., Rodriguez, L. L., et al. (2015). Persistent foot-and-mouth disease virus infection in the nasopharynx of cattle; tissue-specific distribution and local cytokine expression. PLoS One 10:e0125698. doi: 10.1371/journal.pone.0125698
Palm, N. W., Rosenstein, R. K., and Medzhitov, R. (2012). Allergic host defences. Nature 484, 465–472. doi: 10.1038/nature11047
Patch, J. R., Dar, P. A., Waters, R., Toka, F. N., Barrera, J., Schutta, C., et al. (2014). Infection with foot-and-mouth disease virus (FMDV) induces a natural killer (NK) cell response in cattle that is lacking following vaccination. Comp. Immunol. Microbiol. Infect. Dis. 37, 249–257. doi: 10.1016/j.cimid.2014.07.004
Perez-Martin, E., Weiss, M., Diaz-San Segundo, F., Pacheco, J. M., Arzt, J., Grubman, M. J., et al. (2012). Bovine type III interferon significantly delays and reduces the severity of foot-and-mouth disease in cattle. J. Virol. 86, 4477–4487. doi: 10.1128/JVI.06683-11
Piccone, M. E., Rieder, E., Mason, P. W., and Grubman, M. J. (1995). The foot-and-mouth disease virus leader proteinase gene is not required for viral replication. J. Virol. 69, 5376–5382.
Pilipenko, E. V., Pestova, T. V., Kolupaeva, V. G., Khitrina, E. V., Poperechnaya, A. N., Agol, V. I., et al. (2000). A cell cycle-dependent protein serves as a template-specific translation initiation factor. Genes Dev. 14, 2028–2045. doi: 10.1101/gad.14.16.2028
Piñeiro, D., Ramajo, J., Bradrick, S. S., and Martínez-Salas, E. (2012). Gemin5 proteolysis reveals a novel motif to identify L protease targets. Nucleic Acids Res. 40, 4942–4953. doi: 10.1093/nar/gks172
Pintarič, M., Gerner, W., and Saalmüller, A. (2008). Synergistic effects of IL-2, IL-12 and IL-18 on cytolytic activity, perforin expression and IFN-γ production of porcine natural killer cells. Vet. Immunol. Immunopathol. 121, 68–82. doi: 10.1016/j.vetimm.2007.08.009
Polatnick, J., and Wool, S. H. (1983). Association of foot-and-mouth disease virus induced RNA polymerase with host cell organelles. Comp. Immunol. Microbiol. Infect. Dis. 6, 265–272. doi: 10.1016/0147-9571(83)90019-X
Ramirez-Carrozzi, V. R., Braas, D., Bhatt, D. M., Cheng, C. S., Hong, C., Doty, K. R., et al. (2009). A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell 138, 114–128. doi: 10.1016/j.cell.2009.04.020
Rathinam, V. A. K., Vanaja, S. K., and Fitzgerald, K. A. (2012). Regulation of inflammasome signaling. Nat. Immunol. 13, 333–342. doi: 10.1038/ni.2237
Reid, E., Juleff, N., Gubbins, S., Prentice, H., Seago, J., and Charleston, B. (2011). Bovine plasmacytoid dendritic cells are the major source of type I interferon in response to foot-and-mouth disease virus in vitro and in vivo. J. Virol. 85, 4297–4308. doi: 10.1128/jvi.02495-10
Rigden, R. C., Carrasco, C. P., Summerfield, A., and McCullough, K. C. (2002). Macrophage phagocytosis of foot-and-mouth disease virus may create infectious carriers. Immunology 106, 537–548. doi: 10.1046/j.1365-2567.2002.01460.x
Rodríguez Pulido, M., Sánchez-Aparicio, M. T., Martínez-Salas, E., García-Sastre, A., Sobrino, F., and Sáiz, M. (2018). Innate immune sensor LGP2 is cleaved by the leader protease of foot-and-mouth disease virus. PLoS Pathog. 14:e1007135. doi: 10.1371/journal.ppat.1007135
Rodríguez Pulido, M., Serrano, P., Sáiz, M., and Martínez-Salas, E. (2007). Foot-and-mouth disease virus infection induces proteolytic cleavage of PTB, eIF3a,b, and PABP RNA-binding proteins. Virology 364, 466–474. doi: 10.1016/j.virol.2007.03.013
Rodríguez-Pulido, M., Borrego, B., Sobrino, F., and Sáiz, M. (2011). RNA structural domains in noncoding regions of the foot-and-mouth disease virus genome trigger innate immunity in porcine cells and mice. J. Virol. 85, 6492–6501. doi: 10.1128/JVI.00599-11
Ryan, E. L., Hollingworth, R., and Grand, R. J. (2016). Activation of the DNA damage response by RNA viruses. Biomolecules 6, 2–24. doi: 10.3390/biom6010002
Ryan, M. D., King, A. M. Q., and Thomas, G. P. (1991). Cleavage of foot-and-mouth disease virus polyprotein is mediated by residues located within a 19 amino acid sequence. J. Gen. Virol. 72, 2727–2732. doi: 10.1099/0022-1317-72-11-2727
Sáiz, M., Gómez, S., Martínez-Salas, E., and Sobrino, F. (2001). Deletion or substitution of the aphthovirus 3′ NCR abrogates infectivity and virus replication. J. Gen. Virol. 82, 93–101. doi: 10.1099/0022-1317-82-1-93
Samuel, C. E. (1979). Mechanism of interferon action. Kinetics of interferon action in mouse L929 cells: phosphorylation of protein synthesis initiation factor eIF-2 and ribosome-associated protein P1. Virology 93, 281–285. doi: 10.1016/0042-6822(79)90300-3
Sanz-Parra, A., Sobrino, F., and Ley, V. (1998). Infection with foot-and-mouth disease virus results in a rapid reduction of MHC class I surface expression. J. Gen. Virol. 79, 433–436. doi: 10.1099/0022-1317-79-3-433
Satoh, T., Kato, H., Kumagai, Y., Yoneyama, M., Sato, S., Matsushita, K., et al. (2010). LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc. Natl. Acad. Sci. U.S.A. 107, 1512–1517. doi: 10.1073/pnas.0912986107
Schmeisser, H., Bekisz, J., and Zoon, K. C. (2014). New function of type I IFN: induction of autophagy. J. Interferon Cytokine Res. 34, 71–78. doi: 10.1089/jir.2013.0128
Sei, J. J., Waters, R. A., Kenney, M., Barlow, J. W., and Golde, W. T. (2016). Effect of foot-and-mouth disease virus infection on the frequency, phenotype and function of circulating dendritic cells in cattle. PLoS One 11:e0152192. doi: 10.1371/journal.pone.0152192
Serrano, P., Pulido Rodriguez, M., Sáiz, M., and Martínez-Salas, E. (2006). The 3′ end of the foot-and-mouth disease virus genome establishes two distinct long-range RNA-RNA interactions with the 5′ and region. J. Gen. Virol. 87, 3013–3022. doi: 10.1099/vir.0.82059-0
Seth, R. B., Sun, L., Ea, C.-K., and Chen, Z. J. (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122, 669–682. doi: 10.1016/j.cell.2005.08.012
Shaulian, E., and Karin, M. (2002). AP-1 as a regulator of cell life and death. Nat. Cell Biol. 4, E131–E136. doi: 10.1038/ncb0502-e131
Shi, C. S., and Kehrl, J. H. (2008). MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J. Biol. Chem. 283, 33175–33182. doi: 10.1074/jbc.M804478200
Sigal, L. J., Gomez, G., and Braun, M. (1992). Changes in mononuclear peripheral blood cells in cattle with foot-and-mouth disease. Vet. Immunol. Immunopathol. 30, 431–438. doi: 10.1016/0165-2427(92)90111-3
Singh, N., Ramirez-Carvajal, L., de los Santos, T., Golding, M. C., and Long, C. R. (2016). Inhibition of EHMT2 induces a robust antiviral response against foot-and-mouth disease and vesicular stomatitis virus infections in bovine cells. J. Interf. Cytokine Res. 36, 37–47. doi: 10.1089/jir.2015.0006
Šinkora, M., Šinkorová, J., and Holtmeier, W. (2005). Development of γδ thymocyte subsets during prenatal and postnatal ontogeny. Immunology 115, 544–555. doi: 10.1111/j.1365-2567.2005.02194.x
Si-Tahar, M., Blanc, F., Furio, L., Chopy, D., Balloy, V., Lafon, M., et al. (2014). Protective role of LGP2 in influenza virus pathogenesis. J. Infect. Dis. 210, 214–223. doi: 10.1093/infdis/jiu076
Skaug, B., and Chen, Z. J. (2010). Emerging role of ISG15 in antiviral immunity. Cell 143, 187–190. doi: 10.1016/j.cell.2010.09.033
Steinberger, J., and Skern, T. (2014). The leader proteinase of foot-and-mouth disease virus: structure-function relationships in a proteolytic virulence factor. Biol. Chem. 395, 1179–1185. doi: 10.1515/hsz-2014-0156
Steinman, R. M. (2008). Dendritic cells in vivo: a key target for a new vaccine science. Immunity 29, 319–324. doi: 10.1016/j.immuni.2008.08.001
Stenfeldt, C., Arzt, J., Pacheco, J. M., Gladue, D. P., Smoliga, G. R., Silva, E. B., et al. (2018). A partial deletion within foot-and-mouth disease virus non-structural protein 3A causes clinical attenuation in cattle but does not prevent subclinical infection. Virology 516, 115–126. doi: 10.1016/j.virol.2018.01.008
Stenfeldt, C., Eschbaumer, M., Pacheco, J. M., Rekant, S. I., Rodriguez, L. L., and Arzt, J. (2015). Pathogenesis of primary foot-and-mouth disease virus infection in the nasopharynx of vaccinated and non-vaccinated cattle. PLoS One 10:e0143666. doi: 10.1371/journal.pone.0143666
Stenfeldt, C., Eschbaumer, M., Rekant, S. I., Pacheco, J. M., Smoliga, G. R., Hartwig, E. J., et al. (2016a). The foot-and-mouth disease carrier state divergence in cattle. J. Virol. 90, 6344–6364. doi: 10.1128/JVI.00388-16
Stenfeldt, C., Pacheco, J. M., Smoliga, G. R., Bishop, E., Pauszek, S. J., Hartwig, E. J., et al. (2016b). Detection of foot-and-mouth disease virus RNA and capsid protein in lymphoid tissues of convalescent pigs does not indicate existence of a carrier state. Transbound. Emerg. Dis. 63, 152–164. doi: 10.1111/tbed.12235
Stenfeldt, C., Heegaard, P. M., Stockmarr, A., Tjørnehøj, K., and Belsham, G. J. (2011). Analysis of the acute phase responses of serum amyloid A, haptoglobin and type 1 interferon in cattle experimentally infected with foot-and-mouth disease virus serotype O. Vet. Res. 42:66. doi: 10.1186/1297-9716-42-66
Stenfeldt, C., Pacheco, J. M., Rodriguez, L. L., and Arzt, J. (2014). Early events in the pathogenesis of foot-and-mouth disease in pigs; identification of oropharyngeal tonsils as sites of primary and sustained viral replication. PLoS One 9:e106859. doi: 10.1371/journal.pone.0106859
Strebel, K., and Beck, E. (1986). A second protease of foot-and-mouth disease virus. J. Virol. 58, 893–899.
Summerfield, A. (2012). Viewpoint: factors involved in type I interferon responses during porcine virus infections. Vet. Immunol. Immunopathol. 148, 168–171. doi: 10.1016/j.vetimm.2011.03.011
Sun, P., Zhang, S., Qin, X., Chang, X., Cui, X., Li, H., et al. (2018). Foot-and-mouth disease virus capsid protein VP2 activates the cellular EIF2S1-ATF4 pathway and induces autophagy via HSPB1. Autophagy 14, 336–346. doi: 10.1080/15548627.2017.1405187
Swatek, K. N., Aumayr, M., Pruneda, J. N., Visser, L. J., Berryman, S., Kueck, A. F., et al. (2018). Irreversible inactivation of ISG15 by a viral leader protease enables alternative infection detection strategies. Proc. Natl. Acad. Sci. U.S.A. 115, 2371–2376. doi: 10.1073/pnas.1710617115
Sweeney, T. R., Cisnetto, V., Bose, D., Bailey, M., Wilson, J. R., Zhang, X., et al. (2010). Foot-and-mouth disease virus 2C is a hexameric AAA+ protein with a coordinated ATP hydrolysis mechanism. J. Biol. Chem. 285, 24347–24359. doi: 10.1074/jbc.M110.129940
Swiecki, M., and Colonna, M. (2015). The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 15, 471–485. doi: 10.1038/nri3865
Takamatsu, H.-H., Denyer, M. S., Stirling, C., Cox, S., Aggarwal, N., Dash, P., et al. (2006). Porcine gammadelta T cells: possible roles on the innate and adaptive immune responses following virus infection. Vet. Immunol. Immunopathol. 112, 49–61. doi: 10.1016/j.vetimm.2006.03.011
Tesar, M., and Marquardt, O. (1990). Foot-and-mouth disease virus protease 3C inhibits cellular transcription and mediates cleavage of histone H3. Virology 174, 364–374. doi: 10.1016/0042-6822(90)90090-E
Teterina, N. L., Gorbalenya, A. E., Egger, D., Bienz, K., Rinaudo, M. S., and Ehrenfeld, E. (2006). Testing the modularity of the N-terminal amphipathic helix conserved in picornavirus 2C proteins and hepatitis C NS5A protein. Virology 344, 453–467. doi: 10.1016/j.virol.2005.08.044
Toka, F. N., Nfon, C., Dawson, H., and Golde, W. T. (2009). Natural killer cell dysfunction during acute infection with foot-and-mouth disease virus. Clin. Vaccine Immunol. 16, 1738–1749. doi: 10.1128/CVI.00280-09
Vakharia, V. N., Devaney, M. A., Moore, D. M., Dunn, J. J., and Grubman, M. J. (1987). Proteolytic processing of foot-and-mouth disease virus polyproteins expressed in a cell-free system from clone-derived transcripts. J. Virol. 61, 3199–207.
Vázquez-Calvo,Á., Saiz, J. C., McCullough, K. C., Sobrino, F., and Martín-Acebes, M. A. (2012). Acid-dependent viral entry. Virus Res. 167, 125–137. doi: 10.1016/j.virusres.2012.05.024
Venkataraman, T., Valdes, M., Elsby, R., Kakuta, S., Caceres, G., Saijo, S., et al. (2007). Loss of DExD/H Box RNA helicase LGP2 manifests disparate antiviral responses. J. Immunol. 178, 6444–6455. doi: 10.4049/jimmunol.178.10.6444
Wang, D., Fang, L., Li, K., Zhong, H., Fan, J., Ouyang, C., et al. (2012). Foot-and-mouth disease virus 3C protease cleaves NEMO to impair innate immune signaling. J. Virol. 86, 9311–9322. doi: 10.1128/JVI.00722-12
Wang, J., Wang, Y., Liu, J., Ding, L., Zhang, Q., Li, X., et al. (2012). A critical role of N-myc and STAT interactor (Nmi) in foot-and-mouth disease virus (FMDV) 2C-induced apoptosis. Virus Res. 170, 59–65. doi: 10.1016/j.virusres.2012.08.018
Wang, D., Fang, L., Li, P., Sun, L., Fan, J., Zhang, Q., et al. (2011a). The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. J. Virol. 85, 3758–3766. doi: 10.1128/JVI.02589-10
Wang, D., Fang, L., Liu, L., Zhong, H., Chen, Q., Luo, R., et al. (2011b). Foot-and-mouth disease virus (FMDV) leader proteinase negatively regulates the porcine interferon-λ1 pathway. Mol. Immunol. 49, 407–412. doi: 10.1016/j.molimm.2011.09.009
Wang, D., Fang, L., Luo, R., Ye, R., Fang, Y., Xie, L., et al. (2010). Foot-and-mouth disease virus leader proteinase inhibits dsRNA-induced type I interferon transcription by decreasing interferon regulatory factor 3/7 in protein levels. Biochem. Biophys. Res. Commun. 399, 72–78. doi: 10.1016/j.bbrc.2010.07.044
Wang, Y., Zhang, S., Wang, L., Liu, X., Zhang, E., Li, N., et al. (2016). [Protein expression profile of mast cells in response to recombinant VP1-VP4 of foot-and-mouth disease viru]. Sheng Wu Gong Cheng Xue Bao 32, 1194–1203.
Weber, M., Sediri, H., Felgenhauer, U., Binzen, I., Bänfer, S., Jacob, R., et al. (2015). Influenza virus adaptation PB2-627K modulates nucleocapsid inhibition by the pathogen sensor RIG-I. Cell Host Microbe 17, 309–319. doi: 10.1016/j.chom.2015.01.005
White, J. P., Cardenas, A. M., Marissen, W. E., and Lloyd, R. E. (2007). Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2, 295–305. doi: 10.1016/j.chom.2007.08.006
White, J. P., and Lloyd, R. E. (2011). Poliovirus unlinks TIA1 aggregation and mRNA stress granule formation. J. Virol. 85, 12442–12484. doi: 10.1128/JVI.05888-11
Wijngaard, P. L., Metzelaar, M. J., MacHugh, N. D., Morrison, W. I., and Clevers, H. C. (1992). Molecular characterization of the WC1 antigen expressed specifically on bovine CD4-CD8- gamma delta T lymphocytes. J. Immunol. 149, 3273–3277.
Windsor, M. A., Carr, B. V., Bankowski, B., Gibson, D., Reid, E., Hamblin, P., et al. (2011). Cattle remain immunocompetent during the acute phase of foot-and-mouth disease virus infection. Vet. Res. 42:108. doi: 10.1186/1297-9716-42-108
Wu, J., and Chen, Z. J. (2014). Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 32, 461–488. doi: 10.1146/annurev-immunol-032713-120156
Wu, Q., Brum, M. C. S., Caron, L., Koster, M., and Grubman, M. J. (2003). Adenovirus-mediated type I interferon expression delays and reduces disease signs in cattle challenged with foot-and-mouth disease virus. J. Interferon Cytokine Res. 23, 359–368. doi: 10.1089/107999003322226014
Xu, Y., and Eissa, N. T. (2010). Autophagy in innate and adaptive immunity. Proc. Am. Thorac. Soc. 7, 22–28. doi: 10.1513/pats.200909-103JS
Xue, M., Wang, H., Li, W., Zhou, G., Tu, Y., and Yu, L. (2012). Effects of amino acid substitutions in the VP2 B-C loop on antigenicity and pathogenicity of serotype Asia1 foot-and-mouth disease virus. Virol. J. 9:191. doi: 10.1186/1743-422X-9-191
Yang, X., Hu, Z., Fan, S., Zhang, Q., Zhong, Y., Guo, D., et al. (2018). Picornavirus 2A protease regulates stress granule formation to facilitate viral translation. PLoS Pathog. 14:e1006901. doi: 10.1371/journal.ppat.1006901
Ye, X., Pan, T., Wang, D., Fang, L., Ma, J., Zhu, X., et al. (2018). Foot-and-mouth disease virus counteracts on internal ribosome entry site suppression by G3BP1 and inhibits G3BP1-mediated stress granule assembly via post-translational mechanisms. Front. Immunol. 9:1142. doi: 10.3389/fimmu.2018.01142
Yoneyama, M., Kikuchi, M., Matsumoto, K., Imaizumi, T., Miyagishi, M., Taira, K., et al. (2005). Shared and unique functions of the DExD/H-Box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 175, 2851–2858. doi: 10.4049/jimmunol.175.5.2851
Yoneyama, M., Kikuchi, M., Natsukawa, T., Shinobu, N., Imaizumi, T., Miyagishi, M., et al. (2004). The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5, 730–737. doi: 10.1038/ni1087
Yoneyama, M., Onomoto, K., Jogi, M., Akaboshi, T., and Fujita, T. (2015). Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol. 32, 48–53. doi: 10.1016/j.coi.2014.12.012
Zhang, Z., Ahmed, R., Paton, D., and Bashiruddin, J. B. (2009). Cytokine mRNA responses in bovine epithelia during foot-and-mouth disease virus infection. Vet. J. 179, 85–91. doi: 10.1016/j.tvjl.2007.08.012
Zhang, Z. D., and Kitching, R. P. (2001). The localization of persistent foot and mouth disease virus in the epithelial cells of the soft palate and pharynx. J. Comp. Pathol. 124, 89–94. doi: 10.1053/jcpa.2000.0431
Zheng, W., Li, X., Wang, J., Li, X., Cao, H., Wang, Y., et al. (2014). A critical role of interferon-induced protein IFP35 in the type I interferon response in cells induced by foot-and-mouth disease virus (FMDV) protein 2C. Arch. Virol. 159, 2925–2935. doi: 10.1007/s00705-014-2147-7
Zhou, Y., He, C., Wang, L., and Ge, B. (2017). Post-translational regulation of antiviral innate signaling. Eur. J. Immunol. 47, 1414–1426. doi: 10.1002/eji.201746959
Zhou, Z., Mogensen, M. M., Powell, P. P., Curry, S., and Wileman, T. (2013). Foot-and-mouth disease virus 3C protease induces fragmentation of the golgi compartment and blocks intra-golgi transport. J. Virol. 87, 11721–11729. doi: 10.1128/JVI.01355-13
Zhu, J., Weiss, M., Grubman, M. J., and de los Santos, T. (2010). Differential gene expression in bovine cells infected with wild type and leaderless foot-and-mouth disease virus. Virology 404, 32–40. doi: 10.1016/j.virol.2010.04.021
Zhu, Z., Li, C., Du, X., Wang, G., Cao, W., Yang, F., et al. (2017). Foot-and-mouth disease virus infection inhibits LGP2 protein expression to exaggerate inflammatory response and promote viral replication. Cell Death Dis. 8:e2747. doi: 10.1038/cddis.2017.170
Keywords: FMDV, evasion innate immunity, apoptosis, autophagy, IFN, cattle/swine, ubiquitin/ISG, virulence factors
Citation: Medina GN, Segundo FD-S, Stenfeldt C, Arzt J and de los Santos T (2018) The Different Tactics of Foot-and-Mouth Disease Virus to Evade Innate Immunity. Front. Microbiol. 9:2644. doi: 10.3389/fmicb.2018.02644
Received: 02 August 2018; Accepted: 17 October 2018;
Published: 12 November 2018.
Edited by:
Tong-Qing An, Harbin Veterinary Research Institute (CAAS), ChinaReviewed by:
Encarna Martinez-Salas, Centro de Biología Molecular Severo Ochoa (CSIC-UAM), SpainCopyright © 2018 Medina, Segundo, Stenfeldt, Arzt and de los Santos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gisselle N. Medina, Z2lzc2VsbGUubWVkaW5hQGFycy51c2RhLmdvdg== Teresa de los Santos, dGVyZXNhLmRlbG9zc2FudG9zQGFycy51c2RhLmdvdg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.