 Roshan Khadka
Roshan Khadka Lindsay Clothier1
Lindsay Clothier1 Lin Wang
Lin Wang Chee Kent Lim
Chee Kent Lim Martin G. Klotz
Martin G. Klotz Peter F. Dunfield
Peter F. Dunfield
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 29 October 2018
Sec. Evolutionary and Genomic Microbiology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.02493
Copper membrane monooxygenases (CuMMOs) oxidize ammonia, methane and some short-chain alkanes and alkenes. They are encoded by three genes, usually in an operon of xmoCAB. We aligned xmo operons from 66 microbial genomes, including members of the Alpha-, Beta-, and Gamma-proteobacteria, Verrucomicrobia, Actinobacteria, Thaumarchaeota and the candidate phylum NC10. Phylogenetic and compositional analyses were used to reconstruct the evolutionary history of the enzyme and detect potential lateral gene transfer (LGT) events. The phylogenetic analyses showed at least 10 clusters corresponding to a combination of substrate specificity and bacterial taxonomy, but with no overriding structure based on either function or taxonomy alone. Adaptation of the enzyme to preferentially oxidize either ammonia or methane has occurred more than once. Individual phylogenies of all three genes, xmoA, xmoB and xmoC, closely matched, indicating that this operon evolved or was consistently transferred as a unit, with the possible exception of the methane monooxygenase operons in Verrucomicrobia, where the pmoB gene has a distinct phylogeny from pmoA and pmoC. Compositional analyses indicated that some clusters of xmoCAB operons (for example, the pmoCAB in gammaproteobacterial methanotrophs and the amoCAB in betaproteobacterial nitrifiers) were compositionally very different from their genomes, possibly indicating recent lateral transfer of these operons. The combined phylogenetic and compositional analyses support the hypothesis that an ancestor of the nitrifying bacterium Nitrosococcus was the donor of methane monooxygenase (pMMO) to both the alphaproteobacterial and gammaproteobacterial methanotrophs, but that before this event the gammaproteobacterial methanotrophs originally possessed another CuMMO (Pxm), which has since been lost in many species.
The copper membrane monooxygenase (CuMMO) enzyme family includes ammonia monooxygenase (AMO), particulate methane monooxygenase (pMMO) and a few short-chain alkane and alkene monooxygenases (Tavormina et al., 2011). AMO and pMMO perform the initial oxygenation steps in the aerobic catabolism of methane (CH4) and ammonia (NH3), which produce toxic intermediates (methanol, formaldehyde, and/or hydroxylamine) that constitute the internal sources of energy and reductant for these microbes (Klotz and Stein, 2008, 2011; Simon and Klotz, 2013). Organisms with these enzymes therefore play key roles in the global carbon and nitrogen cycles. Substrates other than methane and ammonia are also targeted by some CuMMOs: some Actinobacteria possesses a putative CuMMO that acts as a butane monooxygenase (BMO) (Coleman et al., 2011, 2012; Sayavedra-Soto et al., 2011), and ethylene-assimilating Gammaproteobacteria belonging to the genus Haliea also possess CuMMOs (Suzuki et al., 2012). Genomics projects are also beginning to find operons encoding CuMMOs of unknown function in bacterial genomes, such as Solimonas aquatica and Bradyrhizobium sp. ERR11 (Kyrpides et al., 2014; Whitman et al., 2015). The known diversity of the CuMMO enzyme family is therefore expanding. CuMMOs are promiscuous and co-oxidize several structurally similar substrates, although they usually show clear specialization to one particular substrate (Bedard and Knowles, 1989; Semrau et al., 2010). For example, pMMO will oxidize ammonia and some alkanes, but usually with a lower affinity and lower reaction rates than for methane (Bedard and Knowles, 1989; Nyerges and Stein, 2009). This co-oxidation of ammonia and the resulting production of toxic hydroxylamine and nitrite leads to an inhibitory effect of ammonia on methanotrophs (Bodelier and Laanbroek, 2004; Nyerges and Stein, 2009).
Copper membrane monooxygenase enzymes are encoded by three genes (Klotz et al., 1997), here referred to collectively as xmoC, xmoA, and xmoB (amo specifically refers to genes encoding AMO, and pmo specifically refers to genes encoding pMMO). A clear homology of amoAB/pmoAB genes was first shown by Klotz and Norton (1998), demonstrating that AMO and pMMO were evolutionarily related. In bacteria, the xmoAB genes are always clustered with an xmoC, although xmoC genes sometimes occur as singletons in addition to operonal copies (Klotz et al., 1997; Arp et al., 2007; Op den Camp et al., 2009; Dam et al., 2012). In ammonia oxidizing bacteria, AMO is encoded by an amoCAB operon, but in Thaumarchaeota the amoA, amoB, and amoC gene are not always clustered together. A pMMO is encoded in the genomes of nearly all known aerobic methane oxidizing bacteria as a pmoCAB operon. Another ortholog present in some proteobacterial methanotrophs has been dubbed pxmABC, and the encoding operon is uniquely organized in the ABC order, instead of the more common CAB (Tavormina et al., 2011).
Phylogenies of amoA/pmoA match closely to 16S rRNA gene phylogenies, at least to the extent that individual genera, families, classes, and phyla can be clearly identified via comparative sequence analysis (Knief, 2015; Alves et al., 2018). Recovery of pmoA and amoA genes from natural samples has therefore been used extensively to identify species of methanotrophs and ammonia oxidizers in diverse environments (Rotthauwe et al., 1997; McDonald et al., 2008; Tavormina et al., 2013; Dumont, 2014; Knief, 2015). The first published primer set developed to target pmoA, A189f/A682r (Holmes et al., 1995), is still widely used as a broad-spectrum primer set for detecting known methanotrophs and discovering new ones (Knief et al., 2003). However, it has become clear via cultivation and genomics studies that these supposed universal primers do not target all xmoA genes in nature, for example universal pmoA primers do not amplify genes from verrucomicrobial methanotrophs (Op den Camp et al., 2009).
Aerobic methanotrophy is a rare trait, limited to a few monophyletic clusters of bacteria within the Alphaproteobacteria, Gammaproteobacteria, Verrucomicrobia and candidate phylum NC10 (Ettwig et al., 2009; Op den Camp et al., 2009). Dissimilatory ammonia oxidation is similarly rare, known in only two clusters of bacteria within the classes Betaproteobacteria and Gammaproteobacteria, some Nitrospira, and some Thaumarcheota (Kuypers et al., 2018). This mosaic nature of ammonia and methane-oxidizing taxa, which are constrained to a few monophyletic clusters (based on 16S rRNA gene phylogeny) scattered across several different phyla, indicates that there have been only a few key lateral gene transfer (LGT) events of xmo genes. For example, Tamas et al. (2014) suggested that all Alphaproteobacteria methanotrophs arose from a single common methylotrophic ancestor that had obtained pmoCAB via a single LGT event from a gammaproteobacterium. Gammaproteobacteria and Verrucomicrobia methanotrophs also appear to be monophyletic based on 16S rRNA genes.
However, some Gammaproteobacteria (Tavormina et al., 2011), Alphaproteobacteria (Tchawa Yimga et al., 2003; Wartiainen et al., 2006; Vorobev et al., 2014) and Verrucomicrobia (Dunfield et al., 2007; Op den Camp et al., 2009) methanotrophs have multiple divergent copies of the pmo operon, indicating a more complex history of gene duplication, evolution, and LGT. Two pmoCAB operons in Methylocystis sp. strain SC2 (59–66% amino acid sequence identity) are suspected to have different methane affinities (Dunfield et al., 2002; Baani and Liesack, 2008). Methylacidiphilum spp. have three phylogenetic distinct pmoCAB operons as little as 50% amino acid identity to each other, and these show different expression patterns (Op den Camp et al., 2009; Erikstad et al., 2012; Khadem et al., 2012), although the functional differences among the encoded enzymes are not yet known with certainty. A pxmABC operon is also found in addition to pmoCAB in some proteobacterial methanotrophs. Its function is not clear yet, but transcript levels of pxmA increased in Methylomicrobium album strain BG8 (Kits et al., 2015a) and Methylomonas denitrificans strain FJG1 (Kits et al., 2015b) under nitrite rich and oxygen limited conditions. In addition to utilizing CuMMOs with different substrate affinities and preferences, some Gammaproteobacteria and Betaproteobacteria with multiple nearly identical copies of xmoCAB operons employ differential regulation to preferentially express individual copies upon environmental cues (Stolyar et al., 1999, 2001; Berube et al., 2007; Berube and Stahl, 2012).
Although phylogenetic constructions using xmoA are extensively used to delineate species (Knief, 2015; Alves et al., 2018), the evolutionary history of the genes has not been well-elucidated. In this study we assembled a database of xmoCAB operons from microbial genomes, and performed in depth compositional and phylogenetic analyses of the genes and their genomes to better understand their evolutionary history.
All genes and genomes analyzed in this study were downloaded from the JGI1, NCBI2, or RAST3 genomic databases. The database contained 66 genomes, including 4 Thaumarchaeota, 50 Alpha-, Beta-, and Gamma-proteobacteria, and 13 non-proteobacterial genomes (Verrucomicrobia, Nitrospira, NC10, and Actinobacteria), all of which contained CuMMO-encoding operons. These genomes were chosen based on published papers and BLAST sequences of JGI/NCBI databases. Our aim was to include only cultured bacteria for which physiological data and complete genomes are available. The analyses therefore did not include metagenomic sequences. Some bacterial genomes containing xmo genes were also ignored if they appeared to be the result of private sequencing projects with no published reports to date.
We constructed Bayesian, Maximum likelihood (ML) and Neighbor-Joining (NJ) phylogenies based on derived XmoA, XmoB, XmoC and concatenated XmoCAB protein sequences. The alignment of derived amino acids was done using ClustalW. Seaview version 4.4.12 (Gouy et al., 2010) was used for NJ (with a Poisson evolutionary distance model) and ML (with a Le and Gascuel model) constructions. Bayesian inference methods were calculated with BEAST (Bouckaert et al., 2014), both with strict clock and relaxed clock log normal models. Bayesian analysis employed a Blosum62 substitution model and Gamma site heterogeneity model under a strict clock, or a Gamma site heterogeneity model with four gamma categories with a relaxed clock log-normal model. The inferred tree topology was assessed with 100 bootstrap replications for NJ and ML methods, and 10,000,000 iterations for Bayesian phylogenies minus a burn-in of 20% of the total.
Four programs were used to detect possible LGT of xmo operons based on compositional genome biases: TETRA, Alien Hunter, CodonW, and Island Viewer.
FASTA files containing DNA sequences were uploaded into the TETRA program (Teeling et al., 2004b)4. Each xmoCAB operon and the genome from the respective organism was uploaded separately. Before running the program, the sequences were extended by their reverse complement, and tetra-nucleotide usage patterns were calculated by the program. The program generated a table consisting of all possible 256 tetra-nucleotide combinations with their frequencies in each gene or genome.
The CodonW program (Angellotti et al., 2007), requires coding sequences for calculation. Therefore, the FeatureExtract 1.2 server5 was used to extract the coding sequences of a genome from GenBank (.gbk) files. Then the protein coding genes of each xmoCAB operon and a genome from each organism were uploaded simultaneously into the CodonW database6. The sequences of all coding genes (operons and genomes, separately) were concatenated. The output result contained all the 64 codon bias values. The frequency of each codon was calculated as ‘(codon value)/64’ for each gene or genome separately.
Genomes were uploaded into the Alien Hunter program (Vernikos and Parkhill, 2006) using Linux. The predictions were visualized using Artemis. The program predicts LGT in a moving 2500-bp window, using the Kullback-Leibler (KL) divergence statistic (Kullback and Leibler, 1951) for combined 2-mers to 8-mers.
This equation gives numeric values between 0 and 1 as output. Values close to one indicate that the region has a much different compositional bias compared to the genome, and likely underwent horizontal transfer into its genome (Overbeek et al., 2005; Becq et al., 2010). Values close to zero indicate the region has similar kmer composition to its genome (no detectable LGT). Alien Hunter selects a significance KL threshold to determine whether the composition of the operon differs from the genome. The loci of the xmoCAB operon(s) in the genomes were used to determine whether these were located within predicted “alien” regions.
For TETRA and CodonW, the KL statistic was also calculated comparing the xmoCAB operon(s) to the genome. For TETRA and CodonW we simply report and compare KL values of the xmoCAB operon (on average 2730 nucleotides) compared to the composition of the entire genome, without a presumed significance threshold. Individual genes were not tested as they contained insufficient information to accurately predict k-mer frequencies.
Genomes were also examined via Island Viewer (Bertelli et al., 2017), which combines compositional analyses with examination of mobility genes to identify genomic islands.
Several primers that are specific to detect methanotrophs have been developed (Dumont, 2014; Wang et al., 2017). We have analyzed the sensitivity of primers to different groups of methanotrophs (Supplementary Table S2) that are used in this study. We used the Arb platform to detect the number of mismatches in the existing primer sets (Ludwig et al., 2004).
Highly resolved phylogenies of concatenated inferred XmoCAB sequences were constructed via Bayesian inference methods (strict clock vs. relaxed clock log-normal model) (Figure 1), as well as ML and NJ methods (Supplementary Figures S1A,B). We constructed phylogenies of XmoCAB both with and without thaumarcheotal XmoCAB sequences included, since a large number of sequence gaps/insertions when comparing bacterial and thaumarchaeotal sequences reduce the amount of information available for phylogenetic calculations. Results were similar in each case.
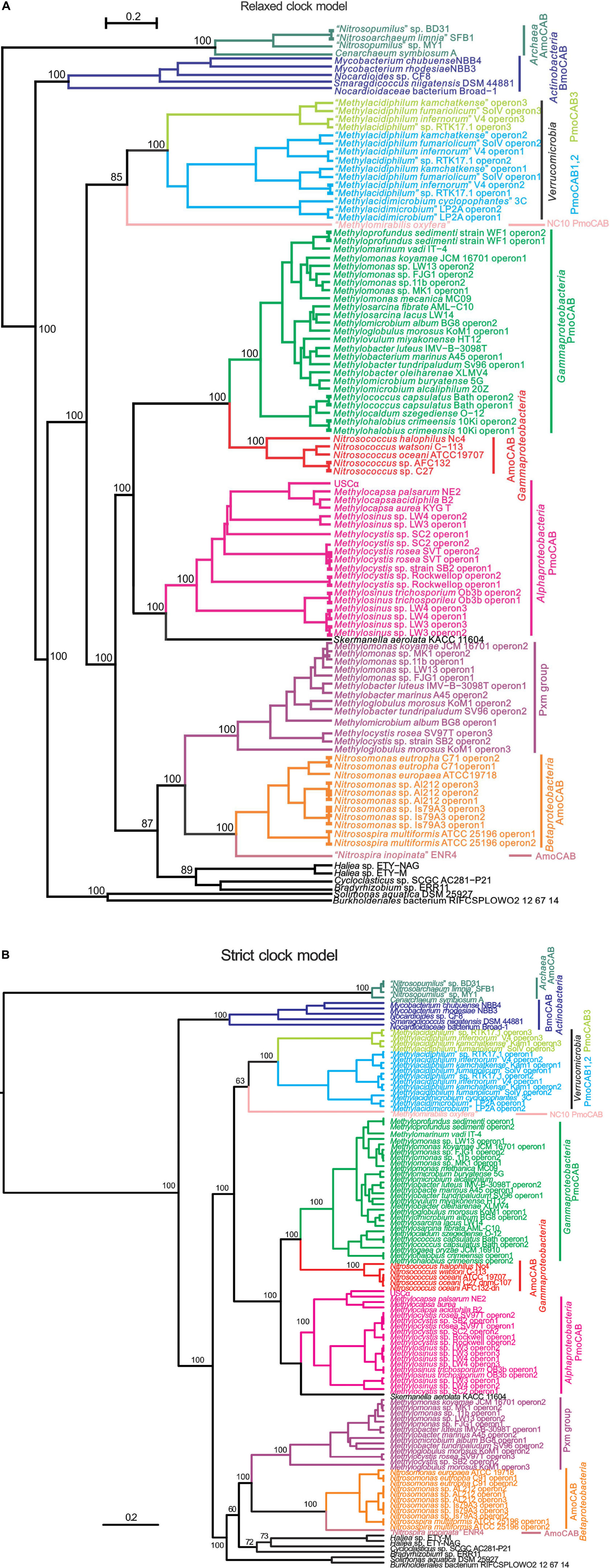
FIGURE 1. Phylogenetic trees based on concatenated inferred XmoCAB sequences (minimum 910 amino acids). The tree was constructed using Bayesian analysis employing: (A) a gamma site heterogeneity model with four gamma categories with a relaxed clock log normal model and (B) a Blosum62 substitution model with gamma site heterogeneity model under a strict clock. Node values are based on 10,000,000 iterations, minus a burn-in of 20% of the total. The scale bar represents 0.2 changes per amino acid position. Colors indicate coherent functional and taxonomic groups. The protein accession numbers for the operons are given in Supplementary Table S3.
The different construction methods largely agreed, with AMO from Thaumarchaeota the most distant group (and a logical outgroup based on total evolutionary distance), and a highly supported primary node separating the AMO of Thaumarchaeota, the BMO of Actinobacteria, and all others. All constructions generally agreed upon at least 10 well-supported monophyletic clusters corresponding to a combination of function (e.g., pMMO vs. AMO) and taxonomic affiliation. These clusters are summarized by the different colors in Figure 1. Support values for these taxonomic/functional clusters are very high in all constructions, reaching 100% posterior probabilities in Bayesian analyses. Relationships across these groups were slightly variable in the different constructions. Most notable was a variable placement of the Verrucomicrobia and NC10. In the Bayesian relaxed clock model and the NJ tree these methanotroph groups were monophyletic with the proteobacterial methanotrophs, in the Bayesian strict clock model and the ML model they were not.
Most of the groups indicated in Figure 1 also represent coherent monophyletic clusters based on 16S rRNA phylogeny, but they are separated by vast numbers of species that do not have xmoCAB genes in their genomes. The most parsimonious scenario for the distribution of xmoCAB is therefore that a single common ancestor of each of the groups indicated in Figure 1 received xmoCAB via a single LGT event. Possible instances where recent LGT has disrupted coherent taxonomic groups include: (i) the pxmABC of the alphaproteobacterium Methylocystis rosea, which clusters within a group of Gammaproteobacteria, and (ii) several sequences of unknown function (indicated in black) belonging to Alpha- Beta-, and Gamma-proteobacteria, particularly near the base of Figure 1. However, while the sequences indicated in black are included to support tree construction, we prefer not to discuss them in detail because: (1) in most cases the function of the respective Xmo enzymes is unknown; (2) most of the branches are deeply rooted and contain a single sequence, and the exact positioning of such branches is problematic due to plesiomorphic character states, and (3) there are still too few of these sequences to draw solid conclusions. Therefore, we focus our analysis on the better known groups of methane and ammonia oxidizers.
Phylogenies of individual inferred XmoA, XmoB, and XmoC polypeptides were constructed using Bayesian inference (strict clock vs. relaxed clock log normal model, Figure 2), ML, and NJ methods (Supplementary Figures S2, S4A–C, respectively). When included, thaumarchaeotal sequences always formed the most distant group. However removing thaumarchaeotal sequences in some of the individual gene trees showed better support values because the large number of sequence gaps/insertions in the thaumarchaeotal genes greatly reduce the amount of information available for phylogenetic calculations. Only trees without thaumarchaeotal sequences are therefore presented. The Bayesian phylogenies are summarized in Figure 2 in a manner to stress the similarities of the three individual XmoA XmoB and XmoC trees. Generally, the same taxonomic/functional groups are identified in the individual trees as in the concatenated tree, suggesting that the three genes/polypeptides have mostly parallel evolutionary histories and are evolved or transferred as a coherent operon.
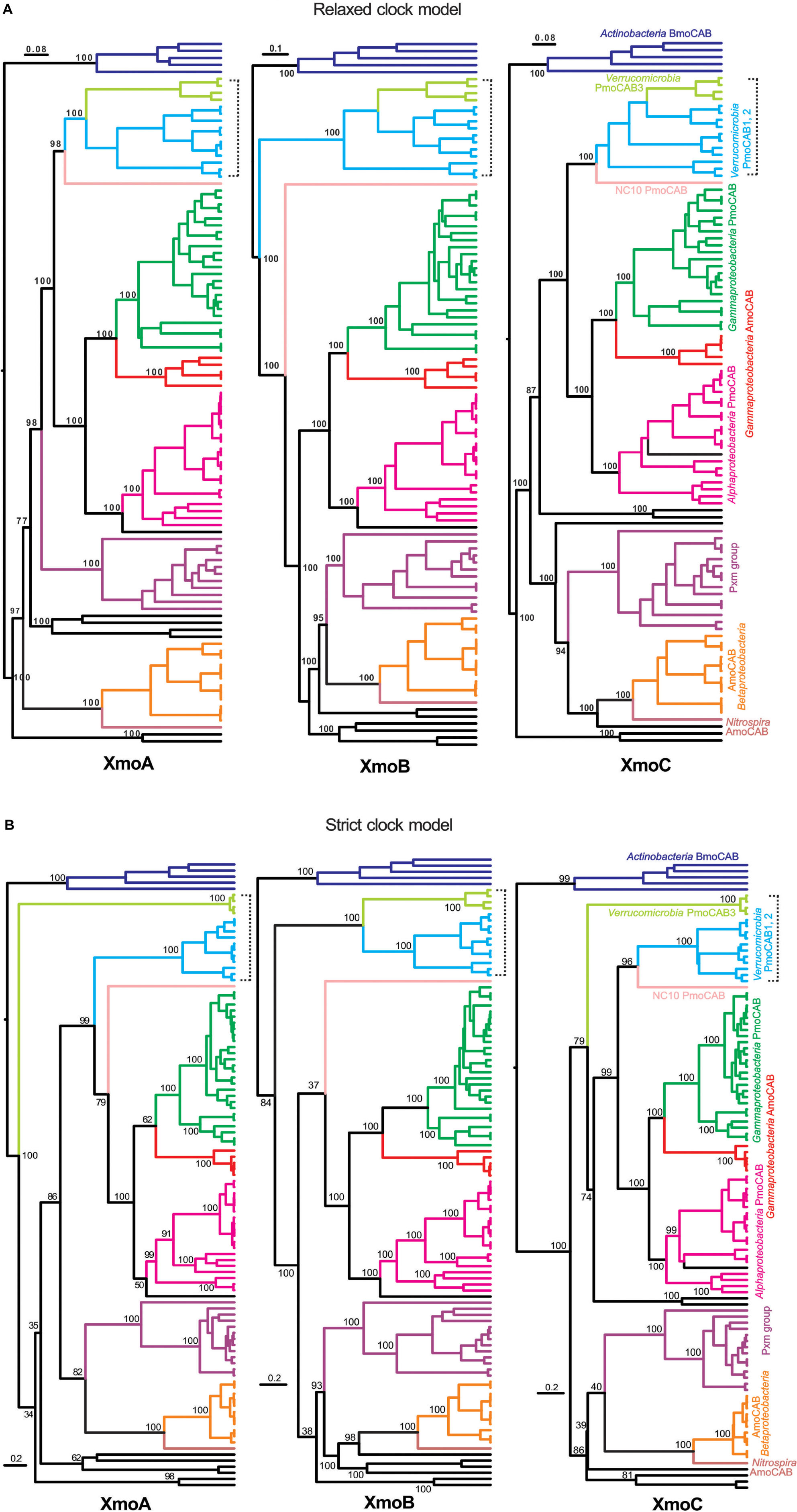
FIGURE 2. Phylogenetic trees based on inferred XmoA, B, and C sequences. Tree was constructed using Bayesian analysis employing: (A) a Gamma site heterogeneity model with four gamma categories with a relaxed clock log normal model and (B) a Blosum62 substitution model with gamma site heterogeneity model under a strict clock. Node values are based on 10,000,000 iterations after a burn-in of 20% of the total trees. The scale bars represents 0.08, 0.1, and 0.08 (A) and 0.2 (B) changes per amino acid position. Lineages are colored and labeled as in Figure 1. The bracket stresses the differences between verrucomicrobial methanotrophs across the individual trees.
One major inconsistency in the individual trees is the placement of the three homologs found in Verrucomicrobia. In the Bayesian (strict molecular clock model) constructions (Figure 2B) the XmoC3 and XmoA3 homologs separate out from the respective XmoC1/XmoC2 and XmoA1/XmoA2 homologs. However this separation is highly dependent on the construction method and often not well-supported (Supplementary Figures S2A–C, S4A–C). It is not seen in the Bayesian (relaxed molecular clock model) (Figure 2A) where the all three homologs in the Verrucomicrobia are always monophyletic.
Another inconsistency is that for XmoB, the entire cluster of Verrucomicrobia is more ancestral in the tree than it is for XmoA and XmoC (Figure 2). XmoA and XmoC of Verrucomicrobia are monophyletic with the corresponding homologs in proteobacterial methanotrophs, but XmoB is not. The uniqueness of the XmoB phylogeny compared to XmoA and C is seen in all four methods (Bayesian strict clock, Bayesian relaxed clock, ML and NJ) (Figure 2 and Supplementary Figures S3A–C).
The KL divergences calculated with Alien Hunter, TETRA, and CodonW are summarized in Figure 3 and Supplementary Table S1. Higher values indicate greater compositional differences between the xmoCAB operon and host genome. Alien Hunter found evidence of significant compositional differences in most of the main clusters we identified in Figure 1. This supports the hypothesis that each group arose via an ancestral LGT event, and the composition of many xmo operons has not yet normalized to the composition of the host organisms.
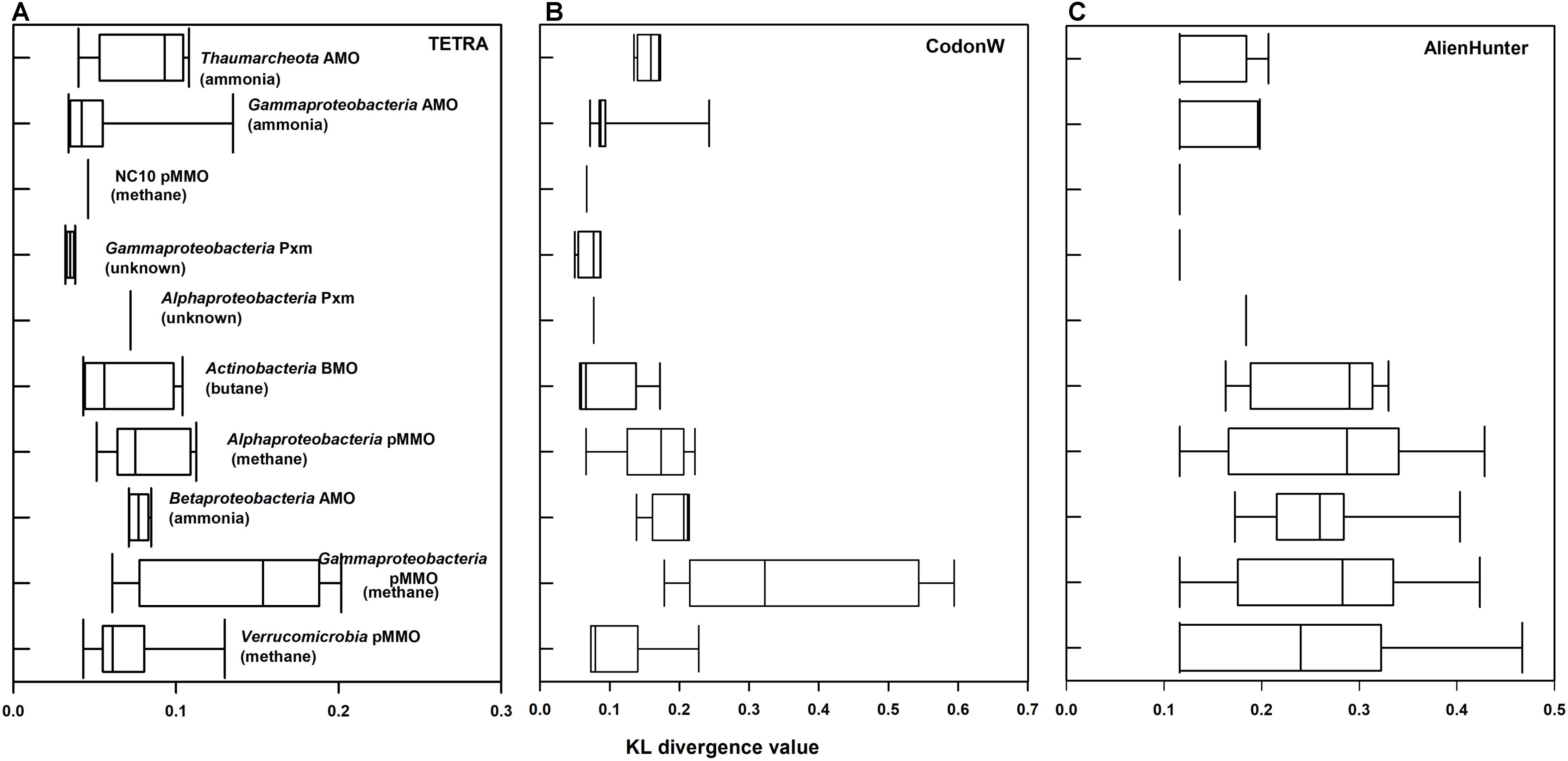
FIGURE 3. KL divergence measure of xmoCAB versus the entire genome in different taxonomic/functional groups of xmoCAB-possessing bacteria. Values on the x axes represent calculated KL values (0–1) by three different analysis: (A) TETRA, (B) CodonW, and (C) Alien Hunter. Larger values indicate stronger compositional differences in xmoCAB operons compared to their genomes and may suggest lateral gene transfer (LGT).
The notable exceptions showing little or no compositional bias in the xmo operon were the gammaproteobacterial Pxm cluster, the gammaproteobacterial AMO (Nitrosococcus spp.), and the NC10 pMMO. These were below the significance threshold using Alien Hunter (Supplementary Table S1) and also showed the lowest KL values of all the groups in other compositional analyses (Figure 3). These KL values calculated using CodonW and TETRA differ absolutely from those of Alien Hunter, but generally agree on the operons with the highest compositional differences to their genomes (Figure 3). Although the acclimatization rates of foreign DNA to a genome may vary, we speculate that these three groups may represent those that have possessed the operon for the longest evolutionary time, and their ancestors were likely donors of xmo to the other groups.
On the other hand, several clusters of xmoCAB show extremely high compositional differences to their host genomes: particularly those encoding the pMMO in Gammaproteobacteria and the AMO in Betaproteobacteria, but also to a lesser extent the pMMO in Alphaproteobacteria and the BMO in Actinobacteria. These may represent more recent LGT acquisitions of xmoCAB. This hypothesis is supported by the analysis using IslandViewer 4, a tool for detection and visualization of genomic islands (Bertelli et al., 2017), which shows that xmoCAB operons in some betaproteobacterial nitrifiers, actinobacterial butane oxidizers, and gammaproteobacterial methanotrophs can still be detected on genomic islands likely transferred from a foreign genome (Supplementary Table S1).
Based on compositional analysis (Supplementary Table S1), Methylacidiphilum sp. xmoCAB3 operons have high KL values for Alien Hunter (0.47), TETRA (0.13), and CodonW (0.23) compared to the other two operons in the Verrucomicrobia. This suggests that they are compositionally foreign. They are also placed variably in different phylogenies.
Longer branch lengths to the xmoCAB3 genes were also observed in some phylogenies. Long branch lengths are associated with accelerated mutation rates. In order to quantify this we used Bayesian inference phylogeny with a relaxed clock log-normal model to estimate the evolutionary rate at each branch in the phylogenetic trees (Bouckaert et al., 2014; Ho and Duchene, 2014). The evolutionary rate of verrucomicrobial pmoCAB3 genes was higher than the evolutionary rates of the pmoCAB1 and pmoCAB2 operon copies, with rates of 2.66, 2.64, and 3.46 for XmoA, XmoB, and XmoC phylogenies respectively (Supplementary Figures S5A–C). Evolutionary rates were mostly below 0.99 for most of the other lineages. This high rate suggests that verrucomicrobial pmoCAB operon three genes are mutating at high rate.
We tested specificity of different primers that have been used to amplify xmoA genes from environmental DNA extracts. These included several “universal” and group-specific primers for methanotroph pmoA genes. The results are summarized in Supplementary Table S2. The commonly used primer sets for methanotrophs 189f/682r (Holmes et al., 1995) and 189f/661 (Costello and Lidstrom, 1999) both have apparent biases and may miss many species, not only the exotic newly described methanotrophs such as the Verrucomicrobia and NC10, but also some proteobacteria like Methylocapsa. Recently, the 189f forward primer in combination with newly developed reverse primer HD616 was used to amplify CuMMO genes with high coverage (Wang et al., 2017). The HD616 primer is more universal than any other primer tested in our database (Supplementary Table S2), but the large number of primer degeneracies may result in non-specific amplifications. However, the primer does not cover specific groups like the methanotrophic USCγ group, and thaumarcheotal amoA genes (Wang et al., 2017).
Methane and ammonia oxidizing microorganisms are well-studied in different environments because of their importance in biogeochemical cycling of carbon and nitrogen. Studies based on 16S rRNA genes and functional marker genes like amoA and pmoA have been used to interpret their diversity and abundance in the environment (Knief, 2015). By using an extensive copper monooxygenase-encoding gene database from fully sequenced genomes, we constructed robust CuMMO phylogenies and performed comparative genomics to elucidate the phylogenetic history of the encoding genes.
All phylogenies showed at least 10 distinct groups based on a combination of substrate and taxonomy. Phylogenies of individual XmoA, B, and C products also showed similar patterns (with the exception of the Verrucomicrobia, discussed below). This suggests that the operon has evolved and been transferred primarily as a single unit. The evolutionary history of Xmo is therefore a mixture of functional evolution and a few LGT events of the entire operon, at minimum one event to each distinct group shown in Figure 1. Although to some extent CuMMOs can each oxidize all the relevant substrates (ammonia, methane, short chain alkanes and alkenes), they tend to prefer one substrate, indicating specialization to different substrates (Bedard and Knowles, 1989). The integration of CuMMOs into catabolic pathways targeting particular substrates has occurred multiple times. For example, in all phylogenies, the AMOs of Thaumarchaeota, Betaproteobacteria, and Gammaproteobacteria formed highly supported, distinct clusters, indicating that integration of CuMMO enzymes into an ammonia oxidizing metabolism occurred multiple times. The most parsimonious scenario based on the Bayesian tree (ignoring the Xmo’s of unknown function in Figure 1, and assuming Pxm is a methane monooxygenase) begins with either a pMMO or AMO ancestor followed by six changes of function.
Despite the coherent functional/taxonomic groups, there is no overriding structuring of the tree based on either function or taxonomy alone. Taxonomically, for example, Gammaproteobacteria clusters are scattered about the phylogenetic tree. Functionally, AMOs of Thaumarchaeota, Betaproteobacteria, and Gammaproteobacteria each form coherent, highly supported clusters, but these three do not together form a monophyletic group, indicating that incorporation of ammonia-specific CuMMO into metabolic pipelines capable of utilizing toxic hydroxylamine for catabolic gain (Simon and Klotz, 2013) has evolved multiple times. Individual pMMO clusters of Alphaproteobacteria, Verrucomicrobia, NC10, and Gammaproteobacteria methanotrophs can be discerned. These four groups all cluster closely together, however methanotrophy as a function is paraphyletic, with a group of ammonia-oxidizers (Nitrosococcus spp.) and the non-methanotrophic Skermanella nested within the larger group of methanotrophs. The pxmABC homologs of pmoCAB in methanotrophs are also not monophyletic with the known pmoCAB operons. Experiments with M. denitrificans FJG1 suggested that Pxm is an alternate methane monooxygenase that cells express particularly under microoxic conditions (Kits et al., 2015b), but its function is still not completely clear. Nevertheless, if it indeed acts as a methane monooxygenase, this pattern indicates the adaptation of CuMMOs to catabolizing methane also evolved multiple times.
The major inconsistencies in different phylogenetic constructions (both in the concatenated trees and in the individual gene trees) were in the placement of the Verrucomicrobia. Verrucomicrobia methanotrophs contain up to three homologous but distinct pmoCAB operons, with the most divergent of these usually named pmoCAB3 (Op den Camp et al., 2009). Some constructions showed the three operons of Verrucomicrobia to be polyphyletic, with XmoC3 and XmoA3 separate from the corresponding XmoC1/2 and XmoA1/2 (Figure 2). However, this was most evident in construction methods that are easily violated by variable mutation rates in different lineages (strict-clock Bayesian and NJ trees). When the clock assumption was relaxed in ML or Bayesian relaxed clock models, the three verrucomicrobial homologs were always monophyletic (Figure 2 and Supplementary Figures S2B, S3B, S4B). Long branch lengths to the xmoCAB3 genes were observed in most phylogenies, suggesting that an accelerated mutation rate of this third operon, which probably made some phylogenetic constructions inconsistent. A Bayesian estimation of evolutionary rates suggested that the xmoCAB3 is indeed evolving faster than other lineages in the phylogenetic tree. We therefore propose that the three operons in the Verrucomicrobia are monophyletic and have arisen from two lineage-specific duplications, but that operon 3 is under accelerated, relaxed selection. This is consistent with expression studies suggesting that this operon is only weakly expressed under methanotrophic growth conditions (Erikstad et al., 2012; Khadem et al., 2012; Anvar et al., 2014).
The other oddity regarding the Verrucomicrobia is that their XmoB gene shows a different phylogeny than XmoA and XmoC (Figure 2). In XmoA and XmoC trees, all methanotrophs including the Proteobacteria, Verrucomicrobia, and NC10 form a single cluster with only the Nitrosococcus AMO group as a nested group. However, in the XmoB trees the methanotrophs are more polyphyletic. The proteobacterial methanotrophs, NC10 and Nitrosococcus remain monophyletic in the XmoB tree, but the Verrucomicrobia are not monophyletic with them, instead branching nearer the base on the tree. This pattern is consistently seen regardless of the construction method used. It strongly suggests a distinct evolutionary history of XmoB compared to XmoA and XmoC in this phylum. A possible evolutionary scenario to explain this pattern is that the pMMO evolved only once, as indicated by XmoA and XmoC (and XmoCAB) phylogenies, but that the Verrucomicrobia at one time exchanged their xmoB gene(s) for that from another (unknown) donor organism. Crystal structure of pMMOs from Methylococcus capsulatus, Methylosinus trichosporium OB3b, Methylocystis sp. strain M and Methylocystis sp. Rockwell all predict a copper binding site at the N-terminus of the PmoB subunit (Sirajuddin and Rosenzweig, 2015). This copper binding site is conserved in all methanotrophs except for the Verrucomicrobia (Op den Camp et al., 2009). It is possible that the Verrucomicrobia have adopted a more distant PmoB to alter the metal profile of their enzymes (Culpepper and Rosenzweig, 2012).
Detecting LGT generally relies on phylogenetic incongruence between different gene trees (Delwiche and Palmer, 1996; Daubin et al., 2003; Lerat et al., 2003; Beiko et al., 2005; Adato et al., 2015), and on the compositional biases of genes or operons compared to genomes (Garcia-Vallve et al., 2000; Karlin, 2001; Teeling et al., 2004a). We used both phylogenetic interference and compositional analysis methods to detect possible LGT events. Compositional analysis showed evidence of extensive LGTs, while phylogenies demonstrated that these were limited to few (minimum 10) total events, followed by diversification of functional/taxonomic groups. From the phylogenetic and compositional analyses, we postulated several possible routes of LGT into different taxa and lineages, summarized in Figure 4. We restrict our speculation to clusters containing many sequences, and therefore prefer not to speculate greatly on the NC10 and Nitrospira, although the history of LGT from or to these groups would also be illuminating.
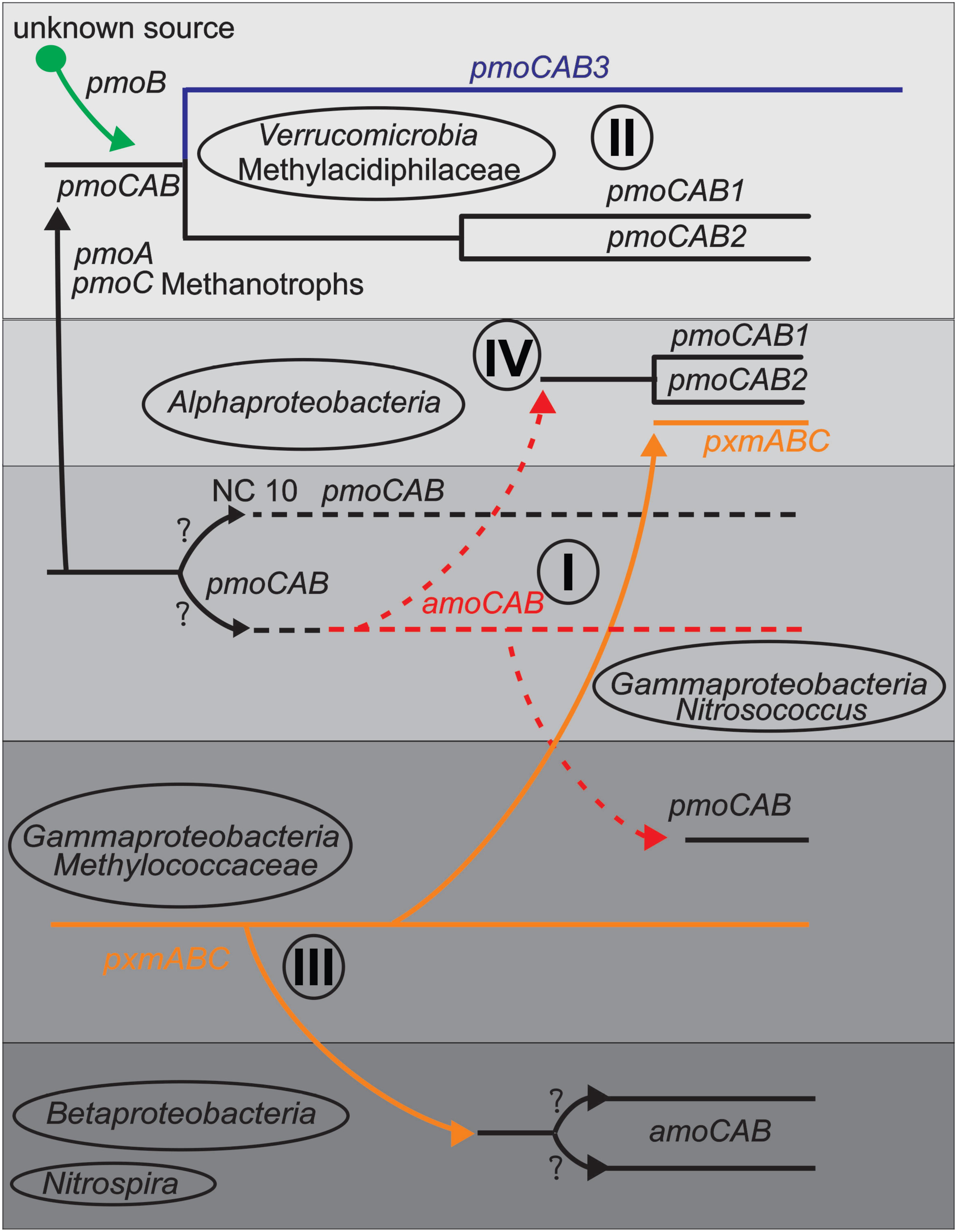
FIGURE 4. Schematic diagram indicating some predicted LGT events (arrows) between taxa or lineages based on phylogeny and compositional analysis items I-IV discussed in the text. Shaded boxes represent taxa and lines represents gene lineages.
(I) The Nitrosococcus amoCAB cluster is nested within a large cluster of methanotrophic pmoCABs, but compositional analysis indicates that it is more native to its genomes than the pmoCABs are to the methanotrophs. pmoCAB operons in Alphaproteobacteria and Gammaproteobacteria each show strong evidence of LGT. We therefore propose that the ancestor of the gammaproteobacterial Nitrosococcus group was a methanotroph with a pmoCAB operon. This Nitrosococcus lineage first transferred its pmoCAB genes to Alphaproteobacteria methanotrophs and then later to the gammaproteobacterial methanotrophs. This fits with recent analyses showing that some gammaproteobacterial nitrifiers: specifically Nitrosococcus wardiae (Wang et al., 2016) and Nitrosococcus halophilus (Campbell et al., 2011a), encode a complete methane-oxidation pipeline, including methanol dehydrogenase, a tetrahydromethanopterin-based formaldehyde oxidation pathway, and formate dehydrogenase (Chistoserdova, 2016; Wang, 2016). These findings may indicate that ancestors of extant Nitrosococcus once functioned primarily as methanotrophs. The AMO from Nitrosococcus oceani also does not exhibit substrate preference for ammonia or methane (Lontoh et al., 2000).
This pattern of lateral transfer of genes implicated in ammonia catabolism from ancestors of Nitrosococcus to the ancestors of extant gammaproteobacterial methanotrophs has also been proposed based on an analysis of the gene cluster encoding hydroxylamine dehydrogenase (Wang, 2016). Hydroxylamine dehydrogenase detoxifies and extracts catabolic electrons from hydroxylamine, the product of ammonia oxidation by CuMMO, and is co-expressed in ammonia-catabolic bacteria with the cytochrome c-based conduit that channels the extracted electrons from the periplasm into the Quinone-pool (Klotz and Stein, 2011; Simon and Klotz, 2013). Interestingly, the hydroxylamine detox capacity has been retained only in methanotrophs that are exposed to fluctuating ammonium concentrations in the environment (Nyerges and Stein, 2009; Nyerges et al., 2010; Campbell et al., 2011b; Stein and Klotz, 2011) but lost in those that typically exist in low ammonium environments (Tamas et al., 2014).
(II) The “Methylacidiphilum” group possesses two similar (pmoCAB1 and pmoCAB2) and one divergent pmoCAB3 operon. All are monophyletic, but the phylogeny of the pmoB differs from that of pmoC and pmoA. We propose that the ancestor of “Methylacidiphilum” obtained pmoCAB genes from an ancestral methanotroph (perhaps the NC10 lineage or the Nitrosococcus ancestor), but replaced that original pmoB gene with an xmoB gene from another, unknown source. The three operons were created by two successive lineage specific duplications. High evolutionary rates are seen along the verrucomicrobial pmoCAB3 operon, and make some phylogenetic constructions inconsistent.
(III) In the Gammaproteobacteria methanotrophs, the pxmABC operon is compositionally indistinguishable from the genomes. Their pmoCAB operons, on the other hand, are compositionally very distinct from most genomes. This suggests that these bacteria first possessed the pxmABC operon, and only later obtained the pmoCAB operon via LGT (from the ancestor of Nitrosococcus). It has been speculated that gammaproteobacterial pxm genes were horizontally transferred (Tavormina et al., 2011); however, above analyses support the opposite evolutionary scenario. The Gammaproteobacteria pxmABC operon may also have been transferred to some Alphaproteobacteria methanotrophs, and to the Betaproteobacteria nitrifiers.
(IV) As previously proposed (Tamas et al., 2014), a methylotrophic ancestor of all alphaproteobacterial methanotrophs in the families Methylocystaceae and Beijerinckiaceae obtained pMMO via a single LGT event from a gammaproteobacterial ancestor. This operon underwent a duplication and divergence into two lineages in the Methylocystaceae, and was lost in many Beijerinckiaceae. A second transfer event of the pxmABC operon from the Gammaproteobacteria has also occurred.
We stress that the evolution of a methanotrophic or ammonia-oxidizing phenotype is more complicated than the simple acquisition of a CuMMO. The downstream metabolic machinery to detoxify toxic alcohol and aldehyde intermediates such as methanol, formaldehyde, and hydroxylamine, while simultaneously feeding catabolic electrons into the Q-pool, are as critical as the monooxygenase step (Klotz and Stein, 2008, 2011; Simon and Klotz, 2013). Hence, the acquisition of the genetic basis for a functional CuMMO (xmoCAB) has to be preceded by the existence of genes that encode detoxification and electron extraction modules paired with a respective quinone-reactive protein (QRP) in order to create an efficient electron-flow pipeline that will integrate the CuMMO to the genome. Hence, it has been proposed for example that the original alphaproteobacterium that obtained pmoCAB via LGT must have been a methylotroph, a proposition strongly supported by phylogenetic examination of methylotrophy genes (Tamas et al., 2014). In this study we have concentrated on the critical first step of the process, and elucidated some of the steps in the evolution, transfer, duplication, and adaptation of CuMMOs.
RK, LC, LW, CL, MK, and PD designed the experiments. RK conducted the majority of work including phylogenetic and compositional analysis of genomes. LW, CL, and MK generated unpublished Nitrosococcus genome sequences. LC initiated compositional analysis work including TETRA, CodonW and Alien Hunter and RK continued further analysis. RK and PD wrote the manuscript. All authors provided input on the final manuscript.
This work was funded via an NSERC (Natural Sciences and Engineering Research Council of Canada) Discovery Grant (Grant No. 2014-05067).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We wish to thank the many colleagues who participated in the various genome sequencing projects that created the raw data for this work, especially Mette Svenning, the OMEGA group and the Joint Genome Institute.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02493/full#supplementary-material
Adato, O., Ninyo, N., Gophna, U., and Snir, S. (2015). Detecting horizontal gene transfer between closely related taxa. PLoS Comput. Biol. 11:e1004408. doi: 10.1371/journal.pcbi.1004408
Alves, R. J. E., Minh, B. Q., Urich, T., Von Haeseler, A., and Schleper, C. (2018). Unifying the global phylogeny and environmental distribution of ammonia-oxidising archaea based on amoA genes. Nat. Commun. 9:1517. doi: 10.1038/s41467-018-03861-1
Angellotti, M. C., Bhuiyan, S. B., Chen, G., and Wan, X. F. (2007). CodonO: codon usage bias analysis within and across genomes. Nucleic Acids Res. 35, W132–W136. doi: 10.1093/nar/gkm392
Anvar, S. Y., Frank, J., Pol, A., Schmitz, A., Kraaijeveld, K., Den Dunnen, J. T., et al. (2014). The genomic landscape of the verrucomicrobial methanotroph Methylacidiphilum fumariolicum SolV. BMC Genomics 15:914. doi: 10.1186/1471-2164-15-914
Arp, D. J., Chain, P. S., and Klotz, M. G. (2007). The impact of genome analyses on our understanding of ammonia-oxidizing bacteria. Annu. Rev. Microbiol. 61, 503–528. doi: 10.1146/annurev.micro.61.080706.093449
Baani, M., and Liesack, W. (2008). Two isozymes of particulate methane monooxygenase with different methane oxidation kinetics are found in Methylocystis sp. strain SC2. Proc. Natl. Acad. Sci. U.S.A. 105, 10203–10208. doi: 10.1073/pnas.0702643105
Becq, J., Churlaud, C., and Deschavanne, P. (2010). A benchmark of parametric methods for horizontal transfers detection. PLoS One 5:e9989. doi: 10.1371/journal.pone.0009989
Bedard, C., and Knowles, R. (1989). Physiology, biochemistry, and specific inhibitors of CH4, NH4 +, and CO oxidation by Methanotrophs and Nitrifiers. Microbiol. Rev. 53, 68–84.
Beiko, R. G., Harlow, T. J., and Ragan, M. A. (2005). Highways of gene sharing in prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 102, 14332–14337. doi: 10.1073/pnas.0504068102
Bertelli, C., Laird, M. R., Williams, K. P., Simon Fraser University Research Computing Group., Lau, B. Y., Hoad, G., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Berube, P. M., Samudrala, R., and Stahl, D. A. (2007). Transcription of all amoC copies is associated with recovery of Nitrosomonas europaea from ammonia starvation. J. Bacteriol. 189, 3935–3944. doi: 10.1128/JB.01861-06
Berube, P. M., and Stahl, D. A. (2012). The divergent amoC3 subunit of ammonia monooxygenase functions as part of a stress response system in Nitrosomonas europaea. J. Bacteriol. 194, 3448–3456. doi: 10.1128/jb.00133-12
Bodelier, P. L., and Laanbroek, H. J. (2004). Nitrogen as a regulatory factor of methane oxidation in soils and sediments. FEMS Microbiol. Ecol. 47, 265–277. doi: 10.1016/s0168-6496(03)00304-0
Bouckaert, R., Heled, J., Kuhnert, D., Vaughan, T., Wu, C. H., Xie, D., et al. (2014). BEAST 2: a software platform for bayesian evolutionary analysis. PLoS Comput. Biol. 10:e1003537. doi: 10.1371/journal.pcbi.1003537
Campbell, M. A., Chain, P. S., Dang, H., El Sheikh, A. F., Norton, J. M., Ward, N. L., et al. (2011a). Nitrosococcus watsonii sp. nov., a new species of marine obligate ammonia-oxidizing bacteria that is not omnipresent in the world’s oceans: calls to validate the names ‘Nitrosococcus halophilus’ and ‘Nitrosomonas mobilis’. FEMS Microbiol. Ecol. 76, 39–48. doi: 10.1111/j.1574-6941.2010.01027.x
Campbell, M. A., Nyerges, G., Kozlowski, J. A., Poret-Peterson, A. T., Stein, L. Y., and Klotz, M. G. (2011b). Model of the molecular basis for hydroxylamine oxidation and nitrous oxide production in methanotrophic bacteria. FEMS Microbiol. Lett. 322, 82–89. doi: 10.1111/j.1574-6968.2011.02340.x
Chistoserdova, L. (2016). Wide distribution of genes for Tetrahydromethanopterin/Methanofuran-linked C1 transfer reactions argues for their presence in the common ancestor of Bacteria and Archaea. Front. Microbiol. 7:1425. doi: 10.3389/fmicb.2016.01425
Coleman, N. V., Le, N. B., Ly, M. A., Ogawa, H. E., Mccarl, V., Wilson, N. L., et al. (2012). Hydrocarbon monooxygenase in Mycobacterium: recombinant expression of a member of the ammonia monooxygenase superfamily. ISME J. 6, 171–182. doi: 10.1038/ismej.2011.98
Coleman, N. V., Yau, S., Wilson, N. L., Nolan, L. M., Migocki, M. D., Ly, M. A., et al. (2011). Untangling the multiple monooxygenases of Mycobacterium chubuense strain NBB4, a versatile hydrocarbon degrader. Environ. Microbiol. Rep. 3, 297–307. doi: 10.1111/j.1758-2229.2010.00225.x
Costello, A. M., and Lidstrom, M. E. (1999). Molecular characterization of functional and phylogenetic genes from natural populations of methanotrophs in lake sediments. Appl. Environ. Microbiol. 65, 5066–5074.
Culpepper, M. A., and Rosenzweig, A. C. (2012). Architecture and active site of particulate methane monooxygenase. Crit. Rev. Biochem. Mol. Biol. 47, 483–492. doi: 10.3109/10409238.2012.697865
Dam, B., Kube, M., Dam, S., Reinhardt, R., and Liesack, W. (2012). Complete sequence analysis of two methanotroph-specific repABC-containing plasmids from Methylocystis sp. strain SC2. Appl. Environ. Microbiol. 78, 4373–4379. doi: 10.1128/AEM.00628-12
Daubin, V., Moran, N. A., and Ochman, H. (2003). Phylogenetics and the cohesion of bacterial genomes. Science 301, 829–832. doi: 10.1126/science.1086568
Delwiche, C. F., and Palmer, J. D. (1996). Rampant horizontal transfer and duplication of rubisco genes in eubacteria and plastids. Mol. Biol. Evol. 13, 873–882. doi: 10.1093/oxfordjournals.molbev.a025647
Dumont, M. G. (2014). “Primers: functional marker genes for methylotrophs and methanotrophs,” in Hydrocarbon and Lipid Microbiology Protocols, eds T. J. Mcgenity, K. N. Timmis and B. Nogales (Berlin: Springer-Verlag), 57–77.
Dunfield, P. F., Yimga, M. T., Dedysh, S. N., Berger, U., Liesack, W., and Heyer, J. (2002). Isolation of a Methylocystis strain containing a novel pmoA-like gene. FEMS Microbiol. Ecol. 41, 17–26. doi: 10.1111/j.1574-6941.2002.tb00962.x
Dunfield, P. F., Yuryev, A., Senin, P., Smirnova, A. V., Stott, M. B., Hou, S., et al. (2007). Methane oxidation by an extremely acidophilic bacterium of the phylum Verrucomicrobia. Nature 450, 879–882. doi: 10.1038/nature06411
Erikstad, H. A., Jensen, S., Keen, T. J., and Birkeland, N. K. (2012). Differential expression of particulate methane monooxygenase genes in the verrucomicrobial methanotroph ‘Methylacidiphilum kamchatkense’ Kam1. Extremophiles 16, 405–409. doi: 10.1007/s00792-012-0439-y
Ettwig, K. F., Van Alen, T., Van De Pas-Schoonen, K. T., Jetten, M. S., and Strous, M. (2009). Enrichment and molecular detection of denitrifying methanotrophic bacteria of the NC10 phylum. Appl. Environ. Microbiol. 75, 3656–3662. doi: 10.1128/AEM.00067-09
Garcia-Vallve, S., Romeu, A., and Palau, J. (2000). Horizontal gene transfer in bacterial and archaeal complete genomes. Genome Res. 10, 1719–1725. doi: 10.1101/gr.130000
Gouy, M., Guindon, S., and Gascuel, O. (2010). SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224. doi: 10.1093/molbev/msp259
Ho, S. Y., and Duchene, S. (2014). Molecular-clock methods for estimating evolutionary rates and time scales. Mol. Ecol. 23, 5947–5965. doi: 10.1111/mec.12953
Holmes, A.J., Costello, A., Lidstrom, M.E., and Murrell, J.C. (1995). Evidence that participate methane monooxygenase and ammonia monooxygenase may be evolutionarily related. FEMS Microbiol. Lett. 132, 203–208. doi: 10.1111/j.1574-6968.1995.tb07834.x
Karlin, S. (2001). Detecting anomalous gene clusters and pathogenicity islands in diverse bacterial genomes. Trends Microbiol. 9, 335–343. doi: 10.1016/S0966-842X(01)02079-0
Khadem, A. F., Pol, A., Wieczorek, A. S., Jetten, M. S., and Op Den Camp, H. J. (2012). Metabolic regulation of “Ca. Methylacidiphilum Fumariolicum” SolV cells grown under different nitrogen and oxygen limitations. Front. Microbiol. 3:266. doi: 10.3389/fmicb.2012.00266
Kits, K. D., Campbell, D. J., Rosana, A. R., and Stein, L. Y. (2015a). Diverse electron sources support denitrification under hypoxia in the obligate methanotroph Methylomicrobium album strain BG8. Front. Microbiol. 6:1072. doi: 10.3389/fmicb.2015.01072
Kits, K. D., Klotz, M. G., and Stein, L. Y. (2015b). Methane oxidation coupled to nitrate reduction under hypoxia by the gammaproteobacterium Methylomonas denitrificans, sp. nov. type strain FJG1. Environ. Microbiol. 17, 3219–3232. doi: 10.1111/1462-2920.12772
Klotz, M. G., and Norton, J. M. (1998). Multiple copies of ammonia monooxygenase (amo) operons have evolved under biased AT/GC mutational pressure in ammonia-oxidizing autotrophic bacteria. FEMS Microbiol. Lett. 168, 303–311. doi: 10.1111/j.1574-6968.1998.tb13288.x
Klotz, M., and Stein, L. (2011). Genomics of Ammonia-Oxidizing Bacteria and Insights to Their Evolution. Washington, DC: ASM Press.
Klotz, M. G., Alzerreca, J., and Norton, J. M. (1997). A gene encoding a membrane protein exists upstream of the amoA/amoB genes in ammonia oxidizing bacteria: a third member of the amo operon? FEMS Microbiol. Lett. 150,65–73.
Klotz, M. G., and Stein, L. Y. (2008). Nitrifier genomics and evolution of the nitrogen cycle. FEMS Microbiol. Lett. 278, 146–156. doi: 10.1111/j.1574-6968.2007.00970.x
Knief, C. (2015). Diversity and habitat preferences of cultivated and uncultivated aerobic methanotrophic bacteria evaluated based on pmoA as molecular marker. Front. Microbiol. 6:1346. doi: 10.3389/fmicb.2015.01346
Knief, C., Lipski, A., and Dunfield, P. F. (2003). Diversity and activity of methanotrophic bacteria in different upland soils. Appl. Environ. Microbiol. 69, 6703–6714. doi: 10.1128/AEM.69.11.6703-6714.2003
Kullback, S., and Leibler, R. A. (1951). On information and sufficiency. Ann. Math. Stat. 22, 79–86. doi: 10.1214/aoms/1177729694
Kuypers, M. M. M., Marchant, H. K., and Kartal, B. (2018). The microbial nitrogen-cycling network. Nat. Rev. Microbiol. 16, 263–276. doi: 10.1038/nrmicro.2018.9
Kyrpides, N. C., Woyke, T., Eisen, J. A., Garrity, G., Lilburn, T. G., Beck, B. J., et al. (2014). Genomic encyclopedia of type strains, phase I: the one thousand microbial genomes (KMG-I) project. Stand. Genomic Sci. 9, 1278–1284. doi: 10.4056/sigs.5068949
Lerat, E., Daubin, V., and Moran, N. A. (2003). From gene trees to organismal phylogeny in prokaryotes: the case of the gamma-Proteobacteria. PLoS Biol. 1:E19. doi: 10.1371/journal.pbio.0000019
Lontoh, S., Dispirito, A. A., Krema, C. L., Whittaker, M. R., Hooper, A. B., and Semrau, J. D. (2000). Differential inhibition in vivo of ammonia monooxygenase, soluble methane monooxygenase and membrane-associated methane monoxygenase by phenylacetylene. Environ. Microbiol. 2, 485–494. doi: 10.1046/j.1462-2920.2000.00130.x
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Yadhukumar Buchner, A., et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371. doi: 10.1093/nar/gkh293
McDonald, I. R., Bodrossy, L., Chen, Y., and Murrell, J. C. (2008). Molecular ecology techniques for the study of aerobic methanotrophs. Appl. Environ. Microbiol. 74, 1305–1315. doi: 10.1128/AEM.02233-07
Nyerges, G., Han, S. K., and Stein, L. Y. (2010). Effects of ammonium and nitrite on growth and competitive fitness of cultivated methanotrophic bacteria. Appl. Environ. Microbiol. 76, 5648–5651. doi: 10.1128/aem.00747-10
Nyerges, G., and Stein, L. Y. (2009). Ammonia cometabolism and product inhibition vary considerably among species of methanotrophic bacteria. FEMS Microbiol. Lett. 297, 131–136. doi: 10.1111/j.1574-6968.2009.01674.x
Op den Camp, H. J., Islam, T., Stott, M. B., Harhangi, H. R., Hynes, A., Schouten, S., et al. (2009). Environmental, genomic and taxonomic perspectives on methanotrophic Verrucomicrobia. Environ. Microbiol. Rep. 1, 293–306. doi: 10.1111/j.1758-2229.2009.00022.x
Overbeek, R., Begley, T., Butler, R. M., Choudhuri, J. V., Chuang, H. Y., Cohoon, M., et al. (2005). The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 33, 5691–5702. doi: 10.1093/nar/gki866
Rotthauwe, J. H., Witzel, K. P., and Liesack, W. (1997). The ammonia monooxygenase structural gene amoA as a functional marker: molecular fine-scale analysis of natural ammonia-oxidizing populations. Appl. Environ. Microbiol. 63, 4704–4712.
Sayavedra-Soto, L. A., Hamamura, N., Liu, C. W., Kimbrel, J. A., Chang, J. H., and Arp, D. J. (2011). The membrane-associated monooxygenase in the butane-oxidizing Gram-positive bacterium Nocardioides sp. strain CF8 is a novel member of the amo/pmo family. Environ. Microbiol. Rep. 3, 390–396. doi: 10.1111/j.1758-2229.2010.00239.x
Semrau, J. D., Dispirito, A. A., and Yoon, S. (2010). Methanotrophs and copper. FEMS Microbiol. Rev. 34, 496–531. doi: 10.1111/j.1574-6976.2010.00212.x
Simon, J., and Klotz, M. G. (2013). Diversity and evolution of bioenergetic systems involved in microbial nitrogen compound transformations. Biochim. Biophys. Acta 1827, 114–135. doi: 10.1016/j.bbabio.2012.07.005
Sirajuddin, S., and Rosenzweig, A. C. (2015). Enzymatic oxidation of methane. Biochemistry 54, 2283–2294. doi: 10.1021/acs.biochem.5b00198
Stein, L. Y., and Klotz, M. G. (2011). Nitrifying and denitrifying pathways of methanotrophic bacteria. Biochem. Soc. Trans. 39, 1826–1831. doi: 10.1042/bst20110712
Stolyar, S., Costello, A. M., Peeples, T. L., and Lidstrom, M. E. (1999). Role of multiple gene copies in particulate methane monooxygenase activity in the methane-oxidizing bacterium Methylococcus capsulatus bath. Microbiology 145(Pt 5), 1235–1244. doi: 10.1099/13500872-145-5-1235
Stolyar, S., Franke, M., and Lidstrom, M. E. (2001). Expression of individual copies of Methylococcus capsulatus bath particulate methane monooxygenase genes. J. Bacteriol. 183, 1810–1812. doi: 10.1128/jb.183.5.1810-1812.2001
Suzuki, T., Nakamura, T., and Fuse, H. (2012). Isolation of two novel marine ethylene-assimilating bacteria, Haliea species ETY-M and ETY-NAG, containing particulate methane monooxygenase-like genes. Microbes Environ. 27, 54–60. doi: 10.1264/jsme2.ME11256
Tamas, I., Smirnova, A. V., He, Z., and Dunfield, P. F. (2014). The (d)evolution of methanotrophy in the Beijerinckiaceae-a comparative genomics analysis. ISME J. 8, 369–382. doi: 10.1038/ismej.2013.145
Tavormina, P. L., Orphan, V. J., Kalyuzhnaya, M. G., Jetten, M. S., and Klotz, M. G. (2011). A novel family of functional operons encoding methane/ammonia monooxygenase-related proteins in gammaproteobacterial methanotrophs. Environ. Microbiol. Rep. 3, 91–100. doi: 10.1111/j.1758-2229.2010.00192.x
Tavormina, P. L., Ussler, W. III, Steele, J. A., Connon, S. A., Klotz, M. G., and Orphan, V. J. (2013). Abundance and distribution of diverse membrane-bound monooxygenase (Cu-MMO) genes within the Costa Rica oxygen minimum zone. Environ. Microbiol. Rep. 5, 414–423. doi: 10.1111/1758-2229.12025
Tchawa Yimga, M., Dunfield, P. F., Ricke, P., Heyer, J., and Liesack, W. (2003). Wide distribution of a novel pmoA-like gene copy among type II methanotrophs, and its expression in Methylocystis strain SC2. Appl. Environ. Microbiol. 69, 5593–5602. doi: 10.1128/aem.69.9.5593-5602.2003
Teeling, H., Meyerdierks, A., Bauer, M., Amann, R., and Glockner, F. O. (2004a). Application of tetranucleotide frequencies for the assignment of genomic fragments. Environ. Microbiol. 6, 938–947. doi: 10.1111/j.1462-2920.2004.00624.x
Teeling, H., Waldmann, J., Lombardot, T., Bauer, M., and Glockner, F. O. (2004b). TETRA: a web-service and a stand-alone program for the analysis and comparison of tetranucleotide usage patterns in DNA sequences. BMC Bioinformatics 5:163. doi: 10.1186/1471-2105-5-163
Vernikos, G. S., and Parkhill, J. (2006). Interpolated variable order motifs for identification of horizontally acquired DNA: revisiting the Salmonella pathogenicity islands. Bioinformatics 22, 2196–2203. doi: 10.1093/bioinformatics/btl369
Vorobev, A., Jagadevan, S., Jain, S., Anantharaman, K., Dick, G. J., Vuilleumier, S., et al. (2014). Genomic and transcriptomic analyses of the facultative methanotroph Methylocystis sp. strain SB2 grown on methane or ethanol. Appl. Environ. Microbiol. 80, 3044–3052. doi: 10.1128/AEM.00218-14
Wang, J. G., Xia, F., Zeleke, J., Zou, B., Rhee, S. K., and Quan, Z. X. (2017). An improved protocol with a highly degenerate primer targeting copper-containing membrane-bound monooxygenase genes for community analysis of methane- and ammonia-oxidizing bacteria. FEMS Microbiol. Ecol. 93:fiw244. doi: 10.1093/femsec/fiw244
Wang, L. (2016). Comparative Analysis of Nitrosococcus Genomes; Isolation, Physiological and Genomic Characterization of the New Ammonia-Oxidizing Species Nitrosococcus wardiae, Strain D1FHST; and Molecular Studies Towards a Better Understanding of Ammonia-Dependent Chemolithotrophy. Ph.D. dissertation, University of North Carolina at Charlotte, Charlotte, NC.
Wang, L., Lim, C. K., Dang, H., Hanson, T. E., and Klotz, M. G. (2016). D1FHS, the type strain of the ammonia-oxidizing bacterium Nitrosococcus wardiae spec. nov.: enrichment, isolation, phylogenetic, and growth physiological characterization. Front. Microbiol. 7:512. doi: 10.3389/fmicb.2016.00512
Wartiainen, I., Hestnes, A. G., Mcdonald, I. R., and Svenning, M. M. (2006). Methylocystis rosea sp. nov., a novel Methanotrophic bacterium from Arctic wetland soil, Svalbard, Norway (78 degrees N). Int. J. Syst. Evol. Microbiol. 56, 541–547. doi: 10.1099/ijs.0.63912-0
Keywords: methane oxidation, ammonia oxidation, copper membrane monooxygenase, lateral gene transfer, phylogenetics
Citation: Khadka R, Clothier L, Wang L, Lim CK, Klotz MG and Dunfield PF (2018) Evolutionary History of Copper Membrane Monooxygenases. Front. Microbiol. 9:2493. doi: 10.3389/fmicb.2018.02493
Received: 27 June 2018; Accepted: 28 September 2018;
Published: 29 October 2018.
Edited by:
Frank T. Robb, University of Maryland, Baltimore, United StatesReviewed by:
Sukhwan Yoon, Korea Advanced Institute of Science & Technology (KAIST), South KoreaCopyright © 2018 Khadka, Clothier, Wang, Lim, Klotz and Dunfield. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter F. Dunfield, cGZkdW5maWVAdWNhbGdhcnkuY2E=
†Present address: Lin Wang, Center for Microbial Pathogenesis, Institute for Biomedical Sciences, Georgia State University, Atlanta, GA, United States; Chee Kent Lim, School of Energy and Environment, City University of Hong Kong, Kowloon, Hong Kong
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.