 Mark Burton
Mark Burton Chidinma Abanobi
Chidinma Abanobi Madeline E. Rasche
Madeline E. Rasche- Department of Chemistry and Biochemistry, Center for Applied Biotechnology Studies, California State University, Fullerton, Fullerton, CA, United States
Methane-producing archaea and methylotrophic bacteria use tetrahydromethanopterin (H4MPT) and/or tetrahydrofolate (H4F) as coenzymes in one-carbon (C1) transfer pathways. The α-proteobacterium Methylobacterium extorquens AM1 contains a dihydromethanopterin reductase (DmrA) and two annotated dihydrofolate reductases (DfrA and DfrB). DmrA has been shown to catalyze the final step of H4MPT biosynthesis; however, the functions of DfrA and DfrB have not been examined biochemically. Moreover, sequence alignment (BLAST) searches have recognized scores of proteins that share up to 99% identity with DmrA but are annotated as diacylglycerol kinases (DAGK). In this work, we used bioinformatics and enzyme assays to provide insight into the phylogeny and substrate specificity of selected Dfr and DmrA homologs. In a phylogenetic tree, DmrA and homologs annotated as DAGKs grouped together in one clade. Purified histidine-tagged versions of the annotated DAGKs from Hyphomicrobium nitrativorans and M. nodulans (respectively, sharing 69 and 84% identity with DmrA) showed only low activity in phosphorylating 1,2-dihexanoyl-sn-glycerol when compared with a commercial DAGK from Escherichia coli. However, the annotated DAGKs successfully reduced a dihydromethanopterin analog (dihydrosarcinapterin, H2SPT) with kinetic values similar to those determined for M. extorquens AM1 DmrA. DfrA and DfrB showed little or no ability to reduce H2SPT under the conditions studied; however, both catalyzed the NADPH-dependent reduction of dihydrofolate. These results provide the first evidence that DfrA and DfrB function as authentic dihydrofolate reductases, while DAGKs with greater than 69% identity to DmrA may be misannotated and are likely to function in H4MPT biosynthesis.
Introduction
In the facultative methylotroph Methylobacterium extorquens AM1, growth on single-carbon (C1) substrates involves the use of both tetrahydromethanopterin (H4MPT) and tetrahydrofolate (H4F) (Chistoserdova et al., 1998). H4MPT was initially thought to be exclusive to methanogenic archaea and sulfur-dependent hyperthermophilic archaea (Achenbach-Richter et al., 1987; DiMarco et al., 1990; Gorris et al., 1991). However, the discovery of H4MPT-linked C1 transfer enzymes in the Bacteria domain has provided evidence for the use of H4MPT beyond a methane-generating pathway (Chistoserdova et al., 1998; Vorholt et al., 1999; Chistoserdova et al., 2004; Chistoserdova, 2016). In the aerobic α-proteobacterium M. extorquens AM1, methylotrophy involves the use of dephospho-H4MPT in a series of oxidative steps to catabolize reduced C1 compounds to CO2 (Chistoserdova et al., 1998); this is in contrast to the reduction of CO2 to methane in the anaerobic metabolism of methanogenic archaea (DiMarco et al., 1990). The use of methylotrophs in biotechnology has gained interest because of its application to the microbial production of useful industrial chemicals starting with C1 compounds as an alternative to glucose and other conventional sugar or acid substrates (Schrader et al., 2009; Ochsner et al., 2015).
In the pathways of H4MPT and H4F biosynthesis, the last step requires the activity of dihydromethanopterin reductase (Dmr) or dihydrofolate reductase (Dfr). M. extorquens AM1 contains one dihydromethanopterin reductase (DmrA) and two putative dihydrofolate reductases, DfrA and DfrB, that, respectively, share 26% identity (41% similarity) and 34% identity (53% similarity) with DmrA. The dmrA gene was first discovered using transposon mutagenesis (Marx et al., 2003) and later deletion mutagenesis which produced a phenotype similar to that of mutants with deletions in H4MPT biosynthesis genes (Marx et al., 2003; Rasche et al., 2004; Chistoserdova et al., 2005). Homology of DmrA to dihydrofolate reductases led to the proposal that DmrA evolved from an ancestral dihydrofolate reductase following horizontal transfer of H4MPT biosynthesis genes from anaerobic archaea to aerobic bacteria (Marx et al., 2003). A driving force for the evolution of DmrA from dihydrofolate reductase may have been the lack of archaea-specific electron donors such as Factor-420 in the recipient bacteria. Absence of a corresponding archaeal electron donor could render the dihydromethanopterin reductase useless in bacteria, providing selective pressure to modify the substrate specificity of an NADPH-dependent dihydrofolate reductase to reduce dihydromethanopterin (Marx et al., 2003; Caccamo et al., 2004).
DmrA has been shown to catalyze the final step of H4MPT biosynthesis in M. extorquens AM1 (Caccamo et al., 2004) (Figure 1A); however, DmrA shares no sequence homology with the FMN-containing dihydromethanopterin reductase discovered in archaea (DmrX) or related archaeal-like flavoproteins (AfpA and DmrB) from β-proteobacteria (Kalyuzhnaya et al., 2005; McNamara et al., 2014; Wang et al., 2014). The FMN prosthetic groups of DmrX and AfpA/DmrB appear to be critical for electron transfer (McNamara et al., 2014; Wang et al., 2014) and may contribute to the absence of homology with the NADPH-dependent DmrA, which lacks flavin cofactors.
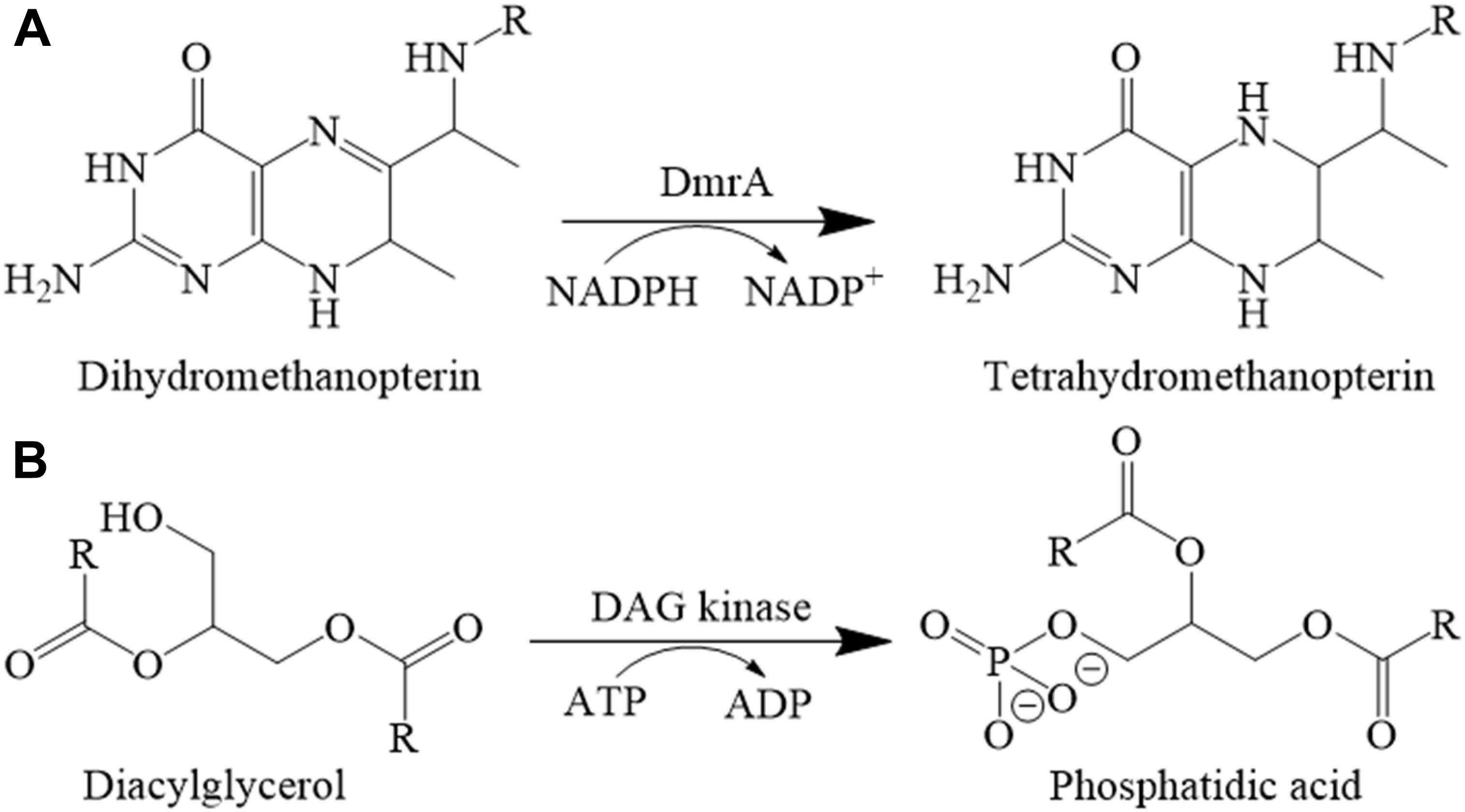
FIGURE 1. Reactions catalyzed by (A) DmrA and (B) DAGK.
In M. extorquens, the dihydrofolate reductase homologs DfrA and DfrB have not been examined biochemically. When originally discovered, a role for DfrA in the synthesis of H4F was proposed based on its 50% sequence identity to dihydrofolate reductase from Lactobacillus casei (Marx et al., 2003) and the genomic location of dfrA near the H4F synthesis genes folC and folE in M. extorquens (Chistoserdova et al., 2003). Furthermore, the dfrA gene is located directly downstream of a gene encoding a putative H4F-dependent thymidylate synthase (Marx et al., 2003).
Little is known about the function of DfrB. When we conducted a BLAST search using M. extorquens DfrB as the sequence alignment query, only a few homologs with high sequence identity could be identified. In a phylogenetic tree, these clustered together as a single group (Figure 2). Among the more distantly related homologs, one clade included DfrA and numerous annotated dihydrofolate reductases (30–48% identical to DfrB). The last clade consisted of a few known DmrA sequences (34–42% identical to DfrB) and a large number of proteins annotated as diacylglycerol kinases (DAGKs) but sharing 60–99% identity with DmrA from M. extorquens. This is curious because DAGKs function in phosphorylation reactions rather than in the reduction of pterins, as shown in Figure 1B. To provide insight into possible roles of the DmrA, DfrA, and DfrB homologs, we have used bioinformatics to assess phylogenetic relationships among the homologs and enzyme assays to probe biochemical function.
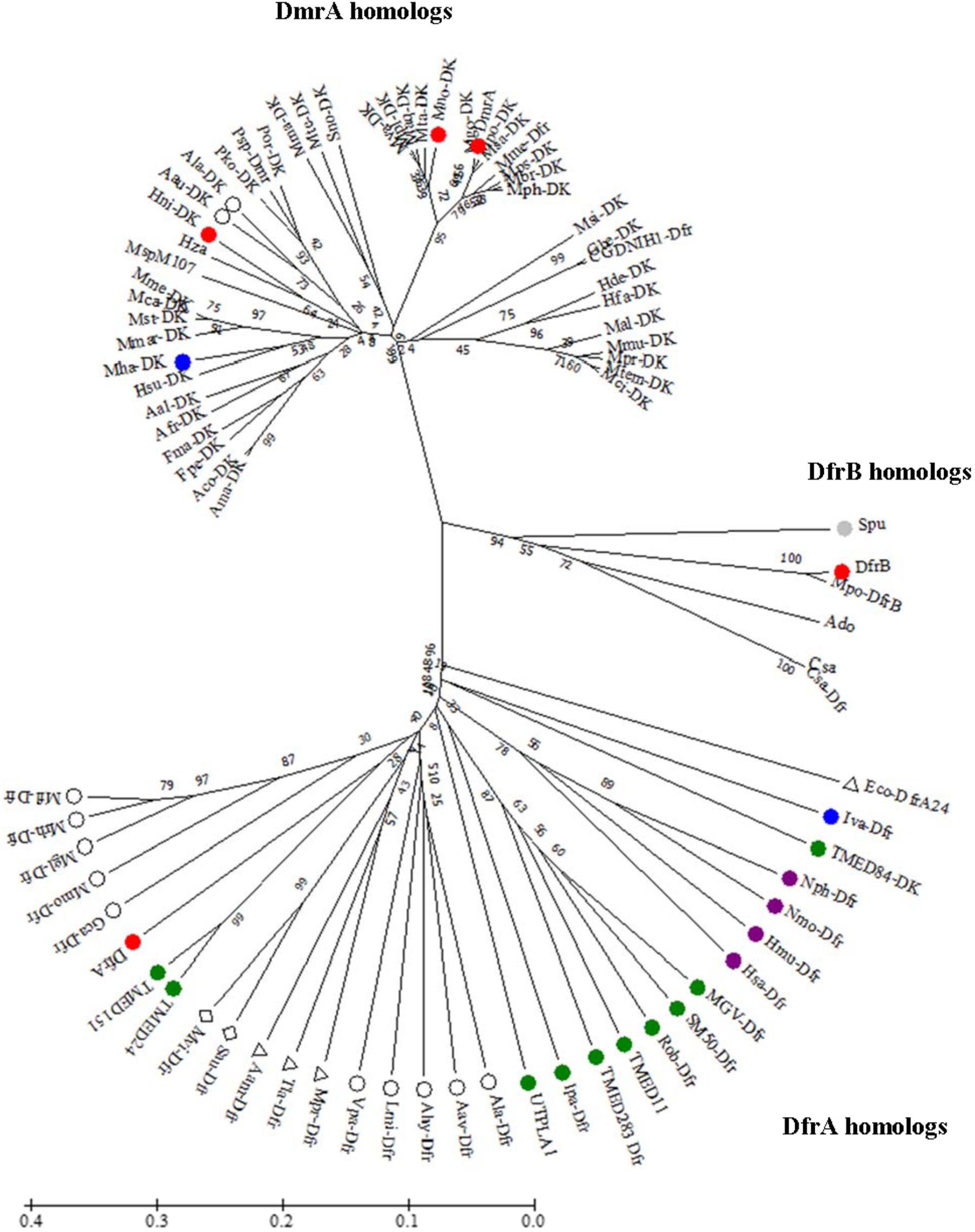
FIGURE 2. Unrooted phylogenetic tree showing position of DfrB (AY093433) relative to orthologs including DfrA (AY093432) and DmrA (AY093431). Sequences were aligned using EMBL-EBI OMEGA bioinformatics tools with default parameters. Phylogenetic analysis was performed by using Maximum Likelihood method with 10,000 bootstrap replicates (Felsenstein, 1985) within MEGA7 software (Kumar et al., 2016). Abbreviation DAGK (DK). Color designations are Firmicute (blue), Actinobacteria (gray), Planctomycetes (green), Euryarchaeota (purple), β-proteobacteria (open circle), γ-proteobacteria (triangle), and δ-proteobacteria (diamond). DfrA, DfrB, DmrA, Mno-DK, and Hni-DK (red). The bar denotes 1 estimated substitution per 100 amino acid positions.
Materials and Methods
Bioinformatics
The DfrB nucleotide sequence (GenBank no. AY093433) (Marx et al., 2003) was used as the query in a non-redundant database BLASTx (translated nucleotide to protein) in the National Center for Biotechnology Information Database (NCBI) using default algorithm patterns with the exception of limiting to 5,000 maximum target sequences (Altschul et al., 1997). Similar results were obtained using the DfrB protein sequence in BLASTp. Sequences were aligned using the Clustal Omega program (Sievers et al., 2011; McWilliam et al., 2013; Li et al., 2015). Aligned sequences were analyzed for phylogenetic relationships and unrooted tree construction (Kumar et al., 2016). The String v10.5 database was used to assess the gene/protein-protein relationships of the neighboring genes to dfrA and dmrA (Snel et al., 2000; von Mering et al., 2003, 2005, 2007; Jensen et al., 2009; Szklarczyk et al., 2011, 2015, 2017; Franceschini et al., 2013, 2016) and BPROM operon predictive method was applied to these genes (Li, 2011). Neighboring genes without any clear annotations in subsequent sequence alignment searches were analyzed using the Protein Homology/analogy Recognition Engine v2.0 database (Kelley et al., 2015).
Chemicals
Luria-Bertani/Miller broth (LB) (Becton, Dickinson and Company, Franklin Lakes, NJ) was purchased from Thermo Fisher Scientific (Waltham, MA, United States). Tris(hydroxymethyl)aminomethane (Tris), 1,4-piperazinediethanesulfonic acid (PIPES), dibasic sodium phosphate (Na2HPO4), monobasic potassium phosphate (KH2PO4), D-(+)-glucose, magnesium sulfate (MgSO4), β-mercaptoethanol (2-ME), kanamycin sulfate, imidazole, sodium ascorbate, magnesium acetate, and ammonium chloride (NH4Cl) were also from Thermo Fisher. Isopropyl-β-D-thiogalactopyranoside (IPTG) was from Ubiquitin-Proteasome Biotechnologies (UBP-Bio, Aurora, CO). N-[Tris(hydroxyl)methyl]-2-aminoethanesulfonic acid (TES), 3-(morpholino)propanesulfonic acid (MOPS), sodium acetate, dihydrofolate (H2F), NADH, NADPH, ethylenediaminetetraacetic acid (EDTA), ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), phosphoenolpyruvate (PEP), adenosine 5′-triphosphate (ATP), lithium chloride (LiCl), and deoxyribonuclease I (DNase I) bovine were from Sigma-Aldrich (St. Louis, MO, United States). 1,2-dihexanoyl-sn-glycerol was from Cayman Chemical (Ann Arbor, MI, United States). Gasses were from Airgas (Placentia, CA, United States). Unless otherwise noted, all other chemicals were purchased from Thermo Fisher Scientific.
Gene Synthesis and Transformation
The dfrA and dfrB genes were subcloned with an N-terminal six-histidine (H6) tag into the NdeI and BamHI sites of the pET-41a(+) expression vector (Novagen, Madison, WI, United States) by GenScript (Piscataway, NJ, United States). For production of H6-DfrA or H6-DfrB, the corresponding plasmid was transformed into chemically competent BL21(DE3) cells (Stratagene, La Jolla, CA, United States). Similarly, cell lines were created to produce DmrA (BL21 + pET41a: H6-DmrA or pET41a:DmrA-H4) and the annotated DAGKs from Hyphomicrobium nitrativorans and M. nodulans (BL21 + pET41a: Hni-DAGK-H6) or BL21 + pET41a:Mno-DAGK-H6). The estimated molecular masses of the corresponding histidine-tagged proteins are 19.3 kDa for H6-DfrA, 19.4 kDa for H6-DfrB, 15.8 kDa for DmrA-H4, and 16.2 kDa for both Hni-DAGK-H6 and Mno-DAGK-H6.
Cell Growth and Gene Induction
For the production of H6-DmrA and DmrA-H4, an overnight culture of BL21 cells with pET41a: H6-DmrA or pET41a: DmrA-H4 was used to inoculate 1 liter of a modified M9 minimal medium (Sambrook and Russell, 2001) containing 48 mM Na2HPO4, 22 mM KH2PO4, 19 mM NH4Cl, and 17 mM NaCl [pH 7.4], supplemented with 0.4% (w/v) D-(+)-glucose, 2 mM MgSO4, and kanamycin (50 μg/ml). Cells were grown at 37°C with shaking (180 rpm). When the optical density at 600 nm reached approximately 0.4, the cells were transferred to another platform shaker previously equilibrated to 15°C (180 rpm) for approximately 45 min. When the optical density at 600 nm reached 0.55–0.60, gene expression was induced with IPTG to 1 mM. The cells were grown at 15°C for 16 h, and then the cell suspension was centrifuged (5,000 × g, 15 min, 4°C). The cell pellet was washed in 30 ml of 50 mM TES [pH 8.0], collected by centrifugation (7,000 ×g, 15 min, 4°C), and stored at -20°C.
To produce tagged DfrA, DfrB, M. nodulans DAGK, and H. nitrativorans DAGK proteins (H6-DfrA, H6-DfrB, Mno-DAGK-H6, and Hni-DAGK-H6), overnight cultures of BL21 cells with the appropriate plasmid were used to inoculate 1 L of LB medium (Bertani, 1951) containing kanamycin (50 μg/ml). Cells were grown at 37°C with shaking (180 rpm). For H6-DfrA, H6-DfrB, and Hni-DAGK-H6, when the optical density at 600 nm reached approximately 0.6, gene expression was induced with IPTG to 1 mM. The culture was transferred to a platform shaker at 20°C, and cells were grown for 16 h with shaking (180 rpm). For Mno-DAGK-H6, after induction, the culture was grown at 30°C for 6 h with shaking (180 rpm). All cells were collected by centrifugation (5,000 ×g, 15 min, 4°C), washed with 30 ml of 50 mM TES, pH 8, centrifuged (7,000 ×g, 15 min, 4°C), and stored at -20°C.
Cell Lysis and Protein Purification
All cells were lysed at 20,000 lb/in2 by one pass through a cold French Press cell (Thermo Fisher Corporation, Waltham, MA, United States) at 4°C in 50 mM Tris, 200 mM NaCl, 20 mM imidazole, 15 mM 2-ME [pH 8.0], and 2 μL of DNase I. Lysed cells were centrifuged for 1 h at 4°C (32,000 ×g). The supernatant (cell-free extract, CFE) was removed and centrifuged for an additional 15 min. The CFE was incubated with 1-part Nickel Nitrilotriacetic acid resin (NiNTA, Qiagen, Germantown, MD, United States) to 4 parts CFE for 2 h with DmrA-H4 or 1 h with H6-DfrA, H6-DfrB, Hni-DAGK-H6, and Mno-DAGK-H6. The CFE-NiNTA slurry was poured into a 10-ml polypropylene column (Bio-Rad Laboratories, Inc., Hercules, CA, United States) and washed three times with 5 ml of 50 mM Tris pH 8, 200 mM NaCl, 30 mM imidazole, 15 mM 2-mercaptoethanol (2-ME). Elution buffers consisted of 50 mM Tris pH 8, 200 mM NaCl, 15 mM 2-ME with 100 mM imidazole or 250 mM imidazole. Buffers were added to the column at room temperature (approximately 23°C) to minimize fluctuations in pH within the column.
Protein concentrations were determined by the Bradford procedure (Bradford, 1976) using bovine serum albumin (Pierce Biotechnology, Rockford, IL, United States) as the standard. The efficiency of protein purification and protein purity were analyzed using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) stained with Coomassie brilliant blue G-250 (Bio-Rad, Hercules, CA, United States) (Garfin, 1990). All histidine-tagged proteins were shown to be greater than 95% pure.
Preparation of Dihydrosarcinapterin (H2SPT) From Methanogen Cell Extract
The H4MPT analog tetrahydrosarcinapterin (H4SPT) was obtained from the methanogen Methanosarcina thermophila TM-1 grown on acetate (Scott and Rasche, 2002) and purified by a previously developed method (Caccamo et al., 2004). Approximately 5 g of cells were removed from liquid nitrogen and sealed in a 37-ml amber anaerobic vial. Cells were purged with hydrogen gas for 10 min and transferred to an anaerobic chamber (Coy Products, Inc., Grass Lake, MI, United States) in 97% nitrogen and 3% hydrogen (Praxair, Inc., Danbury, CT, United States). An anoxic solution (10 ml) of 30 mM sodium acetate pH 4.3 and 200 mM 2-ME was added to re-suspend the cells. The vial containing the cells was sealed with a rubber stopper and aluminum crimp seal, boiled for 15 min (Precision Scientific, Chicago, MI, United States), allowed to cool, and then transferred into the anaerobic chamber. The boiled cell lysate was transferred in aliquots (1 mL) into 2-ml microcentrifuge tubes and centrifuged for 20 min at 13,000 ×g (Eppendorf Minispin plus, Hauppauge, NY, United States). During centrifugation, a 2-ml column of Sephadex A-25 diethylaminoethane (DEAE) was prepared in a 10-ml polypropylene column (Bio-Rad Laboratories, Inc., Hercules, CA, United States) and equilibrated with two column volumes of 50 mM MOPS, 1 M NaCl, 150 mM 2-ME [pH 6.8], followed by two column volumes of 50 mM MOPS, 150 mM 2-ME [pH 6.8]. The column was wrapped in aluminum foil to limit light exposure within the column. An aliquot of boiled CFE (200 μL) was removed for use in methylene-H4MPT reductase (MtdB) assays. The remaining boiled CFE was mixed with one volume of 50 mM MOPS, pH 6.8, 150 mM 2-ME. This mixture was added to the DEAE column. Fractions were collected from a step gradient of 50 mM MOPS, pH 6.8, 150 mM 2-ME with 0–1.0 M NaCl. Two 2-ml aliquots per NaCl step (0, 200, 400, 500, 600, and 1,000 mM) were collected. The highest concentration of H4SPT was found in the first 500 mM fraction of NaCl, as determined by the MtdB enzymatic assay (Rasche et al., 2004). Fractions were sealed in 10-ml anaerobic vials wrapped in foil and stored at -80°C.
To partially oxidize the H4SPT to H2SPT, the first 500 mM NaCl DEAE fraction or second 400 mM NaCl DEAE fraction was exposed to air for 100 s, by gently swirling for 80 s (approximately 2 swirls/s) at 30-s intervals. Oxidation of H4SPT was followed monitoring the increase in absorbance at 280 and 342 nm ( 342[Methanopterin] = 7.4 mM-1cm-1) (van Beelen et al., 1984) and the decrease in absorbance at 302 nm (302[H4MPT] = 15.2 mM-1cm-1) (Escalante-Semerena et al., 1984). Enzymatic assays were used to monitor levels of H4SPT (Rasche et al., 2004) and H2SPT (Caccamo et al., 2004) in addition to the wavelengths mentioned above. The oxidized 500 mM-1 fraction was transferred into an anaerobic chamber and aliquoted (100 μL) into 0.5-ml microcentrifuge tubes and sealed in a 10-ml anaerobic vial wrapped in aluminum foil. Aliquots were stored at -80°C.
342[Methanopterin] = 7.4 mM-1cm-1) (van Beelen et al., 1984) and the decrease in absorbance at 302 nm (302[H4MPT] = 15.2 mM-1cm-1) (Escalante-Semerena et al., 1984). Enzymatic assays were used to monitor levels of H4SPT (Rasche et al., 2004) and H2SPT (Caccamo et al., 2004) in addition to the wavelengths mentioned above. The oxidized 500 mM-1 fraction was transferred into an anaerobic chamber and aliquoted (100 μL) into 0.5-ml microcentrifuge tubes and sealed in a 10-ml anaerobic vial wrapped in aluminum foil. Aliquots were stored at -80°C.
Dfr Assay
Reactions were prepared in an anaerobic chamber (97% N2 and 3% H2) in sealed 2-ml quartz masked cuvettes (Starna, Atascadero, CA, United States). The initial reaction mixtures (1 ml) consisted of about 3.6 μg of protein in an anoxic solution of 500 mM Tris (pH 7.5), 20 mM sodium ascorbate, 15 mM 2-ME, 50 μM H2F, and 0.1 mM NADPH. The reaction was initiated with the injection of protein using a 25-μL gas-tight syringe (Hamilton, Reno, NV, United States) that was purged with anoxic double-deionized water containing 20 mM 2-ME. The cuvette was gently inverted and placed back into the spectrophotometer. The oxidation of NADPH was monitored at 340 nm on a DU-800 spectrophotometer (Beckman Coulter, Brea, CA, United States) using a combined extinction coefficient for NADPH and H4F (340[NADPH + H4F] of 12.3 mM-1cm-1. The effect of pH was analyzed as described above in 200 mM sodium phosphate for pH levels 5.8–8.0 and 200 mM sodium acetate buffer for pH 5.3. The effect of temperatures were tested over the range from 15 to 37°C. The cuvettes were covered and equilibrated in a water bath for 10 min at varying temperatures prior to addition of protein.
DmrA Assay
The DmrA assay of Caccamo et al. (2004) was used based on modifications to a Dfr assay. Reactions were prepared in an anaerobic chamber in sealed 2-ml quartz masked cuvettes. The reaction mixture (250 μL or 1 ml) consisted of about 3.6 μg of enzyme in an anoxic solution containing 500 mM sodium acetate (pH 5.3), 20 mM sodium ascorbate, 1 mM EDTA, 15 mM 2-ME, 80 μM H2SPT, and 0.1 mM NADPH. The reaction was initiated with the injection of protein with a 25-μL gas-tight syringe, purged with anoxic double-deionized water containing 20 mM 2-ME. The cuvette was gently inverted and placed back into the spectrophotometer. The oxidation of NADPH was monitored at 340 nm (340[NADPH] = 6.22 mM-1cm-1) (Dawson, 1986) on a DU-800 spectrophotometer.
Specific Activity for Dfr, DmrA, and DAGK Assays and Kinetics Analysis for DmrA Assays
Rate calculations using the molar extinction coefficient for NAD(P)H were used to measure specific activity, where 1 unit is defined as 1 μmol of NAD(P)H oxidized per min per mg of protein for all assays. Enzyme kinetic constants (Km and Vmax values) were determined with a non-linear regression model fit to the Michaelis-Menten equation using GraphPad Prism v7.03 for Windows (GraphPad Software, La Jolla, CA, United States1)
DAGK Assay
Diacylglycerol kinases activity was assayed by coupling the oxidation of NADH to the production of phosphatidic acid (Badola and Sanders, 1997) (Figure 1B). The headspace of the DAG analog substrate (1,2-dihexanoyl-sn-glycerol in 50% ethanol) was purged under a gentle stream of nitrogen to evaporate the ethanol solvent until an oil residue remained. The sealed residue was transferred to an anaerobic chamber and reconstituted in an anoxic solution of 60 mM PIPES, 50 mM LiCl, 0.1 mM EDTA, 0.1 mM EGTA [pH 6.8] (150-μL) to a final concentration of 50 mg/ml (the approximate solubility of 1,2-dihexanoyl-sn-glycerol in phosphate buffered saline (PBS), pH 7.2.) Lactate dehydrogenase (LDH) (Roche, Mannheim, Germany), pyruvate kinase (PK) (Sigma-Aldrich, St. Louis, MO, United States), DAGK from Escherichia coli (Enzo Life Sciences, Farmingdale, NY, United States), and annotated DAGK from M. nodulans and H. nitrativorans were prepared by transferring 100 μL of each enzyme to 3-ml anaerobic vials and purging the headspace with a gentle stream of nitrogen for approximately 5 min on ice. The reaction was initiated with approximately 3.6 μg of protein in an anoxic reaction mixture (60 mM PIPES, pH 6.8, 50 mM LiCl, 0.1 mM EDTA, 0.1 mM EGTA), 1 mM phosphoenolpyruvate, 3 mM ATP, 2.6 mM 1,2-dihexanoyl-sn-glycerol, 20 mM magnesium acetate, 0.1 mM NADH, and 20 units each of LDH and PK. The oxidation of NADH was monitored using the molar absorption coefficient at 340 nm on a DU-800 spectrophotometer.
Protein Computational Modeling
Conformational modeling of DmrA and DfrB was performed by Andrew Orry (Molsoft, San Diego, CA, United States) using the ICM package. The modeling template was the crystal structure of Mycobacterium avium dihydrofolate reductase co-crystallized with NADPH and trimethoprim (pdb 2w3v). The modeling method is based on the Internal Coordinates (IC) representation of molecular objects, which naturally reflects covalent bond geometry of molecule (Abagyan and Totrov, 1994; Abagyan et al., 1994). After initial placement of the aligned polypeptide chain onto the template structure, the side-chain torsion angles were predicted by simultaneous global optimization of the energy for all non-identical residues. Conformational modeling of protein side chains and loops involved internal coordinate definition of the molecular object combined with computationally efficient ICM Biased Probability Monte Carlo (BPMC) optimization. Optimization of the structures were done in an extended force field (Arnautova et al., 2011), which includes surface terms, electrostatics, and side chain entropy terms. The quality of the 3D model was assessed by an ICM procedure called Protein Health.
Results
In M. extorquens AM1, three genes sharing similarity to dihydrofolate reductase (dfrA, dfrB, and dmrA) have been previously identified (Marx et al., 2003). Prior to the current work, only the protein encoded by the (dmrA) gene had been characterized biochemically (Caccamo et al., 2004; Rasche et al., 2004). In the current study, we used a bioinformatics approach to assess phylogenetic relationships among M. extorquens DfrA, DfrB, and DmrA, and homologs from other organisms. We also employed enzyme assays to assess the biochemical activities of DfrA, DfrB, DmrA, and two DmrA orthologs currently annotated as DAGKs.
Sequence Alignment Searches of DfrB Orthologs Resulted in Three Distinct Clades
DfrB orthologs obtained in a BLASTx search from a non-redundant database in NCBI were used to construct an unrooted phylogenetic tree using a maximum-likelihood method with bootstrap analyses (Dawson, 1986; Kumar et al., 2016) (Figure 2). The resulting tree yielded three clades. Each clade contained either DfrA, DfrB, or DmrA from M. extorquens AM1. The sequences surrounding DfrA were from either Euryarchaeota, Plantomycete, Proteobacteria, or a Firmicute. The small clade containing DfrB revealed sequences from Proteobacteria and Actinobacteria. The largest clade (DmrA) contained homologs from Proteobacteria and a Firmicute.
DfrA Is Closely Related to Annotated Dfr Orthologs From Bacteria
In the phylogenetic tree, the M. extorquens DfrA sequence was located among orthologs from planctomycetes and β-, γ-, and δ-proteobacteria (Figure 2). Two of the proteins in the DfrA clade have been previously crystallized as dihydrofolate reductases: a DfrA homolog from Moritella profunda (Mpr-Dfr) (Hay et al., 2009), and a trimethoprim-resistant ortholog from E. coli (Eco-DfrA24). This observation provides support for the hypothesis that M. extorquens DfrA may function as a standard dihydrofolate reductase All planctomycete homologs in the phylogenetic tree grouped with DfrA. Interestingly, one planctomycete sequence was annotated as a DAGK (TMED84-DK, Figure 2) in the DfrA clade. However, this ortholog was found to be only 14% identical to a known DAGK from E. coli using a percent identity matrix generated in Clustal Omega (Sievers et al., 2011; McWilliam et al., 2013; Li et al., 2015).
To further investigate connections to folate metabolism, genes in the neighborhood of dfrA were analyzed using the STRING v10.5 database, and a gene/protein interaction network module was constructed with dfrA biosynthesis (data not shown). In various genomes, genes with connection to dfrA included thyA, folC, fhs, gcvT, glyA, metH, purH, purN, fmt, and MexAM1_META1p0830 (fmt-like), many of which are associated with folate-requiring pathways of coenzyme, amino acid synthesis, and purine.
One Clade Contained a Small Group of Orthologs Sharing 45–95% Identity With DfrB
Out of the vast number of DfrB orthologs identified by the BLAST search, only five sequences grouped tightly with and are closely related to M. extorquens DfrB in the phylogenetic tree (Figure 2). One ortholog was from an actinobacterium (Streptomyces purpurogeneiscleroticus) and the remaining were from α-proteobacteria. Of these five, two orthologs were annotated as Dfr and the remaining three were labeled as hypothetical proteins. The highest identity to DfrB (95%) was an ortholog annotated as Dfr from M. populi (Mpo-DfrB, Figure 2). This organism has been renamed as M. extorquens strain BJ001 (Marx et al., 2012).
Of the orthologs in the DfrB clade, only DfrB was located on a plasmid. Two genes located upstream of DfrB were a putative transposase and a protein of unknown function. The highest confidence for a homology match in the Phyre2 database for the protein of unknown function resulted in a riboflavin synthase domain-like superfamily (ferredoxin reductase FAD-binding domain-like family), with a reductase/isomerase/elongation factor common domain (30% identity, coverage).
The DmrA Clade Included Annotated DAGKs From Various Bacteria
Homologs in the DmrA clade had identities ranging from 60 to 99% when compared to DmrA from M. extorquens AM1. The DmrA clade contained three homologs from α-proteobacteria annotated as dihydrofolate reductases (Dfr) (M. mesophililcum, Granulibacter bethesdensis CGDNIH1, and M. populi), and a large number of homologs annotated as DAGKs. Many of the annotated DAGKs contained amino acid regions predicted in NCBI to reduce dihydrofolate to H4F using NADPH as a cofactor. Thus, we tested whether some of these annotated DAGKs might function as dihydrofolate reductases or DmrA enzymes.
Most of the putative dihydromethanopterin reductases were from α-proteobacteria. The exceptions were two sequences from the β-proteobacteria Azohydromonas australica and A. lata) (respectively, 67 and 65% identity to M. extorquens DmrA). This is interesting because it is the first evidence of DmrA homologs in β-proteobacteria. In other β-proteobacteria, the proposed dihydromethanopterin reductases are not homologous to DmrA but instead resemble an archaeoflavoprotein (AfpA) found to restore a C1 growth phenotype in M. extorquens following dmrA knockout and complementation (Kalyuzhnaya et al., 2005). The AfpA group in β-proteobacteria has been renamed as dihydromethanopterin reductase B (DmrB). The crystal structure of DmrB points to the role of FMN cofactors in electron or hydride transfer to H2MPT (McNamara et al., 2014). It is intriguing that A. australica and A. lata contain homologs of both DmrA (Figure 2) and DmrB (with identities of 69 and 68%, respectively, to Burkholderia xenovorans DmrB). This raises the evolutionary question of why both forms of dihydromethanopterin reductase (DmrA and DmrB) might coexist in these organisms.
M. extorquens DfrA and DfrB Enzyme Activities
To test the hypotheses that DfrA and DfrB function as dihydrofolate reductases, the enzymes were initially assayed in the presence of 50 μM H2F. Under these initial conditions, H6-DfrA and H6-DfrB reduced H2F with specific activities of 18.5 and 3.13 U/mg, respectively (Figure 3 and Table 1). These values were within 2.5-fold of the rate obtained using a known dihydrofolate reductase from E. coli (7.3 U/mg) (Figure 3 and Table 1). When DfrA and DfrB were tested for dihydromethanopterin reductase activity, only a trace of H2SPT reduction activity was observed for both enzymes (Figure 4, diamond and triangle; Table 1, column 3). This activity was only about 1% of the H2SPT reduction activity of DmrA-H4 measured at pH 5.3 (Table 1). Some caution should be taken in interpreting these data due to the histidine tags, which lacked a protease cut site and could not be removed. However, since the activities of H6-DfrA and H6-DfrB resembled that of untagged DfrB from E. coli, these data provide biochemical support that DfrA and DfrB are likely to function in converting dihydrofolate to H4F in M. extorquens cells.
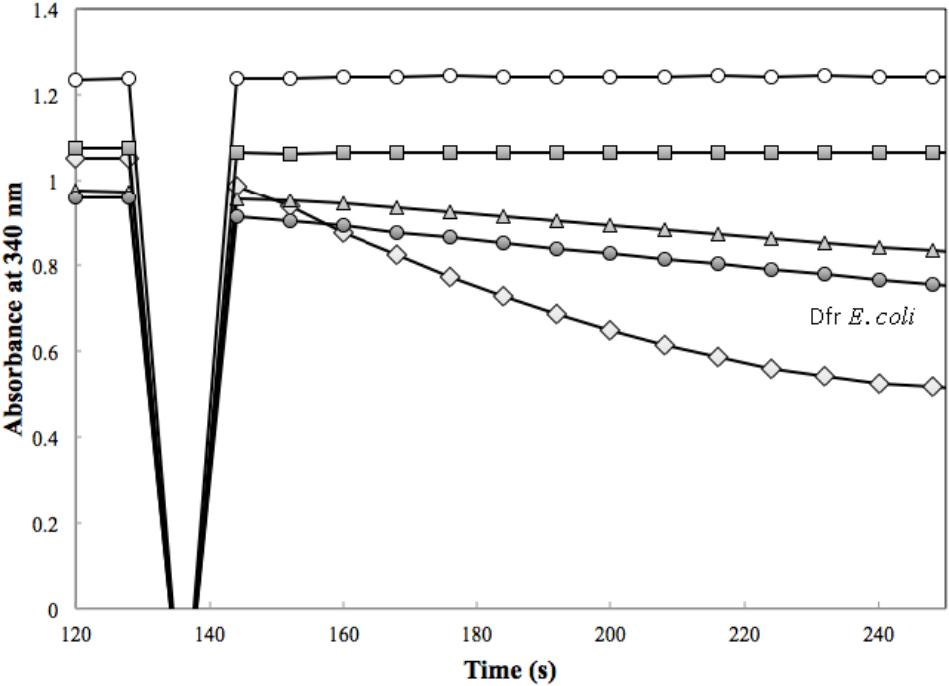
FIGURE 3. Dfr activity was assessed in the presence of 50 μM H2F with 1.7 μg of H6-DfrA (open diamond), 1.9 μg of H6-DfrB (triangle), 3.6 μg of Mno-DAGK-H6 (open circle), 3.6 μg of Hni-DAGK-H6 (square), and 1.0 μg of Dfr from Escherichia coli (closed circle).
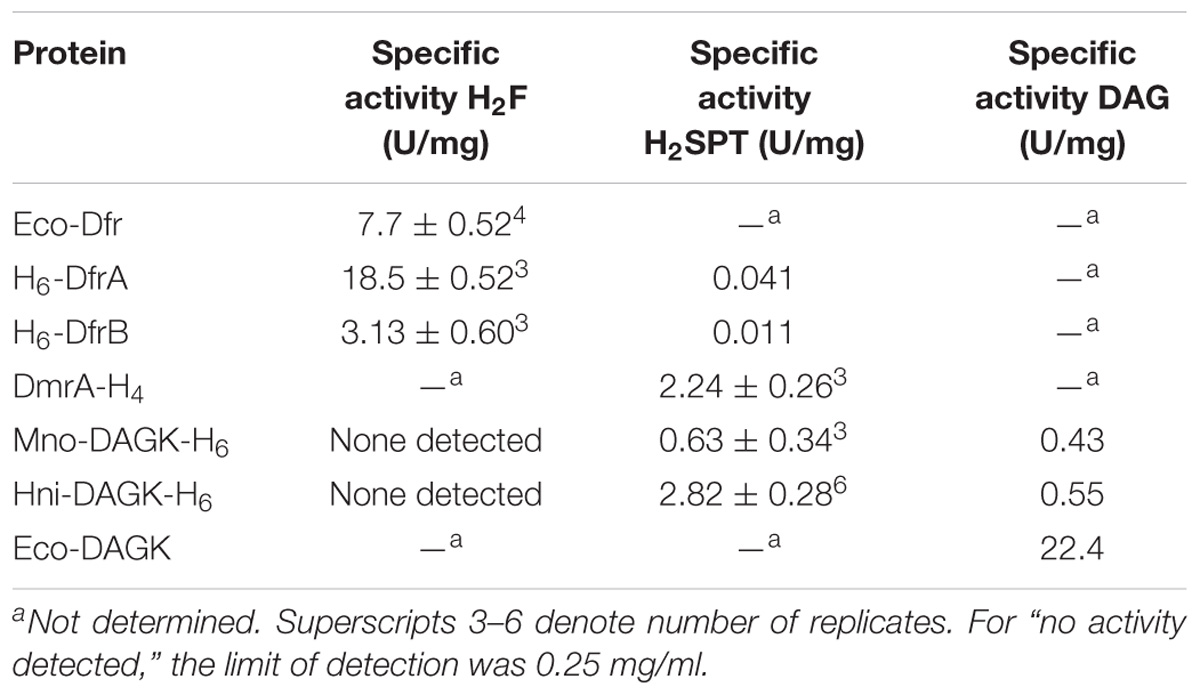
TABLE 1. Initial tests of enzyme activity in the presence of pterin and DAG substrates.
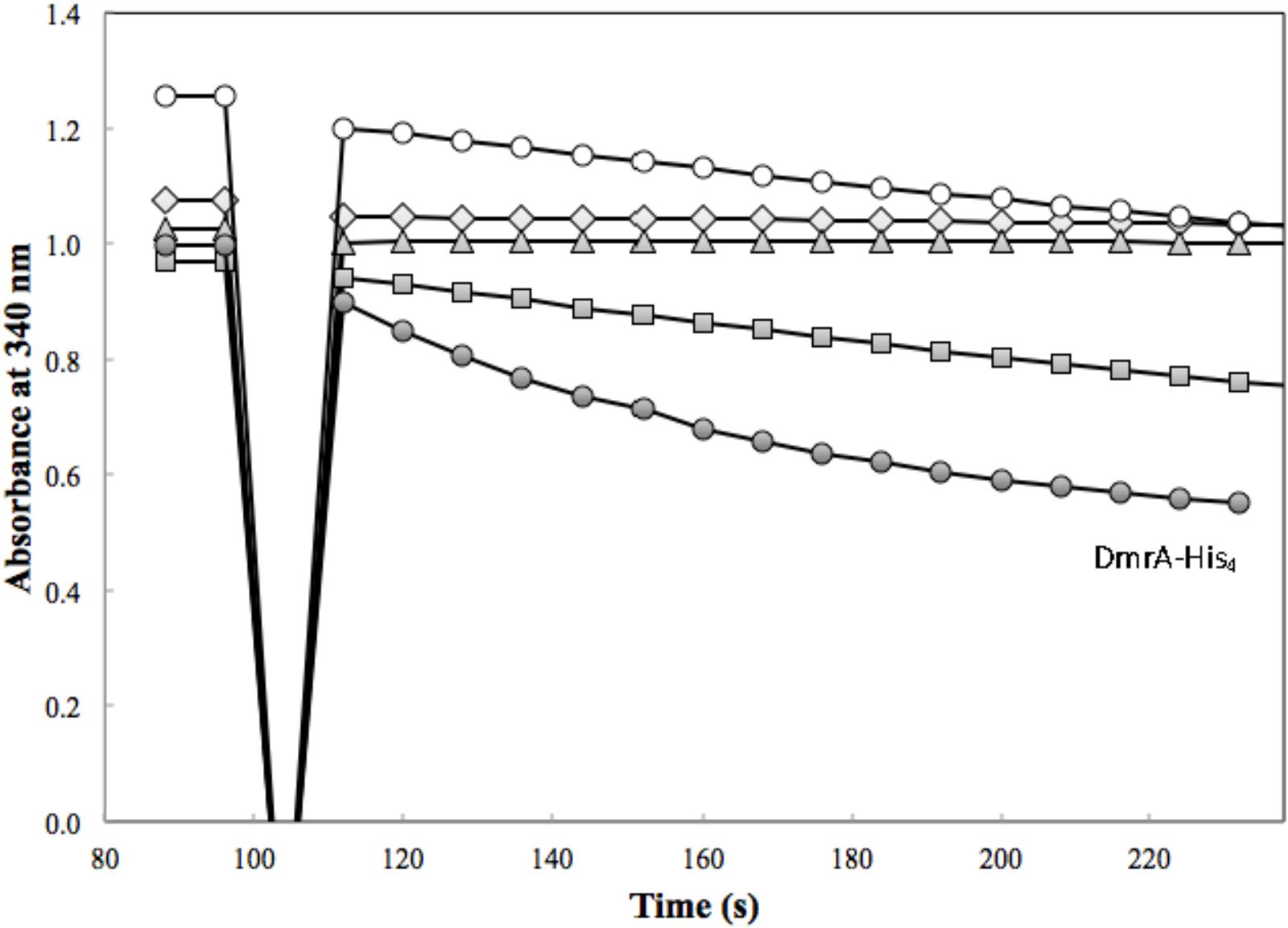
FIGURE 4. DmrA activity was assessed in the presence of 83 μM H2SPT with 3.4 μg of H6-DfrA (open diamond), 4.1 μg of H6-DfrB (triangle), 3.6 μg of Mno-DAGK-H6 (open circle), 0.72 μg of Hni-DAGK-H6 (square), and 4.3 μg of DmrA-H4 (closed circle).
The effect of pH, temperature, and enzyme concentration were studied for H6-DfrA and H6-DfrB in preparation for kinetics studies. Over the pH range tested (5.3–8.0), the highest reaction rates for both enzymes were obtained from pH 6.8 to 7.0 (data not shown). For the temperatures tested (15–37°C or 40°C), H6-DfrA showed a broad temperature optimum from about 23–40°C, while H6-DfrB showed near constant reaction rates between 15 and 37°C. Thus, pH 6.8 and room temperature were used for kinetics measurements. DfrA activity showed a linear response to increasing enzyme concentration up to 1.8 μg per assay (0.093 μM H6-DfrA), while DfrB activity was linear up to 3.6 μg per assay (0.18 μM H6-DfrB).
To estimate kinetic values, the concentration of dihydrofolate was tested over the range from 0 to 150 μM (Figure 5). When fit to the Michaelis-Menten equation for a hyperbola, the estimated KM values were similar (14 ± 3.0 μM dihydrofolate for DfrA and 18 ± 8.9 μM for DfrB). The estimated Vmax for DfrA was 52 ± 2.8 U/mg, corresponding to a kcat of 17/s. The Vmax for DfrB was about 2.5 times lower (22.5 ± 4.2 U/mg, kcat of 7.2/s).
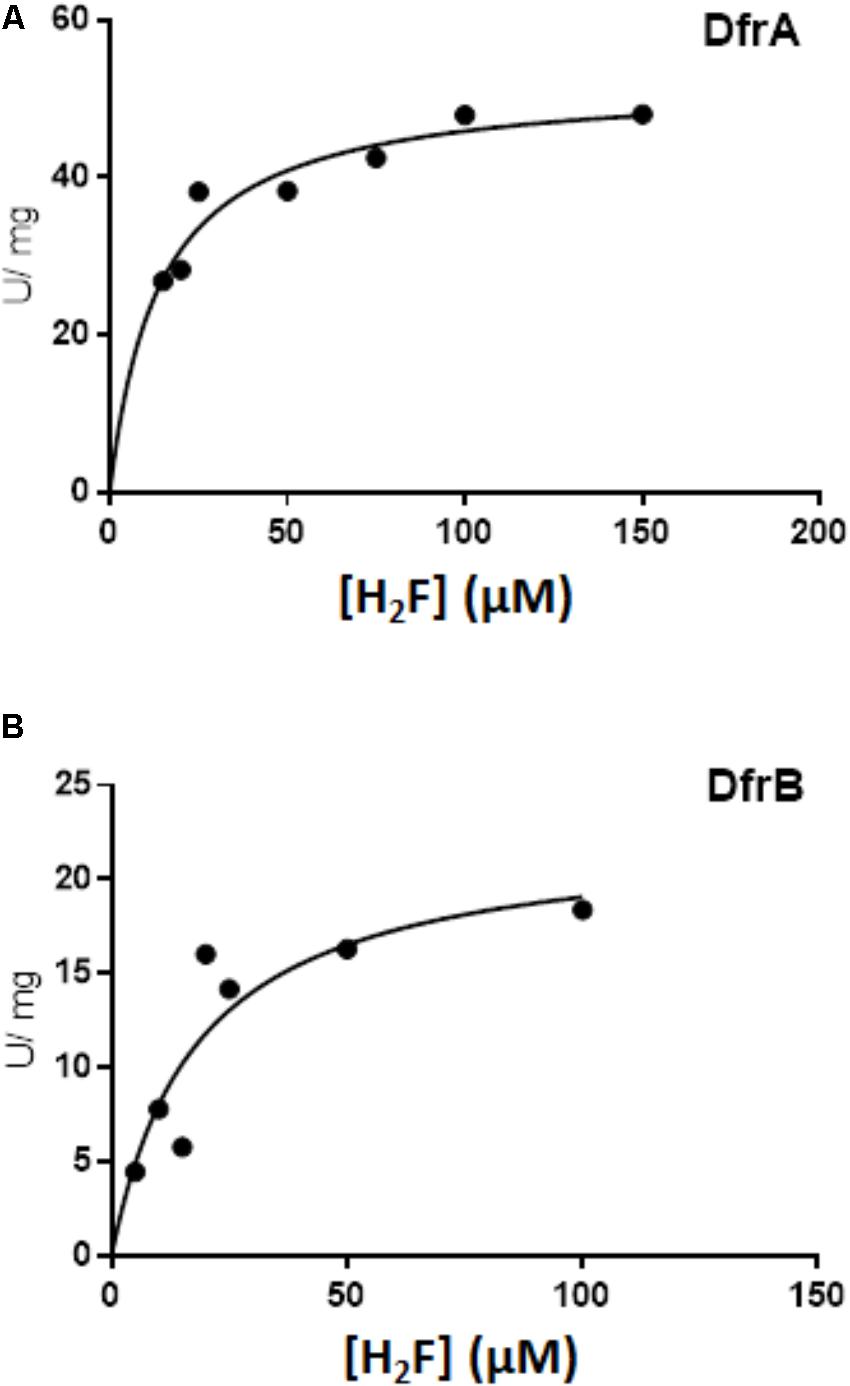
FIGURE 5. Kinetics of DfrA and DfrB with dihydrofolate as the substrate. Dihydrofolate reductase activity was measured as described in Section “Materials and Methods” in the presence of 100 μM NADPH and either DfrA (A) or DfrB (B). Data were fit to the equation for a hyperbola using non-linear least-squares fit with GraphPad Prism 7.04 Software. The estimated KM and Vmax values are presented in the Section “Results.”
Enzyme Activity Assays for Orthologs Within the DmrA Clade
The activity of M. extorquens DmrA-H4 was compared with two annotated DAGKs sharing different degrees of identity with M. extorquens DmrA. The M. nodulans homolog (Mno-DAGK- H6) was 84% identical to DmrA, and the H. nitrativorans homolog (Hni-DAGK-H6) was 69% identical to DmrA. We first tested whether these enzymes showed NADPH-dependent dihydrofolate reductase activity, but no activity was detected with the addition of either the Mno or Hni enzyme (Figure 3 and Table 1).
Annotated DAGKs from M. nodulans and H. nitrativorans were both capable of reducing H2SPT (Figure 4). Under the initial screening conditions, the specific activity of the Hni-DAGK-H6 was about the same as that of DmrA-H4, while the rate for Mno-DAGK-H6 was 3–4 times lower (Table 1). The lower activity of Mno-DAGK-H6 in the initial screening studies may be explained by the higher KM values obtained later in the kinetics studies (Table 2). The Mno-DAGK-H6 appeared to have lower affinity for H2SPT (apparent KM of 695 μM H2SPT) compared to the apparent KM values for M. extorquens DmrA-H4 and Hni-DAGK-H6 (193 and 102 μM H2SPT, respectively). Despite the differences in KM values, the Vmax estimates for the three enzymes were similar, differing only by a factor of two (Table 2).

TABLE 2. Kinetic values (apparent KM, Vmax, and kcat) for DmrA and DmrA orthologs.
DAGK Assays for Homologs Within the DmrA Clade
To test the alternative hypothesis that the DmrA homologs might contain the annotated DAGK activity, a modified DAGK assay was performed (Figure 6A). In these studies, the DAG analog, 1,2-dihexanoyl-sn-glycerol was used, but β-octyl glucoside (OG) and dimyristoyl phosphatidylcholine (DMPC) were excluded. To show that the modified assay was functioning properly in our lab, DAGK from E. coli was tested as a control (Figure 6B). Under the conditions used, the specific activity of the E. coli enzyme (1 μg of protein) was 24.2 U/mg (Table 1), which is comparable to the published value of 22.0 U/mg (Badola and Sanders, 1997).
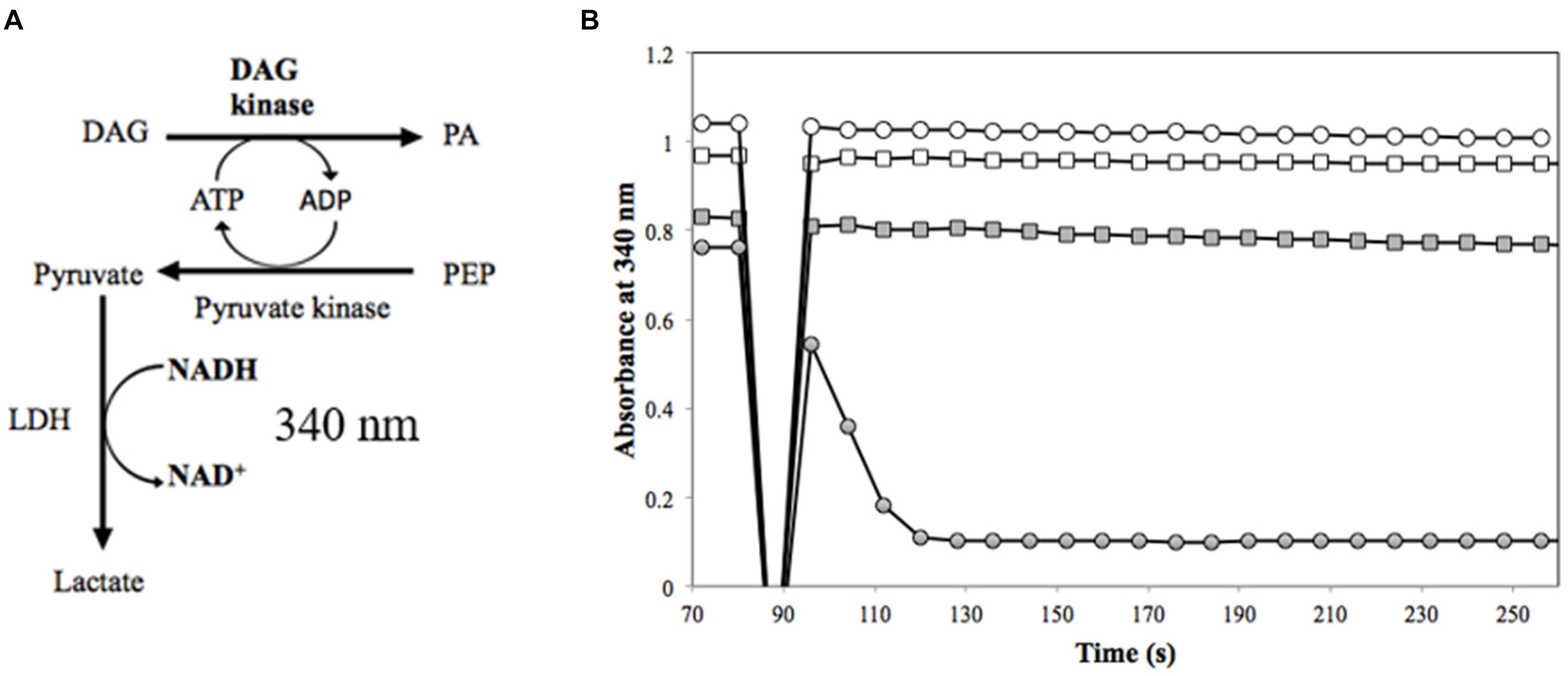
FIGURE 6. Measurement of DAGK activity. (A) In the DAGK assay (Badola and Sanders, 1997), the conversion of DAG to phosphatidic acid (PA) produces ADP, which is used in the pyruvate kinase reaction to convert phosphoenolpyruvate (PEP) to pyruvate. This reaction is coupled to the reduction of pyruvate to lactate by lactate dehydrogenase (LDH). The oxidation of NADH is monitored as a loss in absorbance at 340 nm for DAGK activity. There is a 1:1 ratio of one mole of PA produced to one mole NADH oxidized. (B) DAGK activity of 3.5 μg of Mno-DAGK-H6 (open circle), 3.6 μg of Hni-DAGK-H6 (square), boiled Hni-DAGK-H6 (closed square) compared to DAGK control from E. coli (closed circle).
In the DAGK assay, the addition of 3.5 μg of Mno-DAGK-H6 or Hni-DAGK-H6 produced specific activities of 0.43 and 0.55 U/mg, respectively (Figure 6B and Table 1). This rate was only 1–2% of the activity of commercially purchased Eco-DAGK when comparable amounts of enzyme were used. The slow rate of Hni-DAGK-H6 activity proceeded constantly over a course of 5 min, in contrast with that of the boiled enzyme control, which yielded no activity (Figure 6B).
Discussion
The results of the current study may be interpreted in the context of the previously published model predicting that the M. extorquens DmrA protein evolved from an ancestral dihydrofolate reductase (Dfr) following transfer of H4MPT biosynthesis genes from archaea to bacteria (Marx et al., 2003). This hypothesis is based on the sequence similarity of DmrA to known dihydrofolate reductase sequences, combined with the observation that disruption of the M. extorquens dmrA gene produces a phenotype similar to that of deletion mutants in H4MPT biosynthesis genes (Marx et al., 2003; Rasche et al., 2004; Chistoserdova et al., 2005). Due to the absence of archaeal redox cofactors in bacteria, archaeal oxidoreductases like dihydromethanopterin reductases may have been non-functional in bacteria. To resolve this issue, two separate lineages of bacterial dihydromethanopterin reductases appear to have evolved: one of bacterial origin (DmrA) found almost exclusively in α-proteobacteria, and a second lineage (AfpA/DmrB) derived from an archaeal flavoprotein called DmrX.
The results of the current study are consistent with a bacterial origin for DmrA in α-proteobacteria. The phylogenetic tree in Figure 2 places M. extorquens DfrA, DfrB, and DmrA in separate clades. Duplication of a dfr gene followed by mutations that changed specificity for the pterin substrate would account for the presence of both dihydrofolate reductase and dihydromethanopterin reductase activities in extant α-proteobacteria (Table 1).
Methylobacterium extorquens DfrA has been proposed to function as a standard dihydrofolate reductase based on co-localization of dfrA with genes encoding H4F biosynthesis and H4F-dependent enzymes and additional gene neighborhood analysis of multiple genomes (Chistoserdova et al., 2003; Marx et al., 2003; this study). Prokaryotic genes of related functions often occur together in operons or gene clusters, as demonstrated by the large cluster of proteobacterial genes related to H4MPT-dependent metabolism (Chistoserdova et al., 1998; Kalyuzhnaya et al., 2005). In the current study, the dihydrofolate reductase activities of DfrA and DfrB were demonstrated biochemically for the first time and were comparable to the activity of a known Dfr from E. coli (Table 1 and Figure 5). The evolutionary potential for altering substrate specificity from dihydrofolate to dihydromethanopterin is also supported to some extent by enzymatic assays in which traces of H2SPT reduction activity were detected (Table 1). Conversely, M. extorquens DmrA has been shown to reduce H2SPT at relatively high rates and dihydrofolate at low rates (Caccamo et al., 2004), possibly representing a vestige of an ancestral dihydrofolate reductase activity.
Protein computational modeling also demonstrates the potential for changing the specificity of dihydrofolate reductase toward affinity for dihydromethanopterin. Molecular models of M. extorquens DmrA and DfrB were constructed by Andrew Orry (Molsoft L.L.C., San Diego, CA, United States) (Figure 7) and predict that DmrA (Figure 7, yellow ribbon structure) and DfrB (green ribbon structure) share a similar overall protein fold consisting of primarily parallel β-sheets connected by α-helices. In particular, secondary structural features are conserved in the NADPH binding domain, which includes DmrA residues 59 to 85. This would account for the conserved use of NADPH as an electron donor by both DmrA and DfrA. Unique structural features of DmrA occur in the active site region distant from the NADPH binding domain, where the pterin substrate is presumed to bind. The DmrA model shows an insertion of 7 amino acids (residues 25–31) forming a loop that is absent in the models of DfrB and Mycobacterium dihydrofolate reductase (Figure 7). Another difference is that DmrA also lacks two of the C-terminal β-strands found in the dihydrofolate reductase structures. Although the DmrA model could not predict the details of the DmrA pterin binding site with confidence, the insertion of a DmrA loop and the loss of two β-strands over evolutionary time might have served to accommodate the structural differences between dihydromethanopterin and dihydrofolate. A crystal structure of DmrA would be needed to create a detailed model of the dihydromethanopterin binding site.
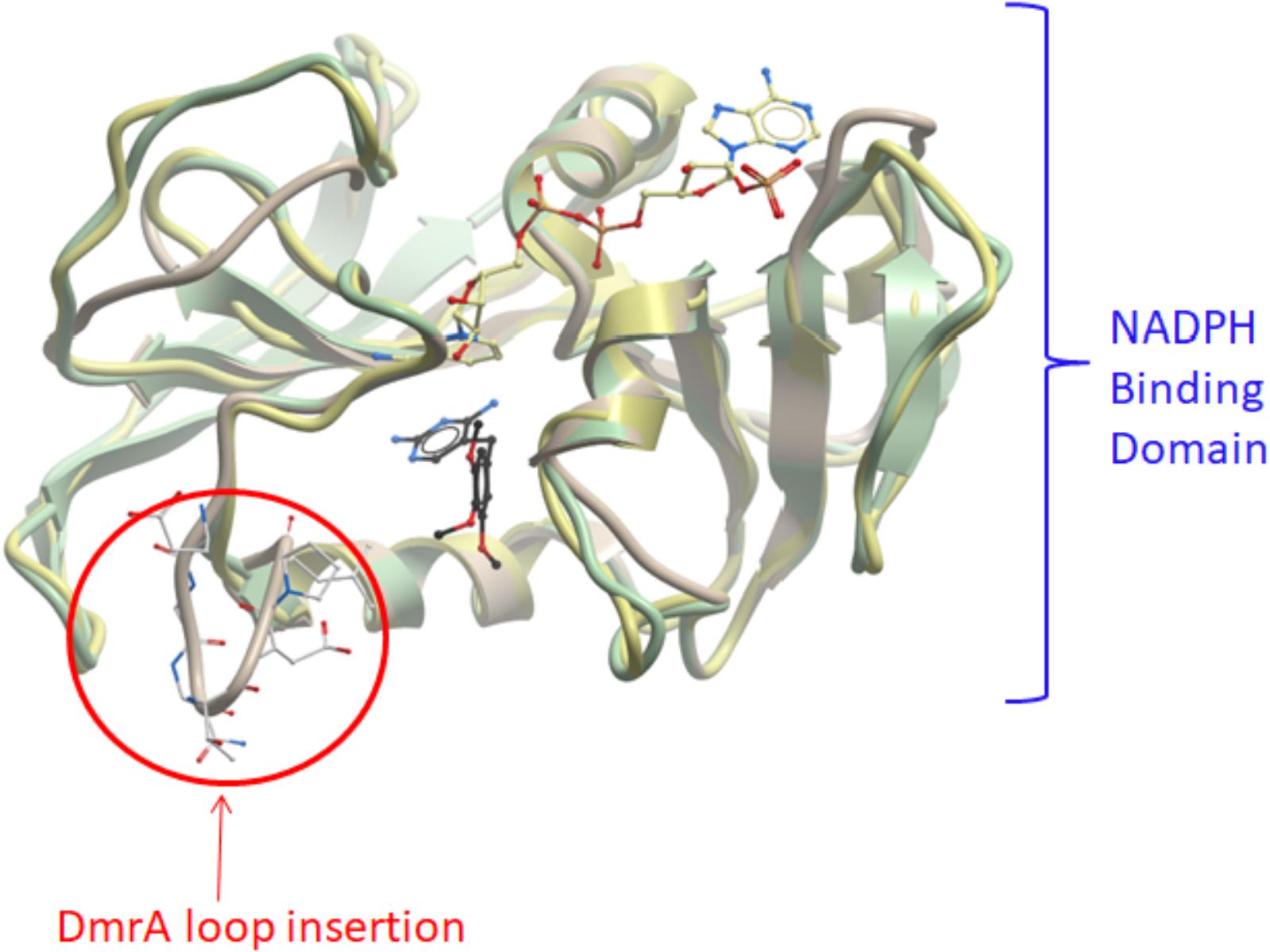
FIGURE 7. Computational models of DmrA and DfrB. 3D models of DmrA and DfrB were constructed at described in Section “Materials and Methods” using ICM-Pro (Molsoft, San Diego, CA, United States) with Mycobacterium avium dihydrofolate reductase as the template (pdb 2w3v). The NADPH binding sites appeared conserved between the two protein models (blue label) with the position of NADPH predicted by analogy to the template structure. An insertion of 7 amino acids in DmrA (residues 25–31) formed a unique loop (circled in red). The DmrA model also predicted the deletion of two C-terminal β-strands found in both DfrB and the template.
The function of the second dihydrofolate reductase in M. extorquens (DfrB) remains a mystery. The estimated KM values for DfrA and DfrB were similar, while the Vmax for DfrB appeared lower than that of DfrA (Figure 5). The presence of a second Dfr is not uncommon in bacteria. Multiple copies of dihydrofolate reductase can provide varying sensitivities to folate competitors such as trimethoprim (Huovinen, 1987). The M. extorquens dfrB gene is located on a megaplasmid, and the protein has 47% similarity to a trimethoprim-resistant dihydrofolate reductase encoded on E. coli plasmid pCJ001, leading to a hypothesis for a role of DfrB in antimicrobial resistance (Jansson and Skold, 1991; Marx et al., 2003). Another proposed role for M. extorquens DfrB as an intermediate in the evolution of DmrA may be inferred from the closer sequence identity between DfrB and DmrA (34% identical) compared that of DfrA (26% identical to DmrA) (Marx et al., 2003) and the phylogenetic position of the DfrB clade between DfrA and DmrA (Figure 2). While this might be possible, the similar low rates of H2SPT reduction by DfrA and DfrB (Table 1) do not seem to favor DfrB as a preferred intermediate in the evolution toward DmrA.
For the DmrA clade, the current annotation of many orthologs as DAGKs was surprising based on sequence alignments. For example, while the annotated DAGKs from M. nodulans and H. nitrativorans are 84 and 69% identical to M. extorquens DmrA, they share only 15 and 12% identity, respectively, with a known DAGK from E. coli. Dihydromethanopterin reductase activity measured at pH 5.3 was observed for Mno-DAGK-H6 and Hni-DAG-kinase-H6 (Figure 4 and Tables 1, 2), while dihydrofolate reductase activity was not detectable for either enzyme under the conditions studied (Table 1 and Figure 3). The DAGK activities of the two enzymes were also very low compared to the activity of a known E. coli DAGK under the same conditions (1–2%) (Table 1 and Figure 6). At this time, we cannot rule out the possibility that the annotated DAGKs in this study may be bifunctional enzymes with both DmrA and DAGK activities playing a role in methylotroph cells. However, given the low sequence identity to characterized DAGKs, the low DAGK activities (Figure 6), and kinetics values similar to those of DmrA (Table 2), we propose that DAGKs sharing at least 69% identity with M. extorquens AM1 DmrA should be renamed as dihydromethanopterin reductases.
An explanation for the large apparent KM difference between Mno-DAGK-H6 and DmrA-H4 might be attributed to either the protein structural health following purification through nickel affinity chromatography or the physical and chemical environment in which M. nodulans is found in nature. M. nodulans exhibits both nitrogen-fixation and specific nodulation of Crotalaria species. These features have not been observed in the Methylobacterium species that have been tested thus far (Sy et al., 2001). In the nodule environment, high amounts of methanol and methylotrophic activity have been observed (Jourand et al., 2005). A high apparent KM for H2MPT may enable M. nodulans to regulate dihydromethanopterin reductase activity to accommodate large influxes of methanol.
Another point of interest is the finding of a DmrA homolog in A. lata and A. australica. These two species of β-proteobacteria also contain an archaea-like dihydromethanopterin reductase with a redox-active FMN cofactor (AfpA/DmrB) (Ding and Ferry, 2004; Kalyuzhnaya et al., 2005). The presence of two phylogenetically diverse forms of dihydromethanopterin reductase (DmrA and DmrB) in a single organism invites additional studies of the evolutionary history and differential roles of the two enzymes in the C1 metabolism of these cells.
Methylotrophic microorganisms are valuable in biotechnology processes that use methanol as an alternative to sugars as a carbon substrate for the biosynthesis of industrial products such as biofuels and biopolymers (Schrader et al., 2009). Additional benefits include the potential to synthesize polyhydroxybutyrates and uncommon dicarboxylic acids or polyketides using the ethylmalonyl-CoA pathway of Methylobacterium species. The ability to grow on minimal media might also simplify product recovery compared to the separations required with rich media, such as Luria broth (Ochsner et al., 2015). Using a natural methylotroph, as opposed to bioengineering E. coli, could eliminate the need to engineer methods to alleviate a potential buildup of formaldehyde as a toxic intermediate during methanol oxidation. The relatively high KM for DmrA (DAGK) from M. nodulans might allow responsiveness at higher concentrations of H2SPT to help accommodate increases in methanol concentration. The ability of methylotrophs to process large extracellular concentrations of methanol, combined with the metabolic machinery to transform chemicals while avoiding formaldehyde bioaccumulation, could provide advantages for methanol-based biotechnology in the future.
Dataset Statement
All relevant data is either contained within the manuscript or will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
MB designed the principle experiments based on an original research idea by CA. MB, CA, KW, and YM executed the experiments and interpreted the data. MB, YM, and MR prepared the manuscript.
Funding
This research was supported by National Science Foundation grant CHE-1508801 and a 2014 Development Grant from the California State University Program for Education and Research in Biotechnology (CSUPERB).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Dr. Andrew Orry (Molsoft, San Diego, CA, United States) for creating the computational models of DmrA and DfrB.
Footnotes
References
Abagyan, R., and Totrov, M. (1994). Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 235, 983–1002. doi: 10.1006/jmbi.1994.1052
Abagyan, R., Totrov, M., and Kuznetsov, D. (1994). ICM—A new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 15, 488–506. doi: 10.1002/jcc.540150503
Achenbach-Richter, L., Stetter, K. O., and Woese, C. R. (1987). A possible biochemical missing link among archaebacteria. Nature 327, 348–349. doi: 10.1038/327348a0
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Arnautova, Y. A., Abagyan, R. A., and Totrov, M. (2011). Development of a new physics-based internal coordinate mechanics force field and its application to protein loop modeling. Proteins 79, 477–498. doi: 10.1002/prot.22896
Badola, P., and Sanders, C. R. (1997). Escherichia coli diacylglycerol kinase is an evolutionarily optimized membrane enzyme and catalyzes direct phosphoryl transfer. J. Biol. Chem. 272, 24176–24182. doi: 10.1074/jbc.272.39.24176
Bertani, G. (1951). Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 62, 293–300.
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. doi: 10.1016/0003-2697(76)90527-3
Caccamo, M. A., Malone, C. S., and Rasche, M. E. (2004). Biochemical characterization of a dihydromethanopterin reductase involved in tetrahydromethanopterin biosynthesis in Methylobacterium extorquens AM1. J. Bacteriol. 186, 2068–2073. doi: 10.1128/JB.186.7.2068-2073.2004
Chistoserdova, L. (2016). Wide distribution of genes for tetrahydromethanopterin /methanofuran-linked C1 transfer reactions argues for their presence in the common ancestor of bacteria and archaea. Front. Microbiol. 7:1425. doi: 10.3389/fmicb.2016.01425
Chistoserdova, L., Chen, S. W., Lapidus, A., and Lidstrom, M. E. (2003). Methylotrophy in Methylobacterium extorquens AM1 from a genomic point of view. J. Bacteriol. 185, 2980–2987. doi: 10.1128/JB.185.10.2980-2987.2003
Chistoserdova, L., Jenkins, C., Kalyuzhnaya, M. G., Marx, C. J., Lapidus, A., Vorholt, J. A., et al. (2004). The enigmatic planctomycetes may hold a key to the origins of methanogenesis and methylotrophy. Mol. Biol. Evol. 21, 1234–1241. doi: 10.1093/molbev/msh113
Chistoserdova, L., Rasche, M. E., and Lidstrom, M. E. (2005). Novel Dephosphotetrahydromethanopterin biosynthesis genes discovered via mutagenesis in Methylobacterium extorquens AM1. J. Bacteriol. 187, 2508–2512. doi: 10.1128/JB.187.7.2508-2512.2005
Chistoserdova, L., Vorholt, J. A., Thauer, R. K., and Lidstrom, M. E. (1998). C1 transfer enzymes and coenzymes linking methylotrophic bacteria and methanogenic Archaea. Science 281, 99–102. doi: 10.1126/science.281.5373.99
DiMarco, A. A., Bobik, T. A., and Wolfe, R. S. (1990). Unusual coenzymes of methanogenesis. Annu. Rev. Biochem. 59, 355–394. doi: 10.1146/annurev.bi.59.070190.002035
Ding, Y. H., and Ferry, J. G. (2004). Flavin mononucleotide-binding flavoprotein family in the domain Archaea. J. Bacteriol. 186, 90–97. doi: 10.1128/JB.186.1.90-97.2004
Escalante-Semerena, J. C., Rinehart, K. L. Jr., and Wolfe, R. S. (1984). Tetrahydromethanopterin, a carbon carrier in methanogenesis. J. Biol. Chem. 259, 9447–9455.
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x
Franceschini, A., Lin, J., von Mering, C., and Jensen, L. J. (2016). SVD-phy: improved prediction of protein functional associations through singular value decomposition of phylogenetic profiles. Bioinformatics 32, 1085–1087. doi: 10.1093/bioinformatics/btv696
Franceschini, A., Szklarczyk, D., Frankild, S., Kuhn, M., Simonovic, M., Roth, A., et al. (2013). STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41, D808–D815. doi: 10.1093/nar/gks1094
Garfin, D. E. (1990). One-dimensional gel electrophoresis. Methods Enzymol. 182, 425–441. doi: 10.1016/0076-6879(90)82035-Z
Gorris, L. G., Voet, A. C., and van der Drift, C. (1991). Structural characteristics of methanogenic cofactors in the non-methanogenic archaebacterium Archaeoglobus fulgidus. Biofactors 3, 29–35.
Hay, S., Evans, R. M., Levy, C., Loveridge, E. J., Wang, X., Leys, D., et al. (2009). Are the catalytic properties of enzymes from piezophilic organisms pressure adapted? Chembiochem 10, 2348–2353. doi: 10.1002/cbic.200900367
Huovinen, P. (1987). Trimethoprim resistance. Antimicrob. Agents Chemother. 31, 1451–1456. doi: 10.1128/AAC.31.10.1451
Jansson, C., and Skold, O. (1991). Appearance of a new trimethoprim resistance gene, dhfrIX, in Escherichia coli from swine. Antimicrob. Agents Chemother. 35, 1891–1899. doi: 10.1128/AAC.35.9.1891
Jensen, L. J., Kuhn, M., Stark, M., Chaffron, S., Creevey, C., Muller, J., et al. (2009). STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 37, D412–D416. doi: 10.1093/nar/gkn760
Jourand, P., Renier, A., Rapior, S., Miana de Faria, S., Prin, Y., Galiana, A., et al. (2005). Role of methylotrophy during symbiosis between Methylobacterium nodulans and Crotalaria podocarpa. Mol. Plant Microbe Interact. 18, 1061–1068. doi: 10.1094/MPMI-18-1061
Kalyuzhnaya, M. G., Korotkova, N., Crowther, G., Marx, C. J., Lidstrom, M. E., and Chistoserdova, L. (2005). Analysis of gene islands involved in methanopterin-linked C1 transfer reactions reveals new functions and provides evolutionary insights. J. Bacteriol. 187, 4607–4614. doi: 10.1128/JB.187.13.4607-4614.2005
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi: 10.1038/nprot.2015.053
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Li, R. W. (2011). Metagenomics and its Applications in Agriculture, Biomedicine, and Environmental Studies. Hauppauge, NY: Nova Science Publishers.
Li, W., Cowley, A., Uludag, M., Gur, T., McWilliam, H., Squizzato, S., et al. (2015). The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 43, W580–W584. doi: 10.1093/nar/gkv279
Marx, C. J., Bringel, F., Chistoserdova, L., Moulin, L., Farhan Ul Haque, M., Fleischman, D. E., et al. (2012). Complete genome sequences of six strains of the genus Methylobacterium. J. Bacteriol. 194, 4746–4748. doi: 10.1128/JB.01009-12
Marx, C. J., O’Brien, B. N., Breezee, J., and Lidstrom, M. E. (2003). Novel methylotrophy genes of Methylobacterium extorquens AM1 identified by using transposon mutagenesis including a putative dihydromethanopterin reductase. J. Bacteriol. 185, 669–673. doi: 10.1128/JB.185.2.669-673.2003
McNamara, D. E., Cascio, D., Jorda, J., Bustos, C., Wang, T. C., Rasche, M. E., et al. (2014). Structure of dihydromethanopterin reductase, a cubic protein cage for redox transfer. J. Biol. Chem 289, 8852–8864. doi: 10.1074/jbc.M113.522342
McWilliam, H., Li, W., Uludag, M., Squizzato, S., Park, Y. M., Buso, N., et al. (2013). Analysis tool web services from the EMBL-EBI. Nucleic Acids Res. 41, W597–W600. doi: 10.1093/nar/gkt376
Ochsner, A. M., Sonntag, F., Buchhaupt, M., Schrader, J., and Vorholt, J. A. (2015). Methylobacterium extorquens: methylotrophy and biotechnological applications. Appl Microbiol. Biotechnol. 99, 517–534. doi: 10.1007/s00253-014-6240-3
Rasche, M. E., Havemann, S. A., and Rosenzvaig, M. (2004). Characterization of two methanopterin biosynthesis mutants of Methylobacterium extorquens AM1 by use of a tetrahydromethanopterin bioassay. J. Bacteriol. 186, 1565–1570. doi: 10.1128/JB.186.5.1565-1570.2004
Sambrook, J., and Russell, D. W. (2001). Molecular Cloning: a Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Schrader, J., Schilling, M., Holtmann, D., Sell, D., Filho, M. V., Marx, A., et al. (2009). Methanol-based industrial biotechnology: current status and future perspectives of methylotrophic bacteria. Trends Biotechnol. 27, 107–115. doi: 10.1016/j.tibtech.2008.10.009
Scott, J. W., and Rasche, M. E. (2002). Purification, overproduction, and partial characterization of beta-RFAP synthase, a key enzyme in the methanopterin biosynthesis pathway. J. Bacteriol. 184, 4442–4448. doi: 10.1128/JB.184.16.4442-4448.2002
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Snel, B., Lehmann, G., Bork, P., and Huynen, M. A. (2000). STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 28, 3442–3444. doi: 10.1093/nar/28.18.3442
Sy, A., Giraud, E., Samba, R., de Lajudie, P., Gillis, M., and Dreyfus, B. (2001). Nodulation of certain legumes of the genus Crotalaria by the new species Methylobacterium. Can. J. Microbiol. 47, 503–508. doi: 10.1139/w01-044
Szklarczyk, D., Franceschini, A., Kuhn, M., Simonovic, M., Roth, A., Minguez, P., et al. (2011). The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 39, D561–D568. doi: 10.1093/nar/gkq973
Szklarczyk, D., Franceschini, A., Wyder, S., Forslund, K., Heller, D., Huerta-Cepas, J., et al. (2015). STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452. doi: 10.1093/nar/gku1003
Szklarczyk, D., Morris, J. H., Cook, H., Kuhn, M., Wyder, S., Simonovic, M., et al. (2017). The string database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362–D368. doi: 10.1093/nar/gkw937
van Beelen, P., Stassen, A. P., Bosch, J. W., Vogels, G. D., Guijt, W., and Haasnoot, C. A. (1984). Elucidation of the structure of methanopterin, a coenzyme from Methanobacterium thermoautotrophicum, using two-dimensional nuclear-magnetic-resonance techniques. Eur. J. Biochem. 138, 563–571. doi: 10.1111/j.1432-1033.1984.tb07951.x
von Mering, C., Huynen, M., Jaeggi, D., Schmidt, S., Bork, P., and Snel, B. (2003). STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 31, 258–261. doi: 10.1093/nar/gkg034
von Mering, C., Jensen, L. J., Kuhn, M., Chaffron, S., Doerks, T., Kruger, B., et al. (2007). STRING 7–recent developments in the integration and prediction of protein interactions. Nucleic Acids Res. 35, D358–D362. doi: 10.1093/nar/gkl825
von Mering, C., Jensen, L. J., Snel, B., Hooper, S. D., Krupp, M., Foglierini, M., et al. (2005). STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 33, D433–D437. doi: 10.1093/nar/gki005
Vorholt, J. A., Chistoserdova, L., Stolyar, S. M., Thauer, R. K., and Lidstrom, M. E. (1999). Distribution of tetrahydromethanopterin-dependent enzymes in methylotrophic bacteria and phylogeny of methenyl tetrahydromethanopterin cyclohydrolases. J. Bacteriol. 181, 5750–5757.
Keywords: methylotrophic bacteria, dihydrofolate reductase, dihydromethanopterin reductase, methanopterin, one-carbon transfer
Citation: Burton M, Abanobi C, Wang KT-C, Ma Y and Rasche ME (2018) Substrate Specificity Analysis of Dihydrofolate/Dihydromethanopterin Reductase Homologs in Methylotrophic α-Proteobacteria. Front. Microbiol. 9:2439. doi: 10.3389/fmicb.2018.02439
Received: 07 June 2018; Accepted: 24 September 2018;
Published: 11 October 2018.
Edited by:
Marina G. Kalyuzhanaya, San Diego State University, United StatesReviewed by:
Ludmila Chistoserdova, University of Washington, United StatesNorma Cecilia Martinez-Gomez, Michigan State University, United States
Copyright © 2018 Burton, Abanobi, Wang, Ma and Rasche. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Madeline E. Rasche, bWVyYXNjaGVAZnVsbGVydG9uLmVkdQ==