 Qiang Zheng
Qiang Zheng Qi Chen1,2
Qi Chen1,2 Yongle Xu
Yongle Xu Curtis A. Suttle
Curtis A. Suttle
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 27 June 2018
Sec. Virology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.01418
In recent metagenomic studies, single-stranded DNA (ssDNA) viruses that infect bacteria have been shown to be diverse and prevalent in the ocean; however, there are few isolates of marine ssDNA phages. Here, we report on a cultivated ssDNA phage (vB_Cib_ssDNA_P1) that infects Citromicrobium bathyomarinum RCC1878 (family Sphingomonadaceae), and other members of the genus. This is the first ssDNA phage reported to infect marine alphaproteobacteria, and represents a newly recognized lineage of the Microviridae infecting members of Sphingomonadaceae, the Amoyvirinae. The ∼26 nm diameter polyhedral capsid contains a 4,360 bp genome with 6 open reading frames (ORFs) and a 59.3% G+C content. ORF1 encodes the capsid protein and ORF3 encodes the replication initiator protein. The replication cycle is ∼5 h, followed by a burst releasing about 180 infectious particles. The closest relative of vB_Cib_ssDNA_P1 is a prophage within the genome of Novosphingobium tardaugens strain NBRC16725. Phylogenetic analysis indicates that the vB_Cib_ssDNA_P1 phage and two related prophages, as well as an environmental sequence, form a novel group within the Microviridae. Our results indicate that this is a previously unknown lineage of ssDNA viruses which also supplies a new model system for studying interactions between ssDNA phages and marine bacteria.
Viruses are the most abundant biological entities in the ocean and can influence microbial population dynamics through infection and cell lysis (Fuhrman, 1999; Weinbauer and Rassoulzadegan, 2004; Suttle, 2005), with consequent effects on nutrient cycling (Wommack and Colwell, 2000; Suttle, 2007). Most characterized viruses that infect marine bacteria, by far, are double-stranded DNA (dsDNA) phages. However, recent metagenomic studies demonstrate that single-stranded DNA (ssDNA) phages, such as those in the family Microviridae, are prevalent in diverse environments (Angly et al., 2006; Labonté and Suttle, 2013a; Székely and Breitbart, 2016). The abundances of ssDNA phages in in situ environments are underestimated because of the weak staining capacities with the small size and the single-stranded nature of their genomes (Holmfeldt et al., 2012; Brum et al., 2013). Nevertheless, the recent quantitative viromics suggested that ssDNA phages might be a minor fraction of DNA viral communities (Roux et al., 2016).
Members of the Microviridae infect bacteria, and have ∼4.5–6.1 kB positive-sense circular ssDNA genomes enclosed within small (∼30 nm) icosahedral capsids (Fane et al., 2006). The DNA is replicated using a rolling-circle mechanism, and encodes two relatively conserved genes for a capsid and replication initiator (Brentlinger et al., 2002; Labonté and Suttle, 2013b). Members of the Microviridae are usually classified into either the subfamily Bullavirinae (formally Microviridae including genus Microvirus) or Gokushovirinae, based on the structure of the viral particle and composition of the genome (Brentlinger et al., 2002; Krupovic et al., 2016). Gokushovirinae have genome ranging from 4.4 to 4.9 kB including nine genes, whereas Bullavirinae contain 11 genes with genome size from 5.4 to 6 kB (Fane et al., 2006; Pearson et al., 2016). Previous studies indicated that members of this family have a limited host-range. Viruses in the genus Microvirus infect cells in the family Enterobacteriaceae (e.g., Escherichia coli) (Chipman et al., 1998; Brentlinger et al., 2002) and are typified by the microvirus phiX174. In contrast, members of the subfamily Gokushovirinae are specialized to infect intracellular parasites, such as Chlamydia, Bdellovibrio, and Spiroplasma (Chipman et al., 1998; Brentlinger et al., 2002; Everson et al., 2002; Garner et al., 2004), although recent isolation-independent investigations have revealed gokushoviruses associated with Gammaproteobacteria (Roux et al., 2012a). In addition, Microviridae-like genomes have been discovered in the genomes of bacteria within the Bacteroidetes (Krupovic and Forterre, 2011), suggesting a lysogenic lifestyle.
Phages belonging to the Microviridae were once thought to have a limited distribution; however, metagenomic studies indicate they are likely widespread across diverse habitats. For example, viruses belonging to the Microviridae have been detected in seawater (Angly et al., 2006; Tucker et al., 2011; Holmfeldt et al., 2012; Labonté and Suttle, 2013b), methane seeps (Bryson et al., 2015), freshwater (López-Bueno et al., 2009; Roux et al., 2012a,b; Zhong et al., 2015), Sphagnum-dominated peatland (Quaiser et al., 2015), sewage and sediment (Hopkins et al., 2014), the human gut and feces (Roux et al., 2012b), and stromatolites (Desnues et al., 2008). Only eight ssDNA phages have been isolated for marine heterotrophic bacteria (Holmfeldt et al., 2012), and all infect Cellulophaga spp. (Bacteroidetes). In addition, a microvirus-like phage has been induced from a marine Synechococcus isolate (McDaniel et al., 2006). Therefore, although there is a broad distribution of ssDNA viruses in the sea, there are few representative isolates.
Members of the genus Citromicrobium are marine aerobic anoxygenic photoheterotrophs (AAPs) in the family Sphingomonadaceae and the class Alphaproteobacteria (Yurkov et al., 1999; Zheng et al., 2016). Citromicrobium-like AAPs mainly occur in the oligotrophic ocean and are more abundant in the upper twilight zone (150–200 m depth) than in subsurface waters (5 and 25 m) of the western Pacific Ocean (Zheng et al., 2015). Prophage are widely present in Citromicrobium strains, as revealed by a comparison of eight Citromicrobial genomes that share identical 16S rRNA sequences but were isolated from diverse environments (Zheng et al., 2014, 2016). Despite the importance of viruses as agents of mortality, and in nutrient cycling in the oceans (Fuhrman, 1999; Suttle, 2005, 2007), there are still relatively few model systems for viruses infecting marine bacteria; this is especially true for photoheterotrophs. Moreover, the limited number of viruses that have been isolated, which infect marine photoheterotrophs (members of genera Roseobacter and Dinoroseobacter), have been tailed phages belonging to the order Caudovirales (Zhang and Jiao, 2009; Ji et al., 2015). Here, we report the first ssDNA phage found to infect marine alphaproteobacterial photoheterotrophs (Citromicrobium spp.). It represents a previously unrecognized lineage of ssDNA viruses (Amoyvirinae) that infect members of the Sphingomonadaceae.
Citromicrobium bathyomarinum RCC1878, an isolate from the Mediterranean Sea, was provided by Dr. Christian Jeanthon at the Roscoff Culture Collection, and used as a host for phage isolation. As well, the following 13 alphaproteobacterial strains were used to test host-range. Four of them were purchased from the Deutsche Sammlung von Mikroorganismen und Zellkulturen: Dinoroseobacter shibae DLF 12, Roseobacter denitrificans DSM 7001, Erythrobacter litoralis DSM 6997, and Erythrobacter longus DSM 8509, as well, Roseibacterium beibuensis JLT1202, Roseibium sp. 1Q1, Porphyrobacter sp. YT40, and six strains of Citromicrobium spp. (JL31, JL89-1, JL477, JL1010, JL1351, and JL2201) from our lab at Xiamen University. The cross-infectivity was tested by double-agar layering and spotting phage suspensions onto a bacterial lawn. All strains were grown in rich organic medium (containing 1.0 g yeast extract, 1.0 g Bacto Peptone, and 1.0 g sodium acetate per liter of artificial seawater with vitamins and trace element) (Yurkov et al., 1999) at 28°C.
Water samples were collected from a marine aquaculture site around Xiamen coast, China, on 24 January 2016 and immediately filtered through 0.2-μm pore-size polycarbonate membrane filters (Millipore, Bedford, MA, United States). A 100 mL of filtrate was added to 150 mL of exponentially growing bacterial culture and incubated overnight at 28°C and 160 rpm in a shaker. Cultures were then centrifuged at 10,000 ×g for 20 min to remove bacteria. A 100 μL subsample of cell-free supernatant was added to 1 mL of exponentially growing culture, and then plaque assayed as described by Suttle and Chen (1992). Each phage isolate was plaque purified at least three times.
One-step growth assays of the phage isolates were performed according to Middelboe et al. (2010). Briefly, the viruses were suspended in SM buffer (10 mM NaCl, 50 mM Tris, 10 mM MgSO4, and 0.1% gelatin, pH 7.5) and added to triplicate exponentially growing cultures (OD = 0.3) at a multiplicity of infection (MOI) of 0.1. After 10 min, cells were removed by centrifugation, leaving the unadsorbed viruses, which were poured off with the supernatant. The bacteria in the pellet were resuspended in medium and 50 μL were transferred to 50 mL of medium and incubated at 28°C. Subsamples were taken every hour for 7 h. The number of plaque forming units (PFUs) was determined by plaque assay. The latent period was determined from the time of phage adsorption to the appearance of the first free phage in the medium during a one-step growth experiment. Concurrently, the burst size was calculated from the number of PFUs (i.e., free viruses) at the end of the first viral rise divided by the PFUs (i.e., infected cells) at the initial time.
The phage suspension was added to 1 L of host culture at an OD600 of 0.2–0.3, and an MOI of 0.01, and incubated overnight at 28°C and 180 rpm in a shaker. Phage particles in lysates were harvested and purified as described by Chen et al. (2006) with some modifications. Briefly, phage lysates were treated with 2 mg DNase I and 2 mg RNase A at room temperature for 1 h. After digestion, the NaCl concentrations of the lysates were adjusted to 1M and incubated at 4°C for 1 h. The treated lysates were centrifuged at 10,000 ×g for 15 min in a Beckman AR centrifuge. Supernatants were collected and filtered through a 0.2-μm pore-size filter to remove the remaining cells and debris at 10,000 ×g, 4°C for 15 min in a Beckman AR centrifuge. The filtrate was then concentrated by polyethylene glycol 8000 precipitation. The precipitate was resuspended in 6 mL SM buffer and incubated overnight at 4°C. Subsequently, the phage suspensions were overlaid onto 1.3–1.6 w/v CsCl gradients at 0.1 intervals and centrifuged for 24 h at 200,000 ×g using a SW41 Ti rotor in a Beckman Coulter Optima L-100 XP Ultracentrifuge. The visible phage bands (in 1.5 w/v CsCl density) were extracted, and the CsCl in the phage suspensions was removed by centrifugal ultrafiltration.
One drop of the CsCl-purified phage suspension was adsorbed to a Formvar/carbon-coated 200-mesh copper grid for 10 min and negatively stained with 0.5% (wt/vol) aqueous uranyl acetate in the dark for 30 s. After drying for 2 h, the grid was examined using a JEM-2100 transmission electron microscope at 80 kV. Images were taken using the GATAN INC CCD image transmission system.
CsCl-purified phage were treated with a proteinase K cocktail (100 μg/mL proteinase K, 50 mM Tris, 25 mM EDTA, and 1% SDS) at 55°C for 3 h. The phage nucleic acid was extracted using the phenol/chloroform method. The ssDNA was then isopropanol-precipitated, washed with ethanol, and dissolved in TE buffer (10 mM Tris, 1 mM EDTA). Phage DNA was amplified using multiple-displacement amplification to convert ssDNA into dsDNA and then resuspended in TE buffer. To identify the nature of the phage genome, nucleic acid digestion using different enzymes was performed. Exonuclease S1 which digests ssDNA and restriction enzymes including Acc I, Alu I, Dpn I which digest dsDNA were used to determine whether the genome was single- or double-stranded. PCR method and BAL-31 nuclease digestion of the phage genome was used to testify whether the genome was linear, circular or circularly permuted (Yoshida et al., 2008).
The genome was sequenced by the Illumina MiSeq system for libraries with insert sizes of ∼395 bp. Paired-end reads of average 250-bp length were assembled using Velvet (v2.8) (Zerbino and Birney, 2008), with coverage >1,000×. Open reading frames (ORFs) were predicted using ORF Finder1 and GeneMarkS (Besemer and Borodovsky, 1999). Translated ORFs were compared with known protein sequences using BLAST (Altschul et al., 1990).
The purified phage particles were treated with STD buffer (4% SDS, 100 mM DTT, 150 mM Tris–HCl pH 8.0) in a water bath at 99°C for 10 min. After centrifugation (16,000 ×g, 4°C, 30 min), the supernatants were stored at -80°C for further proteomic analysis. Protein digestion was performed according to a previously described FASP procedure (Wiśniewski et al., 2009). Proteome analysis was performed on Obitrap Fusion mass spectrometer (Thermo Finnigan, San Jose, CA, United States) that was coupled to an Easy-nLCTM 1000 (Thermo Fisher Scientific, Waltham, MA, United States). MS/MS spectra were searched using a MASCOT engine (Matrix Science, London, United Kingdom; version 2.2) against the phage genome.
The complete major capsid amino acids sequences were used to construct a phylogenetic tree. All sequences collected from the NCBI database were aligned using Clustal X. Columns with gaps were deleted. Maximum-likelihood phylogenetic trees were constructed using RAxML 7.3.0 (Stamatakis, 2006) with the “PROTGAMMALG” model, which assumes that amino acid substitution rates among 289 sites follow a gamma distribution. The phylogenetic trees were supported by bootstrap analysis for the resampling test with 100 replicates.
The GenBank accession number assigned to the complete vB_Cib_ssDNA_P1 is MG214764.
In order to isolate phage of marine photoheterotrophic bacteria, we used C. bathyomarinum strain RCC1878 as a host to screen for viruses. Its genome contains a complete prophage, implying that it is subject to viral infection. Screening against this host resulted in the isolation of a ssDNA virus that belongs to a previously unrecognized lineage of phages (Amoyvirinae) that infect members of the Sphingomonadaceae. A detailed description of this virus is discussed in detail below.
Phage vB_Cib_ssDNA_P1 was isolated from surface water near a coastal aquaculture site using the double-agar layer method, and was found to produce large, clear, round plaques with regular edges. The phage has a polyhedral capsid (∼26 nm in diameter) that is significantly smaller than those of described marine dsDNA phages (podo-, sipho-, and myoviruses), and lacks a tail (Figure 1A). Morphologically, it was similar to members of the Microviridae. According to the nucleic acid digestion result, the phage DNA could be digested by S1 nuclease rather than by restriction enzymes Acc I, Alu I, and Dpn I. This proved its genome was single-strand nucleic acids.
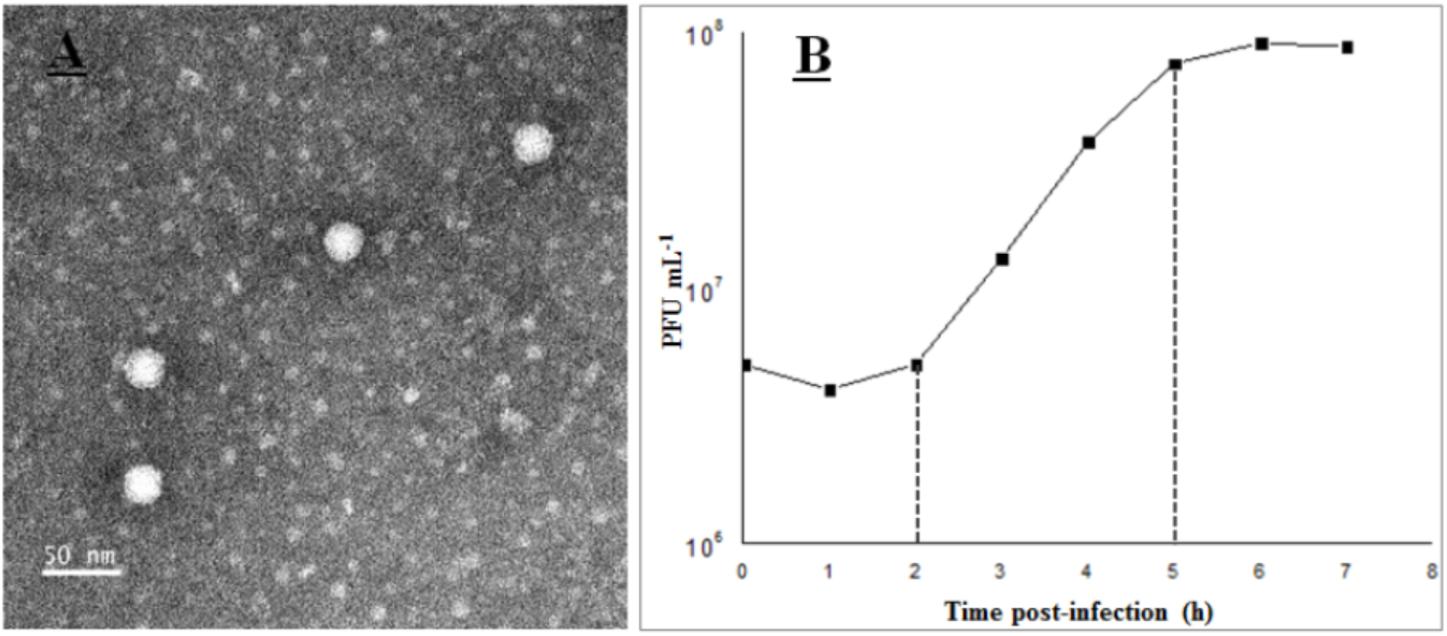
FIGURE 1. Transmission electron microscopy images (A) and one-step curve (B) of phage vB_Cib_ssDNA_P1.
The growth characteristics of vB_Cib_ssDNA_P1 were evaluated by a one-step growth on C. bathyomarinum RCC1878 (Figure 1B). The first 2 h of the ∼5 h replication cycle were classified as the early stage, when almost no viral particles were released. This stage comprises the latent phase, when phages adsorb and inject their genomes into the host cells, and biosynthesis of the viral components occurs, but phage particles are not released. In the subsequent late stage, between ∼3 and 5 h after adsorption, progeny viral particles are assembled and released. The number of released phage particles reached a plateau at 5 h after infection, with a burst size of ∼180 viral particles per infected cell.
To further investigate the new virus, its genome was sequenced. The genome of vB_Cib_ssDNA_P1 is 4,360 bp of circular, ssDNA, which is relatively small among the Microviridae (∼4.5–6.1 kB). The G+C content is 59.3%, lower than the 64.8% of its host, C. bathyomarinum RCC1878 (Zheng et al., 2016). The six identified ORFs in vB_Cib_ssDNA_P1, comprised 92.4% of the genome (Table 1).
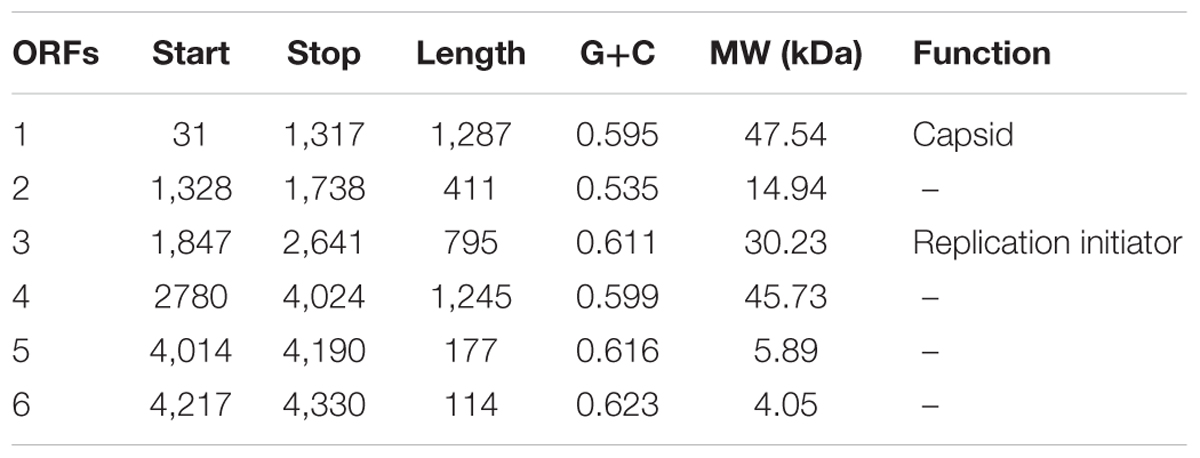
TABLE 1. Predicted ORFs and their annotation.
High-resolution LC-mass spectrometry was used to analyze proteomic characterization of virion particles. Only one protein was identified, and it is the most abundant structural protein with the molecular weight of 47.5 kDa (Table 1 and Supplementary Figure S1). Furthermore, all sequenced peptides matched ORF1. So here, ORF1 was annotated as major capsid protein.
Based on similarity to other sequences in GenBank, ORF3 encodes the replication initiator protein; whereas, ORFs 2, 5, and 6 had no significant matches and a relatively wide range of G+C content from 53.5 to 62.3%. ORF4 is a conserved hypothetical gene.
Similarity analysis demonstrated that the closest matches for ORFs 1, 3, and 4, are with the genome of Novosphingobium tardaugens strain NBRC16725, a bacterium that belongs to the same family as C. bathyomarinum RCC1878 (Sphingomonadaceae) (Figures 2A,B). These results suggest that there is a ssDNA prophage (∼4,207 bp with 57.9% G+C content) in the genome of N. tardaugens NBRC16725 (accession no.: BASZ01000013; position: from 78287 to 82493). Residual gene fragments that display significant amino-acid identities with ORFs 1 (38%, with BLASTP e-value 1e-78) and 4 (35%, 7e-11) are also found in genome of the bacterium, Sphingomonas sp. 67-36, which contains an incomplete ssDNA prophage (accession no.: MKSU01000016; position: from 84561 to 88136). In addition, an environmental ssDNA viral genome (SOG00694, accession no. JX904102) from the Strait of Georgia, British Columbia is 3,926 bp in length with a 58.6% G+C content, and displays ORFs similar to ORFs 1 (42%, 4e-83) and 3 (52%, 1e-55) (Figure 2C) (Labonté and Suttle, 2013a). ORFs 1 and 3 also have homologous genes with related ssDNA viral genomes (Figure 2), while ORF 4 shows gene fragment recombination. The other small genes of vB_Cib_ssDNA_P1 (ORFs 2, 5, and 6) have no homologs within the other two genomes (Figure 2).
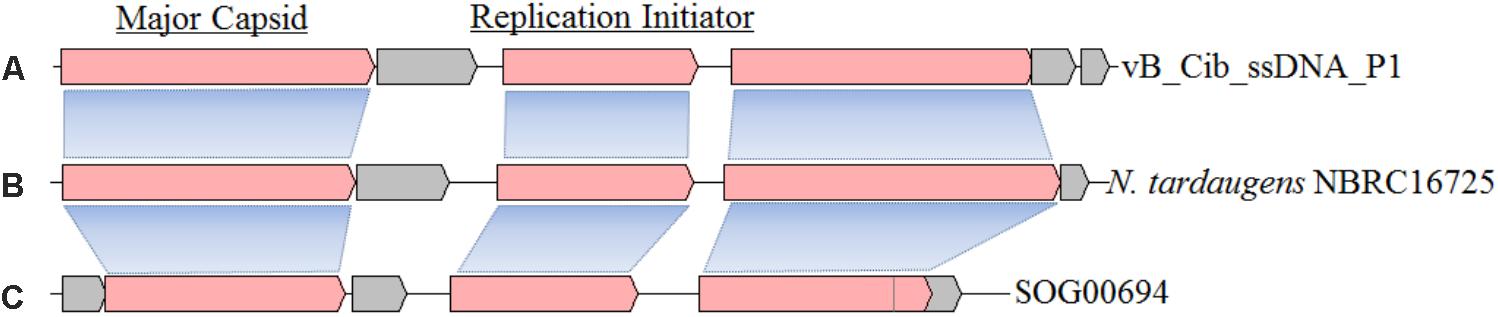
FIGURE 2. The structure and gene compositions of phage vB_Cib_ssDNA_P1 (A) and two other close relatives, including a prophage found in Novosphingobium tardaugens NBRC 16725 (accession no. BASZ01000013) (B) and the environmental ssDNA viral sequence SOG00694 (accession no. JX904102) (C).
To assess the range of bacteria that can serve as hosts for vB_Cib_ssDNA_P1, we applied the virus to fourteen related alphaproteobacterial strains using double-agar layering and spotting phage suspensions. As shown in Table 2, all seven Citromicrobium spp. strains, which shared identical 16S rRNA sequences, were susceptible to vB_Cib_ssDNA_P1 infection; however, no close-relatives belonging to the genus Erythrobacter or other genera could be infected, indicating that this phage has a relatively narrow host-range. Nevertheless, only strain RCC 1878 formed large circular clear plaques (∼1 cm diameter) 24 h after infection. Plaques in the other six Citromicrobium strains after 24 h were relatively small (<0.3 cm diameter), suggesting that strain RCC1878 may be the preferred host.
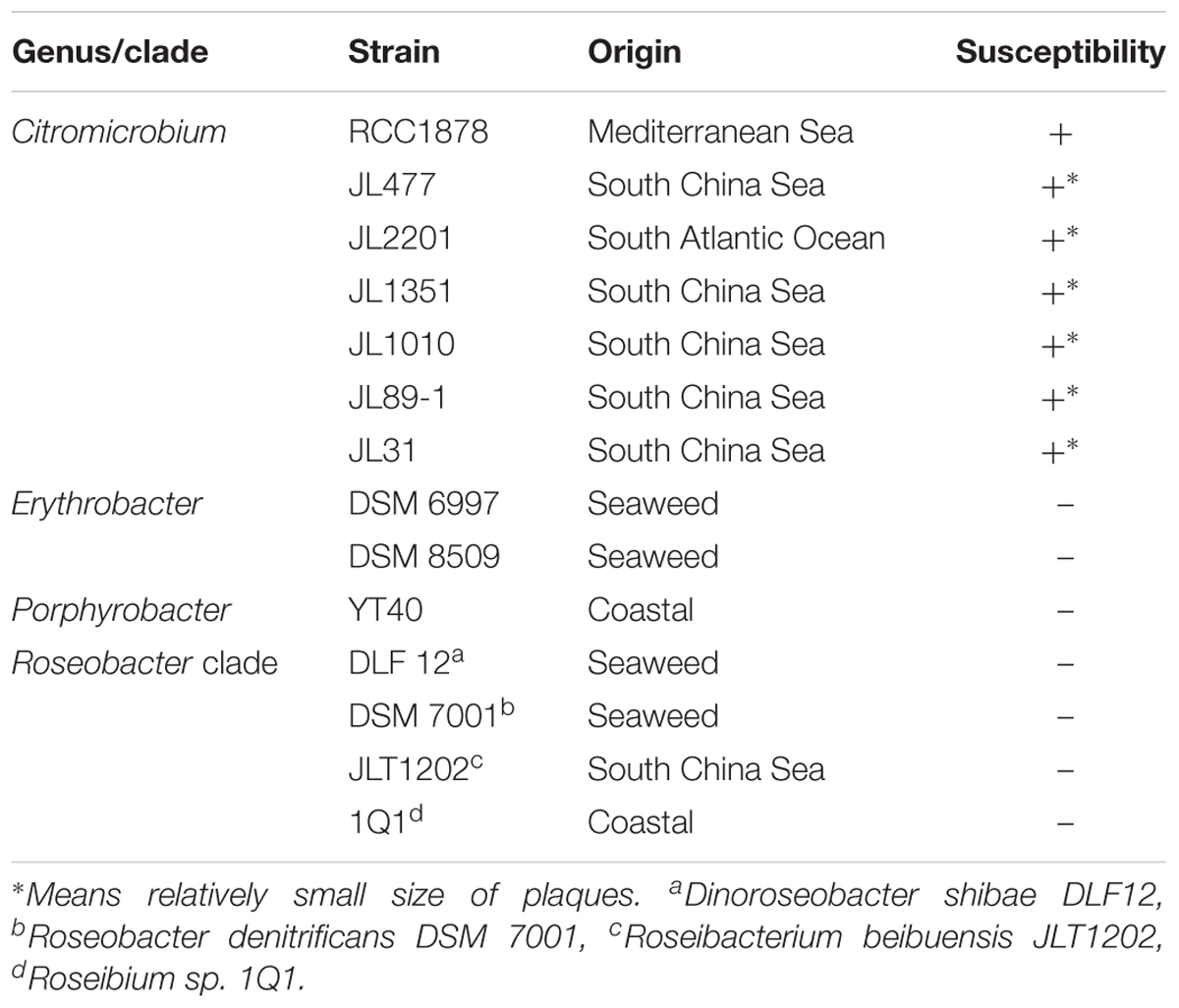
TABLE 2. Bacterial susceptibility to phage vB_Cib_ssDNA_P1.
To explore the evolution and to classify vB_Cib_ssDNA_P1, we performed a phylogenetic analysis. Based on the phylogeny of the major capsid protein sequences, the phage vB_Cib_ssDNA_P1 and its three closest relatives form a novel group within the Microviridae (Figure 3). The host “Citromicrobium” belongs to the family Sphingomonadaceae, while the complete and incomplete related ssDNA prophages are also found in the genomes of other member of the family, N. tardaugens NBRC16725 and Sphingomonas sp. 67-36. To date, the known hosts for Amoyvirinae are members of the Sphingomonadaceae, although the host associated with the environmental metagenomic sequence is unknown. The absence of integrase-related genes in the prophage suggests that Amoyvirinae rely on cellular chromosome dimer resolution machinery (Krupovic and Forterre, 2011).
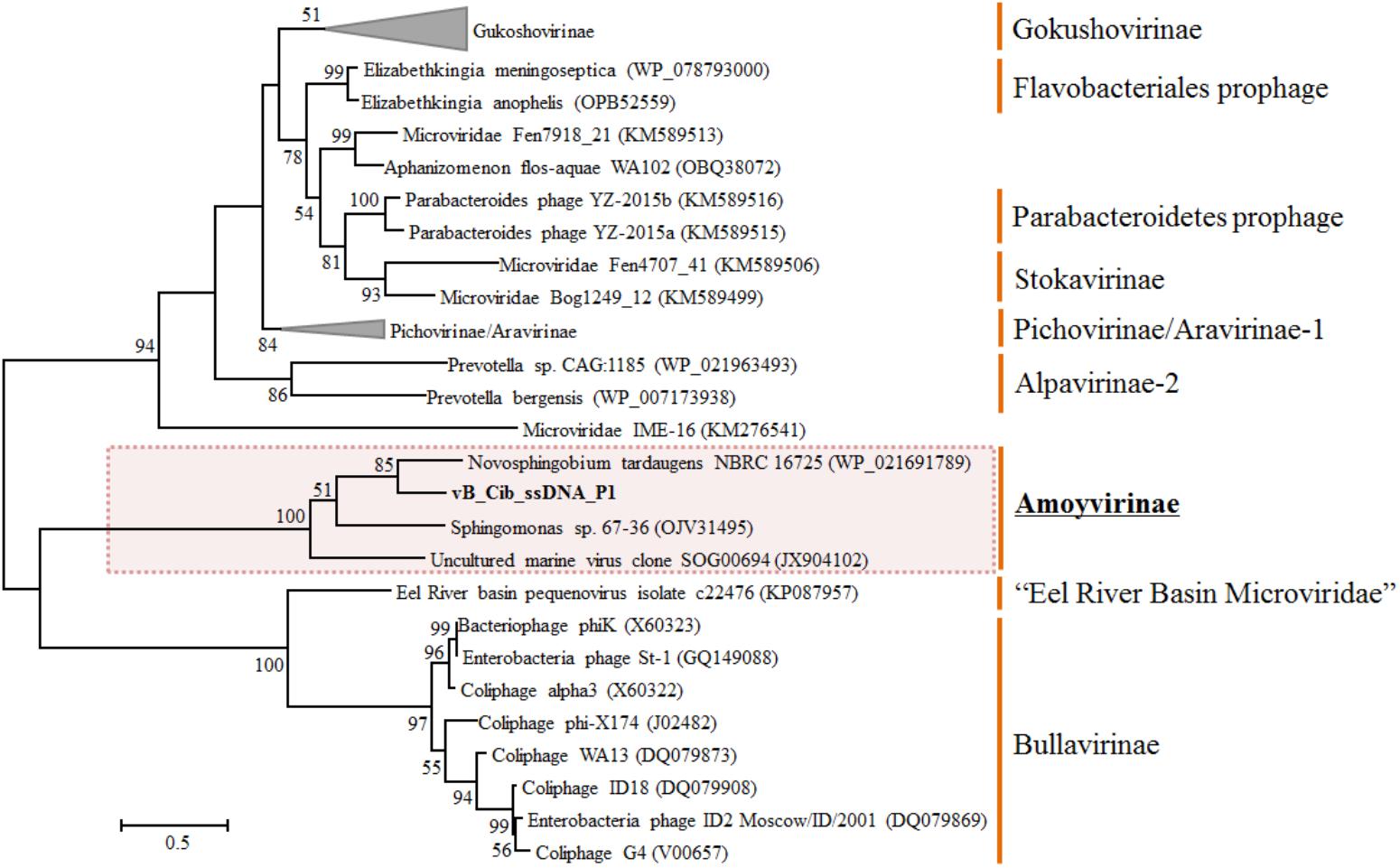
FIGURE 3. Unrooted maximum-likelihood phylogenetic tree based on the full-length major capsid inferred amino-acid sequences in Microviridae genomes. Bootstrap percentages from maximum-likelihood (100 replications) are shown. The scale bar represents a distance of 0.2 substitutions per site.
Based on phylogenetic analyses of the capsid and replication initiator sequences, which display the similar tree topology, Amoyvirinae are phylogenetically related to the Eel River Basin Microviridae (Bryson et al., 2015) (Figure 3 and Supplementary Figure S2). Our data are consistent with Amoyvirinae being a divergent lineage within the Microviridae.
Aerobic anoxygenic photoheterotrophs, as an important functional group, comprise up to 20% of total bacterioplankton in the coastal oceans, and play significant roles in marine carbon and energy cycling (Jiao et al., 2010; Koblížek, 2015). However, only limited number of phages infecting AAPs have been isolated to date, and they are all tailed phages mainly infecting members of genera Roseobacter, Dinoroseobacter, and Erythrobacter (Zhang and Jiao, 2009; Ji et al., 2015; Lu et al., 2017). In addition, an inducible sipho-like bacteriophage was isolated from Citromicrobium sp. JL354 (Zheng et al., 2014). Here, the ssDNA phage vB_Cib_ssDNA_P1 supplies a new model system for studying the interactions between viruses and photoheterotrophs in the ocean.
So far, isolates of Microviridae are strictly lytic (Fane et al., 2006; Holmfeldt et al., 2013); however, Microviridae-related prophages have been found in genomes within the order Bacteroidetes (Krupovic and Forterre, 2011). Here, we present a ssDNA virus (vB_Cib_ssDNA_P1) that infects a marine alphaproteobacterium, and its closely related prophage in N. tardaugens NBRC16725, the first prophage found in an alphaproteobacterium. Collectively, these viruses define a new lineage of ssDNA viruses, the Amoyvirinae, which infect an evolutionary and ecologically important group of bacteria. The significance of these viruses in nature remains to be elucidated.
QZ, CS, and NJ conceived and designed the experiments. QC and YX conducted the experiments. QZ and QC analyzed the data. All of the authors assisted in writing the manuscript, discussed the results, and commented on the manuscript.
This work was supported by the National Key Research Programs (2016YFA0601400), the National Natural Science Foundation of China (NSFC) project (41776145, 41306126, 91751207, and 91428308), the China National Offshore Oil Corp (CNOOC) grants CNOOC-KJ125FZDXM00TJ001-2014, and the Fundamental Research Funds for the Central Universities (20720170107).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would thank Prof. Andrew S. Lang and Dr. Xu Zhong for their useful comments and suggestions and Prof. Rui Zhang for supplying VC samples to this work.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01418/full#supplementary-material
FIGURE S1 | SDS–PAGE of vB_Cib_ssDNA_P1 proteins. Lane M, molecular weight standards; Lane CQ, vB_Cib_ssDNA_P1.
FIGURE S2 | Unrooted maximum-likelihood phylogenetic tree based on the inferred amino-acid sequences of full-length replication initiator in Microviridae genomes. Bootstrap percentages from 100-replicate maximum-likelihood trees are shown. The scale bar represents a distance of 0.2 substitutions per site.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Angly, F. E., Felts, B., Breitbart, M., Salamon, P., Edwards, R. A., Carlson, C., et al. (2006). The marine viromes of four oceanic regions. PLoS Biol. 4:e368. doi: 10.1371/journal.pbio.0040368
Besemer, J., and Borodovsky, M. (1999). Heuristic approach to deriving models for gene finding. Nucleic Acids Res. 27, 3911–3920. doi: 10.1093/nar/27.19.3911
Brentlinger, K. L., Hafenstein, S., Novak, C. R., Fane, B. A., Borgon, R., Mckenna, R., et al. (2002). Microviridae, a family divided: isolation, characterization, and genome sequence of phiMH2K, a bacteriophage of the obligate intracellular parasitic bacterium Bdellovibrio bacteriovorus. J. Bacteriol. 184, 1089–1094. doi: 10.1128/jb.184.4.1089-1094.2002
Brum, J. R., Schenck, R. O., and Sullivan, M. B. (2013). Global morphological analysis of marine viruses shows minimal regional variation and dominance of non-tailed viruses. ISME J. 7, 1738–1751. doi: 10.1038/ismej.2013.67
Bryson, S. J., Thurber, A. R., Correa, A. M., Orphan, V. J., and Vega, T. R. (2015). A novel sister clade to the enterobacteria microviruses (family Microviridae) identified in methane seep sediments. Environ. Microbiol. 17, 3708–3721. doi: 10.1111/1462-2920.12758
Chen, F., Wang, K., Stewart, J., and Belas, R. (2006). Induction of multiple prophages from a marine bacterium: a genomic approach. Appl. Environ. Microbiol. 72, 4995–5001. doi: 10.1128/AEM.00056-06
Chipman, P. R., Agbandjemckenna, M., Renaudin, J., Baker, T. S., and Mckenna, R. (1998). Structural analysis of the Spiroplasma virus, SpV4: implications for evolutionary variation to obtain host diversity among the Microviridae. Structure 6, 135–145. doi: 10.1016/S0969-2126(98)00016-1
Desnues, C., Rodriguezbrito, B., Rayhawk, S., Kelley, S., Tran, T., Haynes, M., et al. (2008). Biodiversity and biogeography of phages in modern stromatolites and thrombolites. Nature 452, 340–343. doi: 10.1038/nature06735
Everson, J. S., Garner, S. A., Fane, B., Liu, B. L., Lambden, P. R., and Clarke, I. N. (2002). Biological properties and cell tropism of Chp2, a bacteriophage of the obligate intracellular bacterium Chlamydophila abortus. J. Bacteriol. 184, 2748–2754. doi: 10.1128/JB.184.10.2748-2754.2002
Fane, B. A., Brentlinger, K. L., Burch, A. D., Chen, M., Hafenstein, S., Moore, E., et al. (2006). ØX174 et al., the Microviridae. The Bacteriophages, 2nd Edn. New York, NY: Oxford University Press, 129–145.
Fuhrman, J. A. (1999). Marine viruses and their biogeochemical and ecological effects. Nature 399, 541–548. doi: 10.1038/21119
Garner, S. A., Everson, J. S., Lambden, P. R., Fane, B. A., and Clarke, I. N. (2004). Isolation, molecular characterisation and genome sequence of a bacteriophage (Chp3) from Chlamydophila pecorum. Virus Genes 28, 207–214. doi: 10.1023/B:VIRU.0000016860.53035.f3
Holmfeldt, K., Solonenko, N., Shah, M., Corrier, K., Riemann, L., Verberkmoes, N. C., et al. (2013). Twelve previously unknown phage genera are ubiquitous in global oceans. Proc. Natl. Acad. Sci. U.S.A. 110, 12798–12803. doi: 10.1073/pnas.1305956110
Holmfeldt, K., Sullivan, M. B., Middelboe, M., and Riemann, L. (2012). Cultivated single-stranded DNA phages that infect marine Bacteroidetes prove difficult to detect with DNA-binding stains. Appl. Environ. Microbiol. 78, 892–894. doi: 10.1128/AEM.06580-11
Hopkins, M., Kailasan, S., Cohen, A., Roux, S., Tucker, K. P., Shevenell, A., et al. (2014). Diversity of environmental single-stranded DNA phages revealed by PCR amplification of the partial major capsid protein. ISME J. 8, 2093–2103. doi: 10.1038/ismej.2014.43
Ji, J., Zhang, R., and Jiao, N. (2015). Complete genome sequence of Roseophage vB_DshP-R1, which infects Dinoroseobacter shibae DFL12. Stand. Genomic Sci. 10, 1–6.
Jiao, N., Zhang, F., and Hong, N. (2010). Significant roles of bacteriochlorophylla supplemental to chlorophylla in the ocean. ISME J. 4, 595–599. doi: 10.1038/ismej.2009.135
Koblížek, M. (2015). Ecology of aerobic anoxygenic phototrophs in aquatic environments. FEMS Microbiol. Rev. 39, 854–870. doi: 10.1093/femsre/fuv032
Krupovic, M., Dutilh, B. E., Adriaenssens, E. M., Wittmann, J., Vogensen, F. K., Sullivan, M. B., et al. (2016). Taxonomy of prokaryotic viruses: update from the ICTV bacterial and archaeal viruses subcommittee. Arch. Virol. 161, 1095–1099. doi: 10.1007/s00705-015-2728-0
Krupovic, M., and Forterre, P. (2011). Microviridae goes temperate: microvirus-related proviruses reside in the genomes of Bacteroidetes. PLoS One 6:e19893. doi: 10.1371/journal.pone.0019893
Labonté, J. M., and Suttle, C. A. (2013a). Previously unknown and highly divergent ssDNA viruses populate the oceans. ISME J. 7, 2169–2177. doi: 10.1038/ismej.2013.110
Labonté, J. M., and Suttle, C. A. (2013b). Metagenomic and whole-genome analysis reveals new lineages of gokushoviruses and biogeographic separation in the sea. Front. Microbiol. 4:404. doi: 10.3389/fmicb.2013.00404
López-Bueno, A., Tamames, J., Velázquez, D., Moya, A., Quesada, A., and Alcamí, A. (2009). High diversity of the viral community from an Antarctic Lake. Science 326, 858–861. doi: 10.1126/science.1179287
Lu, L., Cai, L., Jiao, N., and Zhang, R. (2017). Isolation and characterization of the first phage infecting ecologically important marine bacteria Erythrobacter. Virol. J. 14:104. doi: 10.1186/s12985-017-0773-x
McDaniel, L. D., delaRosa, M., and Paul, J. H. (2006). Temperate and lytic cyanophages from the Gulf of Mexico. J. Mar. Biol. Ass. UK 86, 517–527. doi: 10.1111/1574-6941.12088
Middelboe, M., Chan, A. M., and Bertelsen, S. K. (2010). “Isolation and life-cycle characterization of lytic viruses infecting heterotrophic bacteria and cyanobacteria,” in Manual of Aquatic Viral Ecology, eds S. W. Wilhelm, M. G. Weinbauer, and C. A. Suttle (Waco, TX: ASLO), 118–133.
Pearson, V. M., Caudle, S. B., and Rokyta, D. R. (2016). Viral recombination blurs taxonomic lines: examination of single-stranded DNA viruses in a wastewater treatment plant. PeerJ 4:e2585. doi: 10.7717/peerj.2585
Quaiser, A., Dufresne, A., Ballaud, F., Roux, S., Zivanovic, Y., Colombet, J., et al. (2015). Diversity and comparative genomics of Microviridae in Sphagnum- dominated peatlands. Front. Microbiol. 6:375. doi: 10.3389/fmicb.2015.00375
Roux, S., Enault, F., Robin, A., Ravet, V., Personnic, S., Theil, S., et al. (2012a). Assessing the diversity and specificity of two freshwater viral communities through metagenomics. PLoS One 7:e33641. doi: 10.1371/journal.pone.0033641
Roux, S., Krupovic, M., Poulet, A., Debroas, D., and Enault, F. (2012b). Evolution and diversity of the Microviridae viral family through a collection of 81 new complete genomes assembled from virome reads. PLoS One 7:e40418. doi: 10.1371/journal.pone.0040418
Roux, S., Solonenko, N. E., Dang, V. T., Poulos, B. T., Schwenck, S. M., Goldsmith, D. B., et al. (2016). Towards quantitative viromics for both double-stranded and single-stranded DNA viruses. PeerJ 4:e2777. doi: 10.7717/peerj.2777
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Suttle, C. A. (2007). Marine viruses–major players in the global ecosystem. Nat. Rev. Microbiol. 5, 801–812. doi: 10.1038/nrmicro1750
Suttle, C. A., and Chen, F. (1992). Mechanisms and rates of decay of marine viruses in seawater. Appl. Environ. Microbiol. 58, 3721–3729.
Székely, A. J., and Breitbart, M. (2016). Single-stranded DNA phages: from early molecular biology tools to recent revolutions in environmental microbiology. FEMS Microbiol. Lett. 363:fnw027. doi: 10.1093/femsle/fnw027
Tucker, K. P., Parsons, R., Symonds, E. M., and Breitbart, M. (2011). Diversity and distribution of single-stranded DNA phages in the North Atlantic Ocean. ISME J. 5, 822–830. doi: 10.1038/ismej.2010.188
Weinbauer, M. G., and Rassoulzadegan, F. (2004). Are viruses driving microbial diversification and diversity? Environ. Microbiol. 6, 1–11.
Wiśniewski, J. R., Zougman, A., Nagaraj, N., and Mann, M. (2009). Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362. doi: 10.1038/nmeth.1322
Wommack, K. E., and Colwell, R. R. (2000). Virioplankton: viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 64, 69–114. doi: 10.1128/MMBR.64.1.69-114.2000
Yoshida, T., Nagasaki, K., Takashima, Y., Shirai, Y., Tomaru, Y., Takao, Y., et al. (2008). Ma-LMM01 infecting toxic Microcystis aeruginosa illuminates diverse cyanophage genome strategies. J. Bacteriol. 190, 1762–1772. doi: 10.1128/JB.01534-07
Yurkov, V., Krieger, S., Stackebrandt, E., and Beatty, J. T. (1999). Citromicrobium bathyomarinum, a novel aerobic bacterium isolated from deep-sea hydrothermal vent plume waters that contains photosynthetic pigment-protein complexes. J. Bacteriol. 181, 4517–4525.
Zerbino, D. R., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
Zhang, Y., and Jiao, N. (2009). Roseophage RDJLΦ1, infecting the aerobic anoxygenic phototrophic bacterium Roseobacter denitrificans OCh114. Appl. Environ. Microbiol. 75, 1745–1749. doi: 10.1128/AEM.02131-08
Zheng, Q., Liu, Y., Jeanthon, C., Zhang, R., Lin, W., Yao, J., et al. (2016). The geographic impact on genomic divergence as revealed by comparison of nine Citromicrobial genomes. Appl. Environ. Microbiol. 82, 7205–7216. doi: 10.1128/AEM.02495-16
Zheng, Q., Liu, Y., Steindler, L., and Jiao, N. (2015). Pyrosequencing analysis of aerobic anoxygenic phototrophic bacterial community structure in the oligotrophic western Pacific Ocean. FEMS Microbiol. Lett. 362:fnv034. doi: 10.1093/femsle/fnv034
Zheng, Q., Zhang, R., Xu, Y., White, R. A., Wang, Y., Luo, T., et al. (2014). A marine inducible prophage vB_CibM-P1 isolated from the aerobic anoxygenic phototrophic bacterium Citromicrobium bathyomarinum JL354. Sci. Rep. 4:7118. doi: 10.1038/srep07118
Keywords: Microviridae, Citromicrobium, ssDNA phage, ssDNA prophage, photoheterotrophic bacteria
Citation: Zheng Q, Chen Q, Xu Y, Suttle CA and Jiao N (2018) A Virus Infecting Marine Photoheterotrophic Alphaproteobacteria (Citromicrobium spp.) Defines a New Lineage of ssDNA Viruses. Front. Microbiol. 9:1418. doi: 10.3389/fmicb.2018.01418
Received: 12 April 2018; Accepted: 08 June 2018;
Published: 27 June 2018.
Edited by:
Akio Adachi, Kansai Medical University, JapanReviewed by:
Andrei A. Zimin, Institute of Biochemistry and Physiology of Microorganisms (RAS), RussiaCopyright © 2018 Zheng, Chen, Xu, Suttle and Jiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiang Zheng, emhlbmdxaWFuZ0B4bXUuZWR1LmNu; orcid.org/0000-0002-6836-2310; Nianzhi Jiao, amlhb0B4bXUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.