
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 31 July 2018
Sec. Evolutionary and Genomic Microbiology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.01717
 Shengzhi Yang1†
Shengzhi Yang1† Xin Gao2†
Xin Gao2† Jianghong Meng2
Jianghong Meng2 Anyun Zhang3Yingmin Zhou4Mei Long1Bei Li1Wenwen Deng1Lei Jin1Siyue Zhao1Daifu Wu4Yongguo He4Caiwu Li4
Anyun Zhang3Yingmin Zhou4Mei Long1Bei Li1Wenwen Deng1Lei Jin1Siyue Zhao1Daifu Wu4Yongguo He4Caiwu Li4 Shuliang Liu5Yan Huang4,6*Hemin Zhang4,6*
Shuliang Liu5Yan Huang4,6*Hemin Zhang4,6* Likou Zou1*
Likou Zou1*To obtain full details of gut microbiota, including bacteria, fungi, bacteriophages, and helminths, in giant pandas (GPs), we created a comprehensive microbial genome database and used metagenomic sequences to align against the database. We delineated a detailed and different gut microbiota structures of GPs. A total of 680 species of bacteria, 198 fungi, 185 bacteriophages, and 45 helminths were found. Compared with 16S rRNA sequencing, the dominant bacterium phyla not only included Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria but also Cyanobacteria and other eight phyla. Aside from Ascomycota, Basidiomycota, and Glomeromycota, Mucoromycota, and Microsporidia were the dominant fungi phyla. The bacteriophages were predominantly dsDNA Myoviridae, Siphoviridae, Podoviridae, ssDNA Inoviridae, and Microviridae. For helminths, phylum Nematoda was the dominant. In addition to previously described parasites, another 44 species of helminths were found in GPs. Also, differences in abundance of microbiota were found between the captive, semiwild, and wild GPs. A total of 1,739 genes encoding cellulase, β-glucosidase, and cellulose β-1,4-cellobiosidase were responsible for the metabolism of cellulose, and 128,707 putative glycoside hydrolase genes were found in bacteria/fungi. Taken together, the results indicated not only bacteria but also fungi, bacteriophages, and helminths were diverse in gut of giant pandas, which provided basis for the further identification of role of gut microbiota. Besides, metagenomics revealed that the bacteria/fungi in gut of GPs harbor the ability of cellulose and hemicellulose degradation.
Mammals are metagenomic because they consist of their own gene complements and a large number of microorganisms residing within them (Ley et al., 2008). These microorganisms include bacteria, fungi, protozoa, and viruses and are collectively referred to as microbiota (Kamada et al., 2013). The gut microbiota is an extra organ that regulates the immune system and influences the physiology, health, and nutrition of its host (Maurice et al., 2013; Qin et al., 2014; Zhernakova et al., 2016). It not only makes an essential contribution to physiology and metabolism of host (Bäckhed et al., 2004; Hooper and Macpherson, 2010; Tremaroli and Backhed, 2012) but also serves as an environmental factor that contributes to obesity and its comorbidities (Tremaroli and Backhed, 2012) and protects the gut against invasion of exogenous pathogens (Kamada et al., 2013).
Compared with other members of microbiota, bacteria are abundant and therefore more influential on the gut of mammals (Human Microbiome, and Project, 2012), particularly related to health and disease of host (Kelly et al., 2005). Fungi also play an important role in disease of host (Huffnagle and Noverr, 2013; Sokol et al., 2017) and may be responsible for fiber degradation and fermentative digestion in herbivores (Li and Heath, 1993; Liggenstoffer et al., 2010). Bacteriophages are the most abundant biological group on earth and genetically even more diverse than their bacterial prey/hosts (Reyes et al., 2013). They are the most abundant members of the gut virobiota (Łusiak-Szelachowska et al., 2017) contributing to gut inflammation and bacterial dysbiosis (Norman et al., 2015). Parasitic helminths (PHs) residing in the gut have profound potentials to threaten the stability and persistence of hosts, impair host fitness, and even lead to mortality of hosts. The interactions between gut PHs and commensal bacteria are likely to play a pivotal role in regulating the development of the gut immune system (Cantacessi et al., 2014; Lee et al., 2014).
The giant panda (GP, Ailuropoda melanoleuca) is a rare endemic wild animal found in China and a flagship species for wildlife conservation. GP belongs to the order Carnivora with a typical carnivorous digestive system (Fang et al., 2012; Xue et al., 2015), but almost entirely lives on a bamboo-dominated diet. Its genome encodes all the enzymes necessary for a carnivorous digestive system but lacks those for digesting cellulose and hemicellulose (Fang et al., 2012). However, low digestibility of cellulose and hemicellulose for the GP’s unique bamboo diet may be assisted by microbiota (Zhu et al., 2011; Xue et al., 2015). Necessary enzymes for digestion of cellulose include cellulase (EC 3.2.1.4), cellulose 1,4-β-cellobiosidase (EC 3.2.1.91), and β-glucosidases (EC 3.2.1.21) (Kubicek et al., 2009; Zhu et al., 2011).
To date, most studies have been focusing on diversity of gut bacteria of GPs, and thus little is known about the characteristics of the fungi, bacteriophages, and helminths. The present study was to provide full details of gut microbiota of the GPs, including bacteria, fungi, bacteriophages and helminths, established by metagenomic sequencing. We also provided a detailed characterization of the bacteria and fungi possessing the genes associated with cellulose degradation and glycoside hydrolase (GH) to evaluate the ability of cellulose, hemicellulose and starch hydrolysis of microbiota.
Fecal samples from the captive, semiwild, and wild GPs were collected immediately after defecation, snap frozen, and shipped to the laboratory on dry ice. Four samples from the captive GPs collected from the China Conservation and Research Center for the Giant Panda, six samples of semiwild GPs from Hetaoping Base and Liziping Nature Reserve and three samples from the wild GPs from Wolong Nature Reserve were included. Captive, semiwild, and wild GPs live in different environments. Captive GPs lived in man-made limited space for attracting tourists during visits, whereas semiwild GPs lived in a large area and natural habitat or reintroduced into primeval forest without any disturbance by humans. Wild GPs were born and lived in the wild.
DNA was extracted from fecal samples by using the PowerFecal® DNA Isolation Kit (MO BIO Laboratories, Inc.) following the manufacturer’s instructions. The metagenomic sequencing was performed on an Illumina NextSeq 500 platform in a 2 × 150 paired-end mode.
An in-house comprehensive microbial genome database (CMDB) was created using scaffold-level or chromosome-level genomic data from National Center for Biotechnology Information (NCBI) GenBank/Refseq, which spans 16,574 species including 6,761 bacteria, 1,641 fungi, 456 protozoa, 7,149 viruses, and 567 archaea. The high-quality short reads were aligned against the CMDB. Genomes of PH, including 94 from parasitic nematodes and 29 from parasitic trematodes, were downloaded from wormbase parasite ftp site1 (Howe et al., 2016) (WBPS8). The WBPS8 also includes eight free-living Caenorhabditis worms. Additional genomes, including genomes of human, GP, bamboo, and flies, were included in the analysis for contamination screening and comparison.
Whole metagenome shotgun sequencing reads from GP fecal samples were first subject to quality processing by using the in-house scripts to trim the adaptors, low quality, and duplication reads. Reads with low complexity or length < 90 bp were then removed. In silico decontamination was performed by mapping those processed reads to the potential contamination genomes. The final remaining cleaned reads were used for microbial profiling. If the bacteria, bacteriophages, fungi, and PH genome were uniquely mapped by the >50, 50, 300, and 1800 cleaned reads from a sample, the corresponding bacteria, bacteriophages, fungi, and PH species were considered to be present in this sample. The different cutoffs were selected for fungi and PH because their genomes are generally larger compared with the bacterium/bacteriophage. Abundance of a given genome within a sample was obtained by adding all the mapped reads specific to this genome from the corresponding sample.
These non-redundant reads were performed de novo assembly by using Soapdenovo 2 (Luo et al., 2012) to obtain long contigs and scaffolds. The predicted open reading frames were annotated and compared with BLASTP databases by using MetaGeneMark (Zhu et al., 2010) from the long contigs with a length more than 300 bp. These non-redundant protein sequences were compared in NCBI-NR database with 90% similarity using CD-HIT software (Li and Godzik, 2006). The genes were functionally annotated using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Kanehisa et al., 2017) and Carbohydrate-Active Enzymes (CAZy) database (Cantarel et al., 2009).
The cladogram with circular representations of taxonomic and phylogenetic trees was produced using GraPhlAn (Asnicar et al., 2015). The network displayed correlations between different species by using Cytoscape (Shannon et al., 2003). KEGG metabolic pathways were generated to obtain the most comprehensive image using ipath 2.0 (Yamada et al., 2011).
The 124.6 gigabases (Gbs) of high-quality reads was obtained from all samples, with an average of 9.6 Gbs of each sample (Supplementary Table S1). An average of 10.3 Gbs came from the captive GPs, 7.7 Gbs from semiwild GPs, and 12.5 Gbs from wild GPs. The short reads for each individual sample were performed de novo assembly into long contigs for further analysis and annotation. Totally, 2.3 million contigs and a total sequence length of 1,199 million bps were obtained eventually with an average of 602 contig N50, 7.0% of which (159.1 thousand sequences) were more than 1,000 bp (Supplementary Table S2).
We performed alignment of all high-quantity reads against CMDB and WBPS8 to profile diversity of microbiota in the gut of GPs. After exclusion of the irrelevant reads with bacteria, fungi, bacteriophages, and helminths, 316.2 million reads were obtained. Among them, 231.7 million reads (73.28%) belonged to bacterial genomes, 5.6 million reads (1.77%) to fungal genomes, 3.7 million reads (1.17%) to bacteriophage genomes, and 75.2 million reads (23.78%) to helminthous genomes.
Totally, 680 species of bacteria affiliated with 13 classified phyla, 23 classified classes, 47 classified orders, 88 classified families, 228 classified genera, 1 unclassified phylum, 4 unclassified classes, 5 unclassified orders, 9 unclassified families, and 10 unclassified genera that could not be classified to the corresponding taxonomic level were found (Figure 1A). The two most abundant phyla were Proteobacteria (75.41%) and Firmicutes (23.94%), followed by Bacteroidetes (0.52%), Actinobacteria (0.09%), and Cyanobacteria (0.02%), and others (0.02%) (Supplementary Table S3 and Figure 2A). Within the Proteobacteria, three top abundant classes were the members of Gammaproteobacteria with an average abundance of 72.9%, Betaproteobacteria with 1.7%, and Alphaproteobacteria with 0.8%. Within the Firmicutes, the most abundant class was Bacilli with an average of 23.3%, followed by Clostridia (0.5%) and Erysipelotrichia (0.1%). The phylum Bacteroidetes primarily consisted of Sphingobacteriia (0.27%) and Flavobacteriia (0.25%). Actinobacteria (0.1%) was the only class in the phylum Actinobacteria. Two classes, namely, Gloeobacteria (0.0029%) and Hormogoneae (0.00009%), mainly constituted the phylum Cyanobacteria. At the genus level, 50 most abundant genera are shown in Supplementary Figure S1. The top 10 prevalent genera were Escherichia (41.1%), Streptococcus (15.6%), Pseudomonas (10.7%), Yersinia (8.9%), Lactococcus (4.8%), Acinetobacter (3.5%), Leuconostoc (2.1%), Stenotrophomonas (2.0%), Hafnia (1.7%), and Shigella (1.6%) (Supplementary Table S4). The 50 most abundant species are shown in Figure 3A. The top 10 abundant species were Escherichia coli (40.8%), Yersinia enterocolitica (8.3%), Pseudomonas fluorescens (4.9%), Lactococcus lactis (4.2%), Streptococcus thermophilus (3.5%), Streptococcus infantarius (2.9%), Streptococcus gallolyticus (2.8%), Streptococcus lutetiensis (1.8%), Stenotrophomonas maltophilia (1.7%), and Hafnia alvei (1.7%) (Supplementary Table S5).
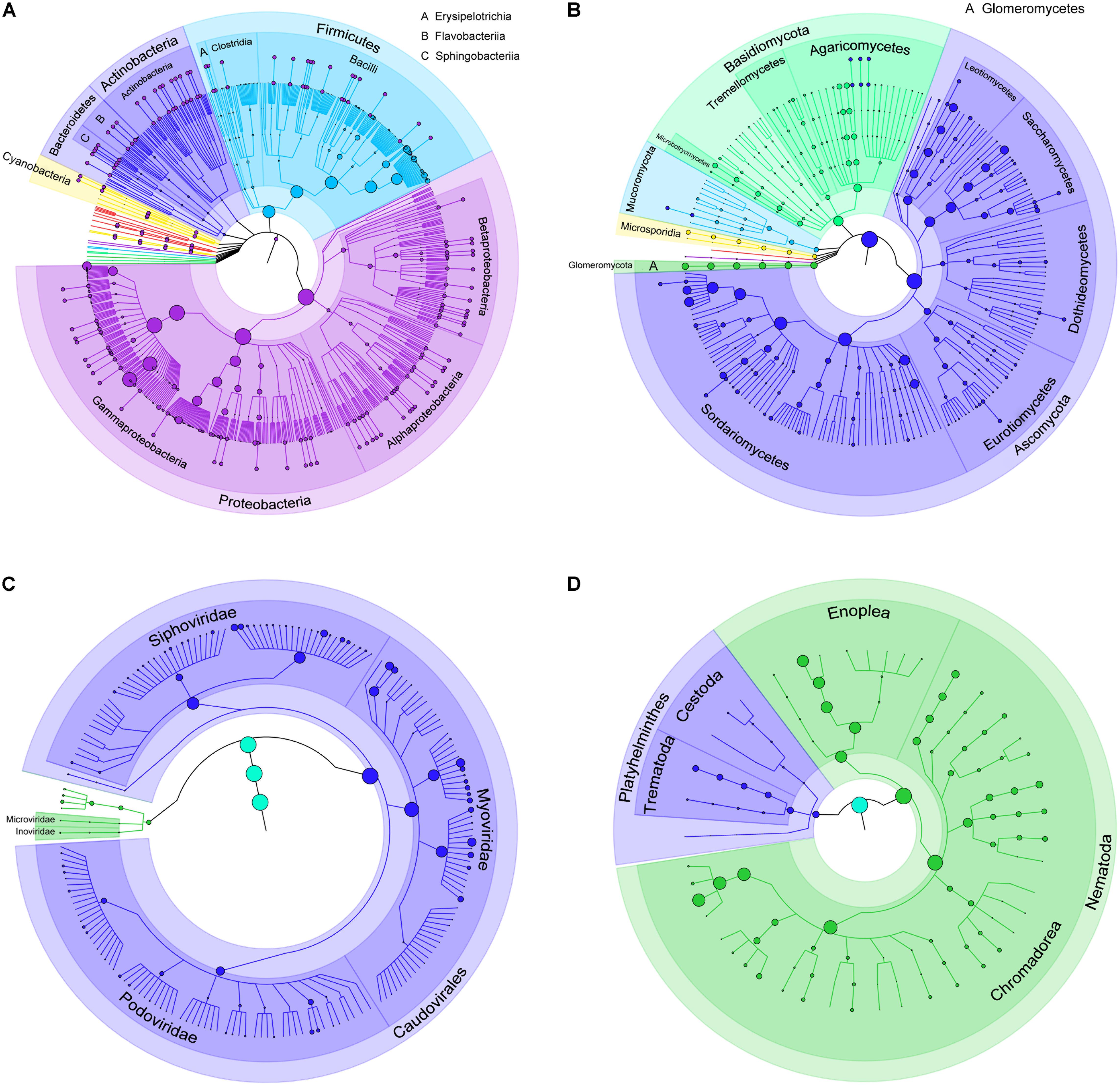
FIGURE 1. Taxonomic tree of bacteria, fungi, bacteriophages, and helminths in the gut of GPs. (A) Bacteria; (B) fungi; (C) bacteriophages; and (D) helminths. From the inner to outer circles, the taxonomic levels range from kingdom to species. The diameter of nodes indicates the abundance at different taxonomic levels, and different colors denote different taxonomic clades.
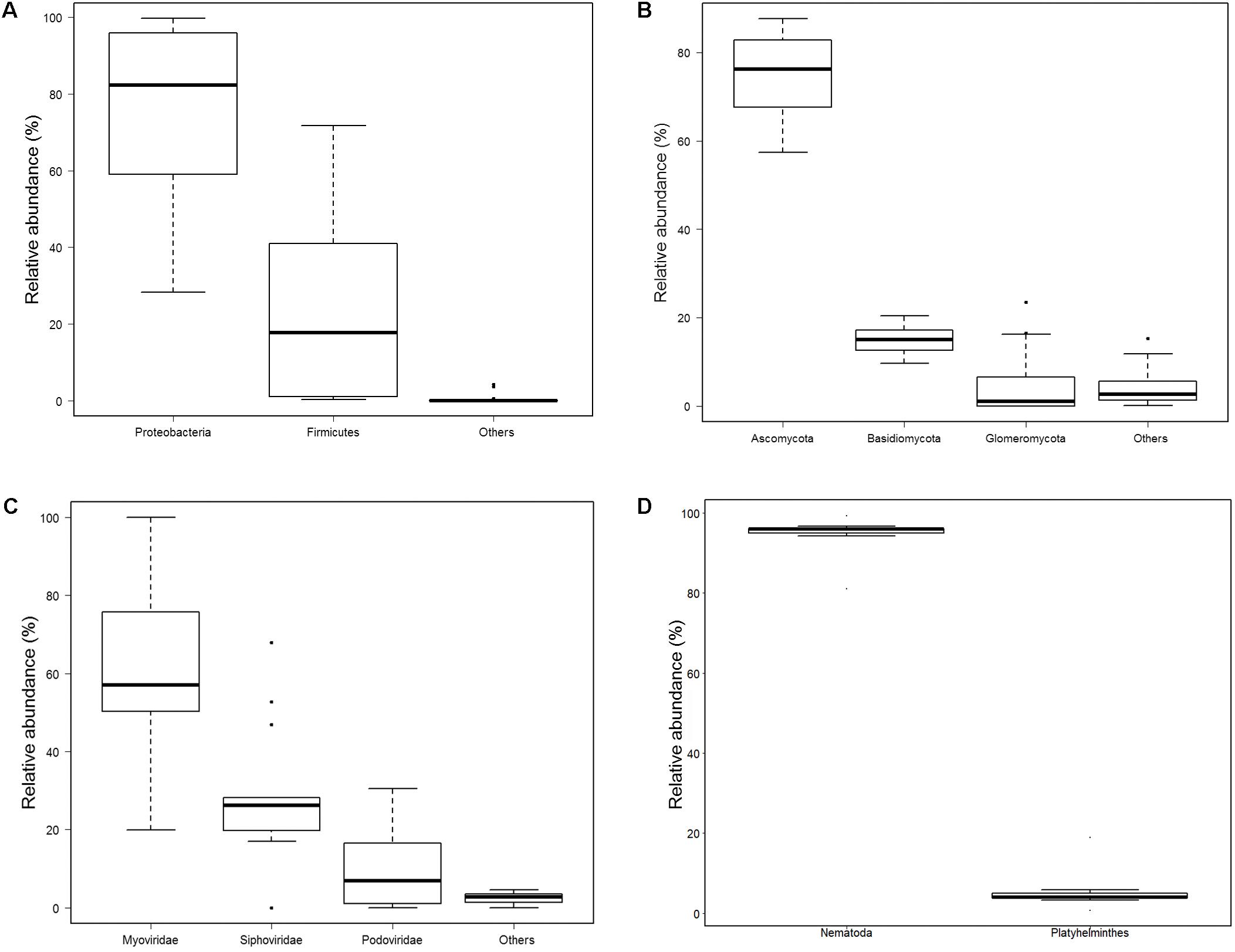
FIGURE 2. Relative abundance of bacteria, fungi, bacteriophages, and helminths at phyla level. (A) Bacteria; others included Bacteroidetes, Actinobacteria, Cyanobacteria, Tenericutes, Candidatus Saccharibacteria, Acidobacteria, Spirochaetes, Deinococcus Thermus, Verrucomicrobia, Fusobacteria, Gemmatimonadetes, and one unclassified phylum. (B) Fungi; others included Mucoromycota, Microsporidia, Chytridiomycota, and Zoopagomycota. (C) Bacteriophages; Others included Microviridae, Inoviridae, and 2 unclassified phyla. (D) helminths.
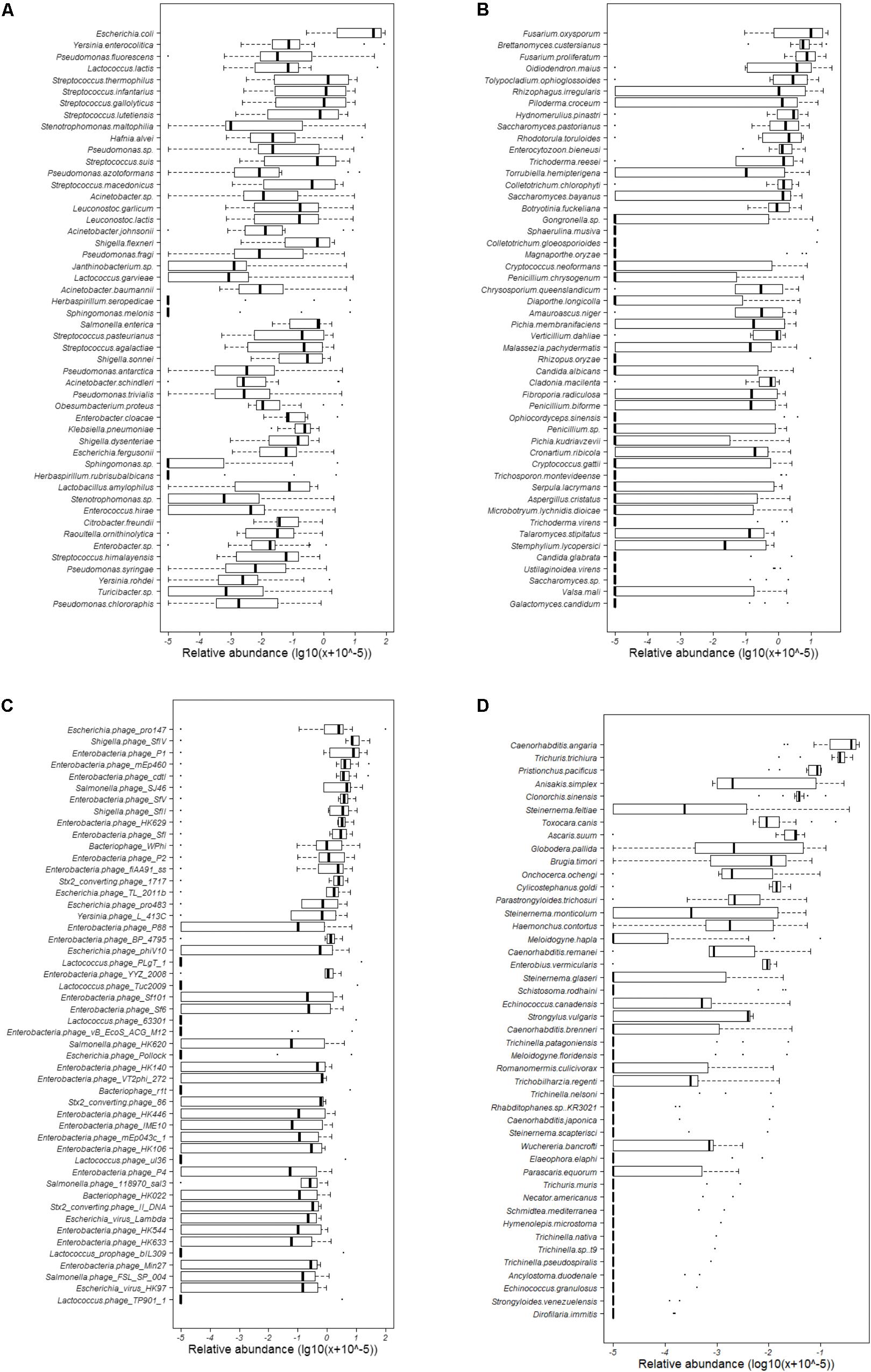
FIGURE 3. Abundance of the 50 most abundant bacterial, fungal, and bacteriophagic species and all helminthous species. (A) Bacteria; (B) fungi; (C) bacteriophages; and (D) helminths.
We also analyzed the correlation of 58 genera with an average abundance of ≥0.01% based on Pearson coefficient of correlation (Supplementary Figure S2). Obviously, these genera were affiliated with the phyla Proteobacteria, Firmicutes, Bacteroidetes, Actinobacteria, and Cyanobacteria. Positive correlations were observed between the majority of genera. The genus Ralstonia showed negative correlations with Escherichia, Shigella, Salmonella, Klebsiella, and an unclassified genus of Enterobacteriaceae. The genus Bacillus had a negative correlation with Serratia and Carnobacterium.
To evaluate the bacterial diversity in the gut of GPs from different environments, we analyzed the average abundance of bacteria at the phylum level (Supplementary Figure S3). The results showed no significant differences in the abundance of Proteobacteria and Firmicutes between GPs from different environments. However, the abundance of both Bacteroidetes and Actinobacteria in the gut of wild GPs were significantly higher than that in the gut of the captive and semiwild GPs (p < 0.05). The phylum Cyanobacteria was only detected in semiwild. Besides, we evaluated the difference at genus level with more than 0.01% among the captive, semiwild, and wild GPs (Supplementary Figure S4). Thirty genera showed high abundance in the wild GPs, and Pseudomonas, Janthinobacterium, and Sphingobacterium were found significantly high (p < 0.05). The abundances of 15 genera were high in the captive GPs, and Raoultella was significantly high (p < 0.05). However, other genera showed high abundance in the semiwild GPs.
In total, 198 species of fungi from 7 classified phyla, 17 classified classes, 44 classified orders, 87 classified families, 130 classified genera, 3 unclassified classes, 3 unclassified orders, and 4 unclassified families were detected (Figure 1B). The most abundant phylum was Ascomycota (75.5%), followed by Basidiomycota (14.7%), Glomeromycota (5.4%), Mucoromycota (2.3%), Microsporidia (2.0%), and others (0.1%) (Figure 2B). Within Ascomycota, five most abundant classes, namely, Sordariomycetes (40.0%), Saccharomycetes (16.7%), Leotiomycetes (10.7%), Eurotiomycetes (5.2%), and Dothideomycetes (2.3%), of more than 1% were observed. Three classes, namely, Agaricomycetes (8.1%), Microbotryomycetes (2.8%), and Tremellomycetes (2.0%), belonged to the phylum Basidiomycota. The Glomeromycetes (5.4%) dominated in Glomeromycota. An unclassified class (2.3%) belonged to Mucoromycota, and an unclassified class (2.0%) belonging to Microsporidia were also found.
At the genus level, 50 most abundant genera are shown in Supplementary Figure S5. The Fusarium (22.6%), Brettanomyces (9.6%), Oidiodendron (9.1%), Tolypocladium (5.5%), Rhizophagus (5.4%), Saccharomyces (4.6%), Piloderma (3.4%), Colletotrichum (3.2%), Hydnomerulius (3.0%), and Rhodotorula (2.5%) were the top 10 genera (Supplementary Table S4). In addition, 50 most abundant species are shown in Figure 3B. The top 10 species were Fusarium oxysporum (17.5%), Fusarium proliferatum (11.2%), Brettanomyces custersianus (11.0%), Oidiodendron maius (9.3%), Rhizophagus irregularis (6.6%), Tolypocladium ophioglossoides (6.0%), Piloderma croceum (3.5%), Hydnomerulius pinastri (3.4%), Rhodotorula toruloides (3.3%), and Saccharomyces pastorianus (3.2%) (Supplementary Table S5).
We also analyzed the correlation of 110 genera with an average abundance ≥0.01% based on Pearson coefficient of correlation (Supplementary Figure S6). Obviously, positive correlations were observed between the majorities of genera. However, negative correlations between several special genera were also found. The Hydnomerulius showed negative correlations with 12 genera. The Rhodotorula showed negative correlations with Hydnomerulius, Oidiodendron, Piloderma, and Tolypocladium.
We also analyzed the correlation of 58 bacteria genera with an average abundance of ≥0.01% and 110 fungi genera with an average abundance ≥0.01% based on Pearson coefficient of correlation (Supplementary Figure S7). Obviously, positive correlations were observed between the majorities of genera. However, several fungi genera, including Brettanomyces, Cladonia, Fusarium, Hydnomerulius, Rhodotorula, Tolypocladium, and Verticillium, had negative correlation with many bacteria. Several bacteria genera, including Streptococcus, Pectobacterium, Lactobacillus, Klebsiella, Eubacterium, Escherichia, Carnobacterium and Anaerostipes, had negative correlation with many fungi.
We found no significant differences between the three groups from different environments at the phylum level (p > 0.05) (Supplementary Figure S8). Subsequently, we analyzed the 57 fungus genera with an average abundance of more than 0.01% among captive, semiwild, and wild GPs (Supplementary Figure S9). The abundance of 18 genera were high in the captive GPs. Eighteen genera were high in the wild GPs, and Gongronella and Ophiocordyceps were significantly high (p < 0.05). Other genera were high in the semiwild GPs, and Rhodotorula was also significantly high (p < 0.05).
A total of 185 bacteriophages from 1 classified and 1 unclassified orders, 5 classified and 2 unclassified families, and 23 classified and 6 unclassified genera were obtained (Figure 1C). Most of bacteriophages (97.9%) came from the order Caudovirales, whereas others came from unclassified order (2.1%). Aside from an unclassified families accounting for 2.1%, the five classified families were Myoviridae (58.9%), Siphoviridae (28.8%), Podoviridae (10.0%), Inoviridae (0.1%), and Microviridae (0.07%) (Figure 2C).
The relative abundance of all 29 genera is shown in Supplementary Figure S10. The top 10 abundant genera were Lambdavirus (22.7%), P2 virus (21.9%), P1 virus (13.8%), Epsilon15 virus (3.3%), P22 virus (1.9%), Nona33 virus (0.7%), Rtp virus (0.6%), G7c virus (0.3%), Tl2011 virus (0.3%), and T4 virus (0.2%), (Supplementary Table S4). Twenty-two species of bacteriophages showed more than 1% abundance (Figure 3C), and 10 most abundant species were Escherichia phage pro147 (10.1%), Shigella phage SfIV (9.9%), Enterobacteria phage P1 (8.9%), Enterobacteria phage mEp460 (6.2%), Enterobacteria phage cdtI (5.8%), Salmonella phage SJ46 (5.0%), Enterobacteria phage SfV (3.9%), Shigella phage SfII (3.8%), Enterobacteria phage HK629 (3.5%), and Enterobacteria phage SfI (3.2%) (Supplementary Table S5). Analyzing the host of bacteriophage showed that most of bacteriophages were from Escherichia (35.7%), followed by Enterobacteria (20.5%), Salmonella (17.8%), Shigella (7.0%), and Lactococcus (4.3%). Two prophages, namely, Lactococcus prophage bIL286 and Lactococcus prophage bIL309, were also found.
We analyzed the correlations of 26 genera based on Pearson coefficient of correlation (Supplementary Figure S11). Positive correlations were observed among all genera. Meanwhile, the family Microviridae was only detected in semiwild GPs, and no significant difference in the abundance of Myoviridae, Siphoviridae, Podoviridae, and Inoviridae among the captive, semiwild, and wild GPs was observed (p > 0.05) (Supplementary Figure S12). Interestingly, the analysis phage genera showed a difference among captive, semiwild, and wild GPs (Supplementary Figure S13). Most genera were more abundant in the gut of captive GP than in semiwild and wild GPs.
A total of 45 helminths were identified from 2 classified phyla, 4 classified classes, 1 unclassified class, 12 classified orders, 23 classified families, and 32 classified genera (Figure 1D). The phylum Nematoda was dominant (94.8%), and Platyhelminthes only accounted for 5.2% (Figure 2D and Supplementary Table S3). The phylum Nematoda contained the class Chromadorea with abundance of 70.6% and Enoplea with 24.2%. The phylum Platyhelminthes had three classes, namely, Trematoda (4.8%), Cestoda (0.3%), and 1 unclassified class (0.01%). At the genus level, all 32 abundant genera are shown in Supplementary Figure S14. The 10 most abundant genera were Caenorhabditis (35.2%), Trichuris (23.7%), Pristionchus (7.5%), Anisakis (5.9%), Steinernema (4.8%), Clonorchis (4.3%), Toxocara (3.0%), Ascaris (2.8%), Globodera (2.3%), and Brugia (1.6%) (Supplementary Table S4).
At the species level, the most dominant helminth was Caenorhabditis angaria (34.0%), followed by Trichuris trichiura (23.7%), Pristionchus pacificus (7.5%), Anisakis simplex (5.9%), Clonorchis sinensis (4.3%), Steinernema feltiae (3.4%), Toxocara canis (3.0%), Ascaris suum (2.8%), Globodera pallida (2.3%), and Brugia timori (1.6%) (Figure 3D). In all 45 helminths, 30 PH were found in mammals or humans (Figure 4 and Table 1).
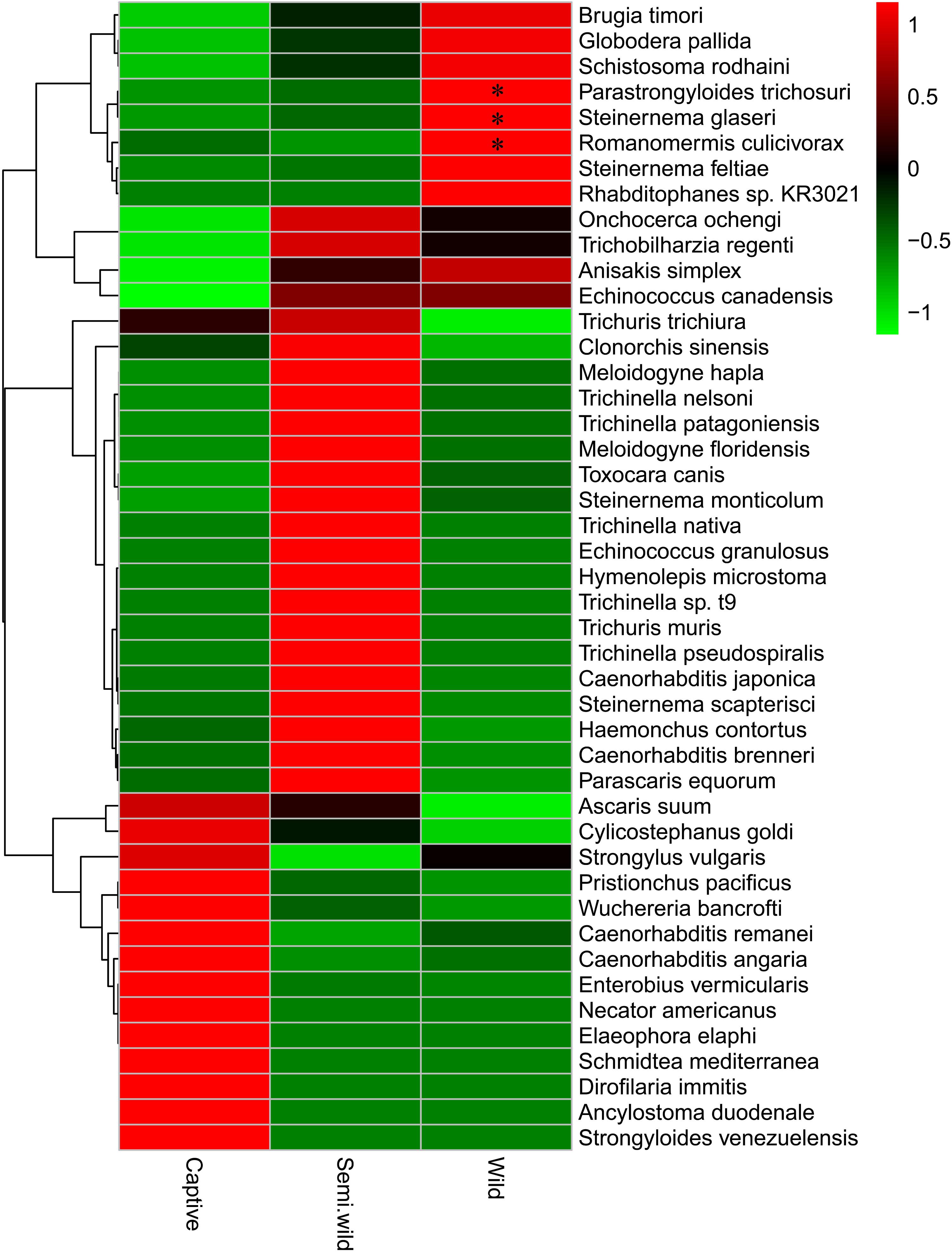
FIGURE 4. Average abundance of all helminths between three different groups. ∗Significant difference (p < 0.05).
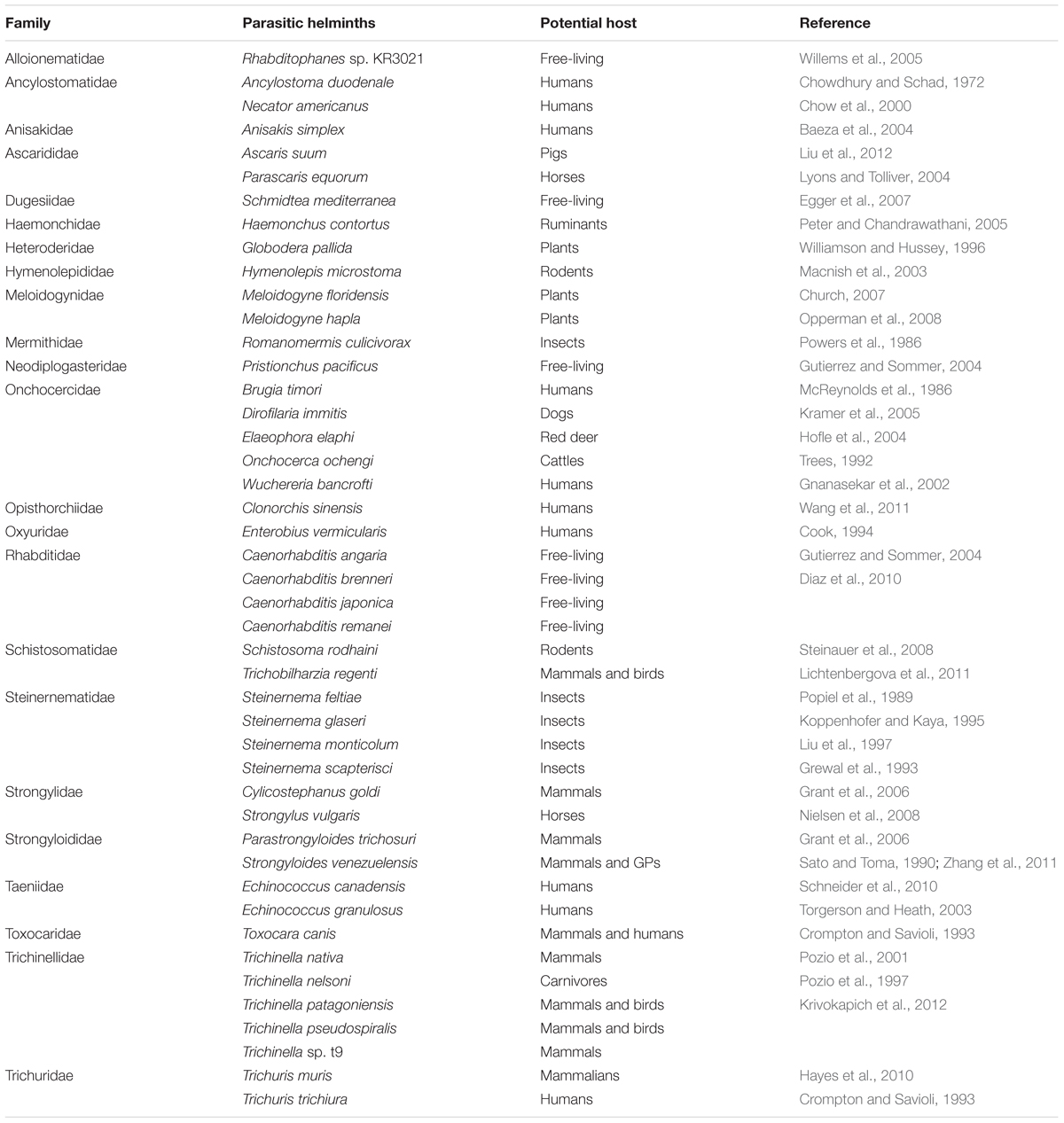
TABLE 1. Species of helminths in the gut of GPs in this study.
No significant differences were observed among the captive, semiwild, and wild GPs at the phylum level (p > 0.05) (Supplementary Figure S15). As shown in Figure 4, 14 species including C. angaria and P. pacificus were found high in captive GPs. Twenty-two species were found high in semiwild GPs. In addition, nine species were found high in wild GPs. Among which, Parastrongyloides trichosuri, Steinernema glaseri, and Romanomermis culicivorax were significantly high (p < 0.05).
We compared the genes present in bacterial and fungal metagenome based on KEGG database and CAZy database to assess the ability of hydrolyzing cellulose and encoding the CAZy. By analyzing 1,446,631 genes from 13,075 KO based on KEGG database, we found 1739 genes possessing homologous sequences to the genes encoding cellulase (EC 3.2.1.4) (n = 364), β-glucosidase (EC 3.2.1.21) (n = 1339), and cellulose 1,4-β-cellobiosidase (EC 3.2.1.91) (n = 36). All of which belonged to 223 different bacterial and 36 fungal genera.
The genes encoding cellulase came from 125 bacteria and 24 fungi. The 125 bacteria belonged to 73 genera. The most abundant bacterium was Bacillus (n = 11), followed by Thermoanaerobacter (n = 7), Pseudomonas (n = 6), Serratia (n = 5), Paenibacillus (n = 5), Xanthomonas (n = 5), and Methylobacterium (n = 5). Among the 24 fungi, 7 members belonged to Aspergillus, 3 belonged to Bipolaris, 2 belonged to Verticillium, and 2 belonged to Neurospora.
The genes encoding β-glucosidase came from 243 bacteria and 50 fungi. The 243 bacteria belonged to 142 genera. The most abundant bacteria belonged to Pseudomonas (n = 21), followed by Bifidobacterium (n = 10), Xanthomonas (n = 8), Paenibacillus (n = 7), and Serratia (n = 6). The 50 fungi belonged to 20 genera, and the most abundant fungi were from Bipolaris (n = 3), Neurospora (n = 2), Aspergillus (n = 2), and Fusarium (n = 2).
The genes encoding cellulose 1,4-β-cellobiosidase came from 11 bacteria and 14 fungi. The 11 bacteria belonged to 8 genera, including Streptomyces (n = 3) and Cellulomonas (n = 2). The 14 fungi belonged to 12 genera, including Bipolaris (n = 2) and Neurospora (n = 2).
In total, we found 128,707 putative GH gene of 117 GH families in all 145 GH families of CAZy database. The 27 GH families were more than 1%, including GH13 (n = 12,475), GH23 (n = 12,355), GH3 (n = 10,253), GH1 (n = 6,655), and GH2 (n = 6,548) (Supplementary Table S6). A total of 30,209 genes encoding the cellulase, β-glucosidase, and cellulose β-1,4-cellobiosidase were also included (Supplementary Table S7).
Kyoto Encyclopedia of Genes and Genome metabolic pathways were generated based on metagenomics to obtain the most comprehensive image of gut microbe metabolism in GPs, suggesting that gut microbiome of GPs features enriched activity for metabolism of carbohydrates, nucleotides, lipids, amino acids, energy, terpenoids, polyketides, glycan, cofactors, vitamins and xenobiotics biodegradation, and biosynthesis of other secondary metabolites (Figure 5).
FIGURE 5. The KEGG pathways determined using iPath of the gut microbiome.
At the phylum level, 14 bacterial groups were identified; this finding was more than six and nine phyla found in GPs using 16S rRNA sequencing (Zhu et al., 2011; Xue et al., 2015). As described previously, Proteobacteria and Firmicutes constituted the vast majority of bacteria in the gut of GPs (Zhu et al., 2011; Fang et al., 2012; Tun et al., 2014; Xue et al., 2015). Unlike the published 16S rRNA sequencing data (Zhu et al., 2011; Fang et al., 2012; Tun et al., 2014; Xue et al., 2015), our metagenomic shotgun sequencing study revealed significantly higher abundance of phyla Proteobacteria than that of Firmicutes. Similarly, the two phyla Firmicutes and Proteobacteria were also the predominant phyla in the gut of both brown bear (Ursus arctos) (Sommer et al., 2016) and Asiatic black bear (Ursus thibetanus) (Song et al., 2017). GP still has a similarity of gut bacteria with other bears in spite of a bamboo diet (Ley et al., 2008). Members of Bacteroidetes were also abundant in the gut of GP, as described in cattle (Shanks et al., 2011) and horse (Dougal et al., 2013). In general, the three phyla, namely, Firmicutes, Bacteroidetes, and Proteobacteria, were numerically the most dominant phyla detected in the gut of mammals (Delsuc et al., 2014). Actinobacteria was another phylum that could be found in the gut of humans (Krogius-Kurikka et al., 2009), cheetahs (Becker et al., 2014), and mice (Murphy et al., 2010); Actinobacteria was generally found in adult GPs and absent in geriatric individuals (Tun et al., 2014). The phylum Cyanobacteria also was detected in the gut of GPs (Zhu et al., 2011; Xue et al., 2015). However, it was only detected in semiwild GP in our study. Living environments may influence the abundance of Cyanobacteria in the gut of GPs. Escherichia was the most predominant genus in GP’s gut microbiota as described previously. Differently, majority members of abundant genera showed different rankings in the study (Zhu et al., 2011; Xue et al., 2015), e.g., Streptococcus, Pseudomonas, Yersinia, and Lactococcus were the most dominant. Besides, bacteria at species level were demonstrated using metagenomic sequencing. As described previously, metagenomic sequencing predicted more microbial species than 16S ribosomal RNA (rRNA) gene sequencing (Zhernakova et al., 2016). Compared with 16S rRNA sequencing technology, we delineated the detailed and different gut bacteria in GPs, especially at species level. As described previously, bacteria belong to phylum Firmicutes possess putative genes coding for cellulose and hemicellulose-digesting enzymes, especially found in species within the Clostridium genus (Zhu et al., 2011). It is clear that gut microbial composition data alone cannot resolve whether gut microorganisms of giant pandas are an adaptation to a bamboo diet and aid in the digestion of cellulose and hemicellulose. Fortunately, we identified other potential cellulolytic and hemicellulolytic bacteria in the gut of giant pandas using metagenomics.
The abundance of Bacteroidetes and Actinobacteria in the gut of wild GPs was significantly higher than that in captive and semiwild GPs. Bacteroidetes and Actinobacteria were also discovered as the dominant phyla in the vagina and uterus of GPs (Yang et al., 2017). High-carbohydrate and low-fat diet in natural environments may contribute to the increase in Bacteroidetes (Ley et al., 2006). Bacteroidetes is well-known for the degradation of high-molecular weight organic matter (Thomas et al., 2011; Wu et al., 2011). Besides, the Bacteroidetes are believed to complement eukaryotic genomes with degradation enzymes targeting resistant dietary polymers (Thomas et al., 2011). Members of Actinobacteria were described in a prior gut study, and they have also been associated with a variety of environments and conditions (D’Argenio and Salvatore, 2015). Diet and environment may lead the wild GPs to obtain these bacteria (Paul et al., 2016; Snijders et al., 2016).
As described previously, the fungal phyla Ascomycota, Basidiomycota, and Mucoromycota were also dominant in the gut of GPs (Tun et al., 2014). Compared with other mammals, the GPs had a more abundant Glomeromycota and Mucoromycota, which were proved to be an important arbuscular mycorrhizal fungi (Hijri et al., 2007; Spatafora et al., 2016). Besides, the phylum Microsporidia has an important proportion in the gut of GPs. Members of Microsporidia are obligate intracellular eukaryotic parasites (Patrick, 2009). At the genus level, most fungi were obviously related with plants. Fusarium (Kistler, 1997; Kolb et al., 2001) and Colletotrichum (O’Connell et al., 2012) served as plant pathogens. Oidiodendron appeared to exist as saprotrophs and could also form ericoid mycorrhizal associations with Ericaceae hosts (Chambers et al., 2008). Rhizophagus is an arbuscular mycorrhizal fungi that form symbiotic relationships with plant (Tisserant et al., 2013). Piloderma is a wood ectomycorrhizal fungus (Hagerberg et al., 2005). Besides, the members of Tolypocladium were widespread as soil-borne insect pathogens and saprotrophs (Mogensen et al., 2011). This discrepancy can be attributed to dietary differences as GPs may consume different species of bamboo, water, and many other foods in nature, which may cause exposure of GPs to these fungi from the diet and environment. Besides, fungi at species level were demonstrated using metagenomic sequencing. Thus, using metagenomic sequencing, we delineated the detailed gut fungi of GPs.
Still now, little was known about the diversity and role of fungi in the gut of giant pandas. Compared to bacteria, the role of fungi within the intestinal microbiota is poorly understood. Fungi also play an important role in disease of host (Huffnagle and Noverr, 2013; Scott et al., 2013; Sokol et al., 2017) or produce a result of interactions that are relevant to health and gut diseases of host with bacteria (Peleg et al., 2010; Mar Rodriguez et al., 2015). In addition to bacteria, fungi in gut of giant pandas harbor the genes coding for starch, cellulose, and hemicellulose-digesting enzymes and could also aid in the digestion of the substances in bamboo (Zhu et al., 2011).
However, most fungus genera were abundant in semiwild and wild GPs than that in captive GPs. The abundance and difference of these fungi can be attributed to diet consumption. Gut fungi may be responsible for fiber degradation and fermentative digestion in herbivores (Li and Heath, 1993; Liggenstoffer et al., 2010). The gut microbiota of herbivore plays a vital role in hydrolyzing carbohydrate components of the plant cell wall including cellulose and xylan components (Brulc et al., 2009; Hooper and Macpherson, 2010; Hess et al., 2011; Wang et al., 2013). In particular, anaerobic fungi are commonly found in the digestive tract of ruminants and monogastric herbivores (Theodorou et al., 1996; Ho et al., 2000; Ljungdahl, 2008; Haitjema et al., 2014) and capable of producing enzymes that efficiently hydrolyze cellulose and hemicelluloses (Ljungdahl, 2008). However, the crucial anaerobic gut fungi, which affiliate with Class Chytridiomycetes, Order Neocallimastigales, and Family Neocallimastigaceae, commonly harbored in many herbivores to assist hydrolysis of cellulose (Theodorou et al., 1996; Ho et al., 2000; Ljungdahl, 2008; Haitjema et al., 2014), were not found in the gut of GPs in this study.
Majority of bacteriophages was found in the gut of GPs, and most of which belonged to the order Caudovirales in nature (Ackermann, 2003; Lim et al., 2016). Previous study demonstrated an increase in the richness of Caudovirales phages in the gut of patients with inflammatory bowel disease (Norman et al., 2015). Clearly, bacteriophages can influence the behavior and pathogenicity of bacteria, and the interaction effect between bacteriophages and bacteria have a significant impact on host health (Bondy-Denomy and Davidson, 2014). The lysis of bacteria by bacteriophages leads to release of proteins, lipids, and nucleic acids, which could induce gut inflammation (Łusiak-Szelachowska et al., 2017). In our study, host of bacteriophages was largely from Enterobacteriaceae. This phenomenon displayed well the symbiotic or parasitic relationships between the bacteriophages and bacteria. Bacteriophages are known as regulators of the bacterial population in the gut (Brown-Jaque et al., 2016), capable to lyse bacterial cell and may control the bacterial population, influencing bacterial diversity and metabolism (Łusiak-Szelachowska et al., 2017). The bactericidal activity of bacteriophages could be used to treat infections gradually as an alternative or a complement to antibiotic therapy in GPs (Sulakvelidze et al., 2001; Merril et al., 2003; Hermoso et al., 2007). Only two prophages, Lactococcus prophage bIL286 and Lactococcus prophage bIL309, were detected in the gut of GPs. Prophage induction may contribute to the dysbiosis of gut microbiota, changing the ratio of symbionts to pathobionts (Mills et al., 2013). Most genera of bacteriophages were more abundant in the gut of captive GPs than that in the gut of semiwild and wild GP. Living environment is an important factor influencing diversity of phages (Łusiak-Szelachowska et al., 2017).
We demonstrated different gut parasites using metagenomic sequencing compared with previous studies. Baylisascaris schroederi is the most common parasitic helminth of GPs (Zhang et al., 2012, 2015; Zhao et al., 2013; Zhou et al., 2013). Meanwhile, Cryptosporidium spp. (Wang et al., 2015), Ancylostoma ailuropodae (Xie et al., 2017), Ogmocotyle sikae, Toxascaris seleactis, and Strongyloides spp. (Zhang et al., 2011) have also been reported to cause parasitic infection in GPs. However, in addition to previously described parasites in GPs, we first found another 44 helminths in GPs in this study which were not described previously (not including Strongyloides spp.). This discrepancy can be attributed to genomes as no data were available in WBPS8 for those parasites found in GPs (B. schroederi, O. sikae, A. ailuropodae, and T. seleactis). It is worth noting that the current PH database is derived from 50 Helminth Genome Project2 (ref), and prominently biased toward the medically important helminth genomes. Therefore, our results should be more sensible at the family level. Among the 45 helminth whose genomes our reads were mapped to, 30 parasitize mammals or human, strongly suggesting that GPs are important reservoir for parasitic helminths. Several major worldwide helminths, such as T. trichiura, Necator americanus, Ancylostoma duodenale, Enterobius vermicularis, and T. canis (Bundy and Cooper, 1989; Crompton and Savioli, 1993; Bethony et al., 2006), found in humans also were detected in GPs. In addition, several free-living nematodes, including C. angaria, C. brenneri, C. japonica, C. remanei, and P. pacificus (Kiontke, 2006; Sommer, 2006; Rödelsperger et al., 2013), were also identified, which were dominant in the gut of GPs. P. pacificus was reported as living in close association with beetles (Herrmann et al., 2006; Kiontke, 2006). Actually, the five genomes of beetle were found highly abundant in GP’s gut metagenome sample, which might be derived from the bamboo feeds. Besides, no significant difference was observed for most helminths between the three groups from different environments, suggesting that the parasitic infection was ubiquitous in captive, semiwild, and wild GPs.
The genes encoding cellulose catabolic enzymes were found in bacteria or fungi of GP’s gut. The number of β-glucosidase gene was higher than that of cellulose and cellulose β-1,4-cellobiosidase, and the number of genes from bacteria was more than fungi. Numerous bacteria and fungi may play a key role in assisting the metabolism of cellulose. A large GH genes were found, implying an active metabolism of carbohydrates in the gut of GPs. Among the GH families found, GH13, GH23, and GH3 were obviously abundant. GH13 was the largest of the CAZYs family, wherein many alpha-glucan active enzymes are found and may be closely related with starch hydrolysis (Turkenburg et al., 2009). Starch is one of the most important polysaccharides in bamboo components (Peng et al., 2011). Family GH23 contains lysozymes and soluble lytic transglycosylase (Tufariello et al., 2006). The lytic transglycosylases are an important class of bacterial enzymes as a class of autolysins (Blackburn and Clarke, 2001; Scheurwater et al., 2008). In addition, the lytic transglycosylases may have a role in pathogenesis of some bacterial species (Boneca, 2005). Family GH3 consists primarily of stereochemistry-retaining β-glucosidases and also contains a subfamily of β-N-acetylglucosaminidases (Macdonald et al., 2015). Importantly, GH1 and GH2 are gene families with hemicellulose degrading activity, including β-glucosidase, β-galactosidase, β-mannosidase, β-glucuronidase,β-xylosidase, glucan 1,3-β-glucosidase, glucan 1,4-β-glucosidase and xylan 1,4-β-xylosidase, etc. Besides, GH1, GH2, and GH3 are also an important family of enzyme with β-glucosidase activity (Kuntothom et al., 2009; Heins et al., 2011; Talens-Perales et al., 2016) associated with cellulose and hemicellulose degrading activity. Therefore, the study indicated that the microbiota in GPs harbor the ability of cellulose and hemicellulose degradation.
We provided full details of microbiota structure, including bacteria, fungi, bacteriophages, and helminths based on metagenomic sequencing, in the gut of GPs. Totally, 680 species of bacteria, 198 species of fungi, 185 bacteriophages, and 45 helminths were found. 44 helminths were first reported in the gut of GPs, among which 30 PH were found in mammals or humans suggesting that GPs are important reservoir for parasitic helminths. 1,739 genes encoding cellulase, β-glucosidase, and cellulose β-1,4-cellobiosidase were responsible for the metabolism of cellulose, and 128,707 putative GH genes were found in numerous bacteria and fungi, revealing that the microbiota in GPs harbor the ability of cellulose, hemicellulose and starch hydrolysis. The abundance of Bacteroidetes and Actinobacteria in wild GPs was significantly higher than that in captive and semiwild GPs (p < 0.05). Only a few of bacteria, fungi, bacteriophages, and helminths at genus or species level were found significantly different between the three groups (p < 0.05). Taken together, the results indicated not only bacteria but also fungi, bacteriophages, and helminths were diverse in gut of giant pandas providing basis for the further identification of role of gut microbiota. Metagenomics revealed that the bacteria/fungi in gut of GPs harbor the ability of cellulose and hemicellulose degradation. Besides species within the Clostridium genus, majority of bacteria and fungi were also found to aid in the digestion of cellulose and hemicellulose in the gut of giant pandas.
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
YH, HZ, and LZ conceived the idea. SY, XG, JM, AZ, YZ, ML, BL, and WD performed the experiments. SY, XG, JM, AZ, YZ, ML, BL, WD, LJ, and SZ performed the statistical analyses. DW, YGH, and CL collected the samples. SY, XG, JM, and AZ wrote the first draft of the manuscript. YH, HZ, SL, and LZ contributed substantially to revisions. All authors read and approved the final manuscript.
This work was supported by the Forestry Department of Sichuan Province and International Cooperation Funding for Giant Pandas, the State Forestry Administration of the People’s Republic of China (YGH, 2017 and YH, 2017).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01717/full#supplementary-material
Ackermann, H. W. (2003). Bacteriophage observations and evolution. Res. Microbiol. 154, 245–251. doi: 10.1016/S0923-2508(03)00067-6
Asnicar, F., Weingart, G., Tickle, T. L., Huttenhower, C., and Segata, N. (2015). Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 3:e1029. doi: 10.7717/peerj.1029
Bäckhed, F., Ding, H., Wang, T., Hooper, L. V., Koh, G. Y., Nagy, A., et al. (2004). The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. U.S.A. 101, 15718–15723. doi: 10.1073/pnas.0407076101
Baeza, M. L., Rodriguez, A., Matheu, V., Rubio, M., Tornero, P., de Barrio, M., et al. (2004). Characterization of allergens secreted by Anisakis simplex parasite: clinical relevance in comparison with somatic allergens. Clin. Exp. Allergy 34, 296–302. doi: 10.1111/j.1365-2222.2004.01883.x
Becker, A. A. M. J., Hesta, M., Hollants, J., Janssens, G. P. J., and Huys, G. (2014). Phylogenetic analysis of faecal microbiota from captive cheetahs reveals underrepresentation of Bacteroidetes and Bifidobacteriaceae. BMC Microbiol. 14:43. doi: 10.1186/1471-2180-14-43
Bethony, J., Brooker, S., Albonico, M., Geiger, S. M., Loukas, A., Diemert, D., et al. (2006). Soil-transmitted helminth infections: ascariasis, trichuriasis, and hookworm. Lancet 367, 1521–1532. doi: 10.1016/S0140-6736(06)68653-4
Blackburn, N. T., and Clarke, A. J. (2001). Identification of four families of peptidoglycan lytic transglycosylases. J. Mol. Evol. 52, 78–84. doi: 10.1007/s002390010136
Bondy-Denomy, J., and Davidson, A. R. (2014). When a virus is not a parasite: the beneficial effects of prophages on bacterial fitness. J. Microbiol. 52, 235–242. doi: 10.1007/s12275-014-4083-3
Boneca, I. G. (2005). The role of peptidoglycan in pathogenesis. Curr. Opin. Microbiol. 8, 46–53. doi: 10.1016/j.mib.2004.12.008
Brown-Jaque, M., Muniesa, M., and Navarro, F. (2016). Bacteriophages in clinical samples can interfere with microbiological diagnostic tools. Sci. Rep. 6:33000. doi: 10.1038/srep33000
Brulc, J. M., Antonopoulos, D. A., Miller, M. E., Wilson, M. K., Yannarell, A. C., Dinsdale, E. A., et al. (2009). Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc. Natl. Acad. Sci. U.S.A. 106, 1948–1953. doi: 10.1073/pnas.0806191105
Bundy, D. A., and Cooper, E. S. (1989). Trichuris and trichuriasis in humans. Adv. Parasitol. 28, 107–173. doi: 10.1016/S0065-308X(08)60332-2
Cantacessi, C., Giacomin, P., Croese, J., Zakrzewski, M., Sotillo, J., McCann, L., et al. (2014). Impact of experimental hookworm infection on the human gut microbiota. J. Infect. Dis. 210, 1431–1434. doi: 10.1093/infdis/jiu256
Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V., and Henrissat, B. (2009). The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 37, D233–D238. doi: 10.1093/nar/gkn663
Chambers, S. M., Curlevski, N. J., and Cairney, J. W. (2008). Ericoid mycorrhizal fungi are common root inhabitants of non-Ericaceae plants in a south-eastern Australian sclerophyll forest. FEMS Microbiol. Ecol. 65, 263–270. doi: 10.1111/j.1574-6941.2008.00481.x
Chow, S. C., Brown, A., and Pritchard, D. (2000). The human hookworm pathogen Necator americanus induces apoptosis in T lymphocytes. Parasite Immunol. 22, 21–29. doi: 10.1046/j.1365-3024.2000.00271.x
Chowdhury, A. B., and Schad, G. A. (1972). Ancylostoma ceylanicum: a parasite of man in Calcutta and environs. Am. J. Trop. Med. Hyg. 21, 300–301. doi: 10.4269/ajtmh.1972.21.300
Church, G. T. (2007). First report of the root-knot nematode Meloidogyne floridensis on tomato (Lycopersicon esculentum) in Florida. Plant Dis. 89, 527–527.
Cook, G. C. (1994). Enterobius vermicularis infection. Gut 35, 1159–1162. doi: 10.1136/gut.35.9.1159
Crompton, D. W., and Savioli, L. (1993). Intestinal parasitic infections and urbanization. Bull. World Health Organ. 71, 1–7.
D’Argenio, V., and Salvatore, F. (2015). The role of the gut microbiome in the healthy adult status. Clin. Chim. Acta 451, 97–102. doi: 10.1016/j.cca.2015.01.003
Delsuc, F., Metcalf, J. L., Wegener Parfrey, L., Song, S. J., Gonzalez, A., and Knight, R. (2014). Convergence of gut microbiomes in myrmecophagous mammals. Mol. Ecol. 23, 1301–1317. doi: 10.1111/mec.12501
Diaz, S. A., Haydon, D. T., and Lindstrom, J. (2010). Sperm-limited fecundity and polyandry-induced mortality in female nematodes Caenorhabditis remanei. Biol. J. Linn. Soc. 99, 362–369. doi: 10.1111/j.1095-8312.2009.01352.x
Dougal, K., de la Fuente, G., Harris, P. A., Girdwood, S. E., Pinloche, E., and Newbold, C. J. (2013). Identification of a core bacterial community within the large intestine of the horse. PLoS One 8:e77660. doi: 10.1371/journal.pone.0077660
Egger, B., Gschwentner, R., and Rieger, R. (2007). Free-living flatworms under the knife: past and present. Dev. Genes Evol. 217, 89–104. doi: 10.1007/s00427-006-0120-5
Fang, W., Fang, Z., Zhou, P., Chang, F., Hong, Y., Zhang, X., et al. (2012). Evidence for lignin oxidation by the giant panda fecal microbiome. PLoS One 7:e50312. doi: 10.1371/journal.pone.0050312
Gnanasekar, M., Rao, K. V., Chen, L., Narayanan, R. B., Geetha, M., Scott, A. L., et al. (2002). Molecular characterization of a calcium binding translationally controlled tumor protein homologue from the filarial parasites Brugia malayi and Wuchereria bancrofti. Mol. Biochem. Parasitol. 121, 107–118. doi: 10.1016/S0166-6851(02)00027-0
Grant, W. N., Skinner, S. J., Newton-Howes, J., Grant, K., Shuttleworth, G., Heath, D. D., et al. (2006). Heritable transgenesis of Parastrongyloides trichosuri: a nematode parasite of mammals. Int. J. Parasitol. 36, 475–483. doi: 10.1016/j.ijpara.2005.12.002
Grewal, P. S., Gaugler, R., Kaya, H. K., and Wusaty, M. (1993). Infectivity of the Entomopathogenic Nematode Steinernema scapterisci (Nematoda: Steinernematidae). J. Invertebr. Pathol. 62, 22–28. doi: 10.1006/jipa.1993.1069
Gutierrez, A., and Sommer, R. J. (2004). Evolution of dnmt-2 and mbd-2-like genes in the free-living nematodes Pristionchus pacificus, Caenorhabditis elegans and Caenorhabditis briggsae. Nucleic Acids Res. 32, 6388–6396. doi: 10.1093/nar/gkh982
Hagerberg, D., Pallon, J., and Wallander, H. (2005). The elemental content in the mycelium of the ectomycorrhizal fungus Piloderma sp. during the colonization of hardened wood ash. Mycorrhiza 15, 387–392. doi: 10.1007/s00572-004-0344-z
Haitjema, C. H., Solomon, K. V., Henske, J. K., Theodorou, M. K., and O’Malley, M. A. (2014). Anaerobic gut fungi: advances in isolation, culture, and cellulolytic enzyme discovery for biofuel production. Biotechnol. Bioeng. 111, 1471–1482. doi: 10.1002/bit.25264
Hayes, K. S., Bancroft, A. J., Goldrick, M., Portsmouth, C., Roberts, I. S., and Grencis, R. K. (2010). Exploitation of the intestinal microflora by the parasitic nematode Trichuris muris. Science 328, 1391–1394. doi: 10.1126/science.1187703
Heins, R. A., Cheng, X., Nath, S., Deng, K., Bowen, B. P., Chivian, D. C., et al. (2011). Phylogenomically guided identification of industrially relevant GH1 β-glucosidases through DNA synthesis and nanostructure-initiator mass spectrometry. Acs Chem. Biol. 9, 2082–2091. doi: 10.1021/cb500244v
Hermoso, J. A., Garcia, J. L., and Garcia, P. (2007). Taking aim on bacterial pathogens: from phage therapy to enzybiotics. Curr. Opin. Microbiol. 10, 461–472. doi: 10.1016/j.mib.2007.08.002
Herrmann, M., Mayer, W. E., and Sommer, R. J. (2006). Nematodes of the genus Pristionchus are closely associated with scarab beetles and the Colorado potato beetle in Western Europe. Zoology 109, 96–108. doi: 10.1016/j.zool.2006.03.001
Hess, M., Sczyrba, A., Egan, R., Kim, T. W., Chokhawala, H., Schroth, G., et al. (2011). Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331, 463–467. doi: 10.1126/science.1200387
Hijri, M., Niculita, H., and Sanders, I. R. (2007). Molecular characterization of chromosome termini of the arbuscular mycorrhizal fungus Glomus intraradices (Glomeromycota). Fungal Genet. Biol. 44, 1380–1386. doi: 10.1016/j.fgb.2007.03.004
Ho, Y. W., Abdullah, N., and Jalaludin, S. (2000). The diversity and taxonomy of anaerobic gut fungi. Fungal Divers. 4, 37–51.
Hofle, U., Vicente, J., Nagore, D., Hurtado, A., Pena, A., de la Fuente, J., et al. (2004). The risks of translocating wildlife. Pathogenic infection with Theileria sp. and Elaeophora elaphi in an imported red deer. Vet. Parasitol. 126, 387–395. doi: 10.1016/j.vetpar.2004.07.026
Hooper, L. V., and Macpherson, A. J. (2010). Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat. Rev. Immunol. 10, 159–169. doi: 10.1038/nri2710
Howe, K. L., Bolt, B. J., Cain, S., Chan, J., Chen, W. J., Davis, P., et al. (2016). WormBase 2016: expanding to enable helminth genomic research. Nucleic Acids Res. 44, D774–D780. doi: 10.1093/nar/gkv1217
Huffnagle, G. B., and Noverr, M. C. (2013). The emerging world of the fungal microbiome. Trends Microbiol. 21, 334–341. doi: 10.1016/j.tim.2013.04.002
Human Microbiome, and Project, C. (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. doi: 10.1038/nature11234
Kamada, N., Seo, S. U., Chen, G. Y., and Nunez, G. (2013). Role of the gut microbiota in immunity and inflammatory disease. Nat. Rev. Immunol. 13, 321–335. doi: 10.1038/nri3430
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, and Morishima, K. (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361. doi: 10.1093/nar/gkw1092
Kelly, D., Conway, S., and Aminov, R. (2005). Commensal gut bacteria: mechanisms of immune modulation. Trends Immunol. 26, 326–333. doi: 10.1016/j.it.2005.04.008
Kistler, H. C. (1997). Genetic diversity in the plant-pathogenic fungus Fusarium oxysporum. Phytopathology 87, 474–479. doi: 10.1094/PHYTO.1997.87.4.474
Kolb, F., Bai, G., Muehlbauer, G., Anderson, J., Smith, K., and Fedak, G. (2001). Host plant resistance genes for Fusarium head blight: mapping and manipulation with molecular markers. Crop Sci. 41, 611–619. doi: 10.2135/cropsci2001.413611x
Koppenhofer, A. M., and Kaya, H. K. (1995). Density-dependent effects on Steinernema glaseri (Nematoda: Steinernematidae) within an insect host. J. Parasitol. 81, 797–799. doi: 10.2307/3283982
Kramer, L. H., Tamarozzi, F., Morchon, R., Lopez-Belmonte, J., Marcos-Atxutegi, C., Martin-Pacho, R., et al. (2005). Immune response to and tissue localization of the Wolbachia surface protein (WSP) in dogs with natural heartworm (Dirofilaria immitis) infection. Vet. Immunol. Immunopathol. 106, 303–308. doi: 10.1016/j.vetimm.2005.03.011
Krivokapich, S. J., Pozio, E., Gatti, G. M., Prous, C. L., Ribicich, M., Marucci, G., et al. (2012). Trichinella patagoniensis n. sp. (Nematoda), a new encapsulated species infecting carnivorous mammals in South America. Int. J. Parasitol. 42, 903–910. doi: 10.1016/j.ijpara.2012.07.009
Krogius-Kurikka, L., Kassinen, A., Paulin, L., Corander, J., Makivuokko, H., Tuimala, J., et al. (2009). Sequence analysis of percent G+C fraction libraries of human faecal bacterial DNA reveals a high number of Actinobacteria. BMC Microbiol. 9:68. doi: 10.1186/1471-2180-9-68
Kubicek, C. P., Mikus, M., Schuster, A., Schmoll, M., and Seiboth, B. (2009). Metabolic engineering strategies for the improvement of cellulase production by Hypocrea jecorina. Biotechnol. Biofuels 2:19. doi: 10.1186/1754-6834-2-19
Kuntothom, T., Luang, S., Harvey, A. J., Fincher, G. B., Opassiri, R., Hrmova, M., et al. (2009). Rice family GH1 glycoside hydrolases with beta-D-glucosidase and beta-D-mannosidase activities. Arch. Biochem. Biophys. 491, 85–95. doi: 10.1016/j.abb.2009.09.004
Lee, S. C., Tang, M. S., Lim, Y. A., Choy, S. H., Kurtz, Z. D., Cox, L. M., et al. (2014). Helminth colonization is associated with increased diversity of the gut microbiota. PLoS Negl. Trop. Dis. 8:e2880. doi: 10.1371/journal.pntd.0002880
Ley, R. E., Hamady, M., Lozupone, C., Turnbaugh, P. J., Ramey, R. R., Bircher, J. S., et al. (2008). Evolution of mammals and their gut microbes. Science 320, 1647–1651. doi: 10.1126/science.1155725
Ley, R. E., Turnbaugh, P. J., Klein, S., and Gordon, J. I. (2006). Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023. doi: 10.1038/4441022a
Li, J., and Heath, I. B. (1993). Chytridiomycetous gut fungi, oft overlooked contributors to herbivore digestion. Can. J. Microbiol. 39, 1003–1013. doi: 10.1139/m93-153
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Lichtenbergova, L., Lassmann, H., Jones, M. K., Kolarova, L., and Horak, P. (2011). Trichobilharzia regenti: host immune response in the pathogenesis of neuroinfection in mice. Exp. Parasitol. 128, 328–335. doi: 10.1016/j.exppara.2011.04.006
Liggenstoffer, A. S., Youssef, N. H., Couger, M. B., and Elshahed, M. S. (2010). Phylogenetic diversity and community structure of anaerobic gut fungi (phylum Neocallimastigomycota) in ruminant and non-ruminant herbivores. ISME J. 4, 1225–1235. doi: 10.1038/ismej.2010.49
Lim, E. S., Wang, D., and Holtz, L. R. (2016). The bacterial microbiome and virome milestones of infant development. Trends Microbiol. 24, 801–810. doi: 10.1016/j.tim.2016.06.001
Liu, G. H., Wu, C. Y., Song, H. Q., Wei, S. J., Xu, M. J., Lin, R. Q., et al. (2012). Comparative analyses of the complete mitochondrial genomes of Ascaris lumbricoides and Ascaris suum from humans and pigs. Gene 492, 110–116. doi: 10.1016/j.gene.2011.10.043
Liu, J., Berry, R. E., and Moldenke, A. F. (1997). Phylogenetic relationships of entomopathogenic nematodes (Heterorhabditidae and Steinernematidae) inferred from partial 18S rRNA gene sequences. J. Invertebr. Pathol. 69, 246–252. doi: 10.1006/jipa.1997.4657
Ljungdahl, L. G. (2008). The cellulase/hemicellulase system of the anaerobic fungus Orpinomyces PC-2 and aspects of its applied use. Ann. N. Y. Acad. Sci. 1125, 308–321. doi: 10.1196/annals.1419.030
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1:18. doi: 10.1186/2047-217X-1-18
Łusiak-Szelachowska, M., Weber-Dąbrowska, B., Jończyk-Matysiak, E., Wojciechowska, R., and Górski, A. (2017). Bacteriophages in the gastrointestinal tract and their implications. Gut Pathog. 9:44. doi: 10.1186/s13099-017-0196-7
Lyons, E. T., and Tolliver, S. C. (2004). Prevalence of parasite eggs (Strongyloides westeri, Parascaris equorum, and strongyles) and oocysts (Emeria leuckarti) in the feces of Thoroughbred foals on 14 farms in central Kentucky in 2003. Parasitol. Res. 92, 400–404. doi: 10.1007/s00436-003-1068-2
Macdonald, S. S., Blaukopf, M., and Withers, S. G. (2015). N-acetylglucosaminidases from CAZy family GH3 are really glycoside phosphorylases, thereby explaining their use of histidine as an acid/base catalyst in place of glutamic acid. J. Biol. Chem. 290, 4887–4895. doi: 10.1074/jbc.M114.621110
Macnish, M. G., Ryan, U. M., Behnke, J. M., and Thompson, R. C. (2003). Detection of the rodent tapeworm Rodentolepis (= Hymenolepis) microstoma in humans. A new zoonosis? Int. J. Parasitol. 33, 1079–1085. doi: 10.1074/jbc.M114.621110
Mar Rodriguez, M., Perez, D., Javier Chaves, F., Esteve, E., Marin-Garcia, P., Xifra, G., et al. (2015). Obesity changes the human gut mycobiome. Sci. Rep. 5:14600. doi: 10.1038/srep14600
Maurice, C. F., Haiser, H. J., and Turnbaugh, P. J. (2013). Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 152, 39–50. doi: 10.1016/j.cell.2012.10.052
McReynolds, L. A., DeSimone, S. M., and Williams, S. A. (1986). Cloning and comparison of repeated DNA sequences from the human filarial parasite Brugia malayi and the animal parasite Brugia pahangi. Proc. Natl. Acad. Sci. U.S.A. 83, 797–801. doi: 10.1016/j.cell.2012.10.052
Merril, C. R., Scholl, D., and Adhya, S. L. (2003). The prospect for bacteriophage therapy in Western medicine. Nat. Rev. Drug Discov. 2, 489–497. doi: 10.1038/nrd1111
Mills, S., Shanahan, F., Stanton, C., Hill, C., Coffey, A., and Ross, R. P. (2013). Movers and shakers: influence of bacteriophages in shaping the mammalian gut microbiota. Gut Microbes 4, 4–16. doi: 10.4161/gmic.22371
Mogensen, J. M., Moller, K. A., von Freiesleben, P., Labuda, R., Varga, E., Sulyok, M., et al. (2011). Production of fumonisins B2 and B4 in Tolypocladium species. J. Ind. Microbiol. Biotechnol. 38, 1329–1335. doi: 10.1007/s10295-010-0916-1
Murphy, E. F., Cotter, P. D., Healy, S., Marques, T. M., O’Sullivan, O., Fouhy, F., et al. (2010). Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut 59, 1635–1642. doi: 10.1136/gut.2010.215665
Nielsen, M. K., Peterson, D. S., Monrad, J., Thamsborg, S. M., Olsen, S. N., and Kaplan, R. M. (2008). Detection and semi-quantification of Strongylus vulgaris DNA in equine faeces by real-time quantitative PCR. Int. J. Parasitol. 38, 443–453. doi: 10.1016/j.ijpara.2007.07.014
Norman, J. M., Handley, S. A., Baldridge, M. T., Droit, L., Liu, C. Y., Keller, B. C., et al. (2015). Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 160, 447–460. doi: 10.1016/j.cell.2015.01.002
O’Connell, R. J., Thon, M. R., Hacquard, S., Amyotte, S. G., Kleemann, J., Torres, M. F., et al. (2012). Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat. Genet. 44, 1060–1065. doi: 10.1038/ng.2372
Opperman, C. H., Bird, D. M., Williamson, V. M., Rokhsar, D. S., Burke, M., Cohn, J., et al. (2008). Sequence and genetic map of Meloidogyne hapla: a compact nematode genome for plant parasitism. Proc. Natl. Acad. Sci. U.S.A. 105, 14802–14807. doi: 10.1073/pnas.0805946105
Patrick, K. (2009). Five questions about Microsporidia. PLoS Pathog. 5:e1000489. doi: 10.1073/pnas.0805946105
Paul, H. A., Bomhof, M. R., Vogel, H. J., and Reimer, R. A. (2016). Diet-induced changes in maternal gut microbiota and metabolomic profiles influence programming of offspring obesity risk in rats. Sci. Rep. 6:20683. doi: 10.1038/srep20683
Peleg, A. Y., Hogan, D. A., and Mylonakis, E. (2010). Medically important bacterial-fungal interactions. Nat. Rev. Microbiol. 8, 340–349. doi: 10.1038/nrmicro2313
Peng, P., Peng, F., Bian, J., Xu, F., and Sun, R. (2011). Studies on the starch and hemicelluloses fractionated by graded ethanol precipitation from bamboo Phyllostachys bambusoides f. shouzhu Yi. J. Agric. Food Chem. 59, 2680–2688. doi: 10.1021/jf1045766
Peter, J. W., and Chandrawathani, P. (2005). Haemonchus contortus: parasite problem No. 1 from tropics - Polar Circle. Problems and prospects for control based on epidemiology. Trop. Biomed. 22, 131–137. doi: 10.1021/jf1045766
Popiel, I., Grove, D. L., and Friedman, J. M. (1989). Infective juvenile formation in the insect parasitic nematode Steinernema feltiae. Parasitology 99, 77–81.
Powers, T. O., Platzer, E. G., and Hyman, B. C. (1986). Large mitochondrial genome and mitochondrial DNA size polymorphism in the mosquito parasite, Romanomermis culicivorax. Curr. Genet. 11, 71–77. doi: 10.1017/S0031182000061047
Pozio, E., Casulli, A., Bologov, V. V., Marucci, G., and La Rosa, G. (2001). Hunting practices increase the prevalence of Trichinella infection in wolves from European Russia. J. Parasitol. 87, 1498–1501. doi: 10.1645/0022-33952001087[1498:HPITPO]2.0.CO;2
Pozio, E., De Meneghi, D., Roelke-Parker, M. E., and La Rosa, G. (1997). Trichinella nelsoni in carnivores from the Serengeti ecosystem, Tanzania. J. Parasitol. 83, 1195–1198. doi: 10.1645/0022-3395(2001)087[1498:HPITPO]2.0.CO;2
Qin, N., Yang, F., Li, A., Prifti, E., Chen, Y., Shao, L., et al. (2014). Alterations of the human gut microbiome in liver cirrhosis. Nature 513, 59–64. doi: 10.1038/nature13568
Reyes, A., Wu, M., McNulty, N. P., Rohwer, F. L., and Gordon, J. I. (2013). Gnotobiotic mouse model of phage-bacterial host dynamics in the human gut. Proc. Natl. Acad. Sci. U.S.A. 110, 20236–20241. doi: 10.1073/pnas.1319470110
Rödelsperger, C., Streit, A., and Sommer, R. J. (2013). Structure, Function and Evolution of the Nematode Genome. Hoboken, NJ: John Wiley & Sons. doi: 10.1073/pnas.1319470110
Sato, Y., and Toma, H. (1990). Strongyloides venezuelensis infections in mice. Int. J. Parasitol. 20, 57–62. doi: 10.1002/9780470015902.a0024603
Scheurwater, E., Reid, C. W., and Clarke, A. J. (2008). Lytic transglycosylases: bacterial space-making autolysins. Int. J. Biochem. Cell Biol. 40, 586–591. doi: 10.1016/j.biocel.2007.03.018
Schneider, R., Gollackner, B., Schindl, M., Tucek, G., and Auer, H. (2010). Echinococcus canadensis G7 (pig strain): an underestimated cause of cystic echinococcosis in Austria. Am. J. Trop. Med. Hyg. 82, 871–874. doi: 10.4269/ajtmh.2010.09-0639
Scott, K. P., Gratz, S. W., Sheridan, P. O., Flint, H. J., and Duncan, S. H. (2013). The influence of diet on the gut microbiota. Pharmacol. Res. 69, 52–60. doi: 10.1016/j.phrs.2012.10.020
Shanks, O. C., Kelty, C. A., Archibeque, S., Jenkins, M., Newton, R. J., McLellan, S. L., et al. (2011). Community structures of fecal bacteria in cattle from different animal feeding operations. Appl. Environ. Microbiol. 77, 2992–3001. doi: 10.1128/Aem.02988-10
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Snijders, A. M., Langley, S. A., Kim, Y. M., Brislawn, J. C., Noecker, C., Zink, E. M., et al. (2016). Influence of early life exposure, host genetics and diet on the mouse gut microbiome and metabolome. Nat. Microbiol. 2:16221. doi: 10.1038/nmicrobiol.2016.221
Sokol, H., Leducq, V., Aschard, H., Pham, H. P., Jegou, S., Landman, C., et al. (2017). Fungal microbiota dysbiosis in IBD. Gut 66, 1039–1048. doi: 10.1136/gutjnl-2015-310746
Sommer, F., Stahlman, M., Ilkayeva, O., Arnemo, J. M., Kindberg, J., Josefsson, J., et al. (2016). The gut microbiota modulates energy metabolism in the hibernating brown bear Ursus arctos. Cell Rep. 14, 1655–1661. doi: 10.1016/j.celrep.2016.01.026
Sommer, R. J. (2006). Pristionchus pacificus. Available at: https://www.ncbi.nlm.nih.gov/books/NBK19735/
Song, C., Wang, B., Tan, J., Zhu, L., Lou, D., and Cen, X. (2017). Comparative analysis of the gut microbiota of black bears in China using high-throughput sequencing. Mol. Genet. Genomics 292, 407–414. doi: 10.1007/s00438-016-1282-0
Spatafora, J. W., Chang, Y., Benny, G. L., Lazarus, K., Smith, M. E., Berbee, M. L., et al. (2016). A phylum-level phylogenetic classification of zygomycete fungi based on genome-scale data. Mycologia 108, 1028–1046. doi: 10.3852/16-042
Steinauer, M. L., Hanelt, B., Mwangi, I. N., Maina, M. G., Agola, L. E., Kinuthia, J. M., et al. (2008). Introgressive hybridization of human and rodent schistosome parasites in western Kenya. Mol. Ecol. 17, 5062–5074. doi: 10.1111/j.1365-294X.2008.03957.x
Sulakvelidze, A., Alavidze, Z., and Jr, J. G. (2001). Bacteriophage therapy. Antimicrob. Agents Chemother. 45, 649–659. doi: 10.1128/AAC.45.3.649-659.2001
Talens-Perales, D., Gorska, A., Huson, D. H., Polaina, J., and Marin-Navarro, J. (2016). Analysis of domain architecture and phylogenetics of family 2 glycoside hydrolases (GH2). PLoS One 11:e0168035. doi: 10.1371/journal.pone.0168035
Theodorou, M. K., Mennim, G., Davies, D. R., Zhu, W. Y., Trinci, A. P., and Brookman, J. L. (1996). Anaerobic fungi in the digestive tract of mammalian herbivores and their potential for exploitation. Proc. Nutr. Soc. 55, 913–926. doi: 10.1371/journal.pone.0168035
Thomas, F., Hehemann, J. H., Rebuffet, E., Czjzek, M., and Michel, G. (2011). Environmental and gut bacteroidetes: the food connection. Front. Microbiol. 2:93. doi: 10.3389/fmicb.2011.00093
Tisserant, E., Malbreil, M., Kuo, A., Kohler, A., Symeonidi, A., Balestrini, R., et al. (2013). Genome of an arbuscular mycorrhizal fungus provides insight into the oldest plant symbiosis. Proc. Natl. Acad. Sci. U.S.A. 110, 20117–20122. doi: 10.1073/pnas.1313452110
Torgerson, P. R., and Heath, D. D. (2003). Transmission dynamics and control options for Echinococcus granulosus. Parasitology 127, S143–S158. doi: 10.1073/pnas.1313452110
Trees, A. J. (1992). Onchocerca ochengi: mimic, model or modulator of O. volvulus? Parasitol. Today 8, 337–339. doi: 10.1017/S0031182003002932
Tremaroli, V., and Backhed, F. (2012). Functional interactions between the gut microbiota and host metabolism. Nature 489, 242–249. doi: 10.1038/nature11552
Tufariello, J. M., Mi, K., Xu, J., Manabe, Y. C., Kesavan, A. K., Drumm, J., et al. (2006). Deletion of the Mycobacterium tuberculosis resuscitation-promoting factor Rv1009 gene results in delayed reactivation from chronic tuberculosis. Infect. Immun. 74, 2985–2995. doi: 10.1128/IAI.74.5.2985-2995.2006
Tun, H. M., Mauroo, N. F., Yuen, C. S., Ho, J. C., Wong, M. T., and Leung, F. C. (2014). Microbial diversity and evidence of novel homoacetogens in the gut of both geriatric and adult giant pandas (Ailuropoda melanoleuca). PLoS One 9:e79902. doi: 10.1371/journal.pone.0079902
Turkenburg, J. P., Brzozowski, A. M., Svendsen, A., Borchert, T. V., Davies, G. J., and Wilson, K. S. (2009). Structure of a pullulanase from Bacillus acidopullulyticus. Proteins 76, 516–519. doi: 10.1002/prot.22416
Wang, L., Hatem, A., Catalyurek, U. V., Morrison, M., and Yu, Z. (2013). Metagenomic insights into the carbohydrate-active enzymes carried by the microorganisms adhering to solid digesta in the rumen of cows. PLoS One 8:e78507. doi: 10.1371/journal.pone.0078507
Wang, T., Chen, Z., Xie, Y., Hou, R., Wu, Q., Gu, X., et al. (2015). Prevalence and molecular characterization of Cryptosporidium in giant panda (Ailuropoda melanoleuca) in Sichuan province, China. Parasit. Vectors 8:344. doi: 10.1186/s13071-015-0953-8
Wang, X., Chen, W., Huang, Y., Sun, J., Men, J., Liu, H., et al. (2011). The draft genome of the carcinogenic human liver fluke Clonorchis sinensis. Genome Biol. 12:R107. doi: 10.1186/gb-2011-12-10-r107
Willems, M., Houthoofd, W., Claeys, M., Couvreur, M., Van Driessche, R., Adriaens, D., et al. (2005). Unusual intestinal lamellae in the nematode Rhabditophanes sp. KR3021 (Nematoda: Alloinematidae). J. Morphol. 264, 223–232. doi: 10.1002/jmor.10332
Williamson, V. M., and Hussey, R. S. (1996). Nematode pathogenesis and resistance in plants. Plant Cell 8, 1735–1745. doi: 10.1105/tpc.8.10.1735
Wu, G. D., Chen, J., Hoffmann, C., Bittinger, K., Chen, Y. Y., Keilbaugh, S. A., et al. (2011). Linking long-term dietary patterns with gut microbial enterotypes. Science 334, 105–108. doi: 10.1126/science.1208344
Xie, Y., Hoberg, E. P., Yang, Z., Urban, J. F. Jr., and Yang, G. (2017). Ancylostoma ailuropodae n. sp. (Nematoda: Ancylostomatidae), a new hookworm parasite isolated from wild giant pandas in Southwest China. Parasit. Vectors 10:277. doi: 10.1186/s13071-017-2209-2
Xue, Z., Zhang, W., Wang, L., Hou, R., Zhang, M., Fei, L., et al. (2015). The bamboo-eating giant panda harbors a carnivore-like gut microbiota, with excessive seasonal variations. mBio 6:e00022-15. doi: 10.1128/mBio.00022-15
Yamada, T., Letunic, I., Okuda, S., Kanehisa, M., and Bork, P. (2011). iPath2.0: interactive pathway explorer. Nucleic Acids Res. 39, W412–W415. doi: 10.1093/nar/gkr313
Yang, X., Cheng, G., Li, C., Yang, J., Li, J., Chen, D., et al. (2017). The normal vaginal and uterine bacterial microbiome in giant pandas (Ailuropoda melanoleuca). Microbiol. Res. 199, 1–9. doi: 10.1016/j.micres.2017.01.003
Zhang, L., Wu, Q., Hu, Y., Wu, H., and Wei, F. (2015). Major histocompatibility complex alleles associated with parasite susceptibility in wild giant pandas. Heredity 114, 85–93. doi: 10.1038/hdy.2014.73
Zhang, L., Yang, X., Wu, H., Gu, X., Hu, Y., and Wei, F. (2011). The parasites of giant pandas: individual-based measurement in wild animals. J. Wildl. Dis. 47, 164–171. doi: 10.7589/0090-3558-47.1.164
Zhang, W., Yie, S., Yue, B., Zhou, J., An, R., Yang, J., et al. (2012). Determination of Baylisascaris schroederi infection in wild giant pandas by an accurate and sensitive PCR/CE-SSCP method. PLoS One 7:e41995. doi: 10.1371/journal.pone.0041995
Zhao, G. H., Xu, M. J., and Zhu, X. Q. (2013). Identification and characterization of microRNAs in Baylisascaris schroederi of the giant panda. Parasit. Vectors 6:216. doi: 10.1186/1756-3305-6-216
Zhernakova, A., Kurilshikov, A., Bonder, M. J., Tigchelaar, E. F., Schirmer, M., Vatanen, T., et al. (2016). Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 352, 565–569. doi: 10.1126/science.aad3369
Zhou, X., Xie, Y., Zhang, Z. H., Wang, C. D., Sun, Y., Gu, X. B., et al. (2013). Analysis of the genetic diversity of the nematode parasite Baylisascaris schroederi from wild giant pandas in different mountain ranges in China. Parasit. Vectors 6:233. doi: 10.1186/1756-3305-6-233
Zhu, L., Wu, Q., Dai, J., Zhang, S., Wei, F., and Wei, F. (2011). Evidence of cellulose metabolism by the giant panda gut microbiome. Proc. Natl. Acad. Sci. U.S.A. 108, 17714–17719. doi: 10.1073/pnas.1017956108
Keywords: bacteria, fungi, bacteriophages, helminths, giant pandas, metagenomic sequencing
Citation: Yang S, Gao X, Meng J, Zhang A, Zhou Y, Long M, Li B, Deng W, Jin L, Zhao S, Wu D, He Y, Li C, Liu S, Huang Y, Zhang H and Zou L (2018) Metagenomic Analysis of Bacteria, Fungi, Bacteriophages, and Helminths in the Gut of Giant Pandas. Front. Microbiol. 9:1717. doi: 10.3389/fmicb.2018.01717
Received: 01 February 2018; Accepted: 10 July 2018;
Published: 31 July 2018.
Edited by:
Rakesh Sharma, Institute of Genomics and Integrative Biology (CSIR), IndiaReviewed by:
Ji-Hoon Lee, Chonbuk National University, South KoreaCopyright © 2018 Yang, Gao, Meng, Zhang, Zhou, Long, Li, Deng, Jin, Zhao, Wu, He, Li, Liu, Huang, Zhang and Zou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Huang, cGFuZGF5YXJkQGhvdG1haWwuY29t Hemin Zhang, d29sb25nX3pobUAxMjYuY29t Likou Zou, em91bGlrb3VAc2ljYXUuZWR1LmNu
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.