 Yuefei Jin
Yuefei Jin Rongguang Zhang
Rongguang Zhang Weidong Wu2
Weidong Wu2 Guangcai Duan
Guangcai Duan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 08 October 2018
Sec. Virology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.02422
This article is part of the Research Topic Innate Immunity Evasion by Picornaviruses View all 9 articles
Enterovirus (EV) infections are a major threat to global public health, and are responsible for mild respiratory illness, hand, foot, and mouth disease (HFMD), acute hemorrhagic conjunctivitis, aseptic meningitis, myocarditis, severe neonatal sepsis-like disease, and acute flaccid paralysis epidemic. Among them, HFMD is a common pediatric infectious disease caused by EVs of the family Picornaviridae including EV-A71, coxsackieviruses (CV)-A2, CV-A6, CV-A10, and CV-A16. Due to lack of vaccines and specific antiviral therapeutics, millions of children still suffer from HFMD. Innate immune system detects foreign invaders by means of a relatively limited number of sensors, such as pattern recognition receptors (PRRs) [e.g., retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), Toll-like receptors (TLRs), and NOD-like receptors (NLRs)] and even some secreted functional proteins. However, a range of research, highlighted in this review, suggest that EV-associated with HFMD have evolved different strategies to avoid detection by innate immunity via different proteases (e.g., 2A, 3C, 2C, and 3D). Ongoing efforts to better understand virus–host interactions that control innate immunity and then distill how that influences HFMD development promises to have real-world significance. In this review, we address this complex topic in nine sections including multiple proteins associated with PRR and type I interferon (IFN) signaling. Recognizing how EVs linked to HFMD evade host innate immune system, we also describe the interactions between them and, finally, suggest future directions to better inform drug development and public health.
Hand, foot, and mouth disease (HFMD) is a common pediatric infectious disease caused by enteroviruses (EVs) of the family Picornaviridae including EV-A71, and coxsackieviruses (CV)-A2, CV-A6, CV-A10, and CV-A16 (Ho et al., 1999; Solomon et al., 2010; Centers for Disease Control and Prevention [CDC], 2012) (see Table 1). Although usually self-limiting, HFMD can lead to severe complications associated with neural infection or fatal respiratory disease (Chang et al., 1998; Lum et al., 1998). Outbreaks that occurred in Malaysia (1997), Taiwan (1998), Vietnam (2011), and Cambodia (2012) led to 702 child deaths (Ho et al., 1999; Chan et al., 2000; Nguyen et al., 2014; Duong et al., 2016). From 2008 to 2017, accumulated incidence and deaths caused by HFMD in mainland China were approximately 14 million and 3.6 thousand, respectively. Inactivated EV-A71 vaccines in mainland China have been demonstrated to be safe in the target population (infants and young children) and confer a high protective rate against EV-A71 infection-related HFMD (Li et al., 2014). However, to date, millions of children across Asia-Pacific countries still suffer from HFMD every year (Koh et al., 2018).
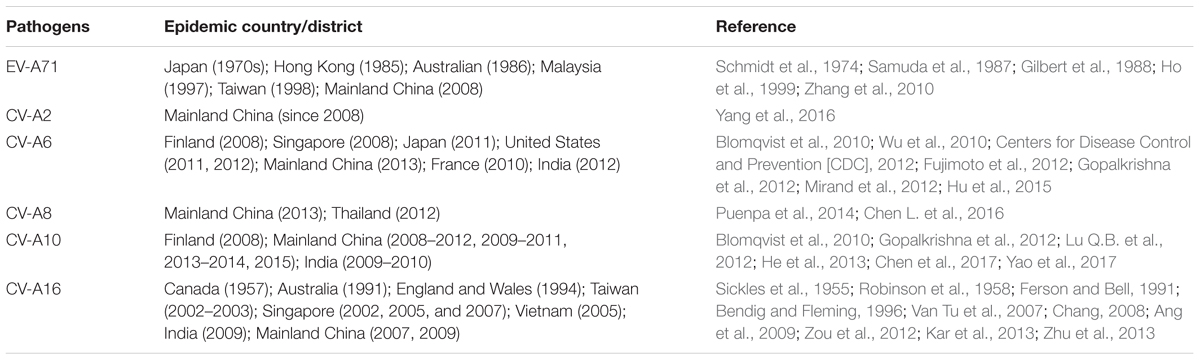
TABLE 1. Summary of Enteroviruses associated with HFMD.
Enteroviruses are small RNA virus identified by a single-stranded, positive-polarity RNA genome of approximately 7.5 kb in size. The viral capsid consists of 60 identical protomers and each contains four different structural proteins, VP1–VP4. By binding the cell surface receptors human scavenger receptor B2 (hSCARB2) and human P-selectin glycoprotein ligand-1 (PSGL-1), EVs invade host cells and subsequently release viral nucleic acids (Yamayoshi et al., 2009; Solomon et al., 2010; Plevka et al., 2012; Lin et al., 2013). As shown in Figure 1, EVs can take advantage of internal ribosome entry site (IRES)-driven translation to subvert host translation machinery (Lee et al., 2017). At different stages, the cleavage of P1–P3 by 2A and 3C proteases results in the synthesis of the capsid proteins VP1–VP4, which subsequently leads to the packaging of the capsid, and seven non-structural proteins: 2A–2C and 3A–3D (Solomon et al., 2010; Plevka et al., 2012). Both sets of these precursors and mature proteins are actively involved with the viral lifecycle, including defense against host innate immunity.
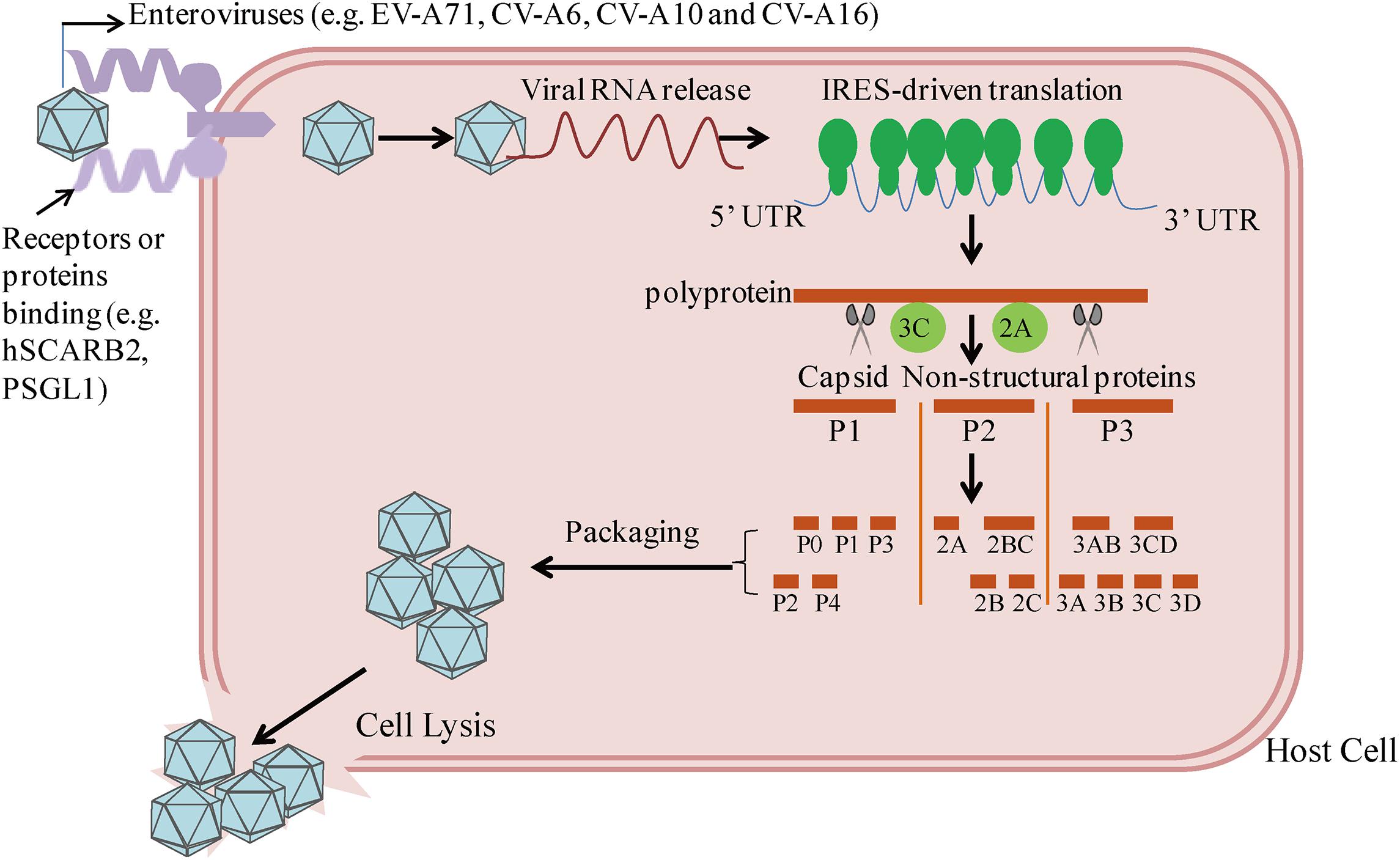
FIGURE 1. Host cell entry and replication by enteroviruses linked to the HFMD epidemic. By binding with receptors or proteins (e.g., hSCARB2, PSGL-1), enteroviruses enter into host cells, followed by viral RNA release. Cap-independent IRES-driven translation produces a single polyprotein followed by proteolytic cleavage into partially processed products and 11 mature products. Among them, VP1-VP4 leads to the packaging of the capsid.
Innate immunity serves as the first line of defense against foreign and dangerous material. Generally, most microbial invaders can be detected and killed within minutes or hours by the body’s defense mechanisms of innate immunity, which do not depend on expansion of antigen-specific lymphocytes but a limited number of secreted proteins and cell-associated receptors (Jensen-Jarolim, 2013). There are at least three broad strategies used by the innate immune system to recognize invading microorganisms (Turvey and Broide, 2010). In the first, innate immunity is equipped with pattern recognition receptors (PRRs) to recognize “microbial non-self” conserved molecular structures [e.g., peptidoglycan, lipopolysaccharide, viral single-stranded RNA (ssRNA) and double-stranded RNA (dsRNA), and viral DNA] termed pathogen-associated molecular patterns (PAMPs). A second fashion equipped by the innate immune system is to monitor dangerous immunologic molecules termed damage-associated molecular patterns (DAMPs) representing common metabolic consequences of infection and inflammation. DAMPs (e.g., unmethylated CpG DNA or pathogen-derived DNA) are upregulated and released during the cell lysis and tissue damage that occurs during infectious and sterile inflammation. For the third strategy, innate immune receptors detect “missing self” molecules expressed by normal healthy cells but not by infected cells or microbes. Recognition of these molecules indicates that “everything is normal,” and an inhibitory signal is followed to suppress activation of the immune response against host tissues.
Enteroviruses have evolved and developed different strategies to evade the innate immune system to facilitate replication inside the host cell. There are abundant studies concerning host innate immunity evasion by EVs. In the present review, we mainly focus on the innate immune evasion by EVs associated with HFMD epidemic.
Pattern recognition receptors residing in membranes and the cytosol of innate immune cells such as macrophages, plasmacytoid dendritic cells (pDCs), and dendritic cells (DCs) have evolved to recognize foreign components essential to microbial pathogenicity (Akira et al., 2006). Several classes of PRRs, including retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), Toll-like receptors (TLRs), and NOD-like receptors (NLRs), are responsible for inducing the production of type I IFNs that are important innate immune regulators during viral infections (Akira et al., 2006). Thus far, three members of RLRs have been identified: RIG-I, MDA5, and Laboratory of Genetics and Physiology 2 (LGP2) which mainly recognize viral dsRNA (Yoneyama et al., 2004). So far, 13 mammalian TLRs have been described, expressed either on the cell surface or in the endosomal compartments. Among them, cell surface TLRs (e.g., TLR1, TLR2, TLR4, TLR5, TLR6, and TLR11) sense lipids, lipoproteins, or peptidoglycans from bacteria, fungi, or protozoa. Endosomal TLR3, TLR7/8, and TLR9 detect viral dsRNA, ssRNA, and endogenous DNA, respectively (Baccala et al., 2007). Recognition of bacterial proteins and viral RNA in the cytoplasm by some NLRs leads to the assembly of inflammasomes followed by the activation of inflammatory caspases. The best known inflammasome-forming NLRs are NLR family pyrin domain-containing protein-1 (NLRP1), NLRP3, and NLR family caspase recruitment domain-containing protein-4 (NLRC4) (Broz and Dixit, 2016). Activation of the above immune receptors controls production of type I interferon (IFN), IFN-α/β, that are secreted by many cell types following a viral infection and can cause neighboring cells to express genes with potential antiviral effects (Muller et al., 1994). Pretreatment with type I IFN can suppress EV-A71 and CV-A16 infection in vivo (Yang et al., 2015; Sun et al., 2016), which can be explained by effective strategies employed by EVs to avoid and/or attenuate production of IFN-α/β and thus their effects on immune responses. Taken together, blockade of signaling by PRR provides a key strategy for evasion of innate immunity employed by EV-A71 and CV-A16.
Retinoic acid-inducible gene I-I is an intracellular dsRNA sensor. After recognizing viral dsRNA, it undergoes conformational alterations and post-translational modification including K63-linked ubiquitination on lysine residues of the CARD and C-terminal domains, and further regulates type I IFN-mediated host antiviral innate immunity (Maelfait and Beyaert, 2012). EV-A71 infection inhibits type I IFN signaling by downregulating RIG-I ubiquitination in human rhabdomyosarcoma (RD) cells. However, upregulation of RIG-I ubiquitination by transfection with HA-Ub vector increases expression of IFN-β and IFN-stimulated genes (ISGs) after EV71 infection (Chen N. et al., 2016). In another study, it was found that EV-A71 3C protein (3Cpro) can inhibit IFN-β expression by targeting the adaptor RIG-I in 293T cells transfected with vectors that can increase amounts of 3Cpro and RIG-I (Lei et al., 2010). MicroRNAs (miRNAs) have critical roles in regulating virus-host interactions (Cui et al., 2010). Previous studies suggested that the ubiquitination status of RIG-I is regulated by CYLD, a tumor suppressor originally identified as a genetic defect in familial cylindromatosis (Bignell et al., 2000). Downregulation of miR-526a by EV-A71 3Cpro impairs RIG-I-mediated type I IFN production through IRF7 cleavage, and downregulation of CYLD in THP-1 cells, while ectopic miR-526a expression inhibits the EV71 replication (Xu et al., 2014). At the same time, pretreatment with all-trans retinoic acid (ATRA) provides antiviral effects through enhancing RIG-I signaling pathway in human monocytic cell line U937 (Chen et al., 2014). However, the mechanism by which ATRA affects RIG-I signaling remains unclear. Together, the lines of evidence described above suggest that inhibition of RIG-I activation by 3Cpro provides a strategy of innate immune evasion employed by EV-A71 (see Figure 2).
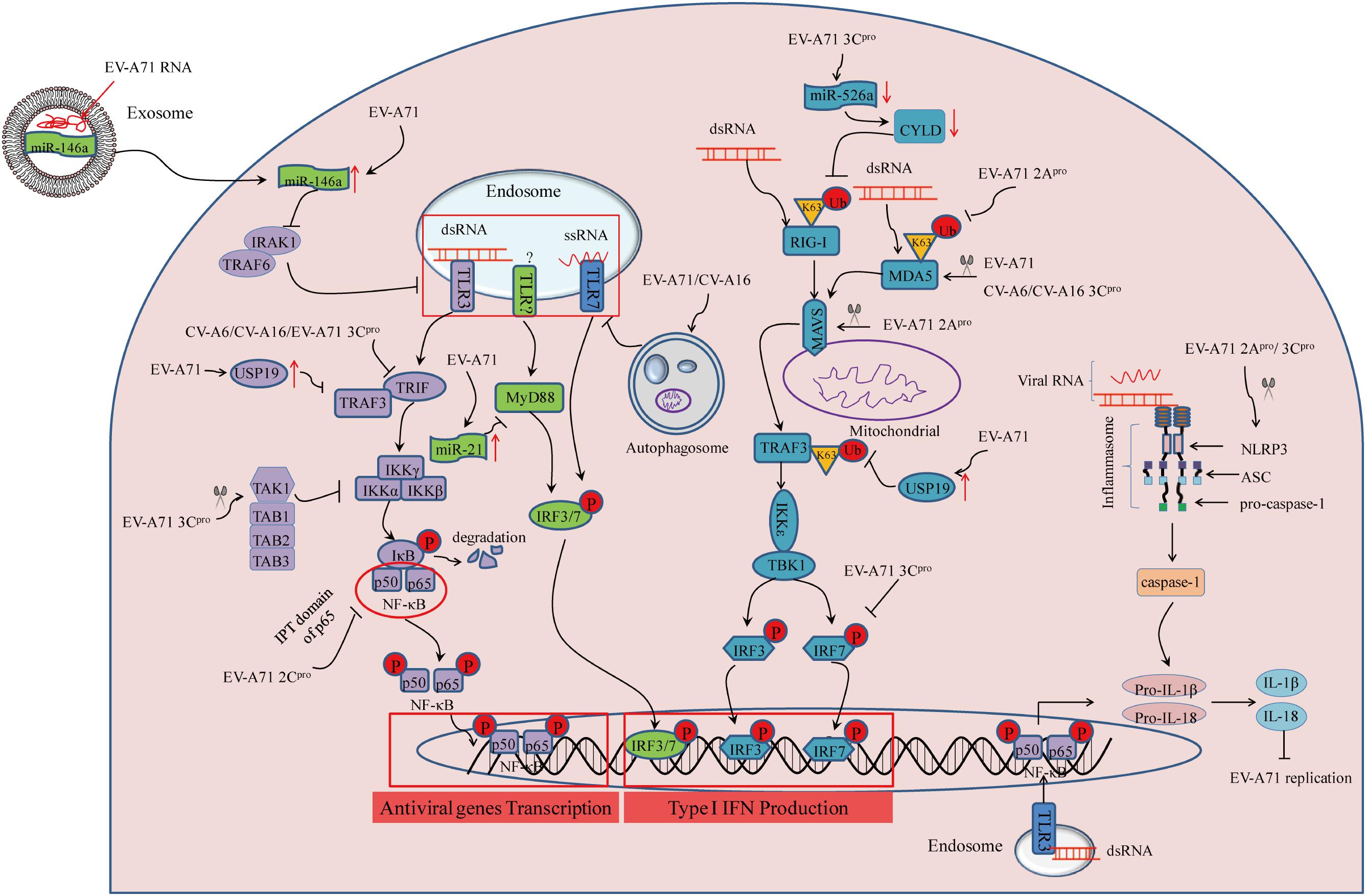
FIGURE 2. Summary of PRR-mediated innate immunity during enterovirus infection linked to the HFMD epidemic Three cytosolic signaling pathways inhibited by viral infection are represented, as follows: (1) TLR activation leads to signaling through TRIF, TRAF3, and IKKα/β to turn on NF-κB-p50/p65 nuclear transport, or MyD88-mediated IRF3/7 activation; (2) RIG-I and MDA5 activation requires binding to dsRNA and subsequent K63-linked ubiquitination. This signals through mitochondrial-bound MAVS, leading to TBK1/IKKε activation to initiate activity of the transcription factors IRF3/7; (3) assembly of NLRP3 inflammasome requires the sensor NLRP3, the adaptor protein ASC, and pro-caspase-1. Recognition of viral RNA leads to NLRP3 activation, which subsequently results in inflammatory caspase-1activation and IL-1β/IL-18 secretion.
MDA5, a member of the RLRs family, senses intracellular dsRNA, has a similar structure as RIG-I (Yoneyama et al., 2004). EV-A71 infection enhances MDA5 degradation, and over-expression of MDA5 can reverse the suppression of type I IFN transcription (Kuo et al., 2013). For further study, Feng et al. (2014) suggested that EV-A71-derived 2A protein (2Apro) counteracts the antiviral type I IFN response by cleaving MDA5 in infected cells. Similar to RIG-I, enhancing K63-linked ubiquitination by ARRDC4 results in activation of the downstream innate signaling pathway of MDA5 during EV-A71 infection (Meng et al., 2017). ARRDC4 functions as an adapter, recruiting ubiquitin-protein ligases to their specific substrates. But unlike EV-A71, 3Cpro of CV-A16, and CV-A6 is responsible for the disruption of MDA5 in 293T cells transfected with relative vectors that can increase amounts of 3Cpro (Rui et al., 2017). Therefore, inhibition of MDA5 activation provides a novel strategy equipped by EV-A71, CV-A16, and CV-A6 to escape host antiviral innate immunity (see Figure 2).
Mitochondrial antiviral-signaling adaptor protein (MAVS) serves as a key adaptor in cellular antiviral innate immunity, residing on the outer membranes of the mitochondria and peroxisomes. MAVS is best known to initiate TANK-binding kinase 1 (TBK1)-dependent and nuclear factor-kappa B (NF-κB)-dependent antiviral gene transcription (Seth et al., 2005). EV-A71 2Apro was confirmed to suppress interferon regulatory factor (IRF)3 signaling through the cleavage of MAVS, resulting in IFN-α/β reduction in HeLa cells (Feng et al., 2014). Meanwhile, another study revealed that EV-A71 infection caused morphologic and functional changes of the mitochondria, whereby in vitro cleavage assay indicated that EV-A71 approached MAVS and led to MAVS cleavage by 2Apro (Wang et al., 2013). Combining the results from the above two publications provides a good explanation for the immune evasion mechanisms employed by EV-A71. For studies of CV-A16 and CV-A6, evidence suggested that the association of adaptor MAVS and MDA5 was disrupted by 3Cpro in a dose dependent manner in 293T cells transfected with plasmids encoding MDA5-N-Myc, MAVS-Flag, and HA-3C (Rui et al., 2017). In considering the above results, it is believed that EV-A71, CV-A16, and CV-A6 share a common immune evasion mechanism by inhibiting MAVS activation (see Figure 2). However, it remains to be definitively shown whether there is direct or indirect interaction between MAVS and these viruses.
TLR signaling is essential to induce type I IFN production during EV-A71 infection (Wang et al., 2016). To escape host innate immunity, EV-A71 3Cpro has been found to block IFN-β production through the adapter protein TIR-domain-containing adapter-inducing interferon-β (TRIF) in response to endosomal TLR3 activation (Lei et al., 2011). Similar to TLR3, TLR7 also resides on endosomes to sense foreign viral ssRNA. Increased EV-A71 and CV-A16 replication induced by autophagy leads to the degradation of endosomes, which further suppresses TLR7-mediated type I IFN responses in human bronchial epithelial (16HBE) cells by using autophage inhibitor (3-MA) and laser confocal (Song et al., 2018). Likewise, upregulation of miR-21 upon EV-A71 infection can suppress myeloid differentiation factor 88 (MyD88) downstream of TLRs, which subsequently blocks type I IFN-mediated antiviral responses in vitro with miR-21 inhibitor and transfection of miRNA and siRNA (Feng et al., 2017). In addition, CV-A16 and CV-A6 3Cpro are proposed to subvert host innate immune responses by suppressing TLR-mediated NF-κB signaling in 293T cells transfected with NF-κB-luc promoter reporter and Flag-TLR3 expression vector alone or with CV-A16 3Cpro or CV-A6 3Cpro or their protease mutation H40D (Rui et al., 2017). Therefore, direct or indirect inhibition of TLR-dependent antiviral signaling are effective mechanisms of immune evasion employed by EV-A71, CV-A16, and CV-A6 (see Figure 2).
The NLRP3 inflammasome complex consists of pro-caspase-1 (casp1), ASC, and NLRP3. Activation of the NLRP3 inflammasome leads to the secretion of interleukin (IL)-1β and IL-18 to provide host protective antiviral effects (Broz and Dixit, 2016). In vivo experiments suggest that pretreatment with recombinant IL-18 can reverse EV-A71 infection-induced pathogenesis (Li et al., 2017). EV-A71 interferes with inflammasome assembly through the cleavage of NLRP3 by 2Apro and 3Cpro in 293T cells transfected with wild-type NLRP3 or NLRP3 mutants. Meanwhile, 3Cpro of EV-A71 interacts with NLRP3 and suppresses IL-1β secretion (Wang H. et al., 2015). By contrast, EV-A71-derived 3D protein (3Dpro) binds with NLRP3 to facilitate the assembly of inflammasome complexes, which results in the secretion of IL-1β in 293T cells transfected with plasmids encoding pro-IL-1β, Flag-pro-caspsase-1, Flag-NLRP3, Flag-ASC, and EV-A71 3Dpro (Wang W. et al., 2017). Together, EV-A71 is able to suppress NLRP3 inflammasome activation as a mode of immune evasion (see Figure 2). Nonetheless, additional studies are warranted to elucidate inconsistent results regarding IL-1β production and determine the mechanism of IL-18 secretion during EV-A71 infection.
Interferon regulatory factor-mediated expression of type I IFN and IFN-inducible genes plays a central role in responses to viral infection (Janeway and Medzhitov, 2002). EV-A71 can suppress ISG expression by blocking IRF3 activation in HeLa cells (Lei et al., 2011). Another study revealed that EV-A71 3Cpro was responsible for the blockade of IRF3 activation and IFN-α/β production in 293T cells transfected with GFP-IRF3 alone or along with Flag-3Cpro (Lei et al., 2010). However, Lei et al. (2013) found that EV-A71 3Cpro was required to cleave IRF7 rather than IRF3 to delay the type I IFN response in vitro with transfection of GFP-3Cpro alone, or IRF7 and GFP-3Cpro along with IFN-β-Luc. Regardless, these lines of evidence suggest that inhibition of IRF activation is an effective mechanism of immune evasion by EV-A71 (see Figure 2).
Type I IFNs (IFN-α/β) and type II IFN (IFN-γ) form the first line of defense against viral infection, and also play a critical role in immunosurveillance for malignant cells. The binding of IFN-α/β and IFN-γ to their specific receptors, type I IFN receptors (IFNAR1 and IFNAR2) and type II IFN receptors (IFNGR1 and IFNGR2), leads to the rapid autophosphorylation and activation of Janus-activated kinase (Jak)/signal transducer and activator of transcription (STAT) pathways which in turn regulates IFN-α/β transcription (Darnell et al., 1994). EV-A71 2Apro blocks STAT1, STAT2, Jak1, and Tyk2 phosphorylation by reducing IFNAR1 expression in 293T cells transfected with 2Apro (Lu J. et al., 2012). In another study, EV-A71 2Apro attenuated IFN-γ-induced serine phosphorylation of STAT1 by blocking ERK signaling in mouse embryonic fibroblasts (MEFs) transfected with 2Apro along with IFN-γ treatment, while EV-A71 3Dpro attenuation of IFN-γ signaling was accompanied by a STAT1 decrease in MEFs transfected with 3Dpro (Wang C. et al., 2015). Furthermore, EV-A71-induced miR-124 can antagonize the antiviral activity of STAT3 (Chang Z. et al., 2017). Based on the above findings, nearly all studies to date provide consistent evidence that EV-A71 2Apro and 3Cpro act as antagonists of cellular type I IFN signaling. In addition, Liu et al. (2014) found that EV-A71 can also inhibit the type I antiviral pathway by downregulating Jak1 expression in vitro, while IFNAR1 expression does not significantly change in infected cells. Likewise, EV-A71 suppresses IFN-β production by blocking Jak/STAT signaling through degradation of karyopherin-α1 (KPNA1), a nuclear localization signal receptor for p-STAT1 in vitro, although this appears to occur independently of EV-A71 2Apro and 3Cpro (Wang C. et al., 2015). These reports suggest EV-A71-mediated inhibition of IFN signaling and Jak/STAT activation suppresses type I IFN production (see Figure 3). However, controversies remain, and more studies will be needed to reveal the precise mechanisms of IFN signaling and Jak/STAT signaling inhibition during EV-A71 infection.
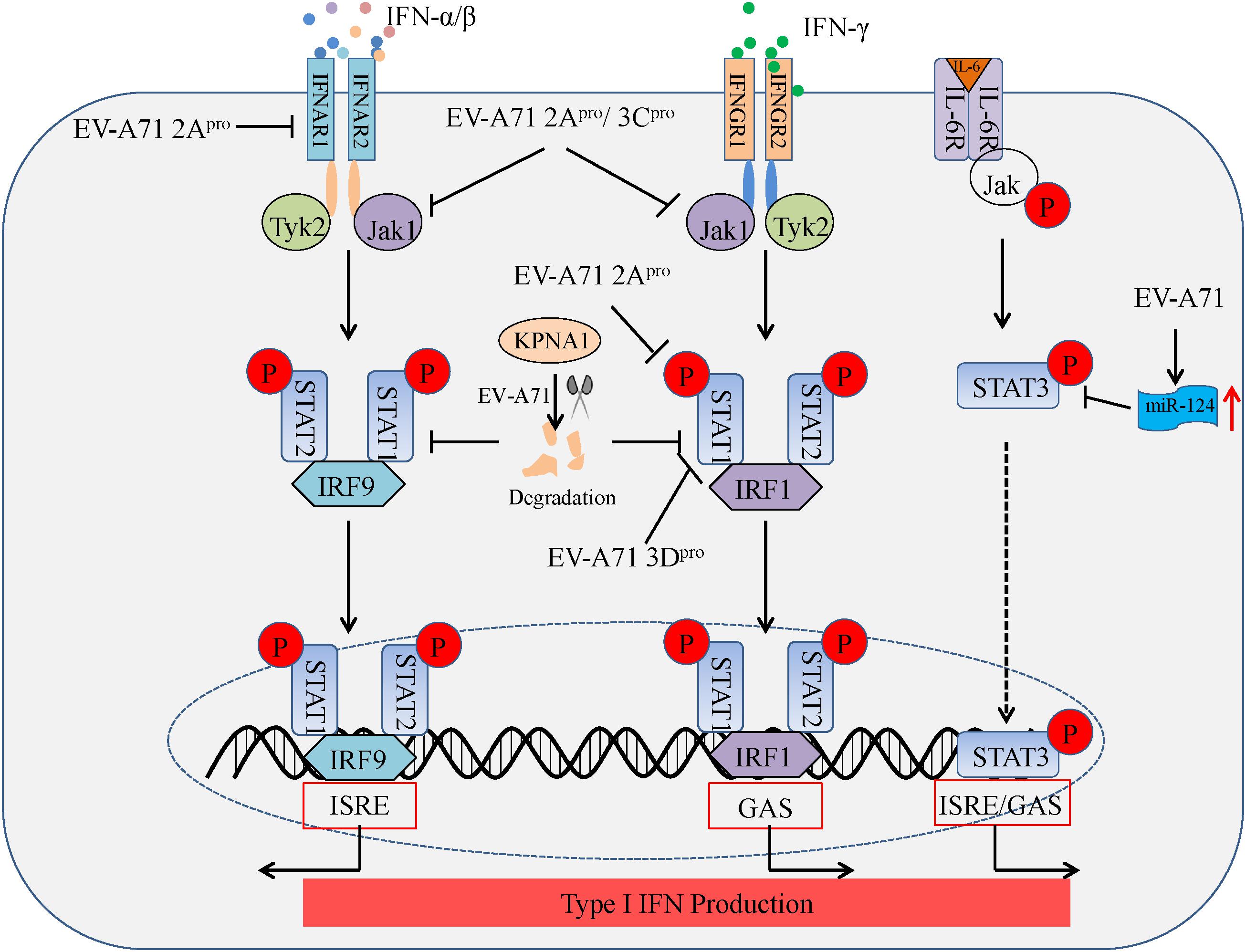
FIGURE 3. Summary of IFN signaling and Jak/STAT signaling suppressed by enteroviruses linked to the HFMD epidemic. Binding of IFNAR by type I IFN (IFN-α/β) triggers downstream kinases, Jak1 and Tyk2. Jak1 and Tyk2 phosphorylate STAT1 and STAT2, which leads to the formation of the STAT1–IRF9–STAT2 complex. This complex translocates to the nucleus and initiates transcription of IFN-stimulated response elements (ISREs) in specific genes. Likewise, binding of IFNGR by type II IFN (IFN-γ) triggers downstream kinases, Jak1 and Tyk2. Jak1 and Tyk2 phosphorylate STAT1 and STAT2, which leads to the formation of the STAT1-IRF1-STAT2 complex. This complex translocates to the nucleus and initiates transcription of IFN-γ-activated sequences (GAS) in additional genes. Nuclear accumulation of phosphorylated STAT3 (p-STAT3) depends on the binding of IL-6R and IL-6, whereby p-STAT3 also initiates transcription via ISRE/GAS elements.
Tumor necrosis factor receptor-associated factor (TRAF) 3 is a crucial adaptor molecule for TLR3- and RLR-mediated type I IFN signaling (Kawai and Akira, 2006; Loo and Gale, 2011). It has been suggested that EV-A71 infection induces ubiquitin-specific protease 19 (USP19) gene expression, which negatively regulates type I IFN signaling by suppressing TRAF3 ubiquitination of K63-linkage in 293T cells transfected with Flag-TRAF3, HA-USP19, and HA-Ub (Gu et al., 2017). This evidence indicates that EV-A71 can escape host innate immunity by suppressing TRAF3 ubiquitination via USP19 induction (see Figure 2). In future studies, elucidating the upstream molecular regulators of TRAF3 will be critical to gain a more complete understanding of the mechanisms of EV-A71 infection-induced innate immune evasion. In addition, targeted inhibition of USP19 may be useful for the treatment of EV-A71 infection-associated HFMD.
The NF-κB p65/p50 heterodimer is the most abundant signaling complex of the NF-κB family, and plays a key role in host defense against viral infection (Rahman and McFadden, 2011). Nuclear transport of p65/p50 heterodimers promotes the secretion of cytokines and chemokines, which are significant for host defense against viral infection through innate immunity (Fagerlund et al., 2005). As reported (Du et al., 2015), by interacting with the IPT domain of p65, EV-A71 2Cpro suppresses the formation of p65/p50 heterodimers in 293T cells transfected with 2Cpro and truncation constructs of p65, which provides another novel strategy employed by EV-A71 to escape innate immunity (see Figure 2). In addition, NF-κB activation can also be inhibited by EV-A71 3Cpro through cleavage of the transforming growth factor-l-activated kinase 1 (TAK1) complex, a TAK1/TAK1-binding protein 1 (TAB1)/TAB2/TAB3 complex, in 293 cells transfected with relative plasmids (Lei et al., 2014). Whether EV-A71 infection affects degradation of NF-κB inhibitor-α (IκBα) as a mechanism to inhibit the NF-κB pathway is essential to be identified in the future.
ZAP, a mammalian host restriction factor, has been demonstrated to suppress RNA virus replication (Zhang et al., 2007). EV-A71 3Cpro was found to induce ZAP cleavage, while over-expression of ZAP can inhibit EV-A71 replication in 293T cells with ZAP transfection (Xie et al., 2018). EV-A71 can control innate immunity by regulating miRNA functions (Ho et al., 2014; Fu et al., 2017). For example, EV-A71 infection upregulates miR-146 expression, which further suppresses TLR signaling and type I IFN production by targeting IL-1 receptor-associated kinase 1 (IRAK1) and TRAF6 in RD cells transfected with the miR-146a over-expressing vector (Ho et al., 2014). The high level miR-146 can be from exosomes secreted by EV-A71-infected cells, which is packaged with exosomal viral RNA, and that in turn facilitates EV-A71 replication in the recipient cells by suppressing type I IFN response (see Figure 2; Fu et al., 2017). IFN-induced dsRNA-activated protein kinase R (PKR), an IFG, acts as a PRR recognizing dsRNA. A short N-terminal PKR fragment originates from PKR cleavage mediated by EV-A71 3Cpro can enhance EV-A71 replication in RD cells transfected with the vector, increasing concentrations of plasmids encoding the PKR (1–188) or PKR (1–188) K64E fragment. Therefore, inhibition of ZAP, miR-146a induction, or PKR cleavage may represent another mechanism equipped by EV-A71 to escape host antiviral responses. These studies provide clues for the design of therapeutic strategies against EV-A71 infection by targeting ZAP, miR-146, and PKR.
Enterovirus infections continue to pose a significant public health threat worldwide, and are associated with the epidemics of mild respiratory illness, HFMD, acute hemorrhagic conjunctivitis, aseptic meningitis, myocarditis, severe neonatal sepsis-like disease, and acute flaccid paralysis. Among these afflictions, HFMD epidemics are serious public health issues for children from Asia-Pacific countries. However, few vaccines and specific antiviral therapeutics are applied for disease control and clinical practice, because of the unique viral structure and mechanisms of innate immunity evasion. The body of evidence presented above suggests that EVs linked to the HFMD epidemic are equipped with various unique strategies to evade multiple arms of the innate immune response by suppressing intracellular antiviral type I IFN signaling, regulating miRNAs, or by modulating functional protein expression (see Table 2). In particular, shielding PRRs recognition becomes a main fashion of innate immunity evasion by 2A, 3C, and 3D proteases derived from EV-A71, CV-A6, or CV-A16. Another mechanism is to block critical intracellular molecules such as NF-κB p65/p50 and ZAP to interfere with antiviral effects. Additionally, more and more studies suggest that miRNAs such as miR-146a, miR-124, miR-21, and miR-526a have a crucial role in modulating innate immune response through PRR signaling and type I IFN signaling. In summary, EVs linked to the HFMD epidemic are relatively efficient at modulating innate immunity, and this property allows these viruses to successfully establish infection in humans.

TABLE 2. Summary of viral proteins and their targets of host proteins or pathways.
Recently, many drugs have been developed and showed protective antiviral effects in cellular and mouse models (Hung et al., 2011; Zhang et al., 2013; Lin et al., 2016). However, most of them still remain unknown for clinical practice. Both in vitro and in vivo studies have indicated that type I IFN administration can suppress EV-A71 replication and increase survival rate (Liu et al., 2005; Yi et al., 2011). A multicenter, randomized, double-blind clinical trial suggested that IFN-α2b spray could rapidly relieved symptoms of HFMD (Lin et al., 2016). Collectively, these layers of evidence indicate that type I IFN should be candidate drug for clinical treatment in the near future. As mentioned earlier, ATRA and ARRDC4 provide antiviral effects through enhancing RIG-I signaling and MDA5 signaling during EV-A71 infection in vitro and in vivo (Chen et al., 2014; Meng et al., 2017). Due to lack of clinical experiment, we cannot say rashly that ATRA and ARRDC4 are useful for the real-world clinical scenarios, but this evidence justifiably matter most to clinicians. Based on above evidence, the earlier event of type I IFN reduction is because of the interactions of viral proteases with PRR signaling or functional proteins. Synthesis of 2A, 3C, and 3D inhibitors should be useful for HFMD treatment. An in vitro study found that combination of 3C inhibitors and IFN-α have synergistic effects on EV-A71 replication (Hung et al., 2011). Another study suggested that siRNAs targeting the 2A region of the EV-A71 genome exerted antiviral effects in vitro (Liu et al., 2016). However, the other viral proteases still can reduce type I IFN production. Therefore, understanding the structural roles of 2A, 3C, and 3D proteases will provide crucial information for the design of a broad spectrum inhibitor. Regulation of miRNAs expression is additional developing therapeutic strategies for HFMD. Synthesis of miR-146a, miR-21, and miR-124 inhibitors and miR-526a intervention will be beneficial for HFMD treatment in the future.
Although previous studies have made significant progress in obtaining new knowledge of the interplay between EVs linked to the HFMD epidemic and innate immunity, several important questions remain to be clarified in the future studies. First, due to minimal evidence suggesting that ssRNA or dsRNA are ligands for PRR stimulation during EVs infection, it is important to identify viral and/or host factors that contribute to the PRR recognition. Second, does mitochondrial damage play any role in RLR-mediated signaling? MAVS residing on outer membrane of mitochondria is downstream of RLR signaling, and a previous study has demonstrated that EV-A71 infection can cause mitochondrial damage (Dang et al., 2017). Therefore, the inhibition of RIG-I, MDA5, and MAVS may be responsible for the disruption of the outer mitochondrial membrane. Third, the mechanism of EV-A71 regulation of NLRP3 assembly is inconsistent to date, and how EV-A71 affects NLRP3 activation and IL-1β secretion should be confirmed in the future. Additionally, the precise targets of EV-A71 in regulating IRF and IFN signaling remain to be fully revealed. Last but not the least, diagnostic meaning of miRNAs during HFMD development should be considered in the future study. As HFMD continues to cause threat to children’s health, identifying the viral factors that antagonize innate immunity will be helpful to develop future therapeutics to restrict the burden of HFMD.
YJ collected the references and wrote this manuscript. RZ, WW, and GD critically reviewed and revised the article. All authors contributed to article’s edits and approved the final manuscript.
This work was funded by the National Natural Science Foundation of China (81172740) and (81573205); Key Scientific Research Projects in Colleges and Universities of Henan Province (15A330003); and The Outstanding Doctoral Thesis Training Fund of Zhengzhou University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Dr. Jonathan Poe of Duke University Medical Center for English editing.
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801. doi: 10.1016/j.cell.2006.02.015
Ang, L. W., Koh, B. K., Chan, K. P., Chua, L. T., James, L., and Goh, K. T. (2009). Epidemiology and control of hand, foot and mouth disease in Singapore, 2001-2007. Ann. Acad. Med. Singapore 38, 106–112.
Baccala, R., Hoebe, K., Kono, D. H., Beutler, B., and Theofilopoulos, A. N. (2007). TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat. Med. 13, 543–551. doi: 10.1038/nm1590
Bendig, J. W., and Fleming, D. M. (1996). Epidemiological, virological, and clinical features of an epidemic of hand, foot, and mouth disease in England and Wales. Commun. Dis. Rep. CDR Rev. 6, R81–R86.
Bignell, G. R., Warren, W., Seal, S., Takahashi, M., Rapley, E., Barfoot, R., et al. (2000). Identification of the familial cylindromatosis tumour-suppressor gene. Nat. Genet. 25, 160–165. doi: 10.1038/76006
Blomqvist, S., Klemola, P., Kaijalainen, S., Paananen, A., Simonen, M. L., Vuorinen, T., et al. (2010). Co-circulation of coxsackieviruses A6 and A10 in hand, foot and mouth disease outbreak in Finland. J. Clin. Virol. 48, 49–54. doi: 10.1016/j.jcv.2010.02.002
Broz, P., and Dixit, V. M. (2016). Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16, 407–420. doi: 10.1038/nri.2016.58
Centers for Disease Control and Prevention [CDC] (2012). Notes from the field: severe hand, foot, and mouth disease associated with coxsackievirus A6 - Alabama, Connecticut, California, and Nevada, November 2011-February 2012. MMWR Morb. Mortal. Wkly. Rep. 61, 213–214.
Chan, L. G., Parashar, U. D., Lye, M. S., Ong, F. G., Zaki, S. R., Alexander, J. P., et al. (2000). Deaths of children during an outbreak of hand, foot, and mouth disease in sarawak, malaysia: clinical and pathological characteristics of the disease. For the outbreak study group. Clin. Infect. Dis. 31, 678–683. doi: 10.1086/314032
Chang, L. Y. (2008). Enterovirus 71 in Taiwan. Pediatr. Neonatol. 49, 103–112. doi: 10.1016/S1875-9572(08)60023-6
Chang, L. Y., Huang, Y. C., and Lin, T. Y. (1998). Fulminant neurogenic pulmonary oedema with hand, foot, and mouth disease. Lancet 352, 367–368. doi: 10.1016/S0140-6736(98)24031-1
Chang, Y. H., Lau, K. S., Kuo, R. L., and Horng, J. T. (2017). dsRNA binding domain of PKR is proteolytically released by enterovirus A71 to facilitate viral replication. Front. Cell. Infect. Microbiol. 7:284. doi: 10.3389/fcimb.2017.00284
Chang, Z., Wang, Y., Bian, L., Liu, Q., and Long, J. E. (2017). Enterovirus 71 antagonizes the antiviral activity of host STAT3 and IL-6R with partial dependence on virus-induced miR-124. J. Gen. Virol. 98, 3008–3025. doi: 10.1099/jgv.0.000967
Chen, L., Yang, H., Wang, C., Yao, X. J., Zhang, H. L., Zhang, R. L., et al. (2016). Genomic characteristics of coxsackievirus A8 strains associated with hand, foot, and mouth disease and herpangina. Arch. Virol. 161, 213–217. doi: 10.1007/s00705-015-2646-1
Chen, N., Li, X., Li, P., Pan, Z., Ding, Y., Zou, D., et al. (2016). Enterovirus 71 inhibits cellular type I interferon signaling by inhibiting host RIG-I ubiquitination. Microb. Pathog. 100, 84–89. doi: 10.1016/j.micpath.2016.09.001
Chen, M., He, S., Yan, Q., Xu, X., Wu, W., Ge, S., et al. (2017). Severe hand, foot and mouth disease associated with Coxsackievirus A10 infections in Xiamen, China in 2015. J. Clin. Virol. 93, 20–24. doi: 10.1016/j.jcv.2017.05.011
Chen, S., Yang, Y., Xu, J., Su, L., and Wang, W. (2014). Effect of all-trans-retinoic acid on enterovirus 71 infection in vitro. Br. J. Nutr. 111, 1586–1593. doi: 10.1017/S0007114513004133
Cui, L., Guo, X., Qi, Y., Qi, X., Ge, Y., Shi, Z., et al. (2010). Identification of microRNAs involved in the host response to enterovirus 71 infection by a deep sequencing approach. J. Biomed. Biotechnol. 2010:425939. doi: 10.1155/2010/425939
Dang, D., Zhang, C., Zhang, R., Wu, W., Chen, S., Ren, J., et al. (2017). Involvement of inducible nitric oxide synthase and mitochondrial dysfunction in the pathogenesis of enterovirus 71 infection. Oncotarget 8, 81014–81026. doi: 10.18632/oncotarget.21250
Darnell, J. E. Jr., Kerr, I. M., and Stark, G. R. (1994). Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264, 1415–1421. doi: 10.1126/science.8197455
Du, H., Yin, P., Yang, X., Zhang, L., Jin, Q., and Zhu, G. (2015). Enterovirus 71 2C protein inhibits NF-kappaB activation by binding to RelA(p65). Sci. Rep. 5:14302. doi: 10.1038/srep14302
Duong, V., Mey, C., Eloit, M., Zhu, H., Danet, L., Huang, Z., et al. (2016). Molecular epidemiology of human enterovirus 71 at the origin of an epidemic of fatal hand, foot and mouth disease cases in Cambodia. Emerg. Microbes Infect. 5:e104. doi: 10.1038/emi.2016.101
Fagerlund, R., Kinnunen, L., Kohler, M., Julkunen, I., and Melen, K. (2005). NF-{kappa}B is transported into the nucleus by importin {alpha}3 and importin {alpha}4. J. Biol. Chem. 280, 15942–15951. doi: 10.1074/jbc.M500814200
Feng, N., Zhou, Z., Li, Y., Zhao, L., Xue, Z., Lu, R., et al. (2017). Enterovirus 71-induced has-miR-21 contributes to evasion of host immune system by targeting MyD88 and IRAK1. Virus Res. 237, 27–36. doi: 10.1016/j.virusres.2017.05.008
Feng, Q., Langereis, M. A., Lork, M., Nguyen, M., Hato, S. V., Lanke, K., et al. (2014). Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J. Virol. 88, 3369–3378. doi: 10.1128/JVI.02712-13
Ferson, M. J., and Bell, S. M. (1991). Outbreak of Coxsackievirus A16 hand, foot, and mouth disease in a child day-care center. Am. J. Public Health 81, 1675–1676. doi: 10.2105/AJPH.81.12.1675
Fu, Y., Zhang, L., Zhang, F., Tang, T., Zhou, Q., Feng, C., et al. (2017). Exosome-mediated miR-146a transfer suppresses type I interferon response and facilitates EV71 infection. PLoS Pathog. 13:e1006611. doi: 10.1371/journal.ppat.1006611
Fujimoto, T., Iizuka, S., Enomoto, M., Abe, K., Yamashita, K., Hanaoka, N., et al. (2012). Hand, foot, and mouth disease caused by coxsackievirus A6, Japan, 2011. Emerg. Infect. Dis. 18, 337–339. doi: 10.3201/eid1802.111147
Gilbert, G. L., Dickson, K. E., Waters, M. J., Kennett, M. L., Land, S. A., and Sneddon, M. (1988). Outbreak of enterovirus 71 infection in Victoria, Australia, with a high incidence of neurologic involvement. Pediatr. Infect. Dis. J. 7, 484–488. doi: 10.1097/00006454-198807000-00007
Gopalkrishna, V., Patil, P. R., Patil, G. P., and Chitambar, S. D. (2012). Circulation of multiple enterovirus serotypes causing hand, foot and mouth disease in India. J. Med. Microbiol. 61(Pt 3), 420–425. doi: 10.1099/jmm.0.036400-0
Gu, Z., Shi, W., Zhang, L., Hu, Z., and Xu, C. (2017). USP19 suppresses cellular type I interferon signaling by targeting TRAF3 for deubiquitination. Future Microbiol. 12, 767–779. doi: 10.2217/fmb-2017-0006
He, Y. Q., Chen, L., Xu, W. B., Yang, H., Wang, H. Z., Zong, W. P., et al. (2013). Emergence, circulation, and spatiotemporal phylogenetic analysis of coxsackievirus a6- and coxsackievirus a10-associated hand, foot, and mouth disease infections from 2008 to 2012 in Shenzhen, China. J. Clin. Microbiol. 51, 3560–3566. doi: 10.1128/JCM.01231-13
Ho, B. C., Yu, I. S., Lu, L. F., Rudensky, A., Chen, H. Y., Tsai, C. W., et al. (2014). Inhibition of miR-146a prevents enterovirus-induced death by restoring the production of type I interferon. Nat. Commun. 5:3344. doi: 10.1038/ncomms4344
Ho, M., Chen, E. R., Hsu, K. H., Twu, S. J., Chen, K. T., Tsai, S. F., et al. (1999). An epidemic of Enterovirus 71 infection in Taiwan. Taiwan Enterovirus epidemic working group. N. Engl. J. Med. 341, 929–935. doi: 10.1056/NEJM199909233411301
Hu, Y. Q., Xie, G. C., Li, D. D., Pang, L. L., Xie, J., Wang, P., et al. (2015). Prevalence of coxsackievirus A6 and Enterovirus 71 in hand, foot and mouth disease in nanjing, China in 2013. Pediatr. Infect. Dis. J. 34, 951–957. doi: 10.1097/INF.0000000000000794
Hung, H. C., Wang, H. C., Shih, S. R., Teng, I. F., Tseng, C. P., and Hsu, J. T. (2011). Synergistic inhibition of enterovirus 71 replication by interferon and rupintrivir. J. Infect. Dis. 203, 1784–1790. doi: 10.1093/infdis/jir174
Janeway, C. A. Jr., and Medzhitov, R. (2002). Innate immune recognition. Annu. Rev. Immunol. 20, 197–216. doi: 10.1146/annurev.immunol.20.083001.084359
Jensen-Jarolim, E. (ed.). (2013). “Common concepts of immune defense: innate immunity, the oldest type of defense,” in Comparative Medicine - Anatomy and Physiology. (Berlin: Springer), 220–242.
Kar, B. R., Dwibedi, B., and Kar, S. K. (2013). An outbreak of hand, foot and mouth disease in Bhubaneswar, Odisha. Indian Pediatr. 50, 139–142. doi: 10.1007/s13312-013-0033-0
Kawai, T., and Akira, S. (2006). Innate immune recognition of viral infection. Nat. Immunol. 7, 131–137. doi: 10.1038/ni1303
Koh, W. M., Badaruddin, H., La, H., Chen, M. I., and Cook, A. R. (2018). Severity and burden of hand, foot and mouth disease in Asia: a modelling study. BMJ Glob. Health 3:e000442. doi: 10.1136/bmjgh-2017-000442
Kuo, R. L., Kao, L. T., Lin, S. J., Wang, R. Y., and Shih, S. R. (2013). MDA5 plays a crucial role in enterovirus 71 RNA-mediated IRF3 activation. PLoS One 8:e63431. doi: 10.1371/journal.pone.0063431
Lee, K. M., Chen, C. J., and Shih, S. R. (2017). Regulation mechanisms of viral IRES-driven translation. Trends Microbiol. 25, 546–561. doi: 10.1016/j.tim.2017.01.010
Lei, X., Han, N., Xiao, X., Jin, Q., He, B., and Wang, J. (2014). Enterovirus 71 3C inhibits cytokine expression through cleavage of the TAK1/TAB1/TAB2/TAB3 complex. J. Virol. 88, 9830–9841. doi: 10.1128/JVI.01425-14
Lei, X., Liu, X., Ma, Y., Sun, Z., Yang, Y., Jin, Q., et al. (2010). The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J. Virol. 84, 8051–8061. doi: 10.1128/JVI.02491-09
Lei, X., Sun, Z., Liu, X., Jin, Q., He, B., and Wang, J. (2011). Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J. Virol. 85, 8811–8818. doi: 10.1128/JVI.00447-11
Lei, X., Xiao, X., Xue, Q., Jin, Q., He, B., and Wang, J. (2013). Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J. Virol. 87, 1690–1698. doi: 10.1128/JVI.01855-12
Li, R., Liu, L., Mo, Z., Wang, X., Xia, J., Liang, Z., et al. (2014). An inactivated enterovirus 71 vaccine in healthy children. N. Engl. J. Med. 370, 829–837. doi: 10.1056/NEJMoa1303224
Li, Z., Wang, H., Chen, Y., Niu, J., Guo, Q., Leng, Q., et al. (2017). Interleukin-18 protects mice from Enterovirus 71 infection. Cytokine 96, 132-137. doi: 10.1016/j.cyto.2017.04.002.
Lin, H., Huang, L., Zhou, J., Lin, K., Wang, H., Xue, X., et al. (2016). Efficacy and safety of interferon-alpha2b spray in the treatment of hand, foot, and mouth disease: a multicenter, randomized, double-blind trial. Arch. Virol. 161, 3073–3080. doi: 10.1007/s00705-016-3012-7
Lin, H. Y., Yang, Y. T., Yu, S. L., Hsiao, K. N., Liu, C. C., Sia, C., et al. (2013). Caveolar endocytosis is required for human PSGL-1-mediated enterovirus 71 infection. J. Virol. 87, 9064–9076. doi: 10.1128/JVI.00573-13
Liu, H., Qin, Y., Kong, Z., Shao, Q., Su, Z., Wang, S., et al. (2016). siRNA targeting the 2Apro genomic region prevents Enterovirus 71 replication in vitro. PLoS One 11:e0149470. doi: 10.1371/journal.pone.0149470
Liu, M. L., Lee, Y. P., Wang, Y. F., Lei, H. Y., Liu, C. C., Wang, S. M., et al. (2005). Type I interferons protect mice against enterovirus 71 infection. J. Gen. Virol. 86(Pt 12), 3263–3269. doi: 10.1099/vir.0.81195-0
Liu, Y., Zhang, Z., Zhao, X., Yu, R., Zhang, X., Wu, S., et al. (2014). Enterovirus 71 inhibits cellular type I interferon signaling by downregulating JAK1 protein expression. Viral Immunol. 27, 267–276. doi: 10.1089/vim.2013.0127
Loo, Y. M., and Gale, M. Jr. (2011). Immune signaling by RIG-I-like receptors. Immunity 34, 680–692. doi: 10.1016/j.immuni.2011.05.003
Lu, J., Yi, L., Zhao, J., Yu, J., Chen, Y., Lin, M. C., et al. (2012). Enterovirus 71 disrupts interferon signaling by reducing the level of interferon receptor 1. J. Virol. 86, 3767–3776. doi: 10.1128/JVI.06687-11
Lu, Q. B., Zhang, X. A., Wo, Y., Xu, H. M., Li, X. J., Wang, X. J., et al. (2012). Circulation of Coxsackievirus A10 and A6 in hand-foot-mouth disease in China, 2009-2011. PLoS One 7:e52073. doi: 10.1371/journal.pone.0052073
Lum, L. C., Wong, K. T., Lam, S. K., Chua, K. B., and Goh, A. Y. (1998). Neurogenic pulmonary oedema and enterovirus 71 encephalomyelitis. Lancet 352:1391. doi: 10.1016/S0140-6736(05)60789-1
Maelfait, J., and Beyaert, R. (2012). Emerging role of ubiquitination in antiviral RIG-I signaling. Microbiol. Mol. Biol. Rev. 76, 33–45. doi: 10.1128/MMBR.05012-11
Meng, J., Yao, Z., He, Y., Zhang, R., Zhang, Y., Yao, X., et al. (2017). ARRDC4 regulates enterovirus 71-induced innate immune response by promoting K63 polyubiquitination of MDA5 through TRIM65. Cell Death Dis. 8:e2866. doi: 10.1038/cddis.2017.257.
Mirand, A., Henquell, C., Archimbaud, C., Ughetto, S., Antona, D., Bailly, J. L., et al. (2012). Outbreak of hand, foot and mouth disease/herpangina associated with coxsackievirus A6 and A10 infections in 2010, France: a large citywide, prospective observational study. Clin. Microbiol. Infect. 18, E110–E118. doi: 10.1111/j.1469-0691.2012.03789.x
Muller, U., Steinhoff, U., Reis, L. F., Hemmi, S., Pavlovic, J., Zinkernagel, R. M., et al. (1994). Functional role of type I and type II interferons in antiviral defense. Science 264, 1918–1921. doi: 10.1126/science.8009221
Nguyen, N. T., Pham, H. V., Hoang, C. Q., Nguyen, T. M., Nguyen, L. T., Phan, H. C., et al. (2014). Epidemiological and clinical characteristics of children who died from hand, foot and mouth disease in Vietnam, 2011. BMC Infect. Dis. 14:341. doi: 10.1186/1471-2334-14-341
Plevka, P., Perera, R., Cardosa, J., Kuhn, R. J., and Rossmann, M. G. (2012). Crystal structure of human enterovirus 71. Science 336:1274. doi: 10.1126/science.1218713
Puenpa, J., Mauleekoonphairoj, J., Linsuwanon, P., Suwannakarn, K., Chieochansin, T., Korkong, S., et al. (2014). Prevalence and characterization of Enterovirus infections among pediatric patients with hand foot mouth disease, herpangina and influenza like illness in Thailand, 2012. PLoS One 9:e98888. doi: 10.1371/journal.pone.0098888
Rahman, M. M., and McFadden, G. (2011). Modulation of NF-kappaB signalling by microbial pathogens. Nat. Rev. Microbiol. 9, 291–306. doi: 10.1038/nrmicro2539
Robinson, C. R., Doane, F. W., and Rhodes, A. J. (1958). Report of an outbreak of febrile illness with pharyngeal lesions and exanthem: toronto, summer 1957; isolation of group A Coxsackie virus. Can. Med. Assoc. J. 79, 615–621.
Rui, Y., Su, J., Wang, H., Chang, J., Wang, S., Zheng, W., et al. (2017). Disruption of MDA5-mediated innate immune responses by the 3C proteins of coxsackievirus A16, coxsackievirus A6, and Enterovirus D68. J. Virol. 91:e00546-17 doi: 10.1128/JVI.00546-17
Samuda, G. M., Chang, W. K., Yeung, C. Y., and Tang, P. S. (1987). Monoplegia caused by Enterovirus 71: an outbreak in Hong Kong. Pediatr. Infect. Dis. J. 6, 206–208. doi: 10.1097/00006454-198702000-00013
Schmidt, N. J., Lennette, E. H., and Ho, H. H. (1974). An apparently new enterovirus isolated from patients with disease of the central nervous system. J. Infect. Dis. 129, 304–309. doi: 10.1093/infdis/129.3.304
Seth, R. B., Sun, L., Ea, C. K., and Chen, Z. J. (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122, 669–682. doi: 10.1016/j.cell.2005.08.012
Sickles, G. M., Mutterer, M., Feorino, P., and Plager, H. (1955). Recently classified types of Coxsackie virus, group A; behavior in tissue culture. Proc. Soc. Exp. Biol. Med. 90, 529–531. doi: 10.3181/00379727-90-22088
Solomon, T., Lewthwaite, P., Perera, D., Cardosa, M. J., McMinn, P., and Ooi, M. H. (2010). Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect. Dis. 10, 778–790. doi: 10.1016/s1473-3099(10)70194-8
Song, J., Hu, Y., Li, J., Zheng, H., Wang, J., Guo, L., et al. (2018). Suppression of the toll-like receptor 7-dependent type I interferon production pathway by autophagy resulting from enterovirus 71 and coxsackievirus A16 infections facilitates their replication. Arch. Virol. 163, 135–144. doi: 10.1007/s00705-017-3592-x
Sun, J., Ennis, J., Turner, J. D., and Chu, J. J. (2016). Single dose of an adenovirus vectored mouse interferon-alpha protects mice from lethal EV71 challenge. Antiviral Res. 134, 207–215. doi: 10.1016/j.antiviral.2016.09.003
Turvey, S. E., and Broide, D. H. (2010). Innate immunity. J. Allergy Clin. Immunol. 125(2 Suppl. 2), S24–S32. doi: 10.1016/j.jaci.2009.07.016
Van Tu, P., Thao, N. T. T., Perera, D., Truong, K. H., Tien, N. T. K., Thuong, T. C., et al. (2007). Epidemiologic and virologic investigation of hand, foot, and mouth disease, southern Vietnam, 2005. Emerg. Infect. Dis. 13, 1733–1741. doi: 10.3201/eid1311.070632
Wang, B., Xi, X., Lei, X., Zhang, X., Cui, S., Wang, J., et al. (2013). Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog. 9:e1003231. doi: 10.1371/journal.ppat.1003231
Wang, C., Ji, L., Yuan, X., Jin, Y., Cardona, C. J., and Xing, Z. (2016). Differential regulation of TLR signaling on the induction of antiviral interferons in human intestinal epithelial cells infected with Enterovirus 71. PLoS One 11:e0152177. doi: 10.1371/journal.pone.0152177
Wang, C., Sun, M., Yuan, X., Ji, L., Jin, Y., Cardona, C. J., et al. (2015). Enterovirus 71 suppresses interferon responses by blocking Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling through inducing karyopherin-alpha1 degradation. J. Biol. Chem. 292, 10262–10274. doi: 10.1074/jbc.M116.745729
Wang, W., Xiao, F., Wan, P., Pan, P., Zhang, Y., Liu, F., et al. (2017). EV71 3D protein binds with NLRP3 and enhances the assembly of inflammasome complex. PLoS Pathog. 13:e1006123. doi: 10.1371/journal.ppat.1006123
Wang, H., Lei, X., Xiao, X., Yang, C., Lu, W., Huang, Z., et al. (2015). Reciprocal regulation between Enterovirus 71 and the NLRP3 inflammasome. Cell Rep. 12, 42–48. doi: 10.1016/j.celrep.2015.05.047
Wang, L. C., Chen, S. O., Chang, S. P., Lee, Y. P., Yu, C. K., Chen, C. L., et al. (2015). Enterovirus 71 proteins 2A and 3D antagonize the antiviral activity of gamma interferon via signaling attenuation. J. Virol. 89, 7028–7037. doi: 10.1128/JVI.00205-15
Wu, Y., Yeo, A., Phoon, M. C., Tan, E. L., Poh, C. L., Quak, S. H., et al. (2010). The largest outbreak of hand; foot and mouth disease in Singapore in 2008: the role of enterovirus 71 and coxsackievirus A strains. Int. J. Infect. Dis. 14, e1076–e1081. doi: 10.1016/j.ijid.2010.07.006
Xie, L., Lu, B., Zheng, Z., Miao, Y., Liu, Y., Zhang, Y., et al. (2018). The 3C protease of enterovirus A71 counteracts the activity of host zinc-finger antiviral protein (ZAP). J. Gen. Virol. 99, 73–85. doi: 10.1099/jgv.0.000982
Xu, C., He, X., Zheng, Z., Zhang, Z., Wei, C., Guan, K., et al. (2014). Downregulation of microRNA miR-526a by enterovirus inhibits RIG-I-dependent innate immune response. J. Virol. 88, 11356–11368. doi: 10.1128/JVI.01400-14
Yamayoshi, S., Yamashita, Y., Li, J., Hanagata, N., Minowa, T., Takemura, T., et al. (2009). Scavenger receptor B2 is a cellular receptor for Enterovirus 71. Nat. Med. 15, 798–801. doi: 10.1038/nm.1992
Yang, J., Yang, C., Guo, N., Zhu, K., Luo, K., Zhang, N., et al. (2015). Type I interferons triggered through the toll-like receptor 3-TRIF pathway control coxsackievirus A16 infection in young mice. J. Virol. 89, 10860–10867. doi: 10.1128/JVI.01627-15
Yang, Q., Zhang, Y., Yan, D., Zhu, S., Wang, D., Ji, T., et al. (2016). Two genotypes of coxsackievirus A2 associated with hand, foot, and mouth disease circulating in China since 2008. PLoS One 11:e0169021. doi: 10.1371/journal.pone.0169021
Yao, X., Bian, L. L., Lu, W. W., Li, J. X., Mao, Q. Y., Wang, Y. P., et al. (2017). Epidemiological and etiological characteristics of herpangina and hand foot mouth diseases in Jiangsu, China, 2013-2014. Hum. Vaccin Immunother. 13, 823–830. doi: 10.1080/21645515.2016.1236879
Yi, L., He, Y., Chen, Y., Kung, H. F., and He, M. L. (2011). Potent inhibition of human Enterovirus 71 replication by type I interferon subtypes. Antivir. Ther. 16, 51–58. doi: 10.3851/IMP1720
Yoneyama, M., Kikuchi, M., Natsukawa, T., Shinobu, N., Imaizumi, T., Miyagishi, M., et al. (2004). The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5, 730–737. doi: 10.1038/ni1087
Zhang, X., Song, Z., Qin, B., Chen, L., Hu, Y., and Yuan, Z. (2013). Rupintrivir is a promising candidate for treating severe cases of enterovirus-71 infection: evaluation of antiviral efficacy in a murine infection model. Antiviral Res. 97, 264–269. doi: 10.1016/j.antiviral.2012.12.029
Zhang, Y., Burke, C. W., Ryman, K. D., and Klimstra, W. B. (2007). Identification and characterization of interferon-induced proteins that inhibit alphavirus replication. J. Virol. 81, 11246–11255. doi: 10.1128/JVI.01282-07
Zhang, Y., Zhu, Z., Yang, W., Ren, J., Tan, X., Wang, Y., et al. (2010). An emerging recombinant human enterovirus 71 responsible for the 2008 outbreak of hand foot and mouth disease in Fuyang city of China. Virol. J. 7:94. doi: 10.1186/1743-422X-7-94
Zhu, J., Luo, Z., Wang, J., Xu, Z., Chen, H., Fan, D., et al. (2013). Phylogenetic analysis of Enterovirus 71 circulating in Beijing, China from 2007 to 2009. PLoS One 8:e56318. doi: 10.1371/journal.pone.0056318
Keywords: innate immunity evasion, enteroviruses, coxsackieviruses, hand-foot-mouth disease, type I IFN signaling
Citation: Jin Y, Zhang R, Wu W and Duan G (2018) Innate Immunity Evasion by Enteroviruses Linked to Epidemic Hand-Foot-Mouth Disease. Front. Microbiol. 9:2422. doi: 10.3389/fmicb.2018.02422
Received: 28 April 2018; Accepted: 21 September 2018;
Published: 08 October 2018.
Edited by:
Zhiyong Li, Lanzhou Veterinary Research Institute (CAAS), ChinaReviewed by:
Siew Pheng Lim, Denka Life Innovation Research (DLIR), SingaporeCopyright © 2018 Jin, Zhang, Wu and Duan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guangcai Duan, Z2NkdWFuQHp6dS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.