 Richard Allen White III1*
Richard Allen White III1* Greg Gavelis2
Greg Gavelis2 Sarah A. Soles3
Sarah A. Soles3 Emma Gosselin4
Emma Gosselin4 Greg F. Slater3Darlene S. S. Lim5,6
Greg F. Slater3Darlene S. S. Lim5,6 Brian Leander5
Brian Leander5 Curtis A. Suttle1,2,4,7
Curtis A. Suttle1,2,4,7- 1Department of Microbiology and Immunology, University of British Columbia, Vancouver, BC, Canada
- 2Department of Zoology, University of British Columbia, Vancouver, BC, Canada
- 3School of Geography and Earth Sciences, McMaster University, Hamilton, ON, Canada
- 4Department of Earth, Ocean and Atmospheric Sciences, The University of British Columbia, Vancouver, BC, Canada
- 5Bay Area Environmental Research Institute, Petaluma, CA, United States
- 6NASA Ames Research Center, Moffett Field, CA, United States
- 7Canadian Institute for Advanced Research, Toronto, ON, Canada
Members of the bacterial genus Agrococcus are globally distributed and found across environments so highly diverse that they include forests, deserts, and coal mines, as well as in potatoes and cheese. Despite how widely Agrococcus occurs, the extent of its physiology, genomes, and potential roles in the environment are poorly understood. Here we use whole-genome analysis, chemotaxonomic markers, morphology, and 16S rRNA gene phylogeny to describe a new isolate of the genus Agrococcus from freshwater microbialites in Pavilion Lake, British Columbia, Canada. We characterize this isolate as a new species Agrococcus pavilionensis strain RW1 and provide the first complete genome from a member of the genus Agrococcus. The A. pavilionensis genome consists of one chromosome (2,627,177 bp) as well as two plasmids (HC-CG1 1,427 bp, and LC-RRW783 31,795 bp). The genome reveals considerable genetic promiscuity via mobile elements, including a prophage and plasmids involved in integration, transposition, and heavy-metal stress. A. pavilionensis strain RW1 differs from other members of the Agrococcus genus by having a novel phospholipid fatty acid iso-C15:1Δ4, β-galactosidase activity and amygdalin utilization. Carotenoid biosynthesis is predicted by genomic metabolic reconstruction, which explains the characteristic yellow pigmentation of A. pavilionensis. Metabolic reconstructions of strain RW1 genome predicts a pathway for releasing ammonia via ammonification amino acids, which could increase the saturation index leading to carbonate precipitation. Our genomic analyses suggest signatures of environmental adaption to the relatively cold and oligotrophic conditions of Pavilion Lake microbialites. A. pavilionensis strain RW1 in modern microbialites has an ecological significance in Pavilion Lake microbialites, which include potential roles in heavy-metal cycling and carbonate precipitation (e.g., ammonification of amino acids and filamentation which many trap carbonate minerals).
Introduction
Microbialites represent the oldest evidence of life on the planet with fossils dating back to around 3.7 billion years ago (Nutman et al., 2016). These structures consist of a specialized microbial mat that lithifies carbonates into two main structural types, (1) thrombolites composed of non-laminated clots, or (2) stromatolites defined by laminated layers (Burne and Moore, 1987; Perry et al., 2007). Microbialites are still present today and represent natural laboratories of early microbial ecosystems, which allow for testing hypotheses around the basic principles of microbial ecology including questions regarding community composition (Wong et al., 2015, 2017), community assembly (Havemann and Foster, 2008), functional traits, and diversity (Breitbart et al., 2009; Saghaï et al., 2015; White et al., 2015, 2016b; Ruvindy et al., 2016; Louyakis et al., 2018) and the discovery of novel taxa (Burns et al., 2012).
While heterotrophs and photoautotrophs—mainly cyanobacteria—have been described and isolated from a range of microbial mats, including microbialites, little work has been done on pigmented heterotrophic bacteria within microbialites. It has been suggested that a variety of pigments could come from non-phototrophic bacteria (Nübel et al., 1999; Lionard et al., 2012). Actinobacteria have also been identified in the pigmented layers in microbial mats (Bottos et al., 2008; Lionard et al., 2012), and it is thought that carotenoids are responsible for their characteristic coloration (Nübel et al., 1999; Mueller et al., 2005; Klassen, 2010). Given that these groups seemingly lack metabolism for oxygenic or anoxygenic photosynthesis, fundamental questions include (1) what is the function of pigmentation in these heterotrophic bacteria? And (2) what are the potential roles of heterotrophic bacteria in cold microbialites or microbial mats? We enriched and isolated >100 pigmented bacteria from microbialites in Pavilion Lake, in southeastern British Columbia, Canada (50.8°N, 121.7°W). Based on their growth in the dark, they were either mixotrophs or heterotrophs. Among our enrichments the one isolate described here; which belongs to the Agrococcus genus and is a Gram-positive member of the phylum Actinobacteria.
Pavilion Lake is a cold, oligotrophic ecosystem (mean total phosphorus, 3.3 μg L-1), with dimictic, circumneutral waters (median pH 8.3; mean calcium carbonate, 182 mg L-1) (White et al., 2016b). Characterization of the limnology of Pavilion Lake is described in detail Lim et al. (2009). Pavilion Lake microbialites are calcium carbonate-based thrombolites with thin (∼5 mm) microbial mats dominated by cyanobacteria that change morphology as a function of lake depth (White et al., 2016b). Characterization of the limnology of Pavilion Lake is described in detail Lim et al. (2009). Our Agrococcus strain was isolated and enriched from a Pavilion microbialite (i.e., a thrombolite) at 20 m depth, where the water temperature remains around 4 to 10°C throughout the year (Lim et al., 2009). Bacteria at this depth should be adapted to cold temperature, low phosphorus, and alkaline conditions.
The genus Agrococcus was described based on two strains of Agrococcus jenensis isolated from soil and the surface of sandstone (Groth et al., 1996). The genus Agrococcus is classified within the family Microbacteriaceae, within the phylum Actinobacteria. All Agrococcus members have diaminobutyric acid within their cell walls (Groth et al., 1996). Diaminobutyric acid may impart the distinctive lemon-yellow color, although its role in pigmentation is unknown (Groth et al., 1996). Agrococcus spp. have been isolated from a wide range of environments, including air (Zlamala et al., 2002), a coal mine (Dhanjal et al., 2011), cheese (Bora et al., 2007), cold-desert soil (Mayilraj et al., 2006), forest soil (Zhang et al., 2010), a medieval wall painting (Wieser et al., 1999), dried seaweed (Lee, 2008), and the phyllosphere of potato plants (Behrendt et al., 2008). There are eight described species of Agrococcus, yet little is known about the genome, metabolism, evolution, or physiology of this genus.
To explore the potential role of this genus in microbialite communities, we herein characterize the new species Agrococcus pavilionensis strain RW1, using both classical bacteriological examination (e.g., chemotaxonomic investigation of its metabolism) along with modern genome-centric approaches. We provide the first complete reference genome (i.e., closed gapless chromosome with two plasmids) from the Agrococcus genus. Our genomic analyses suggest signatures of environmental adaption to the relatively cold and oligotrophic conditions of Pavilion Lake microbialites. Promiscuous mobile elements were found in two plasmids involved in heavy-metal resistance and DNA transposition. The genomes of A. pavilionensis RW1 and A. lahaulensis K22-21 both encode a carotenogenic gene cluster that could be responsible for producing the characteristic lemon-yellow pigmentation found in isolates of Agrococcus spp. We also discuss further the potential roles of A. pavilionensis RW1 in microbialite formation.
Materials and Methods
Isolation, Growth Conditions, Microscopy, Phage Induction, Biochemical and Antibiotic Susceptibility Tests
Agrococcus pavilionensis strain RW1 was isolated by plating 0.5 g of a thrombolytic microbialite collected from a depth of 20 m in Pavilion Lake, British Columbia (50.86677 °N, 121.74191 °W). Plating occurred in lysogeny broth (LB) [1% (w/v) tryptone, 0.5% (w/v) yeast extract, 1% (w/v) NaCl, pH 7], and incubated at 10°C for 1 week as an enrichment culture (Figure 1). The enrichment culture was selected for pigmented single colonies then were streaked for isolation on LB agar plates [solidified with 1.5% (w/v) agar] at 30°C for 3 days. Solidified LB agar was also used for assessing growth at various temperatures (4, 11, 16, 20, 25, 30, 35, 42, and 50°C), at pH 7 and 1% (w/v) NaCl. To assess the effect of pH (5, 6, 6.5, 7, 7.5, 8, 8.5, 9, 10, 10.5, and 11) on growth, standard solidified LB was used at 30°C and at a concentration of 1% (w/v) NaCl. To test the effect of salinity on growth, different amounts of NaCl were added to achieve final salinities of 0–6, 9, 12, 13, and 16% on solidified LB (pH 7). The cultures were grown at 30°C and maintained in LB medium or on agar at 1% NaCl, pH 7 and at 30°C. M-agar medium [0.5% (w/v) tryptone, 0.25% (w/v) yeast extract, 1% (w/v) NaCl, 1.5% (w/v) agar, pH 7]. A diluted version of LB agar medium, was used to determine filamentous growth under carbon-limitation. Strain characteristics, including colony morphology and cell morphologies, were determined by standard methods (Murry et al., 1994). Oxidase tests, biochemical enzyme assays, and carbohydrate utilization tests were completed using API20E (BioMérieux) test strips on cultures re-suspended in sterile water. Antibiotic susceptibility of strain RW1 was determined by the Kirby-Bauer method using antibiotic disks on solidified LB (Collee et al., 1996).

FIGURE 1. Strain RW1 isolation environment, culture plate illustration, and microscopy. (A) Picture of Pavilion Lake microbialite (i.e., thrombolite) taken underwater (20 m depth sample, scale bar: ∼1 m). (B) Pavilion Lake microbialite (i.e., thrombolite) taken at sample station (20 m depth sample, scale bar: ∼15 cm). (C) Culture plate (∼24 h growth, the black scale bar ∼10 mm). (D) Light microscopy (∼72 h growth, scale bar: ∼80 μm); (E) SEM (∼72 h growth, scale bar: ∼5 μm). All microscopy (i.e., SEM/light) and culture plate photo (C-E) were completed at strain RW1 standard growth parameters on solidified LB-agar at 30°C, pH 7 and concentration 1% (w/v) NaCl.
Prophage induction assays were done either by adding 0.2 μg ml-1 of mitomycin C or by heating cultures to 45°C for 5 min, incubating at 30°C for 3–10 h, and monitoring changes in turbidity until a decrease of OD600 to 0.1 or less (starting OD600 = 0.6). At several time points (3, 5, and 10 h), cells were pelleted at 3,250 × g and the supernatant filter-sterilized twice through a 0.22 μm pore-size Millex Durapore PVDF membrane (Millipore) filters before plating using a double agar overlay plaque assay (Kropinski et al., 2009).
Light and scanning electron microscopy (SEM) was completed on exponentially growing cells in LB medium. Cells were harvested after 48 h and viewed under oil immersion at 1,500 times magnification for light microscopy. For SEM, cultures in liquid LB were filtered at 72 h onto a 0.2 μm pore-size Supor polycarbonate membrane (Pall, Port Washington, NY, United States). Cells were fixed in 2.5% glutaraldehyde in phosphate-buffered saline (PBS) [137 mM NaCl, 2.7 mM KCL, 10 mM Na2HPO42H2O, 2 mM KH2PO4, pH 7.4] for 30 min on ice. Cells were washed with PBS and post-fixed in 1% OsO4 for 1 h. Stained cells were passed through a graded ethanol series (25, 50, 70, 95, and 100%) at 10-min intervals, and critical-point dried in 100% EtOH. A sputter coater applied 5 nm of gold/palladium alloy onto the cells before imaging by SEM using a Hitachi S4700 microscope.
Chemotaxonomic Analysis of Phospholipids
Phospholipid fatty acids (PLFAs) were extracted from cultures grown in 25 ml LB medium for 4 days at 22°C using triplicate biological replicates. Cultures were transferred into pre-combusted vials for an overnight solvent extraction in a 1:2:0.8 ratio of dichloromethane (DMC): methanol (MeOH): PBS [137 mM NaCl, 2.7 mM KCL, 10 mM Na2HPO4 2H2O, 2 mM KH2PO4, pH 7.4] solution (Bligh and Dyer, 1959). The extract was filtered through a separatory funnel where DCM and water were added to achieve a mixture of MeOH-:DCM:water of 1:1:0.9 (Bligh and Dyer, 1959). The lower organic phase was removed and purified into polar, neutral, and non-polar fractions using liquid chromatography through silica gel. Phospholipids present in the polar fraction were subjected to mild alkaline methanolysis to produce fatty acid methyl esters (FAMEs) (Guckert et al., 1985). FAMEs were separated, identified, and quantified using gas chromatography mass spectrometry (GC/MS) (Agilent Technologies Inc., Santa Clara, CA, United States) with a DB-5MS capillary column (30 m × 0.32 mm I.D. ×0.25 μm film thickness) at a temperature regime of 50°C (1 min), 20°C min-1 to 130°C, 4°C min-1 to 160°C, and 8°C min-1 to 300°C (5 min). PLFAs were identified by retention time and mass spectra relative to those of reference standards (Bacterial Acid Methyl Ester Mix, Matreya Inc., Pleasant Gap, PA, United States; and Supelco 37 Component FAME Mix, Sigma-Aldrich Co., Bellefonte, PA, United States). A modified picolinyl ester derivatization was used to determine the branching point in unknown compounds (Dowd, 1998; Destaillats and Angers, 2002). Dimethyl disulfide adduct derivatives were prepared to determine the double-bond position in unsaturated fatty acids (Nichols et al., 1986).
DNA Extraction, PCR, and Illumina Library Construction
DNA was extracted from early log-phase colonies of Agrococcus pavilionensis strain RW1 grown on LB agar plates using a QIAamp DNA Mini Kit, followed by MinElute PCR purification cleanup columns (Qiagen Germantown, MD, United States). We amplified 16S rRNA by using the universal primers 27f and 1492r (Lane, 1991), and a second PCR using primers 341f and 907r to obtain sequence overlap between the 27f and 1492r to complete the full-length 16S rRNA gene (Muyzer et al., 1993; Muyzer and Smalla, 1998). PCR products were sequenced using standard Sanger method on an ABI3730 (Applied Biosystems, Foster City, CA, United States). The Illumina MiSeq library was constructed using the NxSeq Library Prep Kit (Lucigen, Middleton, WI, United States) without the final 14-cycle PCR enrichment to avoid PCR bias. Quality control of the resulting library was completed using Agilent high-sensitivity DNA chips and digital droplet PCR (Hindson et al., 2011; White and Suttle, 2013; White et al., 2013a,b).
Phylogenetic Analysis
Sanger sequences obtained from the 27f-1492r and 341f-907r PCR products were merged into a full-length 16S rRNA gene sequence using Consed (Gordon et al., 1998) with manual editing. BLAST analysis of both the full-length PCR product and the whole-genome assembled 16S rRNA gene suggested that our culture was a member of the Agrococcus genus. The phylogenetic position of A. pavilionensis strain RW1 was assessed using the error-corrected whole-genome assembled 16S rRNA gene (∼99% similar to PCR amplified) rather than the PCR amplified sequence.
Multiple locus sequencing typing (MLST) marker analysis was completed by extracting protein sequences from rpoB (β subunit of bacterial RNA polymerase, ∼1156 amino acids), RecA (recombination protein A, ∼352 amino acids), gyrB (DNA gyrase subunit B, ∼679 amino acids), and ppK (Polyphosphate kinase, ∼752 amino acids, from both draft and completed genomes by BLASTP analysis or from prior MLST analysis (A. jenensis strain DSM9580 only, Stackebrandt et al., 2007) then concatenated into a ∼2939 amino acid sequence. All phylogenetic analyses were aligned using muscle-default parameters (-400 gap open with zero gaps extended) then clustered using UPGMB. Trees were then constructed using maximum likelihood with bootstrapping (1000 replicates) and the Jukes-Cantor substitution model for 16S rRNA gene full-length sequences (as default parameters), and Jones-Taylor-Thronton model (as default parameters) for ∼2939 amino acid MLST concatenated sequences in MEGA (Edgar, 2004; version 5.10, Tamura et al., 2011).
Whole-Genome Assembly and Genome Finishing
Read-error correction and Celera assembly (including plasmid pHC-CG425) and read partitioning were done as described (White et al., 2013b). Ray assembly of the bacterial genome using the error-corrected reads and phiX removal were done as described (White et al., 2013b,c).
A method to align two or more genomes, progressiveMauve, was used to find the best representative assembly and contig order, and to complete the genome (Darling et al., 2010). Contigs from Celera and Ray assemblies were pooled, then the remaining gaps were closed by recursive alignments in Mauve. The draft A. lahaulensis genome from NCBI (version ASM42510v1) was used for genome ordering. The ordered and aligned overlapping contigs were merged using the EMBOSS union script, yielding three circular contigs (Rice et al., 2000).
To confirm the three circular contigs as separate circular genomes, read-mapping was used. Error-corrected, phiX-removed reads were mapped back to the genome and plasmids using Bowtie2 (version 2.3.4) with the very sensitive local option (Langmead and Salzberg, 2012). The Bowtie2 read-mapping output file (Sam file) was visually inspected by the Tablet program (Milne et al., 2013).
Annotation was completed on RAST using SEED (Aziz et al., 2008); RAST server parameters used SEED subsystems with FIGfam under the Glimmer 3 option (Meyer et al., 2009). In addition to RAST, metabolic pathways were predicted using MetaPathways, a modular pipeline for gene prediction and annotation that uses pathway tools and the MetaCyc database to construct environmental pathway/genome databases (ePGBDs) (Paley and Karp, 2006; Konwar et al., 2013; Caspi et al., 2014).
Annotations were further analyzed for comparison to the A. lahaulensis strain K22-21 and analyzed for genome synteny, average amino acid identity, and phage lifestyle prediction. The genome circular plot was constructed using CGViewer (Grant and Stothard, 2008). Agrococcus lahaulensis strain K22-21, Celera (k0-k1250), and Ray (k0-k1250v2) assemblies were mapped to the completed genome of the A. pavilionensis strain RW1 using tBLASTx at an Expect (E) value of 1e-3 with 50% identity and 25 bp overlap. Synteny plots were completed in the RAST server module using a BLAST-based dot plot format (Aziz et al., 2008). Average amino acid identity (AAIr) analysis and functional gene similarities were calculated on the RAST server module, then parsed by a web-based tool (Aziz et al., 2008; Krebs et al., 2013). RAST-server annotation predicted a prophage element in the genome, which was analyzed for lifestyle preference (lytic or lysogenic) using the phage classification toolset (PHACTS) (McNair et al., 2012).
FR-hit program was used for metagenomic recruitment for the Agrococcus genomes using default parameters with a minimum identity >70% and an Expect (E) value >1e-5 (Niu et al., 2011). The recruitments were then visualized with the R library ggplot2 (Wickham, 2009).
Data Availability
Agrococcus pavilionensis strain RW1 is listed at NCBI under BioProject accession PRJNA201450. The Celera assembly (k0-k1250) of Agrococcus pavilionensis RW1 is under RefSeq NZ_ASHR00000000.1 and assembly GCF_000400485.1 at NCBI. The Agrococcus lahaulensis strain K22-21 is listed at NCBI under BioProject accession PRJNA188801.
Results and Discussion
Morphology and Growth Characteristics
The cells were coccoid during log phase (∼48 h) and were irregular rod-like or coccoid in stationary phase (∼72 h) (Figure 1). The cell size of A. pavilionensis strain RW1 was 0.5 to 0.7 μm in diameter, which is similar to other described members of the genus (Zhang et al., 2010; Table 1). On solidified LB, colonies were bright yellow, smooth and circular, and were typically 0.5 to 2 mm in diameter after ∼72 h of growth at 30°C.
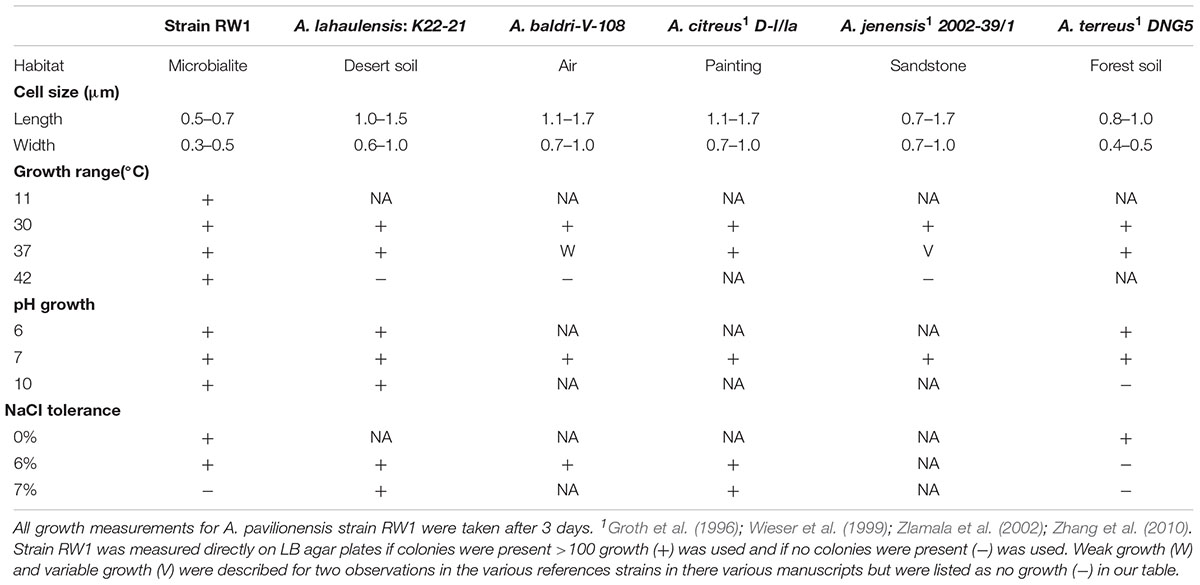
TABLE 1. Comparison of physiological properties of selected strains of Agrococcus spp. – No growth (-), Growth (+), Data not available (NA), Temperature (temp).
The morphology of A. pavilionensis strain RW1 shared features with other members of the genus but had a novel phenotype of filament-like growth. This growth form emerged in low-carbon conditions (i.e., diluted LB or M-agar), and had pale-yellow to white colonies with irregularly branching filaments. These could potentially act as nucleation points for carbonate precipitation within the Pavilion Lake microbialite mat. However, further study of carbonate precipitation and nucleation on Agrococcus pavilionensis strain RW1 is ongoing. While filamentous growth morphology is common among actinobacteria (e.g., isolates of Streptomyces spp.), this phenotype has not been reported for other members of the Microbacteriaceae (Doroghazi and Metcalf, 2013). Further experimentation is needed to confirm whether this is a unique adaptation of A. pavilionensis strain RW1 to microbialites or whether this phenotype is more widely spread across the genus of Agrococcus under low-carbon conditions.
Agrococcus pavilionensis strain RW1 grows on under many conditions. Growth occurred from pH 6 to 10, at 0 to 6% added NaCl, and over a temperature range of 11 to 42°C on LB agar (Table 1). A close relative, A. lahaulensis strain K22-21, has a narrower temperature range of growth (between 30 and 37°C) but can grow at salt concentrations as high as 7% (Mayilraj et al., 2006; Table 1). Contrary to expectations for a cold-water isolate, Agrococcus pavilionensis strain RW1 had the highest reported growth temperature for the genus (Zhang et al., 2010), at 42°C, and had no observed growth at or below 4°C (Table 1), while it exhibited slow growth at 10°C. This suggests that growth of A. pavilionensis RW1 within microbialites may be seasonal. Water temperatures in Pavilion Lake at 20 m range from 4 to 10°C throughout the year (Lim et al., 2009).
PLFA Characterization and Comparative Analysis
Phospholipid fatty acid is commonly used to distinguish bacterial isolates in classical bacterial strain naming. The PLFA composition of A. pavilionensis RW1 was distinct from other strains of Agrococcus spp., including its close relative A. lahaulensis. Agrococcus pavilionensis RW1 had half the amount of iC16:0 but three times as much C16:0 compared to A. lahaulensis (Mayilraj et al., 2006; Table 2). The branched unsaturated PLFA iC15:1Δ4 was 3.5% of the total PLFAs found in A. pavilionensis RW1. It was only found in trace amounts (<1%) in A. versicolor strain K 114/01T (Behrendt et al., 2008; Table 2). Branched monoenoic PLFAs such as iC15:1Δ4 are typically used as biomarkers for anaerobic sulfate-reducing bacteria. Yet A. pavilionensis RW1 grows aerobically and does not reduce sulfate (Kohring et al., 1994). Branched PLFAs found in A. pavilionensis RW1 are known biomarkers for Gram-positive bacteria (Kaur et al., 2005). Although the PLFA profiles between A. pavilionensis RW1 and A. lahaulensis K22-21 are quite similar, the differences support the phylogenetic inference that the two isolates are from different taxonomic groups.
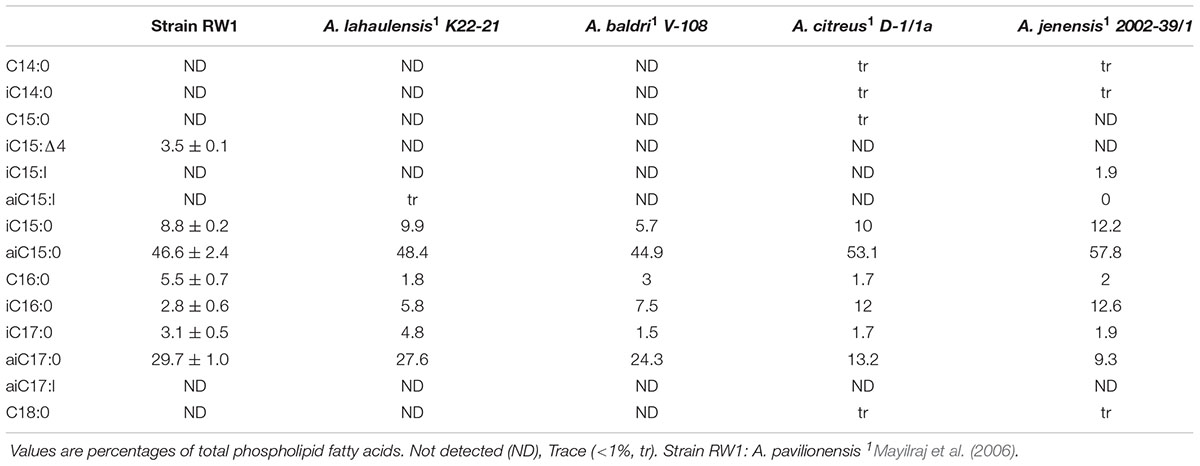
TABLE 2. Selected cellular phospholipid fatty acids of selected strains of Agrococcus spp.
Unsaturated branched PLFAs found in A. pavilionensis RW1 may be a survival adaptation to cold temperatures present in Pavilion Lake since unsaturated fatty acids are used to compensate for a decrease in membrane fluidity found at cold temperatures (Los and Murata, 2004). A. lahaulensis was isolated from cold soil in Lahaul-Spiti Valley in the Indian Himalayas, which also contains unsaturated, branched PLFAs (Mayilraj et al., 2006). These features of unsaturated, branched PLFAs may therefore facilitate the adaption to colder temperatures of both A. pavilionensis and A. lahaulensis.
Evolutionary Placement of Agrococcus pavilionensis Strain RW1
Phylogenetic analysis of the 16S rRNA gene indicates that A. pavilionensis RW1 was most closely related to a clade containing A. lahaulensis K22-21, and an isolate from human-skin (Figure 2). However, a full-length 16S rRNA sequence alone was unable to resolve whether A. pavilionensis RW1 and A. lahaulensis are different species. MLST analysis suggests that A. pavilionensis strain RW1 and A. lahaulensis are in the same clade, but was unable to resolve whether they are separate species (Figure 3). MLST needs a minimum of seven loci to assign a species-level classification of closely related bacterial species. Only four loci are available for the genus Agrococcus (Maiden et al., 2013), so speciation could not be assigned by MLST alone.
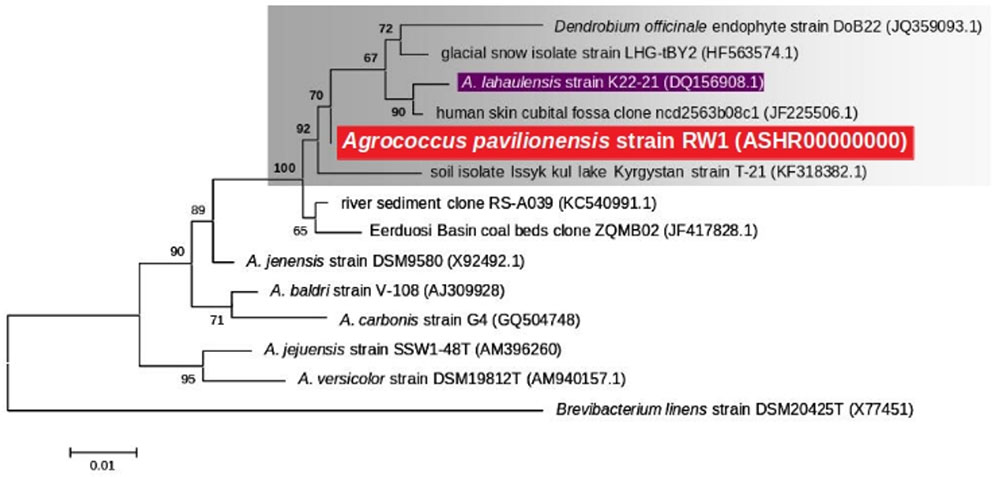
FIGURE 2. Maximum likelihood tree based on 16S rRNA gene sequences (∼1409 bp) showing the phylogenetic relationship among the different isolates of the genus Agrococcus. Bootstrap values greater than 50% are given at nodes. Bar represents a nucleotide substitution rate per 100 nucleotides.
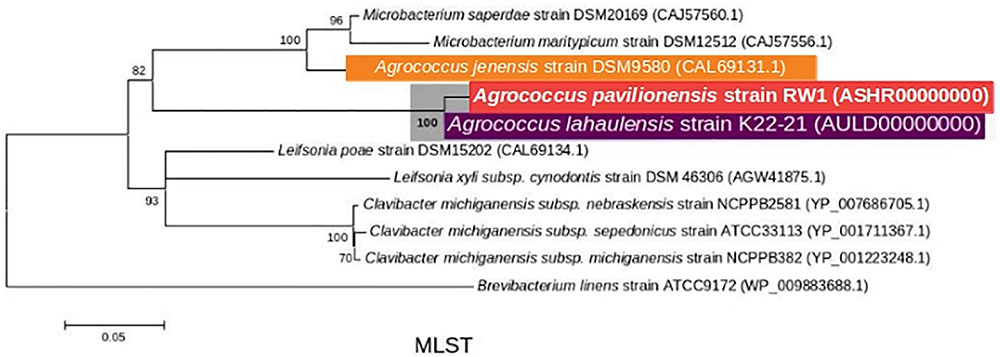
FIGURE 3. Maximum-Likelihood tree based on MLST analysis (∼2939 amino acid) showing the phylogenetic relationship among the different isolates of the genus Agrococcus and deeper relatives of the Microbacteriaceae. Bootstrap values greater than 50% are given at nodes. Bar represents a nucleotide substitution rate per 100 nucleotides.
Fortunately, a draft genome is available for A. lahaulensis, making it possible to infer their relationship based on an analysis of synteny between the two genomes. We mapped the assemblies of A. lahaulensis K22-21 and A. pavilionensis RW1 (both Ray and Celera) against the final circular chromosome of A. pavilionensis RW1 using tBLASTx. Only the A. lahaulensis assembly showed gaps (Figure 4 and Supplementary Table S1). Synteny plots revealed 12 large gaps between the genomes of A. pavilionensis RW1 and A. lahaulensis, K22-21, along with 1752 non-conserved intergenic regions in A. lahaulensis (Figure 5). A comparison of functional gene annotations for A. pavilionensis RW1 and A. lahaulensis K22-21, using both SEED (RAST-based) and MetaCyc (MetaPathways-based), revealed >200 conserved genes, demonstrating that only a small core genome is conserved between the isolates (Figure 5). Thus, while they are closely related, it seems that their subclade within the genus shows high genomic plasticity.
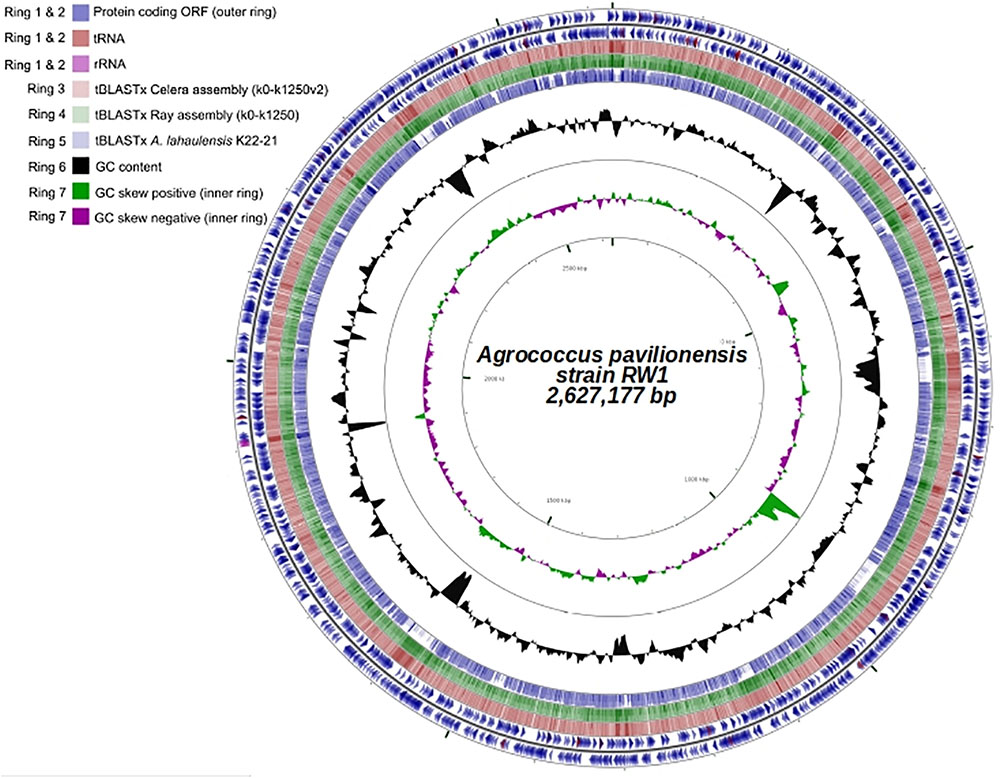
FIGURE 4. Genome plot (∼2.6 Mb) of A. pavilionensis strain RW1. Genome key (left corner): starts with the innermost ring, which is a genome ruler followed by GC skew (purple/green) and ends with two outer rings, which contain protein coding ORFs, tRNAs, and rRNAs.
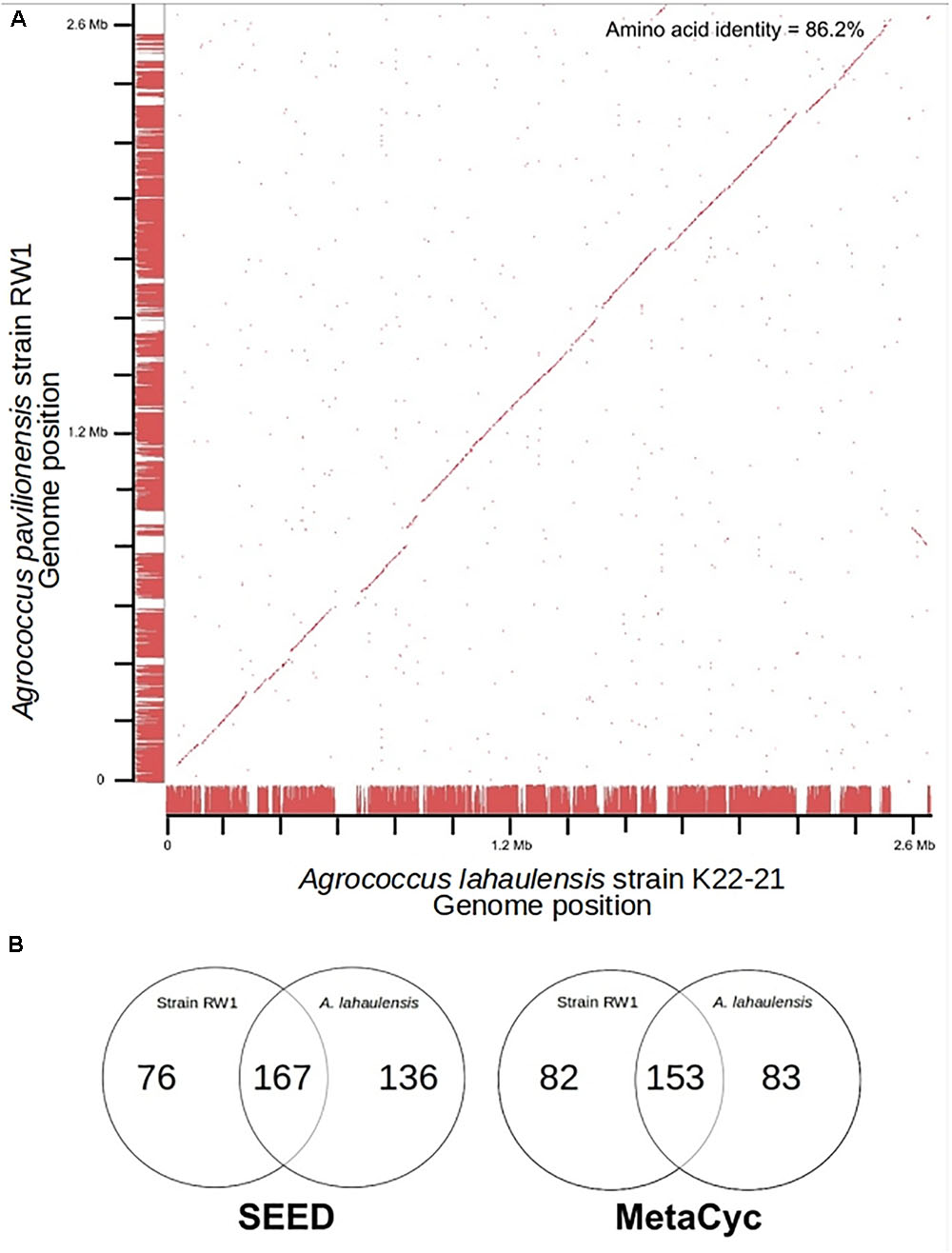
FIGURE 5. Genome synteny and Venn diagrams of A. pavilionensis RW1 vs. A. lahaulensis K22-21. (A) RAST bidirectional BLAST genome synteny dot plot with Progressive Mauve alignments as axes. Red dots are positive BLAST hits based on the RAST genome comparison module. Red lines at the axes are regions of synteny based on Progressive Mauve. Amino acid identity was calculated by RAST functional module with a web-based tool (Krebs et al., 2013). (B) Venn Diagrams based on RAST SEED/FIGfams and Metapathways MetaCyc annotations.
Average amino acid identity between the two genomes supports classifying A. pavilionensis RW1 and A. lahaulensis K22-21 as different species. This is a robust measure for bacterial species classification based on whole-genome sequences and comparable to DNA-DNA hybridization (Konstantinidis and Tiedje, 2005). The standard cutoff to distinguish isolates as different bacterial species is <70% similarity by DNA-DNA hybridization. This corresponds to <95% average amino acid identity (Konstantinidis and Tiedje, 2005). The average amino acid identity for A. pavilionensis RW1 and A. lahaulensis K22-21 was only 86.2%, based on bidirectional whole-genome best-hit protein analysis using RAST annotation. That supports the classification of the isolates as different species (Konstantinidis and Tiedje, 2005; Krebs et al., 2013; Figure 5).
Biochemical Properties and Antibiotic Susceptibility
Agrococcus pavilionensis RW1 shared many biochemical properties with other members of the genus. That included being Gram-positive, but negative for oxidase, arginine dihydrolase, lysine decarboxylase, ornithine decarboxylase, urease, use of citrate and inositol/myo-inositol, production of hydrogen sulfide, and indole and acetoin and positive for catalase (Behrendt et al., 2008). In contrast, A. pavilionensis RW1 was positive for β-galactosidase activity, whereas other isolates have only weak or no activity (Behrendt et al., 2008; Table 3), and A. lahaulensis has no predicted β-galactosidase activity or corresponding genes (Mayilraj et al., 2006; Table 3). Given the diversity of galactosidases in other microbial mat-dwelling heterotrophs, it is possible that β-galactosidase allows A. pavilionensis RW1 to digest exopolysaccharides or other carbohydrates within the mat, though this remains to be tested (Leyn et al., 2017).
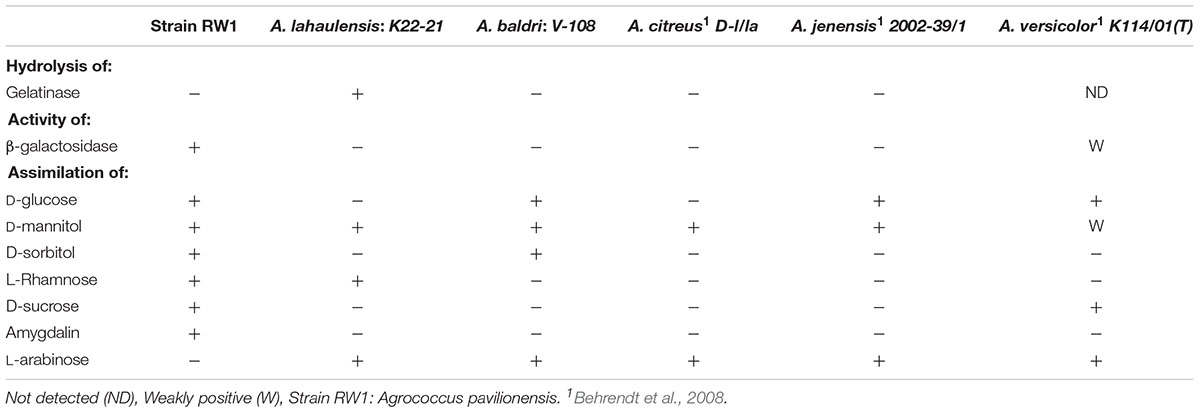
TABLE 3. Biochemical properties of Agrococcus selected strains.
Tests for antibiotic sensitivity in A. pavilionensis RW1 show a pattern that is similar to other isolates of Agrococcus spp., including being sensitive to penicillin, tetracycline, streptomycin, and rifampin (Wieser et al., 1999; Table 4). It is also sensitive to tobramycin, vancomycin, and clindamycin, but resistant to cefixime, sulfisoxazole, oxacillin, trimethoprim and a mixture of sulfamethoxazole/trimethoprim, antibiotics for which patterns of resistance in other strains are less clear, or for which comparable data are not available (Table 4). Agrococcus citreus and A. jenensis strain DSM9580T and DSM9996 are sensitive to oxacillin at 5 μg and weakly sensitive to polymyxin (Wieser et al., 1999); whereas A. pavilionensis RW1 was resistant to oxacillin at 1 μg and sensitive to polymyxin (Table 4). The resistance of A. pavilionensis RW1 to 1 μg of oxacillin, while other Agrococcus spp. are sensitive to doses of 5 μg, suggests that A. pavilionensis RW1 could be sensitive to higher oxacillin concentrations (Wieser et al., 1999). β-lactamase is commonly involved in oxacillin resistance, but evidence for its occurrence was not found in the genomes of either A. pavilionensis RW1 or A. lahaulensis K22-21 (Hou et al., 2007). Although there were no putative antibiotic resistance genes predicted within the genome of A. pavilionensis RW1, pathways were predicted for aromatic compound degradation, including salicylate and gentisate catabolism, which may be involved in resistance. It is conceivable that this antibiotic resistance may be necessitated by exposure to toxic organic molecules produced by cyanobacterial mats, and that it may act as a survival mechanism (Neilan et al., 2013). To better understand antibiotic resistance in Agrococcus spp., more isolates should be tested in future studies.
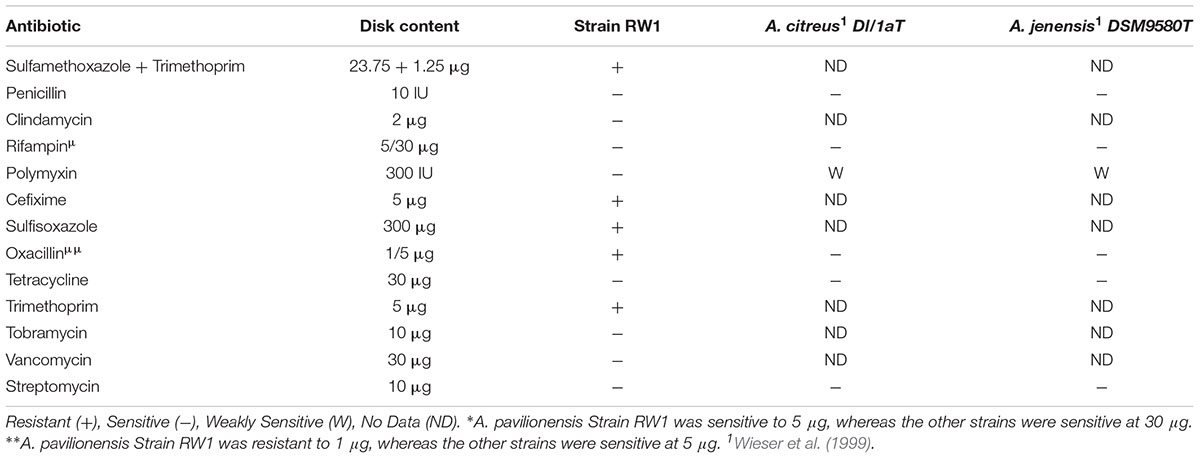
TABLE 4. Antibiotic susceptibility of selected strains of Agrococcus spp.
Mobile DNA and Viral Elements
Mobile DNA elements, which are plasmid-encoded in Agrococcus sp. RW1, are predicted to function in integration, transposition, and heavy-metal resistance. Two plasmids (pHCCG425 and pLC-RRW783) discovered in A. pavilionensis RW1 are involved in integration, transposition, and heavy-metal resistance. The 1,427-bp plasmid pHC-CG425 has a GC content of 67.8% (4.7% less than the main chromosome) and two ORFs. One encodes a putative integrase, and the other encodes a hypothetical protein of unknown function. Meanwhile, pHC-CG425 plasmid shares strong similarities to gene clusters in other members of the phylum Actinobacteria including isolates of Brevibacterium linens and Mycobacterium spp. The second plasmid, pLC-RRW783, is 31,975 bp in length with a GC content of 70.6% (2% less than the main chromosome) and 36 ORFs (Figure 6), including putative coding sequences. Plasmid pLC-RRW783 contained ORFs annotated for mercuric reduction, arsenic resistance, various metal-dependent proteases, peptidases, ATPases, cadmium and unknown transporters, and an unclassified oxidoreductase. Seven ORFs in pLC-RRW783 have no predicted function and are annotated as hypothetical proteins.
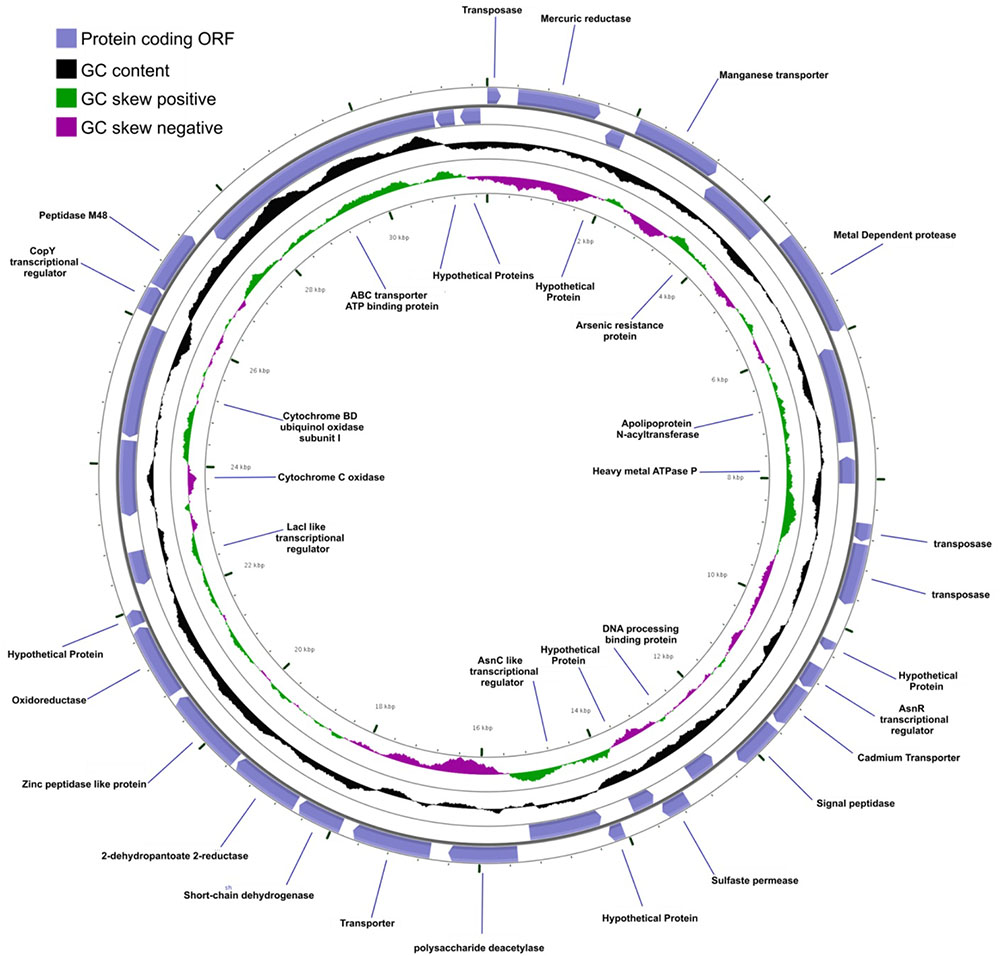
FIGURE 6. Agrococcus pavilionensis strain RW1 LC-RRW783 plasmid genomic plot. The blue lines are for the name of the predicted protein-coding open reading frame.
Annotation of the genome of A. pavilionensis RW1 revealed a 34,174 bp prophage-like element that resembles Siphoviridae prophages found in actinobacteria. It has 43 predicted ORFs and a GC content of 70.1%, which is ∼2% less than the GC content of the genome (Supplementary Figure S1). The addition of heat and mitomycin C did not result in induction, suggesting the prophage is incapable of entering the lytic cycle, or that the treatments were not suitable inducing agents (Zheng et al., 2014). By comparison, no prophage or phage-like genes are predicted in the genome of A. lahaulensis K22-21. The prophage in A. pavilionensis RW1 has a predicted coding sequence for a phage tail-length tape measure protein and a phage-protease gene that are related to sequences found in other phages of Siphoviridae. Those include VWB, phi-c31, and phi-BT1, as well as the Mycobacterium phage Brujita, which were found infecting Streptomyces spp. (Gregory et al., 2003; Van Dessel et al., 2005).
Nitrogen and Phosphorus Metabolism
Agrococcus pavilionensis RW1 encodes an incomplete ammonium utilization pathway that could also be involved in glutamine, glutamate, aspartate, and asparagine biosynthesis. It includes ORFs with similar coding sequences for glutamate-ammonia ligase adenyltransferase and for three NADPH glutamate synthase proteins. Glutamate-ammonia ligase adenyltransferase is conserved across related members in the phylum, including Clavibacter michiganensis and Kocuria rhizophila. It encodes about 1000 amino acids in length, whereas in A. pavilionensis RW1 it is truncated to 113 amino acids and is not predicted to be functional. The genomes of strain K22-21 and RW1 predicted ammonification of amino acids via aspartate, histidine, serine, glutamine, threonine ammonia lyases, and ammonium transporters. While both genomes encode a QacE-family quaternary ammonium compound efflux SMR transporter, strain RW1 encodes an extra copy. Agrococcus pavilionensis RW1 does release ammonium in late log phase or older cultures (via characteristic ammonium odor) Ammonium has been shown to increase carbonate biomineralization via ammonification through the deamination of amino acids (Rodriguez-Navarro et al., 2003). Further experimental confirmation is needed to ascertain if Agrococcus pavilionensis RW1 ammonification leads to carbonate biomineralization.
Genes related to those encoding the phosphate (Pho) regulon for high-affinity uptake of phosphate. Included were the phosphate permease protein (PstA), phosphate regulon sensor protein (PhoR), and the phosphate-regulon transcriptional regulatory protein (PhoB). The phosphate-regulon proteins (PhoR/PhoB) and PstA were not predicted within the A. lahaulensis K22-21 genome. Exopolyphosphatase, a purine metabolism enzyme, is predicted in both A. lahaulensis K22-21 and A. pavilionensis RW1. Polyphosphate glucokinase is only predicted in the A. lahaulensis K22-21 genome. Pavilion Lake is oligotrophic, with low concentrations of total phosphorus (3.3 μg L-1) (Lim et al., 2009). The phosphorus regulon (Pho) in the genome of A. pavilionensis RW1, which may be evidence of adaptation to low phosphate by encoding gene clusters linked to phosphorus regulation and acquisition. This machinery may represent an adaptation to the oligotrophic habitat where A. pavilionensis was found, since phosphate limitation has been found to actively induce the pho regulon in other bacteria (Suzuki et al., 2004). Indeed, genes associated with phosphorus adaptation and scavenging have also been found in metagenomic studies of other freshwater microbialites (Breitbart et al., 2009). Still, the response of the Pho regulon in A. pavilionensis RW1 under phosphorus limitation still has to be experimentally investigated.
Life in a Cold and Oligotrophic Microbialite Mat
Pavilion Lake microbialites exist in water that ranges in temperature from 4 to 10°C (Lim et al., 2009). This frigid environment is reflected in the genome of A. pavilionensis RW1, which has signatures of cold adaptation. These signatures include a single copy of cspA that encodes cold-shock protein A (CspA), a protein that is induced at cold temperatures (<10°C) and which is essential for growth at <10°C. It functions as a sort of molecular chaperone that binds mRNA, preventing secondary structure formation and ensuring translation at low temperatures (Yamanaka and Inouye, 1997). CspA is also expressed during sub-zero temperature growth in members of the genus Exiguobacterium (Rodrigues et al., 2008). A. pavilionensis RW1 also contains a rare branched unsaturated PLFA iC15:1Δ4 and other branched unsaturated PLFAs, which could regulate membrane fluidity to combat the colder temperatures found in Pavilion Lake (Los and Murata, 2004).
Agrococcus pavilionensis RW1 was able to metabolize a wide range of carbon compounds. These include amygdalin (Table 3), which has not been reported for other Agrococcus spp., and is surprising because amygdalin-specific glycosylases were not predicted by the genome. Amygdalin utilization is known for its distant relative Rhodococcus kunmingensis, an actinobacteria isolated from soil (Wang et al., 2008). Analysis of the A. pavilionensis RW1 genome predicts carbohydrate utilization pathways for mannose, fructose, D-gluconate, trehalose, D-ribose, and glycogen, as well as for chitin, lactate, glycerate, deoxyribose, and deoxynucleoside catabolism. Carbohydrate utilization tests for D-glucose, D-mannitol, and D-sucrose validated the metabolic potential of the A. pavilionensis RW1 genome (Table 3). Agrococcus pavilionensis RW1 was able to grow on many more single-carbon sources compared to other members of the genus, possibly allowing access to carbon provided by cyanobacterial mats (Breitbart et al., 2009; Table 3).
Heavy Metal Metabolism and Detoxification
Agrococcus spp. appear to be a component of the Pavilion Lake microbialite community and potentially contribute to the detoxification of heavy metals. Such detoxification potential appears to be, an accessory feature of microbialite communities (Ruvindy et al., 2016; White et al., 2016b; Kurth et al., 2017). Heavy-metal resistance genes, particularly those for arsenic resistance and metabolism, appear to be common in freshwater microbialites in Pavilion Lake (White et al., 2016b), as well as in microbialites from Socompa Lake in the Andes (Kurth et al., 2017) and in the marine stromatolites of Australia’s Shark Bay (Ruvindy et al., 2016). We completed metagenomic-read recruitments from the 20 m Pavilion Lake microbialite metagenome (White et al., 2016b), comparing them to reads from our genome of Agrococcus pavilionensis strain RW1 (i.e., plasmids and chromosome). Using metagenomic-read recruitment, we confirm our Agrococcus pavilionensis strain RW1 is part of the Pavilion Lake microbial community (White et al., 2016b) We also obtained an assignment of previously unclassified actinobacterial sequences as 1% of metagenomic reads from the microbialite from which A. pavilionensis RW1 was isolated (Supplementary Figure S2). Metagenomic sequences recruited with the highest similarity (>95%) to the A. pavilionensis RW1 genome through tBLASTx (1e-3) were predicted to be heavy-metal resistance genes. The metagenomic-read recruitment found hits to heavy-metal resistance genes contained on both the plasmid pLC-RRW783 and in the main chromosome of the A. pavilionensis RW1 genome. Sequences from the Pavilion Lake 20 m metagenome match the mercuric ion reductase and arsenic resistance genes (arsC and arsR) in the pLC-RRW783 plasmid. That means these heavy-metal resistance genes encoded in pLC-RRW783 plasmid could be mobile and potentially could be transferred to other bacteria. Together these data suggest that A. pavilionensis RW1 is a source of the heavy-metal resistance genes within the Pavilion Lake metagenome (White et al., 2016b). That adds new metabolic capabilities linking a cultured isolate directly to the Pavilion Lake microbialite community.
In other microbialite studies, these heavy-metal resistance genes are predominantly recruited to Proteobacteria and Firmicute phyla. However, some sequences were found relating to Corynebacterium, a distant relative of Agrococcus (Kurth et al., 2017). It is possible that members of distant phyla are transferring these heavy-metal resistance genes around by way of the horizontal gene transfer of plasmids (e.g., pLC-RRW783). Pavilion Lake water has undetectable levels of arsenic, cadmium, cobalt, copper, and chromium, along with very low levels of zinc (0.01 to 0.03 mg L-1) (Lim et al., 2009). Thus, it is unclear why an organism from Pavilion Lake would carry gene clusters for heavy-metal resistance. Nevertheless, heavy metal resistance in microbialite communities appears to be an accessory metabolism feature (Ruvindy et al., 2016; White et al., 2016b; Kurth et al., 2017). The pLC-RRW783 plasmid arsenic resistance genes (arsC and arsR) encoded in RW1 are glutathione-dependent, which appears to be common in low-arsenic environments (Escudero et al., 2013). Mercury was actively mined near Pavilion Lake in the 1940s (Stevenson, 1940), and could have been at a higher concentration at one time, suggesting these are vestiges from that era.
Generally, heavy metals including arsenic limit microbial growth which in turn would limit the growth of microbialites. However, we find two examples of thriving modern microbialite ecosystems in the presence of high arsenic, Laguna Brava (Sancho-Tomás et al., 2018) and Socompa Lake microbialites (Kurth et al., 2017). While the remnants of heavy-metal metabolism and detoxification are present in genomes of organisms currently in low heavy-metal environments Pavilion Lake (White et al., 2016b), and marine stromatolites of Shark Bay (Ruvindy et al., 2016).
The extracellular polymeric substances within cyanobacterial microbialite mats and biofilms bind heavy metals, then concentrate and remove them from the water column (Arp et al., 1999). Cyanobacteria are the primary producers in microbialites ecosystems (Dupraz et al., 2009), and are sensitive to heavy metals (Dudkowiak et al., 2011). Any heterotroph that removes and detoxifies heavy metals as a byproduct of their metabolism would be rewarded by substrates for growth (e.g., carbon, nitrogen, phosphorus, and metals) by healthy cyanobacterial mats. The removal and detoxification of these heavy metals would benefit the entire microbial communities within microbialites because high metal concentrations would lead to eventual collapse of microbial population levels. Initially, these genes may have conferred heavy-metal resistance, but now function under other stressors. Or they are still maintained to resist heavy metal, which would limit growth at the sub-micron level in cyanobacterial mats.
However, alternative hypotheses are possible, including that initially heavy-metal resistance genes are now serving alternative functions. Heavy-metal resistance genes can have secondary roles, as in Rhodobacter sphaeroides, where arsenic resistance genes have higher expression under high-salt stress (Tsuzuki et al., 2011). Metagenomic sequencing of Pavilion Lake microbialites revealed accessory metabolic genes related to heavy-metal and antibiotic resistance (White et al., 2016b). The heavy metal resistance genes in strain RW1 are retained in a low heavy-metal environment because they detoxify other substrates (e.g., antibiotics). Heavy metals drive co-selection of antibiotic resistance when aquatic systems are impacted by agriculture or other anthropogenic means (Seiler and Berendonk, 2012). Resistance in heavy metals has conferred resistance to antibiotics in a complex microbiome (e.g., chicken guts) (Nisanian et al., 2014). Further experimental evidence is needed to confirm whether these genes within strain RW1 confer heavy-metal resistance or other functions. In either case, strain RW1 carries these genes on plasmids and may be involved in transferring such accessory genes (e.g., heavy-metal resistance or antibiotic resistance) to the entire microbiome of Pavilion Lake.
Carotenoid Biosynthesis
The pathway responsible for the yellow pigmentation in the genus Agrococcus has not been described, although it has been suggested that diaminobutyric acid within the cell wall could impart the distinctive lemon-yellow colony color (Groth et al., 1996). However, other bacteria (e.g., Cronobacter sakazakii) have yellow colonies in the absence of diaminobutyric acid (Zhang et al., 2014). Actinobacterial isolates form yellow colonies and produce C40 carotenoids (e.g., canthaxanthin and echinenone) and C50 carotenoids (e.g., flavuxanthin) (Tao et al., 2007; Klassen, 2010). Cronobacter sakazakii strain BAA894 is a Gammaproteobacterium that produces yellow-pigmented colonies via a carotenoid biosynthetic pathway. When the Cronobacter carotenoid biosynthetic pathway was reconstructed and expressed in E. coli, the resulting colonies could produce lycopene, β-carotene, and cryptoxanthin, or zeaxanthin (Zhang et al., 2014). The production of zeaxanthin or zeaxanthin glycoside in E. coli changed the colony pigmentation from white to yellow (Zhang et al., 2014).
We screened A. pavilionensis RW1 and A. lahaulensis K22-21 for genes involved in carotenoid production; based on these results we propose a C40/C50 carotenoid biosynthetic pathway (Figure 7). Both A. pavilionensis RW1 and A. lahaulensis K22-21 have the genetic potential to produce lycopene, β-carotene, canthaxanthin, echinenone, and zeaxanthin, or astaxanthin (Figure 7). The carotenoid biosynthetic pathways include predicted genes for phytoene synthase, phytoene dehydrogenase, beta-carotene ketolase, and the second beta carotene-like ketolase that probably converts echinenone to canthaxanthin, which t implies a C40/C50 carotenoid pathway (Tao et al., 2007; Figure 7). Genes within the carotenogenic gene cluster of A. pavilionensis RW1 and A. lahaulensis K22-21 shared similarity to both Brevibacterium linens, a C50 carotenoid producer, and to Cronobacter sakazakii BAA894 (Krubasik and Sandmann, 2000).
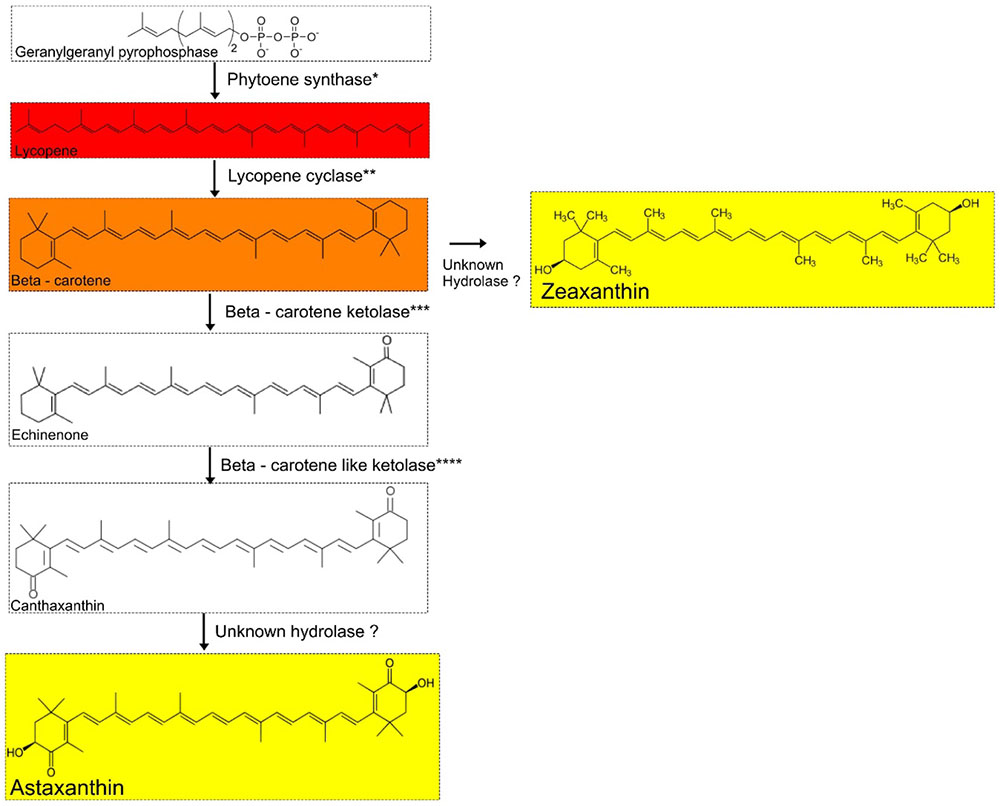
FIGURE 7. Proposed carotenoid biosynthetic pathway for Agrococcus spp. Isolates of Agrococcus spp. have the genetic potential to accumulate C40 carotenoids (canthaxanthin and echinenone). Hydrolases are not predicted in the genomes of A. pavilionensis RW1 or A. lahaulensis K22-21. ∗Phytoene synthase, ∗∗Lycopene cyclase, ∗∗∗Beta-carotene ketolase, ∗∗∗∗Beta-carotene-like ketolase are predicted in the genomes of A. pavilionensis strain RW1 and A. lahaulensis strain K22-21.
The carotenoid biosynthetic pathway in A. pavilionensis RW1 and A. lahaulensis K22-21 is similar to that described for Cronobacter sakazakii BAA894 (Zhang et al., 2014). Zeaxanthin is the most likely pigment responsible for the yellow pigmentation in Agrococcus strains since its genome lacks the hydrolase gene necessary to convert zeaxanthin to other yellow pigments, such as astaxanthin, another yellow carotenoid that could be responsible (Figure 7). No hydrolase coding sequences were found in the genomes of A. pavilionensis RW1 or A. lahaulensis K22-21, which is the only type of enzyme known to convert β-carotene to zeaxanthin or canthaxanthin to astaxanthin (Klassen, 2010; Zhang et al., 2014).
The function of the yellow pigmentation in A. pavilionensis RW1 remains unclear, though we have excluded the possibility of phototrophy since bacteriorhodopsins and xanthorhodopsins were absent from its genome. A bacterial phytopathogen, Pantoea stewartii, has a phytoene synthase similar to strain RW1. It produces a yellow-pigmented carotenoid that has antioxidant properties (i.e., it is less sensitive to hydrogen peroxide stress), and it enables UV radiation protection (Mohammadi et al., 2012). The possibility that they function in photoprotection is a reasonable alternative, since the exceptionally clear waters of Pavilion Lake (due to low dissolved organic carbon) allow for high penetration of solar UV radiation (Lim et al., 2009; Lionard et al., 2012). In this way, the yellow pigment in Agrococcus pavilionensis RW1 may act as a kind of protective sunscreen.
Carotenoids are known signal molecules beyond their role in pigmentation or photoprotection. Carotenoids can inhibit virulence factors in pathogens such as zeaxanthin, which inhibits Pseudomonas aeruginosa quorum-sensing systems and biofilm formation (Gökalsın et al., 2017). Pantoea stewartii yellow carotenoid, while providing both antioxidant properties and UV protection, also makes its carotenoids in a quorum-sensing dependent manner via the EsaI/EsaR system (Mohammadi et al., 2012). As with Pseudomonas aeruginosa, Pantoea stewartii losses virulence when its carotenoids production is limited (Mohammadi et al., 2012). In the non-pathogen Rhodococcus sp. SD-74, carotenoids are rapidly accumulated in biofilms (∼1 week). Bacterial cell aggregation or biofilm formation may trigger their synthesis (Zheng et al., 2013). We speculate that carotenoids in strain RW1 may help to trigger biofilm formation on carbonate minerals, initializing the steps in microbialite formation. Whether the function of carotenoids in strain RW1 is beyond the colony pigmentation presented here is unknown. Further investigations are needed to put strain RW1 into carotenoid roles in photoprotection, antioxidant properties, quorum-sensing, cell aggregation, and biofilm formation.
Conclusion
Our study provides a complete reference genome sequence for the first time from a member of the genus Agrococcus. Strain RW1 was isolated from a modern microbialite and possesses some features that distinguish it from previously characterized members of this genus. The presence of mobile elements, plasmids and a putative prophage in the genome implies much genetic promiscuity and could in part be responsible for high genomic plasticity as revealed by the low-gene similarities between A. pavilionensis RW1 and A. lahaulensis K22-21. The LC-RRW783 plasmid and the chromosome of A. pavilionensis encode genes related to heavy-metal resistance (and confer antibiotic resistance). Signatures of this encoding were also found in the metagenomic data from Pavilion Lake, confirming its presence and a potential role. In addition, the biochemical properties and physiological capabilities of A. pavilionensis RW1 were distinct from other members of the genus. A. pavilionensis RW1 possesses PLFA iC15:1Δ4, has β-galactosidase activity, and uses amygdalin as a sole carbon source.
Phylogenetic analysis using either 16S rRNA gene or MLST could not resolve A. pavilionensis RW1 and A. lahaulensis K22-21 as different species and placed them consistently in the same clade. However, the whole-genome analysis did resolve that A. pavilionensis and A. lahaulensis are separate species based on relatively low functional gene conservation and less than 95% amino acid identity between the genomes. The presence of many non-conserved intergenic regions in A. lahaulensis also supports the classification of A. pavilionensis RW1 and A. lahaulensis K22-21 as separate species.
One of the most surprising aspects of A. pavilionensis was its high growth temperature, which may reflect its descent from a population of durable generalists, as seen in the diverse habitats where the genus can be found. However, the genome of A. pavilionensis also shows characteristics that may reflect adaptations to its present environment (or to conditions in the recent past, as in the case of heavy-metal resistance). These include a lipid profile ostensibly suited for cold climates, the possession of cold-shock proteins, and a low-phosphorous response regulon (Pho), all of which could be of use in a cold, oligotrophic environment. Other features may represent pre-adaptations; such as the carotenogenic gene cluster whose products could provide photoprotection in the clear water column of Pavilion Lake.
Strain RW1 was investigated for its potential role in Pavilion Lake microbialites, including candidate processes (e.g., ammonification and filament-like growth) by which actinobacteria may contribute to microbialite formation. These results provide a blueprint for future efforts to characterize stress response, pigment synthesis, and phage interactions in this widespread genus. Transcriptomics (including single cell) (Gavelis et al., 2015) and proteomics (White et al., 2016a; Callister et al., 2018) should be used in future experiments with strain RW1 to further elucidate functional profiles within the genome. A. pavilionensis strain RW1 represents a model system for further study of non-photosynthetic pigmented heterotrophic bacteria present within modern microbialites and microbial mats. Further exploration of microbialite-associated taxa is crucial to the understanding of these ecosystems and should include not only those driving the formation of the microbialite but also those contributing to the overall development and health of the community.
Author Contributions
RW designed the study, collected and plated the isolate, performed growth studies, extracted DNA, prepared libraries, assembled and annotated the genome, and performed comparative genomic and phylogenetic analysis. SS performed PLFA, with financial support from GS. EG performed culturing experiments. RW preserved cells for scanning electron microscopy, which was imaged by GG. RW and GG wrote the manuscript. All authors participated in the manuscript drafting process.
Funding
Financial support for sample collection was provided by the MARSLIFE Project [9F052-10-0176] funded by the Canadian Space Agency. The laboratory work was supported by a Discovery Grant from the Natural Science and Engineering Council of Canada (CAS), and grants to CS from the Tula Foundation and the Canadian Institute for Advanced Research. Infrastructure support for laboratory work was provided to CS from the Canadian Foundation for Innovation and the British Columbia Knowledge Development Fund.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AL and handling Editor declared their shared affiliation at time of review.
Acknowledgments
Thanks to Donnie Reid and members of the dive team who recovered the microbialite from the lake, Amy M. Chan and Jan Finke who helped with sample collection and processing, and the other individuals associated with the PLRP team. We thank Sugandha Dandekar (Uma) and Hemani Wijesuriya at the UCLA Sequencing & Genotyping Core and the UBC NAPS facility for conducting the sequencing. We are grateful to the Ts’kw’aylaxw First Nation, Linda and Mickey Macri and the Pavilion Community, and British Columbia Parks for their continued support. We thank Dr. Julian Davies for antibiotic disks and Cedric Brimacombe for light microscopy. We thank Dr. Brendan P. Burns for his help editing the final manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02180/full#supplementary-material
FIGURE S1 | Putative prophage genome from RAST annotation. The gray represents genes of hypothetical function. The red gene (label 1) is hypothetical phage gene homologous across strain RW1 and the representative phage/bacterial comparison genomes. The light blue (labeled 16) is phage terminase and tail proteins. The numbers represent genome locations.
FIGURE S2 | FR-Hit recruitment plot of Pavilion Lake 20 m microbialite reads (∼7.5 Million) to A. pavilionensis strain RW1 isolated from 20 m microbialites and to A. lahaulensis strain K22-21.
TABLE S1 | Final assembly statistics. ∗Published in White and Suttle (2013).
References
Arp, G., Reimer, A., and Reitner, J. (1999). Calcification in cyanobacterial biofilms of alkaline salt lakes. Eur. J. Phycol. 34, 393–403. doi: 10.1080/09670269910001736452
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Behrendt, U., Schumann, P., and Ulrich, A. (2008). Agrococcus versicolor sp. nov., an actinobacterium associated with the phyllosphere of potato plants. Int. J. Syst. Bacteriol. 58, 2833–2838. doi: 10.1099/ijs.0.2008/001610-0
Bligh, E. G., and Dyer, W. J. (1959). A rapid method of total lipid extraction and purification. Can. J. Biochem. Phys. 37, 911–917. doi: 10.1139/y59-099
Bora, N., Vancanneyt, M., Gelsomino, R., Swings, J., Brennan, N., Cogan, T. M., et al. (2007). Agrococcus casei sp. nov., isolated from the surfaces of smear-ripened cheeses. Int. J. Syst. Evol. Microbiol. 57, 92–97. doi: 10.1099/ijs.0.64270-0
Bottos, E. M., Vincent, W. F., Greer, C. W., and Whyte, L. G. (2008). Prokaryotic diversity of arctic ice shelf microbial mats. Environ. Microbiol. 10, 950–966. doi: 10.1111/j.1462-2920.2007.01516.x
Breitbart, M., Hoare, A., Nitti, A., Siefert, J., Haynes, M., Dinsdale, E., et al. (2009). Metagenomic and stable isotopic analyses of modern freshwater microbialites in Cuatro Cienegas, Mexico. Environ. Microbiol. 11, 16–34. doi: 10.1111/j.1462-2920.2008.01725.x
Burne, R. V., and Moore, L. S. (1987). Microbialites: organosedimentary deposits of benthic microbial communities. Palaios 2, 241–254. doi: 10.2307/3514674
Burns, B. P., Gudhka, R. K., and Neilan, B. A. (2012). Genome sequence of the halophilic archaeon Halococcus hamelinensis. J. Bacteriol. 194, 2100–2101. doi: 10.1128/JB.06599-11
Callister, S. J., Fillmore, T. L., Nicora, C. D., Shaw, J. B., Purvine, S. O., Orton, D. J., et al. (2018). Addressing the challenge of soil metaproteome complexity by improving metaproteome depth of coverage through two-dimensional liquid chromatography. Soil Biol. Biochem. 125, 290–299. doi: 10.1016/j.soilbio.2018.07.018
Caspi, R., Altman, T., Billington, R., Dreher, K., Foerster, H., Fulcher, C. A., et al. (2014). The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 42, D459–D471. doi: 10.1093/nar/gkt1103
Collee, J. G., Miles, R. S., and Watt, B. (1996). “Tests for the identification of bacteria,” in Mackie and McCartney Practical Medical Microbiology, 14th Edn, eds J. G. Collee, A. G. Fraser, B. P. Marmion, and A. Simmons (London: Churchill Livingstone), 131–149.
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
Destaillats, F., and Angers, P. (2002). One-step methodology for the synthesis of FA picolinyl esters from intact lipids. JAOCS 3, 253–256. doi: 10.1007/s11746-002-0469-7
Dhanjal, A., Kaur, I., Suresh, K., Schumann, P., Cameotra, S. S., Pukall, R., et al. (2011). Agrococcus carbonis sp. nov., isolated from soil of a coal mine. Int. J. Syst. Bacteriol. 61, 1253–1258. doi: 10.1099/ijs.0.024745-0
Doroghazi, J. R., and Metcalf, W. W. (2013). Comparative genomics of actinomycetes with a focus on natural product biosynthetic genes. BMC Genomics 14:611. doi: 10.1186/1471-2164-14-611
Dowd, M. (1998). Identification of the unsaturated heptadecyl fatty acids in the seed oils of Thespesia populnea and Gossypium hirsutum. J. Am. Oil Chem. Soc. 89, 1599–1609. doi: 10.1007/s11746-012-2071-5
Dudkowiak, A., Olejarz, B., Łukasiewicz, J., Banaszek, J., Sikora, J., and Wiktorowicz, K. (2011). Heavy metals effect on cyanobacteria Synechocystis aquatilis study using absorption, fluorescence, flow cytometry, and photothermal measurements. Intern. J. Thermophys. 32, 762–773. doi: 10.1007/s10765-010-0852-3
Dupraz, C., Reid, R. P., Braissant, O., Decho, A. W., Norman, R. S., and Visscher, P. T. (2009). Processes of carbonate precipitation in modern microbial mats. Earth Sci. Rev. 96, 141–162. doi: 10.1016/j.earscirev.2008.10.005
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Escudero, L. V., Casamayor, E. O., Chong, G., Pedrós-Alió, C., and Demergasso, C. (2013). Distribution of microbial arsenic reduction, oxidation and extrusion genes along a wide range of environmental arsenic concentrations. PLoS One 8:e78890. doi: 10.1371/journal.pone.0078890
Gavelis, S., White, R. A. III, Suttle, C. A., Keeling, P. J., and Leander, B. S. (2015). Single-cell transcriptomics using spliced leader PCR: evidence for multiple losses of photosynthesis in polykrikoid dinoflagellates. BMC Genomics 16:528. doi: 10.1186/s12864-015-1636-8
Gökalsın, B., Aksoydan, B., Erman, B., and Sesal, N. C. (2017). Reducing virulence and biofilm of Pseudomonas aeruginosa by potential quorum sensing inhibitor carotenoid: zeaxanthin. Microb. Ecol. 74, 466–473. doi: 10.1007/s00248-017-0949-943
Gordon, D. C., Abajian, C., and Green, P. (1998). Consed: a graphical tool for sequence finishing. Genome Res. 8, 195–202. doi: 10.1101/gr.8.3.195
Grant, J. R., and Stothard, P. (2008). The CGView server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 36, 181–184. doi: 10.1093/nar/gkn179
Gregory, M. A., Till, R., and Smith, M. C. (2003). Integration site for Streptomyces phage phiBT1 and development of site-specific integrating vectors. J. Bacteriol. 185, 5320–5323. doi: 10.1128/JB.185.17.5320-5323.2003
Groth, I., Schumann, P., Weiss, N., Martin, K., and Rainey, F. A. (1996). Agrococcus jenensis gen. nov., sp. nov., a new genus of actinomycetes with diaminobutyric acid in the cell wall. Int. J. Syst. Bacteriol. 46, 234–239. doi: 10.1099/00207713-46-1-234
Guckert, J. B., Antworth, C. P., Nichols, P. D., and White, D. C. (1985). Phospholipid, ester-linked fatty acid profiles as reproducible assays for changes in prokaryotic community structure of estuarine sediments. FEMS Microbiol. Ecol. 31, 147–158. doi: 10.1111/j.1574-6968.1985.tb01143.x
Havemann, S. A., and Foster, J. S. (2008). A comparative characterization of the microbial diversity in an artificial microbialite model and a natural stromatolite. Appl. Environ. Microbiol. 74, 7410–7421. doi: 10.1128/AEM.01710-08
Hindson, B. J., Ness, K. D., Masquelier, D. A., Belgrader, P., Heredia, N. J., Makarewicz, A. J., et al. (2011). High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 83, 8604–8610. doi: 10.1021/ac202028g
Hou, Z., Meng, J. R., Zhao, J. R., Hu, B. Q., Liu, J., Yan, X. J., et al. (2007). Inhibition of beta-lactamase-mediated oxacillin resistance in Staphylococcus aureus by a deoxyribozyme. Acta Pharmacol. Sin. 28, 1775–1782. doi: 10.1111/j.1745-7254.2007.00646.x
Kaur, A., Chaudhary, A., Kaur, A., Choudhary, R., and Kaushik, R. (2005). Phospholipid fatty acid – a bioindicator of environment monitoring and assessment in soil ecosystem. Curr. Sci. 89, 1103–1112.
Klassen, J. L. (2010). Phylogenetic and evolutionary patterns in microbial carotenoid biosynthesis are revealed by comparative genomics. PLoS One 5:e11257. doi: 10.1371/journal.pone.0011257
Kohring, L. L., Ringelberg, D. B., Devereux, R., Stahl, D. A., Mittelman, M. W., and White, D. C. (1994). Comparison of phylogenetic relationships based on phospholipid fatty acid profiles and ribosomal RNA sequence similarities among dissimilatory sulfate–reducing bacteria. FEMS Microbiol. Lett. 119, 303–308. doi: 10.1111/j.1574-6968.1994.tb06905.x
Konstantinidis, K. T., and Tiedje, J. M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 102, 2567–72. doi: 10.1073/pnas.0409727102
Konwar, K. M., Hanson, N. W., Pagé, A. P., and Hallam, S. J. (2013). MetaPathways: a modular pipeline for constructing pathway/genome databases from environmental sequence information. BMC Bioinform. 14:202. doi: 10.1186/1471-2105-14-202
Krebs, J. E., Gale, A. N., Sontag, T. C., Keyser, V. K., Peluso, E. M., and Newman, J. D. (2013). A Web-Based Method to Calculate Average Amino Acid Identity (AAI) between Prokaryotic Genomes. Available at: www.lycoming.edu/newman/aai
Kurth, D., Amadio, A., Ordoñez, O. F., Albarracín, V. H., Gärtner, W., and Farías, M. E. (2017). Arsenic metabolism in high altitude modern stromatolites revealed by metagenomic analysis. Sci. Rep. 7:1024. doi: 10.1038/s41598-017-00896-0
Krubasik, P., and Sandmann, G. (2000). A carotenogenic gene cluster from Brevibacterium linens with novel lycopene cyclase genes involved in the synthesis of aromatic carotenoids. Mol. Gen. Genet. 263, 423–432. doi: 10.1007/s004380051186
Lane, D. J. (1991). “16S/23S rRNA sequencing,” in Nucleic Acid Techniques in Bacterial Systematics, eds E. Stackebrandt and M. Goodfellow (New York, NY: John Wiley and Sons), 115–175.
Langmead, B., and Salzberg, S. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lee, S. D. (2008). Agrococcus jejuensis sp. nov., isolated from dried seaweed. Int. J. Syst. Evol. Microbiol. 58, 2297–2300. doi: 10.1099/ijs.0.65731-0
Leyn, S. A., Maezato, Y., Romine, M. F., and Rodionov, D. A. (2017). Genomic reconstruction of carbohydrate utilization capacities in microbial-mat derived consortia. Front. Microbiol. 8:1304. doi: 10.3389/fmicb.2017.01304
Lim, D. S. S., Laval, B. E., Slater, G., Antoniades, D., Forrest, A. L., Pike, W., et al. (2009). Limnology of Pavilion Lake B.C. - Characterization of a microbialite forming environment. Fundam. Appl. Limnol. 173, 329–351. doi: 10.1127/1863-9135/2009/0173-0329
Lionard, M., Péquin, B., Lovejoy, C., and Vincent, W. F. (2012). Benthic cyanobacterial mats in the high arctic: multi-layer structure and fluorescence responses to osmotic stress. Front. Microbiol. 3:140. doi: 10.3389/fmicb.2012.00140
Los, D. A., and Murata, N. (2004). Membrane fluidity and its roles in the perception of environmental signals. Biochim. Biophys. Acta 2, 142–157. doi: 10.1016/j.bbamem.2004.08.002
Louyakis, A. S., Gourlé, H., Casaburi, G., Bonjawo, R. M. E., Duscher, A. A., and Foster, J. S. (2018). A year in the life of a thrombolite: comparative metatranscriptomics reveals dynamic metabolic changes over diel and seasonal cycles. Environ. Microbiol. 20, 842–861. doi: 10.1111/1462-2920.14029
Maiden, M. C., van Rensburg, M. J., Bray, J. E., Earle, S. G., Ford, S. A., Jolley, K. A., et al. (2013). MLST revisited: the gene-by-gene approach to bacterial genomics. Nat. Rev. Microbiol. 11, 728–736. doi: 10.1038/nrmicro3093
Mayilraj, S., Suresh, K., Schumann, P., Kroppenstedt, R. M., and Saini, H. S. (2006). Agrococcus lahaulensis sp. nov., isolated from a cold desert of the Indian Himalayas. Int. J. Syst. Evol. Microbiol. 56, 1807–1810. doi: 10.1099/ijs.0.64247-0
McNair, K., Bailey, B. A., and Edwards, R. A. (2012). PHACTS, a computational approach to classifying the lifestyle of phages. Bioinformatics 28, 614–618. doi: 10.1093/bioinformatics/bts014
Meyer, F., Overbeek, R., and Rodriguez, A. (2009). FIGfams: yet another set of protein families. Nucleic Acids Res. 37, 6643–6654. doi: 10.1093/nar/gkp698
Milne, I., Stephen, G., Bayer, M., Cock, P. J. A., Pritchard, L., Cardle, L., et al. (2013). Using tablet for visual exploration of second-generation sequencing data. Brief. Bioinform. 14, 193–202. doi: 10.1093/bib/bbs012
Mohammadi, M., Burbank, L., and Roper, M. C. (2012). Biological role of pigment production for the bacterial phytopathogen Pantoea stewartii subsp. stewartii. Appl. Environ. Microbiol. 78, 6859–6865. doi: 10.1128/AEM.01574-1512
Mueller, D. R., Vincent, W. F., Bonilla, S., and Laurion, I. (2005). Extremotrophs, extremophiles and broadband pigmentation strategies in a high arctic shelf ecosystem. FEMS Microbiol. Ecol. 53, 73–87. doi: 10.1016/j.femsec.2004.11.001
Muyzer, G., De Waal, E. C., and Uitterlinden, A. G. (1993). Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 59, 695–700.
Muyzer, G., and Smalla, K. (1998). Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie Van Leeuwenhoek 73, 127–141. doi: 10.1023/A:1000669317571
Neilan, B. A., Pearson, L. A., Muenchhoff, J., Moffitt, M. C., and Dittmann, E. (2013). Environmental conditions that influence toxin biosynthesis in cyanobacteria. Environ. Microbiol. 5, 1239–1253. doi: 10.1111/j.1462-2920.2012.02729.x
Nichols, P. D., Gukert, J. B., and White, D. C. (1986). Determination of monounsaturated fatty acid double bond position and geometry for microbial monocultures and complex consortia by capillary GC-MS of their dimethyl disulfide adducts. J. Microb. Methods 5, 49–55. doi: 10.1016/0167-7012(86)90023-0
Nisanian, M., Holladay, S. D., Karpuzoglu, E., Kerr, R. P., Williams, S. M., Stabler, L., et al. (2014). Exposure of juvenile Leghorn chickens to lead acetate enhances antibiotic resistance in enteric bacterial flora. Poult. Sci. 93, 891–897. doi: 10.3382/ps.2013-03600
Niu, B., Zhu, Z., Fu, L., Wu, S., and Li, W. (2011). FR-HIT, a very fast program to recruit metagenomic reads to homologous reference genomes. Bioinformatics 27, 1704–1705. doi: 10.1093/bioinformatics/btr252
Nübel, U., Garcia-Pichel, F., Kühl, M., and Muyzer, G. (1999). Quantifying microbial diversity: morphotypes, 16S rRNA genes, and carotenoids of oxygenic phototrophs in microbial mats. Appl. Environ. Microbiol. 65, 422–430.
Nutman, A. P., Bennett, V. C., Friend, C. R. L., Van Kranendonk, M. J., and Chivas, A. R. (2016). Rapid emergence of life shown by discovery of 3,700-million-year-old microbial structures. Nature 537, 535–538. doi: 10.1038/nature19355
Paley, S. M., and Karp, P. D. (2006). The Pathway Tools cellular overview diagram and Omics Viewer. Nucleic Acids Res. 34, 3771–3778. doi: 10.1093/nar/gkl334
Perry, R. S., Mcloughlin, N., Lynne, B. Y., Sephton, M. A., Oliver, J. D., Perry, C. C., et al. (2007). Defining biominerals and organominerals: direct and indirect indicators of life. Sed. Geol. 201, 157–179. doi: 10.1016/j.sedgeo.2007.05.014
Rice, P., Longden, I., and Bleasby, A. (2000). EMBOSS: the european molecular biology open software suite. Trends Genet. 16, 276–277. doi: 10.1016/S0168-9525(00)02024-2
Rodrigues, D. F., Ivanova, N., He, Z., Huebner, M., Zhou, J., and Tiedje, J. M. (2008). Architecture of thermal adaptation in an Exiguobacterium sibiricum strain isolated from 3 million-year-old permafrost: a genome and transcriptome approach. BMC Genomics 9:547. doi: 10.1186/1471-2164-9-547
Rodriguez-Navarro, C., Rodriguez-Gallego, M., Ben Chekroun, K., and Gonzalez-Munoz, M. T. (2003). Conservation of ornamental stone by Myxococcus xanthus-induced carbonate biomineralization. Appl. Environ. Microbiol. 69, 2182–2193. doi: 10.1128/AEM.69.4.2182-2193
Ruvindy, R., White, R. A. III, Neilan, B. A., and Burns, B. P. (2016). Unraveling core microbial metabolisms in the hypersaline microbial mats of Shark Bay using high-throughput metagenomics. ISME J. 10, 183–196. doi: 10.1038/ismej.2015.87
Saghaï, A., Zivanovic, Y., Zeyen, N., Moreira, D., Benzerara, K., Deschamps, P., et al. (2015). Metagenome-based diversity analyses suggest a significant contribution of non-cyanobacterial lineages to carbonate precipitation in modern microbialites. Front. Microbiol. 6:797. doi: 10.3389/fmicb.2015.0079
Sancho-Tomás, M., Somogyi, A., Medjoubi, K., Bergamaschi, A., Visscher, P. T., Van Driesschee, E. S. A., et al. (2018). Distribution, redox state and (bio)geochemical implications of arsenic in present-day microbialites of Laguna Brava, Salar de Atacama. Chem. Geol. 490, 13–21. doi: 10.1016/j.chemgeo.2018.04.029
Seiler, C., and Berendonk, U. T. (2012). Heavy metal driven co-selection of antibiotic resistance in soil and water bodies impacted by agriculture and aquaculture. Front. Microbiol. 3:399. doi: 10.3389/fmicb.2012.00399
Stackebrandt, E., Brambilla, E., and Richert, K. (2007). Gene sequence phylogenies of the family Microbacteriaceae. Curr. Microbiol. 55, 42–46. doi: 10.1007/s00284-006-0569-5
Stevenson, J. S. (1940). Mercury Deposits of British Columbia. Bulletin No.5. Victoria, BC: British Columbia department of mines.
Suzuki, S., Ferjani, A., Suzuki, I., and Murata, N. (2004). The SphS-SphR two-component system is the exclusive sensor for the induction of gene expression in response to phosphate limitation in synechocystis. J. Biol. Chem. 279, 13234–13240. doi: 10.1074/jbc.M313358200
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Tao, L., Yao, H., and Cheng, Q. (2007). Genes from a Dietzia sp. for synthesis of C40 and C50 β-cyclic carotenoids. Gene 386, 90–97. doi: 10.1016/j.gene.2006.08.006
Tsuzuki, M., Moskvin, O. V., Kuribayashi, M., Sato, K., Retamal, S., Abo, M., et al. (2011). Salt stress-induced changes in the transcriptome, compatible solutes, and membrane lipids in the facultatively phototrophic bacterium Rhodobacter sphaeroides. Appl. Environ. Microbiol. 77, 7551–7559. doi: 10.1128/AEM.05463-11
Van Dessel, W., Van Mellaert, L., Liesegang, H., Raasch, C., De Keersmaeker, S., Geukens, N., et al. (2005). Complete genomic nucleotide sequence and analysis of the temperate bacteriophage VWB. Virology 331, 325–337. doi: 10.1016/j.virol.2004.10.028
Wang, Y. X., Wang, H. B., Zhang, Y. Q., Xu, L. H., Jiang, C. L., and Li, W. J. (2008). Rhodococcus kunmingensis sp. nov., an actinobacterium isolated from a rhizosphere soil. Int. J. Syst. Evol. Microbiol. 58, 1467–1471. doi: 10.1099/ijs.0.65673-65670
White, R. A. III, Callister, S. J., Moore, R. J., Baker, E. S., and Jansson, J. K. (2016a). The past, present, and future of microbiome analyses. Nat. Protoc. 11, 2049–2053. doi: 10.1038/nprot.2016.148
White, R. A. III, Chan, A. M., Gavelis, G. S., Leander, B. S., Brady, A. L., Slater, G. F., et al. (2016b). Metagenomic analysis suggests modern freshwater microbialites harbor a distinct core microbial community. Front. Microbiol. 6:1531. doi: 10.3389/fmicb.2015.01531
White, R. A. III, Grassa, C. J., and Suttle, C. A. (2013a). Draft genome sequence of Exiguobacterium pavilionensis strain RW-2, with wide thermal, salinity, and pH tolerance, isolated from modern freshwater microbialites. Genome Announc. 1:e597-13. doi: 10.1128/genomeA.00597-13
White, R. A., III Grassa, C. J., and Suttle, C. A. (2013b). First draft genome sequence from a member of the genus Agrococcus, isolated from modern microbialites. Genome Announc. 1:e391-13. doi: 10.1128/genomeA.00391-13
White, R. A., III Power, I. M., Dipple, G. M., Southam, G., and Suttle, C. A. (2015). Metagenomic analysis reveals that modern microbialites and polar microbial mats have similar taxonomic and functional potential. Front. Microbiol. 6:966. doi: 10.3389/fmicb.2015.00966
White, R. A., III and Suttle, C. A. (2013). The draft genome sequence of Sphingomonas paucimobilis strain HER1398 (Proteobacteria), host to the giant PAU phage, indicates that it is a member of the genus Sphingobacterium (Bacteroidetes). Genome Announc. 1:e598-13. doi: 10.1128/genomeA.00598-13
Wickham, H. (2009). ggplot2: Elegant Graphics for Data Analysis. New York, NY: Springer. doi: 10.1007/978-0-387-98141-3
Wieser, M., Schumann, P., Martin, K., Altenburger, P., Burghardt, J., Lubitz, W., et al. (1999). Agrococcus citreus sp. nov., isolated from a medieval wall painting of the chapel of Castle Herberstein (Austria). Int. J. Syst. Bacteriol. 49, 1165–1170. doi: 10.1099/00207713-49-3-1165
Wong, H. L., Smith, D. L., Visscher, P. T., and Burns, B. P. (2015). Niche differentiation of bacterial communities at a millimeter scale in Shark Bay microbial mats. Sci. Rep. 5:15607. doi: 10.1038/srep15607
Wong, H. L., Visscher, P. T., White, R. A. III, Smith, D. L., Patterson, M. M., and Burns, B. P. (2017). Dynamics of archaea at fine spatial scales in Shark Bay mat microbiomes. Sci. Rep. 7:46160. doi: 10.1038/srep46160
Yamanaka, K., and Inouye, M. (1997). Growth-phase-dependent expression of cspD, encoding a member of the CspA family in Escherichia coli. J. Bacteriol. 179, 5126–5130. doi: 10.1128/jb.179.16.5126-5130.1997
Zhang, J. Y., Liu, X. Y., and Liu, S. J. (2010). Agrococcus terreus sp. nov. and Micrococcus terreus sp. nov., isolated from forest soil. Int. J. Syst. Evol. Microbiol. 60, 1897–1903. doi: 10.1099/ijs.0.013235-0
Zhang, W., Hu, X., Wang, L., and Wang, X. (2014). Reconstruction of the carotenoid biosynthetic pathway of Cronobacter sakazakii BAA894 in Escherichia coli. PLoS One 9:e86739. doi: 10.1371/journal.pone.0086739
Zheng, Y. T., Toyofuku, M., Nomura, N., and Shigeto, S. (2013). Correlation of carotenoid accumulation with aggregation and biofilm development in Rhodococcus sp. SD-74. Anal. Chem. 85, 7295–7301. doi: 10.1021/ac401188f
Zheng, Q., Zhang, R., Xu, Y., White, III R. A., Wang, Y., Luo, T., et al. (2014). A marine inducible prophage vB_CibM-P1 isolated from the aerobic anoxygenic phototrophic bacterium Citromicrobium bathyomarinum JL354. Sci. Rep. 4:7118. doi: 10.1038/srep07118
Keywords: actinobacteria, microbialite, cosmopolitan, oligotrophic, metabolic potential
Citation: White RA III, Gavelis G, Soles SA, Gosselin E, Slater GF, Lim DSS, Leander B and Suttle CA (2018) The Complete Genome and Physiological Analysis of the Microbialite-Dwelling Agrococcus pavilionensis sp. nov; Reveals Genetic Promiscuity and Predicted Adaptations to Environmental Stress. Front. Microbiol. 9:2180. doi: 10.3389/fmicb.2018.02180
Received: 05 January 2018; Accepted: 24 August 2018;
Published: 15 October 2018.
Edited by:
Pieter T. Visscher, University of Connecticut, United StatesReviewed by:
Haley M. Sapers, California Institute of Technology, United StatesJamie S. Foster, University of Florida, United States
Artemis S. Louyakis, University of Connecticut, United States
Copyright © 2018 White, Gavelis, Soles, Gosselin, Slater, Lim, Leander and Suttle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard Allen White III, cmF3OTM3QGdtYWlsLmNvbQ==