 Qin Li
Qin Li Shuo Chai1
Shuo Chai1 Yongdi Li
Yongdi Li Jianan Huang
Jianan Huang Yu Luo
Yu Luo Lizheng Xiao
Lizheng Xiao Zhonghua Liu
Zhonghua Liu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 10 July 2018
Sec. Food Microbiology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.01509
This article is part of the Research Topic Microbial Decontamination by Novel Technologies – Mechanisms and Application Concepts View all 10 articles
Primary dark tea is used as raw material for compressed dark tea, such as Fu brick tea, Hei brick tea, Hua brick tea, and Qianliang tea. Pile-fermentation is the key process for the formation of the characteristic properties of primary dark tea, during which the microorganism plays an important role. In this study, the changes of major chemical compounds, enzyme activities, microbial diversity, and their correlations were explored during the pile-fermentation process. Our chemical and enzymatic analysis showed that the contents of the major compounds were decreased, while the activities of polyphenol oxidase, cellulase, and pectinase were increased during this process, except peroxidase activity that could not be generated from microbial communities in primary dark tea. The genera Cyberlindnera, Aspergillus, Uwebraunia, and Unclassified Pleosporales of fungus and Klebsiella, Lactobacillus of bacteria were predominant in the early stage of the process, but only Cyberlindnera and Klebsiella were still dominated in the late stage and maintained a relatively constant until the end of the process. The amino acid was identified as the important abiotic factor in shaping the microbial community structure of primary dark tea ecosystem. Network analysis revealed that the microbial taxa were grouped into five modules and seven keystone taxa were identified. Most of the dominant genera were mainly distributed into module III, which indicated that this module was important for the pile-fermentation process of primary dark tea. In addition, bidirectional orthogonal partial least squares (O2PLS) analysis revealed that the fungi made more contributions to the formation of the characteristic properties of primary dark tea than bacteria during the pile-fermentation process. Furthermore, 10 microbial genera including Cyberlindnera, Aspergillus, Eurotium, Uwebraunia, Debaryomyces, Lophiostoma, Peltaster, Klebsiella, Aurantimonas, and Methylobacterium were identified as core functional genera for the pile-fermentation of primary dark tea. This study provides useful information for improving our understanding on the formation mechanism of the characteristic properties of primary dark tea during the pile-fermentation process.
Dark tea is a unique kind of microbial fermented teas, which is native to many regions of China, including Hunan, Yunnan, and Hubei provinces (Zhang et al., 2013). The manufacturing process of dark tea can be divided into primary processing stage and reprocessing stage. During the primary processing stage, primary dark tea was produced and was mainly used as material for compressed dark tea, including Fu brick tea, Hei brick tea, Hua brick tea, and Qiangliang tea. The manufacturing process of primary dark tea covers the following steps, harvesting fresh tea leaves, fixing, rolling, pile-fermentation, and drying processes (Li et al., 2017). In the above processes, the pile-fermentation, a kind of solid-state fermentation, is the key process in the production of primary dark tea. The pile-fermentation process of primary dark tea is characterized by the growth and succession of microbial communities, and the microorganism plays a key role in the formation of the characteristic properties of primary dark tea during the pile-fermentation process (Wang et al., 1991a).
A comprehensive study of microbial community succession could help to understand that how the sensory quality of primary dark tea was formed during the pile-fermentation process. Some kinds of microbial genera, including Candida sp., Aspergillus niger, and Penicillium in fungal community and Non-spore bacteria, Bacillus, and Coccus in bacterial community, were identified as the dominant microbial genera by the culture-dependent method during the pile-fermentation process of primary dark tea (Wen and Liu, 1991). However, the culture-dependent method is of limited value for identifying the microbial communities due to the limitation of cultural media (Li et al., 2011). Hence, the culture-independent methods, including terminal restriction fragment length polymorphism (T-RFLP), denaturing gradient gel electrophoresis (DGGE), and high-throughput sequencing technology (i.e., Illumina MiSeq sequencing) that use direct analysis of the diversity of ribosomal RNA genes with no culture step (Amann and Ludwig, 2000), were introduced for identifying the structure of microbial communities in compressed dark tea, such as Fu brick tea and Pu-erh tea (Abe et al., 2008; Xu et al., 2011; Zhao et al., 2013; Fu et al., 2016; Li et al., 2017). The genera Aspergillus, Blastobotrys, Bacillus, and Enterobacteriaceae were the major microbial communities in Pu-erh tea (Abe et al., 2008; Zhao et al., 2013), and Aspergillus, Cyberlindnera, Candida, Penicillium, Eurotium, Beauveria, Debaryomyces, Pestalotiopsis, Pichia, Rhizomucor, and Verticillium were the dominant microbial communities in Fu brick tea by PCR-DGGE (Xu et al., 2011) and high-throughput sequencing technology (Li et al., 2017).
During the pile-fermentation process, some new transformation products were generated in tea leaves. Our previous study showed that the power of substance transformation in tea leaves were mainly derived from the microbial enzymes, which were secreted from the microorganism during the pile-fermentation process of primary dark tea (Wang et al., 1991a). These transformations resulted in the formation of the characteristic properties of primary dark tea. The microbial metabolism, which produce the secreted enzymes such as polyphenol oxidase (PPO), cellulase (CEL), and pectinase (PEC), thereby providing the effective biochemical power to oxidation of catechins, decomposition of cellulose, and hydrolyses of pectin and proteins. The microbial enzymatic action played an important role in the formation of the characteristics of primary dark tea (Liu et al., 1991). During the pile-fermentation process, the total contents of polyphenol, catechin, and soluble carbohydrate were decreased, and the catechin composition varied irregularly (Wang et al., 1991c). The content of total amino acid (AA) was decreased, especially theanine and glutamic acid were decreased significantly; by contrast, the content of indispensable AAs, such as lusine, phenylalanine, leucine, isoleucine, methionine, and valine, were increased remarkably. And the levels of caffeine, theobromine, and theophylline were not significantly variated (Wang et al., 1991b).
The succession of microbial community in primary dark tea during pile-fermentation process results in a dynamic taste and aroma of primary dark tea. Variation of major chemical compounds and enzyme activities in fermented tea has been studied (Liu et al., 1991; Wang et al., 1991b,c). The culturable microorganisms have also been identified in primary dark tea during the pile-fermentation process (Wen and Liu, 1991). However, the associations among microbiota and chemical compounds or enzyme activities, and so far the core functional microbiota were not clarified in primary dark tea. Thus, the assembly and dynamics of microbial community were characterized by Illumina MiSeq sequencing during the pile-fermentation of primary dark tea. The changes of major chemical compounds content and enzyme activity were also investigated during this process. Based on these results, the relationship between microbiota and chemical compounds or enzyme activities was investigated by bidirectional orthogonal partial least squares (O2PLS). Finally, the core functional microorganisms were identified by comparison of the comprehensive importance of microorganisms correlated with the formation of characteristic properties of primary dark tea. The knowledge provide a new insight for better understanding on the mechanism of pile-fermentation in primary dark tea.
Sampling of primary dark tea was carried out in Ba Jiao tea factory (Yiyang, Hunan province, China). The manufacturing process of primary dark tea was performed as described by DB43/T660-2011 with a little modification (Liu et al., 2011). Briefly, mature tea leaves (one bud and three to six leaves) were plucked from medium and sprinkled with water then fixing at 280–320°C. The fixing leaves rolled twice by machine. Then, the rolling leaves (about 930 kg) were piled 1.1 m high, with ambient temperature about 28°C and moisture content about 78% in pile-fermentation apparatus for 12 h. Auto drying machine was used for drying the fermentation leaves at 120–130°C twice or drying in the sun. Fifteen samples were collected from three independent batches of primary dark tea at different time of pile-fermentation process: pile-fermentation 0-, 3-, 6-, 9-, and 12-h stages. The samples were packaged in sterile polyethylene bags, transported to the laboratory, and stored at −80°C until required.
The content of water extract (WE) was determined according to the National Standard GB/T 8305-2013 (Tea-Determination of water extracts content) (Zhou et al., 2013). Total polyphenols (TP) and flavonoids (FLA) content were determined by a colorimetric method, as described previously (Li et al., 2013). Free AA content was determined according to the National Standard GB/T 8314-2013 (Tea-Determination of free amino acids content) (Xu et al., 2013). Soluble sugar (SS) content was determined according to sulfuric acid–anthrone colorimetric method, as described previously (DuBois et al., 1956). All analyses were performed three times.
Extraction of enzyme extracts was based on our previous method with slight modifications (Teng et al., 2017). 1.25-g tea leaves were ground with 0.75-g polyvinylpolypyrrolidone (PVPP) in a mortar on ice. The crude enzyme was extracted with 25-mL citrate buffer (pH 5.6) on ice for 12 h. Subsequently, the mixture was centrifuged at 15,000 × g for 30 min at 10°C, and the clear supernatant was collected and filled to 25 mL with citrate buffer. The clear supernatant was used in all enzyme assays. The activities of PPO, peroxidase (POD), CEL, and PEC were determined according to previous studies (Wood and Bhat, 1988; Huang, 1997; Biz et al., 2014). All analyses was performed three times. The detail approaches were described in Supplementary Material 1.
Each tea sample (5 g) was mixed with sterile water (25 mL), stirred thoroughly, filtered through three layers of coarse sterile gauze to remove large particles, and centrifuged at 10,000 × g for 10 min at 4°C. The pellets were used for genomic DNA extraction. DNA from the sample was extracted using PowerSoil® DNA Isolation Kit (Mo Bio Laboratories Inc., Carlsbad, CA, United States), according to the manufacturer’s protocols. The duplicate samples were pooled together for one high-throughput sequencing. The V4 region of 16S was amplified using the primer pair 515F (5′-GTGCCAGCMGCCGCGG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) (Xiong et al., 2012; Fan et al., 2014); the ITS rRNA gene was amplified using the primer pair ITS1F (5′-CTTGGTCATTTAGAGGAAGTAA-3′) and ITS2 (5′-GCTGCGTTCTTCATCGATGC-3′) (Mukherjee et al., 2014). The forward and reverse primers were tagged with adapter, pad, and linker sequences. The reaction mixture (50 μL) contained 25 μL of Taq PCR Master Mix (QIAGEN), 0.2 μM of each primer, and 10 ng DNA templates. Thermal cycling consisted of initial denaturation at 94°C for 4 min, followed by 35 cycles of denaturation at 94°C for 40 s, annealing at 54°C (ITS) and 52°C (16S) for 45 s, elongation at 72°C for 50 s, and finally elongation at 72°C for 10 min. Negative control was treated similarly with the exclusion of template DNA and failed to produce visible PCR products. For each sample, three independent reactions were performed. PCR products were mixed in equimolar ratios and mixture PCR products were purified with GeneJET Gel Next® UltraTM DNA Library Prep Kit for Illumina (NEB, United States) following manufacturer’s recommendations. The library quality was assessed on the Qubit@2.0 Fluorometer (Thermo Scientific) and Agilent Bioanalyzer 2100 system. Finally, the library was sequenced on an Illumina MiSeq platform using paired-end sequencing by Novogene Inc. (Beijing, China).
Raw reads files were demultiplexed, quality filtered, and analyzed using the Quantitative Insights Into Microbial Ecology (QIIME, v1.7.0) pipeline (Caporaso et al., 2010). In brief, reads with less than length 220 bp and ambiguous bases were discarded. Chimeras were removed using UCHIME as implemented in QIIME. The effective tags were binned into operational taxonomic units (OTUs) by UCLUST based on 97% pairwise identity (Edgar et al., 2011). The most abundant sequence from each OTU was selected to represent the OUT. OTUs were classified taxonomically using a QIIME-based wrapper of the Ribosomal Database Project (RDP) classifier against UNITE (User-friendly Nordic ITS Ectomycorrhiza Database) and INSD (International Nucleotide Sequence Databank) database (Caporaso et al., 2010). All identified OTU sequences assigned to chloroplast and mitochondrion origins were removed from the dataset.
The alpha diversity (Shannon index), richness (Chao1 index), Good’s coverage, and rarefaction curve were performed by Mothur (Schloss et al., 2011). In addition, rank abundance curves, principal components analysis (PCA) and non-metric multidimensional scaling (NMDS) were generated by using R package vegan (version 3.4.01). CANOCO software (version 4.52) was used to conduct multivariate analysis using detrended correspondence analysis (DCA) and redundancy analysis (RDA). One-way ANOVA test, Pearson’s correlation coefficients, and P-values were calculated with IBM SPSS 19.0.3 All results were presented as the mean value ± SD. Differences between groups were declared significant at P < 0.05.
The co-occurrence patterns of microbial community in the pile-fermentation process of primary dark tea were constructed and analyzed using the online Molecular Ecological Network Analyses Pipeline (MENAP4; Deng et al., 2012). The OTUs, which presented in at least six samples, were used for pairwise Pearson correlation calculation, for which a proper threshold was identified based on the random matrix theory (RMT) approach. Network topological properties were also calculated using MENAP. O2PLS analysis was performed using the SIMCA-P 14 (Version 14.1.0.2047), according to the previous study (Wang et al., 2016). The X-matrix was designed as the microbiota dataset, and the Y-matrix was designed as the chemical compounds and enzyme activities datasets. Finally, the networks and the correlation among microbiota, chemical compounds, and enzyme activities were visualized using the Cytoscape software (Version 3.5.1).
The contents of major chemical compounds, including WE, TP, FLA, AA, and SS, were analyzed during the pile-fermentation process of primary dark tea. As shown in Figure 1, all the contents of major compounds were significantly decreased after the pile-fermentation process (P < 0.05). The concentrations of WE and AA were significantly decreased beginning at 6-h stage, 5.43 and 47.90% decrease was observed at 12-h stage compared with 0-h stage, respectively. The levels of TP and FLA were slightly decreased from 0-h stage to 6-h stage and sharply decreased at 9-h stage until the end of process, where 31.68 and 18.84% decrease was observed. A slightly increased content of SS was found from 0-h stage to 3-h stage, but a 44.24% decrease of SS was also observed at the end of process (Figure 1).
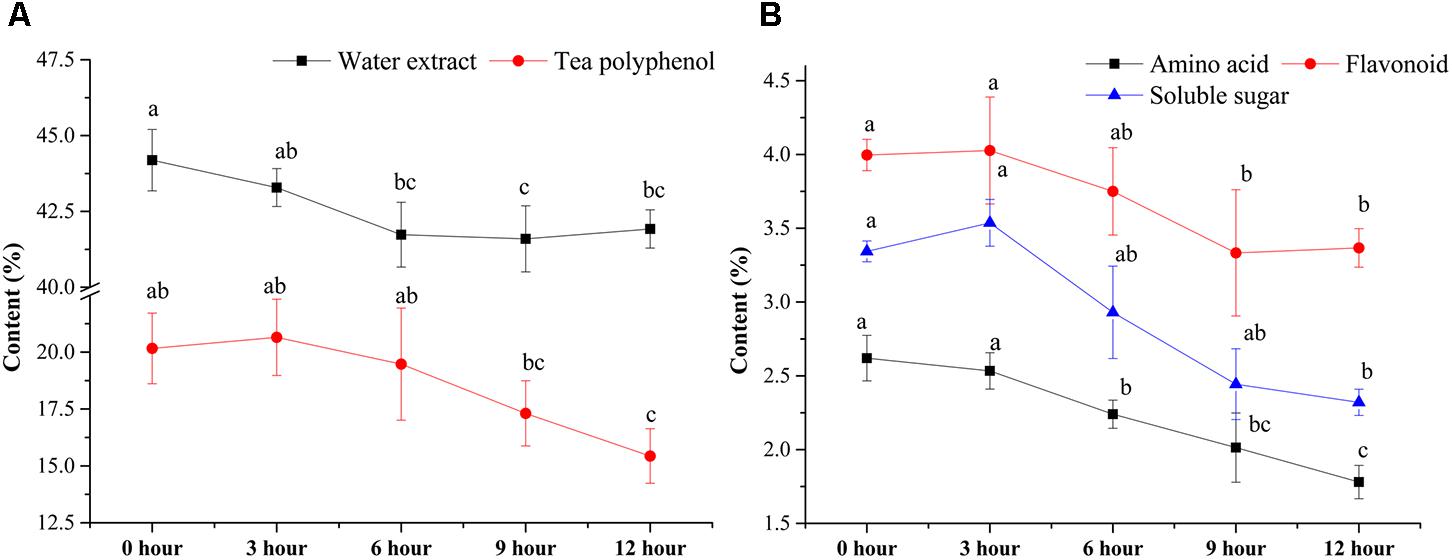
FIGURE 1. Changes in the major chemical compounds during the pile-fermentation process of primary dark tea. Water extract and tea polyphenol (A), amino acid, flavonoid, and soluble sugar (B).
Several enzyme activities, including PPO, POD, CEL, and PEC, were also detected during the pile-fermentation of primary dark tea (Figure 2). The activities of PPO, CEL, and PEC were significantly increased after the fermentation process, but the POD activity was significantly decreased in this process. The PPO activity was significantly increased from 0-h stage (not detected) to 0.92 ± 0.27 U at 6-h stage and remained stable until the end of process. And the levels of CEL and PEC activities were markedly increased from 0-h stage (not detected) to 0.22 ± 0.03 and 0.21 ± 0.05 U at 3-h stage then slightly increased until the end of process. But the POD activity was significantly decreased by 74.56% after pile-fermentation process.
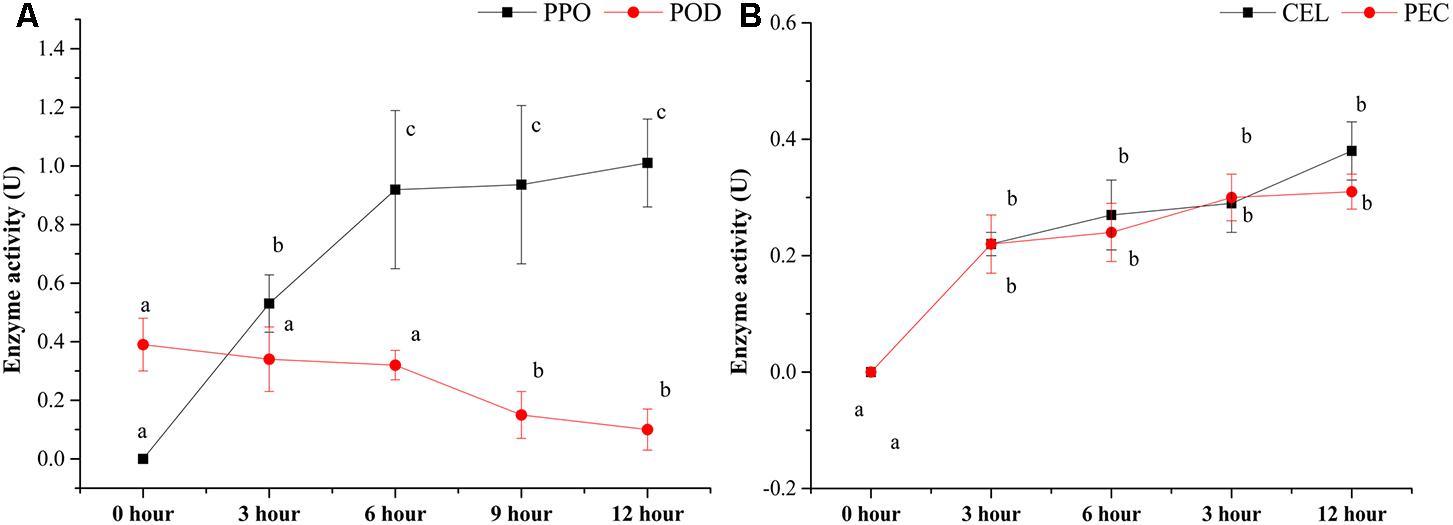
FIGURE 2. Changes in the enzyme activities during the pile-fermentation process of primary dark tea. PPO, polyphenol oxidase; POD, peroxidase (A); CEL, cellulase; PEC, pectinase (B).
In total, 1,401,447 valid fungal sequences and 418,889 valid bacterial sequences across all samples clustered into 297 fungal OTUs and 77 bacterial OTUs after trimming and filtration at 97% sequence identify. The values of Good’s coverage were >99% for all sequences from five groups (Supplementary Table S1). The rarefaction curves for all samples almost reached the saturation phase, suggesting that few new microorganisms would possibly be identified by increasing the sequencing depth, and the majority of microorganisms for the samples had already been captured in the current analysis (Supplementary Figure S1). The Shannon index indicated that the diversity of fungus and bacteria was significantly decreased from 0-h stage to 6-h stage and maintained relatively constant until the end of the process. The Chao1 index indicated that the richness of fungus and bacteria was slightly decreased during the pile-fermentation process, but no significant difference was observed at the end of the process (Figure 3). In addition, the rank abundance curves of fungus and bacteria also showed that a slightly decreased richness and evenness of fungus and bacteria after the pile-fermentation process of primary dark tea (Supplementary Figure S2).
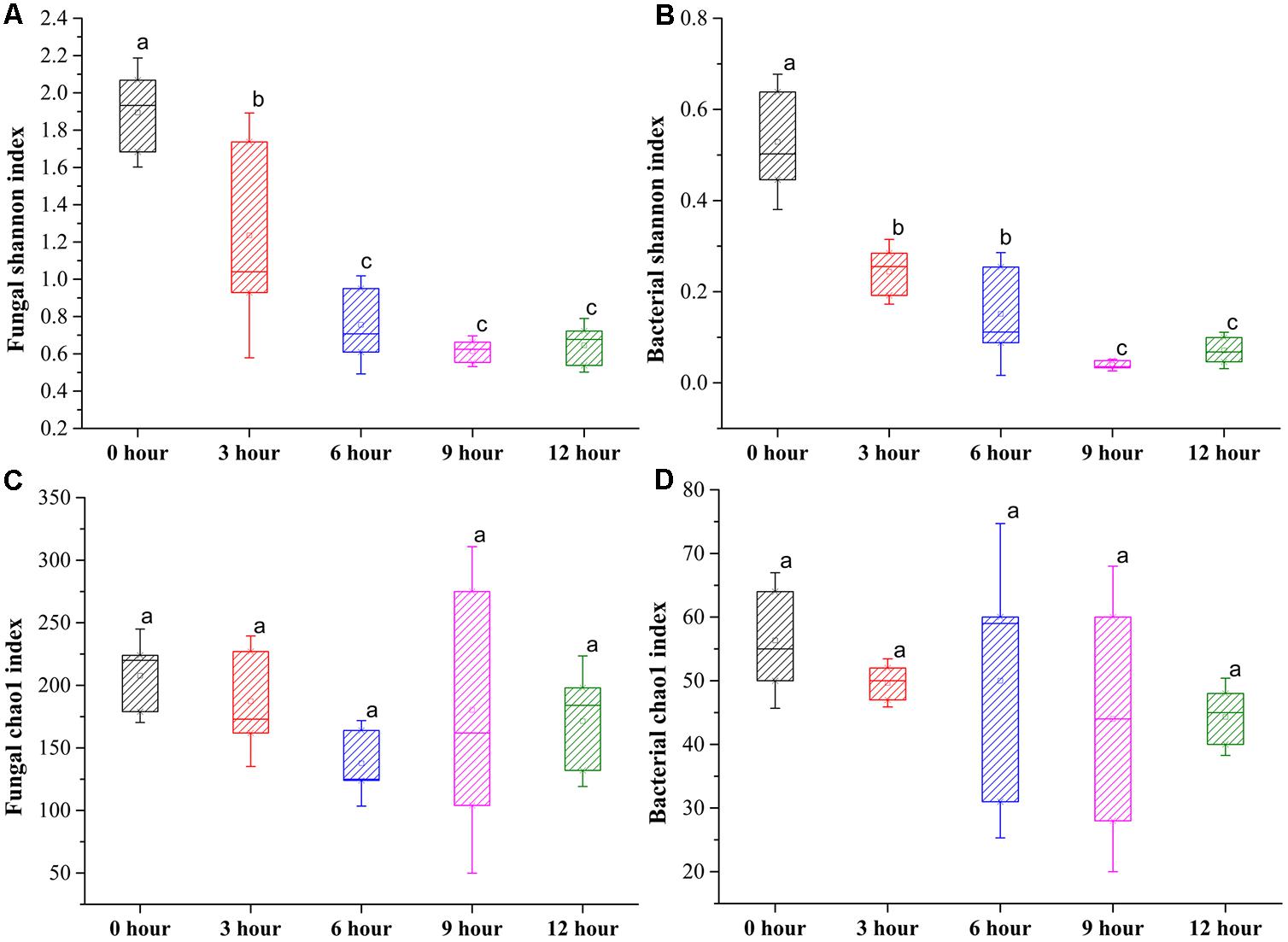
FIGURE 3. Changes in Shannon and Chao1 index of fungal (A,C) and bacterial (B,D) community during the pile-fermentation process of primary dark tea.
To assess the microbial β-diversity, the PCA and NMDS analyses were employed to investigate the overall difference among bacterial and fungal community structure during the pile-fermentation process of primary dark tea. The results indicated that all samples could be separated into two groups with based on Euclidean distance and Bray–Curtis distance, including group I (0-h stage) and group II (3-h stage-12-h stage; Figure 4).
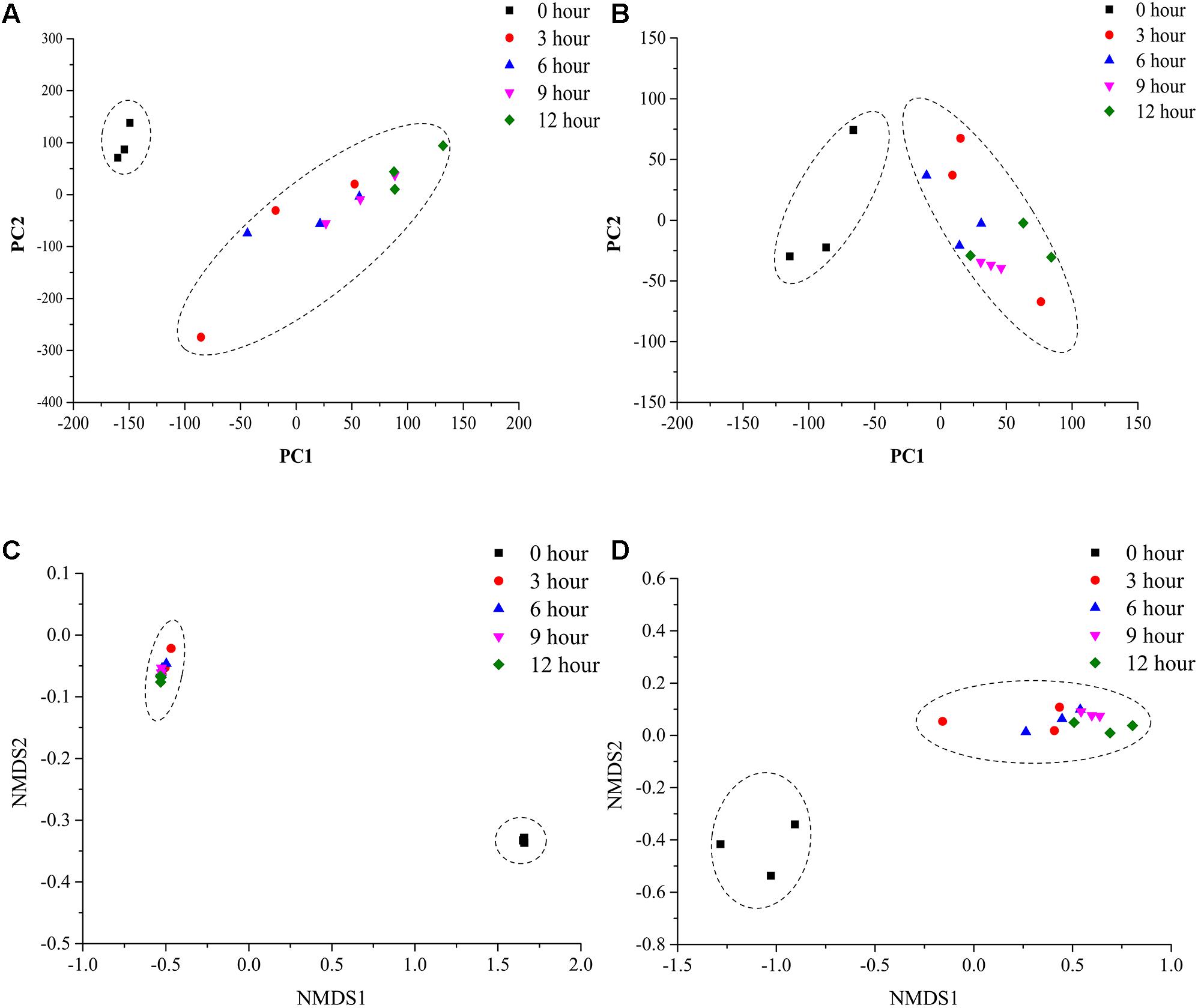
FIGURE 4. PCA and NMDS analysis of microbial communities in sample during the pile-fermentation process of primary dark tea. Fungal PCA (A), bacterial PCA (B), fungal NMDS (C), and bacterial NMDS (D).
For microbial diversity and succession, 4 phyla, 53 classes, 47 orders, 216 families, and 153 genera in fungal community and 5 phyla, 9 classes, 20 orders, 35 families, and 55 genera in bacterial community were identified during the pile-fermentation process of primary dark tea. At the genus level of fungal composition, the four most dominant fungal genera, including Cyberlindnera, Uwebraunia, Aspergillus, and Unclassified Pleosporales constituted 89.91–99.47% of all sequences. Cyberlindnera dominated the whole fermentation process, with the relative abundance dramatically increased from the initial stage (0-h stage) 32.63–95.52% in 12-h stage. However, Uwebraunia dramatically decreased from 16.37% in 0-h stage to 1.64% in 12-h stage. Aspergillus and Unclassified Pleosporales accounted for 11.57 and 29.35% of all sequences in 0-h stage and was significantly decreased to 0.33% in 3-h stage and 5.18% in 6-h stage, then remained relative stable until the end of the pile-fermentation process. Changes in some small proportions in the fungal groups Fusarium (1.45–0.45%), Eurotium (1.44–0.31%), and Peyronellaea (1.11–0.23%) were also observed during the pile-fermentation process (Figures 5A,C,E). For the bacterial composition, the dominant genera Klebsiella and Lactobacillus accounted for 88.87–99.48% during the pile-fermentation process of primary dark tea. Klebsiella dominated the whole fermentation process, with the relative abundance gradually increased from the initial stage (0-h stage) 81.99–99.06% in 12-h stage. However, Lactobacillus dramatically decreased from 6.88% in 0-h stage to 0.41% in 12-h stage. Variations in some small proportions in the bacterial groups Kluyvera (3.31–0.29%), Methylobacterium (2.02–0.04%), and Aurantimonas (1.44–0.02%) were also observed during the pile-fermentation process of primary dark tea (Figures 5B,D,F).
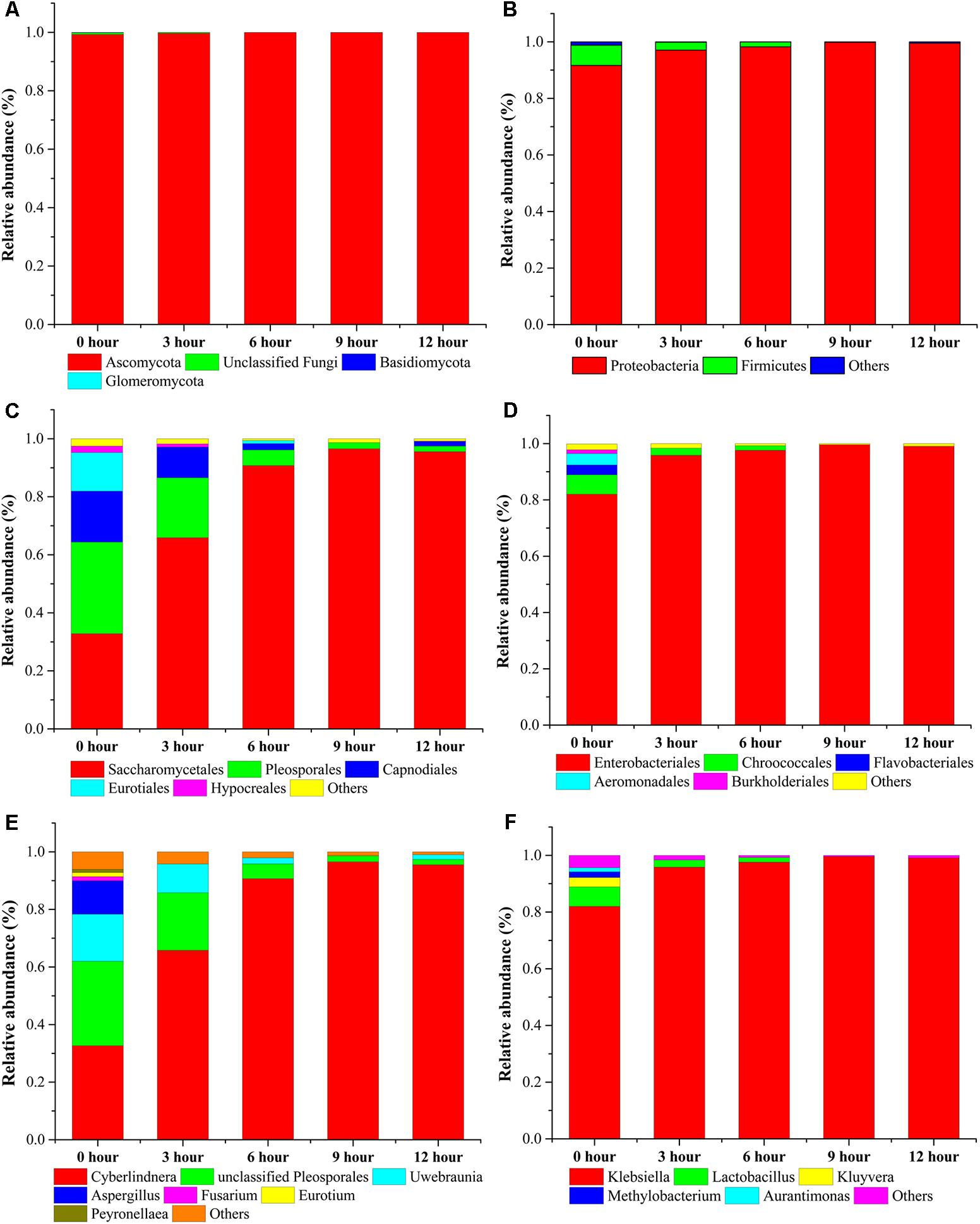
FIGURE 5. Microbial taxonomic compositions showing the microbial successions at phylum: (A) fungus, (B) bacteria; order: (C) fungus, (D) bacteria; and genus: (E) fungus, (F) bacteria level during pile-fermentation process of primary dark tea. The taxonomic abundance <1% were classified into “others.”
To understand the interactions and connectivity within microbial communities during the pile-fermentation process of primary dark tea, we identified molecular ecological networks (MENs) using a novel RMT-based approach (Deng et al., 2012). In RMT analysis, the similarity threshold was determined to be 0.79. A total of 339 pairs of significant and robust correlations (edges) were identified from 124 nodes (28 nodes for bacteria and 96 nodes for fungus) (Figure 6). Specifically, 267 pairs of positive correlations and 72 pairs of negative correlations were identified in the network. Moreover, the value of modularity index for network was 0.446 (>0.4), suggesting the network has a modular structure (Newman, 2006) and the network global properties were showed in Supplementary Table S2.
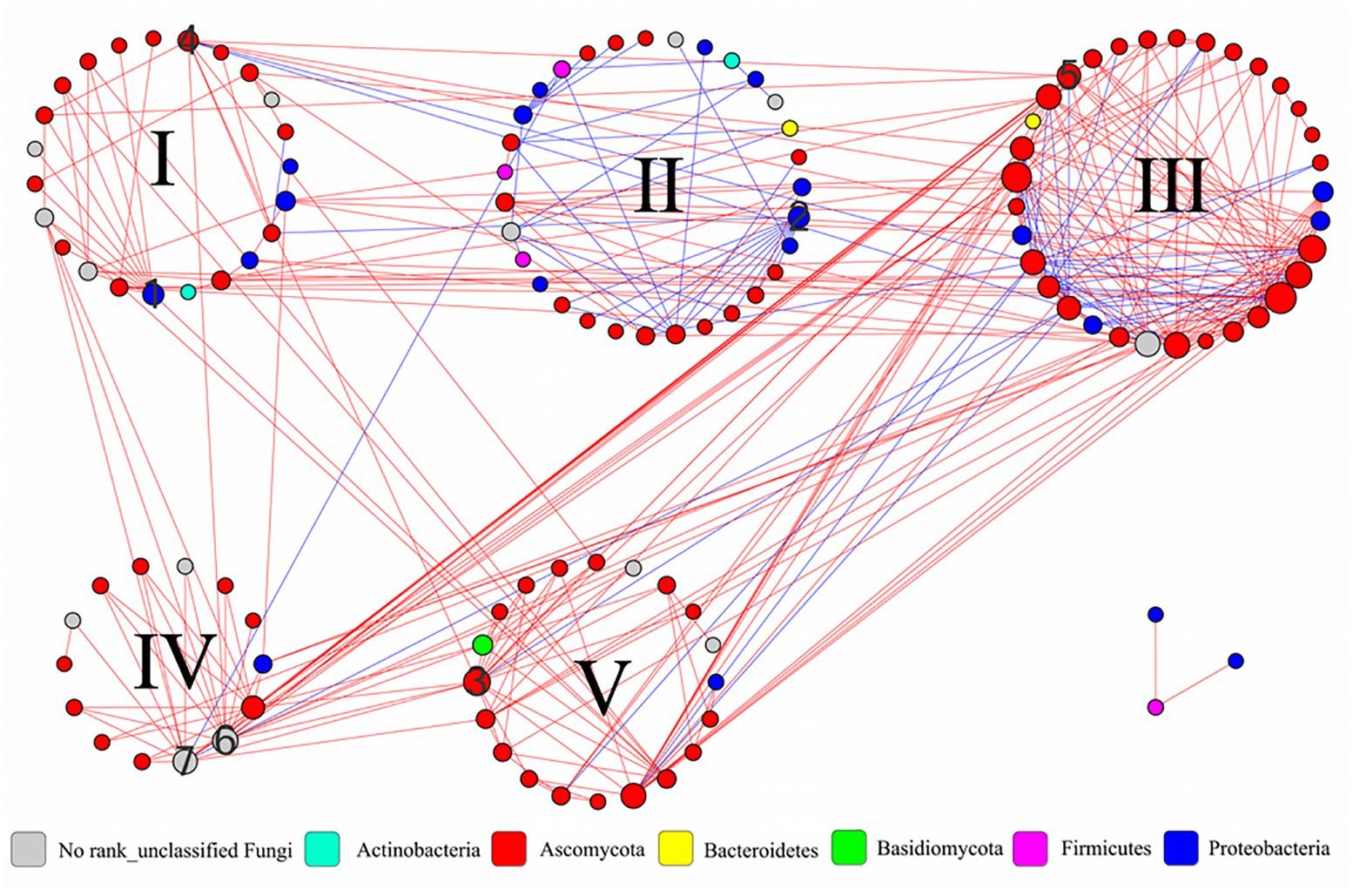
FIGURE 6. Interaction networks in the microbial communities of primary dark tea. The nodes of network were group into five modules. The co-occurring networks are colored by phylum. The size of each node is proportion to the number of connections (that is, degree), and the red edge indicates a positive interaction and the blue edge indicated a negative interaction between two nodes. The numbers are indicated as follows: 1∼3 the module hubs bOTU8, bOTU21, and fOTU7. 4∼7 the connectors fOTU21, fOTU30, fOTU58, and fOTU162. I∼V indicated the number of modules.
All nodes in the network were assigned to seven phyla, including four bacterial phyla (Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria) and three fungal phyla (Ascomycota, Basidiomycota, and Unclassified Fungi); the individual node’s properties were shown in Supplementary Table S3. All nodes could be grouped into five modules and the module compositions were shown in Supplementary Figure S3 at the genus level. The majority of nodes (117, 94.35%) were peripherals with most of their links inside their own modules during the pile-fermentation process of primary dark tea. A total of four nodes were identified as connectors, which were derived from Staninwardia (phyla Ascomycota), Saccharata (phyla Ascomycota), and two Unclassified Fungi nodes. Three nodes were identified as module hubs, which were derived from Aeromonas (phyla Proteobacteria), Acidovorax (phyla Proteobacteria), and Unclassified Pleosporales (phyla Ascomycota) (Supplementary Figure S4). These keystone species served as gatekeepers in the ecological functions of the microbial community, implying that they may play critical roles in maintaining the stability of structure and function of ecological communities during the pile-fermentation of primary dark tea.
The effects of major chemical compounds on the structure of microbial community during the pile-fermentation process of primary dark tea were evaluated by the DCA and RDA. The result of DCA indicated that the gradient length of the axis was less than 3 SD (lengths of gradient 1.543, 0.409, 0.353, 0.861). Therefore, the linear model of RDA was considered to be the appropriate ordination method for direct gradient analysis. As shown in Figure 7, the pattern of microbial communities exhibited a continuous succession along with the time gradient during the pile-fermentation process. A sharply change from 0-h stage to 3-h stage and a gradual variation occurred in from 3-h stage to 9-h stage, then a greater variation occurred in the transitions from 9-h stage to 12-h stage. RDA revealed that there was a significant relationship between the structure of microbial community and major chemical component factors (P = 0.014). All five chemical component factors (i.e., WE, TP, AA, FLA, and SS) explained 74.5% (P < 0.05) of the total variability in the microbial community. The results of Monte Carlo permutation tests showed that the variability of the microbial composition was strongly related to AA (P = 0.026). The first ordination RDA axis (axis 1), which was strongly related with AA and SS, explained 72.6% of the variability in the microbial composition. The second ordination RDA axis (axis 2) was mainly related to WE and explained 1.1% of the total variability in the microbial community.
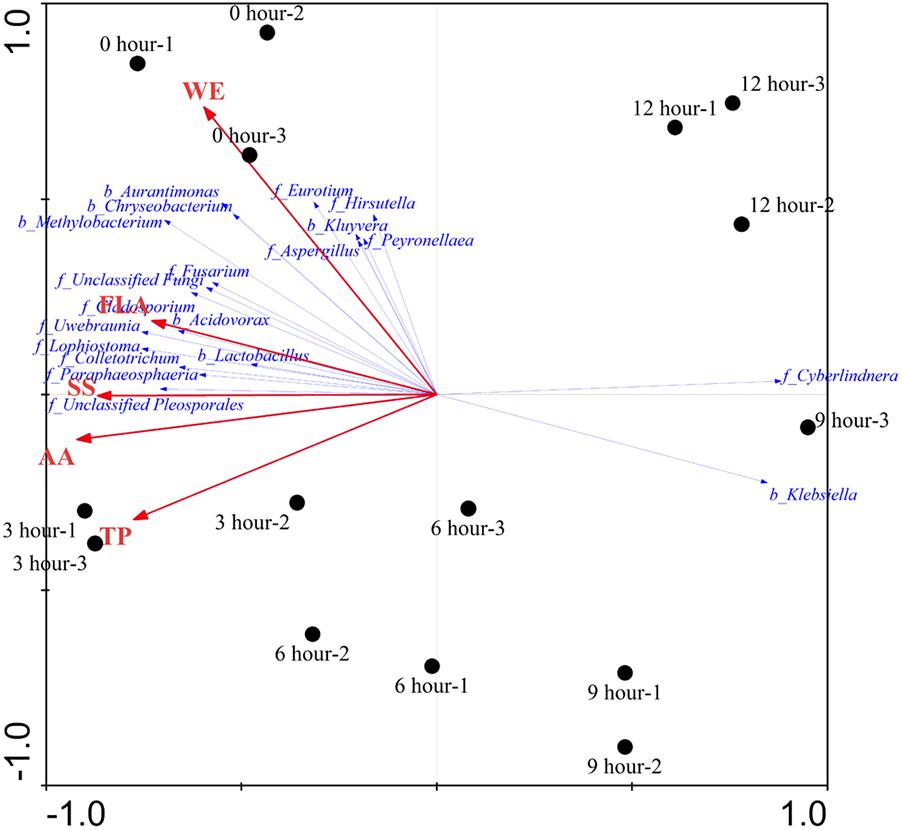
FIGURE 7. Redundancy analysis (RDA) showing the relationship between microbial structure and major chemical compounds concentrations during the pile-fermentation process of primary dark tea. The red arrow indicated the compounds variances; the blue arrow indicated the important genera of microbial communities (relative abundance >0.01%). Filled circles indicated the samples during the pile-fermentation of primary dark tea. WE, water extract; TP, tea polyphenol; AA, amino acid; FLA, flavonoid; SS, soluble sugar.
Bidirectional orthogonal partial least squares method was used to analyze the associations among microbiota, chemical compounds, and enzyme activities during the pile-fermentation process of primary dark tea. It was shown R2 and Q2 of the model was 0.938 and 0.518, respectively, thereby suggesting that O2PLS method was well fitted for analysis and prediction. Three latent variables were identified, with the microbiota (X dataset)-predictive structures accounting for 80.8% of the total variation in the X dataset, the chemical compounds and enzyme activities (Y dataset)-predictive structures accounting for 99.9% of the total variation in the Y dataset. According to the values of predictive component variable importance in the projection (VIPpred) and the correlation coefficient between microbiota and chemical compounds or enzyme activities, 123 OTUs (VIPpred > 1.0 and |r| > 0.6), including 19 bacterial OTUs and 104 fungal OTUs, had important effects on chemical compounds and enzyme activities (Figure 8A). In total, 106 OTUs including 13 bacterial OTUs and 93 fungal OTUs were correlated with chemical compounds. And 83 OTUs including 17 bacterial OTUs and 66 fungal OTUs were correlated with enzyme activities. These results suggested that the fungus were more important for primary dark tea production than bacteria.
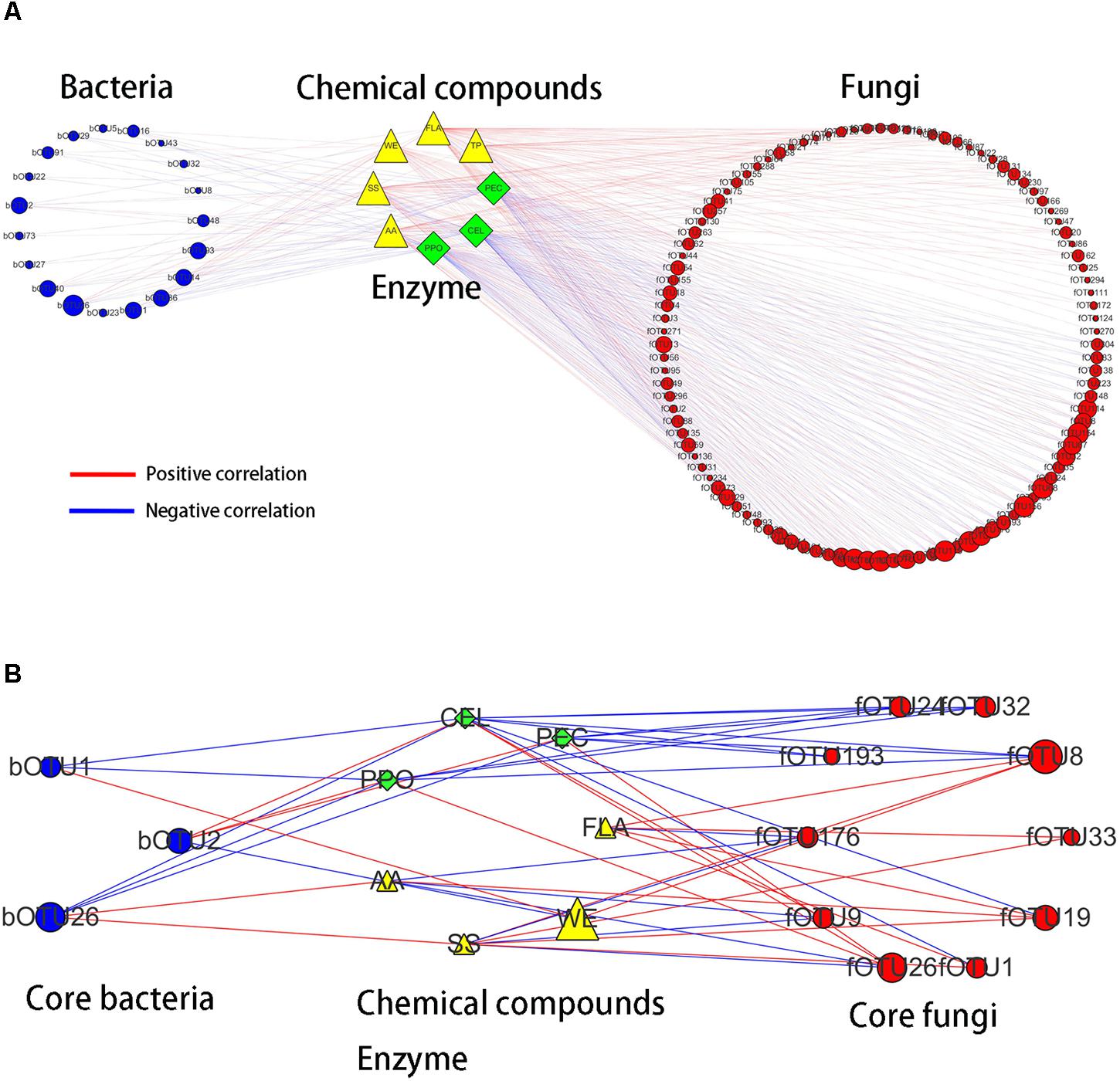
FIGURE 8. Correlation analyses among microbiota, chemical compounds, enzyme activities (A) and the core microorganisms (B) in primary dark tea during pile-fermentation process.
In order to identify the core functional microorganisms in the pile-fermentation process of primary dark tea, several conditions should be considered: (a) detected stably in pile-fermentation process; (b) the VIPpred value must be greater than 1; (c) the correlation coefficient must be greater than 0.7; (d) the number of microbe highly correlated with (|r| > 0.7) chemical compounds and enzyme activity was greater than 1. Based on these, 13 OTUs including Klebsiella (bOTU2), Aurantimonas (bOTU1), and Methylobacterium (bOTU26) of bacterial genera and Cyberlindnera (fOTU176, fOTU26, and fOTU9), Aspergillus (fOTU24), Uwebraunia (fOTU1 and fOTU8), Debaryomyces (fOTU33), Eurotium (fOTU193), Lophiostoma (fOTU19), and Peltaster (fOTU32) of fungal genera were identified as core functional microorganisms for the pile-fermentation process of primary dark tea. As shown in Figure 8B, Aurantimonas was mainly responsible for the changes of CEL, WE, and PPO. Klebsiella was mainly responsible for WE, PPO, PEC, and CEL. Methylobacterium was mainly responsible for AA, SS, PPO, PEC, and CEL. Cyberlindnera was mainly responsible for AA, SS, FLA, PPO, PEC, and CEL. Uwebraunia was mainly responsible for WE, FLA, SS, PPO, PEC, and CEL. Peltaster was mainly responsible for PPO, PEC, and CEL. Lophiostoma was mainly responsible for AA, SS, FLA, and CEL. Eurotium was mainly responsible for PEC and CEL. Aspergillus was mainly responsible for PPO, PEC, and CEL.
Diverse and rich communities of bacteria and fungi with potential health benefits have been identified from various fermented food and beverages (Bourdichon et al., 2012). Microbiota inhabiting in tea leaves is of great importance for the distinctive biochemical characteristics of primary dark tea. To the best of our knowledge, the present study is the first work to extensively investigate the structural differentiation of microbial communities by Illumina MiSeq sequencing and linked with the major chemical compounds or enzyme activity during pile-fermentation process of primary dark tea.
Our chemical and enzymatic analysis showed that all major chemical compounds content were decreased, while the activities of PPO, CEL, and PEC were increased during the pile-fermentation process of primary dark tea, except POD. Previous studies revealed that the decrease of chemical compounds mainly resulted from enzymatic reaction of secretive enzyme of microbial communities during the fermentation process (Wang et al., 1991a). The activities of PPO, CEL, and PEC sourced from tea leave were inactivated by high temperature in fixing process. Very low activity of POD was remained after the fixing process, which is a kind of hyper-thermoresistant enzyme and could not be secreted by microbial communities during the pile-fermentation of primary dark tea (Liu et al., 1991). The activities of PPO, CEL, and PEC were increased along with the growth of microbial communities and provided the effective biochemical power to the biotransformation of chemical compounds in the tea leaves during the pile-fermentation process. The catechin was oxidation by PPO, which resulted in the decrease of TP content during the process. The decrease of SS maybe resulted from the growth of microorganisms, which was used as the most important carbon source for microbial communities (Wang et al., 1991c). The decreased of AA was also used as the mainly nitrogen source for the growth of microorganisms (Wang et al., 1991b). The genera Cyberlindnera, Aspergillus, Uwebraunia, and Unclassified Pleosporales of fungus and Klebsiella and Lactobacillus of bacteria were predominant at 0-h stage of process, but after 3-h stage and 6-h stage, only Klebsiella and Cyberlindnera were still dominated in fungal and bacterial community, and maintained a relatively constant until to the end of pile-fermentation process. This shift pattern of microbial communities might be because of the environment changes in the tea pile during fermentation process. Our RDA result also revealed that AA was the important factor shaping the microbial structure. AA, caffeine, theobromine, and theophylline were the major nitrogenous compounds of primary dark tea, but caffeine, theobromine, and theophylline were heterocyclic compounds, which could hardly be used by microorganisms (Wang et al., 1991b). Therefore, AA is the most important nitrogen source of microorganisms during the pile-fermentation process of primary dark tea. Co-occurrence analysis showed that the OTUs were grouped into five modules, which might perform different functions during the pile-fermentation process of primary dark tea. In this study, most of the dominant genera were distributed in module III (Supplementary Figure S3), such as Cyberlindnera, Aspergillus, and Uwebraunia of fungal communities and Klebsiella of bacterial community, which might be mainly related to carbon and nitrogen metabolism during pile-fermentation process (Bekatorou et al., 2006; Du et al., 2006; Cho et al., 2012).
Bidirectional orthogonal partial least squares approach was used to integrate the microbiota, chemical compounds, and enzyme activities dataset in order to dig into the association among them in primary dark tea during pile-fermentation process. The results suggested that fungus were more important for primary dark tea production than bacteria. In addition, Cyberlindnera, Aspergillus, Debaryomyces, Uwebraunia, Eurotium, Lophiostoma, and Peltaster of fungal genera and Klebsiella, Aurantimonas, and Methylobacterium of bacterial genera were identified as the core microorganisms during the pile-fermentation process of primary dark tea.
The fungal genus Cyberlindnera contains 23 recognized teleomorphic species and 12 Candida species (Chang et al., 2012), in which the Cyberlindnera jadinii was applicated in the production of papaya wine (Pinrou et al., 2010) and prevented the growth of spoilage yeasts in cheese (Liu and Tsao, 2009). This specie is a close relative of Candida utilis (also referred to as Torula yeast), which was cultivated on economical waste liquors and used in the food and feed industries as a nutritious, well-tasting, and safe source of single-cell protein. Cyberlindnera (Williopsis) saturnus was reported that had the ability for producing significant amounts of sweetener xylitol from xylose and corn cob hydrolysate (Kamat et al., 2013). Moreover, some species of Cyberlindnera, which had a high capability of tannin tolerance, were also isolated from Miang (a fermented food product prepared from tea leaves) (Kanpiengjai et al., 2016). Aspergillus is the most important economically fungal genus (Samson et al., 2014), and many species within this genus are used in biotechnology for the production of various metabolites in many food fermentations (Bourdichon et al., 2012), such as soy sauce (Wicklow et al., 2007), miso (Harayama and Yasuhira, 1994), grape pomace (Botella et al., 2007), millet spirits (Takaya et al., 2007), and katsuobushi (Manabe, 2001). In all these cases, Aspergillus species which produce and secrete a variety of enzymes including α-amylases, glucoamylases, CELs, PECs, xylanases and other hemicellulase, and proteases, play an important role in the improvement of taste by decomposing proteins and/or lipids and producing unique flavors (Ward et al., 2005). Eurotium, which adopted the newly established principle “one fungus, one name” (Norvell, 2011), species formerly included in the genus Eurotium are displayed with their Aspergillus name (Samson et al., 2014), such as the dominant fungi Eurotium cristatum in Fu brick tea, which is called Aspergillus cristatum now (Ge et al., 2016; Li et al., 2017). Several indole alkaloids and indole diketopiperazine alkaloid were identified from the culture extract of Aspergillus cristatum and these compounds were proved to possess brine shrimp lethality, antibacterial activity against E. coli, radical scavenging activity against DPPH radicals, and exhibited marginal attenuation of 3T3L1 pre-adipocytes (Du et al., 2012, 2017; Zou et al., 2014). And non-mycotoxigenic strains of Aspergillus repens and Aspergillus rubrum are used as starter cultures in the manufacture of the traditional fermented food katsuobushi, from bonito (Katsuwonus pelamis) (Manabe, 2001). Debaryomyces contains 19 species (Ainsworth, 2014), in which the species Debaryomyces hansenii (anamorph Candida famata) is a significant species in foods and play an important role for the food industry, as it is used for surface ripening of cheese and meat products, over-production of riboflavin (vitamin B2), bioconversion of xylose into the sweetener xylitol, and potential synthesis of arabinitol and pyruvic acid (Satyanarayana and Kunze, 2009). And its enzyme activities improve tea quality by producing the sweet substance xylitol, vitamins, and other organic acids in Fu brick tea (Xu et al., 2011). Uwebraunia is a kind of plant pathogenic fungi, which was a synonym of Dissoconium (Crous et al., 2006), but the genus have not been well described, and the understanding of its roles in pile-fermentation systems of primary dark tea is limited.
The bacterial genus Klebsiella have shown an unbeatable production performance of 2,3-butanediol by fermentation from a wide range of substrates, including pentoses (xylose and arabinose), hexoses (glucose, mannose, and galactose), and disaccharides (sucrose, lactose, cellobiose) (Du et al., 2006; Cho et al., 2012). Klebsiella was also found in the fermentations of Pu-erh tea, soy sauce, and other fermented foods (Puerari et al., 2015; Zhao et al., 2015; Qin et al., 2016; Yang et al., 2017).
In conclusion, a complex microbial community, inhabiting the primary dark tea ecosystem, plays important roles in the formation of quality of primary dark tea during the pile-fermentation process. The dynamic changes of major chemical compounds, enzyme activities, and microbial community structure, as well as the relationships among them, were clarified during the pile-fermentation process of primary dark tea. The results indicated that the fungi made more contributions to the formation of the characteristic properties of primary dark tea than bacteria during the pile-fermentation process. And 10 core functional microbial genera were identified in primary dark tea. These findings improved our understanding on the formation mechanism of the characteristic properties of primary dark tea during the pile-fermentation process.
QL, LX, ZL, and JH designed the study. QL conducted the experiment, analyzed the data, and drafted the manuscript. SC and YOL prepared the sample and conducted the chemical and enzyme analysis. YUL helped to revise the manuscript.
The work was financially supported by the National Natural Science Foundation of China (31471706 and 41671233), National Key Research and Development Program of China (2017YFD0400803), Key Research and Development Program of Hunan Province (2018NK2035), Natural Science Foundation of Hunan Province (13JJ4067 and 2018JJ2181), Technology System Project of Tea Industry in Hunan Province, Scientific Research Fund of Hunan Agricultural University (13YJ13), and the “1515 Talent Project” of Hunan Agricultural University, Open Fund Research Project of Hunan Provincial Key Laboratory of Crop Germplasm Innovation and Utilization (16KFXM11).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Dr. Yuan Chen and Dr. Xianfeng Ma for helping analyzed the data and revising the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01509/full#supplementary-material
FIGURE S1 | Rarefaction curves of fungal OTUs (A) and bacterial OTUs (B) derived from primary dark tea.
FIGURE S2 | Rank abundance curve of fungal OTUs (A) and bacterial OTUs (B) derived from primary dark tea.
FIGURE S3 | Microbial community composition of each co-occurrence network module.
FIGURE S4 | ZP plot showing distribution of OTUs based on their module-based topological roles.
TABLE S1 | Comparison of microbial diversity estimation and coverage at 97% similarity from the sequencing analysis.
TABLE S2 | Topological properties of network during the pile-fermentation process of primary dark tea.
TABLE S3 | Individual node’s properties of network during the pile-fermentation process of primary dark tea.
Abe, M., Takaoka, N., Idemoto, Y., Takagi, C., Imai, T., and Nakasaki, K. (2008). Characteristic fungi observed in the fermentation process for Puer tea. Int. J. Food Microbiol. 124, 199–203. doi: 10.1016/j.ijfoodmicro.2008.03.008
Ainsworth, G. C. (2014). Ainsworth & Bisby’s Dictionary of the Fungi, Vol. 4, 43, 2013. IMA Fungus 5. Wallingford: CAB International.
Amann, R., and Ludwig, W. (2000). Ribosomal RNA-targeted nucleic acid probes for studies in microbial ecology. FEMS Microbiol. Rev. 24, 555–565. doi: 10.1111/j.1574-6976.2000.tb00557.x
Bekatorou, A., Psarianos, C., and Koutinas, A. A. (2006). Production of food grade yeasts. Food Technol. Biotech. 44, 407–415.
Biz, A., Farias, F. C., Motter, F. A., de Paula, D. H., Richard, P., Krieger, N., et al. (2014). Pectinase activity determination: an early deceleration in the release of reducing sugars throws a spanner in the works! PLoS One 9:e109529. doi: 10.1371/journal.pone.0109529
Botella, C., Díaz, A., Ory, I., Webb, C., and Blandino, A. (2007). Xylanase and pectinase production by Aspergillus awamori on grape pomace in solid state fermentation. Process Biochem. 42, 98–101. doi: 10.1016/j.procbio.2006.06.025
Bourdichon, F., Casaregola, S., Farrokh, C., Frisvad, J. C., Gerds, M. L., Hammes, W. P., et al. (2012). Food fermentations: microorganisms with technological beneficial use. Int. J. Food Microbiol. 154, 87–97. doi: 10.1016/j.ijfoodmicro.2011.12.030
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335. doi: 10.1038/nmeth.f.303
Chang, C. F., Liu, Y. R., Chen, S. F., Naumov, G. I., Naumova, E. S., and Lee, C. F. (2012). Five novel species of the anamorphic genus Candida in the Cyberlindnera clade isolated from natural substrates in Taiwan. Antonie Van Leeuwenhoek 102, 9–21. doi: 10.1007/s10482-012-9708-0
Cho, J. H., Rathnasingh, C., Song, H., Chung, B. W., Lee, H. J., and Seung, D. (2012). Fermentation and evaluation of Klebsiella pneumoniae and K. oxytoca on the production of 2,3-butanediol. Bioprocess Biosyst. Eng. 35, 1081–1088. doi: 10.1007/s00449-012-0691-697
Crous, P. W., Wingfield, M. J., Mansilla, J. P., Alfenas, A. C., and Groenewald, J. Z. (2006). Phylogenetic reassessment of Mycosphaerella spp. and their anamorphs occurring on Eucalyptus. II. Stud. Mycol. 55 (Suppl. C), 99–131. doi: 10.3114/sim.55.1.99
Deng, Y., Jiang, Y. H., Yang, Y., He, Z., Luo, F., and Zhou, J. (2012). Molecular ecological network analyses. BMC Bioinformatics 13, 113–113. doi: 10.1186/1471-2105-13-113
Du, C., Yan, H., Zhang, Y., Li, Y., and Cao, Z. (2006). Use of oxidoreduction potential as an indicator to regulate 1,3-propanediol fermentation by Klebsiella pneumoniae. Appl. Microbiol. Biotechnol. 69, 554–563. doi: 10.1007/s00253-005-0001-2
Du, F. Y., Li, X., Li, X. M., Zhu, L. W., and Wang, B. G. (2017). Indolediketopiperazine alkaloids from Eurotium cristatum EN-220, an endophytic fungus isolated from the marine alga Sargassum thunbergii. Mar. Drugs 15, 1–10. doi: 10.3390/md15020024
Du, F. Y., Li, X. M., Li, C. S., Shang, Z., and Wang, B. G. (2012). Cristatumins A-D, new indole alkaloids from the marine-derived endophytic fungus Eurotium cristatum EN-220. Bioorg. Med. Chem. Lett. 22, 4650–4653. doi: 10.1016/j.bmcl.2012.05.088
DuBois, M., Gilles, K. A., Hamilton, J. K., Rebers, P. A., and Smith, F. (1956). Colorimetric method for determination of sugars and related substances. Anal. Chem. 28, 350–356. doi: 10.1021/ac60111a017
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Fan, W., Tang, Y., Qu, Y., Cao, F., and Huo, G. (2014). Infant formula supplemented with low protein and high carbohydrate alters the intestinal microbiota in neonatal SD rats. BMC Microbiol. 14:279. doi: 10.1186/s12866-014-0279-2
Fu, J., Lv, H., and Chen, F. (2016). Diversity and variation of bacterial community revealed by MiSeq sequencing in chinese dark teas. PLoS One 11:e0162719. doi: 10.1371/journal.pone.0162719
Ge, Y., Wang, Y., Liu, Y., Tan, Y., Ren, X., Zhang, X., et al. (2016). Comparative genomic and transcriptomic analyses of the Fuzhuan brick tea-fermentation fungus Aspergillus cristatus. BMC Genomics 17:428. doi: 10.1186/s12864-016-2637-y
Harayama, F., and Yasuhira, H. (1994). Miso fermentation using white mutants derived from Aspergillus sojae. J. Brew. Soc. Jpn 89, 901–905. doi: 10.6013/jbrewsocjapan1988.89.901
Kamat, S., Gaikwad, S., Ravi Kumar, A., and Gade, W. N. (2013). Xylitol production by Cyberlindnera (Williopsis) saturnus, a tropical mangrove yeast from xylose and corn cob hydrolysate. J. Appl. Microbiol. 115, 1357–1367. doi: 10.1111/jam.12327
Kanpiengjai, A., Chui-Chai, N., Chaikaew, S., and Khanongnuch, C. (2016). Distribution of tannin-’tolerant yeasts isolated from Miang, a traditional fermented tea leaf (Camellia sinensis var. assamica) in northern Thailand. Int. J. Food Microbiol. 238, 121–131. doi: 10.1016/j.ijfoodmicro.2016.08.044
Li, Q., Huang, J., Li, Y., Zhang, Y., Luo, Y., Chen, Y., et al. (2017). Fungal community succession and major components change during manufacturing process of Fu brick tea. Sci. Rep. 7:6947. doi: 10.1038/s41598-017-07098-8
Li, Q., Liu, Z. H., Huang, J. A., Luo, G., Liang, Q. L., Wang, D., et al. (2013). Anti-obesity and hypolipidemic effects of Fuzhuan brick tea water extract in high-fat diet-induced obese rats. J. Sci. Food Agric. 93, 1310–1316. doi: 10.1002/jsfa.5887
Li, X. R., Ma, E. B., Yan, L. Z., Meng, H., Du, X. W., Zhang, S. W., et al. (2011). Bacterial and fungal diversity in the traditional Chinese liquor fermentation process. Int. J. Food Microbiol. 146, 31–37. doi: 10.1016/j.ijfoodmicro.2011.01.030
Liu, S. Q., and Tsao, M. (2009). Inhibition of spoilage yeasts in cheese by killer yeast Williopsis saturnus var. saturnus. Int. J. Food Microbiol. 131, 280–282. doi: 10.1016/j.ijfoodmicro.2009.03.009
Liu, Z. H., Huang, J. A., and Shi, Z. P. (1991). Dynamics of the major enzymes during the primary processing of dark green tea. J. Tea Sci. 11 (Suppl. 1), 17–22.
Liu, Z. H., Xiao, L. Z., Li, W. S., Chen, H. Q., Zeng, X. J., Xiao, W. J., et al. (2011). The Technique Specification for Raw Anhua Dark Tea Processing. Changsha: Administration of Quality and Technology Supervision of Hunan Province Press.
Manabe, M. (2001). Fermented foods and mycotoxins. JSM Mycotoxins 51, 25–29. doi: 10.2520/myco.51.25
Mukherjee, P. K., Chandra, J., Retuerto, M., Sikaroodi, M., Brown, R. E., Jurevic, R., et al. (2014). Oral mycobiome analysis of HIV-infected patients: identification of Pichia as an antagonist of opportunistic fungi. PLoS Pathog. 10:e1003996. doi: 10.1371/journal.ppat.1003996
Newman, M. E. J. (2006). Modularity and community structure in networks. Proc. Natl. Acad. Sci. U.S.A. 103, 8577–8582. doi: 10.1073/pnas.0601602103
Pinrou, L., Yuenling, O., Yu, B., Curran, P., and Liu, S. Q. (2010). Evolution of volatile compounds in papaya wine fermented with three Williopsis saturnus yeasts. Int. J. Food Sci. Tech. 45, 2032–2041. doi: 10.1111/j.1365-2621.2010.02369.x
Puerari, C., Magalhães-Guedes, K. T., and Schwan, R. F. (2015). Physicochemical and microbiological characterization of chicha, a rice-based fermented beverage produced by Umutina Brazilian Amerindians. Food Microbiol. 46, 210–217. doi: 10.1016/j.fm.2014.08.009
Qin, H., Sun, Q., Pan, X., Qiao, Z., and Yang, H. (2016). Microbial diversity and biochemical analysis of Suanzhou: a traditional Chinese fermented cereal Gruel. Front. Microbiol. 7:1311. doi: 10.3389/fmicb.2016.01311
Samson, R. A., Visagie, C. M., Houbraken, J., Hong, S. B., Hubka, V., Klaassen, C. H. W., et al. (2014). Phylogeny, identification and nomenclature of the genus Aspergillus. Stud. Mycol. 78, 141–173. doi: 10.1016/j.simyco.2014.07.004
Satyanarayana, T., and Kunze, G. (2009). Yeast Biotechnology: Diversity and Applications. Dordrecht: Springer. doi: 10.1007/978-1-4020-8292-4
Schloss, P. D., Gevers, D., and Westcott, S. L. (2011). Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One 6:e27310. doi: 10.1371/journal.pone.0027310
Takaya, Y., Furukawa, T., Miura, S., Akutagawa, T., Hotta, Y., Ishikawa, N., et al. (2007). Antioxidant constituents in distillation residue of Awamori spirits. J. Agric. Food Chem. 55, 75–79. doi: 10.1021/jf062029d
Teng, J., Gong, Z. H., Deng, Y. L., Chen, L., Li, Q., Shao, Y. Y., et al. (2017). Purification, characterization and enzymatic synthesis of theaflavins of polyphenol oxidase isozymes from tea leaf (Camellia sinensis). LWT Food Sci. Technol. 84 (Suppl. C), 263–270. doi: 10.1016/j.lwt.2017.05.065
Wang, Z. M., Lu, Z. M., Shi, J. S., and Xu, Z. H. (2016). Exploring flavour-producing core microbiota in multispecies solid-state fermentation of traditional Chinese vinegar. Sci. Rep. 6:26818. doi: 10.1038/srep26818
Wang, Z., Shi, Z., Liu, Z., Huang, J., Wen, Q., and Zhu, S. (1991a). Discussion on the mechanism of quality and flavour formation of dark green tea. J. Tea Sci. 11(Suppl. 1), 1–9.
Wang, Z., Tan, H., and Shi, L. (1991b). Dynamics of major nitrogenous compounds during the promary processing of dark green tea. J. Tea Sci. 11(Suppl. 1), 29–33.
Wang, Z., Zhang, Y., Tong, X., and Liu, Z. (1991c). Dynamics of polyphenols and carbohydrates during the primary processing of dark green tea. J. Tea Sci. 11(Suppl. 1), 23–28.
Ward, O. P., Qin, W. M., Dhanjoon, J., Ye, J., and Singh, A. (2005). “Physiology and biotechnology of Aspergillus,” in Advances in Applied Microbiology, eds J. W. B. G. M. G. Allen, I. Laskin, and S. Sima Cambridge: Academic Press), 1–75.
Wen, Q. Y., and Liu, S. C. (1991). Variation of the microorganism groups during the pile-fermentation of dark green tea. J. Tea Sci. 11(Suppl. 1), 10–16.
Wicklow, D. T., McAlpin, C. E., and Yeoh, Q. L. (2007). Diversity of Aspergillus oryzae genotypes (RFLP) isolated from traditional soy sauce production within Malaysia and Southeast Asia. Mycoscience 48, 373–380. doi: 10.1007/s10267-007-0383-3
Wood, T. M., and Bhat, K. M. (1988). Methods for measuring cellulase activities. Methods Enzymol. 160, 87–112. doi: 10.1016/0076-6879(88)60109-1
Xiong, J., Liu, Y., Lin, X., Zhang, H., Zeng, J., Hou, J., et al. (2012). Geographic distance and pH drive bacterial distribution in alkaline lake sediments across Tibetan Plateau. Environ. Microbiol. 14, 2457–2466. doi: 10.1111/j.1462-2920.2012.02799.x
Xu, A., Wang, Y., Wen, J., Liu, P., Liu, Z., and Li, Z. (2011). Fungal community associated with fermentation and storage of Fuzhuan brick-tea. Int. J. Food Microbiol. 146, 14–22. doi: 10.1016/j.ijfoodmicro.2011.01.024
Xu, J. F., Zhou, W. L., Lu, X. L., Wang, J., and Sha, H. T. (2013). Tea-Determination of Free Amino Acids Content. Beijing: Standardization Administration of The People’s Republic of China Press.
Yang, Y., Deng, Y., Jin, Y., Liu, Y., Xia, B., and Sun, Q. (2017). Dynamics of microbial community during the extremely long-term fermentation process of a traditional soy sauce. J. Sci. Food Agric. 97, 3220–3227. doi: 10.1002/jsfa.8169
Zhang, L., Zhang, Z., Zhou, Y., Ling, T., and Wan, X. (2013). Chinese dark teas: postfermentation, chemistry and biological activities. Food Res. Int. 53, 600–607. doi: 10.1016/j.foodres.2013.01.016
Zhao, M., Xiao, W., Ma, Y., Sun, T., Yuan, W., Tang, N., et al. (2013). Structure and dynamics of the bacterial communities in fermentation of the traditional Chinese post-fermented pu-erh tea revealed by 16S rRNA gene clone library. World J. Microbiol. Biotechnol. 29, 1877–1884. doi: 10.1007/s11274-013-1351-z
Zhao, M., Zhang, D. L., Su, X. Q., Duan, S. M., Wan, J. Q., Yuan, W. X., et al. (2015). An integrated metagenomics/metaproteomics investigation of the microbial communities and enzymes in solid-state fermentation of Pu-erh tea. Sci. Rep. 5:10117. doi: 10.1038/srep10117
Zhou, W. L., Xu, J. F., Lu, X. L., Wang, J., and Sha, H. T. (2013). Tea-Determination of Water Extract Content. Beijing: Standardization Administration of The People’s Republic of China Press.
Keywords: primary dark tea, food fermentation, microbial community, biochemical components, Illumina MiSeq sequencing
Citation: Li Q, Chai S, Li Y, Huang J, Luo Y, Xiao L and Liu Z (2018) Biochemical Components Associated With Microbial Community Shift During the Pile-Fermentation of Primary Dark Tea. Front. Microbiol. 9:1509. doi: 10.3389/fmicb.2018.01509
Received: 11 January 2018; Accepted: 18 June 2018;
Published: 10 July 2018.
Edited by:
Henry Jaeger, Universität für Bodenkultur Wien, AustriaReviewed by:
Qingping Zhong, South China Agricultural University, ChinaCopyright © 2018 Li, Chai, Li, Huang, Luo, Xiao and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lizheng Xiao, MTM2OTk0OTA1NkBxcS5jb20= Zhonghua Liu, emhvbmdodWEtbGl1QGhvdG1haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.