
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 02 October 2018
Sec. Aquatic Microbiology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.02353
This article is part of the Research Topic Cyanobacterial Blooms in a Changing World View all 23 articles
 María E. Alcamán-Arias1,2,3
María E. Alcamán-Arias1,2,3 Carlos Pedrós-Alió4
Carlos Pedrós-Alió4 Javier Tamames4
Javier Tamames4 Camila Fernández1,5,6
Camila Fernández1,5,6 Danilo Pérez-Pantoja7
Danilo Pérez-Pantoja7 Mónica Vásquez2
Mónica Vásquez2 Beatriz Díez2,3*
Beatriz Díez2,3*Composition, carbon and nitrogen uptake, and gene transcription of microbial mat communities in Porcelana neutral hot spring (Northern Chilean Patagonia) were analyzed using metagenomics, metatranscriptomics and isotopically labeled carbon (H13CO3) and nitrogen (15NH4Cl and K15NO3) assimilation rates. The microbial mat community included 31 phyla, of which only Cyanobacteria and Chloroflexi were dominant. At 58∘C both phyla co-occurred, with similar contributions in relative abundances in metagenomes and total transcriptional activity. At 66∘C, filamentous anoxygenic phototrophic Chloroflexi were >90% responsible for the total transcriptional activity recovered, while Cyanobacteria contributed most metagenomics and metatranscriptomics reads at 48∘C. According to such reads, phototrophy was carried out both through oxygenic photosynthesis by Cyanobacteria (mostly Mastigocladus) and anoxygenic phototrophy due mainly to Chloroflexi. Inorganic carbon assimilation through the Calvin–Benson cycle was almost exclusively due to Mastigocladus, which was the main primary producer at lower temperatures. Two other CO2 fixation pathways were active at certain times and temperatures as indicated by transcripts: 3-hydroxypropionate (3-HP) bi-cycle due to Chloroflexi and 3-hydroxypropionate-4-hydroxybutyrate (HH) cycle carried out by Thaumarchaeota. The active transcription of the genes involved in these C-fixation pathways correlated with high in situ determined carbon fixation rates. In situ measurements of ammonia assimilation and nitrogen fixation (exclusively attributed to Cyanobacteria and mostly to Mastigocladus sp.) showed these were the most important nitrogen acquisition pathways at 58 and 48∘C. At 66∘C ammonia oxidation genes were actively transcribed (mostly due to Thaumarchaeota). Reads indicated that denitrification was present as a nitrogen sink at all temperatures and that dissimilatory nitrate reduction to ammonia (DNRA) contributed very little. The combination of metagenomic and metatranscriptomic analysis with in situ assimilation rates, allowed the reconstruction of day and night carbon and nitrogen assimilation pathways together with the contribution of keystone microorganisms in this natural hot spring microbial mat.
Microbial mats have been studied for decades as model systems for testing principles of microbial ecology (Stal, 1995; Ward, 2006; Hou et al., 2013; Inskeep et al., 2013). Microbial mats in hot springs are dynamic and relatively simple ecosystems exhibiting spatial and temporal heterogeneity (Ward et al., 1998; Ward, 2006; Bhaya et al., 2007; Klatt et al., 2011; Alcamán et al., 2015). These microenvironments support a diversity of species carrying out a wide range of metabolic processes (Jensen et al., 2011; Kim et al., 2015). The upper few millimeters in alkaline and neutral mats are dominated by oxygenic phototrophic cyanobacteria, such as the unicellular cyanobacterium Synechococcus spp. (Steunou et al., 2006, 2008; Bhaya et al., 2007; Jensen et al., 2011; Klatt et al., 2011), the filamentous non-heterocystous Oscillatoria spp. and the filamentous heterocystous Mastigocladus spp. (Stewart, 1970; Miller et al., 2006; Mackenzie et al., 2013; Alcamán et al., 2015), as well as the filamentous anoxygenic phototrophs (FAPs) Roseiflexus sp. and Chloroflexus sp. (van der Meer et al., 2010; Klatt et al., 2011; Liu et al., 2011; Kim et al., 2015). Several diversity studies in hot spring microbial mats have shown that members of the phototrophic Cyanobacteria and Chloroflexi phyla can co-exist, sometimes in a collaborative manner (Liu et al., 2011). For instance, Synechococcus sp. produces low-molecular weight organic compounds as by-products of its metabolism (as primary producers), that are assimilated photoheterotrophically by FAPs (Sandbeck and Ward, 1981; Anderson et al., 1987; Bateson and Ward, 1988).
During daytime light penetrates only a few millimeters into the microbial mat (Kühl and Jørgensen, 1992), and the upper part of the mat is supersaturated with oxygen photosynthetically produced by Cyanobacteria (Canfield and Des Marais, 1994; Jensen et al., 2011) but the concentration of oxygen rapidly diminishes with depth. On the other hand, anoxygenic photosynthetic microorganisms, such as Chloroflexi, may use sulfide (Madigan and Brock, 1977) or hydrogen (Holo and Sirevåg, 1986) as electron donors and fix CO2, through the 3-hydroxypropionate (3-HP) bi-cycle (Strauss and Fuchs, 1993; Klatt et al., 2007; Zarzycki et al., 2009; Zarzycki and Fuchs, 2011). Moreover, Chloroflexi can simultaneously incorporate inorganic (CO2) and organic carbon such as acetate (generated under anoxic conditions at night) or glycolate (generated by photorespiration under O2 supersaturation during the day) produced by Cyanobacteria (Bateson and Ward, 1988; van der Meer et al., 2005, 2007; Zarzycki and Fuchs, 2011; Bryant et al., 2012). Therefore, the availability and abundance of these inorganic and organic compounds are factors that shape the relative degree to which FAPs behave as heterotrophs, mixotrophs or autotrophs (van der Meer et al., 2005; Zarzycki and Fuchs, 2011; Klatt et al., 2013a; Urschel et al., 2015). However, the dynamics on a daily basis and along the temperature gradient of the different autotrophic strategies for inorganic carbon fixation by thermophiles are still poorly characterized.
The nitrogen cycle also shows a variety of potentially active pathways in thermal mats, such as N2-fixation attributed to Cyanobacteria (such as Synechococcus sp. and Mastigocladus sp.) (Miller et al., 2006; Steunou et al., 2006, 2008; Alcamán et al., 2015), ammonia oxidation by Archaea (Candidatus Nitrosocaldus yellowstonii) (de la Torre et al., 2008; Reigstad et al., 2008; Hamilton et al., 2014; Thiel et al., 2016), and denitrification and dissimilatory nitrate reduction to ammonia (DNRA) attributed to Aquificales and Thermales (Hydrogenobacter sp., Sulfurihydrogenibium sp., Anoxybacillus sp. and Thermus sp.) (Dodsworth et al., 2011b). Hot springs are commonly N-limited systems due to the fast assimilation and turnover of inorganic nitrogen forms (Alcamán et al., 2015; Lin et al., 2015). Thus, N2-fixation carried out by Cyanobacteria has been reported to be the most relevant biological process for the input of exogenous nitrogen to microbial mats in Porcelana (Alcamán et al., 2015). Evidence of a balance between inputs and outputs of N was demonstrated by Hamilton et al. (2014) in Yellowstone National Park (YNP) Perpetual Spouter hot spring (pH 7.03, 86.4°C), where a rapid depletion of bioavailable nitrogen by putatively oxidizing Thaumarchaeota lead to an increase in the activity of the putative nitrogen-fixing bacterium Thermocrinis albus (phylum Aquificae), in the absence of other nitrogen sources. It has also been reported that Archaea are able to carry out different N reductive pathways, including nitrate assimilation, N2 fixation, and dissimilatory reactions (Cabello et al., 2004).
Recent development of high-throughput sequencing techniques has expanded knowledge of taxonomical diversity in hot springs (Bhaya et al., 2007; Klatt et al., 2011; Liu et al., 2011, 2012b; Thiel et al., 2016). Metagenomics and metatranscriptomics are now most effective approaches to target the community structure and the expressed genes to reveal community functions carried out at a specific time (Bhaya et al., 2007; Urich et al., 2008; Liu et al., 2011; Klatt et al., 2013b; Thiel et al., 2016). Some metagenomics studies have revealed new aspects of microbial diversity distribution with temperature in hot springs (Bhaya et al., 2007; Klatt et al., 2013b), as well as the characterization of dominant phototrophic populations and previously unidentified members of the Chloroflexi phylum (Klatt et al., 2011). Also, putative ecotypes adapted to different niches have been found within the undermat, particularly of Roseiflexus spp. (Thiel et al., 2016). In addition, metatranscriptomics has provided important information about temporal patterns of key gene transcription in processes such as N2-fixation (nifH), oxygenic (psaA) and anoxygenic (pufM) photosynthesis (Liu et al., 2011), as well as survival strategies of relevant populations in hot springs (Quaiser et al., 2014).
Most metagenomics and metatranscriptomics studies, however, have concentrated on just one spot per spring or just one diel cycle at a particular point ignoring changes along the temperature gradient (Liu et al., 2011; Klatt et al., 2013a,b; Thiel et al., 2017). Thus, the activities and corresponding gene transcription patterns of all such autotrophic carbon and nitrogen pathways, and the organisms responsible for each one of them, are not well understood along temperature gradients and at different times. In the present study, we used metagenomics, metatranscriptomics, and biogeochemical analyses (isotopic assimilation rates) to clarify the structure and activities of the microbial mat communities along a moderately thermophilic gradient (66, 58, and 48°C) at noon and night in the neutral hot spring of Porcelana (Northern Patagonia, Chile). Within an area of rainforest, the water surfaces close to 70°C, runs downstream to join River Porcelana, cooling down to approximately 40°C in its transit. The pH is close to neutral and the channel walls are heavily colonized by microbial mats, first orange and later green in color. Porcelana hot spring had only received some sporadic visits determining a few physicochemical parameters (Hauser, 1989; Waring, 1965), but no microbiological studies had been carried out until 2013. We analyzed the taxonomic composition of the mats by DGGE and sequencing (Mackenzie et al., 2013) and measured nitrogen fixation in a diel study attributing all the activity to the cyanobacteria Mastigocladus sp. (Alcamán et al., 2015).
Porcelana hot spring is located in Chilean Patagonia (42° 27′ 29.1″S – 72° 27′ 39.3″W) at the end of the Comau Fjord and is part of an area of shallow depth geothermic events whose outflow pours over volcanic rocks originated in the Quaternary Period and is encircled by active Quaternary volcanoes such as the Huequi Volcano (Duhart et al., 2000). It is a slightly acidic pH (∼6.5) system with a maximal temperature of 66°C when was sampled in March 2013 (Figure 1). The channel walls are heavily colonized by microbial mats, first orange and later green in color. Microbial mats (up to 2 cm thick) grow from 70 to 46°C downstream, representing a stable biogeochemical system as previously reported by Mackenzie et al. (2013) and Alcamán et al. (2015). Colorful microbial mats growing at 66 (orange mat), 58 (orange and green mat) and 48°C (green mat) (Figure 1), were sampled using a cork borer with a diameter of 7 mm. Cores 1 cm thick were collected in triplicate at noon (12:00 h), and also at night (23:00 h) at 66, 58, and 48°C. Isotopic nitrogen (15NH4Cl, and K15NO3) and carbon (H13CO3) uptake experiments were performed in microbial mats growing at 58 and 48°C (but not at 66°C), cores were collected in triplicate as explained above. Samples for DNA and RNA were transported in liquid nitrogen and kept at -80°C until analysis. In the laboratory, DNA and RNA for sequencing analyses (metagenomics/metatranscriptomics) were extracted from pooled triplicate and homogenized mat samples.

FIGURE 1. Porcelana hot spring and the pigmented microbial mats growing along the temperature gradient. Sampling sites are indicated by squares: 66°C (orange), 58°C (orange + green) and 48°C (green).
Incubations at 58 and 48°C were run in situ under light and dark conditions. Unfortunately, assays at 66°C were not possible due to logistic problems at the time of sampling. For each sample point, three microbial mat cores (7 mm in diameter and 1 cm thick) were mixed to homogenize potential spatial heterogeneity and transferred to pre-sterilized 10 ml vials with 1 ml of pre-filtered (0.2 μm filter pore) spring water from the same sampling point. Dual 15N/13C uptake experiments were conducted independently by adding 50 μL of 15NH4Cl (500 μmol L-1) plus 500 μl of H13CO3 (500 μmol L-1), and 30 μl K15NO3 (500 μmol L-1) plus 500 μL of H13CO3 (500 μmol L-1) in parallel. These incubations were run in triplicate for biological replication. The 15N tracer additions were generally kept close to 10% of natural abundance of nitrate (which was 3.2 μmol L-1 NO3-) and ammonium (natural abundance was 0.04 μmol L-1 NH4+). The nitrogen fixation (15N2) experiments were carried out simultaneously but have been published separately (see details in Alcamán et al., 2015). Briefly, 1 ml of 15N2 gas (98% atom 15N2 gas; Sigma-Aldrich) was added through a gas-tight syringe into the headspace of each vial. For each temperature analyzed, two replicates without the isotope (15N and 13C) were also incubated in order to determine the natural isotopic composition and to be used as negative controls. Vials were incubated at 58 and 48°C in situ for 6 h. After incubations, mat samples were dried at 70°C for 48 h. Isotopic analyses of 15N and 13C were carried out using an IRMS delta plus Thermo FinniganH Mass Spectrometer (Stable Isotope Laboratory, Granada, Spain). Rates of carbon fixation and nitrogen assimilation were expressed as daily carbon or nitrogen assimilation rates (nmoles cm-2 d-1), and calculated as:
where %Rexc, the excess enrichment of the tracer after inoculation, is calculated using Eq. (1): Vadd (L-1) indicates the volume of isotope added to the sample; Tconc (mol L-1) is the tracer concentration added to the sample; Vinc (L-1) is the sample volume incubated; Natconc (mol L-1) represents the initial amount C or N in the sample after isotope added; Natabundance (mol L-1) corresponds to natural abundance of C or N in the mat. A fixed dissolved inorganic carbon value of 0.8 mM was used based on measurements in the Porcelana mat (Alcamán et al., 2015). The assimilation rate (ρ) was calculated with Eq. (2), where %ATf is the percentage of total atoms after incubation; PON or POC (mg) are the amounts of particulate organic nitrogen or carbon recovered after incubation and measured by mass spectrometry; mg is the microbial mat mass (dry weight) analyzed in mass spectrometry; and Timeinc is the incubation time (h). The final rates were extrapolated per core area (cm-2) and as daily rate (d-1).
Turnover time of ammonium and nitrate, at the time of sampling was estimated using the uptake rate obtained by dividing the in situ ammonium and nitrate natural concentrations by their respective uptake rates, giving units of hours. Concentrations in water did not change over 24 h in our previous studies (i.e., Alcamán et al., 2015).
The whole contents of each previously homogenized mat sample were extracted as previously described in Alcamán et al. (2015). Briefly for DNA, glass beads were added to the sample, which then was homogenized by bead beating three times for 20 s, followed by an organic Phenol–Chloroform extraction. Usually between 0.4 and 0.7 g of mat-samples were used. In the case of RNA samples, the Trizol-mat mixture was subjected to beating only twice. Quality and quantity of the extracted nucleic acids were checked by spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE, United States) and by 0.8% agarose gel DNAse/RNAse-free, and the material was kept at -80°C. Then just one DNA (∼600 ng) and one RNA (1 μg) sample was sent to sequencing for each temperature and time of day using Illumina technology (Research and Testing Laboratory, Lubbock, TX, United States). The metagenomes and metatranscriptomes were sequenced. Briefly, enzymatic fragmentation was done prior to DNA library construction using NEBNext dsFragmentase, then fragmented DNA was cleaned up by column purification. The construction of libraries (Ultra DNA Library Prep Kit for Illumina) was conducted by: End prep, adaptor ligation, size selection of adaptor-ligated DNA, PCR amplification and product purification. Finally, fragment size was checked by Fragment Analyzer (Advanced Analytical Technologies, Ankeny, IA, United States). For metatranscriptomes, total RNA (1 μg) was cleaned up of rRNA prior to library construction by Ribo-Zero rRNA Removal Kit Bacteria (Illumina, San Diego, CA, United States) according the manufacturer’s instruction followed by clean up using Agencourt RNAClean XP Kit (Beckman Coulter, Indianapolis, IN, United States). A gel-free or low input small RNA library prep kit with reduced bias for Illumina sequencing was used. The specific kit (NEXTflexTM Illumina Small RNA Sequencing Kit v3) and all previous mentioned protocols are available at http://www.biooscientific.com/next-gen-sequencing/nextflex-illumina-small-rna-seq-library-prep-kits (Bioo Scientific, Austin, TX, United States). Unfortunately, it was not possible to obtain a metatranscriptome of sufficient quality for the 66°C sample collected at night. All read and transcript sequences, corresponding to metagenomes and metatranscriptomes, obtained from this study have been deposited in the Sequence Read Archive under the accession number SRP104009.
To ensure feasibility of downstream analysis, FastQC1 was used to assess the quality of the sequence data. To correct the quality issues found Cutadapt2 was used, leaving only mappable sequences longer than 30 bp (-m 30), with a 3′ end trimming for bases with quality below 28 (-q 28), a hard clipping of the first five leftmost bases (-u 5), and finally a perfect match of at least 10 bp (-O 10) against the standard Illumina adaptors. This procedure reduced the total number of sequences from 452.7 to 394.7 million (Supplementary Table S1).
For the classification of 16S rRNA gene sequences, the rRNA reads in DNA and RNA high quality samples were identified and separated using Ribopicker (Schmieder et al., 2012) with the non-redundant rRNA database that combines Silva, Greengenes, RDP-II, NCBI archaeal/bacterial, HMP and Rfam databases. Ribosomal RNA sequences represented 4.2% of the total, with 378.2 million of reads related to functional genes (Supplementary Table S1). The BLAST parameters used were: e-value 1e-10, min-score 200 and min query coverage of 0.95.
Taxonomical assignment of the non-rRNA high quality reads was conducted by Diamond (Buchfink et al., 2015) with default parameters and by the NCBI non-redundant (NR) database, previously filtered by bacterial sequences (ids obtained from gbbct database3) and added with new bacterial genomes not incorporated yet from JGI4. The NCBI taxonomic identifiers (taxid) for each match were manually added as a new column in Diamond’s output (csv file), using an in-house bash script based on Unix command line programs that take each GI code and match it with the corresponding taxid from the NCBI taxonomy tree (Sayers et al., 2009). Subsequently, the results were parsed using the lowest common ancestor algorithm trough MEGAN 5 (Huson et al., 2007) under default parameters (score = 50). The total number of non-redundant hits was 114.2 million. On average around 86.6% of the reads could be taxonomically assigned in most samples. Only in the 66°C cDNA day sample, the percentage was near 20% (Supplementary Table S1).
Taxonomic assignment of reads is a process subject to biases and errors. This is particularly the case when the organisms studied have few reference genomes in databases. In these cases, taxonomic assignment will tend to assign reads to genomes in the database, even if the match is not very good. Thus, we also used the rDNA sequences extracted with riboPicker for taxonomic assignment of the most relevant organisms in the mat, both in metagenomes and metatranscriptomes. In this case, other problems appear, for example the difference in copy number among different microorganisms will alter the relative abundances in an unknown way. For the present analysis, we will use the metagenomic assignments (Supplementary Table S2), because our interest is to analyze them together with metatranscriptomes, which will logically have similar biases to metagenomes. However, we will use the 16S rDNA sequences to confirm the identity of the main microorganisms discussed (Supplementary Table S3), accession numbers SUB4537948.
To identify the specific pathways of photosynthesis, CO2 fixation and nitrogen metabolisms, the three daytime metagenomes (66, 58, and 48°C) were co-assembled using the Spades software (Bankevich et al., 2012). Key genes from pathways of interest were used as indicators of the importance of each pathway (Supplementary Table S4). This was done using the normalized abundance of reads corresponding to each relevant gene in either metagenomes or metatranscriptomes. The raw counts were normalized to fragments per kilobase and million reads using a custom script. Prodigal software (Hyatt et al., 2010) was used to predict genes for taxonomic and functional annotations. A homology search with the amino acid translations of the genes was done using RapSearch2 (Zhao et al., 2012) against the GenBank non-redundant protein database. Functional annotation in KEGG and COG codes was done using the fun3 software, as described in Guazzaroni et al. (2013). The abundance of genes in each sample was determined by mapping the corresponding reads to the co-assembled metagenomic contigs. We used Bowtie2 (Langmead and Salzberg, 2012) for that purpose, and quantified the number of mapped reads to each gene using HTSeq (Anders et al., 2015). The raw counts were normalized to fragments per kilobase and million reads using a custom script. Metatranscriptomic reads were also mapped against metagenomic contigs to obtain the abundance of transcripts for each gene. The mapping was conducted with the BBMap software using a minimum identity of 95%, k = 9, and other default parameters (Bushnell, 2014). We assumed that the transcripts that did not map to the reference contigs of the metagenome correspond to highly expressed genes of rare species that could not be assembled because of their low abundance in the metagenome. To include these genes in the analysis, we extracted the unmapped transcripts from the three metatranscriptomes and assembled them using Spades. The resulting new contigs from the metatranscriptomes (composed often by a single gene, but also of polycistrons corresponding to operons) were added to the metatranscriptomics contig set and treated as described above (taxonomic and functional assignment, mapping and quantification of transcripts).
The same 36 phyla (31 bacterial and 5 archaeal) were present at the three temperatures sampled regardless of time of day (Supplementary Table S2). Two bacterial phyla, Cyanobacteria (green mat) and Chloroflexi (orange mat), accounted for most of the reads in both metagenomes and metatranscriptomes (Figure 2A). Chloroflexi showed higher relative number of reads in metagenomes and metatranscriptomes at 66°C, decreasing at 48°C. On the contrary, Cyanobacteria reached their maximal abundance and activity (up to 80% of all reads) at 48°C at daytime. At the intermediate temperature of 58°C, Chloroflexi and Cyanobacteria were represented in similar abundance both in metagenomic and day and night metatranscriptomic libraries (Figure 2A). Notoriously, Proteobacteria showed their highest transcriptional activity (>50%) at 58 and 48°C during periods of darkness, unlike Cyanobacteria and Chloroflexi that showed more transcripts during daytime (Figure 2A).
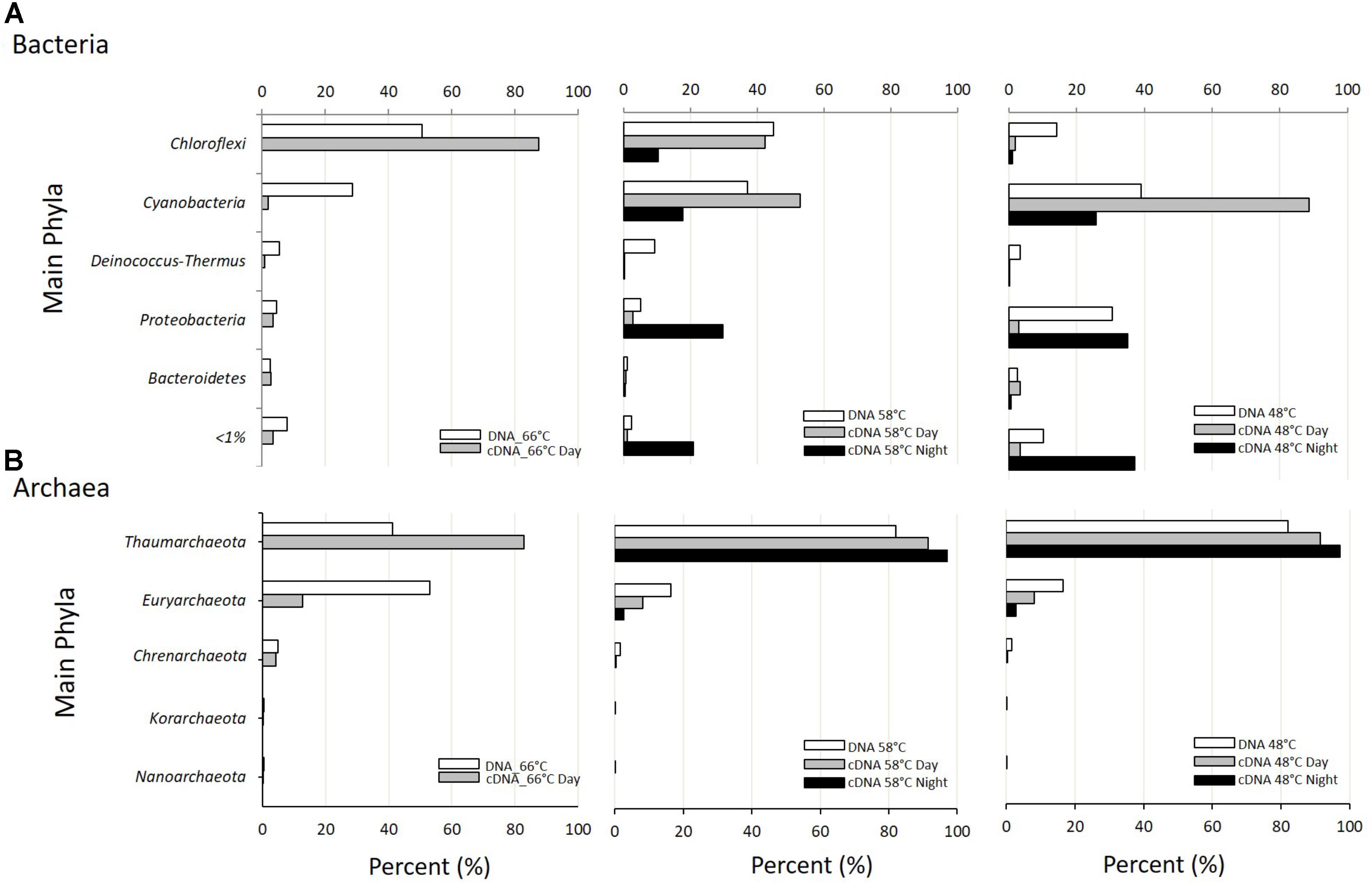
FIGURE 2. Taxonomic assignment at the Phylum level of metagenomic (DNA; white bars) and metatranscriptomic reads (cDNA day: gray bars; cDNA night: black bars) from samples at the three temperatures studied. (A) Percent of Bacteria reads assigned to the most abundant Phyla. (B) Percent of Archaea reads assigned to the most abundant Phyla.
Most filamentous anoxygenic bacteria (FAPs) of the phylum Chloroflexi, belonged to the order Chloroflexales (Supplementary Figure S1a). Members of Order Chloroflexales dominated at all temperatures and time of day studied and contributed with more than 90% to the total diurnal transcriptional activity. Most Chloroflexales reads belonged to two genera: Chloroflexus and Roseiflexus. The 16S rDNA sequences retrieved showed 96.9% and 98.9% similarity to Chloroflexus aurantiacus J-10-fl and Roseiflexus sp. RS-1, respectively (Supplementary Table S3).
Oscillochloris trichoides (99.6% similarity with strain DG-6 OSCT) was also present but in very low abundance (Supplementary Figure S1b). The novel member of Chloroflexi, Ca. Chloranaerofilum corporosum found in Mushroom Spring (Thiel et al., 2016), was specifically searched for in our metagenomes but we did not find it.
Chloroflexus sp. showed maximal abundance and transcriptional activity at 58°C, but transcriptional activity was also high at 66°C representing more than 70% of the transcriptional activity at this temperature. Roseiflexus sp., in turn, had maximal abundance at 66°C but represented ≤30% of the Chloroflexi transcriptional activity, whereas its transcriptional activity was highest (>90%) at 48°C at both day and night, compared to Chloroflexus (Supplementary Figure S1b). Roseiflexus sequences were less abundant at the intermediate temperature of 58°C in the metagenomes and metatranscriptomes. This might be due to different strains adapted to different temperature ranges. In effect, Nübel et al. (2002) found three Roseiflexus-like clades with temperature optima at 60 to 70°C, 47 to 53°C, 35 to 41°C. Similarly, our results indicate the presence of two populations with different temperature optima.
Klatt et al. (2011) mentioned an Anaerolinea-like chlorophototroph, later named Ca. Roseilinea gracile (Tank et al., 2017) in microbial mats in YNP. However, the sequences found in Porcelana were closer to Anaerolinea thermophila (NC_014960.1) than to Ca. Roseilinea gracile (Supplementary Table S3). To double check the presence of Roseilinea, its genome was used to recruit sequences from our metagenomes (with 95% minimal coverage) finding only around 490 sequences (with 95–100% identity). Therefore, Roseilinea would be found in barely detectable amounts (less than 0.001% of the abundance). In any case, the Anaerolinea sequences represented a very minor fraction of both metagenomes and metatranscriptomes (Supplementary Figure S1).
The phylum Cyanobacteria was represented by unicellular members of subsection II (Order Chroococcales), filamentous non-heterocystous members of subsection III (Order Oscillatoriales), and by filamentous heterocystous members of subsections IV (Order Nostocales) and V (Order Stigonematales) (Supplementary Figure S2a). Apart from the few reads of Oscillatoriaceae found at higher temperatures, most cyanobacterial reads corresponded to the single genus Mastigocladus (Order Stigonematales), a group of branching filamentous heterocystous cyanobacteria. This was confirmed by looking at the 16S rRNA genes retrieved from the metagenomes that showed 99.9% similarity to Fischerella sp. NIES-3754 (Supplementary Table S3). Dominance by this cyanobacterium has also been found in many hot springs around the world (Finsinger et al., 2008; Kaštovský and Johansen, 2008; McGregor and Rasmussen, 2008; Lukavsky et al., 2011), although not always in such high abundance (Miller et al., 2007; McGregor and Rasmussen, 2008) as in Porcelana. At the lowest temperatures, the Cyanobacteria contributed most of the total transcripts detected (>90%) during the day (Figure 2A), due mostly to the Stigonematales that, in turn, were largely (>90%) represented by members of the genus Mastigocladus sp. (Supplementary Figure S2b). Nostocales were also abundant (<40%) but with low (<10%) representation in metatranscriptomes at 48°C (Supplementary Figure S2a). At 66°C, Oscillatoriales were the most transcriptionally active members of cyanobacteria (>75%) (Supplementary Figure S2a). It must be remembered, however, that cyanobacteria at this temperature were a smaller percent of the total reads and, thus, most of the cyanobacterial abundance and transcriptional activity were those of Mastigocladus at the two lower temperatures (Supplementary Figure S2b). During the night, Oscillatoriales and Chroococcales contributed in a similar level to the cyanobacterial transcriptional activity at 58°C. At 48°C, in turn Chroococcales and Stigonematales were the most active (based on normalized abundance of reads in metatranscriptomes, Supplementary Figure S2a).
Dominance by Cyanobacteria and FAPs is common in most hot spring mats at the range of temperatures covered here. For example, in the slightly alkaline Mushroom and Octopus springs in YNP, Cyanobacteria and Chloroflexi dominated the temperature range between 30 and 70°C (Miller et al., 2009; Liu et al., 2011; Klatt et al., 2013a; Thiel et al., 2016). Whereas in Porcelana and YNP the same main Chloroflexi (Chloroflexus and Roseiflexus) were recovered, Cyanobacteria were rather different. In YNP, Synechococcus spp. dominated above 60°C and Phormidium-like filamentous cyanobacteria were present between 30 and 60°C. A similar importance of Synechococcus spp. has been found in other hot spring mats such as those in northern Thailand (pH 7.3 to 9.1, Sompong et al., 2005) or in Tibet (pH 7.0 to 8.1, Wang et al., 2013). However, in Porcelana we did not identify a significant number of Synechococcus spp. reads, neither did we find more than 0.5% of reads when the 16S rRNA fragments were analyzed. This confirmed our own previous studies using DGGE in Porcelana (Mackenzie et al., 2013) where presence of Synechococcus and Thermosynechococcus sequences was marginal. Cyanobacteria are typically absent from very acidic hot springs (i.e., Bohórquez et al., 2012), and particularly Synechococcus is commonly present in springs that tend to be slightly alkaline (pH 8). This might explain why this cyanobacterium was absent from Porcelana since it is slightly acidic (pH 6.5).
Proteobacteria were the third phylum in importance (Figure 2A). Their relative abundance in metagenomes increased from 5 to 30% as temperature decreased from 66 to 48°C. Interestingly, the contribution of Proteobacteria to metatranscriptomes was negligible at 66°C but very significant at the two lower temperatures during the night but not during the day (Figure 2A).
Proteobacteria belonged to the alpha-Proteobacteria (mostly Rhodospirillales), beta-Proteobacteria (mostly Burkholderiales), gamma-Proteobacteria (mostly Legionellales) and some delta- and epsilon-Proteobacteria. These data were confirmed by looking at the 16S rRNA reads retrieved from the metagenomes (Supplementary Figure S3). Most alpha-Proteobacteria reads could be assigned to the Rhodospirillales genus Elioraea, whose 16S rDNA sequences were 98.8% similar to Elioraea tepidiphila DSM 17972 (Supplementary Table S3). This bacterium was initially isolated from a hot spring at 45–50°C. More recently a second member of the genus, Ca. Elioraea thermophila was identified in YNP. Both of them contain genes for bacteriochlorophyll a synthesis (Tank et al., 2017). We did detect pufM genes from Elioraea sp. both in metagenomes and metatranscriptomes and this will be discussed in the next section. Our sequence was 96.7% similar to the 16S rDNA gene of Ca. Elioraea thermophila. Therefore, the Elioraea sp. from Porcelana is different from that from YNP.
Among the beta-Proteobacteria, a Burkholderia (97% similar to Cupriavidus sp. PIC4; Supplementary Table S3) was the most represented, but we also found sequences assigned to Nitrosospira (100% similar to Nitrosospira sp.; Supplementary Table S3).
Members belonging to phyla Deinococcus–Thermus and Bacteroidetes contributed less than 6% each to the total reads in metagenomes (Figure 2A). The former phylum was more abundant at 58°C, while the Bacteroidetes were more or less uniformly distributed. These results were consistent with the 16S rRNA reads extracted from the metagenomes, where we found 16S rDNA sequences 98.1 and 97.1% similar to Thermus and Meiothermus, respectively, among the Deinococcus–Thermus, as well as sequences 99.9% and 100% similar to Rhodothermus and Sphingobacterium, respectively, among the Bacteroidetes (Supplementary Table S3). Both phyla showed very little contribution to the reads in metatranscriptomes although we could recover some activity of the genera Rhodothermus and Sphingobacterium (both in phylum Bacteroidetes) in the nitrogen cycle (see below).
The remaining bacterial phyla found (i.e., Chlorobi, Planctomycetes, Actinobacteria, etc.) accounted for less than 1% each of the total abundance and transcriptional activity in the mat at all temperatures, both during day and night (Supplementary Table S2). Among these minor components of the mats, Chlorobi showed more transcriptional activity at 48°C during the night, while Planctomycetes were largely inactive at that temperature and time of the day, with most activity during the day at 66°C (Supplementary Table S2). The presence of Chlorobi has also been described previously in many hot springs (Schmid et al., 2003; Jaeschke et al., 2009; Bryant et al., 2012; Klatt et al., 2013a), although the particular members can be different in different springs. Our Chlorobi sequences accounted for 0.38, 0.07, and 1.96% of all the metagenomic reads at 48, 58, and 66°C, respectively. Most of them showed between 96 and 100% (average 99.3%) similarity to sequences in the genome of the phototroph Candidatus Thermochlorobacteriaceae bacterium GBChlB (Supplementary Table S3), as the closest relative. In order to confirm that most sequences belonged to Ca. Thermochlorobacteriaceae bacterium GBChlB we did an additional search: metagenomically retrieved 16S rRNA and fmoA were compared to those of Ca. Thermochlorobacteriaceae bacterium GBChlB and were found to be 98.9% and 94.3% similar, respectively (at 48°C and 66°C, with no signal at 58°C) confirming the identity of our sequences.
Recently Ca. Thermochlorobacter aerophilum was identified in YNP (Liu et al., 2012b). We checked whether this interesting organism was present in Porcelana. When looking for potential similarity to the Ca. Thermochlorobacter sequence, only 10 sequences showed a similarity close to 95%, but they had higher similarity to Chlorobium. We also used the genome of Ca. Thermochlorobacter to recruit sequences from the metagenomes. Only between 100 and 300 reads were retrieved from each metagenome with similarities above 80%, and all of them showed higher similarities to Chlorobium. Therefore, we think it safe to state that the sequence that we recovered was representative of another genus than Ca. Thermochlorobacter, which was either absent or present at concentrations undetectable with our sequencing depth. It is interesting to remark that both Ca. Thermochlorobacteriaceae bacterium GBChlB and Ca. Thermochlorobacter aerophilum seem to be aerobic and unable to oxidize sulfur (Stamps et al., 2014) unlike the previously known phototrophic Chlorobi. The organism found in Porcelana would likely belong to the Thermochlorobacteriaceae family.
Among the Planctomycetes we detected the presence of sequences 96.6% similar in 16S rDNA to Ca. Scalindua marina (Supplementary Table S3). Ca. Scalindua brodae has been reported as an anaerobic ammonium oxidizing bacterium (Annamox) (Schmid et al., 2003), and its presence up to 52°C has been reported in other springs such as those in California and Nevada, where anammox bacteria such as Ca. Brocadia fulgida, Ca. Brocadia anammoxidans, and Ca. Kuenenia stuttgartiensis have been found (Jaeschke et al., 2009). Therefore, the anammox process might be present in the Porcelana mat, but more direct evidence is necessary (see section on nitrogen below).
Archaea are an important group in some hot springs participating in methanogenesis (Merkel et al., 2015), ammonia oxidation (Dodsworth et al., 2011b; Chen et al., 2016), denitrification, DNRA (Dodsworth et al., 2011a), and other major biochemical processes (Offre et al., 2013). Archaea present in Porcelana showed transcriptional activity in the microbial mat (Supplementary Table S2), but they reached on average only 1.9% and 0.4% of the reads at day and night, respectively. The low representation of this domain could be due to the relatively low temperatures registered in Porcelana with respect to other hot springs, since high abundance and activity of Archaea have been only reported from 70 to 95°C (de la Torre et al., 2008; Reigstad et al., 2008; Hamilton et al., 2014). However, the presence of the amoA gene attributed to this group in metatranscriptomes suggests that they may have an important role as ammonia oxidizers. Specifically, Thaumarchaeota (Nitrososphaera sp.) were the most active Archaea at the three temperatures at day and night (Figure 2B). At the upper temperature of 66°C, Thaumarchaeota and Euryarchaeota showed similar abundances, but most of the diurnal transcriptional activity was still due to Thaumarchaeota. During the night, members of Thaumarchaeota were the only active archaeal representatives at 58 and 48°C. We found 16S rDNA sequences in metagenomes assigned to Nitrososphaera (16S rDNA 99.7% similar to Crenarchaeote enrichment culture clone OREC-B1045) and Nitrosopumilus (99.7% similar to Nitrosopumilus sp. DDS1) (Supplementary Table S3).
Several genes essential for light utilization were chosen as representative of photosystems I and II (psaA and psbA, respectively), reaction centers (RC) 1 and 2 (pscA and pufM, respectively), a bacteriochlorophyll a binding protein found in phototrophs that contain RC type-1 and chlorosomes (fmoA), and bacteriochlorophyllide a synthesis (bchC) (Supplementary Table S4). The two first genes are associated with the oxygenic photosynthesis carried out by Cyanobacteria, while the latter four are involved in anoxygenic photosynthesis or just phototrophy carried out by a variety of bacteria, including Chloroflexi and Proteobacteria (with RC type-2) and Chlorobi and Heliobacterium (with RC type-1). The transcriptional activity of these genes is shown in Figure 3 and Supplementary Figure S4. Figure 3 provides a synthetic picture, while actual transcripts for each process are presented in Supplementary Figure S4. Marker genes for both photosystems I and II were mostly expressed at the lower temperatures of 58 and 48°C, essentially during daytime (Figure 3A), with Mastigocladus sp. being the genus responsible for most of these phototrophic activity, as expected given its previously reported optimal growth temperature within this range (Ionescu et al., 2010; Alcamán et al., 2017). In addition to Mastigocladus, a minor contribution of Oscillatoriales of the LPP group (such as Leptolyngbya sp.) was detected at 66°C. These genes showed very little transcriptional activity at 66°C during the day (Figure 3A), in fact, it was almost two orders of magnitude lower than at the two lower temperatures (Supplementary Figure S4a). During the night, transcriptional activity was lower than during the day at the two lower temperatures (Figure 3A). Most transcripts were assigned to Mastigocladus, an organism that was mostly active at the two lower temperatures (Supplementary Figure S2). Moreover, down-regulation of photosystem and CO2 fixation genes in the dark has been shown for Cyanobacteria both in the laboratory (Hernández-Prieto et al., 2016) and in the field (Ottesen et al., 2014). At temperatures higher that 58°C, a diel study in Mushroom Spring effluent channel (60–64°C) by Steunou et al. (2008) attributed transcripts of psbB gene exclusively to Synechococcus. And in a more detailed study of phototrophy in the same spring, Liu et al. (2011) observed again that Synechococcus was the most important organism carrying out oxygenic photosynthesis using chlorophyll a.
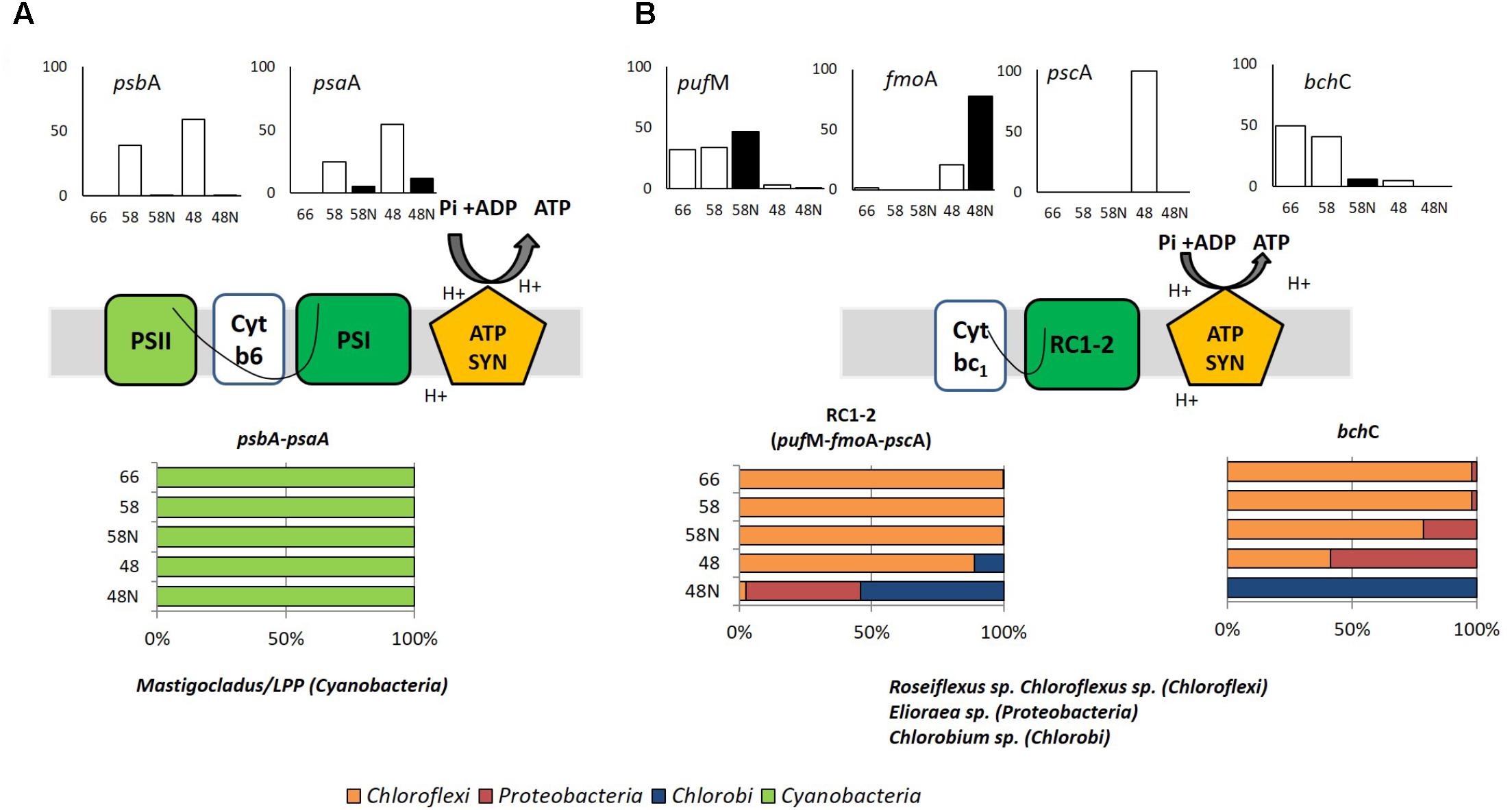
FIGURE 3. Phototrophy in Porcelana mat. (A) Oxygenic (psbA and psaA genes) and (B) Anoxygenic (pufM, fmoA, pscA, and bchC genes) photosynthesis. Cartoons in the middle part show the selected photosynthetic genes. Bar graphs show the percent of the total transcripts of each gene found in each sample, during the day (white bars) and night (black bars) at each temperature. The lower horizontal bars show the percent contribution of the main taxa to transcription for each process at each temperature. The genes considered are shown above the bars.
In Porcelana, some of the transcripts found were also assigned to anoxygenic phototrophy (Figure 3B). These genes showed very low transcriptional activity compared to those of photosystems I and II, between one and two orders of magnitude lower (compare Supplementary Figures S4a,b). Most of this activity could be attributed to Chloroflexus/Roseiflexus at the two higher temperatures, while Elioraea sp. and Ca. Thermochlorobacteriaceae contributed significantly at 48°C, especially during the night (Figure 3B). The bchC gene was transcribed at the two upper temperatures during the day and essentially all this activity was due to Chloroflexi. At 48°C there was ten times lower activity and it was due to Chlorobi. Klatt et al. (2013b) showed that Chloroflexus and Roseiflexus expressed pufM and bchC genes preferentially in the dark in YNP, which is in accordance with the results reported here at 58°C for pufM but not for bchC. In summary in our study, the anoxygenic phototrophy shows a minor activity for the whole mat compared to the full oxygenic photosynthesis (Figure 3 and Supplementary Figure S4). However, it was significant at the highest temperature. It was carried out by Chloroflexi at 66 and 58°C. At the lower temperature Elioraea sp. (an alpha-Proteobacteria with reaction center-2; 98.8% similar to E. tepidiphila as explained above) and a Chlorobium sp. (99.3% similar to Ca. Thermochlorobacteriaceae bacterium GBChlB as mentioned above) showed some activity. Tank et al. (2017) found Ca. Elioraea termophilum to be a photoheterotroph in YNP. Thus, the Elioraea in Porcelana must be metabolically similar to the YNP strain.
Although –considering transcriptomic data– light utilization was mostly due to the oxygenic autotroph Mastigocladus (Figure 3A), a range of light harvesting strategies was present along the temperature gradient. Our study demonstrates the importance of considering several temperatures along the gradient, since light was used by different mechanisms depending on microbial composition as modulated by temperature. This will be essential to generate a complete understanding of all potential functions carried out by the whole mat ecosystem.
The genes chosen as representative of different carbon fixation pathways are shown in Supplementary Table S4. These genes were searched for in metagenomes and metatranscriptomes as in the previous section. Most of the transcription of genes involved in autotrophic CO2 fixation, occurred at intermediate and lower temperatures, during the day (Figure 4A and Supplementary Figure S5). The Calvin–Benson–Bassham cycle (CBB) for CO2 fixation, based on the large number of rbcL (ribulose-bisphosphate carboxylase large chain) gene transcripts (Figure 4A), was attributed mostly (up to 90% at the lowest temperatures) to Mastigocladus sp. and a small proportion to the LPP group (5%) (Lyngbya – Phormidium – Plectonema) at 58 and 48°C at daytime. At night, a basal signal (<0.1%) of transcriptional activity was recorded for Cyanobacteria (Supplementary Figure S5), mostly associated with Oscillatoriophycideae (LPP group). Proteobacteria (Bradyrhizobium sp.) had the CBB genes transcribed at 66°C (66.8%), but this was extremely low at 58°C (1.1%) and 48°C (2.8%) during the day (Figure 4A), without any signal at night. Proteobacteria were more abundant and active at the lower temperatures considering all the reads (Figure 2). However, the Proteobacteria CBB genes were more transcribed at 66°C (Figure 4A). Since total CBB transcriptional activity at this temperature was low (Supplementary Figure S5), the contribution to total CBB of Proteobacteria was minor. Some Bradyrhizobium strains like B. japonicum are known to be able to fix CO2 through the CBB (Franck et al., 2008). Some other Bradyrhizobium strains have the genes to synthesize bacteriochlorophyll and carry out a photoheterotrophic metabolism (Okubo et al., 2012; Hanada, 2016). Analysis of 16S rRNA fragments in metagenomes corresponding to Bradyrhizobium were 100% similar to Bradyrhizobium sp. ORS278, one of these aerobic anoxygenic phototrophs. However, it was not possible to detect the pufM and bchC genes for these organisms in the metagenomes. Therefore, the Proteobacteria (alpha-Proteobacteria such as Bradyrhizobium) in Porcelana show gene transcription compatible with some CO2 fixation activity but do not appear to have pigments to use light. It is also important to mention that transcripts related to Proteobacteria at 66°C, represent just ∼2% of the Cyanobacteria transcripts at the two lower temperatures. As already mentioned, therefore, the CO2 fixation associated to Proteobacteria was very small compared to that of Cyanobacteria.
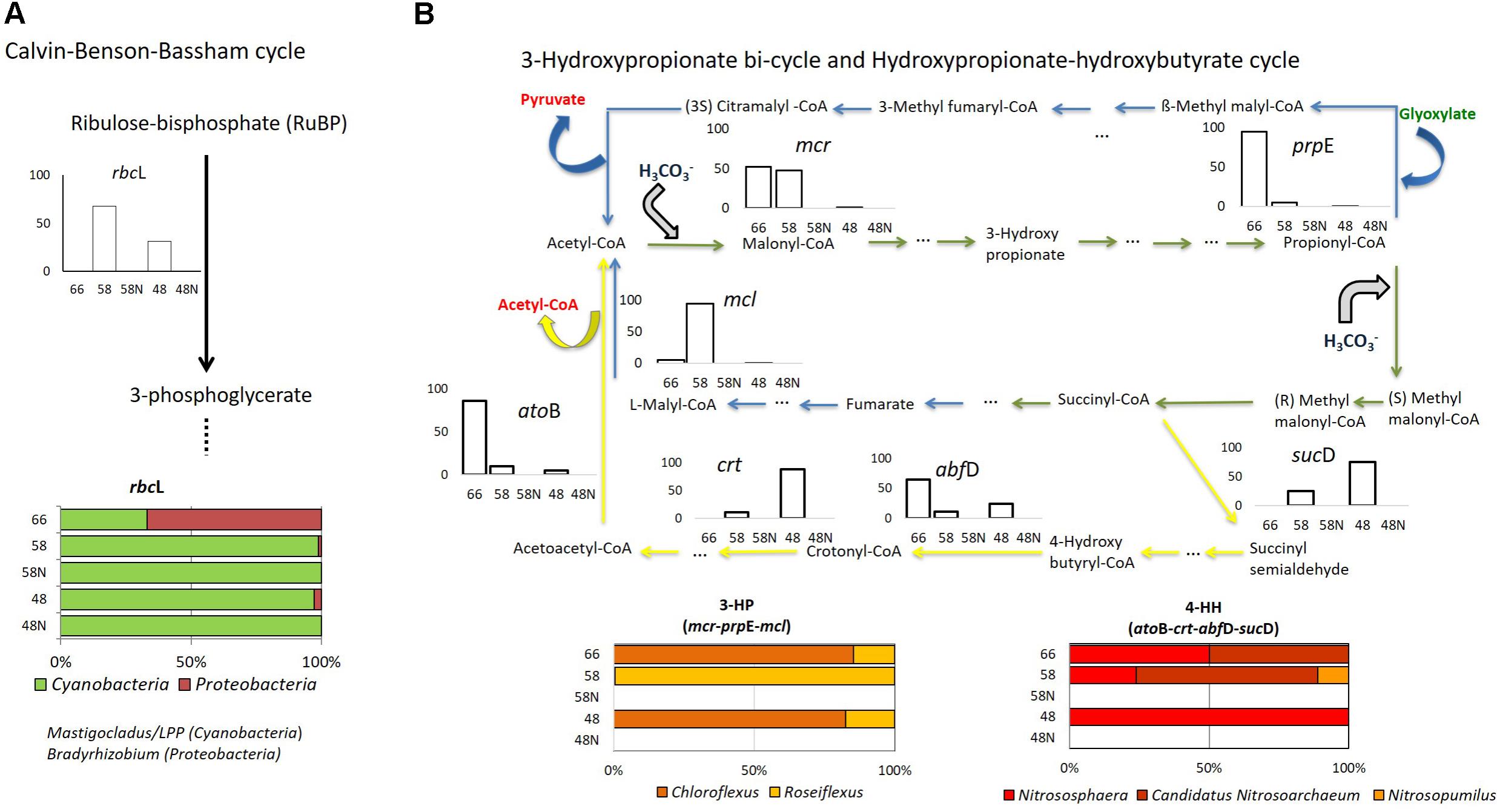
FIGURE 4. Autotrophy in Porcelana mat. (A) Calvin–Benson–Bassham cycle (rbcL gene); (B) 3-hydroxypropionate bi-cycle (blue arrows: mcl, mcr, and prpE genes), and hydroxypropionate-hydroxybutyrate cycle (yellow arrows: atoB, crt, abfD, sucD genes). Green arrows show the steps shared by the two pathways in Bacteria and Archaea. Bar graphs show the percent of the total transcripts of each gene found in each sample, during the day (white bars) and night (black bars) at each temperature. The lower horizontal bars show the percent contribution of the main taxa to transcription for each process at each temperature.
The 3-hydroxypropionate bi-cycle (3-HP), characteristic of Chloroflexi (Hügler et al., 2002) was followed through the three key genes mcl, mcr, and prpE (Supplementary Table S4). These genes showed active transcription at 66 and 58°C, while at 48°C transcription was orders of magnitude lower (Figure 4B and Supplementary Figure S6a). We could not detect transcription of these genes at night. Transcripts associated with the 3-HP bi-cycle were all assigned to Chloroflexus sp. and Roseiflexus sp. (Figure 4B). The total number of transcripts was similar to that of the CBB cycle at 66°C but it was two and four orders of magnitude lower than the CBB at 58 and 48°C, respectively (Supplementary Figure S5).
Several steps involved in this cycle (noted by the green arrows in Figure 4B) are shared with the carbon fixation performed by Archaea through the hydroxypropionate-hydroxybutyrate (HH) cycle, while several other steps are exclusively found in Archaea (yellow arrows in Figure 4B). We looked for four of these genes: atoB, crt, abfD, and sucD (Supplementary Table S4). Transcripts of these genes were assigned to Thaumarchaeota, including Nitrososphaera sp., Candidatus Nitrosoarchaeum, and Nitrosopumilus sp., and they were found exclusively during the day (Figure 4B). More transcripts were found at 66°C and 48°C than at 58°C (Supplementary Figure S6b). However, the HH cycle only represented 0.2% of the total C-uptake pathways found in the mat (Supplementary Figure S5), thus accounting for a very low portion of CO2 fixation.
Swingley et al. (2012) found the same carbon fixation pathways at the lower temperature range (57 and 65.5°C) similar to that analyzed here (48 and 58°C) in Bison Pool (YNP). In contrast, at 67°C these authors could only detect heterotrophic metabolisms, while in Porcelana both presence and transcriptional activity of genes associated with CO2 fixation assigned to Chloroflexi and Thaumarchaeota were still recorded. Swingley et al. (2012) also found the reverse tricarboxylic acid (rTCA), and the acetyl-CoA cycles at higher temperatures (84 and 90°C). Thiel et al. (2017) found evidence not only of CBB and 3-HP bi-cycle, but also of the reductive TCA and Wood–Ljungdahl pathways in Mushroom Spring mat at 60°C. However, none of these previous studies presented the relative contributions of each of these processes to the ecosystem at different temperatures.
To corroborate that the transcription of genes in C-pathways found actually resulted in CO2 fixation, in situ daily rates of bicarbonate (H13CO3) assimilation were measured at 58 and 48°C, under light and dark conditions (Supplementary Figure S7). The high rates found on average (534 nmoles C cm-2 d-1) under light compared to (7.95 nmoles C cm-2 d-1) dark conditions, are consistent with contributions by the different CO2 fixation processes detected in metatranscriptomes. However, based on the number of transcripts (Supplementary Figure S5), most of this assimilation was referred to oxygenic photosynthesis carried out by the cyanobacterium Mastigocladus sp. Therefore, at lower temperatures where Cyanobacteria and Chloroflexi co-occur (Liu et al., 2011), Chloroflexi biomass was probably supported by its heterotrophic metabolism (Sandbeck and Ward, 1981; Anderson et al., 1987; Bateson and Ward, 1988) using by-products of cyanobacterial primary producers (van der Meer et al., 2005; Zarzycki and Fuchs, 2011; Bryant et al., 2012; Klatt et al., 2013a), although some degree of mixotrophy cannot be excluded.
Three other carbon fixation pathways are known in prokaryotes. We looked for the key genes (Supplementary Table S4) but the number of reads found was several orders (from 10e-1 to 10e-32) of magnitude lower than for the three cycles already described.
Since nitrogen is the most common limiting nutrient in Porcelana microbial mat (Alcamán et al., 2015) as well as in other circumneutral hot spring mats (Hamilton et al., 2014), pathways and genes involved in nitrogen acquisition and transformations were evaluated (Figure 5).
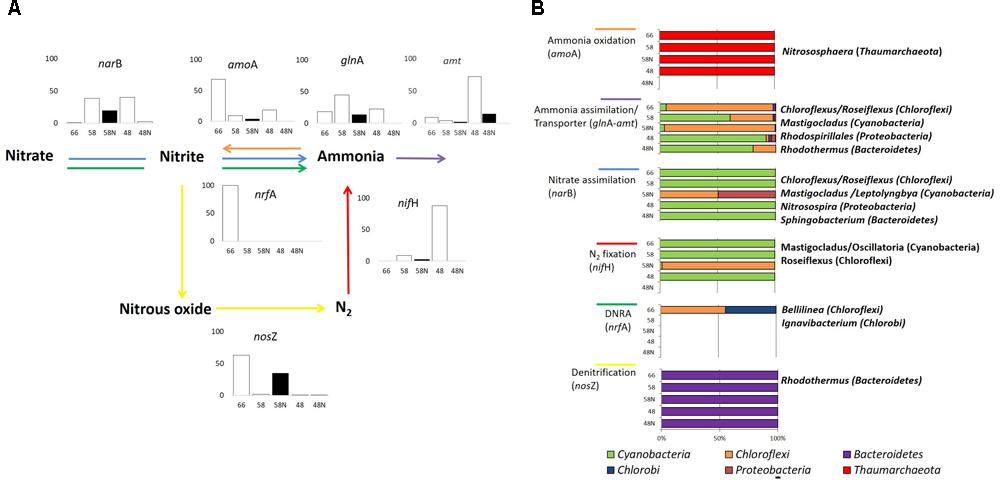
FIGURE 5. Nitrogen cycle in Porcelana mat. (A) Scheme of the reactions, representative genes, and transcriptional activity for each gene at the specific pathway. Bar graphs show the percent of the total transcripts of each gene found in each sample, during the day (white bars) and night (black bars) at each temperature. (B) Percent contribution of the main Phyla to transcription for each gene is shown by the horizontal bars. The most important genera in each case are noted to the right.
According to isotopic assimilation rates (Supplementary Figure S7), nitrogen fixation was the most important mechanism for the incorporation of new nitrogen to the system (Alcamán et al., 2015), and 16S rRNA and metatranscriptomics data showed that this fixation was entirely due to Cyanobacteria at daytime and particularly to Mastigocladus sp. This cyanobacterium has been recently isolated from Porcelana, and has been physiologically characterized as a thermotolerant diazotrophic cyanobacterium (Alcamán et al., 2017).
The largest numbers of nifH gene transcripts associated with Mastigocladus sp. were registered mostly at the lowest temperature (only during the day), with few transcripts at 66°C, involving sequences assigned to Oscillatoria sp. (96.2% similarity with Oscillatoriales cyanobacterium JSC-12) in addition to those of Mastigocladus (Figure 5A and Supplementary Table S3). The large signal of nifH genes correlated positively with the high nitrogen fixation rates, and also with nitrogenase activity previously reported at lower temperatures in Porcelana mat (Alcamán et al., 2015). Steunou et al. (2008) found that nifH activity was highest in the evening and in the early morning in Mushroom Spring. The difference with present results is due to the fact that Synechococcus was the abundant nitrogen fixer in YNP, and this cyanobacterium needs to separate in time oxygen production through photosynthesis and nitrogen fixation. Mastigocladus, on the other hand, has heterocysts and, therefore, both activities can occur at the same time.
During the night, and specifically at 58°C, Roseiflexus sp. expressed nifH gene (Figure 5B). It is still not clear what could be the function of the nifHDK-like genes in Roseiflexus spp. given the fact that many other accessory proteins required for the nitrogenase maturation are not present in any available Roseiflexus spp. genome (Thiel et al., 2017). Similarly, under controlled conditions Roseiflexus castenholzii is unable to grow on N2 as sole nitrogen source (Thiel et al., 2017).
Ammonia and nitrate assimilation rates at 58 and 48°C were three orders of magnitude lower than nitrogen fixation (Supplementary Figure S7). Bacteroidetes such as Rhodothermus (96.8% similarity to 16S rRNA of Rhodothermus marinus SG0.5JP17-172) and Sphingobacterium spp. (87.3% similarity with Sphingobacterium sp. TM-2) and Proteobacteria like Rhodospirillales and Nitrosospira sp. (100% similarity with Nitrosospira sp., Supplementary Table S3) also had small contributions to both processes. The large amount of glnA and amt gene transcripts, mostly associated with Chloroflexi at high temperatures and with Cyanobacteria at the lowest, indicated rather active ammonia assimilation in the mat during day and night periods. However, the number of gene transcripts of amt transporter were lower than those associated to glnA at all temperatures (Supplementary Figure S8). This could be tightly correlated with the very low 15NH4Cl assimilation rates recorded in Porcelana at this time (Supplementary Figure S7). The low natural concentrations of ammonia or nitrate measured in the system suggest that the microbial community is forced to maintain a low gene transcription rate necessary for the acquisition of these nutrients, and the preference to utilize energy in order to obtain nitrogen by N2 fixation.
The turnover time estimated resulted in about 1 h for ammonia. In contrast, nitrate apparently remained more time in the mat with a turnover between 12 and 34 h, evidencing that ammonia was preferred as nitrogen source in the community. High nutrient turnover rates were also found in other hot springs such as Perpetual Spring (YNP, Hamilton et al., 2014).
Thaumarchaeota transcripts of the amoA gene were detected at all temperatures, especially at 66°C (Figure 5B and Supplementary Figure S8). This indicated Thaumarchaeota were active as ammonia oxidizers as in other springs (Dodsworth et al., 2011b; Chen et al., 2016). This transcription was assigned particularly to Nitrososphaera sp. relatives (97.9% similarity with Crenarchaeote enrichment culture clone OREC-B1045 (Supplementary Table S3 and Figure 5B). A similar pattern has been found in a biofilm growing at 63°C in a thermal artesian spring at YNP, where Thaumarchaeota dominated among other ammonia oxidizers (Marks et al., 2012). Other studies have also documented Archaea in hot springs with very low pH and high temperature (Hou et al., 2013), as well as at high temperatures in circumneutral springs (Cole et al., 2013). In fact, Nitrososphaera was the most abundant and active Thaumarchaeota at all temperatures, decreasing its dominance in metagenomes and metatranscriptomes with decreasing temperature. An opposite pattern was observed for other less abundant and active Thaumarchaeota such as Nitrosopumilus sp. (99.7% similarity with Nitrosopumilus sp. DDS1) that increased slightly with decreasing temperature. In fact, it has been reported that the thermophilic Thaumarchaeota Nitrosophaera sp. can grow under low ammonium concentrations such as those found in Porcelana (0.01 μmol L-1), with the potential to carry out CO2 fixation under culture conditions (Hatzenpichler et al., 2008). The widespread distribution of putative archaeal amo genes and their numerical dominance over their bacterial counterparts in most marine and terrestrial environments suggest that ammonia oxidizing Archaea play a major role in global nitrification (Francis et al., 2005; Zhang et al., 2008). Some studies by Dodsworth et al. (2011b) in Great Boiling Spring and Sandy’s Spring West (United States Great Basin) reported the autotrophic ammonia-oxidizing archaeon Ca. Nitrosocaldus yellowstonii and identified ammonia oxidation as a major source of energy fueling primary production in these extreme environments.
Finally, it was also possible to detect in the different metatranscriptomes, genes related to the principal enzymes involved in the two nitrogen dissimilatory processes: nitrate reduction to ammonium (DNRA) (nrfA) and denitrification (nosZ) (Figure 5, Supplementary Table S4, and Supplementary Figure S8). DNRA was detected only at 66°C and was taxonomically associated with Chloroflexi (16S rDNA 93.8% similar to Bellilinea caldifistulae) and Chlorobi (97.8% similar to Ignavibacterium album JCM 16511, Supplementary Table S3 and Figure 5B). Bellilinea and Ignavibacterium have been previously found in hot springs. Ignavibacterium was isolated from Yumata hot spring (Japan), and it contains the two genes (nrfA as nrfH) as indicators of nitrite reductase (Liu et al., 2012a). The nosZ gene was also more actively expressed at 66°C and 58°C at night (Figure 5A) and was primarily attributed to heterotrophic organisms (i.e., Rhodothermus sp. 99.9% 16S rRNA similarity, Supplementary Table S3), whose denitrification activity has been shown to take place typically under low oxygen conditions (Zumft, 1997). Even though active genes of DNRA and denitrification were found at high temperatures in the mat, they both quantitatively represented very low number of reads in comparison to the other nitrogen transformation processes (Supplementary Figure S8).
As mentioned above, the presence of Ca. Scalindua (Planctomycetes) (16S rDNA sequence 99.8% similar, Supplementary Table S3) suggests the potential anammox activity of this species in the mat (Schmid et al., 2003). However, the absence of the enzyme hydrazine synthase (hzsA gene) in our metagenomic data and the low contribution of the Planctomycetes phylum (only reached <1% of reads in metatranscriptomes) at high temperatures, prevent determining the relevance of this process in Porcelana. Further studies in Porcelana mats at night when oxygen is depleted, are now needed to better understand the relevance of this and other anaerobic processes.
In summary, nitrogen fixation was the most active nitrogen uptake process at intermediate and low temperatures, and it was mostly due to Cyanobacteria (Mastigocladus sp.). At the highest and intermediate temperatures, however, ammonia oxidation and assimilation by members of Chloroflexi and some Thaumarchaeota might have a more significant contribution to the system. Probably at higher temperatures (>70°C), the input of nitrogen could be due to the contribution of others microbial members of Cyanobacteria by nitrogen fixation (Loiacono et al., 2012) or by Archaea through nitrification (Reigstad et al., 2008). More speculative, in Porcelana spring the nitrogen could be supplied by fumarole water inputs. Now more time-sampling points at the temperature gradient are necessary to have a more complete view of the microbial community activity inhabiting Porcelana hot spring.
Combining isotopic rate measurements with metagenomics and metatranscriptomics analyses allowed the reconstruction of the main carbon and nitrogen pathways, as well as light utilization forms, over a temperature gradient between 48 and 66°C in Porcelana, with the identification of main microbial players in each of these pathways (Figure 6). It was demonstrated that the most important taxon in Porcelana thermal mat was the cyanobacterium Mastigocladus, who was present at all temperatures but mostly active at the lower temperatures in the gradient. It has been shown that this cyanobacterium has an optimal temperature for growth and nitrogenase activity at 45–50°C (Alcamán et al., 2017). In culture the maximal temperature for growth was 58°C, thus, growth at 66°C is not expected. The cyanobacteria found in Porcelana at the latter temperature are likely the result of past growth when temperature distribution along the stream might have been lower. Moreover, this cyanobacterium was responsible for most of the primary production in this system through its oxygenic photosynthesis and nitrogen fixation activities.
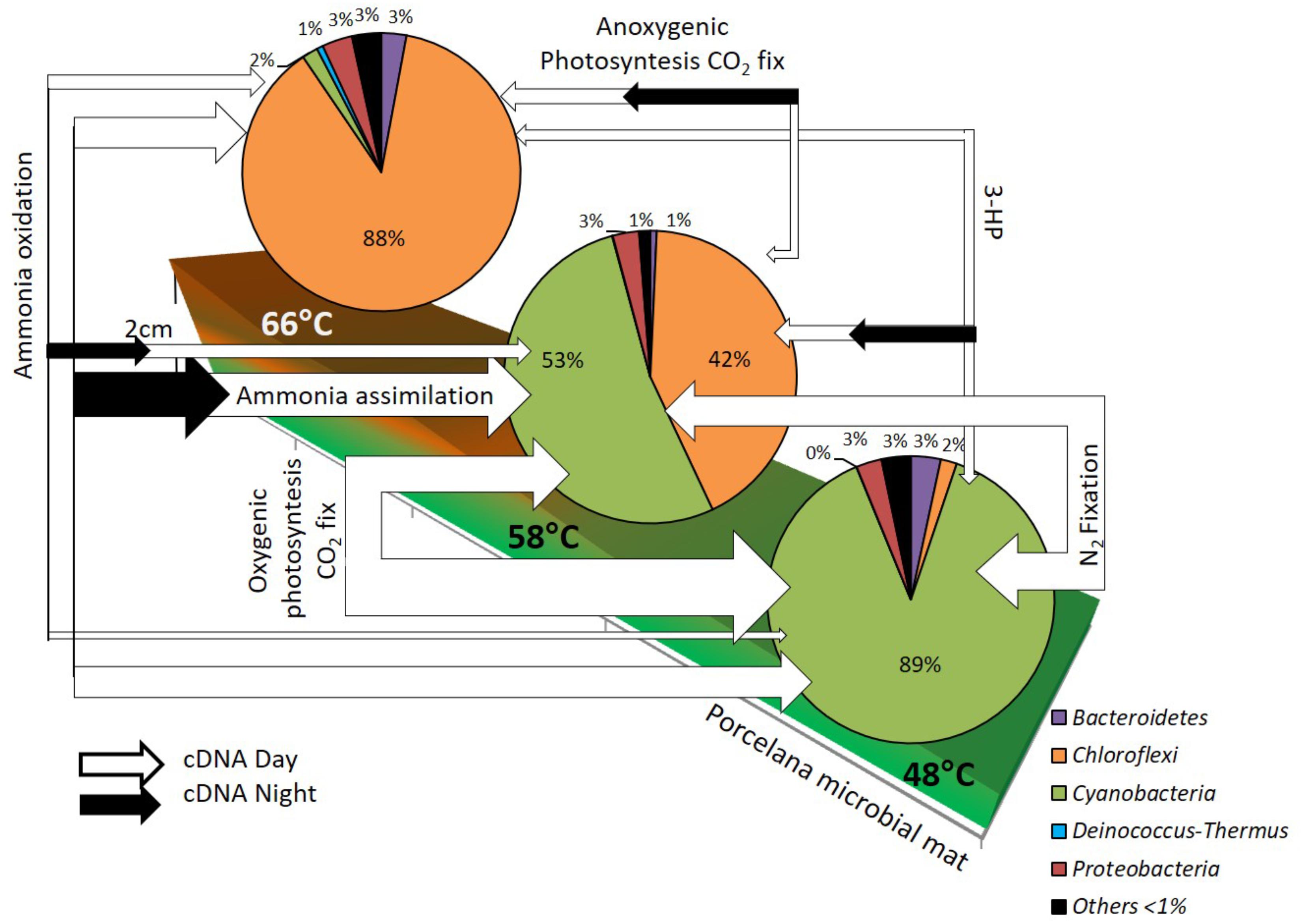
FIGURE 6. Reconstruction of the diurnal changes in active carbon and nitrogen pathways, based in metatranscriptomics analysis, along the temperature gradient in Porcelana microbial mat dominated by Chloroflexi and Cyanobacteria (pie charts). The white (day) and black (night) arrow width represent the total gene transcripts to each pathway and indicates the importance of these at each temperature.
Another relevant guild was the FAPs, represented mostly by Roseiflexus and Chloroflexus. Particularly, Chloroflexus can use light as a source of energy via bacteriochlorophyll c, carrying out anoxygenic photosynthesis according to our transcriptomics data as well as be active in CO2 fixation through the 3-HP bi-cycle. Roseiflexus might carry out the same metabolism, although no autotrophic Roseiflexus strain has been described so far. Since the photosynthesis genes of these bacteria are usually active during the evening or night, a role in photosynthesis at 66°C and in the other temperatures is likely. However, FAPs seem to contribute very little to the total carbon fixation in the mat although their contribution is significant at the highest temperature. On the other hand, this group was responsible for most of the ammonia uptake at the highest temperature of 66°C.
Other taxa in the mat were also noteworthy, such as the alpha-Proteobacteria anoxygenic phototroph Elioraea. This bacterium was mostly responsible for the transcription of anoxygenic phototrophic genes at 48°C during the night. In this respect, Candidatus Elioraea thermophila has also been found to be phototrophic in YNP hot spring mats (Thiel et al., 2016, 2017; Tank et al., 2017). Regarding ammonia oxidation to nitrate, all the amoA gene transcripts found were attributed to Thaumarchaeota, which were active at the three temperatures, likely growing chemolithoautotrophically and fixing CO2 via the HH cycle. Although their abundance was relatively low, and their contribution to primary production was probably minor compared to that of Cyanobacteria, their contribution to nitrification was likely very important in the ecosystem, particularly at the highest temperature, where there was no nitrogen fixation by Cyanobacteria, and nitrification could act as a new source of nitrate to the community.
One final point was the frequent decoupling between abundance and total transcriptional activity of the microorganisms at different temperatures such as those observed here for Chloroflexi members and Mastigocladus. This decoupling might be explained by changes in the temperature of the water source that would shift the distance to the source where the optimal temperature of each microorganism would be found. If temperature increased, for example, Mastigocladus would stop growing at that point where a large biomass would remain for a while. The cyanobacterium would then start growing actively further away from the source at lower temperatures, where biomass would be temporarily low. This emphasizes the importance of determining both abundance and activity (both measured by incorporation and fixation rates as well as transcriptional activity) along the temperature gradient to fully understand the ecology of hot spring microbial mats.
BD and CP-A designed the study. MA-A and BD did the in situ experiments. MA-A and CF carried out the analysis of isotope experiments. JT and DP-P did the bioinformatics analyses. MA-A, CP-A, and BD wrote the draft of manuscript. JT, CF, DP-P, and MV revised the manuscript critically and provided substantial contributions. All authors read and approved the final version of the article.
This work was financially supported by Ph.D. scholarship CONICYT N° 21110900, the French Embassy scholarship and LIA MORFUN for Ph.D. mobility (Chile), and the following grants funded by FONDECYT N° 1110696, 1161232, and 1150171; FONDAP No15110009, 15110027, and PFB-31. Sequencing and work in Madrid were funded by grant CTM2016-80095-C2-1-R from the Spanish Ministerio de Economía y Competitividad.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to Huinay Research Station for logistical assistance, Sebastian Espinoza for his assistance with sample collection, and Gonzalo Sepúlveda, Germán Marchandón, and Tomás Alarcón for bioinformatics assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02353/full#supplementary-material
FIGURE S1 | Taxonomic assignment of Chloroflexi by metagenomic (DNA; white bars) and metatranscriptomic reads (cDNA day: gray bars; cDNA night: black bars) at the Order (a) and genus (b) levels at the three temperatures studied.
FIGURE S2 | Taxonomic assignment of Cyanobacteria by metagenomic (DNA; white bars) and metatranscriptomic reads (cDNA day: gray bars; cDNA night: black bars) at the three temperatures studied. (a) Proportion of cyanobacterial reads in different Orders. (b) Proportion of Stigonematales reads in different genera.
FIGURE S3 | Total reads assigned to Proteobacteria by 16S rRNA sequences recovered from metagenomes. Rhodospirillales (alphaproteobacteria) and Burkholderiales (betaproteobacteria) were the most abundant orders at the lowest temperature. Note that the vertical scale is logarithmic.
FIGURE S4 | Total transcripts (cDNA) assigned to phototrophic processes. (a) Transcripts associated with photosystems PSI (psaA gene) and PSII (psbA gene) in oxygenic photosynthesis. (b) Transcripts (pufM, fmoA, pscA and bchC genes) associated with anoxygenic photosynthesis. Each bar represents a different temperature and daily period. Transcripts were normalized by RPKM.
FIGURE S5 | Total transcripts (cDNA) associated with the autotrophic carbon fixation pathways. CC, Calvin–Benson–Basham cycle (rbcL gene); 3HP, 3-hydroxypropionate bi-cycle (mcr gene); HH, hydroxypropionate-hydroxybutyrate cycle (atoB gene). Transcripts were normalized by RPKM.
FIGURE S6 | Total transcripts (cDNA) associated with specific carbon fixation pathways. (a) 3-Hydroxypropionate bi-cycle (mcr, prpE, and mcl genes) and (b) hydroxypropionate-hydroxybutyrate atoB, crt, abfD, sucD genes) cycle. Transcripts were normalized by RPKM.
FIGURE S7 | Diurnal carbon and nitrogen assimilation rates recorded at 58 and 48°C in Porcelana microbial mats. Nitrogen fixation data from Alcamán et al. (2015). The black bars represent the C or N assimilation rates in the dark.
FIGURE S8 | Total transcripts (cDNA) assigned to nitrogen cycle pathways: ammonia transporter (amt gene), nitrate assimilation (narB gene), ammonia assimilation (glnA gene), ammonia oxidation (amoA gene), N2 fixation (nifH gene), DNRA (nrfA gene), and denitrification (nosZ gene). Transcripts were normalized by RPKM.
TABLE S1 | Results of high throughput sequencing: raw reads, quality reads, percentage of reads corresponding to ribosomal RNA, non-ribosomal genes, non-redundant (NR) hits and percentage of the latter that could be taxonomically assigned. Numbers of sequences are shown in millions.
TABLE S2 | Total percentage of Bacteria and Archaea phyla present in the entire temperature gradient of Porcelana hot spring at noon and night (N) periods.
TABLE S3 | Best hits and % similarity in the SILVA database of the most relevant organisms found in Porcelana mat.
TABLE S4 | Process and pathways analyzed with principal genes associated and encoding protein. All this was used as diagnostic for phototrophy, autotrophy, and nitrogen cycle transformations.
Alcamán, M. E., Alcorta, J., Bergman, B., Vásquez, M., and Díez, B. (2017). Physiological and gene expression responses to nitrogen regimes and temperatures in Mastigocladus sp. strain CHP1, a predominant thermotolerant cyanobacterium of hot springs. Syst. Appl. Microbiol. 40, 103–113. doi: 10.1016/j.syapm.2016.11.007
Alcamán, M. E., Fernández, C., Delgado, A., Bergman, B., and Díez, B. (2015). The cyanobacterium Mastigocladus fulfills the nitrogen demand of a terrestrial hot spring microbial mat. ISME J. 9, 2290–2303. doi: 10.1038/ismej.2015.63
Anders, S., Pyl, P. T., and Huber, W. (2015). HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1093/bioinformatics/btu638
Anderson, K. L., Timothy, A. T., and Ward, D. M. (1987). Formation and fate of fermentation products in hot spring cyanobacterial mats. Appl. Environ. Microbiol. 53, 2343–2352.
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bateson, M. M., and Ward, D. M. (1988). Photoexcretion and fate of glycolate in a hot spring cyanobacterial mat. Appl. Environ. Microbiol. 54, 1738–1743.
Bhaya, D., Grossman, A. R., Steunou, A., Khuri, N., Cohan, F. M., Hamamura, N., et al. (2007). Population level functional diversity in a microbial community revealed by comparative genomic and metagenomic analyses. ISME J. 1, 703–713. doi: 10.1038/ismej.2007.46
Bohórquez, L. C., Delgado-Serrano, L., López, G., Osorio-Forero, C., Klepac-Ceraj, V., Kolter, R., et al. (2012). In-depth characterization via complementing culture-independent approaches of the microbial community in an acidic hot spring of the colombian andes. Microbiol. Ecol. 63, 103–115. doi: 10.1007/s00248-011-9943-3
Bryant, D. A., Klatt, C. G., Frigaard, N. U., Liu, Z., Li, T., Zhao, F., et al. (2012). “Comparative and functional genomics of anoxygenic green bacteria from the taxa Chlorobi, Chloroflexi, and Acidobacteria,” in Functional Genomics and Evolution of Photosynthetic Systems, Vol. 3, eds R. L. Burnap and W. Vermaas (Dordrecht: Springer), 47–102.
Buchfink, B., Xie, C. H., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–63. doi: 10.1038/nmeth.3176
Bushnell, B. (2014). BBMap: A Fast, Accurate, Splice-Aware Aligner. United States. Available at: https://www.osti.gov/servlets/purl/1241166
Cabello, P., Dolores, M., and Moreno-Vivián, C. (2004). Nitrate reduction and the nitrogen cycle in archaea. Microbiology 150, 3527–3546. doi: 10.1099/mic.0.27303-0
Canfield, D. E., and Des Marais, D. J. (1994). Cycling of carbon, sulfur, oxygen and nutrients in a microbial mat. Science 251, 1471–1473. doi: 10.1126/science.11538266
Chen, S., Peng, X., Xu, H., and Ta, K. (2016). Nitrification of archaeal ammonia oxidizers in a hightemperature hot spring. Biogeosciences 13, 2051–2060. doi: 10.5194/bg-13-2051-2016
Cole, J. K., Peacock, J. P., Dodsworth, J. A., Williams, A. J., Thompson, D. B., Dong, H., et al. (2013). Sediment microbial communities in great boiling spring are controlled by temperature and distinct from water communities. ISME J. 7, 718–729. doi: 10.1038/ismej.2012.157
de la Torre, J. R., Walker, C. B., Ingalls, A. E., Könneke, M., and Stahl, D. A. (2008). Cultivation of a thermophilic ammonia oxidizing archaeon synthesizing crenarchaeol. Environ. Microbiol. 10, 810–818. doi: 10.1111/j.1462-2920.2007.01506.x
Dodsworth, J. A., Hungate, B. A., de la Torre, J. R., Jiang, H., and Hedlund, B. P. (2011a). Measuring nitrification, denitrification, and related biomarkers in terrestrial geothermal ecosystems. Methods Enzymol. 486, 172–198. doi: 10.1016/B978-0-12-381294-0.00008-0
Dodsworth, J. A., Hungate, B. A., and Hedlund, B. P. (2011b). Ammonia oxidation, denitrification and dissimilatory nitrate reduction to ammonium in two US Great Basin hot springs with abundant ammonia-oxidizing archaea. Environ. Microbiol. 13, 2371–2386. doi: 10.1111/j.1462-2920.2011.02508.x
Duhart, P., Crignola, P., Ordonez, A., and Munoz, J. (2000). Franjas metalogenicas en Chiloe Continental (41–44S). Congreso Geologico Chileno. 9, 201–205.
Finsinger, K., Scholz, I., Serrano, A., Morales, S., Uribe-Lorio, L., Mora, M., et al. (2008). Characterization of true-branching cyanobacteria from geothermal sites and hot springs of Costa Rica. Environ. Microbiol. 10, 460–473. doi: 10.1111/j.1462-2920.2007.01467.x
Francis, C. A., Roberts, K. J., Beman, J. M., Santoro, A. E., and Oakley, B. B. (2005). Ubiquity and diversity of ammoniaoxidizing archaea in water columns and sediments of the ocean. Proc. Natl. Acad. Sci. U.S.A. 102, 14683–14688. doi: 10.1073/pnas.0506625102
Franck, W. L., Chang, W., Qi, J., Sugawara, M., Sadowsky, M. J., Smith, S. A., et al. (2008). Whole-genome transcriptional profiling of Bradyrhizobium japonicum during chemoautotrophic growth. J. Bacteriol. 190, 6697–6705. doi: 10.1128/JB.00543-08
Guazzaroni, M. E., Herbst, F. A., Lores, I., Tamames, J., Peláez, A. I., López-Cortés, N., et al. (2013). Metaproteogenomic insights beyond bacterial response to naphthalene exposure and bio-stimulation. ISME J. 7, 122–136. doi: 10.1038/ismej.2012.82
Hamilton, T. L., Koonce, E., Howells, A., Havig, J. R., Jewell, T., de la Torre, J. R., et al. (2014). Competition for ammonia influences the structure of chemotrophic communities in geothermal springs. Appl. Environ. Microbiol. 80, 653–661. doi: 10.1128/AEM.02577-13
Hanada, S. (2016). Anoxygenic photosynthesis —a photochemical reaction that does not contribute to oxygen reproduction—. Microbes Environ. 31, 1–3. doi: 10.1264/jsme2.ME3101rh
Hatzenpichler, R., Lebedeva, E. V., Spieck, E., Stoecker, K., Richter, A., Daims, H., et al. (2008). A moderately thermophilic ammonia-oxidizing crenarchaeote from a hot spring. Proc. Natl. Acad. Sci. U.S.A. 105, 2134–2139. doi: 10.1073/pnas.0708857105
Hauser, A. (1989). Fuentes termales y minerales en torno a la carretera Austral, Regiones X-XI, Chile. Rev. Geol. Chile 16, 229–239.
Hernández-Prieto, M. A., Semeniuk, T. A., Giner-Lamia, J., and Futschik, M. E. (2016). The transcriptional landscape of the photosynthetic model cyanobacterium Synechocystis sp. PCC6803. Sci. Rep. 6:22168. doi: 10.1038/srep22168
Holo, H., and Sirevåg, R. (1986). Autotrophic growth and CO2 fixation in Chloroflexus aurantiacus. Arch. Microbiol. 145, 173–180. doi: 10.1007/BF00446776
Hou, W., Wang, S. H., Dong, H., Jiang, H., Briggs, B. R., Peacock, J. P., et al. (2013). A comprehensive census of microbial diversity in hot springs of tengchong, yunnan province china using 16SrRNA gene pyrosequencing. PLoS One 8:e53350. doi: 10.1371/journal.pone.0053350
Hügler, M., Menendez, C., Schägger, H., and Fuchs, G. (2002). Malonyl-coenzyme a reductase from Chloroflexus aurantiacus, a Key Enzyme of the 3-hydroxypropionate cycle for autotrophic CO2 fixation. J. Bacteriol. 184, 2404–2410. doi: 10.1128/JB.184.9.2404-2410.2002
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386. doi: 10.1101/gr.5969107
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 8:119. doi: 10.1186/1471-2105-11-119
Inskeep, W. P., Jay, Z. J., Tringe, S. G., Herrgård, M. J., Rusch, D. B., and Ynp Metagenome Project Steering Committee and Working Group Members (2013). The YNP metagenome project: environmental parameters responsible for microbial distribution in the Yellowstone geothermal ecosystem. Front. Microbiol. 4:67. doi: 10.3389/fmicb.2013.00067
Ionescu, D., Hindiyeh, M., Malkawi, H., and Oren, A. (2010). Biogeography of thermophilic cyanobacteria: insights from the Zerka Ma’in hot springs (Jordan). FEMS Microbiol. Ecol. 72, 103–113. doi: 10.1111/j.1574-6941.2010.00835.x
Jaeschke, A., Op den Camp, H. J., Harhangi, H., Klimiuk, A., Hopmans, E. C., and Jetten, M. S. (2009). 16S rRNA gene and lipid biomarker evidence for anaerobic ammonium-oxidizing bacteria (anammox) in California and Nevada hot spring. FEMS Microbiol. Ecol. 67, 343–350. doi: 10.1111/j.1574-6941.2008.00640.x
Jensen, S. I., Steunou, A. S., Bhaya, D., Kühl, M., and Grossman, A. R. (2011). In situ dynamics of O2, pH and cyanobacterial transcripts associated with CCM, photosynthesis and detoxification of ROS. ISME J. 5, 317–328. doi: 10.1038/ismej.2010.131
Kaštovský, J., and Johansen, J. (2008). Mastigocladus laminosus (Stigonematales, Cyanobacteria): phylogenetic relationship of strains from thermal springs to soil-inhabiting genera of the order and taxonomic implications for the genus. Phycologia 43, 307–320. doi: 10.2216/PH07-69.1
Kim, Y. M., Nowack, S. P., Olsen, M. T., Becraft, E. D., Wood, J. M., Thiel, V., et al. (2015). Diel metabolomics analysis of a hot spring chlorophototrophic microbial mat leads to new hypotheses of community member metabolisms. Front. Microbiol. 6:209. doi: 10.3389/fmicb.2015.00209
Klatt, C. G., Bryant, D. A., and Ward, D. M. (2007). Comparative genomics provides evidence for the 3-hydroxypropionate autotrophic pathway in filamentous anoxygenic phototrophic bacteria and in hot spring microbial mats. Environ. Microbiol. 9, 2067–2078. doi: 10.1111/j.1462-2920.2007.01323.x
Klatt, C. G., Inskeep, W. P., Herrgard, M. J., Jay, Z. J., Rusch, D. B., Tringe, S. G., et al. (2013a). Community structure and function of high-temperature chlorophototrophic microbial mats inhabiting diverse geothermal environments. Front. Microbiol. 4:106. doi: 10.3389/fmicb.2013.00106
Klatt, C. G., Liu, Z., Ludwig, M., Kühl, M., Jensen, S. I., Bryant, D. A., et al. (2013b). Temporal metatranscriptomic patterning in phototrophic Chloroflexi inhabiting a microbial mat in a geothermal spring. ISME J. 7, 1775–1789. doi: 10.1038/ismej.2013.52
Klatt, C. G., Wood, J. M., Rusch, D. B., Bateson, M. M., Hamamura, N., Heidelberg, J. F., et al. (2011). Community ecology of hot spring cyanobacterial mats: predominant populations and their functional potential. ISME J. 5, 1262–1278. doi: 10.1038/ismej.2011.73
Kühl, M., and Jørgensen, B. B. (1992). Spectral light measurements in microbenthic phototrophic communities with a fiber-optic microprobe coupled to a sensitive diode array detector. Limnol. Oceanogr. 37, 1813–1823. doi: 10.4319/lo.1992.37.8.1813
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lin, K. H., Liao, B. Y., Chang, H. W., Huang, S. W., Chang, T. Y., Yang, C. Y., et al. (2015). Metabolic characteristics of dominant microbes and key rare species from an acidic hot spring in Taiwan revealed by metagenomics. BMC Genomics 16:1029. doi: 10.1186/s12864-015-2230-9
Liu, Z., Frigaard, N., Vogl, K., Iino, T., Ohkuma, M., Overmann, J., et al. (2012a). Complete genome of Ignavibacterium album, a metabolically versatile, flagellated, facultative anaerobe from the phylum Chlorobi. Front. Microbiol 3:185. doi: 10.3389/fmicb.2012.00185
Liu, Z., Klatt, C. G., Ludwig, M., Rusch, D. B., Jensen, S. I., Kühl, M., et al. (2012b). ‘Candidatus Thermochlorobacter aerophilum:’ an aerobic chlorophotoheterotrophic member of the phylum Chlorobi defined by metagenomics and metatranscriptomics. ISME J. 6, 1869–1882. doi: 10.1038/ismej.2012.24
Liu, Z., Klatt, C. G., Wood, J. M., Rusch, D. B., Ludwig, M., Wittekindt, N., et al. (2011). Metatranscriptomic analyses of chlorophototrophs of a hot-spring microbial mat. ISME J. 5, 1279–1290. doi: 10.1038/ismej.2011.37
Loiacono, S. T., Meyer-Dombard, D. R., Havig, J. R., Poret-Peterson, A. T., Hartnett, H. E., and Shock, E. L. (2012). Evidence for high-temperature in situ nifH transcription in an alkaline hot spring of Lower Geyser Basin, Yellowstone National Parkemi. Environ. Microbiol. 14, 1272–1283. doi: 10.1111/j.1462-2920.2012.02710.x
Lukavsky, J., Furnadzhieva, S., and Pilarski, P. (2011). Cyanobacteria of the thermal spring at Pancharevo, Sofia, Bulgaria. Acta Bot. Croat. 70, 191–208. doi: 10.2478/v10184-010-0015-4
Mackenzie, R., Pedrós-Alió, C., and Díez, B. (2013). Bacterial composition of microbial mats in hot springs in Northern Patagonia: variations with seasons and temperature. Extremophiles 17, 123–136. doi: 10.1007/s00792-012-0499-z
Madigan, M. T., and Brock, T. D. (1977). Adaptation by hot spring phototrophs to reduced light intensities. Arch. Microbiol. 113, 111–120. doi: 10.1007/BF00428590
Marks, C. R., Stevenson, B. S., Rudd, S., and Lawson, P. A. (2012). Nitrospira-dominated biofilm within a thermal artesian spring: a case for nitrification-driven primary production in a geothermal setting. Geobiology 10, 457–466. doi: 10.1111/j.1472-4669.2012.00335.x
McGregor, G. B., and Rasmussen, J. P. (2008). Cyanobacterial composition of microbial mats from an Australian thermal spring: a polyphasic evaluation. FEMS Microbiol. Ecol. 63, 23–35. doi: 10.1111/j.1574-6941.2007.00405.x
Merkel, A. Y., Podosokorskaya, O. A., Cheynyh, N. A., and Bonch-Osmolovckaya, E. A. (2015). Occurrence, diversity, and abundance of methanogenic archaea in terrestrial hot springs of Kamchatka and Saõ Miguel Island. Microbiology 84, 577–583. doi: 10.1134/S002626171504013X
Miller, S., Williams, C., Strong, A., and Carvey, D. (2009). Ecological specialization in a spatially structured population of the thermophilic cyanobacterium Mastigocladus laminosus. Appl. Environ. Microbiol. 75, 729–734. doi: 10.1128/AEM.01901-08
Miller, S. R., Castenholz, R. W., and Pedersen, D. (2007). Phylogeography of the thermophilic cyanobacterium Mastigocladus laminosus. Appl. Environ. Microbiol. 73, 4751–4759. doi: 10.1128/AEM.02945-06
Miller, S. R., Purugganan, M., and Curtis, S. E. (2006). Molecular population genetics and phenotypic diversification of two populations of the thermophilic cyanobacterium Mastigocladus laminosus. Appl. Environ. Microbiol. 72, 2793–2800. doi: 10.1128/AEM.72.4.2793-2800.2006
Nübel, U., Bateson, M. M., Vandieken, V., Kühl, M., and Ward, D. M. (2002). Microscopic examination of distribution and phenotypic properties of phylogenetically diverse Chloroflexaceae-related bacteria in hot spring microbial mats. Appl. Environ. Microbiol. 68, 4593–4603. doi: 10.1128/AEM.68.9.4593-4603.2002
Offre, P., Spang, A., and Schleper, C. H. (2013). Archaea in biogeochemical cycles. Annu. Rev. Amicrobiol. 67, 437–457. doi: 10.1146/annurev-micro-092412-155614
Okubo, T., Tsukui, T., Maita, H., Okamoto, S., Oshima, K., Fujisawa, T., et al. (2012). Complete genome sequence of Bradyrhizobium sp. S23321: insights into symbiosis evolution in soil oligotrophs. Microbes Environ. 27, 306–315. doi: 10.1264/jsme2.ME11321
Ottesen, E. T., Young, C. R., Gifford, S. M., Eppley, J. M., Marin, R. III, Schuster, S. C., et al. (2014). Multispecies diel transcriptional oscillations in open ocean heterotrophic bacterial assemblages. Science 345, 207–212. doi: 10.1126/science.1252476
Quaiser, A., Bodi, X., Dufresne, A., Naquin, D., Francez, A. J., Dheilly, A., et al. (2014). Unraveling the stratification of an iron-oxidizing microbial mat by metatranscriptomics. PLoS One 9:e102561. doi: 10.1371/journal.pone.0102561
Reigstad, L. J., Richter, A., Daims, H., Urich, H., Schwark, L., and Schleper, S. (2008). Nitrification in terrestrial hot springs of Iceland and Kamchatka. FEMS Microbiol. Ecol. 64, 167–174. doi: 10.1111/j.1574-6941.2008.00466.x
Sandbeck, K. A., and Ward, D. M. (1981). Fate of immediate methane precursors in low-sulfate hot spring algal-bacterial mats. Appl. Environ. Microbiol. 41, 775–782.
Sayers, E. W., Barrett, T., Benson, D. A., Bryant, S. H., Canese, K., Chetvernin, V., et al. (2009). Database resources of the national center for biotechnology information. Nucleic Acids Res. 37, 5–15. doi: 10.1093/nar/gkn741
Schmid, M., Walsh, K., Webb, R., Rijpstra, W. I., van, de Pas-Schoonen K, Verbruggen, M. J., et al. (2003). Candidatus ”Scalindua brodae”, sp. nov., Candidatus ”Scalindua wagneri”, sp. nov., two new species of anaerobic ammonium oxidizing bacteria. Syst. Appl. Microbiol. 26, 529–538. doi: 10.1078/072320203770865837
Schmieder, R., Lim, Y. W., and Edwards, R. (2012). Identification and removal of ribosomal RNA sequences from metatranscriptomes. Bioinformatics 28, 433–435. doi: 10.1093/bioinformatics/btr669
Sompong, U., Hawkins, P. R., Besley, C., and Peerapornpisal, Y. (2005). The distribution of cyanobacteria across physical and chemical gradients in northern Thailand. FEMS Microbiol. Ecol. 52, 365–376. doi: 10.1016/j.femsec.2004.12.007
Stal, L. J. (1995). Physiological ecology of cyanobacteria in microbial mats and other communities. New Phytol. 131, 1–32. doi: 10.1111/j.1469-8137.1995.tb03051.x
Stamps, B. W., Corsetti, F. A., Spear, J. R., and Stevenson, B. S. (2014). Draft genome of a novel Chlorobi member assembled by tetranucleotide binning of a hot spring metagenome. Genome Announc. 2:e00897-14. doi: 10.1128/genomeA.00897-14
Steunou, A. S., Bhaya, D., Bateson, M. M., Melendrez, M., Ward, D., Brecht, E., et al. (2006). In situ analysis of nitrogen fixation and metabolic switching in unicellular thermophilic cyanobacteria inhabiting hot spring microbial mats. Proc. Natl. Acad. Sci. U.S.A. 103, 2398–2403. doi: 10.1073/pnas.0507513103
Steunou, A. S., Jensen, S. I., Brecht, E., Becraft, E. D., Bateson, M. M., Kilian, O., et al. (2008). Regulation of nif gene expression and the energetics of N2 fixation over the diel cycle in a hot spring microbial mat. ISME J. 2, 364–378. doi: 10.1038/ismej.2007.117
Stewart, W. (1970). Nitrogen fixation by blue-green algae in Yellowstone thermal areas. Phycologia 9, 261–268. doi: 10.2216/i0031-8884-9-3-261.1
Strauss, G., and Fuchs, G. (1993). Enzymes of a novel autotrophic CO2 fixation pathway in the phototrophic bacterium Chloroflexus aurantiacus, the 3-hydroxypropionate cycle. Eur. J. Biochem. 215, 633–643. doi: 10.1111/j.1432-1033.1993.tb18074.x
Swingley, W. D., Meyer-Dombard, D. R., Shock, E. L., Alsop, E. B., Falenski, H. D., Havig, J. R., et al. (2012). Coordinating environmental genomics and geochemistry reveals metabolic transitions in a hot spring ecosystem. PLoS One 7:e38108. doi: 10.1371/journal.pone.0038108
Tank, M., Thiel, V., Ward, D. M., and Bryant, D. A. (2017). “A Panoply of Phototrophs: An Overview of the Thermophilic Chlorophototrophs of the Microbial Mats of Alkaline Siliceous Hot Springs in Yellowstone National Park, WY, USA,” in Modern Topics in the Phototrophic Prokaryotes, ed. P. Hallenbeck (Cham: Springer), doi: 10.1007/978-3-319-46261-5_3
Thiel, V., Hügler, M., Ward, D. M., and Bryant, D. A. (2017). The Dark Side of the Mushroom spring microbial mat: life in the shadow of chlorophototrophs. II. Metabolic functions of abundant community members predicted from metagenomic analyses. Front. Microbiol. 8:943. doi: 10.3389/fmicb.2017.00943
Thiel, V., Wood, J. M., Olsen, M. T., Tank, M., Klatt, C. G., Ward, D. M., et al. (2016). The dark side of the mushroom spring microbial mat: life in the shadow of chlorophototrophs. I. microbial diversity based on 16S rRNA gene amplicons and metagenomic sequencing. Front. Microbiol. 7:919. doi: 10.3389/fmicb.2016.00919
Urich, T., Lanzén, A., Qi, J., Huson, D. H., Schleper, C., and Schuster, S. C. (2008). Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS One 3:e2527. doi: 10.1371/journal.pone.0002527
Urschel, M. R., Kubo, M. D., Hoehler, T. M., Peters, J. W., and Boyd, E. S. (2015). Carbon source preference in chemosynthetic hot spring communities. Appl. Environ. Microbiol. 81, 3834–3847. doi: 10.1128/AEM.00511-15
van der Meer, M. T., Klatt, C. G., Wood, J., Bryant, D. A., Bateson, M. A., Lammerts, L., et al. (2010). Cultivation and genomic, nutritional, and lipid biomarker characterization of roseiflexus strains closely related to predominant in situ populations inhabiting yellowstone hot spring microbial mats. J. Bacteriol. 12, 3033–3042. doi: 10.1128/JB.01610-09
van der Meer, M. T., Schouten, S., Bateson, M. M., Nübel, U., Wieland, A., Kühl, M., et al. (2005). Diel variations in carbon metabolism by green nonsulfur-like bacteria in alkaline siliceous hot spring microbial mats from Metatranscriptomics of Chloroflexi. Appl. Environ. Microbiol. 71, 3978–3986. doi: 10.1128/AEM.71.7.3978-3986.2005
van der Meer, M. T., Schouten, S., Damsté, J. S., and Ward, D. M. (2007). Impact of carbon metabolism on 13C signatures of cyanobacteria and green nonsulfur-like bacteria inhabiting a microbial mat from an alkaline siliceous hot spring in Yellowstone National Park (USA). Environ. Microbiol. 9, 482–491. doi: 10.1111/j.1462-2920.2006.01165.x
Wang, S., Hou, W., Dong, H., Jiang, H., and Huang, L. (2013). Control of temperature on microbial community structure in hot springs of the Tibetan Plateau. PLoS One 8:e62901. doi: 10.1371/journal.pone.0062901
Ward, D. M. (2006). Microbial diversity in natural environments: focusing on fundamental questions. Antonie Van Leeuwenhoek 90, 309–324. doi: 10.1007/s10482-006-9090-x
Ward, D. M., Ferris, M. J., Nold, S. C., and Bateson, M. M. (1998). A natural view of microbial biodiversity within hot spring cyanobacterial mat communities. Microbiol. Mol. Biol. Rev. 62, 1353–1370.
Waring, G. A. (1965) “Thermal springs of the United States and other countries of the world—a summary,” in Geological Survey Professional Paper, eds R. R. Blankeship and R. Bentall (Washington DC: US Government PrintingOffice)
Zarzycki, J., Brecht, V., Müller, M., and Fuchs, G. (2009). Identifying the missing steps of the autotrophic 3-hydroxypropionate CO2 fixation cycle in Chloroflexus aurantiacus. Proc. Natl. Acad. Sci. U.S.A. 106, 21317–21322. doi: 10.1073/pnas.0908356106
Zarzycki, J., and Fuchs, G. (2011). Coassimilation of organic substrates via the autotrophic 3-Hydroxypropionate Bi-Cycle in Chloroflexus aurantiacus. Appl. Environ. Microbiol. 77, 6181–6188. doi: 10.1128/AEM.00705-11
Zhang, C. L., Ye, Q., Huang, Z. Y., Li, W. J., Chen, J. Q., Song, Z. Q., et al. (2008). Global occurrence of archaeal amoA genes in terrestrial hot springs. Appl. Environ. Microbiol. 74, 6417–6426. doi: 10.1128/AEM.00843-08
Zhao, Y., Tang, H., and Ye, Y. (2012). RAPSearch2: a fast and memory-efficient protein similarity search tool for next-generation sequencing data. Bioinformatics 28, 125–126. doi: 10.1093/bioinformatics/btr595
Keywords: Cyanobacteria, carbon and nitrogen assimilation, neutral hot spring, metagenomics, metatranscriptomics, microbial mat, photosynthesis
Citation: Alcamán-Arias ME, Pedrós-Alió C, Tamames J, Fernández C, Pérez-Pantoja D, Vásquez M and Díez B (2018) Diurnal Changes in Active Carbon and Nitrogen Pathways Along the Temperature Gradient in Porcelana Hot Spring Microbial Mat. Front. Microbiol. 9:2353. doi: 10.3389/fmicb.2018.02353
Received: 22 November 2017; Accepted: 13 September 2018;
Published: 02 October 2018.
Edited by:
Ana Beatriz Furlanetto Pacheco, Universidade Federal do Rio de Janeiro, BrazilReviewed by:
Eric Daniel Becraft, University of North Alabama, United StatesCopyright © 2018 Alcamán-Arias, Pedrós-Alió, Tamames, Fernández, Pérez-Pantoja, Vásquez and Díez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Beatriz Díez, YmRpZXpAYmlvLnB1Yy5jbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.