 Marie E. Kroeger1
Marie E. Kroeger1 Tom O. Delmont2
Tom O. Delmont2 A. M. Eren2,3
A. M. Eren2,3 Kyle M. Meyer4
Kyle M. Meyer4 Jiarong Guo5
Jiarong Guo5 Kiran Khan1
Kiran Khan1 Jorge L. M. Rodrigues6
Jorge L. M. Rodrigues6 Brendan J. M. Bohannan4
Brendan J. M. Bohannan4 Susannah G. Tringe7
Susannah G. Tringe7 Clovis D. Borges8
Clovis D. Borges8 James M. Tiedje5
James M. Tiedje5 Siu M. Tsai8
Siu M. Tsai8 Klaus Nüsslein1*
Klaus Nüsslein1*- 1Department of Microbiology, University of Massachusetts Amherst, Amherst, MA, United States
- 2Department of Medicine, University of Chicago, Chicago, IL, United States
- 3Josephine Bay Paul Center, Marine Biological Laboratory, Woods Hole, MA, United States
- 4Institute of Ecology and Evolution, University of Oregon, Eugene, OR, United States
- 5Center for Microbial Ecology, Michigan State University, East Lansing, MI, United States
- 6Department of Land, Air, and Water Resources, University of California, Davis, Davis, CA, United States
- 7DOE Joint Genome Institute, Walnut Creek, CA, United States
- 8Centro de Energia Nuclear na Agricultura, University of São Paulo, Piracicaba, Brazil
Deforestation in the Brazilian Amazon occurs at an alarming rate, which has broad effects on global greenhouse gas emissions, carbon storage, and biogeochemical cycles. In this study, soil metagenomes and metagenome-assembled genomes (MAGs) were analyzed for alterations to microbial community composition, functional groups, and putative physiology as it related to land-use change and tropical soil. A total of 28 MAGs were assembled encompassing 10 phyla, including both dominant and rare biosphere lineages. Amazon Acidobacteria subdivision 3, Melainabacteria, Microgenomates, and Parcubacteria were found exclusively in pasture soil samples, while Candidatus Rokubacteria was predominant in the adjacent rainforest soil. These shifts in relative abundance between land-use types were supported by the different putative physiologies and life strategies employed by the taxa. This research provides unique biological insights into candidate phyla in tropical soil and how deforestation may impact the carbon cycle and affect climate change.
Introduction
The Amazon Basin contains the largest continuous tropical rainforest on the planet (Laurance et al., 2001) and is home to unparalleled levels of plant and animal diversity (Da Silva et al., 2005). Rainforests in this region are under threat from human activities, with the largest single threat being conversion to agriculture (Soares-Filho et al., 2006). Several studies have illustrated that human activities in the tropics such as land use change have adverse effects on macro-organismal biodiversity (Dale et al., 1994; McKinney and Lockwood, 1999; Sala et al., 2000), and more recently it became apparent that belowground microbial communities are altered as well (Rodrigues et al., 2013; Paula et al., 2014; Ranjan et al., 2015; Meyer et al., 2017). Soil microbial biodiversity in the Amazonian tropics is primarily known through 16S rRNA and ITS gene sequencing surveys (Borneman and Triplett, 1997; Peixoto et al., 2002; Jesus et al., 2009; Tripathi et al., 2012; Mueller et al., 2014), with only limited genomic and metagenomic surveys (Navarrete et al., 2015; Meyer et al., 2017).
Phylogenetic analyses based on 16S rRNA gene sequences organize bacterial and archaeal life into more than 60 phyla. Half of these phyla have no cultured representatives, yet these ‘candidate’ phyla represent a substantial portion of the tree of life (Hugenholtz, 2002; Rinke et al., 2013; Hug et al., 2016). Until recently, these microorganisms have only been detected through surveys of 16S rRNA gene surveys of environmental sequencing (Dojka et al., 2000; Hedlund et al., 2014), which provided very little direct information about their ecology or metabolic capabilities. With the development of shotgun metagenomics, insights into the diversity and functional potential of microbial communities have drastically increased. Expanding on that advance, genome-resolved metagenomics (Alneberg et al., 2014; Imelfort et al., 2014; Eren et al., 2015; Kang et al., 2015; Sangwan et al., 2016) and single-cell genome sequencing (Gawad et al., 2016) were developed, and can now be used to characterize the genomic content of microbial populations without the need for cultivation. Metagenome-assembled genomes (MAGs) have been characterized from a variety of environments, including recently from soil (Wrighton et al., 2012; Brown et al., 2015; Delmont et al., 2015; Sekiguchi et al., 2015; Butterfield et al., 2016; Hu et al., 2016; White et al., 2016). By integrating large sequencing datasets from environmental samples into reconstructed genomic composition, MAGs have increased the resolution at which microbial ecologists can understand a microorganism’s functional potential.
The use of MAGs has increased our understanding of candidate phyla in the environment (Siegl et al., 2011; Wrighton et al., 2012; Kantor et al., 2013; McLean et al., 2013; Probst et al., 2017). A study by Brown et al. (2015) identified a distinct radiation from known bacteria that altered our view of the tree of life and was termed the candidate phyla radiation (CPR) (Hug et al., 2016). Altogether, the CPR lineages might encompass over 70 phyla and two superphyla, Microgenomates, and Parcubacteria (Anantharaman et al., 2016). CPR genomes are generally streamlined (<1.5 Mb) with limited biosynthetic capabilities suggesting that they rely on other organisms (micro- or macro-) for their essential or additional metabolic needs (Rinke et al., 2013; Kantor et al., 2013; Brown et al., 2015; Probst et al., 2017). Until now, most CPR genomes have been characterized from within subsurface environments, limiting our understanding of this major bacterial group in highly heterogeneous environments such as soil.
Soil is regarded as one of the most diverse ecosystems on Earth, and characterizing even the most abundant MAGs has been considered an outstanding challenge due to the exceptional levels of soil biodiversity (Gans et al., 2005; Howe et al., 2014; Prosser, 2015; White et al., 2016). Few studies have successfully performed genome-resolved metagenomics on soil and among those, some have perturbed the community composition prior to sampling to lower the complexity of the microbial communities (Delmont et al., 2015; Yeoh et al., 2015), while others have assembled MAGs without manipulation (Butterfield et al., 2016; White et al., 2016; Tas et al., 2018). Despite these recent metagenomic accomplishments, the genomic content of most microbial populations inhabiting soil from the Amazon basin and elsewhere have yet to be characterized.
Here, we analyzed ten extensive tropical soil metagenomes (five from pristine rainforest soil and five from an adjacent cattle pasture) to understand the microbial community composition and how deforestation may affect these communities. In addition, we also characterized 28 MAGs from the rare soil biosphere for their functional potential in these tropical rainforest soils, because there is no knowledge of the metabolic capabilities of these groups either in any soil (Microgenomates and Parcubacteria) or in tropical soils (Rokubacteria). We also described shifts in the soil chemistry, and in the soil microbial functional potential. The sequencing depth of our dataset (6.4 billion shotgun metagenomic reads) was critical in the successful reconstruction of microbial genomes from this highly complex biological system. We were able to characterize MAGs encompassing 10 phyla, including CPR superphyla Microgenomates, and Parcubacteria, and from ubiquitous soil bacteria, Acidobacteria. Using comparative genomic approaches, we elucidated the unique features and potential metabolic capabilities of various tropical soil microbial populations. Not only does our study begin to depict the genomic and functional potential of poorly understood phyla, but it also increases our understanding of understudied tropical microbial biodiversity.
Materials and Methods
Site Description, Sampling, and Soil Chemistry
This study was performed at the Amazon Rainforest Microbial Observatory site, established in 2009 (10°10′5″ S and 62°49′27″ W). Sample design and soil chemical analyses were performed as specified previously (Rodrigues et al., 2013; Meyer et al., 2017). Briefly, in April 2010 soil cores collected from primary rainforest and a 38-year-old converted pasture (five each). The pasture site was initially a pristine rainforest. After selective logging of timber trees all remaining vegetation was cut at the end of the raining season and left to dry. At the end of the 3 months long dry season the remaining biomass was burned. The pasture site was then aerially seeded with Urochloa brizantha. No herbicides, tillage, or chemical fertilizers are used in this process. Weeds are controlled by fire when necessary (roughly every 2 years). There are many soil studies at Fazenda Nova Vida ranch in Rondonia and overall the pasture soil has an increase in soil bulk density especially in the top 0–5 cm of soil. The soil type for both pasture and forest sites are Ultisols (yellow–red podzolic soils) (de Moraes et al., 1996). Soil cores were sampled to a depth of 10 cm using 5 cm diameter PVC tubes and homogenized. Soil cores were frozen on the spot, transported on dry ice to the laboratory and stored at −80°C until further processing for DNA extraction. Sub-samples of each soil, stored at 4°C, were also analyzed for pH, total C and N, and other elemental analyses as described previously (Rodrigues et al., 2013).
Nucleic Acid Extraction and Sequencing
Soil DNA was extracted following the same protocol as Rodrigues et al. (2013). DNA was sequenced as described in Meyer et al. (2017) at the United States Department of Energy – Joint Genome Institute (JGI) on the Illumina HiSeq platform across 21 lanes to produce 6,366,557,730 paired-end reads of 150 bp.
Bioinformatic and Statistical Analysis of Metagenomes
Raw reads were uploaded to MG-RAST (Meyer et al., 2008). Then, functional and taxonomic annotations were obtained as described in detail by Meyer et al. (2017), except that in this study the sequences were not rarefied. Instead, the raw annotation counts for both function and taxonomy were analyzed using DESeq2 package in R version 3.4.2 (Love et al., 2014; R Core Team, 2016). Microbial community cluster analysis and visualization were done using the vegan R package (R Core Team, 2016; Oksanen et al., 2018).
Metagenomic Assembly
All raw reads of metagenome samples were quality trimmed using fastq-mcf (Aronesty, 2011) and paired-end reads assembled using FLASH (Magoč and Salzberg, 2011) as described in Guo et al. (2016). The reads were pooled and assembled with MEGAHIT (Li et al., 2015) (−m 1.38e12 −t 32 −l 400 –k-mins 35 –k-max 75 –cpu-only), which used 42.4% of all reads resulting in 36,786,646 contigs with a total length of ∼27 Gbp. For the genome reconstruction, we only used contigs longer than 5 kb decreasing the total contigs to 79,226 and total nucleotides to 627 Mb. The number of reads used to map each sample to the filtered metagenome co-assembly is detailed in Supplementary Table S1.
Genome Reconstruction and Annotation
All genome reconstructions followed the protocol outlined in detail by Eren et al. (2015). Briefly, quality filtered reads for each metagenome were mapped to the co-assembly of short reads (>5 kb) using bowtie2 with default parameters (Langmead and Salzberg, 2012). The sequence alignment map (SAM) files were converted to a binary format (BAM) that were then imported into the Anvi’o 2.0 pipeline along with the metagenomic co-assembly for genomic binning using sequence composition and differential mean coverage (Li et al., 2009; Eren et al., 2015). An initial binning using CONCOCT (40 clusters) was performed (Alneberg et al., 2014) followed by human-guided manual binning and curation of MAGs using the anvi’o interactive interface (see Eren et al., 2015 for details). MAGs were annotated using PATRIC (Wattam et al., 2014). The completion of the genome and the redundancy percentage (i.e., more than one single copy gene is present) for the genomic bins was determined using the 139 single copy genes used in anvi’o except for the MAGs designated as Microgenomates or Parcubacteria, which used the 44 single copy genes outlined by Brown et al. (2015). The unique features found in each MAG were determined using the PATRIC FIGFam protein family sorter. The features were then checked against reference genomes to ensure they could not be found. For each phylum or order, there is a different size set of reference genomes; 186 for Acidobacteria, 55 for Melainabacteria, 75 for Rokubacteria, 859 for Microgenomates, 4 for Pacebacteria, and 236 for Parcubacteria. The potential metabolism of the organisms based on the MAG annotations was determined by the PATRIC comparative pathway tool, by searching for specific functional annotations, and using BLAST (Altschul et al., 1997). All NCBI genome comparisons were completed in PATRIC by searching for the genomes, creating a genome group, and comparing the genomes in the protein family sorter with FIGFams and the comparative pathway tool. The new genome reporting standards and metadata for MAGS as outlined by Bowers et al. (2017) can be found in Supplementary Table S2. tRNAscan-SE v.2.0 was used to determine the number of tRNAs in all MAGs, except Amazon_FNV_2010_0_2_1 and Amazon_FNV_2010_0_4 (Lowe and Chan, 2016). These two MAGs used Aragorn to check for tRNAs due to large file sizes that tRNAscan-SE online could not process (Laslett and Canback, 2004). CheckM was used to determine if 16S or 18S was present in each MAG (Parks et al., 2015). Lateral gene transfer (lgt) in the Amazon Rokubacteria MAG was predicted using CompareM lgt-di function with default settings1. The heatmap visualizing the absolute abundance of each MAG in each metagenome sample was made using the heatmap.2 from the gplots package in R (R Core Team, 2016; Warnes et al., 2016).
Pangenome Analysis
All pangenomic analyses were performed in the anvi’o 2.0 workflow for microbial pangenomes using the NCBI-blast analysis2. The “core” included protein clusters detected in at least 66% of the genomes. Protein clusters only detected in one genome were considered “unique.”
Phylogenetic Analysis
All phylogenetic analyses were performed using MEGA 7.0 (Kumar et al., 2016) using a modified protocol from Brown et al. (2015). Briefly, concatenated gene trees were generated by aligning each gene in MEGA using MUSCLE (Edgar, 2004). Then, the gene alignments were trimmed using phyutility (−clean 0.95) and concatenated (−concat) (Smith and Dunn, 2008). Maximum likelihood phylogenetic trees with 500 bootstrap replications were made from the concatenated gene alignments using the Poisson model with default parameters. The phylogenetic trees were visualized in ITOL (Letunic and Bork, 2016). CompareM was used to determine the mean amino acid identity between related microbial genomes (see footnote 1).
Geochemical Data Statistics
Geochemical data from the soil cores used for metagenome sequencing were analyzed using a MANOVA test in R version 3.2.4 (R Core Team, 2016).
Data Availability
Contigs for all MAGs are available on figshare3 and NCBI BioProject PRJNA432584. The raw sequence FASTA files for the ten metagenomes are available at JGI (Supplementary Table S1).
Results
Metagenomes of Rainforest and Cattle Pasture Soil Exhibit Distinct Taxonomic and Functional Profiles
With land-use change from rainforest to pasture there was a significant change in soil geochemistry with increases in acidic potential (total soil acidity), and in organic matter, copper, iron, and zinc concentrations in the cattle pasture, while boron and aluminum concentrations were significantly higher in rainforest soil (Supplementary Table S3). The dominant phyla in both the rainforest and cattle pasture soil metagenomes were Proteobacteria, Actinobacteria, Firmicutes, Acidobacteria, and Verrucomicrobia. Beyond this similarity, the microbial community composition changes drastically (Figure 1) with the abundance of 13 out of 34 identified microbial phyla significantly changing between rainforest and cattle pasture soils. Thaumarchaeota, a known ammonia-oxidizing archaeon, almost disappeared during rainforest-to-pasture conversion (loss of 99.5%). Other phyla significantly decreased by deforestation were Crenarchaeota, Nitrospirae, Gemmatimonadetes, Fusobacteria, Aquificae, Lentisphaerae, and Korarchaeota (Supplementary Figure S1 and Supplementary Table S4).
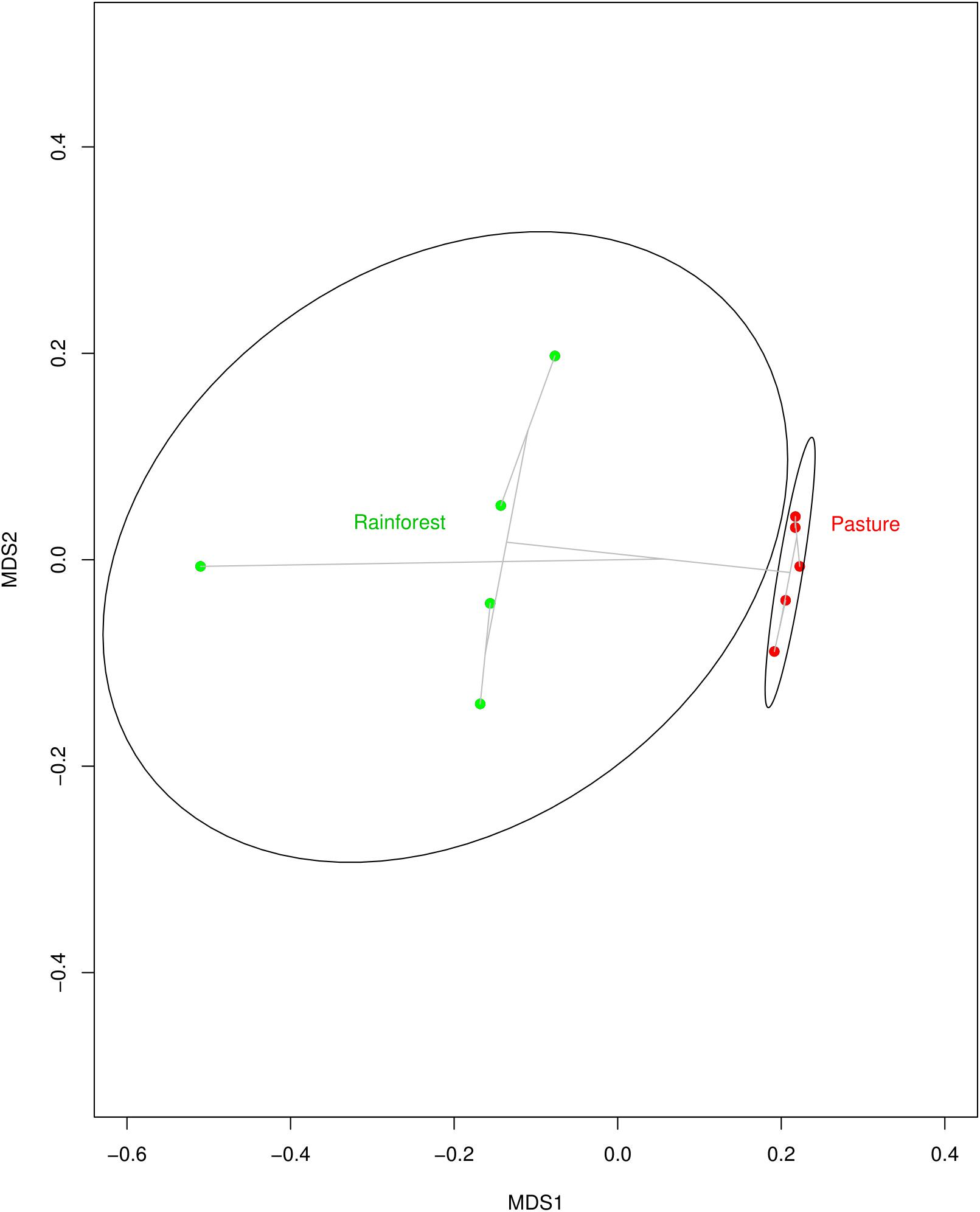
FIGURE 1. Multidimensional scaling (MDS) plot indicating how similar soil microbial communities from Rainforest samples (green dots) cluster together, but away from communities of the Pasture samples (red dots). The clustering is visualized with gray lines connecting samples with similar communities. The MDS plot was created using Bray–Curtis dissimilarity among samples and an average-linkage algorithm for community clustering.
In addition to taxonomic shifts in the microbial community, there were significant alterations to functional groups between rainforest and pasture soil. Genes related to carbohydrate metabolism, dormancy and sporulation, and regulation and cell signaling were all significantly increased in the pasture compared to rainforest, while genes related to transcription and vitamin production were significantly higher in rainforest soil (Figure 2). Additionally, significant shifts were found in carbon cycling genes. Lignin degradation genes including a superoxide dismutase were significantly higher in rainforest soils. All methyl coenzyme M reductase genes that are essential for methanogenesis were significantly higher in pasture while the particulate methane monooxygenase genes important to methane oxidation were significantly higher in rainforest. Additionally, key genes involved carbon assimilation by methanotrophs using the serine-glyoxylate cycle were significantly higher in rainforest and the ribulose monophosphate pathway genes were significantly higher in pasture soils. Overall, there was a predominance of significantly higher genes involved in monosaccharide, oligosaccharide, and polysaccharide metabolism, along with fermentation in pasture (Supplementary Table S5).
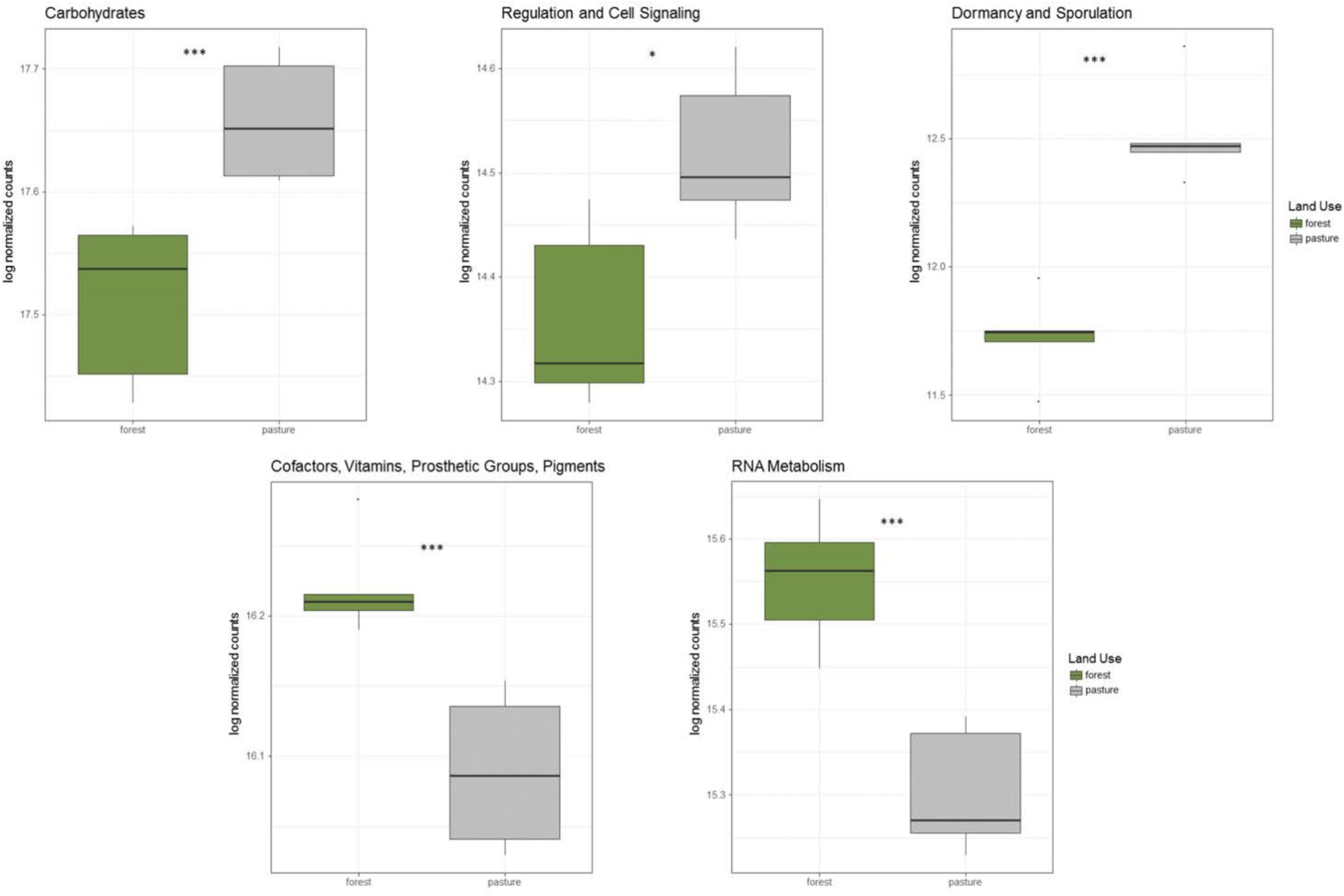
FIGURE 2. Significantly altered abundancies of functional gene groups between rainforest and pasture soils. These log-normalized counts were determined using DESeq2 (p < 0.05∗, p < 0.01∗∗, p < 0.001∗∗∗).
Genome-Resolved Metagenomics Exposed Novel Genomes From the Soil Rare Biosphere
Using contigs longer than 5 kb from each metagenome, we characterized 28 MAGs representing three of the dominant phyla (Proteobacteria, Acidobacteria, Verrucomicrobia) as well as other lineages (TM6, Bacteroidetes, Melainabacteria, Chloroflexi, Chlamydiae, Microgenomates, Parcubacteria, Rokubacteria) identified as members of the rare biosphere (<0.01%) based on iTag sequence annotations (Supplementary Table S6). The genome length ranged from 0.96 to 7.44 Mb with an average of 2.5 Mb due to many streamlined genomes from candidate phyla MAGs. The GC content ranged from 30.77 to 67.72% covering a large portion of known bacterial GC content (Table 1 and Supplementary Table S2). If we consider genomes that are shorter than 2 Mb to be streamlined, then the average GC content is 38.12% with a range of 30.77–43.41%. Genomes longer than 2 Mb have an average GC content of 54.26% with a range of 42.76–67.72%. Although there are not many replicates at the MAG level, we observed a clear effect of land-use change on the abundance of specific taxonomic groups. MAGs found only in the pasture soils belong to the Microgenomates, Parcubacteria, Verrucomicrobia, Chlamydiae, and Melainabacteria, while MAGs of Rokubacteria and Chloroflexi were in much higher abundance in rainforest soils. Acidobacteria MAGs were found in both land-use types, with subdivisions 3 and 4 only found in pasture sites (Figure 3). Beyond the new insights we gained regarding the effects of land usage on the community composition of Amazon soils, the MAGs we have characterized correspond to many understudied bacterial lineages that deserve detailed descriptions of their unique functional features in the context of relevant genomic databases.
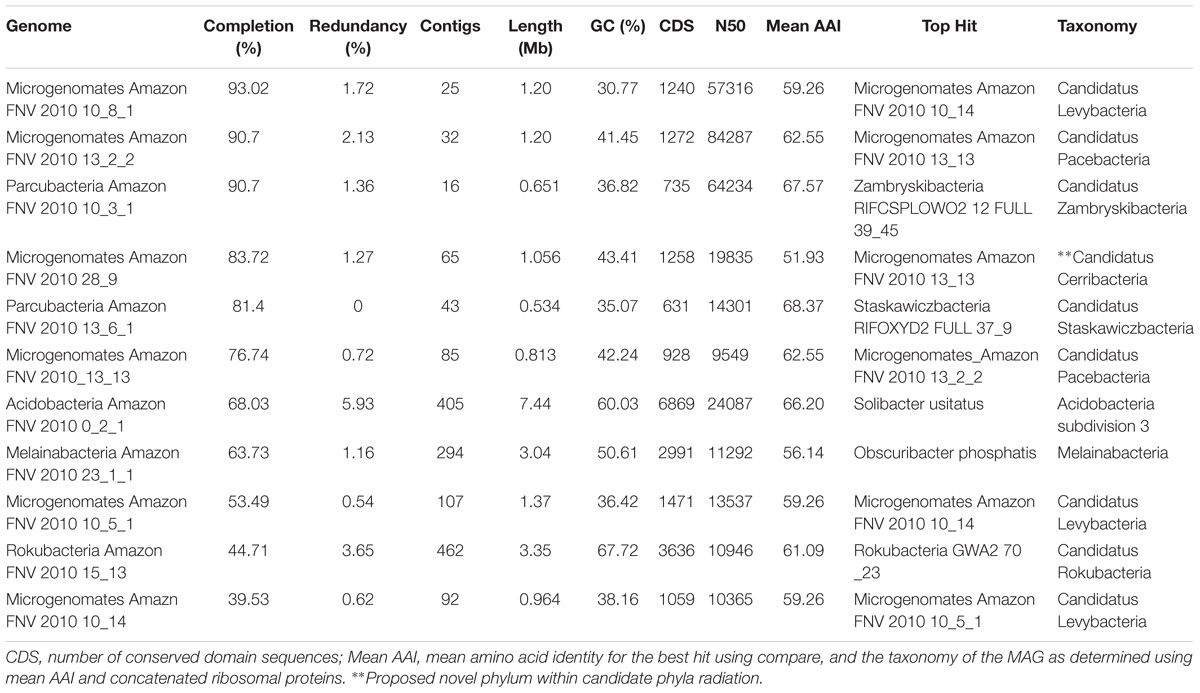
TABLE 1. Genome characteristics for the 11 metagenome-assembled genomes (MAGs) discussed thoroughly in this study.
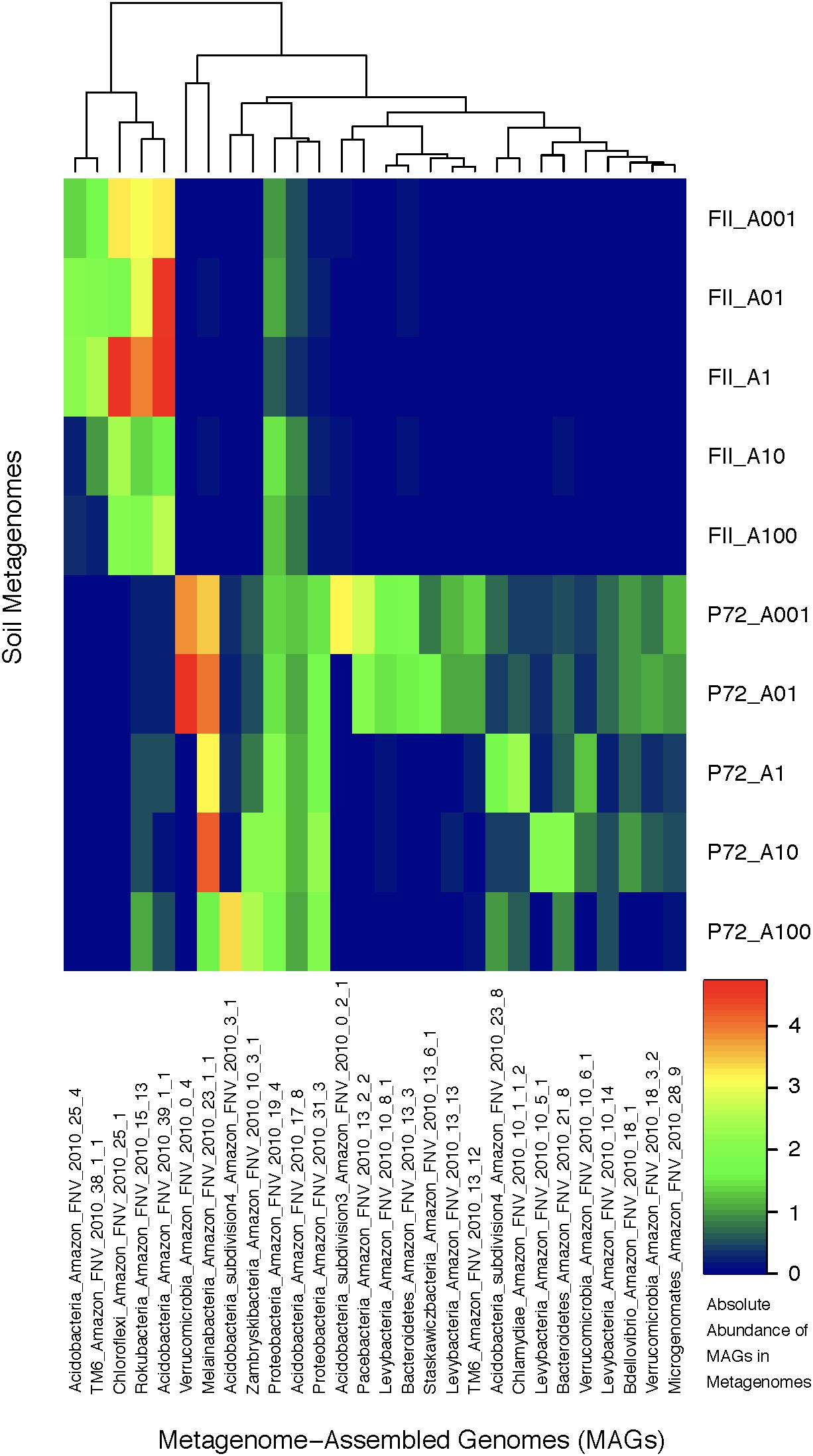
FIGURE 3. A heatmap of the absolute abundance of MAGs in each metagenome sample from this study. Absolute abundance means the number of the specific genome found in each soil metagenome; an absolute abundance of four would mean that four instances of that genome are found in that specific metagenome. The x-axis depicts the MAGs labeled with the following scheme: Taxonomic association, location of sampling (Amazon FNV), year of sampling (2010), and bin number from anvi’o. The y-axis are the soil metagenome samples for rainforest (FII) or cattle pasture (P72), differentiated by the location on a 100 m transect indicating the distance from the starting location in meters. The clustering at the top of the graph is based on hierarchical clustering.
Amazon Acidobacteria Involved in Pasture Hydrocarbon Metabolism
The six Acidobacteria MAGs represented five of the currently 26 described subdivisions (Supplementary Figure S2). Amongst these are MAGs from three of the most well characterized subdivisions 1, 3, and 4, and from the lesser known subdivision 13, which has no cultured representatives. Many Acidobacteria subdivisions do not currently have cultured representatives; therefore, molecular marker genes are used to determine the different subdivisions. For Acidobacteria the 16S rRNA gene is used, which were not detected in many of the Acidobacteria MAGs from this study. Unfortunately, concatenated ribosomal proteins are not able to provide clarity, which leaves some of the Acidobacteria MAGs from this study in undetermined subdivisions. Only Acidobacteria with confirmed subdivisions were analyzed for unique potential functions. An interesting feature of the Amazon Acidobacteria is that five out of six have a 4-hydroxybenzoate transporter found in only nine other Acidobacteria MAGs (out of 186 other MAGs publicly available on NCBI analyzed). The Amazon Acidobacteria MAG from subdivision 3 (Bin_0_2_1; 68% completion; 5.93% redundancy) exhibited the highest completion and was further characterized. This MAG has the functional potential to metabolize many carbon substrates including aromatic compounds, starch, glycogen, cellobiose, glucose, fructose, sucrose, maltose, and trehalose. Furthermore, this Amazon Acidobacterium has genes involved in nitrogen and sulfur cycling with the potential to reduce nitrite, nitric-oxide, sulfate, and sulfite.
Melainabacteria Involved in Pasture Carbon Cycling
One Melainabacteria MAG corresponded to a newly discovered lineage within the candidate order Obscuribacter (Supplementary Figure S3 and Table 1). This MAG contains 72 unique FIGFams (i.e., protein encoding genes) when compared to the 55 Melainabacteria available in NCBI. Some features of interest were a nitric-oxide reductase (quinol-dependent) and a ubiquinol-cytochrome c reductase only found in Candidatus Obscuribacter phosphatis and the Amazon Melainabacteria. Similar to O. phosphatis, the Amazon Melainabacteria has the genetic potential to metabolize simple carbon sources including fructose, glucose, starch, glycogen, and maltose. However, only the Amazon Melainabacteria MAG contains genes to synthesize cellulose from UDP-alpha-D-glucose and can potentially metabolize cellobiose, maltose, and xylose. This Amazon Melainabacteria MAG also contains genes for both aerobic and anaerobic respiration including a complete respiratory chain like O. phosphatis.
Amazon Rokubacteria Potentially Oxidize Methane in Rainforest Soil
The Amazon Rokubacteria MAG (Amazon-R-15-13) contains 103 unique FIGFams when compared to the 75 available Rokubacteria MAGs from NCBI and the recently published single cell genomes from Becraft et al. (2017) (Supplementary Table S7). Among the features unique to Amazon-R-15-13 is a complete particulate methane monooxygenase (pmmo) necessary for methane oxidation to methanol. A phylogenetic analysis of the pmmo alpha subunit (pmoA) found the Amazon-R-15-13 pmoA to be unique and most closely related to pmoA genes from Streptomyces thermoautotrophicus, Nocardioides luteus, and Smaragdicoccus niigatensis (Figure 4). The lateral gene transfer (LGT) analysis found that LGT-predicted genes are spread across many contigs supporting proper binning and none of the annotated genes were related to methane cycling (Supplementary Table S8).
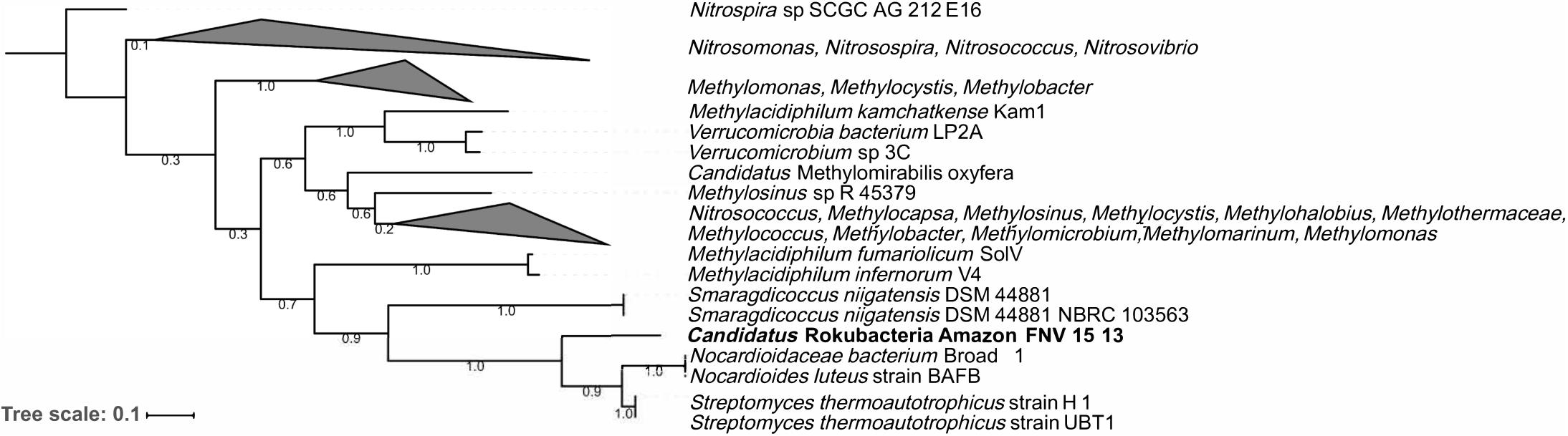
FIGURE 4. Phylogenetic tree of the particulate methane monooxygenase alpha subunit (pmoA). The pmoA sequences identified with PATRIC were used to create a maximum likelihood tree with 500 bootstrap replications. The pmoA from this study’s Amazon Rokubacteria MAG is indicated in bold. The numbers on each node represent the bootstrap support.
A meta-analysis of all 52 Rokubacteria MAGs available in NCBI and of our Amazon Rokubacteria MAG revealed several genomic features supporting the metabolic potential to metabolize larger hydrocarbons. The majority of soil Rokubacteria MAGs (17 out of 22) have P450 enzymes necessary for the oxidation of large hydrocarbons. Rokubacteria MAGs (16 out of 53) contain genes for alkane 1-monooxygenase enzymes to oxidize shorter hydrocarbons. Following hydrocarbon metabolism, Rokubacteria MAGs have the potential for beta-oxidation of fatty acids. These functional features suggest that soil Rokubacteria have the potential to metabolize hydrocarbons followed by β-oxidation of fatty acids, which then feeds into the TCA cycle and the electron transport chain (Supplementary Table S9).
Enabled by the expanded dataset of 53 Rokubacteria MAGs, a conceptual metabolic model was formed based on annotated genes (Figure 5 and Supplementary Table S10). This model is based on gene annotations without gap-filling and extends our knowledge of phylum Candidatus Rokubacteria. Rokubacteria MAGs have many metabolic capabilities including hydrocarbon metabolism, but the hydrocarbon methane can follow a different fate. Methane can be oxidized through three enzymatic steps to formate, which can then follow several paths: (1) it can be converted to acetyl-CoA using the tetrahydrofolate (H4F) pathway, (2) it can be oxidized to carbon dioxide via formate dehydrogenase, or (3) it can be oxidized to carbon dioxide and hydrogen using a formate hydrogen lyase (Figure 5). When methane is not converted to acetyl-CoA via the H4F pathway, Rokubacteria MAGs have the functional potential to utilize the serine cycle for carbon assimilation (Supplementary Table S9). Rokubacteria MAGs have the genomic potential to produce acetate and ethanol via the Wood–Ljungdahl pathway. There are genes for an electron transport Rnf complex in 20 Rokubacteria MAGs that is involved in energy generation. This phylum has the genomic potential to use all three central metabolic pathways and although there are signs of aerobic respiration, such as cytochrome c oxidase, there are also indications of anaerobic respiration including the potential to denitrify nitrate to nitrous oxide (Supplementary Table S9). Another possible electron acceptor is sulfate using dissimilatory sulfate reduction; although, there are also a few MAGs with assimilatory sulfate reduction genes (Supplementary Table S9). The wide variety of carbon substrates, energy sources, and electron acceptors may help this phylum survive in varying environmental conditions.
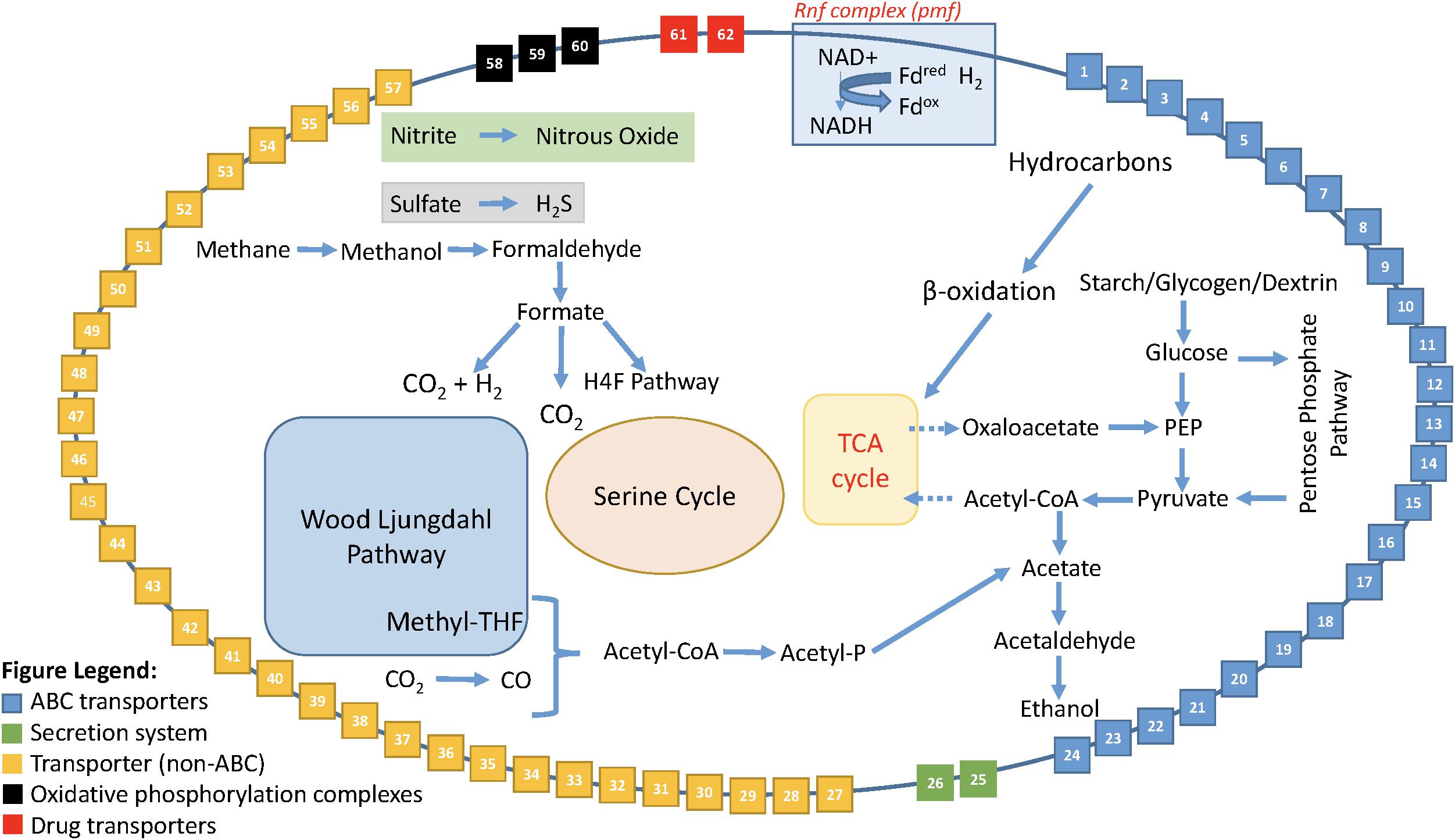
FIGURE 5. Model of the metabolic reconstruction of Candidatus Rokubacteria based on PATRIC annotations. Indicated are the major metabolic processes including central carbon metabolism and energy generation in the center of the cell. The cell wall includes the various transporters, secretion systems, and electron transport complexes labeled by number. All red asterisks indicate that a particular function is found only in the Amazon Rokubacteria MAG when compared to the other 52 MAGs available for Rokubacteria. The legend for the cell wall box numbers is found in Supplementary Table S10.
Amazon Microgenomates Retain DNA Mismatch Repair Machinery
Our database contains six MAGs from the CPR superphylum Microgenomates, which correspond to the first soil genomic representatives of this large branch of the tree of life. Two of these MAGs (Amazon-Pacebacteria-13-2-2 and Amazon-Levybacteria-10-8-1) are over 90% complete (Table 1). Phylogenetic analyses uncovered that the three genomes Amazon-Levybacteria-10-8-1, Amazon-Levybacteria-10-5-1, and Amazon-Levybacteria-10-14 are most closely related to each other, and fall within Candidatus Levybacteria. MAGs Amazon-Pacebacteria-13-2-2, Amazon-Pacebacteria-13-13, and Amazon-Microgenomates-28-9 are most closely related to each other with Amazon-Pacebacteria-13-2-2 and Amazon-Pacebacteria-13-13 forming a basal monophyletic clade with Candidatus Pacebacteria. Amazon-Microgenomates-28-9 likely represents a novel candidate phylum within the superphylum Microgenomates based on its low mean amino acid identity compared to known phyla and its phylogenetic placement between Pacebacteria and Chisholmbacteria. Thus, we propose the name Candidatus Cerribacteria (Table 1 and Supplementary Figure S4).
An extensive analysis across all Microgenomates MAGs (n = 859) available on NCBI revealed only 13 genomes with the essential DNA mismatch repair machinery (mutS, mutL). In contrast, we found mutS, mutL, and uvrD in all but one of our Amazon Microgenomates MAGs (Supplementary Table S11). Although almost none of the Microgenomates MAGs (13 out of 859) analyzed have mutS/L, uvrD is found in all the phyla within the superphylum Microgenomates. Genes involved in DNA replication initiation (DnaA, DnaB) are still present, which supports that these organisms can possibly still replicate independently from a host.
Amazon Pacebacteria Have Expanded Carbohydrate Metabolism
An additional three genomes reconstructed from Amazon soil are most closely related to Candidatus Pacebacteria, one of which is thought to represent a novel phylum for which we propose the name Candidatus Cerribacteria. A pangenomic analysis of the four available Pacebacteria MAGs together with the three novel Amazon MAGs revealed a “core” of gene clusters. This “core” encodes for proteins involved in mannose metabolism, one-carbon pool by folate, glycolysis, and an ATP synthase (Supplementary Figure S5). This “core” provides a base understanding of their metabolism, which can utilize basic sugars like glucose and mannose via glycolysis and the pentose phosphate pathway to produce lactic acid. This phylum completely lacks the TCA cycle and is unable to produce acetyl-CoA or oxaloacetate to feed into the cycle. All genomes contain genes to use the one carbon pool by folate pathway, but none of them contain genes to perform folate biosynthesis except Amazon-Pacebacteria-13-2-2. The three Amazon Microgenomates MAGs most closely related to Pacebacteria formed a grouping of their own with 180 gene calls found in at least two genomes including genes for proteins essential for starch/glycogen metabolism. Additionally, Amazon-Pacebacteria-13-2-2 encoded a cellulase protein necessary for cellulose metabolism.
Amazon Parcubacteria Are Phylogenetically Distinct, but Functionally Redundant
Two Amazon MAGs fall within the superphylum Parcubacteria and are the first representative genomes of this lineage in soil (Supplementary Figure S6 and Table 1). Most of the unique genes from these MAGs encode hypothetical proteins. Nevertheless, some functional annotations unique to Amazon Parcubacteria MAGs (compared to 236 genomes available on NCBI) are a hyaluronan synthase and cadherin-like proteins (RTX toxins). Amazon Parcubacteria have reduced metabolic capabilities, but potentially can break down various simple sugars to pyruvate using glycolysis and the pentose phosphate shunt, which is then converted to D-lactate. Amazon-Parcubacteria-13-6-1 is also capable of synthesizing trehalose from glucose-1-phosphate. Overall, the functional potential of soil Parcubacteria MAGs are in accordance with other studies, but Amazon Parcubacteria are phylogenetically distinct (Supplementary Figure S6).
Discussion
In this study, we investigated the impact of deforestation on the genomic diversity and functional potential of soil microorganisms using metagenomics and MAGs. Previous studies have examined how forest-to-pasture conversion in the Brazilian Amazon affects soil microbial communities (Jesus et al., 2009; Rodrigues et al., 2013; Navarrete et al., 2015; Meyer et al., 2017) and discovered that land-use change is accompanied by significant shifts in the geochemistry and the microbial community of the soil. Jesus et al. (2009) determined that pasture soil microbial communities were significantly different from rainforest soil communities driven by pH, copper, and iron concentrations. Our study concurs with Jesus et al. (2009) that as the soil geochemistry changes the microbial community shifts as well with pH, iron, copper, and zinc significantly increasing in pasture soil. Previous work by Meyer et al. (2017) identified key shifts in methane cycling taxa and life history traits between rainforest and pasture samples. This study expands on work from Meyer et al. (2017) by focusing more broadly on genes related to the degradation of carbohydrates. On average, we have observed higher soil C content (g kg−1 soil) in the pasture sites (2.93 ± 0.65; mean ± 95% CI) than in the forest sites (1.52 ± 0.42), which suggests that higher soil C content might have selectively favored copiotrophic microorganisms with diverse carbohydrate metabolism. Furthermore, the two fast growing grass species grown in the pastures have been reported to secrete high quantities of labile C (Reis et al., 2001). Lignin is a highly aromatic, recalcitrant carbon compound produced by plants (Novaes et al., 2010). Previous work by Lammel et al. (2015) found a significantly higher abundance of lignin in rainforest compared to pasture soils, while the pasture samples had significantly higher amounts of hemicellulose. Using metagenomics, we found a differential abundance of genes related to lignin degradation in the rainforest and an increase in genes related to hemicellulose and cellulose degradation in the pasture soil. This is not surprising due to the drastic change in plant species from a diverse rainforest of large tropical trees with lignin contents around 40% to a monoculture of Urochloa brizantha (and a few occurrences of Panicum maximum) with ∼5–10% lignin (Herrero et al., 2001; Novaes et al., 2010; Mauri et al., 2015). As the carbon sources available in each land-use type change, we see significant alterations to the microbial community composition with a higher abundance of Firmicutes in the pasture. This is not unexpected since previous research has found that Firmicutes respond to increased organic matter (Cleveland et al., 2006). We also observed an almost complete loss of the ammonia-oxidizing archaea Thaumarchaeota in the pasture soil, confirming earlier studies (Hamaoui et al., 2016). A study by Subbarao et al. (2009) identified that the grass used in Brazilian cattle pastures (Urochloa) produces biological nitrification inhibitors (BNIs), specifically a cyclic diterpene termed brachialactone, that inhibits both the ammonia monooxygenase (amo) and hydroxylamine oxidoreductase (hao). These Urochloa grasses that produce BNIs have been shown to inhibit both ammonia-oxidizing bacteria and archaea (Moreta et al., 2014).
By assembling MAGs, we have gained a new depth of insight into the putative life strategies of some Amazonian soil microorganisms that is not possible with only metagenomics. In pastures we see a change in carbon metabolism coherent with results from our metagenomic data. These observed increases in fermentation and diversified carbon sources may have long-term consequences on carbon storage and greenhouse gas emissions. The Acidobacteria MAG from subdivision 3 (Bin_0_2_1) have the genetic potential to degrade a variety of carbon substrates similar to many related Acidobacteria (Kielak et al., 2016) but has the unique genetic setup to degrade aromatic compounds. The abundance of Acidobacteria subdivisions have been previously found to significantly change between land-use types (Navarrete et al., 2013, 2015) with subdivisions 4, 7, 10, and 17 increasing in pasture, and subdivisions 2 and 13 increasing in rainforest soils. Due to Acidobacteria’s predominance in soils, it has been previously proposed this group may be involved in the degradation of aromatic compounds in soil and with continued efforts to sequence and isolate these organisms we will have a better understanding of their role in soil carbon cycling (Pérez-Pantoja et al., 2010). Melainabacteria (Bin_23_1_1) appears to encode the ability to breakdown a variety of carbon substrates as well, including cellobiose. This group of non-photosynthetic fermenters have been found to metabolize cellulose and cellobiose in soil using stable isotope probing experiments (Soo et al., 2014; Pepe-Ranney et al., 2016; Juottonen et al., 2017). Both Amazon Acidobacteria subdivision 3 and Melainabacteria were found only in the pasture soil metagenomes likely playing an important role in metabolizing polycyclic aromatic hydrocarbons and plant-derived carbon deposited from the slash and burn deforestation practice. While we do not have the MAG replication to definitely test whether Acidobacteria subdivisions 3 and 4 and Melainabacteria are changing in abundance, we only detect their presence in one of the land uses, suggesting they are affected by land use change. Since Acidobacteria are considered ubiquitous in soils it is important to understand how the different subdivisions may be affected by land-use change. All Microgenomates and Parcubacteria MAGs were found only in the pasture, suggesting these groups are also influenced by land use change. This study described the first Microgenomates MAGs from soil with one MAG representing a novel phylum for which we proposed the name Candidatus Cerribacteria. Although the potential physiology of these candidate phyla MAGs was similar to what has previously been described (Wrighton et al., 2012; Rinke et al., 2013; Brown et al., 2015), the Amazon Microgenomates MAGs retained essential DNA mismatch repair genes (mutS, mutL) that are highly conserved in bacteria likely due to their role in providing genomic stability (Fukui, 2010). With 859 genomes examined, there is strong evidence that selection has or is occurring either for the loss of DNA mismatch repair in subsurface Microgenomates or for our Amazon soil Microgenomates to retain such machinery. These mismatch repair proteins provide DNA stability under the stress of environmental fluctuations, which in the case of cattle pasture may be temperature, moisture, and UV radiation (Denamur et al., 2000). In pathogenic or endosymbiotic bacteria, the loss of DNA repair and recombination proteins increases mutation rates which is thought to cause AT bias in small genomes (Moran and Wernegreen, 2000).
In rainforest soil we found a differential abundance of genes related to methanotrophy as has been previously reported by Meyer et al. (2017). The Rokubacteria MAG (Bin_15_13) was found predominantly in the rainforest soil metagenomes, which is not surprising due to the rainforests wide array of potential carbon sources, and in particular its ability to oxidize more methane than it produces. Rokubacteria have been described to oxidize methanol (Butterfield et al., 2016), but this is the first study to describe this group’s potential role in methane oxidation. This discovery led to an investigation into potential hydrocarbon metabolism that appears to be wide-spread in the soil MAGs. An electron transport Rnf complex was found in many of the Rokubacteria MAGs, which is suggested to be essential for ATP synthesis in some acetogens when grown autotrophically on H2 and CO2 by creating a proton motive force (Biegel et al., 2011; Tremblay et al., 2012). Our expanded analyses of the Rokubacteria MAGs indicate a diverse metabolism within this phylum including a wide variety of carbon sources ranging from gaseous substrates to complex hydrocarbons, many possible energy sources, and electron acceptors.
The combination of metagenome and MAG analyses allowed for increased understanding of the potential biological functions altered by land-use change. Carbon cycling in both datasets appeared to be significantly altered by deforestation, especially related to complex carbon sources like lignin and polycyclic aromatic hydrocarbons. This study provides the basis for continuing research into how deforestation affects soil microbial communities, biogeochemical cycling, and for the first-time tropical soil candidate phyla.
Author Contributions
MK contributed all metagenome and MAG analyses, all statistical analyses, drafted the manuscript, and drafted the figures and tables. JR, BB, CB, JT, SMT, and KN contributed sampling collection. JG contributed metagenome co-assembly and critical review of manuscript. KM provided raw sequence annotations from MG-RAST and critical review of manuscript. ST and KK contributed in revising of manuscript. TD, AE, and KN contributed in writing and revising of manuscript.
Funding
This project was supported by the National Science Foundation – Dimensions of Biodiversity (DEB 14422214), NSF-FAPESP 446 (2014/50320-4), the Agriculture and Food Research Initiative Competitive Grant 2009-447 35319-05186 from the United States Department of Agriculture National Institute of Food and Agriculture, and by the United States Department of Energy Joint Genome Institute through the Office of Science of the United States Department of Energy under Contract DE-AC02-442 05CH11231.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the owners and staff of Agropecuaria Nova Vida for logistical support and permission to work on their property. We are grateful to Rebecca Mueller, Fabiana Paula, and Wagner Piccinini for assistance with fieldwork.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01635/full#supplementary-material
Footnotes
- ^ https://github.com/dparks1134/CompareM
- ^ http://merenlab.org/2016/11/08/pangenomics-v2/
- ^ https://doi.org/10.6084/m9.figshare.5151571.v1
References
Alneberg, J., Bjarnason, B. S., de Bruijn, I., Schirmer, M., Quick, J., Ijaz, U. Z., et al. (2014). Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146. doi: 10.1038/nmeth.3103
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Anantharaman, K., Brown, C. T., Hug, L. A., Sharon, I., Castelle, C. J., Probst, A. J., et al. (2016). Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat. Commun. 7:13219. doi: 10.1038/ncomms13219
Aronesty, E. (2011). ea-utils: Command-line Tools for Processing Biological Sequencing Data. Available at: https://github.com/ExpressionAnalysis/ea-utils
Becraft, E. D., Woyke, T., Jarett, J., Ivanova, N., Godoy-Vitorino, F., Pulton, N., et al. (2017). Rokubacteria: genomic giants among the uncultured bacterial phyla. Front. Microbiol. 8:2264. doi: 10.3389/fmicb.2017.02264
Biegel, E., Schmidt, S., González, J. M., and Müller, V. (2011). Biochemistry evolution and physiological function of the Rnf complex a novel ion-motive electron transport complex in prokaryotes. Cell. Mol. Life Sci. 68, 613–634. doi: 10.1007/s00018-010-0555-8
Borneman, J., and Triplett, E. W. (1997). Molecular microbial diversity in soils from eastern Amazonia: evidence for unusual microorganisms and microbial population shifts associated with deforestation. Appl. Environ. Microbiol. 63, 2647–2653.
Bowers, R. M., Kyrpides, N. C., Stepanauskas, R., Harmon-Smith, M., Doud, D., Reddy, T. B. K., et al. (2017). Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35, 725–731. doi: 10.1038/nbt.3893
Brown, C. T., Hug, L. A., Thomas, B. C., Sharon, I., Castelle, C. J., Singh, A., et al. (2015). Unusual biology across a group comprising more than 15% of domain bacteria. Nature 523, 208–211. doi: 10.1038/nature14486
Butterfield, C. N., Li, Z., Andeer, P. F., Spaulding, S., Thomas, B. C., Singh, A., et al. (2016). Proteogenomic analyses indicate bacterial methylotrophy and archaeal heterotrophy are prevalent below the grass root zone. PeerJ 4:e2687. doi: 10.7717/peerj.2687
Cleveland, C. C., Nemergut, D. R., Schmidt, S. K., and Townsend, A. R. (2006). Increases in soil respiration following labile carbon additions linked to rapid shifts in soil microbial community composition. Biogeochemistry 82, 229–240. doi: 10.1007/s10533-006-9065-z
Da Silva, J. M., Rylands, A. B., and Da Fonseca, G. A. B. (2005). The fate of the Amazonian areas of endemism. Conserv. Biol. 19, 689–694. doi: 10.1111/j.1523-1739.2005.00705.x
Dale, V. H., Pearson, S. M., Offerman, H. L., and O’Neill, R. V. (1994). Relating patterns of land-use change to faunal biodiversity in the central Amazon. Conserv. Biol. 8, 1027–1036. doi: 10.1046/j.1523-1739.1994.08041027.x
de Moraes, J. F. L., Volkoff, B., Cerri, C. C., and Bernoux, M. (1996). Soil properties under Amazon forest and changes due to pasture installation in Rondônia, Brazil. Geoderma 70, 63–81. doi: 10.1016/0016-7061(95)00072-0
Delmont, T. O., Eren, A. M., Maccario, L., Prestat, E., Esen, Ö. C., Pelletier, E., et al. (2015). Reconstructing rare soil microbial genomes using in situ enrichments and metagenomics. Front. Microbiol. 6:358. doi: 10.3389/fmicb.2015.00358
Denamur, E., Lecointre, G., Darlu, P., Tenaillon, O., Acquaviva, C., Sayada, C., et al. (2000). Evolutionary implications of the frequent horizontal transfer of mismatch repair genes. Cell 103, 711–721. doi: 10.1016/S0092-8674(00)00175-6
Dojka, M. A., Harris, J. K., and Pace, N. R. (2000). Expanding the known diversity and environmental distribution of an uncultured phylogenetic division of bacteria. Appl. Environ. Microbiol. 66, 1617–1621. doi: 10.1128/AEM.66.4.1617-1621.2000
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Eren, A. M., Esen, Ö., Quince, C., Vineis, J. H., Morrison, H. G., Sogin, M. L., et al. (2015). Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3:e1319. doi: 10.7717/peerj.1319
Fukui, K. (2010). DNA mismatch repair in eukaryotes and bacteria. J. Nucleic Acids 2010:260512. doi: 10.4061/2010/260512
Gans, J., Wolinsky, M., and Dunbar, J. (2005). Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science 309, 1387–1390. doi: 10.1126/science.1112665
Gawad, C., Koh, W., and Quake, S. R. (2016). Single-cell genome sequencing: current state of the science. Nat. Rev. Genet. 17, 175–188. doi: 10.1038/nrg.2015.16
Guo, J., Cole, J. R., Zhang, Q., Brown, C. T., and Tiedje, J. M. (2016). Microbial community analysis with ribosomal gene fragments from shotgun metagenomes. Appl. Environ. Microbiol. 82, 157–166. doi: 10.1128/AEM.02772-15
Hamaoui, G. S. Jr., Rodrigues, J. L. M., Bohannan, B. J. M., Tiedje, J. M., and Nüsslein, K. (2016). Land-use change drives abundance and community structure alterations of thaumarchaeal ammonia oxidizers in tropical rainforest soils in Rondônia, Brazil. Appl. Soil Ecol. 107, 48–56. doi: 10.1016/j.apsoil.2016.05.012
Hedlund, B. P., Dodsworth, J. A., Murugapiran, S. K., Rinke, C., and Woyke, T. (2014). Impact of single-cell genomics and metagenomics on the emerging view of extremophile ‘microbial dark matter’. Extremophiles 18, 865–875. doi: 10.1007/s00792-014-0664-7
Herrero, M., do Valle, C. B., Hughes, N. R. G., de, O., Sabatel, V., and Jessop, N. S. (2001). Measurements of physical strength and their relationship to the chemical composition of four species of Brachiaria. Anim. Feed Sci. Technol. 92, 149–158. doi: 10.1016/S0377-8401(01)00261-9
Howe, A. C., Jansson, J. K., Malfatti, S. A., Tringe, S. G., Tiedje, J. M., and Brown, C. T. (2014). Tackling soil diversity with the assembly of large complex metagenomes. Proc. Natl. Acad. Sci. U.S.A. 111, 4904–4909. doi: 10.1073/pnas.1402564111
Hu, P., Tom, L., Singh, A., Thomas, B. C., Baker, B. J., Piceno, Y. M., et al. (2016). Genome-resolved metagenomic analysis reveals roles for candidate phyla and other microbial community members in biogeochemical transformations in oil reservoirs. mBio 7:e01669-15. doi: 10.1128/mBio.01669-15
Hug, L. A., Baker, B. J., Anantharaman, K., Brown, C. T., Probst, A. J., Castelle, C. J., et al. (2016). A new view of the tree of life. Nat. Microbiol. 1:16048. doi: 10.1038/nmicrobiol.2016.48
Hugenholtz, P. (2002). Exploring prokaryotic diversity in the genomic era. Genome Biol. 3:REVIEWS0003. doi: 10.1186/gb-2002-3-2-reviews0003
Imelfort, M., Parks, D., Woodcroft, B. J., Dennis, P., Hugenholtz, P., and Tyson, G. W. (2014). GroopM: an automated tool for the recovery of population genomes from related metagenomes. PeerJ 2:e603. doi: 10.7717/peerj.603
Jesus, E. C., Marsh, T. L., Tiedje, J. M., de, S., and Moreira, F. M. (2009). Changes in land use alter the structure of bacterial communities in Western Amazon soils. ISME J. 3, 1004–1011. doi: 10.1038/ismej.2009.47
Juottonen, H., Eiler, A., Biasi, C., Tuittila, E. S., Yrjälä, K., and Fritze, H. (2017). Distinct anaerobic bacterial consumers of cellobiose-derived carbon in boreal fens with different CO2/CH4 production ratios. Appl. Environ. Microbiol. 83:e02533-16. doi: 10.1128/AEM.02533-16
Kang, D. D., Froula, J., Egan, R., and Wang, Z. (2015). MetaBAT an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3:e1165. doi: 10.7717/peerj.1165
Kantor, R. S., Wrighton, K. C., Handley, K. M., Sharon, I., Hug, L. A., Castell, C. J., et al. (2013). Small genomes and sparse metabolisms of sediment-associated bacteria from four candidate phyla. mBio 4:e00708-13. doi: 10.1128/mBio.00708-13
Kielak, A. M., Barreto, C. C., Kowalchuk, G. A., van Veen, J. A., and Kuramae, E. E. (2016). The ecology of acidobacteria: moving beyond genes and genomes. Front. Microbiol. 7:744. doi: 10.3389/fmicb.2016.00744
Kumar, S., Stecher, G., and Koichiro, T. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lammel, D. R., Feigl, B. J., Cerri, C. C., and Nusslein, K. (2015). Specific microbial gene abundances and soil parameters contribute to C, N, and greenhouse gas process rates after land use change in Southern Amazonian Soils. Front. Microbiol. 6:1057. doi: 10.3389/fmicb.2015.01057
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Laslett, D., and Canback, B. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16. doi: 10.1093/nar/gkh152
Laurance, W. F., Cochrane, M. A., Bergen, S., Fearnside, P. M., Delamônica, P., Barber, C., et al. (2001). The future of the Brazilian Amazon. Science 291, 438–439. doi: 10.1126/science.291.5503.438
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Li, D., Luo, R., Liu, C. M., Leung, C. M., Ting, H. F., Sadakane, K., et al. (2015). MEGAHIT v10: a fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102, 3–11. doi: 10.1016/j.ymeth.2016.02.020
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25,2078–2079. doi: 10.1093/bioinformatics/btp352
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lowe, T. M., and Chan, P. P. (2016). tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57. doi: 10.1093/nar/gkw413
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Mauri, J., Technio, V. H., Davide, L. C., Periera, D. L., Sobrinho, F. S., and Periera, F. J. (2015). Forage quality in cultivars of Brachiaria spp.: association of lignin and fibers with anatomical characteristics. Aust. J. Crop Sci. 9, 1148–1153.
McKinney, M. L., and Lockwood, J. L. (1999). Biotic Homogenization: a few winners replacing many losers in the next mass extinction. Trends Ecol. Evol. 14, 450–453. doi: 10.1016/S0169-5347(99)01679-1
McLean, J. S., Lombardo, M. J., Badger, J. H., Edlund, A., Novotny, M., Yee-Greenbaum, J., et al. (2013). Candidate phylum TM6 genome recovered from a hospital sink biofilm provides genomic insights into this uncultivated phylum. Proc. Natl. Acad. Sci. U.S.A. 110, E2390–E2399. doi: 10.1073/pnas.1219809110
Meyer, F., Paarmann, D., D’Souza, M., Olson, R., Glass, E. M., Kuban, M., et al. (2008). The metagenomics RAST server – a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9:386. doi: 10.1186/1471-2105-9-386
Meyer, K. M., Klein, A. M., Rodrigues, J. L. M., Nüsslein, K., Tringe, S. G., Mirza, B. S., et al. (2017). Conversion of Amazon rainforest to agriculture alters community traits of methane-cycling organisms. Mol. Ecol. 26, 1547–1556. doi: 10.1111/mec.14011
Moran, N. A., and Wernegreen, J. J. (2000). Lifestyle evolution in symbiotic bacteria: insights from genomics. Trends Ecol. Evol. 15, 321–326. doi: 10.1016/S0169-5347(00)01902-9
Moreta, D. E., Arango, J., Sotelo, M., Vergara, D., Rincón, A., Ishitani, M., et al. (2014). Biological nitrification inhibition (BNI) in Brachiaria pastures: a novel strategy to improve eco-efficiency of crop-livestock systems and to mitigate climate change. Trop. Grassl. Forrajes Trop. 2, 88–91. doi: 10.17138/TGFT(2)88-91
Mueller, R. C., Paula, F. S., Mirza, B. S., Rodrigues, J. L. M., Nusslein, K., and Bohannan, B. J. M. (2014). Links between plant and fungal communities across a deforestation chronosequence in the Amazon rainforest. ISME J. 8, 1548–1550. doi: 10.1038/ismej.2013.253
Navarrete, A. A., Kuramae, E. E., de Hollander, M., Pijl, A. S., van Veen, J. A., and Tsai, S. M. (2013). Acidobacterial community responses to agricultural management of soybean in Amazon forest soils. FEMS Microbiol. Ecol. 83, 607–621. doi: 10.1111/1574-6941.12018
Navarrete, A. A., Tsai, S. M., Mendes, L. W., Faust, K., de Hollander, M., Cassman, N. A., et al. (2015). Soil microbiome responses to the short-term effects of Amazonian deforestation. Mol. Ecol. 24, 2433–2448. doi: 10.1111/mec.13172
Novaes, E., Kirst, M., Chiang, V., Winter-Sederoff, H., and Sederoff, R. (2010). Lignin and biomass: a negative correlation for wood formation and lignin content in trees. Plant Physiol. 154, 555–561. doi: 10.1104/pp.110.161281
Oksanen, J. F., Blanchet, G., Friendly, M., Kindt, R., Legendre, P., McGlinn, D., et al. (2018). vegan: Community Ecology Package. R Package Version 2.4-6. Available at: https://CRAN.R-project.org/package=vegan
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes from isolates, single cells and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Paula, F. S., Rodrigues, J. L. M., Zhou, J., Wu, L., Mueller, R. C., Mirza, B. S., et al. (2014). Land use change alters functional gene diversity composition and abundance in Amazon forest soil microbial communities. Mol. Ecol. 23, 2988–2999. doi: 10.1111/mec.12786
Peixoto, R. S., da Costa Coutinho, H. L., Rumjanek, N. G., Macrae, A., and Rosado, A. S. (2002). Use of rpoB and 16S rRNA genes to analyze bacterial diversity of a tropical soil using PCR and DGGE. Lett. Appl. Microbiol. 35, 316–320. doi: 10.1046/j.1472-765X.2002.01183.x
Pepe-Ranney, C., Campbell, A. N., Koechli, C. N., Berthrong, S., and Buckley, D. H. (2016). Unearthing the ecology of soil microorganisms using a high resolution DNA-SIP approach to explor cellulose and xylose metabolism in soil. Front. Microbiol. 7:703. doi: 10.3389/fmicb.2016.00703
Pérez-Pantoja, D., González, B., and Pieper, D. H. (2010). “Aerobic degradation of aromatic hydrocarbons,” in Handbook of Hydrocarbon and Lipid Microbiology, ed. K. N. Timmis (Berlin: Springer), 799–837. doi: 10.1007/978-3-540-77587-4_60
Probst, A. J., Castelle, C. J., Singh, A., Brown, C. T., Anantharaman, K., Sharon, I., et al. (2017). Genomic resolution of a cold subsurface aquifer community provides metabolic insights for novel microbes adapted to high CO2 concentrations. Environ. Microbiol. 19, 459–474. doi: 10.1111/1462-2920.13362
Prosser, J. I. (2015). Dispersing misconceptions and identifying opportunities for the use of ‘omics’ in soil microbial ecology. Nat. Rev. Microbiol. 13, 439–446. doi: 10.1038/nrmicro3468
R Core Team (2016). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Ranjan, K., Paula, F. S., Mueller, R. C., Jesus, E. C., Cenciani, K., and Bohannan, B. (2015). Forest-to-pasture conversion increases the diversity of the phylum Verrucomicrobia in Amazon rainforest soils. Front. Microbiol. 6:779. doi: 10.3389/fmicb.2015.00779
Reis, V. M., dos Reis, F. B., Quesada, D. M., de Oliveira, O. C. A., Alves, B. J. R., Urquiaga, S., et al. (2001). Biological nitrogen fixation associated with tropical pasture grasses. Aust. J. Plant Physiol. 28, 837–844.
Rinke, C., Schwientek, P., Sczyrba, A., Ivanoca, N. N., Anderson, I. J., Cheng, J. F., et al. (2013). Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–437. doi: 10.1038/nature12352
Rodrigues, J. L. M., Pellizari, V. H., Mueller, R., Baek, K., Jesus, E. C., Paula, F. S., et al. (2013). Conversion of the Amazon rainforest to agriculture results in biotic homogenization of soil bacterial communities. Proc. Natl. Acad. Sci. U.S.A. 110, 988–993. doi: 10.1073/pnas.1220608110
Sala, O. E., Chapin, F. S., Armesto, J. J., Berlow, E., Bloomfield, J., Dirzo, R., et al. (2000). Global biodiversity scenarios for the year 2100. Science 287, 1770–1774. doi: 10.1126/science.287.5459.1770
Sangwan, N., Xia, F., and Gilbert, J. A. (2016). Recovering complete and draft population genomes from metagenome datasets. Microbiome 4:8. doi: 10.1186/s40168-016-0154-5
Sekiguchi, Y., Ohashi, A., Parks, D. H., Yamauchi, T., Tyson, G. W., and Hugenholtz, P. (2015). First genomic insights into members of a candidate bacterial phylum responsible for wastewater bulking. PeerJ 3:e740. doi: 10.7717/peerj.740
Siegl, A., Kamke, J., Hochmuth, T., Piel, J., Richter, M., Liang, C., et al. (2011). Single-cell genomics reveals the lifestyle of Poribacteria a candidate phylum symbiotically. ISME J. 5, 61–70. doi: 10.1038/ismej.2010.95
Smith, S. A., and Dunn, C. W. (2008). Phyutility: a phyloinformatics tool for trees alignments and molecular data. Bioinformatics 24, 715–716. doi: 10.1093/bioinformatics/btm619
Soares-Filho, B. S., Nepstad, D. C., Curran, L. M., Cerqueira, G. C., Garcia, R. A., Ramos, C. A., et al. (2006). Modelling conservation in the Amazon basin. Nature 440, 520–523. doi: 10.1038/nature04389
Soo, R. M., Skennerton, C. T., Sekiguchi, Y., Imelfort, M., Paech, S. J., Dennis, P. G., et al. (2014). An expanded genomic representation of the phylum cyanobacteria. Genome Biol. Evol. 6, 1031–1045. doi: 10.1093/gbe/evu073
Subbarao, G. V., Nakahara, K., Hurtado, M. P., Ono, J., Moreta, D. E., Salcedo, A. F., et al. (2009). Evidence for biological nitrification inhibition in Brachiaria pastures. Proc. Natl. Acad. Sci. U.S.A. 106, 17302–17307. doi: 10.1073/pnas.0903694106
Tas, N., Prestat, E., Wang, S., Wu, Y., Ulrich, C., Kneafsey, T., et al. (2018). Landscape topography structures the soil microbiome in arctic polyconal tundra. Nat. Commun. 9:777. doi: 10.1038/s41467-018-03089-z
Tremblay, P. L., Zhang, T., Dar, S. A., Leang, C., and Lovley, D. R. (2012). The Rnf complex of Clostridium ljungdahlii is a proton-translocating ferredoxin:NAD+ oxidoreductase essential for autotrophic growth. mBio 4:e00406-12. doi: 10.1128/mBio.00406-12
Tripathi, B. M., Kim, M., Singh, D., Lee-Cruz, L., Lai-Hoe, A., Ainuddin, A. N., et al. (2012). Tropical soil bacterial communities in Malaysia: Ph dominates in the equatorial tropics too. Microb. Ecol. 64, 474–484. doi: 10.1007/s00248-012-0028-8
Warnes, G. R., Bolker, B., Bonebakker, L., Gentleman, R., Huber, W., Liaw, A., et al. (2016). gplots: Various R Programming Tools for Plotting Data. R Package Version 3.0.1. Available at: https://CRAN.R-project.org/package=gplots
Wattam, A. R., Abraham, D., Dalay, O., Disz, T. L., Driscoll, T., Gabbard, J. L., et al. (2014). PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 42, D581–D591. doi: 10.1093/nar/gkt1099
White, R. A. III, Bottos, E. M., Chowdhury, T. R., Zucker, J. D., Brislawn, C. J., Nicora, C. D., et al. (2016). Moleculo long-read sequencing facilitates assembly and genomic binning from complex soil metagenomes. mSystems 1:e00045-16. doi: 10.1128/mSystems.00045-16
Wrighton, K. C., Thomas, B. C., Sharon, I., Miller, C. S., Castelle, C. J., VerBerkmoes, N. C., et al. (2012). Fermentation hydrogen and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337, 1661–1665. doi: 10.1126/science.1224041
Keywords: Amazon rainforest soil, land-use change, metagenome assembled genomes, rare biosphere, soil metagenomics
Citation: Kroeger ME, Delmont TO, Eren AM, Meyer KM, Guo J, Khan K, Rodrigues JLM, Bohannan BJM, Tringe SG, Borges CD, Tiedje JM, Tsai SM and Nüsslein K (2018) New Biological Insights Into How Deforestation in Amazonia Affects Soil Microbial Communities Using Metagenomics and Metagenome-Assembled Genomes. Front. Microbiol. 9:1635. doi: 10.3389/fmicb.2018.01635
Received: 16 April 2018; Accepted: 30 June 2018;
Published: 23 July 2018.
Edited by:
Etienne Yergeau, Institut National de la Recheriche Scientifique (INRS), CanadaReviewed by:
Carl-Eric Wegner, Friedrich-Schiller-Universität Jena, GermanyTerrence H. Bell, Pennsylvania State University, United States
Erick Cardenas Poire, Microbiome Insights, Canada
Copyright © 2018 Kroeger, Delmont, Eren, Meyer, Guo, Khan, Rodrigues, Bohannan, Tringe, Borges, Tiedje, Tsai and Nüsslein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Klaus Nüsslein, bnVzc2xlaW5AbWljcm9iaW8udW1hc3MuZWR1