 Mianzhi Wang1,2
Mianzhi Wang1,2 Wenguang Xiong1,2
Wenguang Xiong1,2 Peng Liu1,2Xiying Xie1,2Jiaxiong Zeng1,2
Peng Liu1,2Xiying Xie1,2Jiaxiong Zeng1,2 Yongxue Sun1,2*
Yongxue Sun1,2* Zhenling Zeng1,2*
Zhenling Zeng1,2*- 1National Risk Assessment Laboratory for Antimicrobial Resistance of Animal Original Bacteria, South China Agricultural University, Guangzhou, China
- 2Guangdong Provincial Key Laboratory of Veterinary Pharmaceutics Development and Safety Evaluation, College of Veterinary Medicine, South China Agricultural University, Guangzhou, China
In this study, we examined the types of antibiotic resistance genes (ARGs) possessed by bacteria and bacteriophages in swine feedlot wastewater before and after treatment using a metagenomics approach. We found that the relative abundance of ARGs in bacterial DNA in all water samples was significantly higher than that in phages DNA (>10.6-fold), and wastewater treatment did not significantly change the relative abundance of bacterial- or phage-associated ARGs. We further detected the distribution and diversity of the different types of ARGs according to the class of antibiotics to which they confer resistance, the tetracycline resistance genes were the most abundant resistance genes and phages were more likely to harbor ATP-binding cassette transporter family and ribosomal protection genes. Moreover, the colistin resistance gene mcr-1 was also detected in the phage population. When assessing the contribution of phages in spreading different groups of ARGs, β-lactamase resistance genes had a relatively high spreading ability even though the abundance was low. These findings possibly indicated that phages not only could serve as important reservoir of ARG but also carry particular ARGs in swine feedlot wastewater, and this phenomenon is independent of the environment.
Introduction
The over-reliance and over-use of antibiotics in humans, animals and agriculture has resulted in widespread dissemination of antibiotic resistant bacteria (ARB) and associated antibiotic resistance genes (ARGs). Especially troublesome is the “superbugs” ESKAPE group of pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.) and newly discovered ARGs types NDM-1 and mcr-1 (Boucher et al., 2009; Walsh et al., 2011; Liu et al., 2016). Obviously, the emergence and distribution of these pathogens and ARGs highly compromises the clinical treatment and presents a high-risk to public health. Moreover, the ARB and ARGs could also be horizontal transferred by air and water throughout the multiple transmission mechanisms such as transformation, conjugation, and transduction (Andersson and Hughes, 2010; McEachran et al., 2015; Rodriguez-Mozaz et al., 2015; Wang et al., 2015), these processes will further led to an indirect adverse effect to the surroundings ecosystems.
In these transmission processes, the most studied modes are transformation and conjugation, which are mediated by transposons, integrons and plasmids (Harmer et al., 2014; Gillings et al., 2015; Sun et al., 2016). However, the role for bacteriophage (or phage) transduction in HGT has most likely been underestimated. On the other hand, more and more studies have reported that phage genomes originating from animal sources, such as animal farm sludge samples, wastewater samples, soils samples and river water and sediments samples, contain a large reservoir of ARGs (Colomer-Lluch et al., 2014b; Ross and Topp, 2015; Calero-Caceres and Muniesa, 2016; Calero-Caceres et al., 2017; Lekunberri et al., 2017a,b). Therefore, as an important reservoir of ARGs, a thorough insight into phage contribution to antibiotic resistance will be of great importance in animal-associated environments, especially the water samples related to swine feedlot wastewater treatment that may have a significant impact on the surrounding environment.
In this study, we examined treated wastewater and pond water in swine feedlots from farms in Guangdong Province, China. We used whole genome sequencing to determine whether phage genomes carried ARGs and performed a comprehensive analysis of whether ARG-carrying phages contribute to antibiotic resistance.
Materials and Methods
Site Selection and Water Collection
We examined 16 wastewater samples that included bacterial and phage DNA from the following locations: influent water (INF-B and INF-P), aerobic-facultative intermediate water (AA-B and AA-P), effluent water (EF-B and EF-P), and pond water (PD-B and PD-P). Among these processes, the dissolved oxygen in the aerobic tanks was controlled at 2–4 mg/L and in the facultative tank was controlled at 0.5–1 mg/L (Figure 1).
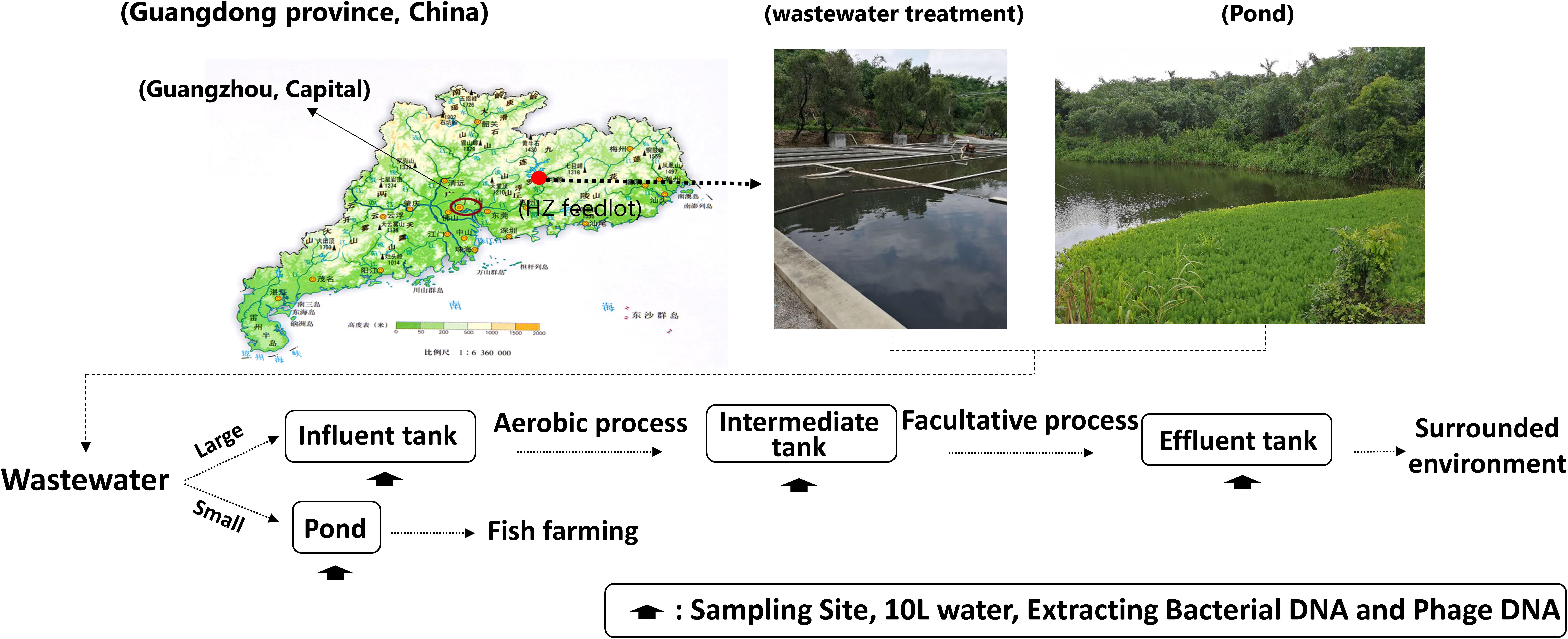
FIGURE 1. Geographical locations and schematic illustration of water sampling sites.
Samples were collected from Huizhou pig feedlots during May 2017. The detailed sampling strategy was adopted from Wang with a few modifications (Wang et al., 2016). In brief: approximately 10 L of surface water were collected; to avoid sampling site fluctuations, water samples were collected four times at 1-h intervals from 9:00 AM to 12:00 AM (a total of 4 h), and then pooled into one composite sample for each respective site (Figure 1).
Bacterial Acquisition and DNA Extraction
Water samples (10 L) were obtained and divided into two aliquots under aseptic conditions. One aliquot was used for DNA extraction from bacteria and bacteriophage and the other was stored at -80°C.
From the 5 L water, 0.5 L was used for bacterial DNA extraction. One hundred and fifty milliliters water was filtered through a 0.22-μm membrane filter (GTTP04700, Millipore, United States) using a vacuum filtration apparatus. The membrane filter was then used to extract DNA using an EZNA water DNA kit (Omega Bio-Tek, United States) according to the manufacturer’s instructions. Then, DNA concentrations were determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop, United States).
Bacteriophage Acquisition and DNA Extraction
The remaining 4.5 L of each wastewater sample was used for phage DNA extraction. Because of phages could pass through the 0.22-μm filtrate while bacteria are trapped on the membrane surface, we centrifuged the original wastewater of 1.5 L at 3,000 g for 15 min to remove solid matter and retained the supernatant. Then, the supernatant was filtered through a sequential series of membranes with pore sizes of 5, 1.2, and 0.45 μm (Midwest Great Technology, Beijing, China) and a final filtration through a 0.22 μm membrane (Millex-GP, Millipore).
After that, all the filtrate was concentrated using a 100 kDa Amicon Ultra centrifugal filter units (Millipore, United States) to a final volume of about 2 mL following the instructions provided by the manufacturer (Supplementary Figure S1). Phage DNA extraction was performed according to a published method (Thurber et al., 2009). Briefly, 100 U of DNase I was added to each milliliter of sample and the samples were incubated for 2 h at 37°C. Then, 0.1 volume of 2 M Tris–HCl (pH 8.5)/0.2 M EDTA, 0.01 volume of 0.5 M EDTA (pH 8.0), 1 volume of formamide and 2 volume of 100% ethanol were added to each sample, followed by a centrifugation for 20 min at 8,000 g at 4°C to pellet virions. The pellets were then treated with 3 μL of 20 mg/mL proteinase K. Finally, CTAB/NaCl solution, phenol/chloroform/isoamyl alcohol (25:24:1), and 0.7 volumes of isopropanol were used to acquire phages DNA precipitates. The DNA precipitates were then re-suspended in 30 μL of TE (10 mM Tris–HCl, pH 7.5, 1 mM EDTA) for use.
The absence of non-packaged DNA was verified using controls as previously described (Colomer-Lluch et al., 2014a). In brief, an aliquot of each sample was used for the conventional PCR detection of two pairs of different eubacterial 16S rDNA after DNase treatment but before desencapsidation. Only negative samples were used for the subsequent sequencing analysis and others were discarded. The eubacterial 16S rDNA primers were listed in Supplementary Table S1. DNA concentrations were measured by UV spectroscopy with a ND-1000 spectrophotometer (NanoDrop, Wilmington, DE, United States).
Metagenome Sequencing of Water Samples
Bacterial and phage DNA were sequenced using an Illumina HiSeqTM X Ten platform (Illumina Inc., San Diego, CA, United States). Approximately 40 million Illumina sequencing reads were generated (Table 1). 16S rRNA gene sequences in each phage DNA fraction were determined using METAXA2 to assess potential bacterial DNA contamination (Bengtsson-Palme et al., 2015). The Comprehensive Antibiotic Resistance Database (CARD) was used for ARG annotation and for ARG grouping according to resistance mechanism (Jia et al., 2017). ARG relative abundance was calculated in relation to the total number of reads in each metagenome.
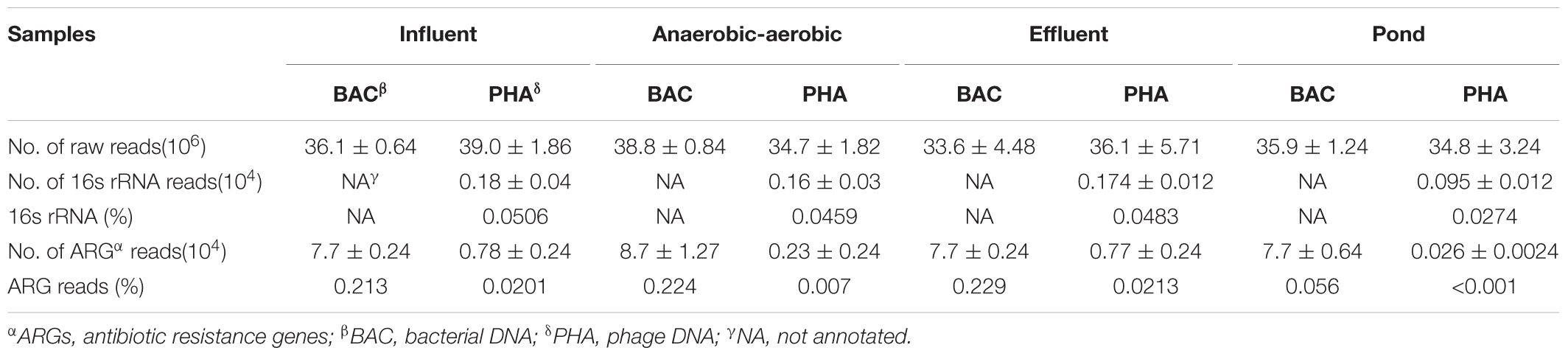
TABLE 1. Summary of data generated from the metagenomes.
Statistical Analysis
Usually, phage transduction depends upon possession of a cognate receptor from the target. If an ARG can be transferred via phage in the environment, the communication frequency between phage and bacteria is an indirect measure of the phage contribution to antibiotic resistance and serves as an assessment of antibiotic resistance’s spreading ability (Wang et al., 2018). In this study, the communication frequency was defined as a “spreading ability” value of a phage to spread an ARG by transduction (Eq. 1).
All statistical tests were analyzed in this study were performed with SPSS software (Version 20.0, SPSS, IBM, United States). One-way ANOVA was based on a 5% significance level. A two-tailed Pearson’s bivariate correlation analysis was used to compare bacterial and phage DNA ARG abundance for each antimicrobial. The raw data are uploaded to the database of MG-RAST1 and the accession number and detailed information were listed in the Supplementary Table S2 or access website2.
Results and Discussion
Abundance of Antibiotic Resistance Genes in Water Samples
According to previous studies, ARG abundance in phages has most likely been overestimated due to the bacterial DNA contamination (Roux et al., 2013; Lekunberri et al., 2017a). Therefore, we screened all unassembled reads from each of the phage DNA samples for 16S rRNA gene. The results showed that all selected influent water, aerobic-facultative intermediate water, effluent water and pond water viromes had low levels of bacterial DNA contamination (i.e., <0.02%) (Table 1). These results indicate that downstream analysis of ARG abundance could be further conducted; a relative comparison of ARG types and abundance would reflect phage ARG types. Also, after an annotation to CARD, the relative abundance of ARGs in four kinds of water samples showed some differences.
In the wastewater treatment system, the relative abundance of bacterial ARGs in all water samples was always 10- to 33.4-fold greater than that of phages. Surprisingly, though we found a significant decrease in phage ARG abundance between influent and intermediate water samples from 0.0201 to 0.00712%, the ARG elimination effect was not observed when the influent and effluent water was compared and stayed relatively constant at 0.213 and 0.229% for bacterial DNA and 0.0201–0.0203% for phage ARGs, respectively (Figure 2 and Table 1). Moreover, as an adjunct wastewater treatment system, the relative phage ARG abundance (0.001%) in the pond water was 55.7-fold lower than in the bacterial DNA fractions (0.0557%) (Figure 2). These results indicate that the swine feedlot wastewater treatment system did not significantly decrease the abundance of ARGs. Meanwhile, phages probably were an important ARG reservoir and play a major role in the spread of antibiotic resistance (Colomer-Lluch et al., 2014a; Subirats et al., 2016; Lekunberri et al., 2017a).
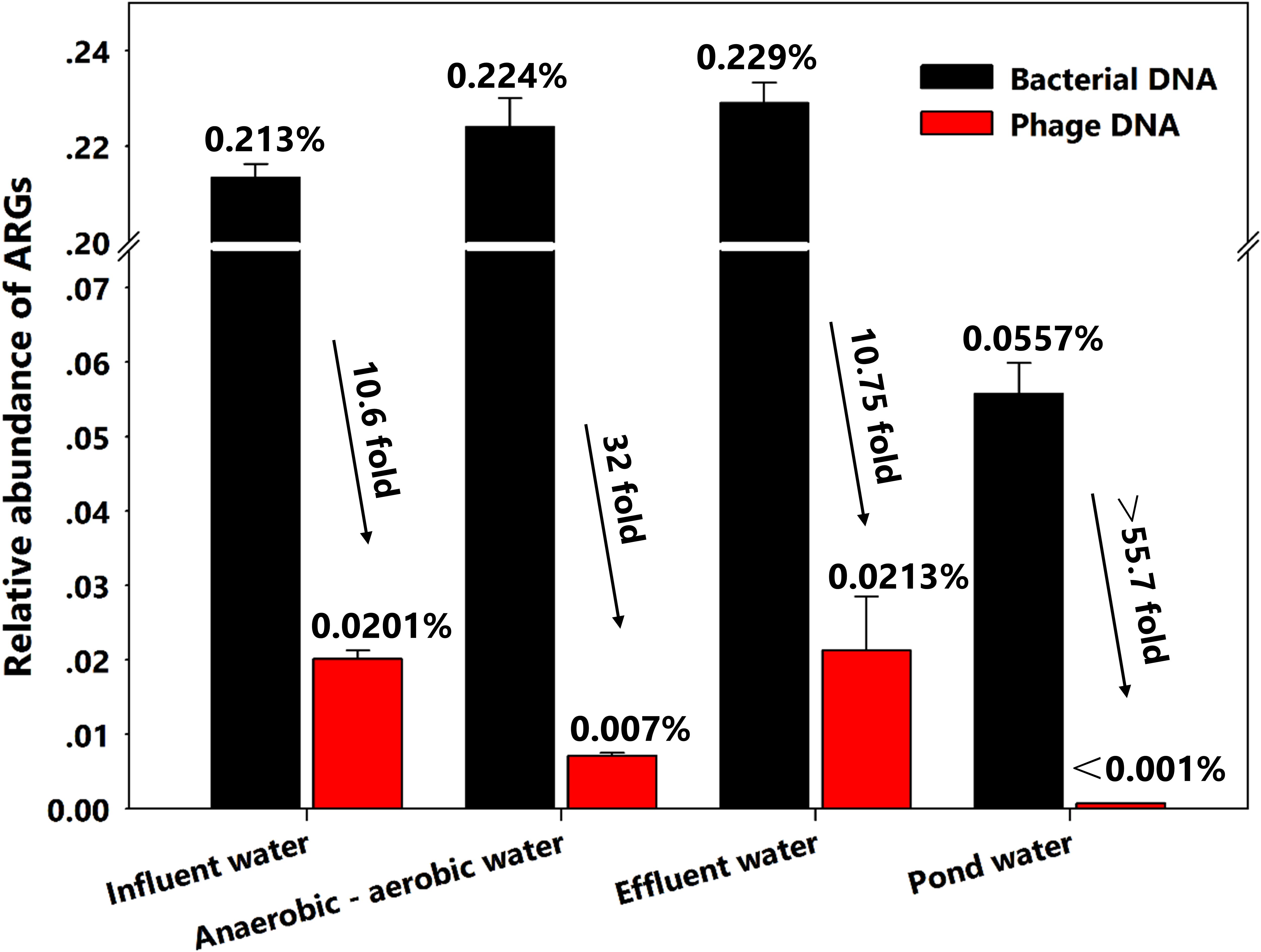
FIGURE 2. Relative abundance of all antibiotic resistance genes (ARGs) in bacterial and phage DNA from different water samples.
On the other hand, a possible explanation for the higher increase (55.7-fold) in pond water probably is that the low diversity both in bacterial and viral communities may have led to the co-selection of certain bacteria and phages. Yang et al. (2017) have found that ratios of bacteria to virus-like particles in municipal wastewater treatment systems were increased up to 17.5-fold even with low diversity of these communities. This might suggest a co-selection for certain phage types plays a dominant role in the spread of antibiotic resistance. Moreover, phage numbers are estimated to be >10-fold greater than their bacterial hosts in the environment (Pawluk, 2017). The imbalance in the bacteria-phage ratio in wastewater samples might lead to greater numbers of ARGs in phages compared with pond water samples.
However, more experimental and directed data are still needed to support our hypothesis. In addition, the lack of knowledge of the classification and identification of the phages from viral genomes is still a roadblock and it requires more in-depth studies.
Distribution and Diversity of Antibiotic Resistance Genes in Water Samples
Further analysis of the distribution and diversity for different types of ARGs according to the class of antibiotics that they confer resistance to in these two genetic pools, bacteria and virus DNA is needed. Results showed that a total of 13 different types of ARGs between bacterial and phage DNA were detected and tended to a fixed hierarchy of tetracycline ∼ aminoglycoside ∼ macrolide>phenicol ∼ sulphonamide > β-lactams ∼ trimethoprim ∼ quinolone > vancomycin ∼ nitroimidazole ∼ rifampicin > fosfomycin ∼ colistin (Figures 3, 4A). These results were similar to findings from another study using raw sewage (Lekunberri et al., 2017a). However, we were surprised that colistin resistance gene mcr-1 was present in influent water. Colistin and tigecycline are among last resort of antimicrobials in the treatment of infections caused by carbapenemase-producing Enterobacteriaceae (Pitout et al., 2015). The emergence of mcr-1 might imply that apart from the plasmid-mediated horizontal genes transfer, phage transduction is probably another driving force for HGT in the environment.
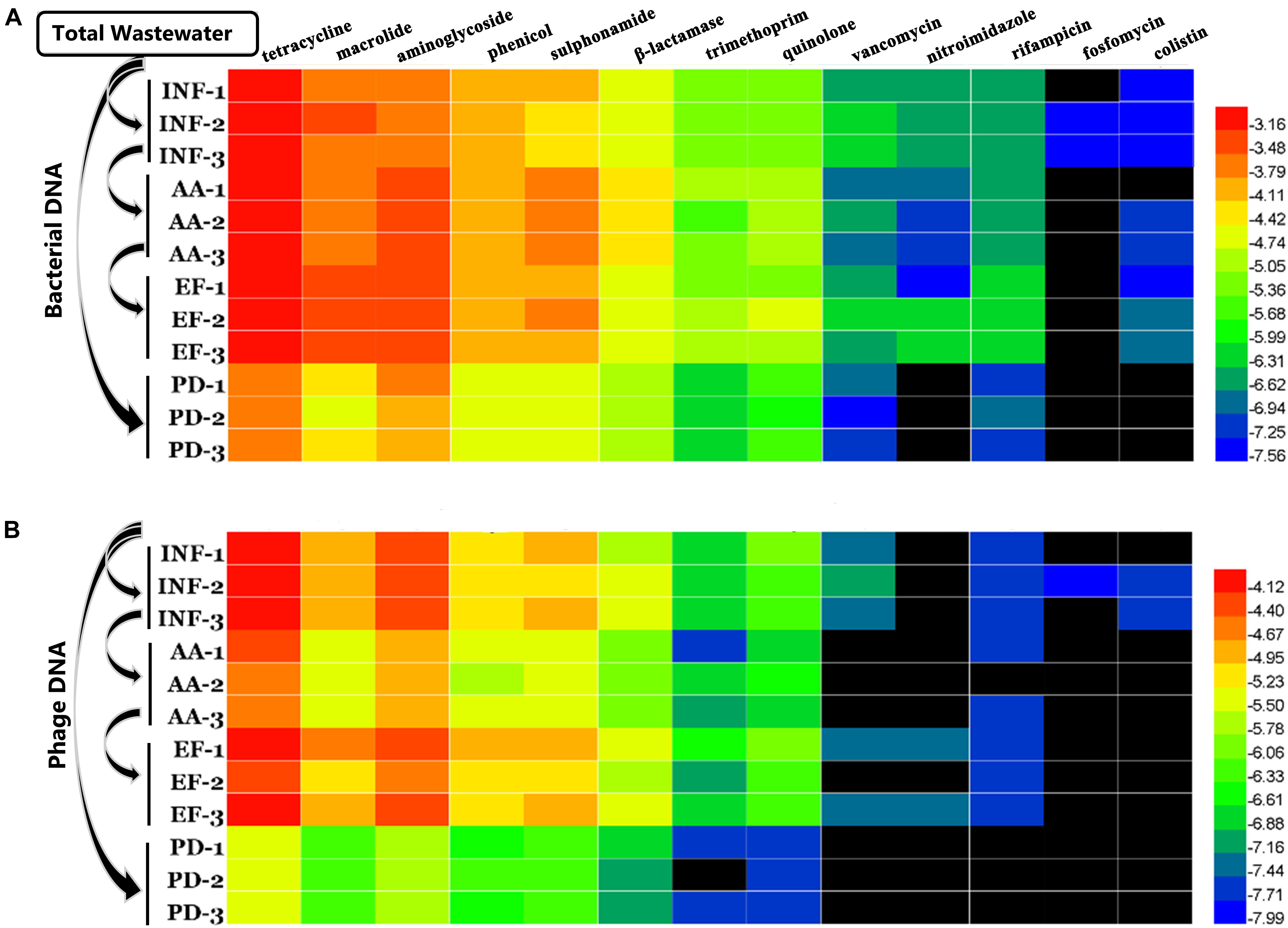
FIGURE 3. Variations of relative abundance of various ARG types at different locations. (A) Bacterial DNA and (B) phage DNA. The data were log10 transformed. The black bars indicate that the genes were not detected.
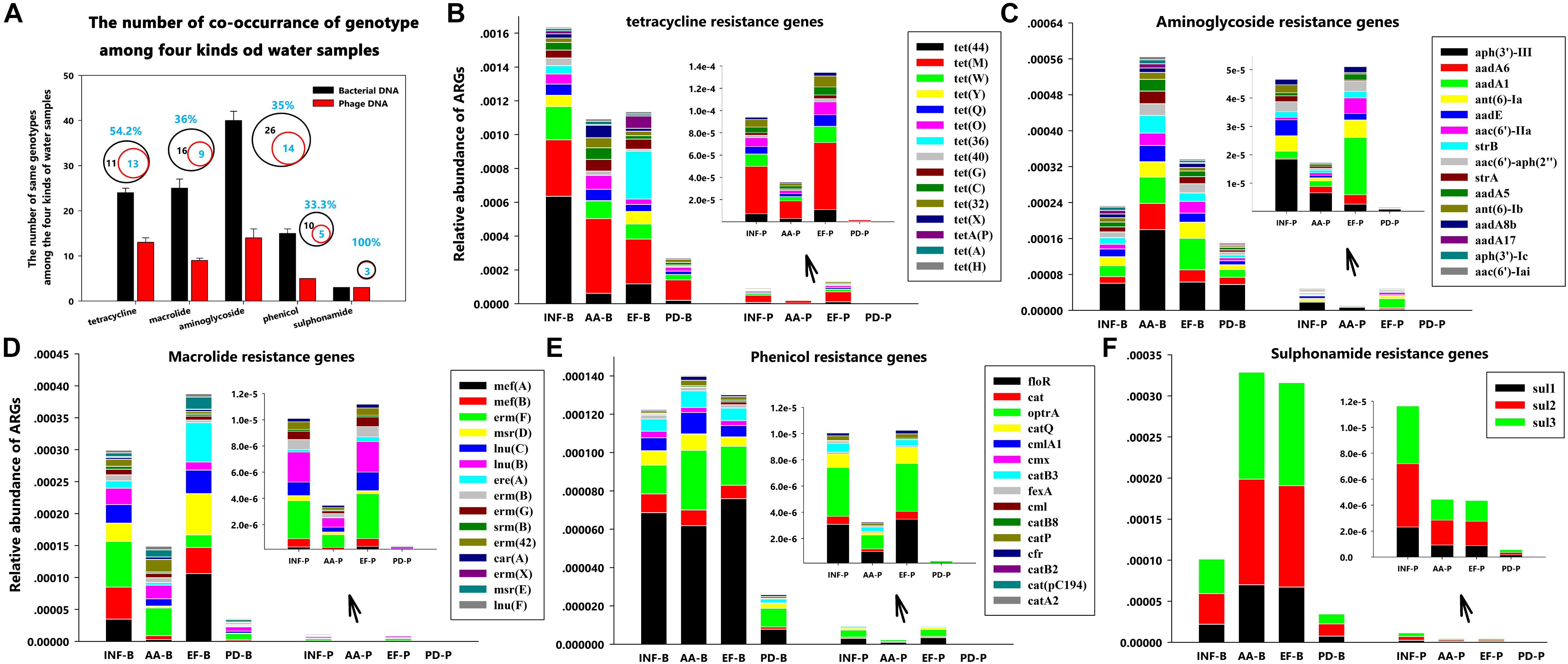
FIGURE 4. Distribution patterns of ARG types at different sampling locations. (A) The number of co-occurrence of genotypes for each antibiotic group among the four kinds of water samples; (B) Tetracycline resistance genes; (C) Aminoglycoside resistance genes; (D) Macrolide resistance genes; (E) Phenicol resistance genes; and (F) Sulphonamide resistance gene.
Moreover, since we found the same trend for ARG distribution in bacterial and phage DNA fractions, we conducted a Pearson correlation analysis between these two samples based on ARG co-occurrence. All ARG types had significant correlations (p < 0.05) except for macrolide resistance genes in effluent water (Table 2). These results not only directly demonstrate an intimate correlation between phages and bacteria but also suggest a prediction that the higher the abundance of ARGs in bacteria is, the more the ARGs in phage are. This also implies a greater threat to human health due to greater persistence characteristics of phages (Calero-Caceres and Muniesa, 2016). Thus, for example, mcr-1, though we now detected phage-associated mcr-1 in influent water at a relatively low abundance, this may spread and become problematic for therapeutic treatments in long run.
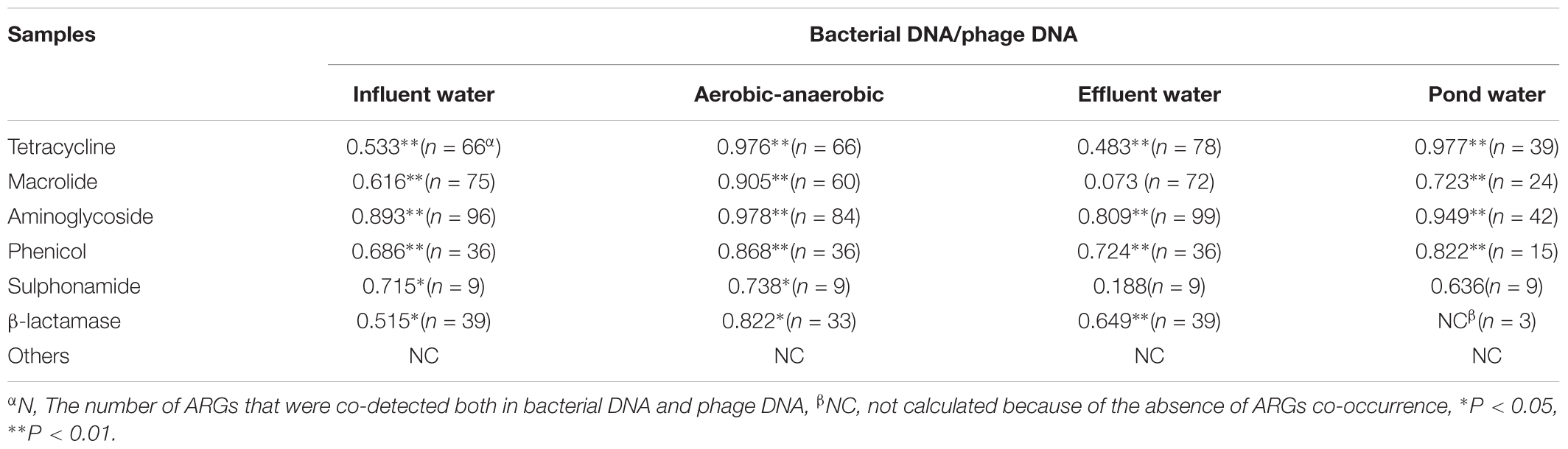
TABLE 2. Pearson correlations between ARG types in bacterial and phage DNA for each antibiotic class at four water-sampling sites.
Furthermore, during careful screening of independent genotypes for each antibiotic group, we abandoned the downstream genotype analysis for vancomycin, nitroimidazole, rifampicin, fosfomycin and colistin groups due to absence of ARGs in phage DNA. We found that, though tetracycline resistance genes had the highest abundance (Figure 3), the highest ARG diversity was the aminoglycoside group (40 different genotypes in bacterial DNA and 14 different genotypes in phage DNA among four kinds of water samples) (Figure 4A and Table 3). Among these antibiotic groups, tet(44), tet(M), tet(W), tet(Q), and tet(O) encoding resistance to tetracycline (Figure 4B), aph(3)-III, aadA6, and aadA1 encoding resistance to aminoglycoside (Figure 4C), mef(A), mef(B), and erm(F) encoding resistance to macrolide (Figure 4D), floR and optrA encoding resistance to phenicol (Figure 4E) and three sul resistance genes (Figure 4F) were the most frequently identified genotypes.
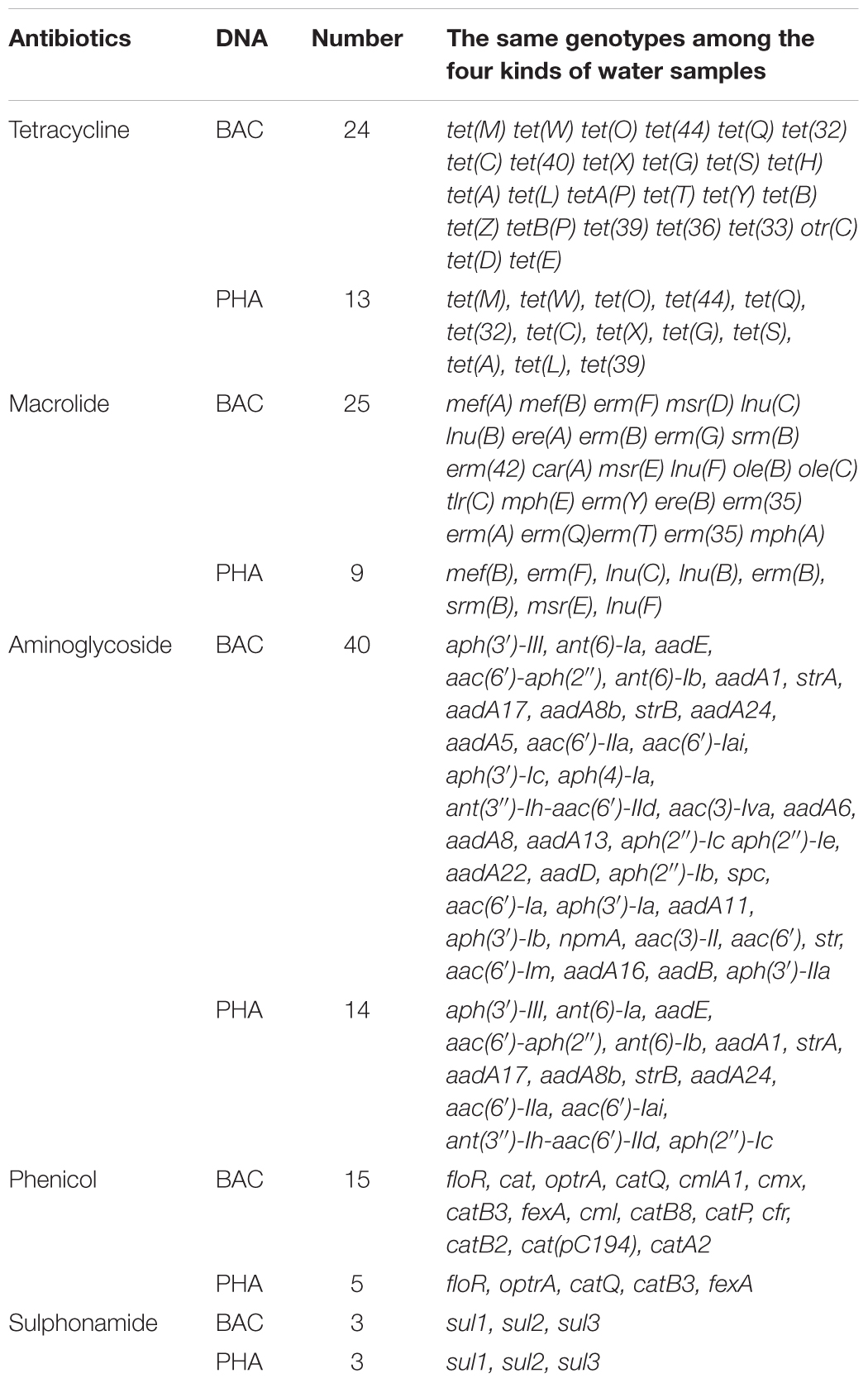
TABLE 3. The number of co-occurrence of genotypes among the four kinds of water samples.
With a careful analysis of these genes we found that all of these highly abundant ARGs have similar properties according to their mechanisms of action. Genes encoding major facilitator superfamily (MFS) [including floR, mef(A), mef(B), and erm(F)] (Coyne et al., 2011), genes encoding ATP-binding cassette (ABC) transporter family (including optrA gene that mediated antibiotic resistance through the ribosomal protection) (Sharkey et al., 2016; Wilson, 2016), and genes encoding ribosomal protection [including genes tet(44), tet(M), tet(W), tet(Q), and tet(O)] (Chopra and Roberts, 2001) were the most identified ARG groups. These results were similar to previous studies using raw sewage samples obtained from (i) Pittsburgh, Pennsylvania, United States, (ii) Barcelona, Spain; and (iii) Addis Ababa, Ethiopia (Cantalupo et al., 2011; Lekunberri et al., 2017a). Obviously, we clearly observed that their presence and distribution in phages and bacteria had geographic independence and the levels of these genes were also not fundamentally changed by wastewater treatment or pond natural degradation. Thus, whether the phages are more likely to carry these types of resistance genes in the contribution to antibiotic resistance in environment remains an open question.
On the other hand, according to a previous study that though phages rarely encode ARGs in the human-associated viromes, a relative high abundance of genes encoding ribosomal protection protein in tetracycline resistance genes group was detected in the human feces (Lekunberri et al., 2017a), which showed a similar trend to our results. Interestingly, in this study, we precisely identified these genes as the tet(44), tet(M), tet(W), tet(Q), and tet(O). As for the highly abundant and gene groups simultaneously occurring between an animal-related environment and a human-related environment, our alarming findings may indicates that phages serve as a bridge to spread the ARGs from animals to human.
In addition, the β-lactamase, quinolone and trimethoprim resistance genes, blaOXA-347, aac(6)-Ib-cr, and dfrA1, were the most abundant in their own groups, even though their abundance was less than that of the top five antibiotic resistance groups, but they should be of particular concern to address future research (Supplementary Figures S2–S4). The gene blaOXA groups were the most identified sub-groups D belonging to the β-lactamases (Subirats et al., 2016). The aac(6′)-Ib-cr is a variant gene, similar to the widespread aminoglycoside N-acetyltransferase aac(6′)-Ib (Vetting et al., 2008). Gene dfrA1 has previously been associated with classes 1 and 2 integrons and co-occurred with aadA1 and sul1 (van Essen-Zandbergen et al., 2009; Kehrenberg and Schwarz, 2011; Roth and Breaker, 2013). These findings suggest that popular genotypes and integrons may be an important connector for phages to spread antibiotic resistance and therefore may accelerate the evolution of these genes in the process of communication with bacteria.
Assessing the Contribution of Phages to Antibiotic Resistance Genes
We next assessed the contribution of phages to transfer antibiotic resistance in swine feedlot wastewater and used three important indices: ARG abundance in bacteria, ARG abundance phage and their ratio values (RA values). The RA value was used as an estimator of “spreading ability.” In this study, we consider that the spreading ability depends on the abundance as well as the mutual exposure and exchange probabilities or communication frequencies between phage and bacteria. Therefore, a simple measure of ARG abundance does not necessarily correlate with a greater probability of ARG transfer by transduction.
For RA value measurements, we chose the five most abundant genotypes from the top six most abundant antibiotic resistance groups. The RA values were drawn as three dimensional scatter plots to better represent the transduction abilities of different ARG types (Figure 5). Figure 5 shows that the ARGs were divided into three categories from the top to the bottom. However, the ARGs in wastewater treatment water samples showed a diversity feature for ARGs (Figures 5A–C) whereas they gather together in the pond water samples ARGs (Figure 5D). On the other hand, the SA values in pond water samples were lower than that in the wastewater treatment water samples. These results again indirectly demonstrate that animal feedlot wastewater treatment is an important ARG reservoir and is an important contributor to the spread of antibiotic resistance.
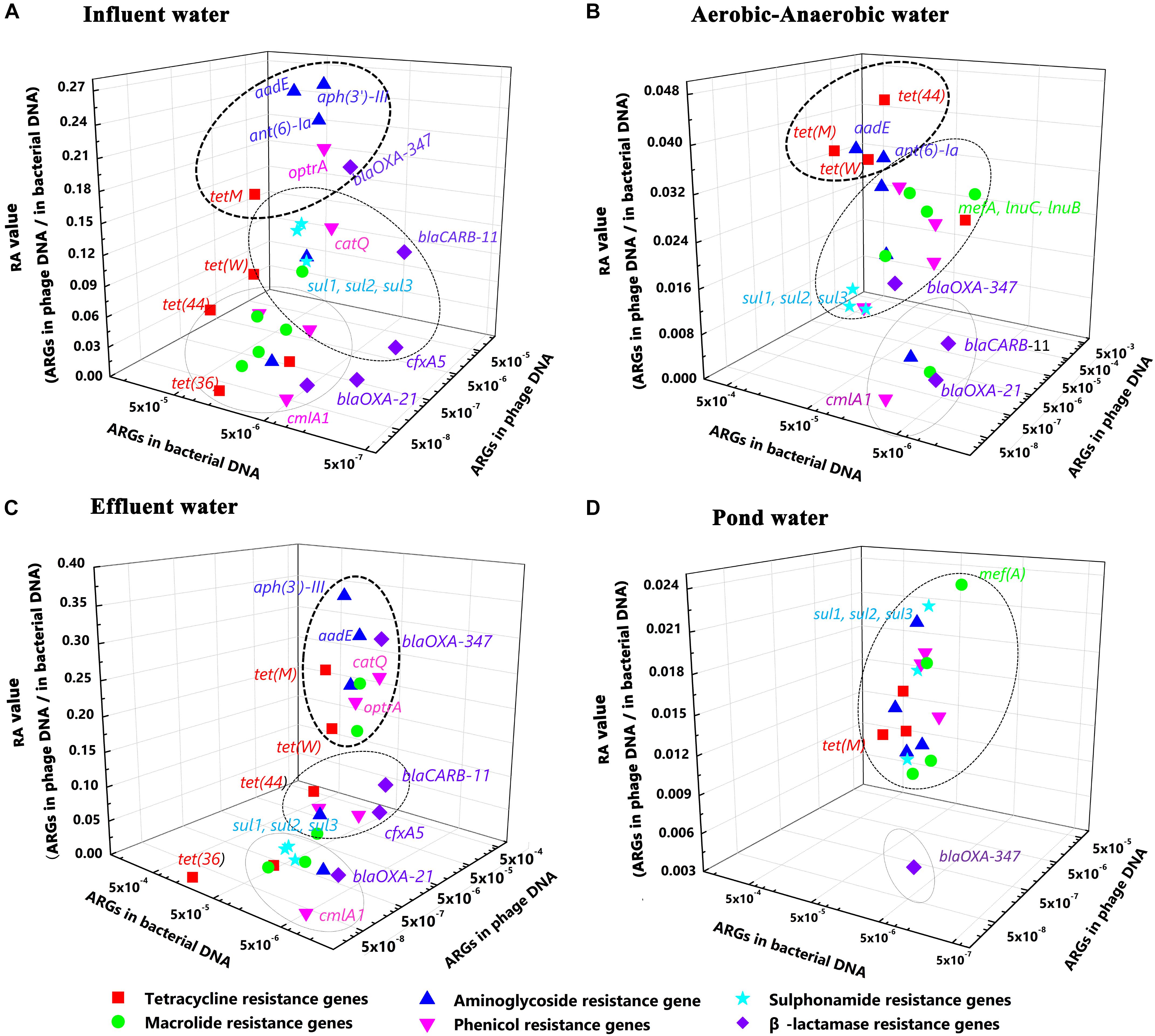
FIGURE 5. The five most abundant genotypes of the six most abundant antibiotic groups in three dimensional scatter plots. The X-axis represents the relative ARG abundance in bacterial DNA. The Y-axis represents relative ARG abundance in phage DNA. The Z-axis represents the Ratio, (A) Influent water; (B) Aerobic-Anaerobic water; (C) Effluent water; (D) Pond water.
Among the three water samples from the wastewater treatment system, we found that the intermediate water and effluent water representing aerobic and facultative digestion procedures did not significantly alter ARG distribution and composition characteristics (Figures 5A–C). Meanwhile, the aminoglycoside resistance genes aadE and aph(3′)-III, the tetracycline [tet(M)] and phenicol (optrA) resistance genes always possessed the highest RA values. At the same time, these genes were also the most abundant in bacterial and phage DNA. Obviously, these genes should be of high priority in future studies.
Surprisingly, though the relative abundance of β-lactamase genes was lower compared with the other antibiotic groups, some of the β-lactamase ARGs still possessed relatively high RA values, indicating a potential risk especially for blaOXA-347 that was detected with a very high RA value. On the other hand, considering combining the high abundance results of β-lactamase ARG in the animal-related environmental samples using qPCR method from other studies, future studies for assessing the contribution of phages to antibiotic resistance should also include screening for β-lactamase genes rather than only for aadE, aph(3′)-III, tet(M), and optrA.
Conclusion
We present the first study to use metagenomics to analyze the contribution of phages to antibiotic resistance in water samples from swine feedlot wastewater. Our data showed that aerobic and facultative digestions did not significantly decrease the abundance of ARGs in the phage DNA fractions. At the same time, ARGs from phage DNA could be an important reservoir of ARGs and play a major role in the spread of antibiotic resistance by transduction. Moreover, ARG distributions in phages and bacteria were independent of the geographical location and their trends were not fundamentally changed by wastewater treatment or natural degradation in pond water. Tetracycline resistance genes were always the most abundant ARG group and phages were more likely to harbor ABC transporter family and ribosomal protection genes. Surprisingly, the colistin resistance gene mcr-1 was detected in our samples, which may imply that phages are another driving force for the horizontal genes transfer in the environment and have a potential risk to humans. Besides, further analysis of the spreading ability of different ARGs, aadE, aph(3′)-III, tet(M), optrA, and blaOXA-347 should be given a high priority in future research.
Author Contributions
ZZ and YS designed the study and prepared the manuscript. MW, PL, XX, and JZ performed the experiments. WX analyzed the high-throughput sequencing data. All authors discussed the results. MW analyzed qPCR data and drafted the manuscript.
Funding
This work was supported by the Major Scientific Research Projects of Guangdong Provincial (2017KZDXM006), Graduate Student Overseas Study Program from South China Agricultural University (2018LHPY006), and National Natural Science Foundation of China (31772803).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02474/full#supplementary-material
Abbreviations
ARB, antibiotic resistant bacteria; ARGs, antibiotic resistance genes; HGT, horizontal gene transfer.
Footnotes
References
Andersson, D. I., and Hughes, D. (2010). Antibiotic resistance and its cost: is it possible to reverse resistance? Nat. Rev. Microbiol. 8, 260–271. doi: 10.1038/nrmicro2319
Bengtsson-Palme, J., Hartmann, M., Eriksson, K. M., Pal, C., Thorell, K., Larsson, D. G., et al. (2015). METAXA2: improved identification and taxonomic classification of small and large subunit rRNA in metagenomic data. Mol. Ecol. Resour. 15, 1403–1414. doi: 10.1111/1755-0998.12399
Boucher, H. W., Talbot, G. H., Bradley, J. S., Edwards, J. E., Gilbert, D., Rice, L. B., et al. (2009). Bad bugs, no drugs: no ESKAPE! an update from the infectious diseases Society of America. Clin. Infect. Dis. 48, 1–12. doi: 10.1086/595011
Calero-Caceres, W., Mendez, J., Martin-Diaz, J., and Muniesa, M. (2017). The occurrence of antibiotic resistance genes in a Mediterranean river and their persistence in the riverbed sediment. Environ. Pollut. 223, 384–394. doi: 10.1016/j.envpol.2017.01.035
Calero-Caceres, W., and Muniesa, M. (2016). Persistence of naturally occurring antibiotic resistance genes in the bacteria and bacteriophage fractions of wastewater. Water Res. 95, 11–18. doi: 10.1016/j.watres.2016.03.006
Cantalupo, P. G., Calgua, B., Zhao, G., Hundesa, A., Wier, A. D., Katz, J. P., et al. (2011). Raw sewage harbors diverse viral populations. mBio 2:e180-11. doi: 10.1128/mBio.00180-11
Chopra, I., and Roberts, M. (2001). Tetracycline antibiotics: mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 65, 232–260. doi: 10.1128/MMBR.65.2.232-260.2001
Colomer-Lluch, M., Calero-Caceres, W., Jebri, S., Hmaied, F., Muniesa, M., and Jofre, J. (2014a). Antibiotic resistance genes in bacterial and bacteriophage fractions of Tunisian and Spanish wastewaters as markers to compare the antibiotic resistance patterns in each population. Environ. Int. 73, 167–175. doi: 10.1016/j.envint.2014.07.003
Colomer-Lluch, M., Jofre, J., and Muniesa, M. (2014b). Quinolone resistance genes (qnrA and qnrS) in bacteriophage particles from wastewater samples and the effect of inducing agents on packaged antibiotic resistance genes. J. Antimicrob. Chemother. 69, 1265–1274. doi: 10.1093/jac/dkt528
Coyne, S., Courvalin, P., and Perichon, B. (2011). Efflux-mediated antibiotic resistance in Acinetobacter spp. Antimicrob. Agents Chemother. 55, 947–953. doi: 10.1128/AAC.01388-10
Gillings, M. R., Gaze, W. H., Pruden, A., Smalla, K., Tiedje, J. M., and Zhu, Y. G. (2015). Using the class 1 integron-integrase gene as a proxy for anthropogenic pollution. ISME J. 9, 1269–1279. doi: 10.1038/ismej.2014.226
Harmer, C. J., Moran, R. A., and Hall, R. M. (2014). Movement of IS26-associated antibiotic resistance genes occurs via a translocatable unit that includes a single IS26 and preferentially inserts adjacent to another IS26. mBio 5:e01801-14. doi: 10.1128/mBio.01801-14
Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., et al. (2017). CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–D573. doi: 10.1093/nar/gkw1004
Kehrenberg, C., and Schwarz, S. (2011). Trimethoprim resistance in a porcine Pasteurella aerogenes isolate is based on a dfrA1 gene cassette located in a partially truncated class 2 integron. J. Antimicrob. Chemother. 66, 450–452. doi: 10.1093/jac/dkq461
Lekunberri, I., Subirats, J., Borrego, C. M., and Balcazar, J. L. (2017a). Exploring the contribution of bacteriophages to antibiotic resistance. Environ. Pollut. 220, 981–984. doi: 10.1016/j.envpol.2016.11.059
Lekunberri, I., Villagrasa, M., Balcázar, J. L., and Borrego, C. M. (2017b). Contribution of bacteriophage and plasmid DNA to the mobilization of antibiotic resistance genes in a river receiving treated wastewater discharges. Sci. Total Environ. 60, 206–209. doi: 10.1016/j.scitotenv.2017.05.174
Liu, Y.-Y., Wang, Y., Walsh, T. R., Yi, L.-X., Zhang, R., Spencer, J., et al. (2016). Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect. Dis. 16, 161–168. doi: 10.1016/S1473-3099(15)00424-7
McEachran, A. D., Blackwell, B. R., Hanson, J. D., Wooten, K. J., Mayer, G. D., Cox, S. B., et al. (2015). Antibiotics, bacteria, and antibiotic resistance genes: aerial transport from cattle feed yards via particulate matter. Environ. Health Perspect. 123, 337–343. doi: 10.1289/ehp.1408555
Pawluk, A. (2017). Tiny answers to big questions. Cell 170, 215–217. doi: 10.1016/j.cell.2017.06.033
Pitout, J. D., Nordmann, P., and Poirel, L. (2015). Carbapenemase-producing Klebsiella pneumoniae, a key pathogen set for global nosocomial dominance. Antimicrob. Agents Chemother. 59, 5873–5884. doi: 10.1128/AAC.01019-15
Rodriguez-Mozaz, S., Chamorro, S., Marti, E., Huerta, B., Gros, M., Sanchez-Melsio, A., et al. (2015). Occurrence of antibiotics and antibiotic resistance genes in hospital and urban wastewaters and their impact on the receiving river. Water Res. 69, 234–242. doi: 10.1016/j.watres.2014.11.021
Ross, J., and Topp, E. (2015). Abundance of antibiotic resistance genes in bacteriophage following soil fertilization with dairy manure or municipal biosolids, and evidence for potential transduction. Appl. Environ. Microbiol. 81, 7905–7913. doi: 10.1128/AEM.02363-15
Roth, A., and Breaker, R. R. (2013). Integron attI1 sites, not riboswitches, associate with antibiotic resistance genes. Cell 153, 1417–1418. doi: 10.1016/j.cell.2013.05.043
Roux, S., Krupovic, M., Debroas, D., Forterre, P., and Enault, F. (2013). Assessment of viral community functional potential from viral metagenomes may be hampered by contamination with cellular sequences. Open Biol. 3:130160. doi: 10.1098/rsob.130160
Sharkey, L. K., Edwards, T. A., and O’Neill, A. J. (2016). ABC-F proteins mediate antibiotic resistance through ribosomal protection. mBio 7:e01975-15. doi: 10.1128/mBio.01975-15
Subirats, J., Sanchez-Melsio, A., Borrego, C. M., Balcazar, J. L., and Simonet, P. (2016). Metagenomic analysis reveals that bacteriophages are reservoirs of antibiotic resistance genes. Int. J. Antimicrob. Agents 48, 163–167. doi: 10.1016/j.ijantimicag.2016.04.028
Sun, J., Yang, R. S., Zhang, Q., Feng, Y., Fang, L. X., Xia, J., et al. (2016). Co-transfer of blaNDM-5 and mcr-1 by an IncX3-X4 hybrid plasmid in Escherichia coli. Nat Microbiol. 1:16176. doi: 10.1038/nmicrobiol.2016.176
Thurber, R. V., Haynes, M., Breitbart, M., Wegley, L., and Rohwer, F. (2009). Laboratory procedures to generate viral metagenomes. Nat. Protoc. 4, 470–483. doi: 10.1038/nprot.2009.10
van Essen-Zandbergen, A., Smith, H., Veldman, K., and Mevius, D. (2009). In vivo transfer of an incFIB plasmid harbouring a class 1 integron with gene cassettes dfrA1-aadA1. Vet. Microbiol. 137, 402–407. doi: 10.1016/j.vetmic.2009.02.004
Vetting, M. W., Park, C. H., Hegde, S. S., Jacoby, G. A., Hooper, D. C., and Blanchard, J. S. (2008). Mechanistic and structural analysis of aminoglycoside N-acetyltransferase AAC(6′)-Ib and its bifunctional, fluoroquinolone-active AAC(6′)-Ib-cr variant. Biochemistry 47, 9825–9835. doi: 10.1021/bi800664x
Walsh, T. R., Weeks, J., Livermore, D. M., and Toleman, M. A. (2011). Dissemination of NDM-1 positive bacteria in the New Delhi environment and its implications for human health: an environmental point prevalence study. Lancet Infect. Dis. 11, 355–362. doi: 10.1016/S1473-3099(11)70059-7
Wang, F. H., Qiao, M., Chen, Z., Su, J. Q., and Zhu, Y. G. (2015). Antibiotic resistance genes in manure-amended soil and vegetables at harvest. J. Hazard. Mater. 299, 215–221. doi: 10.1016/j.jhazmat.2015.05.028
Wang, M., Liu, P., Zhou, Q., Tao, W., Sun, Y., and Zeng, Z. (2018). Estimating the contribution of bacteriophage to the dissemination of antibiotic resistance genes in pig feces. Environ. Pollut. 238, 291–298. doi: 10.1016/j.envpol.2018.03.024
Wang, M., Sun, J., Zhong, W., Xiong, W., Zeng, Z., and Sun, Y. (2016). Presence and distribution of Macrolides-Lincosamide-Streptogramin resistance genes and potential indicator ARGs in the university ponds in Guangzhou, China. Environ. Sci. Pollut. Res. Int. 23, 22937–22946. doi: 10.1007/s11356-016-7521-4
Wilson, D. N. (2016). The ABC of ribosome-related antibiotic resistance. mBio 7:e598-16. doi: 10.1128/mBio.00598-16
Keywords: bacteriophages, metagenomics, antimicrobial resistance genes, swine feedlot, wastewater
Citation: Wang M, Xiong W, Liu P, Xie X, Zeng J, Sun Y and Zeng Z (2018) Metagenomic Insights Into the Contribution of Phages to Antibiotic Resistance in Water Samples Related to Swine Feedlot Wastewater Treatment. Front. Microbiol. 9:2474. doi: 10.3389/fmicb.2018.02474
Received: 15 March 2018; Accepted: 27 September 2018;
Published: 16 October 2018.
Edited by:
Fiona Walsh, Maynooth University, IrelandReviewed by:
Jose Luis Balcazar, Catalan Institute for Water Research, SpainFriederike Hilbert, Veterinärmedizinische Universität Wien, Austria
Copyright © 2018 Wang, Xiong, Liu, Xie, Zeng, Sun and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongxue Sun, c3VueXhAc2NhdS5lZHUuY24= Zhenling Zeng, emx6ZW5nQHNjYXUuZWR1LmNu