
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 08 June 2018
Sec. Infectious Agents and Disease
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.01220
 Paulina A. Fernández†
Paulina A. Fernández† Felipe Velásquez†
Felipe Velásquez† Héctor Garcias-Papayani
Héctor Garcias-Papayani Fernando A. Amaya
Fernando A. Amaya Jaime Ortega
Jaime Ortega Sebastián Gómez
Sebastián Gómez Carlos A. Santiviago
Carlos A. Santiviago Sergio A. Álvarez*
Sergio A. Álvarez*Lipid A is the bioactive component of lipopolysaccharide, and presents a dynamic structure that undergoes modifications in response to environmental signals. Many of these structural modifications influence Salmonella virulence. This is the case of lipid A hydroxylation, a modification catalyzed by the dioxygenase LpxO. Although it has been established that oxygen is required for lipid A hydroxylation acting as substrate of LpxO in Salmonella, an additional regulatory role for oxygen in lpxO expression has not been described. The existence of this regulation could be relevant considering that Salmonella faces low oxygen tension during infection. This condition leads to an adaptive response by changing the expression of numerous genes, and transcription factors Fnr and ArcA are major regulators of this process. In this work, we describe for the first time that lipid A hydroxylation and lpxO expression are modulated by oxygen availability in Salmonella enterica serovar Enteritidis (S. Enteritidis). Biochemical and genetic analyses indicate that this process is regulated by Fnr and ArcA controlling the expression of lpxO. In addition, according to our results, this regulation occurs by direct binding of both transcription factors to specific elements present in the lpxO promoter region. Altogether, our observations revealed a novel role for oxygen acting as an environment signal controlling lipid A hydroxylation in S. Enteritidis.
Salmonella enterica serovar Enteritidis (S. Enteritidis) is considered the leading cause of food-borne salmonellosis worldwide (Roberts and Sockett, 1994; Guard-Petter, 2001). In humans, this pathogen causes a self-limited gastroenteritis characterized by diarrhea, fever, and abdominal pain (Guard-Petter, 2001). Virulence mechanisms used by S. Enteritidis are poorly understood, and most knowledge on the subject is based on studies carried out in S. Typhimurium, which also causes gastroenteritis in humans. In spite of this, it is known that S. Enteritidis lipopolysaccharide (LPS) plays a major role in pathogenicity, as genes involved in the synthesis of this macromolecule are required for systemic colonization (Silva et al., 2012; Coward et al., 2013), resistance to serum (Bravo et al., 2008), and survival in egg albumen (Coward et al., 2013).
Lipid A is the bioactive component of LPS and constitutes the anchor portion of this molecule to the outer membrane (Raetz and Whitfield, 2002). When particular structures of lipid A are recognized by the TLR-4/MD2 complex, an inflammatory response is triggered in macrophages (Park et al., 2009). In response to environmental cues, lipid A can be covalently modified by different enzymes in a process that is essential for bacterial adaptation to its host (Raetz et al., 2007). These modifications are implicated in cationic antimicrobial peptides (CAMPs) resistance and in avoiding its recognition by TLR-4.
LpxO is a Fe2+/α-ketoglutarate-dependent dioxygenase that catalyzes the hydroxylation of lipid A by modifying the 3’-secondary myristoyl chain to generate S-2-hydroxymiristate in S. Typhimurium (Gibbons et al., 2000, 2008). Studies addressing the role played by this modification in Salmonella virulence are scarce. Despite this, it has been reported that an appropriate lipid A hydroxylation level is crucial for S. Typhimurium replication in macrophages and for systemic colonization in mice (Moreira et al., 2013). Also, a role for lipid A hydroxylation on reduction of inflammatory response and CAMP resistance has been described in Klebsiella pneumoniae (Llobet et al., 2015; Mills et al., 2017).
The expression of most genes encoding lipid A-modifying enzymes is regulated by two-component systems PhoP/PhoQ and PmrA/PmrB in S. Typhimurium (Raetz, 2001). PhoP/PhoQ responds to environmental signals encountered by S. Typhimurium and S. Enteritidis during infection and is essential for virulence of these bacteria in mice (Dalebroux and Miller, 2014). In addition, PhoP/PhoQ and PmrA/PmrB systems are functionally linked (Gunn et al., 2000). Although little is known on the mechanisms that regulate lpxO expression in Salmonella, a study confirmed that this gene is not regulated by PhoP/PhoQ (Gibbons et al., 2005).
Even though it has been established that oxygen is required for lipid A hydroxylation acting as a substrate of LpxO (Gibbons et al., 2008), a regulatory role for oxygen on lpxO expression has not been described. The existence of such regulatory role could be relevant as low oxygen tension is one of the conditions that Salmonella encounters in the distal ileum and within host cells (Altier, 2005; Rhen and Dorman, 2005). Transcription factors Fnr and ArcA are the major regulators involved in adaptation to oxygen availability (Compan and Touati, 1994; Kiley and Beinert, 1998; Sawers, 1999; Fink et al., 2007; Ravcheev et al., 2007). In addition to regulating the expression of several genes involved in energy metabolism, Fnr also regulates the expression of virulence genes in S. Typhimurium (Fink et al., 2007). On the other hand, it has also been reported that ArcA contributes to S. Enteritidis virulence in the murine model (Silva et al., 2012). Previously, we have shown that Fnr and ArcA regulate O-antigen (OAg) chain-length distribution in an oxygen-dependent fashion in S. Enteritidis (Silva-Valenzuela et al., 2016). In the present study, we describe for the first time that lipid A hydroxylation and lpxO expression are regulated by oxygen availability in S. Enteritidis, and that Fnr and ArcA participate in this process by direct interaction with elements present in the lpxO promoter region.
All bacterial strains and plasmids used in this study are listed in Table 1. Bacteria were routinely grown in Luria-Bertani (LB) medium (10 g/L tryptone, 5 g/L yeast extract, 5 g/L NaCl) at 37°C with agitation (180 rpm). For assays under aerobic and anaerobic conditions, overnight cultures in LB were diluted 1:100 in minimal E medium (0.2 g/L MgSO4 × 7H2O, 2 g/L citric acid monohydrate, 13.1 g/L K2HPO4 × 3H2O, 3.3 g/L NaNH4HPO4 × 4H2O, pH 7.0), supplemented with 0.2% glucose as sole carbon source. Aerobic cultures were obtained by incubation at 37°C with agitation (180 rpm) and anaerobic cultures were obtained by incubation at 37°C in an anaerobic jar with the AnaeroGen system (Oxoid). When required, media were supplemented with ampicillin (Amp, 100 mg/L), chloramphenicol (Cam, 20 mg/L), or kanamycin (Kan, 75 mg/L). Media were solidified by the addition of agar (15 g/L).

TABLE 1. Bacterial strains and plasmids used in this study.
Salmonella Enteritidis mutant strains ΔlpxO::kan and Δfnr::cam were constructed by the Red-swap method (Datsenko and Wanner, 2000) with modifications (Santiviago et al., 2009), using plasmid pCLF4 (KanR, GenBank accession number EU629214) or pCLF3 (CamR, GenBank accession number EU629213) as template. Correct allelic replacement in these mutants was confirmed by PCR amplification using primers flanking the substitution site. All primers for PCR amplifications are listed in Supplementary Table S1. Next, the mutations were transduced into the wild-type background using P22 HT105/1 int-201 (Schmieger, 1972) as described (Maloy, 1990). To obtain non-polar deletion mutants (i.e., ΔlpxO::FRT and Δfnr::FRT), the antibiotic-resistance genes were removed by transforming the insertion mutants with pCP20 (Cherepanov and Wackernagel, 1995; Datsenko and Wanner, 2000).
An lpxO–lacZ transcriptional fusion was constructed using the Flp-mediated site-specific recombination method (Ellermeier et al., 2002). To do this, the ΔlpxO::FRT mutant harboring pCP20 was transformed with pKG136, a derivative of the lacZ transcriptional fusion plasmid pCE36. This procedure resulted in the stable integration of pKG136 in the chromosome and the loss of pCP20. The single-copy integration of pKG136 in the ΔlpxO::FRT allele was confirmed by PCR amplification as described (Ellermeier et al., 2002). Finally, the Δfnr::cam allele was transduced into the mutant harboring the lpxO–lacZ fusion using P22 HT105/1 int-201.
Lipopolysaccharide was extracted by the TRIzol method as described (Yi and Hackett, 2000), with modifications. Bacteria from cultures grown to an OD600 0.3 under aerobic or anaerobic conditions in 50 mL of minimal E medium were harvested by centrifugation. Each pellet was suspended in 2 mL of TRIzol (Life Technologies) and 400 μL of chloroform were added to the suspension. The mixture was vigorously vortexed until complete homogenization and then incubated during 30 min at room temperature. The resulting mixture was centrifuged at 3500 × g during 10 min at room temperature, and the aqueous phase was recovered in a clean centrifuge tube. Next, 1 mL of distilled water was added to the organic phase, the extraction procedure was repeated, and the new aqueous phase was recovered in a clean centrifuge tube. Both recovered aqueous phases were combined and lyophilized. Further LPS purification was achieved by the cold ethanol-magnesium precipitation procedure (Darveau and Hancock, 1983) followed by two rounds of Folch extraction (Folch et al., 1957). LPS integrity in our preparations was evaluated by Tricine-SDS-PAGE in 12% polyacrylamide gels (Marolda et al., 2006) and silver staining (Tsai and Frasch, 1982).
Lipid A was isolated from LPS preparations by mild acid hydrolysis as described (Caroff and Novikov, 2011), with modifications. After addition of hydrolysis solution (1% SDS in 10 mM sodium acetate pH 4.5), LPS samples were allowed to dissolve during 30 min at room temperature. Then, samples were incubated during 2 h at 100°C and the hydrolysis products were lyophilized. The sediment was suspended in 800 μL of chloroform/water/methanol (10:9:1 v/v/v) and centrifuged during 5 min at 2100 × g. Finally, the organic phase containing purified lipid A was recovered and the organic solvents were evaporated at room temperature. The absence of non-hydrolyzed LPS in our lipid A preparations was evaluated by Tricine-SDS-PAGE in 12% polyacrylamide gels (Marolda et al., 2006) and silver staining (Tsai and Frasch, 1982).
Lipid A samples were dissolved in 90 μL of chloroform/methanol (1:1 v/v) and this solution was mixed with one volume of matrix (10 mg/mL 2.5-dihydroxybenzoic acid in 100 mM citric acid). Spectra were obtained in the negative reflection mode using a Microflex LRF mass spectrometer (Bruker Daltonics Inc., MA, United States). flexControl 3.0 (Bruker Daltonik GmbH, Germany) and mMass version 5.5.01 software packages were used for spectrometer control and spectra analysis, respectively.
RNA isolation was performed as described (Kroger et al., 2012) from at least three independent cultures grown in minimal E medium. Briefly, aerobic or anaerobic cultures were grown to an OD600 of 0.3 and total RNA was extracted using the “SV Total RNA Isolation System” (Promega). All samples were treated with RNase-free DNaseI (Qiagen) at 25°C for 45 min and kept at -20°C until further use.
For qRT-PCR assays, 1 μg of total RNA was reverse transcribed using random hexamers and SuperScript II (Life Technologies) following manufacturer’s instructions. For each gene evaluated, a specific DNA fragment (∼300 bp) was amplified using the “KAPA SYBR FAST qPCR kit Master Mix (2x) Universal” (Kapa Biosystems) as recommended by the manufacturer in an Mx3000P qPCR System with MxPro software (Agilent Technologies, Inc.). Ct values were normalized using rpoD as housekeeping gene.
Salmonella Enteritidis strains carrying the lpxO–lacZ fusion were grown without antibiotics under aerobic or anaerobic conditions at 37°C in minimal E medium to an OD600 0.3–0.5. β-galactosidase activity was measured as described (Miller, 1972). Briefly, 100 μL of culture was suspended in Z buffer pH 7.0 (0.6 M Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol) to a final volume of 1 mL. Bacteria were permeabilized with 10 μL of chloroform, 10 μL of 0.1% SDS, and vortexed for 20 s. After incubation at 30°C for 10 min, 200 μL of 4 mg/mL ONPG was added to the mix. Reactions were stopped by the addition of 500 μL of 1 M Na2CO3. Each assay was made in duplicate and repeated at least three times, in different days. β-galactosidase activity was expressed in Miller units.
For overexpression of ArcA, a DNA fragment containing the arcA gene (including the promoter region) was amplified from the genome of S. Enteritidis NCTC13349 using primers listed in Supplementary Table S1. The PCR product was purified from 1% agarose gels and cloned into pBAD-TOPO using the “pBAD-TOPO TA Expression Kit” (Invitrogen). The presence and orientation of the insert in the recombinant plasmid generated (parcA) was confirmed by PCR amplification using combinations of primers listed in Supplementary Table S1. S. Enteritidis strains were transformed by electroporation with plasmid parcA for ArcA overexpression assays.
For expression and purification of ArcA and FnrD154A, a DNA fragment containing the open reading frame (ORF) of arcA or fnrD154A was amplified from the genome of S. Enteritidis NCTC13349 and plasmid pPK822 (Lazazzera et al., 1993), respectively, using primers listed in Supplementary Table S1. Each PCR product was purified from 1% agarose gels and cloned into pBAD-TOPO to generate a 6 × His-tagged protein (C-terminal) using the “pBAD-TOPO TA Expression Kit” (Invitrogen). The presence and orientation of each insert in the recombinant plasmids generated (pBAD-arcA and pBAD-fnrD154A) were confirmed by PCR amplification using combinations of primers listed in Supplementary Table S1. Finally, Escherichia coli TOP10 was transformed with plasmid pBAD-arcA or pBAD-fnrD154A for expression and purification of ArcA and FnrD154A.
Expression of ArcA and FnrD154A was induced during 4 h by adding 0.02% or 0.2% L-arabinose to cultures (OD600 ∼0.4) of E. coli TOP10 harboring pBAD-arcA or pBAD-fnrD154A, respectively. Bacteria were harvested and suspended in lysis buffer (50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 500 μM DTT), supplemented with “cOmplete Mini, EDTA-free” protease inhibitor cocktail (Roche) according to manufacturer’s recommendations. Next, the mixture was supplemented with lysozyme (1 mg/mL final concentration) and MgCl2 (1 mM final concentration). After 30 min of incubation at 37°C, the mixture was subjected to five cycles of sonication (20 s of sonication, 1 min of recess) on ice. The lysate was centrifuged at 3500 × g during 5 min at 4°C, and the supernatant was recovered and further centrifuged at 21,000 × g during 30 min at 4°C. This clarified supernatant was recovered and supplemented with imidazole (30 mM final concentration). To purify each recombinant protein, the clarified lysate was applied to a 1 mL HisTrap FF column (GE Healthcare) previously equilibrated with 12 volumes of binding buffer (50 mM Tris-HCl pH 7.4, 500 mM NaCl, 30 mM imidazole). Then, the column was washed with 20 volumes of binding buffer. The recombinant proteins were eluted with a linear imidazole gradient (30–500 mM) in binding buffer. Purity of protein samples was checked by SDS-PAGE in 12% polyacrylamide gels stained with Coomassie Brilliant Blue G-250 (Supplementary Figure S1). Fractions containing the purified proteins were dialyzed overnight at 4°C against the corresponding reaction buffer (FnrD154A: 10 mM sodium phosphate pH 7.5, 100 mM sodium glutamate, 1 mM EDTA, 50 mM DTT, 5% glycerol; ArcA: 100mM Tris-HCl pH 8.0, 10 mM MgCl2, 125 mM KCl, 2 mM DTT, 10% glycerol). Protein concentration in each preparation was determined by the Bradford assay using the Coomassie Protein Assay Reagent (Thermo Scientific).
Electrophoretic mobility shift assays (EMSA) were performed to evaluate protein–DNA interaction. For EMSA carried out in the presence of FnrD154A, a 480 bp fragment including the complete lpxO promoter region was amplified from the genome of S. Enteritidis NCTC13349 using primers listed in Supplementary Table S1. This fragment (120 ng) was incubated with different amounts of FnrD154A for 30 min at 37°C in reaction buffer supplemented with 50 μg BSA and 120 ng of a 261 bp fragment of non-specific competitor DNA corresponding to the polylinker region of pGEM-T Easy (Promega). Heat-denatured FnrD154A was used as negative control.
For EMSA carried out in the presence of ArcA, the complete lpxO promoter region was used along with internal fragments of 253, 162, and 109 bp (amplified from S. Enteritidis genomic DNA using primers listed in Supplementary Table S1) including unique putative ArcA binding sites (ABS) predicted in the promoter. Each fragment (120 ng) was incubated with different amounts of in vitro phosphorylated ArcA (P-ArcA) for 30 min at 25°C in reaction buffer. P-ArcA was obtained as described (Lynch and Lin, 1996). Non-phosphorylated ArcA and heat-denatured P-ArcA were used as negative controls.
In all cases, reaction mixtures were loaded in 6% polyacrylamide gels and resolved by electrophoresis in 0.5X TBE buffer. Gels were stained with GelRed (Thermo Fisher Scientific) 3X solution. DNA and protein–DNA complexes were detected and captured using a Syngene Dyversity Imaging System (Cambridge, United Kingdom).
In order to determine the effect of oxygen availability on S. Enteritidis lipid A covalent modifications, the wild-type strain was grown under aerobic or anaerobic conditions and lipid A was purified and analyzed by MALDI-TOF mass spectrometry. Under aerobic conditions, the negative-ion spectrum of lipid A showed four major peaks at m/z 1797.5, 1813.3, 2036.1, and 2051.8 (Figure 1, upper left panel), which were interpreted as canonic hexaacylated lipid A, its hydroxylated counterpart, heptaacylated lipid A and its hydroxylated counterpart, respectively (Figure 2). Thus, hydroxylated species were more abundant than non-hydroxylated species under aerobic conditions. In contrast, non-hydroxylated species were more abundant than their hydroxylated counterparts when S. Enteritidis was grown under anaerobic conditions (Figure 1, lower left panel). Of note, during anaerobic growth, the signal corresponding to the heptaacylated hydroxylated species was undetectable. These results show that lipid A hydroxylation in S. Enteritidis is modulated by oxygen availability, as described in S. Typhimurium (Gibbons et al., 2000).
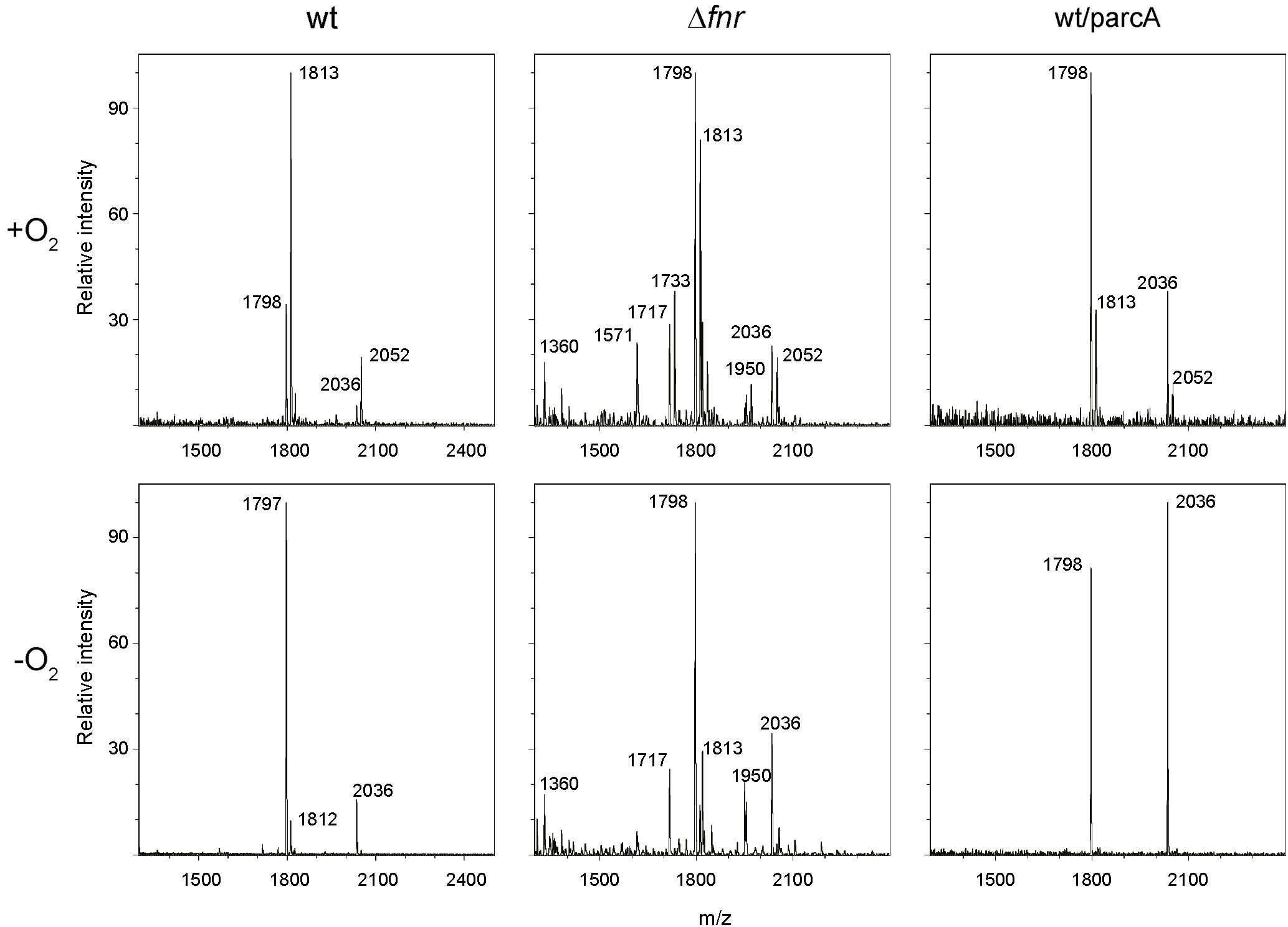
FIGURE 1. Mass spectrometry analysis of lipid A species produced by S. Enteritidis strains. Lipid A samples were obtained from cultures of the wild-type strain (left panels), its isogenic Δfnr mutant (middle panels), and the wt/parcA strain (right panels) grown under aerobic (upper panels) or anaerobic (lower panels) conditions. MALDI-TOF mass spectrometry analysis of lipid A preparations was performed and experimental m/z values are indicated.
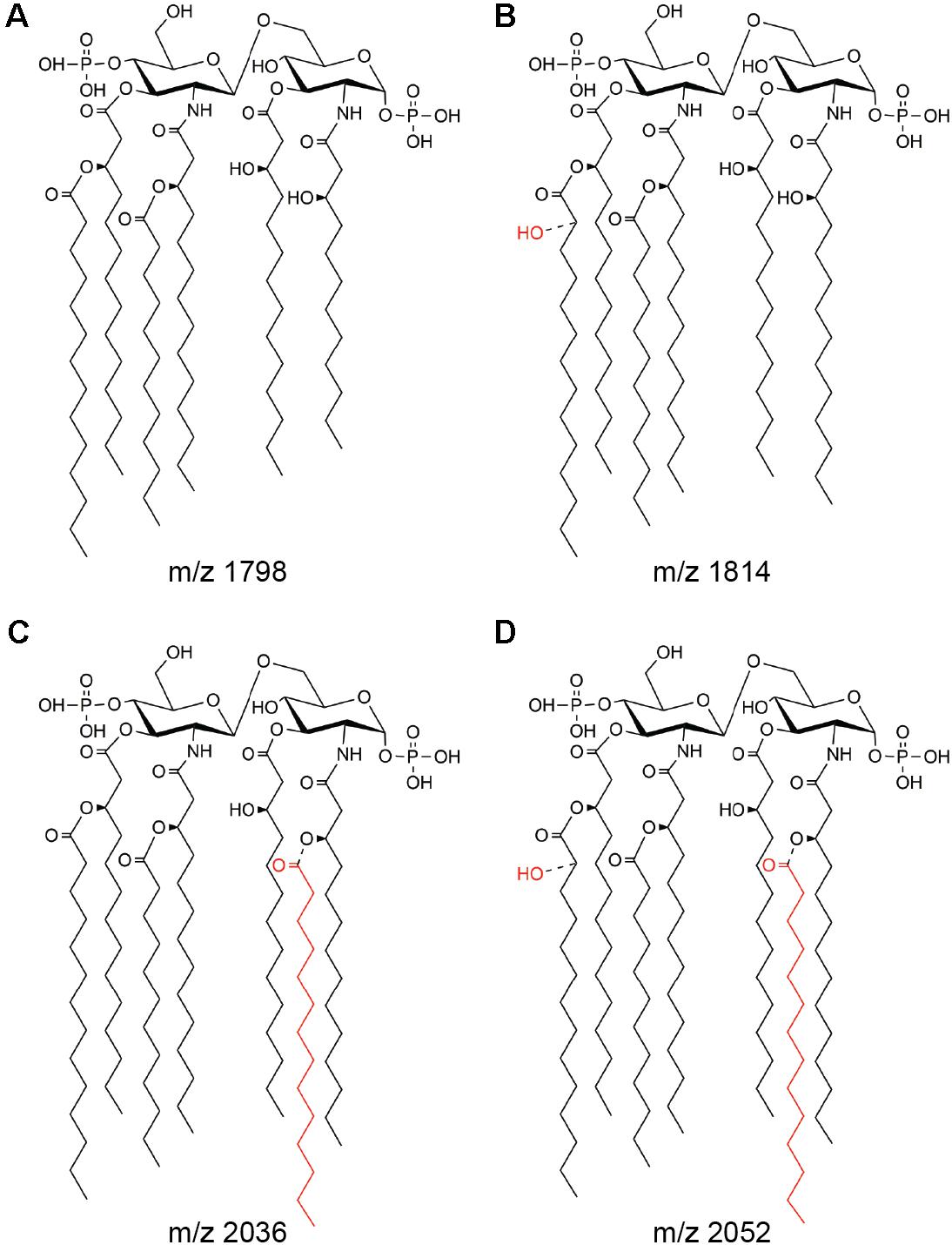
FIGURE 2. Main lipid A species produced by S. Enteritidis. The structures correspond to hexaacylated lipid A (A), its hydroxylated counterpart (B), heptaacylated lipid A (C), and its hydroxylated counterpart (D). m/z values associated to each species (Kawasaki et al., 2007) are indicated.
As mentioned, oxygen is required as substrate for LpxO during lipid A hydroxylation in S. Typhimurium (Gibbons et al., 2000). However, a regulatory role for oxygen (acting as environmental signal) has not been described in this process. To address this possibility, we evaluated the participation of global regulators ArcA and Fnr on lipid A modification. Deletion of the fnr gene dramatically changed the lipid A species profile under both aerobic and anaerobic conditions, as spectra showed a higher number of species not detected in the wild-type stain (m/z 1360.1, 1717.4, 1733.2, and 1950.3; Figure 1, middle panels). These species were inferred according to published data. Thus, a peak at m/z 1360.1 is consistent with 3’-O-deacylated lipid A produced by LpxR (Reynolds et al., 2006). Peaks at m/z 1717.4 and 1733.2 were interpreted as lipid A dephosphorylated by UgtL (Shi et al., 2004) and its counterpart hydroxylated by LpxO (Kong et al., 2012), respectively. Although genetic evidence supports the former assignments, a peak at m/z 1718 (similar to 1717.4) has been interpreted as lipid A hydroxylated by LpxO, 3-O-deacylated by PagL, and modified with an aminoarabinose by ArnT (Guo et al., 1998). Finally, a peak at m/z 1950.3 is consistent with a highly modified lipid A that has been 3-O-deacylated by PagL, palmitoylated by PagP, hydroxylated by LpxO, and including a phosphoethanolamine attached by EptA (Kawasaki et al., 2007).
Unlike what was observed in the wild-type strain, the relative abundance of all hydroxylated lipid A species was comparable to the abundance of their non-hydroxylated counterparts when the Δfnr mutant was grown under aerobic conditions (Figure 1, compare upper middle and upper left panels). However, under anaerobiosis, the relative abundance of hydroxylated and non-hydroxylated lipid A in the Δfnr mutant was similar to that observed in the wild-type strain (Figure 1, compare lower middle and lower left panels). These results revealed for the first time that Fnr plays a role in the regulation of lipid A modification in S. Enteritidis.
As previously described, the S. Enteritidis ΔarcA mutant exhibits impaired growth in minimal medium under anaerobic conditions (Silva-Valenzuela et al., 2016), preventing the use of this strain to determine a role for ArcA on oxygen-modulated lipid A hydroxylation. Therefore, we decided to characterize the lipid A species profile when the gene dose of arcA is increased. To this end, we transformed the wild-type strain with a plasmid harboring arcA gene (parcA). Under aerobic conditions, ArcA overexpression led to an increase of non-hydroxylated lipid A species (m/z 1797.2 and 2035.8) together with a decrease of the hydroxylated counterparts (m/z 1812.9 and 2051.7; Figure 1, upper right panel). Of note, this distribution of lipid A species resembles that exhibited by the wild-type strain grown under anaerobic conditions (Figure 1, compare upper right and lower left panels). On the other hand, only non-hydroxylated lipid A species were detected when ArcA was overexpressed under anaerobic conditions (Figure 1, lower right panel). These results indicate that ArcA modulates lipid A hydroxylation in S. Enteritidis. Thus, the involvement of Fnr and ArcA transcription regulators in lipid A modification reveals a novel role for oxygen as a regulatory cue controlling this process in S. Enteritidis.
The above results led us to hypothesize that lpxO expression is regulated by oxygen availability through the activity of transcription regulators ArcA and/or Fnr. To address this notion, we measured the relative abundance of lpxO transcript by qRT-PCR in the wild-type strain, the Δfnr mutant, and the wild-type strain overexpressing ArcA (wt/parcA) (Figure 3A). In addition, we analyzed the activity of the lpxO promoter using a chromosomal transcriptional fusion to lacZ in the same genetic backgrounds (Figure 3B). Both qRT-PCR and β-galactosidase assays revealed that lpxO expression in the wild-type strain is reduced under anaerobic conditions (Figure 3). This regulation could account for the reduction in lipid A hydroxylation levels observed in anaerobiosis (Figure 1, lower left panel). These results indicate that oxygen availability regulates lpxO expression.
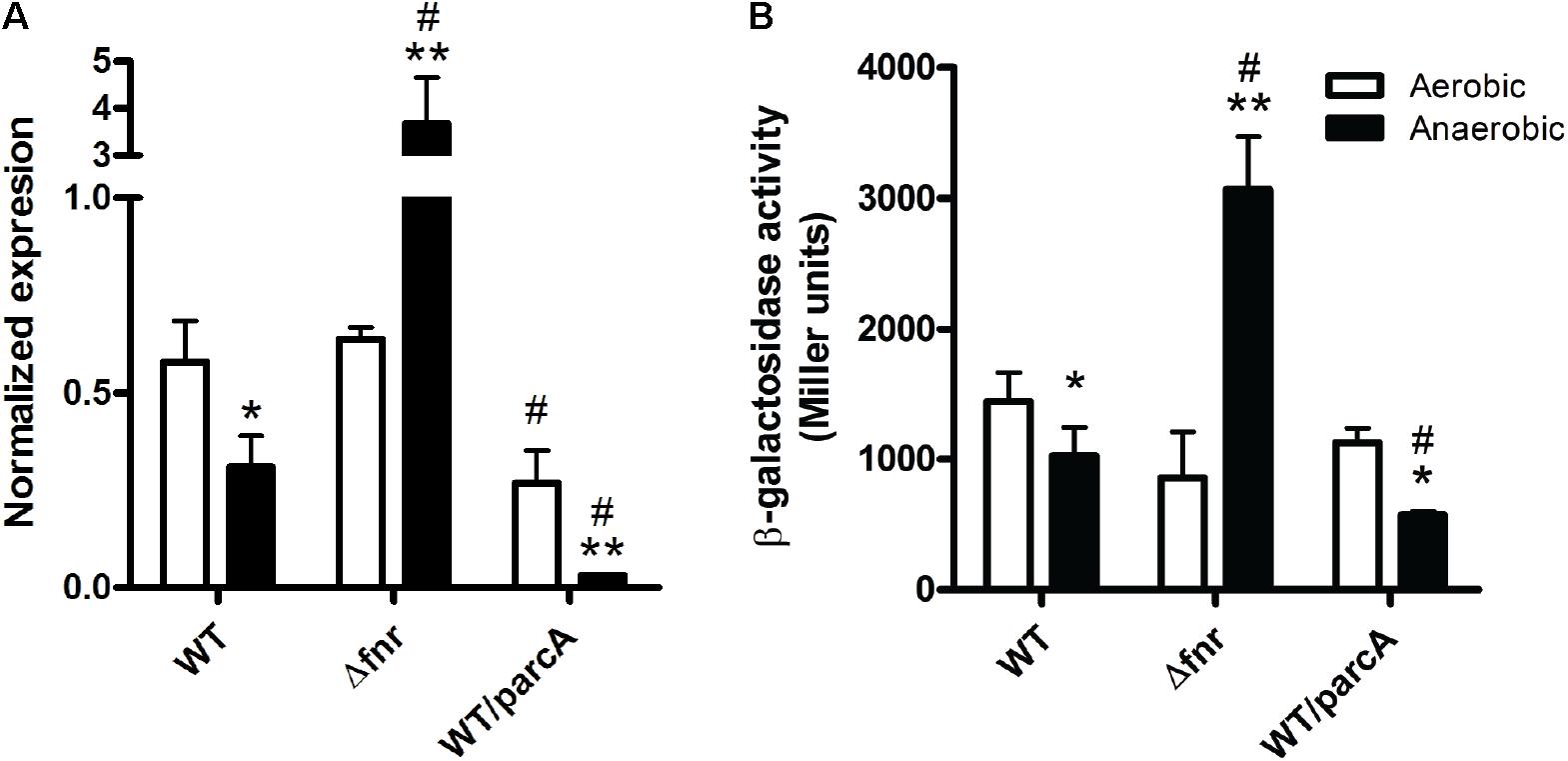
FIGURE 3. Expression of lpxO in S. Enteritidis strains. Expression levels were determined by qRT-PCR and normalized using rpoD as housekeeping gene (A) or by measuring β-galactosidase activity using strains carrying an lpxO–lacZ transcriptional fusion (B). Bars represent mean values from three independent replicates. Error bars denote standard deviation. Statistical significance of observed differences was determined using a two-tailed Student’s t-test (∗p < 0.05 vs. the same strain grown aerobically; ∗∗p < 0.01 vs. the same strain grown aerobically; #p < 0.05 vs. the wild-type strain grown under the same condition).
On the contrary, lpxO expression in the Δfnr mutant was increased under anaerobic conditions (Figure 3). Thus, the oxygen-dependent regulation of lpxO expression described for the wild-type strain is lost when fnr is deleted, indicating that Fnr is a negative regulator of lpxO expression under anaerobic conditions. This higher lpxO expression under anaerobiosis was not reflected on an increment in the relative abundance of lipid A hydroxylated species in this mutant grown under the same conditions (Figure 1, lower middle panel). This was expected considering that oxygen is a substrate of the LpxO-catalyzed reaction (Gibbons et al., 2000).
As in the case of the wild type, lpxO expression in the wt/parcA strain is strongly reduced in anaerobiosis (Figure 3), reflecting the absence of hydroxylated lipid A species detected by mass spectrometry analysis (Figure 1, lower right panel). Of note, arcA overexpression also accounts for lower lpxO expression under anaerobic conditions in the wt/parcA strain when compared to the wild-type strain (Figure 3). These observations suggest that ArcA is a negative regulator of lpxO expression under anaerobic conditions.
Our results indicate that Fnr and ArcA have a role controlling lipid A hydroxylation by modulating lpxO expression. However, it remains unknown whether the regulatory effect of these transcription factors is exerted directly or indirectly on the lpxO promoter. A bioinformatic analysis using the PRODORIC tool (Munch et al., 2005) allowed us to identify one putative Fnr binding site (FBS) and three putative ABS in the lpxO promoter region (Figure 4), suggesting a direct effect for both regulators. To confirm this notion, we performed electrophoretic mobility shift assays (EMSA) using a DNA fragment including the lpxO promoter and FnrD154A, a constitutively active variant of Fnr (Lazazzera et al., 1993), or P-ArcA. Our results showed that both FnrD154A and P-ArcA bound to the lpxO promoter in a dose-dependent fashion (Figure 5). When incubated in the presence of FnrD154A, the intensity of a highly retarded band on top of the gel increased considerably with increasing amounts of FnrD154 (Figure 5A). The mobility of this DNA–protein complex was similar to an unspecific band present in each lane. Concomitantly, we observed a pronounced decrease in the intensity of the band corresponding to free DNA probe. No binding of FnrD154A to a non-specific DNA competitor present in each reaction mix was detected. On the other hand, both P-ArcA and ArcA bound to the lpxO promoter, but generated different band-shift patterns (Figure 5B). The same behavior has been described for other ArcA-controlled promoters in E. coli (Lynch and Lin, 1996; Mika and Hengge, 2005). Of note, only in the presence of high amounts of P-ArcA (0.9 and 1.2 μg), the interaction with the lpxO promoter seems to be more stable and produced a complex with the slowest mobility (Figure 5B), which is consistent with P-ArcA multimerization (Jeon et al., 2001). As expected, no binding was observed to any DNA probe when either protein was heat-inactivated (Figures 5A,B). These results indicate that Fnr and ArcA regulate lpxO expression through direct binding to its promoter region.

FIGURE 4. Nucleotide sequence of the lpxO promoter region in S. Enteritidis. The genome sequence of strain NCTC13349 was obtained from EMBL (accession number AM933172). Underlined sequences represent transcriptional start site (+1), –10, and –35 elements predicted by Softberry BPROM tool http://www.softberry.com/berry.phtml. The locations of FBS and ABS predicted by PRODORIC Virtual Footprint 3.0 server http://www.prodoric.de/vfp/ are shown in boxes.
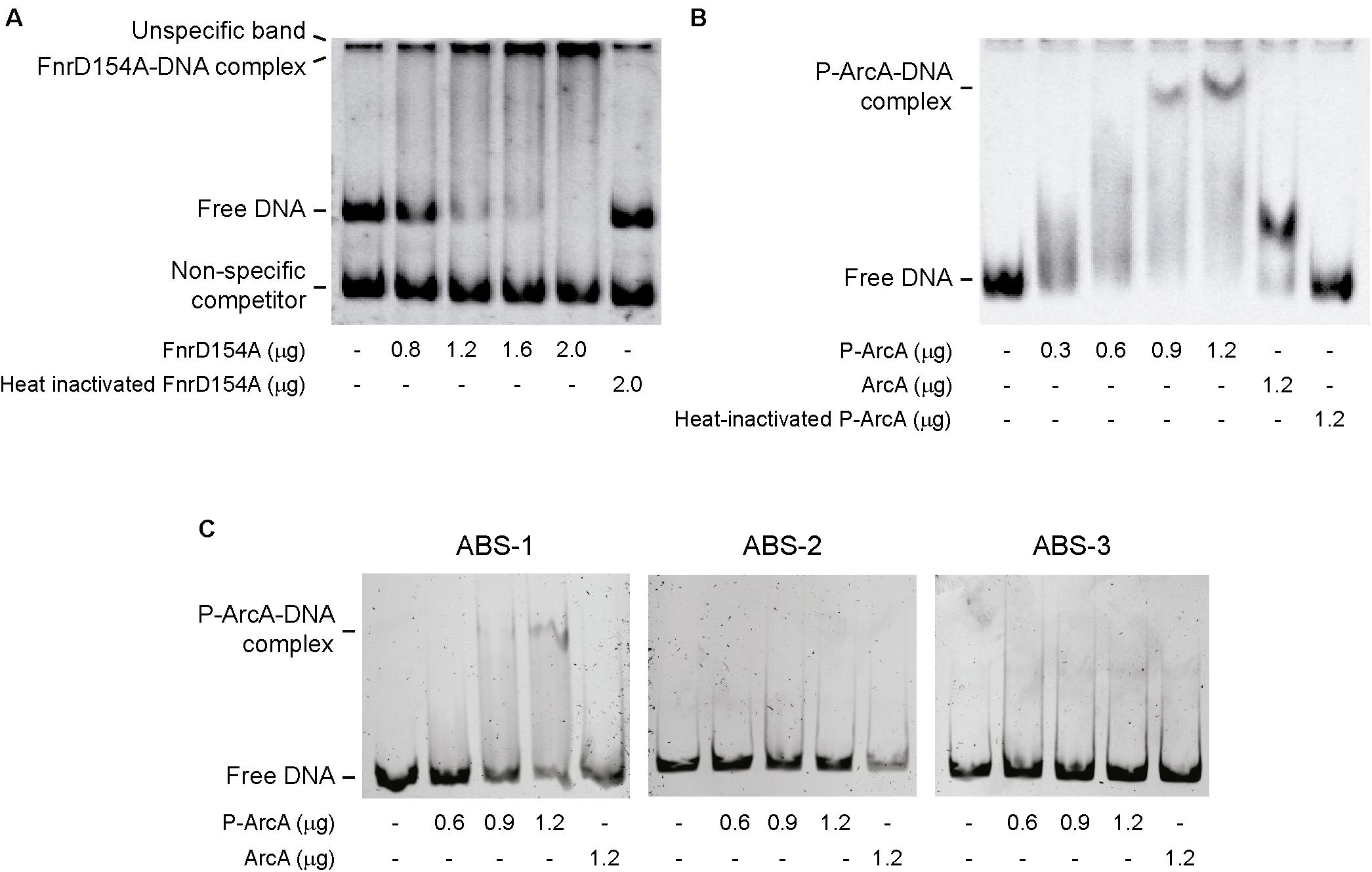
FIGURE 5. Binding of Fnr and ArcA to S. Enteritidis lpxO promoter region. EMSA were conducted using a 480 bp fragment carrying the complete lpxO promoter and purified FnrD154A (A) or P-ArcA (B) proteins. EMSA were also performed incubating purified P-ArcA with fragments of the lpxO promoter region containing ABS-1, ABS-2, or ABS-3 (C). In all cases, EMSA included heat-inactivated proteins and/or non-phosphorylated ArcA as negative controls. When indicated, a 261 bp DNA fragment was used as non-specific competitor.
Because the lpxO promoter only presents a unique FBS, this element should be responsible for Fnr binding to the promoter. To determine which putative ABS in the lpxO promoter is responsible for P-ArcA binding, the promoter region was dissected in three regions each one containing only one putative ABS. Our results showed that P-ArcA only bound to the lpxO promoter fragment containing ABS-1 (Figure 5C). In addition, no bound was detected when either fragment was incubated with non-phosphorylated ArcA. Noteworthy, ABS-1 is located downstream of the putative -10 element in a position that likely represses transcription.
In this work, we showed that oxygen is not only a substrate for LpxO, the enzyme that catalyzes lipid A hydroxylation, but also acts as an environmental cue that regulates this process in S. Enteritidis. In addition, we also demonstrate that global regulators Fnr and ArcA control the oxygen-dependent lipid A hydroxylation by directly regulating the expression of lpxO. To the best of our knowledge, this is the first time that a regulatory role for oxygen and Fnr and ArcA on lipid A covalent modifications has been described in Salmonella. Taking into consideration that oxygen is essential for lipid A hydroxylation, it makes perfect sense the oxygen-dependent regulation of lpxO expression in order to limit the production of LpxO during anaerobiosis, where one of its substrates is absent.
As described in S. Typhimurium (Gibbons et al., 2000), our data showed that lipid A hydroxylation is dependent on oxygen availability in S. Enteritidis (Figure 1). These observations are in line with the described role played by this molecule as LpxO substrate. Noteworthy, our results also revealed that lpxO expression is regulated by oxygen availability (Figure 3). Furthermore, this regulation correlates with the relative levels of hydroxylated and non-hydroxylated lipid A species produced by wild-type S. Enteritidis during aerobiosis and anaerobiosis (Figure 1). In contrast, oxygen-dependent regulation of lipid A hydroxylation was lost in the Δfnr mutant and in the wt/parcA strain (Figure 1), which correlates with the altered expression of lpxO observed in these genetic backgrounds (Figure 3). Of note, the lipid A species distribution shown by strain wt/parcA grown aerobically resembles the one shown by the wild-type strain grown anaerobically (Figure 1). These observations further support the notion that oxygen is not only an LpxO substrate, but also constitutes a regulatory signal controlling lipid A hydroxylation.
Together, these findings suggested that the role played by Fnr and ArcA in lipid A hydroxylation most probably occurs by controlling lpxO expression. This idea was supported by our bioinformatics analyses that revealed the presence of a putative FBS centered at position -379, and three putative ABS centered at positions 12, -47, and -237 with respect to the predicted transcription initiation site of lpxO (Figure 4). Several genes regulated by oxygen availability in E. coli, such as cydAB, cyoABCDE (Salmon et al., 2005), ydbN, and ydeJ (Partridge et al., 2006), also possess similarly positioned ABS and/or FBS. Our results also showed that both Fnr and ArcA are able to bind in vitro to the promoter regions of lpxO (Figure 5), strongly suggesting a direct role for these regulators in the control of lpxO transcription. After assessing the functionality of each ABS in the lpxO promoter by EMSA, we observed that ArcA binds to ABS-1, but not to ABS-2 and ABS-3. Since each ABS was evaluated individually, we cannot rule out a cooperative interaction among these sites for ArcA binding to the lpxO promoter in vivo.
The regulation of lpxO expression has been scarcely studied. Just one work reported that a S. Typhimurium ΔphoP mutant presents wild-type levels of lpxO expression, indicating that this gene is not regulated by PhoP/PhoQ (Gibbons et al., 2005). In spite of this, the same study reported that lpxO expression was increased in a strain carrying the pho-24 allele encoding a constitutively active version of PhoP. According to our bioinformatic analyses (data not shown), the lpxO promoter region does not contain putative PhoP binding sequences, suggesting that the observations made in the pho-24 background are due to an indirect effect on lpxO expression.
Our data also indicate that Fnr not only controls lipid A hydroxylation, since several lipid A species not observed in the wild-type strain were detected in the Δfnr mutant (Figure 1). Similarly, some of these species were also detected in a ΔarcA strain grown aerobically (Supplementary Figure S2). This indicates that Fnr and/or ArcA also control the expression of genes encoding other enzymes involved in lipid A covalent modifications. Supporting this idea, our unpublished bioinformatic analyses revealed putative binding sites for both regulators in the promoter regions of genes eptA, lpxR, and pagP (data not shown). Furthermore, we recently demonstrated an oxygen-dependent regulation of OAg chain-length distribution in S. Enteritidis (Silva-Valenzuela et al., 2016). This process was dependent on the regulation exerted by Fnr and ArcA on the expression of genes wzzSE and wzzfepE, which encode the OAg chain length regulators. Thus, it is tempting to speculate that oxygen availability modulates LPS plasticity through these transcription regulators, contributing to the adaptation of S. Enteritidis to environmental challenges.
Although we can only speculate on the physiological role of the described oxygen-dependent control of lipid A hydroxylation, it has been established that most lipid A covalent modifications have an impact on the virulence of Gram-negative bacteria (reviewed in Needham and Trent, 2013). For instance, K. pneumoniae produces hydroxylated lipid A in vivo during lung infection in mice (Llobet et al., 2015). In addition, stimulation of alveolar and bone marrow-derived murine macrophages in vitro with hydroxylated lipid A from K. pneumoniae triggers a lower TLR-4-dependent inflammatory response than non-hydroxylated lipid A (Llobet et al., 2015; Mills et al., 2017). Furthermore, lipid A hydroxylation has been linked to K. pneumoniae resistance to CAMPs such as colistin and polymyxin B (Llobet et al., 2015; Mills et al., 2017). All these observations indicate that lipid A hydroxylation contributes to K. pneumoniae pathogenicity by facilitating evasion of the innate immune response.
Regarding S. Typhimurium, it has been reported that lipid A hydroxylation levels impact on epithelial cell invasion, intracellular survival in macrophages, and fitness during infection in mice (Moreira et al., 2013). Thus, these observations strongly suggest that appropriate levels of hydroxylated lipid A are essential for Salmonella virulence. However, it is important to mention that these observations were obtained from in vitro experiments and we still do not know whether Salmonella produces or not hydroxylated lipid A in vivo. In spite of this, it is known that non-typhoidal Salmonella serovars (including S. Enteritidis and S. Typhimurium, among others) trigger a strong inflammatory response in the gut during infection. This process generates a beneficial niche containing specific substrates that the pathogen can use in order to outgrowth the gut microbiota. Furthermore, the host inflammatory response enables the transmission of the pathogen by the fecal–oral route (revised in Rivera-Chávez and Bäumler, 2015). Taking into consideration that hydroxylation reduces the pro-inflammatory effects of lipid A in K. pneumoniae, we hypothesize that non-typhoidal Salmonella serovars mainly produce non-hydroxylated lipid A in response to anaerobic conditions in the gut, increasing the pro-inflammatory properties of the bacterial envelope in order to colonize the host efficiently. Further studies are required to confirm this hypothesis.
In addition to Salmonella and Klebsiella, many Gram-negative bacterial pathogens such as Bordetella bronchiseptica, Pseudomonas aeruginosa, Legionella pneumophila, Acinetobacter baumannii, and Vibrio cholerae also produce a lipid A modified with hydroxylated secondary acyl chains (Kulshin et al., 1991; Zähringer et al., 1995; Gibbons et al., 2000; Beceiro et al., 2011; Hankins et al., 2011; MacArthur et al., 2011; Llobet et al., 2015). In V. cholera, this modification has been associated with resistance to polymyxin B (Hankins et al., 2011). In the case of P. aeruginosa, it has been reported that modifications of lipid A structure, including hydroxylation, are required to alter the innate immune responses in order to achieve bacterial persistence and chronic infections, as in the case of cystic fibrosis patients (Moskowitz and Ernst, 2010). Thus, lipid A hydroxylation seems to be a conserved mechanism exploited by bacterial pathogens in order to modulate the innate immune response of the host during infection.
Overall, our results support a model in which Fnr and ArcA control the expression of lpxO in S. Enteritidis to achieve a fine-tuning of lipid A hydroxylation levels. This regulatory mechanism could modulate the pro-inflammatory properties of the bacterial envelope in response to variations in oxygen levels during infection.
PF, FV, CS, and SÁ conceived and designed the experiments. PF, FV, HG-P, FA, JO, and SG performed the experiments. PF, FV, HG-P, FA, CS, and SÁ analyzed the data. PF, CS, and SÁ contributed with reagents, materials, and analysis tools, and wrote the paper. All authors read and approved the final manuscript.
This work was supported by FONDECYT grant 1130225 (to SÁ). CS was supported by FONDECYT grant 1140754 and 1171844. PF was supported by CONICYT fellowship 21140692. FA was supported by CONICYT fellowship 22151395.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank James Slauch for generous gift of strains and plasmids required for generation of chromosomal lacZ fusions and Patricia Kiley for generous gift of plasmid pPK822. We are indebted to Mauricio Báez for technical support and assistance during purification of proteins.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01220/full#supplementary-material
Altier, C. (2005). Genetic and environmental control of Salmonella invasion. J. Microbiol. 43, 85–92.
Beceiro, A., Llobet, E., Aranda, J., Bengoechea, J. A., Doumith, M., Hornsey, M., et al. (2011). Phosphoethanolamine modification of lipid A in colistin-resistant variants of Acinetobacter baumannii mediated by the pmrAB two-component regulatory system. Antimicrob. Agents Chemother. 55, 3370–3379. doi: 10.1128/AAC.00079-11
Bravo, D., Silva, C., Carter, J. A., Hoare, A., Álvarez, S. A., Blondel, C. J., et al. (2008). Growth-phase regulation of lipopolysaccharide O-antigen chain length influences serum resistance in serovars of Salmonella. J. Med. Microbiol. 57(Pt 8), 938–946. doi: 10.1099/jmm.0.47848-0
Caroff, M., and Novikov, A. (2011). Micromethods for lipid A isolation and structural characterization. Methods Mol. Biol. 739, 135–146. doi: 10.1007/978-1-61779-102-4_12
Cherepanov, P. P., and Wackernagel, W. (1995). Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158, 9–14. doi: 10.1016/0378-1119(95)00193-A
Compan, I., and Touati, D. (1994). Anaerobic activation of arcA transcription in Escherichia coli: roles of Fnr and ArcA. Mol. Microbiol. 11, 955–964. doi: 10.1111/j.1365-2958.1994.tb00374.x
Coward, C., Sait, L., Cogan, T., Humphrey, T. J., and Maskell, D. J. (2013). O-antigen repeat number in Salmonella enterica serovar Enteritidis is important for egg contamination, colonisation of the chicken reproductive tract and survival in egg albumen. FEMS Microbiol. Lett. 343, 169–176. doi: 10.1111/1574-6968.12143
Dalebroux, Z. D., and Miller, S. I. (2014). Salmonellae PhoPQ regulation of the outer membrane to resist innate immunity. Curr. Opin. Microbiol. 17, 106–113. doi: 10.1016/j.mib.2013.12.005
Darveau, R. P., and Hancock, R. E. (1983). Procedure for isolation of bacterial lipopolysaccharides from both smooth and rough Pseudomonas aeruginosa and Salmonella typhimurium strains. J. Bacteriol. 155, 831–838.
Datsenko, K. A., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645. doi: 10.1073/pnas.120163297
Ellermeier, C. D., Janakiraman, A., and Slauch, J. M. (2002). Construction of targeted single copy lac fusions using lambda Red and FLP-mediated site-specific recombination in bacteria. Gene 290, 153–161. doi: 10.1016/S0378-1119(02)00551-6
Fink, R. C., Evans, M. R., Porwollik, S., Vázquez-Torres, A., Jones-Carson, J., Troxell, B., et al. (2007). FNR is a global regulator of virulence and anaerobic metabolism in Salmonella enterica serovar Typhimurium (ATCC 14028s). J. Bacteriol. 189, 2262–2273. doi: 10.1128/JB.00726-06
Folch, J., Lees, M., and Sloane Stanley, G. H. (1957). A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226, 497–509.
Gibbons, H. S., Kalb, S. R., Cotter, R. J., and Raetz, C. R. (2005). Role of Mg2+ and pH in the modification of Salmonella lipid A after endocytosis by macrophage tumour cells. Mol. Microbiol. 55, 425–440. doi: 10.1111/j.1365-2958.2004.04409.x
Gibbons, H. S., Lin, S., Cotter, R. J., and Raetz, C. R. (2000). Oxygen requirement for the biosynthesis of the S-2-hydroxymyristate moiety in Salmonella typhimurium lipid A. Function of LpxO, a new Fe2+/α-ketoglutarate-dependent dioxygenase homologue. J. Biol. Chem. 275, 32940–32949. doi: 10.1074/jbc.M005779200
Gibbons, H. S., Reynolds, C. M., Guan, Z., and Raetz, C. R. (2008). An inner membrane dioxygenase that generates the 2-hydroxymyristate moiety of Salmonella lipid A. Biochemistry 47, 2814–2825. doi: 10.1021/bi702457c
Guard-Petter, J. (2001). The chicken, the egg and Salmonella enteritidis. Environ. Microbiol. 3, 421–430. doi: 10.1046/j.1462-2920.2001.00213.x
Gunn, J. S., Ryan, S. S., Van Velkinburgh, J. C., Ernst, R. K., and Miller, S. I. (2000). Genetic and functional analysis of a PmrA-PmrB-regulated locus necessary for lipopolysaccharide modification, antimicrobial peptide resistance, and oral virulence of Salmonella enterica serovar Typhimurium. Infect. Immun. 68, 6139–6146. doi: 10.1128/IAI.68.11.6139-6146.2000
Guo, L., Lim, K. B., Poduje, C. M., Daniel, M., Gunn, J. S., Hackett, M., et al. (1998). Lipid A acylation and bacterial resistance against vertebrate antimicrobial peptides. Cell 95, 189–198. doi: 10.1016/S0092-8674(00)81750-X
Hankins, J. V., Madsen, J. A., Giles, D. K., Childers, B. M., Klose, K. E., Brodbelt, J. S., et al. (2011). Elucidation of a novel Vibrio cholerae lipid A secondary hydroxy-acyltransferase and its role in innate immune recognition. Mol. Microbiol. 81, 1313–1329. doi: 10.1111/j.1365-2958.2011.07765.x
Jeon, Y., Lee, Y. S., Han, J. S., Kim, J. B., and Hwang, D. S. (2001). Multimerization of phosphorylated and non-phosphorylated ArcA is necessary for the response regulator function of the Arc two-component signal transduction system. J. Biol. Chem. 276, 40873–40879. doi: 10.1074/jbc.M104855200
Kawasaki, K., China, K., and Nishijima, M. (2007). Release of the lipopolysaccharide deacylase PagL from latency compensates for lack of lipopolysaccharide aminoarabinose modification-dependent resistance to the antimicrobial peptide polymyxin B in Salmonella enterica. J. Bacteriol. 189, 4911–4919. doi: 10.1128/JB.00451-07
Kiley, P. J., and Beinert, H. (1998). Oxygen sensing by the global regulator, FNR: the role of the iron-sulfur cluster. FEMS Microbiol. Rev. 22, 341–352. doi: 10.1111/j.1574-6976.1998.tb00375.x
Kong, Q., Six, D. A., Liu, Q., Gu, L., Wang, S., Alamuri, P., et al. (2012). Phosphate groups of lipid A are essential for Salmonella enterica serovar Typhimurium virulence and affect innate and adaptive immunity. Infect. Immun. 80, 3215–3224. doi: 10.1128/IAI.00123-12
Kroger, C., Dillon, S. C., Cameron, A. D., Papenfort, K., Sivasankaran, S. K., Hokamp, K., et al. (2012). The transcriptional landscape and small RNAs of Salmonella enterica serovar Typhimurium. Proc. Natl. Acad. Sci. U.S.A. 109, E1277–E1286. doi: 10.1073/pnas.1201061109
Kulshin, V. A., Zähringer, U., Lindner, B., Jäger, K. E., Dmitriev, B. A., and Rietschel, E. T. (1991). Structural characterization of the lipid A component of Pseudomonas aeruginosa wild-type and rough mutant lipopolysaccharides. Eur. J. Biochem. 198, 697–704. doi: 10.1111/j.1432-1033.1991.tb16069.x
Lazazzera, B. A., Bates, D. M., and Kiley, P. J. (1993). The activity of the Escherichia coli transcription factor FNR is regulated by a change in oligomeric state. Genes Dev. 7, 1993–2005. doi: 10.1101/gad.7.10.1993
Llobet, E., Martinez-Moliner, V., Moranta, D., Dahlstrom, K. M., Regueiro, V., Tomas, A., et al. (2015). Deciphering tissue-induced Klebsiella pneumoniae lipid A structure. Proc. Natl. Acad. Sci. U.S.A. 112, E6369–E6378. doi: 10.1073/pnas.1508820112
Lynch, A. S., and Lin, E. C. (1996). Transcriptional control mediated by the ArcA two-component response regulator protein of Escherichia coli: characterization of DNA binding at target promoters. J. Bacteriol. 178, 6238–6249. doi: 10.1128/jb.178.21.6238-6249.1996
MacArthur, I., Jones, J. W., Goodlett, D. R., Ernst, R. K., and Preston, A. (2011). Role of pagL and lpxO in Bordetella bronchiseptica lipid A biosynthesis. J. Bacteriol. 193, 4726–4735. doi: 10.1128/JB.01502-10
Marolda, C. L., Lahiry, P., Vinés, E., Saldías, S., and Valvano, M. A. (2006). Micromethods for the characterization of lipid A-core and O-antigen lipopolysaccharide. Methods Mol. Biol. 347, 237–252. doi: 10.1385/1-59745-
Mika, F., and Hengge, R. (2005). A two-component phosphotransfer network involving ArcB, ArcA, and RssB coordinates synthesis and proteolysis of sigmaS (RpoS) in E. coli. Genes Dev. 19, 2770–2781. doi: 10.1101/gad.353705
Miller, J. (1972). Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Mills, G., Dumigan, A., Kidd, T., Hobley, L., and Bengoechea, J. A. (2017). Identification and characterization of two Klebsiella pneumoniae lpxL lipid A late acyltransferases and their role in virulence. Infect. Immun. 85, e68–e17. doi: 10.1128/IAI.00068-17
Moreira, C. G., Herrera, C. M., Needham, B. D., Parker, C. T., Libby, S. J., Fang, F. C., et al. (2013). Virulence and stress-related periplasmic protein (VisP) in bacterial/host associations. Proc. Natl. Acad. Sci. U.S.A. 110, 1470–1475. doi: 10.1073/pnas.1215416110
Moskowitz, S. M., and Ernst, R. K. (2010). The role of Pseudomonas lipopolysaccharide in cystic fibrosis airway infection. Subcell. Biochem. 53, 241–253. doi: 10.1007/978-90-481-9078-2_11
Munch, R., Hiller, K., Grote, A., Scheer, M., Klein, J., Schobert, M., et al. (2005). Virtual footprint and PRODORIC: an integrative framework for regulon prediction in prokaryotes. Bioinformatics 21, 4187–4189. doi: 10.1093/bioinformatics/bti635
Needham, B. D., and Trent, M. S. (2013). Fortifying the barrier: the impact of lipid A remodelling on bacterial pathogenesis. Nat. Rev. Microbiol. 11, 467–481. doi: 10.1038/nrmicro3047
Park, B. S., Song, D. H., Kim, H. M., Choi, B. S., Lee, H., and Lee, J. O. (2009). The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458, 1191–1195. doi: 10.1038/nature07830
Partridge, J. D., Scott, C., Tang, Y., Poole, R. K., and Green, J. (2006). Escherichia coli transcriptome dynamics during the transition from anaerobic to aerobic conditions. J. Biol. Chem. 281, 27806–27815. doi: 10.1074/jbc.M603450200
Raetz, C. R. (2001). Regulated covalent modifications of lipid A. J. Endotoxin. Res. 7, 73–78. doi: 10.1177/09680519010070010201
Raetz, C. R., Reynolds, C. M., Trent, M. S., and Bishop, R. E. (2007). Lipid A modification systems in Gram-negative bacteria. Annu. Rev. Biochem. 76, 295–329. doi: 10.1146/annurev.biochem.76.010307.145803
Raetz, C. R., and Whitfield, C. (2002). Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71, 635–700. doi: 10.1146/annurev.biochem.71.110601.135414
Ravcheev, D. A., Gerasimova, A. V., Mironov, A. A., and Gelfand, M. S. (2007). Comparative genomic analysis of regulation of anaerobic respiration in ten genomes from three families of gamma-proteobacteria (Enterobacteriaceae, Pasteurellaceae, Vibrionaceae). BMC Genomics 8:54. doi: 10.1186/1471-2164-8-54
Reynolds, C. M., Ribeiro, A. A., McGrath, S. C., Cotter, R. J., Raetz, C. R., and Trent, M. S. (2006). An outer membrane enzyme encoded by Salmonella typhimurium lpxR that removes the 3′-acyloxyacyl moiety of lipid A. J. Biol. Chem. 281, 21974–21987. doi: 10.1074/jbc.M603527200
Rhen, M., and Dorman, C. J. (2005). Hierarchical gene regulators adapt Salmonella enterica to its host milieus. Int. J. Med. Microbiol. 294, 487–502. doi: 10.1016/j.ijmm.2004.11.004
Rivera-Chávez, F., and Bäumler, A. J. (2015). The pyromaniac inside you: Salmonella metabolism in the host gut. Annu. Rev. Microbiol. 69, 31–48. doi: 10.1146/annurev-micro-091014-104108
Roberts, J. A., and Sockett, P. N. (1994). The socio-economic impact of human Salmonella enteritidis infection. Int. J. Food Microbiol. 21, 117–129. doi: 10.1016/0168-1605(94)90205-4
Salmon, K. A., Hung, S. P., Steffen, N. R., Krupp, R., Baldi, P., Hatfield, G. W., et al. (2005). Global gene expression profiling in Escherichia coli K12: effects of oxygen availability and ArcA. J. Biol. Chem. 280, 15084–15096. doi: 10.1074/jbc.M414030200
Santiviago, C. A., Reynolds, M. M., Porwollik, S., Choi, S. H., Long, F., Andrews-Polymenis, H. L., et al. (2009). Analysis of pools of targeted Salmonella deletion mutants identifies novel genes affecting fitness during competitive infection in mice. PLoS Pathog. 5:e1000477. doi: 10.1371/journal.ppat.1000477
Sawers, G. (1999). The aerobic/anaerobic interface. Curr. Opin. Microbiol. 2, 181–187. doi: 10.1016/S1369-5274(99)80032-0
Schmieger, H. (1972). Phage P22-mutants with increased or decreased transduction abilities. Mol. Gen. Genet. 119, 75–88. doi: 10.1007/BF00270447
Shi, Y., Latifi, T., Cromie, M. J., and Groisman, E. A. (2004). Transcriptional control of the antimicrobial peptide resistance ugtL gene by the Salmonella PhoP and SlyA regulatory proteins. J. Biol. Chem. 279, 38618–38625. doi: 10.1074/jbc.M406149200
Silva, C. A., Blondel, C. J., Quezada, C. P., Porwollik, S., Andrews-Polymenis, H. L., Toro, C. S., et al. (2012). Infection of mice by Salmonella enterica serovar Enteritidis involves additional genes that are absent in the genome of serovar Typhimurium. Infect. Immun. 80, 839–849. doi: 10.1128/iai.05497-11
Silva-Valenzuela, C. A., Velásquez, F., Peñailillo, J., Garcias-Papayani, H., Fernández, P., Tobar, P., et al. (2016). O-antigen chain-length distribution in Salmonella enterica serovar Enteritidis is regulated by oxygen availability. Biochem. Biophys. Res. Commun. 477, 563–567. doi: 10.1016/j.bbrc.2016.06.074
Tsai, C. M., and Frasch, C. E. (1982). A sensitive silver stain for detecting lipopolysaccharides in polyacrylamide gels. Anal. Biochem. 119, 115–119. doi: 10.1016/0003-2697(82)90673-X
Yi, E. C., and Hackett, M. (2000). Rapid isolation method for lipopolysaccharide and lipid A from Gram-negative bacteria. Analyst 125, 651–656. doi: 10.1039/b000368i
Keywords: Salmonella, LPS, lipid A hydroxylation, anaerobiosis, LpxO, Fnr, ArcA
Citation: Fernández PA, Velásquez F, Garcias-Papayani H, Amaya FA, Ortega J, Gómez S, Santiviago CA and Álvarez SA (2018) Fnr and ArcA Regulate Lipid A Hydroxylation in Salmonella Enteritidis by Controlling lpxO Expression in Response to Oxygen Availability. Front. Microbiol. 9:1220. doi: 10.3389/fmicb.2018.01220
Received: 30 March 2018; Accepted: 18 May 2018;
Published: 08 June 2018.
Edited by:
George Grant, University of Aberdeen, United KingdomReviewed by:
Uwe Mamat, Forschungszentrum Borstel (LG), GermanyCopyright © 2018 Fernández, Velásquez, Garcias-Papayani, Amaya, Ortega, Gómez, Santiviago and Álvarez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sergio A. Álvarez, c2FsdmFyZXpAdWNoaWxlLmNs
†These authors have contributed equally to this study.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.