
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 08 June 2018
Sec. Terrestrial Microbiology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.01188
This article is part of the Research Topic Soil and Plant-Associated Microbiome to Promote Ecological Resource Use Efficiency in Agro-Ecosystems View all 10 articles
 Chih-Ying Lay1
Chih-Ying Lay1 Terrence H. Bell1,2
Terrence H. Bell1,2 Chantal Hamel3
Chantal Hamel3 K. Neil Harker4
K. Neil Harker4 Ramona Mohr5
Ramona Mohr5 Charles W. Greer6
Charles W. Greer6 Étienne Yergeau6,7
Étienne Yergeau6,7 Marc St-Arnaud1*
Marc St-Arnaud1*Canola is one of the most economically important crops in Canada, and the root and rhizosphere microbiomes of a canola plant likely impact its growth and nutrient uptake. The aim of this study was to determine whether canola has a core root microbiome (i.e., set of microbes that are consistently selected in the root environment), and whether this is distinct from the core microbiomes of other crops that are commonly grown in the Canadian Prairies, pea, and wheat. We also assessed whether selected agronomic treatments can modify the canola microbiome, and whether this was associated to enhanced yield. We used a field experiment with a randomized complete block design, which was repeated at three locations across the canola-growing zone of Canada. Roots and rhizosphere soil were harvested at the flowering stage of canola. We separately isolated total extractable DNA from plant roots and from adjacent rhizosphere soil, and constructed MiSeq amplicon libraries for each of 60 samples, targeting bacterial, and archaeal 16S rRNA genes and the fungal ITS region. We determined that the microbiome of the roots and rhizosphere of canola was consistently different from those of wheat and pea. These microbiomes comprise several putative plant-growth-promoting rhizobacteria, including Amycolatopsis sp., Serratia proteamaculans, Pedobacter sp., Arthrobacter sp., Stenotrophomonas sp., Fusarium merismoides, and Fusicolla sp., which correlated positively with canola yield. Crop species had a significant influence on bacterial and fungal assemblages, especially within the roots, while higher nutrient input or seeding density did not significantly alter the global composition of bacterial, fungal, or archaeal assemblages associated with canola roots. However, the relative abundance of Olpidium brassicae, a known pathogen of members of the Brassicaceae, was significantly reduced in the roots of canola planted at higher seeding density. Our results suggest that seeding density and plant nutrition management modified the abundance of other bacterial and fungal taxa forming the core microbiomes of canola that are expected to impact crop growth. This work helps us to understand the microbial assemblages associated with canola grown under common agronomic practices and indicates microorganisms that can potentially benefit or reduce the yield of canola.
Canada is one of the world's main canola-producing countries, ranking third in oil production (USDA, 2017). It is therefore important for Canada to optimize agronomic treatments in this key agro-industry. Different approaches, such as increasing seeding density, adapting fertilization regimes, and selecting optimal rotation crops and rotation sequences, were previously shown to potentially enhance canola yield or reduce disease outbreaks (Harker et al., 2003, 2012, 2015a; Guo et al., 2005; Hwang et al., 2009).
The microorganisms inhabiting plant root environments are essential in facilitating nutrient uptake, preventing colonization by pathogens, mitigating the impact of abiotic stressors, and modulating the levels of plant hormones (Yang et al., 2009; Dodd et al., 2010; Berendsen et al., 2012). Previous research revealed that canola-root-associated microbiomes are largely determined by the season and plant developmental stage (Smalla et al., 2001; Dunfield and Germida, 2003; de Campos et al., 2013). Rhizoctonia solani could affect the emergence and early development of canola seedlings, with impacts on the overall fungal assemblage associated with canola roots, especially in winter crops (Neupane et al., 2013a). Another study attributed a reduction in canola yield to root infection by zoospores of Olpidium brassicae, a known parasite of the Brassicaceae (Hartwright et al., 2010; Hilton et al., 2013). However, the correlations of the yields of canola and the structure of the canola root and rhizosphere microbiome have rarely been thoroughly studied. In particular, the interactions between bacterial, fungal, and archaeal communities have yet to be examined. In order to identify putatively important microorganisms in the canola root environment, we applied the concept of the core microbiome, as defined by Vandenkoornhuyse et al. (2015). In this context, the “core” microbiome of a plant species is the collection of operational taxonomic units (OTUs) that are present in any condition, while the OTUs that associate with the plant in a given condition but not under all conditions (e.g., that depend on a specific environment) are called the “eco” microbiome. Agronomic treatments that influence root-associated microbial assemblages can increase or reduce disease risks in crop production (Gomiero et al., 2011; Chaparro et al., 2012).
The aims of this work were (1) to identify and define the canola microbiome in the Canadian Prairies and to compare the bacterial, fungal, and archaeal communities associated with canola with those of reference crops; (2) to assess the effect of increased seeding density and fertilization on the canola-associated microbiomes and putative interactions between taxa from different communities; and (3) to assess the links between microbial composition and productivity metrics, including crop yield, weed counts, and seedling emergence rates. To achieve these objectives, we sampled canola roots and rhizosphere soil from an ongoing field experiment set up at three locations across western Canada (Brandon, Manitoba; Beaverlodge, Alberta; and Lacombe, Alberta). In 2014, plots were seeded with canola or with either wheat or pea as a comparison crop. The treatments applied to the canola plots included canola grown as recommended, canola fertilized at 150% of the recommended rate, and canola seeded at 150% of the recommended rate. Higher seeding density (Harker et al., 2003, 2012, 2015a,b) and higher nutrient supply (especially N) (Grant et al., 2012) were previously shown to increase canola yield in the Canadian Prairies. To our knowledge, this is the first study to comprehensively reveal the composition of all three major microbial communities of the canola microbiome using high-throughput sequencing, and to describe the core microbiome of canola. The results identify potentially important soil microorganisms for plant productivity and provide new insight into the effect of agricultural practices on canola-root-associated microbiomes.
The present study used a subset of the plots of an experiment established in 2008 at three sites across the canola-producing area of western Canada. The sites in Lacombe (Alberta), and Brandon, (Manitoba), are in the Black soil zone, while the site in Beaverlodge (Alberta), is in the Dark Gray soil zone (Grant and Wu, 2008). At each site, the experiment is arranged in a randomized complete block design with four blocks. The initial soil characteristics are given in Table S1, and information on site management is given in Harker et al. (2015b) and Supplementary Information.
In 2014, five of the 14 treatments of the experiment were used for our study. Three of the treatments followed six years of canola monoculture, namely, (1) canola grown at 100% of the recommended rate (Can_RE), (2) canola fertilized at 150% of the recommended rate (Can_HF), and (3) canola seeded at 150% of the recommended rate (Can_HD), and the other two treatments were (4) wheat or (5) pea following canola and wheat in crop rotation systems involving canola. In 2013, the previous year, all plots had been planted with the same canola cultivar (Table 1). In the study year (2014), the plots under Can_RE were planted at the recommended seeding rate for canola, which was 100 seeds m−2 and received the recommended N, P, K, and S fertilization rate for canola, on the basis of soil tests. The plots under Can_HF were planted at 100 seeds m−2 and received 150% of the recommended fertilization rate, and the plots under Can_HD were planted at 150 seeds m−2 and received 100% of the recommended fertilizer rate (Table 1 and Supplementary Information). The canola cultivar was InVigor L135C (Bayer CropScience, Calgary, AB), a clubroot-resistant canola (Brassica napus) cultivar with resistance to the herbicide glufosinate. The wheat cultivar was Stettler, a doubled haploid hard red spring wheat (Triticum aestivum L.), and the pea cultivar was CDC Meadow, a yellow cotyledon field pea (Pisum sativum L.). Field pea and wheat were planted at the recommended rates of 100 and 300 seeds m−2, respectively, and were fertilized and maintained according to best management practices.
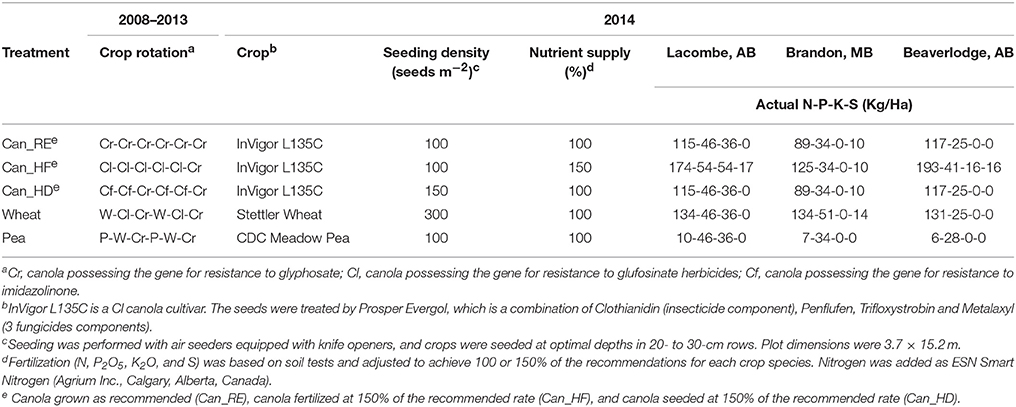
Table 1. Description of the crops and the treatments applied to the fields.
Root and rhizosphere soil samples were collected in the fourth week of July 2014, corresponding to growth stage 6 of canola (spanning from first to last flower bud opening), stage 4 of wheat development (flag leaf), and stage 6 of pea (mid-bloom) on the BBCH scale (Weber and Bleiholder, 1990). Randomly choosen plants were excavated, and fine roots with adhering soil were harvested from three or four plants at each of four random locations within each plot. In total, there was 60 plots (3 locations × 4 blocks × 5 treatments = 60 plots, including 36 canola, 12 wheat, and 12 pea plots). The roots were pooled into one composite sample per plot. In the field, immediately after collection, the root samples were placed in 15-mL Falcon tubes containing RNAlater Stabilization Solution (Ambion, Foster City, CA, USA) for preservation. Each sample consisted of 8 mL of fine roots plus adhering soil material per plot, as measured by displacement of the RNAlater solution. During harvest, the tubes were placed in a cooler on freezer packs. The samples were kept at 4°C before they were shipped to Swift Current, Saskatchewan, and then placed at −20°C until they were shipped by air cargo from Swift Current to Montreal, Quebec, in a Styrofoam cooler on freezer packs. Upon reception, the roots and rhizosphere soil were separated using an ethanol-sterilized sieve and scoop and were preserved at −80°C until DNA extraction. For each plot, crop emergence, crop maturity, and weed counts were recorded as described in Supplementary Information.
To isolate total genomic DNA, roots and soil samples were recovered from the RNAlater solution and cleaned by rinsing with sterilized water. The roots were then ground using a mortar and pestle with liquid nitrogen. Total root DNA was extracted from 350 mg of root material using the NucleoSpin Soil DNA isolation kit (Macherey-Nagel, Düren, Germany) according to the manufacturer's instructions with modifications (see Supplementary Information). The final DNA yield ranged from 2 to 6 μg in 100 μL of elution solution.
Microbial genomic DNA from rhizosphere soil was isolated using the RNA PowerSoil Total RNA Isolation Kit plus the RNA PowerSoil DNA Elution Accessory Kit (Mo Bio Laboratories Inc., Carlsbad, CA, USA). The accessory kit was used because RNA had to be extracted for use in another study, and the kit allows DNA to be extracted at the same time. For each sample, 1.5–2 g of preserved rhizosphere soil was added to 2-mL microcentrifuge tubes. To reduce increases in salt content caused by the RNAlater solution, the samples were washed in a DEPC (diethypyrocarbonate)–treated PBS (phosphate-buffered saline) solution three times before being added to the bead-beating tubes. The samples were prepared according to the manufacturer's instructions with some modifications (see Supplementary Information). The DNA products were eluted using 100 μL of elution buffer from the accessory kit and visualized on a 1% agarose gel using electrophoresis. The final DNA yield ranged from 2 to 6 μg in 100 μL of elution solution. If the appropriate band size for genomic DNA appeared, the extracts were considered usable for further analysis.
We constructed amplicon libraries for bacterial and archaeal 16S ribosomal RNA (rRNA) genes and fungal internal transcribed spacer (ITS) sequences by using target-specific PCR primers attached to Illumina overhang sequences for Nextera preparation. The primer pairs used were S-D-Bact-0341-b-S-17 with S-D-Bact-0785-a-A-21 for bacteria (Klindworth et al., 2013), ARCH517F-Illu with ARCH909R-Illu for archaea (Burggraf et al., 1997; Baker et al., 2003), and ITS1F-Illu with 58A2R-Illu for fungi (Martin and Rygiewicz, 2005; Manter and Vivanco, 2007). For each 25-μL PCR reaction of bacteria and fungi, the reaction buffer consisted of 0.5 μL each of forward and reverse primer, along with 10 μL of H2O, 0.5 μL of 25 mM MgCl2, 12.5 μL of KAPA HiFi HotStart ReadyMix (Kapa Biosystems, Cape Town, South Africa), and 1 μL of sample DNA. The 25-μL PCR reaction buffer for archaeal PCR consisted of 0.5 μL of forward and reverse primer, respectively, with 10.5 μL H2O, 12.5 μL KAPA HiFi HotStart ReadyMix, and a 1-μL sample of DNA. Reaction conditions are given in Supplementary Information.
Dual Nextera indexes were then attached to PCR products on the basis of the suggested protocol titled “16S Metagenomic Sequencing Library Preparation,” provided by Illumina, Inc. (San Diego, CA, USA), with certain modifications. The concentrated product was quantified by Qubit Fluorometric Quantitation (ThermoFisher Scientific, Waltham, MA, USA) and run on 1.5% agarose gels, and then the bands of the proper size were gel-extracted using the PureLink Quick Gel Extraction Kit (Invitrogen, Löhne, Germany) with a final elution volume of 50 μL. The gel-extraction products were quantified and then sequenced on an Illumina MiSeq sequencer using the 600-cycle MiSeq Reagent Kit v.3, according to the manufacturer's recommendations. The concentrations of samples were 33.33 nM for bacteria, 13.25 nM for archaea, and 40.00 nM for fungi. Details of the PCR reactions and indexing are available in Supplementary Information.
The preliminary processing of bacterial and archaeal 16S rRNA gene libraries was performed using mothur v.1.34.4 (Schloss et al., 2009) to join the paired ends. To reduce the file size for bacterial 16S rRNA gene sequences in order to accommodate the size limit of the 32-bit version of USEARCH (Edgar, 2010), we discarded singletons using the “unique.seqs” and “split.abund” commands with a cutoff of 1. This method replaces the equivalent procedure within USEARCH that is recommended by the Brazilian Microbiome Project (BMP) bacterial 16S pipeline (Pylro et al., 2014a,b). The database for aligning and classifying the bacterial and archaeal reads was downloaded from GreenGenes (gg_13_8_otus). Paired-end fungal reads were also joined similarly into contigs using mothur as described above, and the reads were then processed using the BMP fungal ITS pipeline (Pylro et al., 2014a,b) using QIIME (Caporaso et al., 2010), USEARCH, ITSx (Bengtsson-Palme et al., 2013), FASTA formatter (http://hannonlab.cshl.edu/fastx_toolkit/index.html), and the scripts written by the members of the BMP, with the UNITE ITS database (sh_qiime_release_s_01.08.2015) (Kõljalg et al., 2013). The identity threshold for the OTUs was set at 97%. Since the database was poorly representative of archaea, we constructed an archaeal phylogenetic tree, based on the OTUs of the members of the archaeal core microbiomes, which we had computed previously using the “compute_core_microbiome.py” script with Phylogeny.fr (Dereeper et al., 2008). The phylogenetic tree improved the identification of OTUs using reference sequences selected from GenBank. More details on bioinformatic processing are available in Supplementary Information.
The MiSeq sequencing data generated in this work were deposited in the Sequence Read Archive (SRA) database of NCBI. The NCBI Bioproject accession numbers are PRJNA383339 (Bacteria), PRJNA383353 (Fungi), and PRJNA383350 (Archaea).
We used the QIIME script “core_diversity_analyses.py” to estimate α-diversity (Chao1, Simpson's reciprocal, and evenness indices). The effect of increased fertilization or seeding density, crop identity, and sample biotope (roots or rhizosphere soil) on the indices was tested by analysis of variance (ANOVA) with Tukey's post-hoc analyses using the “HSD.test” function of the “agricolae” package in R (https://www.r-project.org). The effect of crops and canola treatments on microbial community structure was assessed by permutational multivariate analysis of variance (PERMANOVA) using the “adonis” function of the “vegan” package in R. Two groups of comparisons were conducted: (1) the effect of crops (canola, wheat, and pea) on root and rhizosphere communities of bacteria, fungi, and archaea, and (2) the effect of canola treatments (Can_RE, Can_HF, and Can_HD) on root and rhizosphere microbial communities. To assess the clustering of samples in each group of comparisons, principal coordinates analysis (PCoA) plots were drawn using the “vegan” package in R based on the OTU matrices generated by the QIIME script “core_diversity_analyses.py” (see Supplementary Information for details).
In order to identify potentially important microorganisms in our system, we used the concept of core microbiome, as defined by Vandenkoornhuyse et al. (2015). The core microbiome associated with each crop and the eco microbiomes associated with each treatment were identified using the QIIME script “compute_core_microbiome.py”. Only the OTUs that formed more than 1% of the assemblage for at least one of the combinations of treatment and location were used. The criteria for selection and screening OTUs are detailed in Supplementary Information.
To determine whether OTUs belong to core or eco microbiomes, we compared them across crops and canola treatments, without considering location, using the QIIME script “group_significance.py” to assess their association with canola. Kruskal–Wallis tests and Benjamini–Hochberg FDR (false-discovery-rate-corrected) P-values (Goeman and Solari, 2014) were used to evaluate the significance of the treatment effect. When the frequency of an OTU exceeded a threshold in all samples from the three canola treatments and locations, the OTU was considered part of the canola core microbiome. If an OTU was present in one or two treatments but not in all, the OTU was considered part of the eco microbiome. The remaining OTUs constituted the unshared fraction and were not considered further. Details on the definition of the core and eco microbiomes are given in Supplementary Information. Bacterial, fungal, and archaeal OTUs of the core and eco microbiomes were analyzed across two different subgroups, (1) roots and (2) rhizospheres, and contrasted between all canola plots with reference crops (canola, wheat, and pea) and between canola treatments (Can_RE, Can_HF, and CAN_HD).
To estimate the similarity of the core microbiomes of canola, wheat, and pea, and the similarity of the core microbiomes of canola as influenced by seeding rate and fertilization, we computed the Sørensen indices using the QIIME script “beta_diversity.py” with the “binary_sorensen_dice” option. We used canola crops as the baseline for comparison when comparing the crops and Can_RE as the baseline for comparison when comparing the canola treatments. The values of the Sørensen index were tested by ANOVA with Tukey's HSD post-hoc analyses using the “HSD.test” function of the “agricolae” package in R.
The links between the bacterial and fungal core microbiomes from roots and from rhizosphere soil were separately analyzed with co-inertia analyses (CoIAs) using the “ade4” package in R. The input OTU tables were Hellinger transformed using the ‘deconstand’ function of the “vegan” package. The transformed matrices were used as input data for the principal component analysis (PCA) using the “dudi.pca” function from “ade4.” Outputs of “dudi.pca” were run with the function “coinertia” from “ade4” to generate the co-intertia analysis. The results were tested using the Monte Carlo method to obtain the simulated P value.
Redundancy analysis (RDA) was used to correlate post-spray weed counts and emergence counts with the bacterial and fungal root and rhizosphere core microbiomes and plant yields in order to assess their association with these field variables. The matrices were Hellinger-transformed using the “decostand” function followed by the “rda” function of the “vegan” package in R. The significance of the RDA model was tested by ANOVA and the R2 values were generated by the “RsquareAdj” function in R. The significance of the whole model, the axes, and each factor were also tested by ANOVA.
To identify the microorganisms correlated with yields, the association of each member of the core microbiomes of canola roots and rhizospheres with yield was tested using Spearman's correlation. The matrices were standardized using the “decostand” function of the “vegan” package in R, followed by the “rcorr” function in the “Hmisc” package. Only the members with significant correlations were included in the results.
After the reads from different data sets were filtered for quality, the bacterial gene data set allowed us to retrieve a total of 3,230,956 sequences (ranging from 3,574 to 72,331 reads across the 120 samples; Table S2) that were assigned to 6,376 OTUs after subsampling to 3,574 reads per sample. For the fungal data set, 1,112,137 sequences (ranging from 97 to 59,199 reads across 120 samples) were obtained and assigned to 679 OTUs after subsampling to 123 reads while discarding the sample with <100 reads. In the archaeal data set, we obtained a total of 2,936,314 sequences (ranging from 329 to 203,921 reads across 120 samples) that were assigned to 49 OTUs after subsampling to 329 reads per sample.
Sixteen bacterial phyla with four classes from the phylum Proteobacteria were identified. In the canola roots (Figure 1A), the most abundant phyla/classes were Gammaproteobacteria (up to 40%), Actinobacteria (up to 50%), and Betaproteobacteria (up to 20%). In the canola rhizosphere, Gammaproteobacteria (up to 30%) and Actinobacteria (up to 30%) were still abundant (although lower than in the roots), followed by Planctomycetes (up to 15%), Alphaproteobacteria (up to 10%), and Bacteroidetes (about 5%). In comparison with canola, the reference crop pea showed a very different pattern, with Alphaproteobacteria forming up to 96% of bacterial taxa within the roots (Figure 1A). Wheat roots also showed a different trend in bacterial community composition at the phylum or class level, with a decrease in Gammaproteobacteria and an increase in Actinobacteria in comparison with canola. The fungal ITS data set was composed mostly of Chytridiomycota (up to 85% in canola roots on average) and Ascomycota (more in wheat or pea roots, up to 50%) (Figure 1B). Zygomycota were more abundant in the rhizosphere (up to 15%) than in the roots. For most of the samples, <10% of the fungal sequences were assigned to unclassified fungi. More than 99% of the archaeal reads belonged to the phylum Thaumarchaeota. The archaeal members that we detected in the samples were mostly unidentified, but the core members were mostly close to Nitrocosmicus spp. according to the phylogenetic analysis (Figure S1). Rarefaction curves showed that read abundances were close to saturation for most of the samples, and Good's coverage values ranked between 0.83 and 0.98 (Bacteria), 0.62 and 1 (Fungi), and 0.97 and 1 (Archaea) (Figure S2).
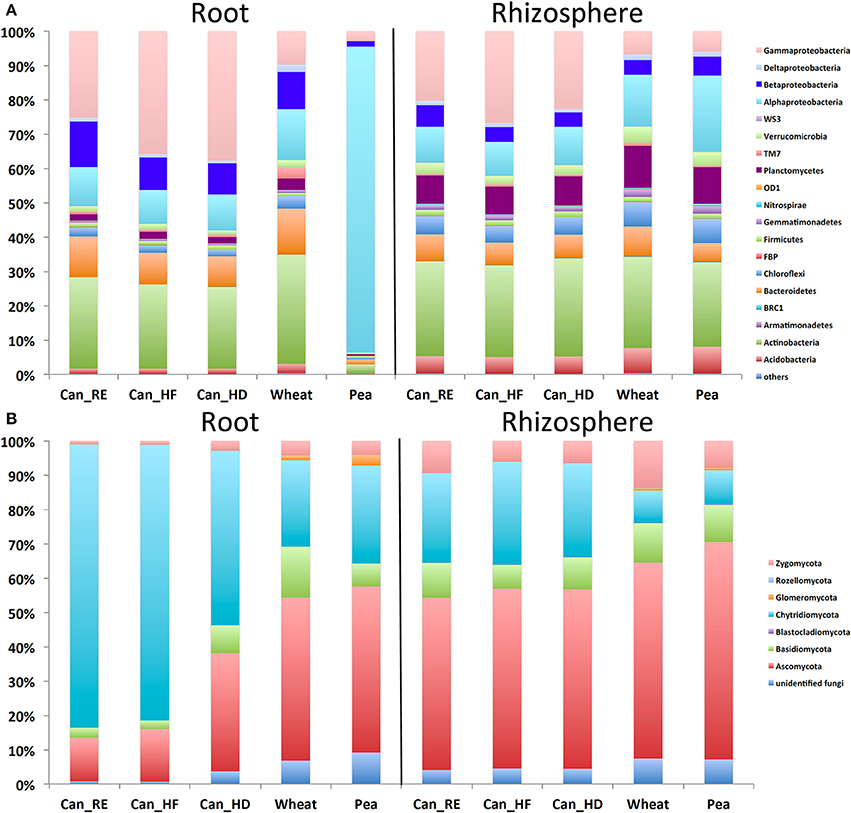
Figure 1. Mean (n = 12) relative abundances of (A) bacterial taxa based on the 16S ribosomal RNA (rRNA) gene fragments, and (B) fungal taxa based on the internal transcribed spacer (ITS) sequences in the roots and rhizospheres of canola, wheat, and pea. For canola, three different treatments were applied: canola grown as recommended (Can_RE), canola fertilized at 150% of the recommended rate (Can_HF), and canola seeded at 150% of the recommended rate (Can_HD).
The diversity of bacterial, fungal, and archaeal assemblages was estimated using the Chao1 index, Simpson's reciprocal index, and OTU evenness, and compared between the different crops and canola treatments. For bacterial assemblages, these diversity indices were all significantly influenced by biotope (Table S3). For the archaeal community, only the Chao1 index was significantly different between biotopes. For bacteria, OTU richness was significantly higher in the rhizosphere soils than in the roots (Figure S3), irrespective of crop or treatment (Figure S3). The major differences in microbial richness were found between the root samples of the different crops, while richness was similar among the canola treatments (Figure S3). Simpson's reciprocal index and the evenness index differed between the crops in both the roots and the rhizospheres for bacteria, and the pea values were much lower than those of the other two crops (Figure S3).
For fungal assemblages, the three OTU richness indices were all significantly influenced by biotope (Table S3). In the roots, the three indices of canola significantly differed from those of wheat and pea (Figure S4). Similar trends were observed in the rhizospheres (Figure S4). However, when the three canola treatments were compared, only Simpson's reciprocal index and evenness were significantly modified in the roots (Figure S4), with higher seeding density showing higher diversity than the other treatments did. Although the biotopes had significant influence on the archaeal assemblages (Table S3), the crop types and canola treatments had no significant effect on the Chao1, Simpson's reciprocal, and evenness indices of archaeal assemblages (Figure S5).
We found that bacterial, fungal, and archaeal community compositions were significantly different between crops and biotopes, with a significant interaction between crop and biotope for bacterial and fungal communities but not for archaeal communities (Table 2). The results indicate that crop type and biotope were indeed affecting microbial community composition. The pairwise comparison (Table 3) showed that bacterial communities were all significantly different either in the roots or in the rhizosphere soil between canola and the other crops. The same effects were found on the fungal community compositions (Table 3). Significant differences in archaeal community compositions occurred between canola roots and pea roots and between wheat roots and pea roots but not between canola and wheat, which shared a similar archaeal community in their roots. In the rhizosphere soils, the archaeal community composition was not different between the crops. The PCoA projections showed support for the PERMANOVA tests (Figure 2). Bacterial and fungal samples were relatively clearly grouped within crops or biotopes, especially for the pea samples, which stood out considerably from the other two crops. Since the archaeal projections are distributed in a smaller range of Bray-Curtis values, the significant effects found with PERMANOVA could not be easily visualized in the PCoA.
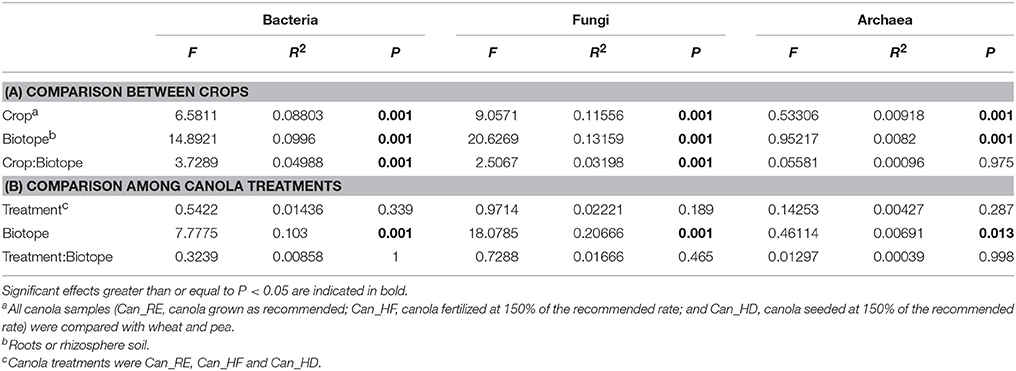
Table 2. (A) Effect of crop identity and biotopes on the microbial community compositions at the operational taxonomic unit (OTU) level and (B) effect of canola treatments and biotopes on the microbial community compositions at the OTU level, as determined by permutational multivariate analysis of variance (PERMANOVA).
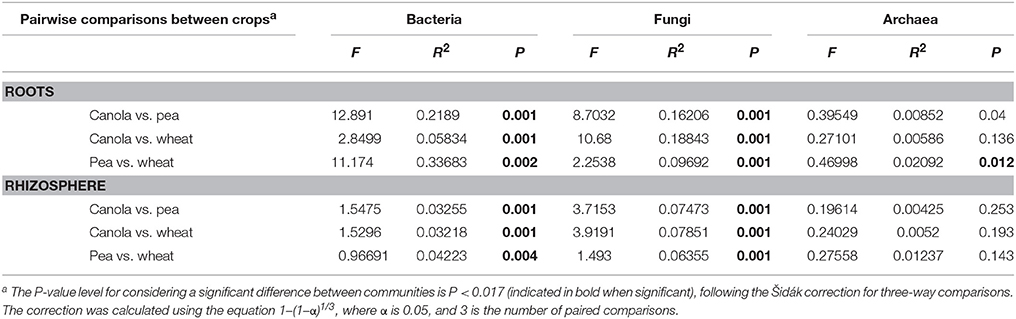
Table 3. Pairwise comparisons of the compositions of canola microbial communities with the compositions of those associated with wheat or pea, as determined by permutational multivariate analysis of variance (PERMANOVA).
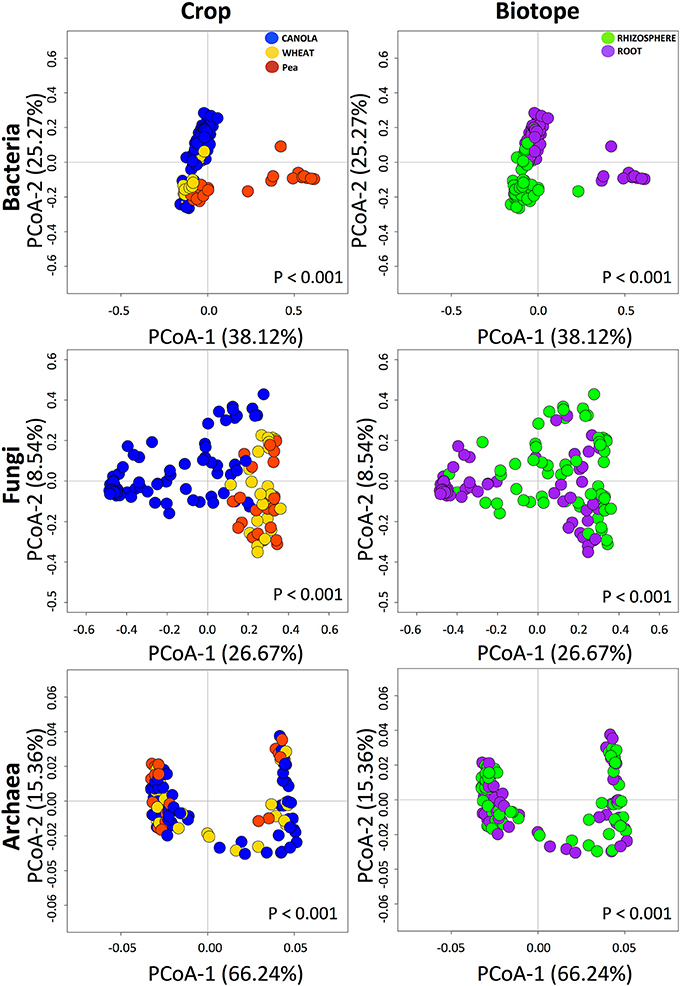
Figure 2. Principal coordinates analyses of the bacterial, fungal, and archaeal operational taxonomic units (OTUs), showing the grouping based on crops and biotopes. The percentages represent the variance explained by each axis. Notice that the projection range based on Bray-Curtis values of the archaeal samples was smaller than those of bacteria and fungi.
In a second step, the microbial community structures were compared among the different canola treatments and biotopes (Table 2). The agronomic treatments applied to the fields were not found to affect bacterial, fungal, or archaeal community composition. There was a significant difference in microbial communities between biotopes, but there was no interaction between treatment and biotope.
There were 17 bacterial OTUs highly associated with canola roots (Table S4). Among those, 14 OTUs belonged to the core microbiome of canola (marked with “C” in Table S4), and were associated with the genera: Streptomyces, Cryocola, Arthrobacter, Flavobacterium, Janthinobacterium, Serratia, Kaistobacter, Pseudomonas, Pedobacter, Agrobacterium, Burkholderia, Acidovorax, Erwinia, and Stenotrophomonas. When we compared these core members of canola roots with the reference crops, we found that wheat and pea roots harbored fewer core members than canola did (wheat had 12 OTUs and pea had 1 OTU with abundance ≥ 1%; Table 4). Furthermore, wheat shared many more root core members with canola than with pea, as indicated by the lower Sørensen index of dissimilarity between canola and wheat root microbiomes (0.33) than between canola and pea root microbiomes (0.88). However, these common members still showed significant differences in abundance between crops (FDR P-value < 0.001; Table 4). The higher difference between canola and pea is most likely related to the presence of nodules on pea roots and to the high dominance of Rhizobium leguminosarum in this crop. In the rhizosphere soil, the canola bacterial core microbiome was formed of seven OTUs. Surprisingly, the bacterial core microbiome in the canola rhizosphere was more similar to the one found in the pea rhizosphere than to the one found in the wheat rhizosphere (Sørensen index = 0.34 with pea and 0.37 with wheat). It seems that the rhizosphere was a less selective environment than the roots.
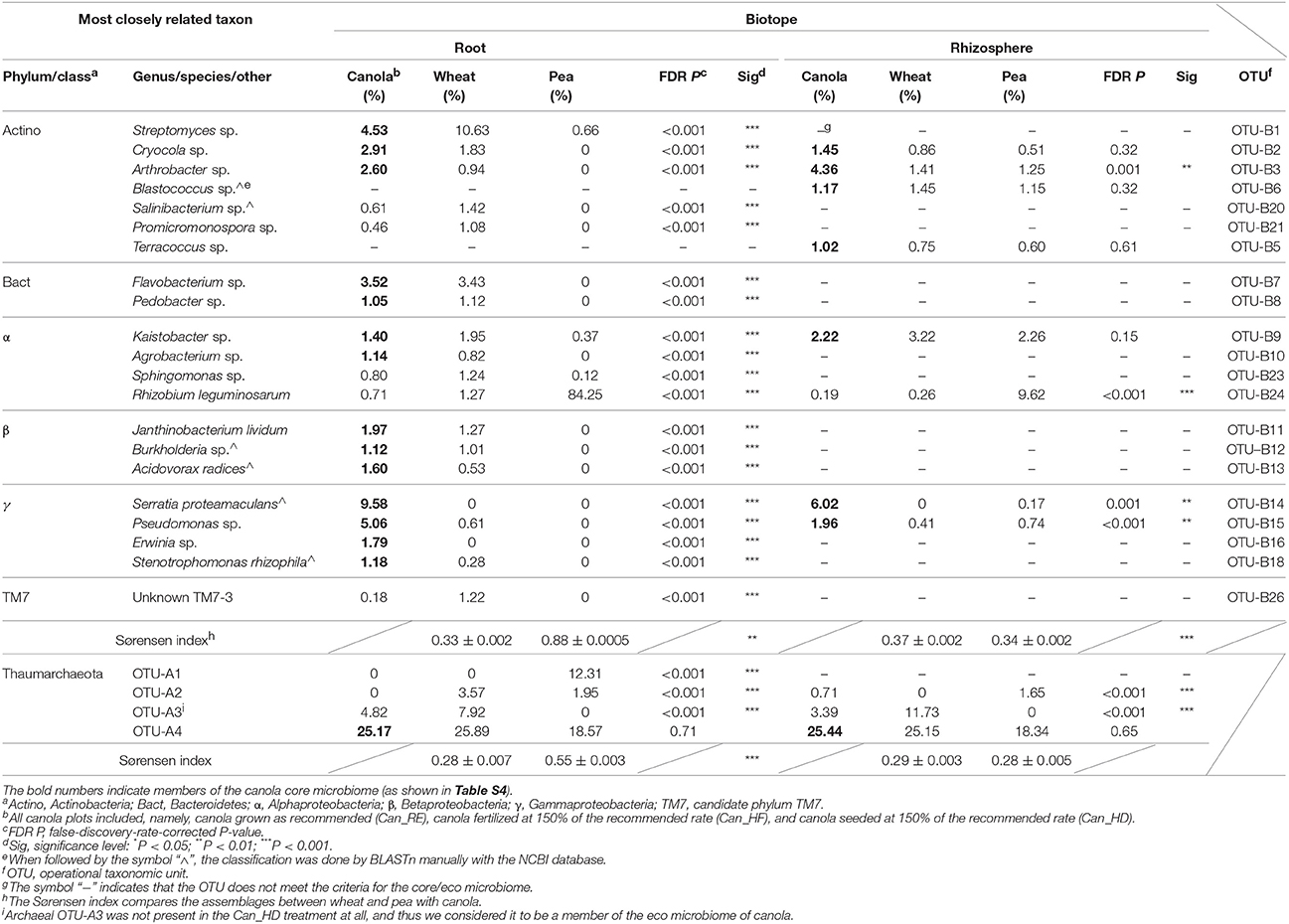
Table 4. Comparison of the abundances of members of the canola, wheat, and pea core microbiomes.
The only archaeal member of the core microbiome of canola roots was OTU-A4 (Table S4), while OTU-A3 was found to be a member of the root eco microbiome since that OTU was not found in the Can_HD treatment (Table S4). OTU-A1 was detected only in pea roots, and OTU-A2 was detected in wheat and pea roots but not in canola roots. OTU-A4 was also present in wheat and pea roots, with similar proportions in each crop (25–26% in canola and wheat, and 19% in pea). OTU-A4 was also present in the rhizosphere of all three crops, with proportions similar to those found in roots, while OTU-A2 and OTU-A3 were detected only in canola from the Can_HF treatment. OTU-A2 was also found in the pea rhizosphere and OTU-A3 in the wheat rhizosphere. These four OTUs from the Canadian Prairies grouped closely with Nitrocosmicus spp. (Figure S1), including Nitrocosmicus oleophilus (from Korean soil samples), and Nitrocosmicus exaquare (from a wastewater treatment plant in Guelph, Ontario, Canada) (Sauder et al., 2017) and with the archaeal OTUs (SCA1145 and SCA1170), identified from Wisconsin agricultural soil in the United States (Bintrim et al., 1997). OTU-A2 grouped closely with SCA1170, and OTU-A1, OTU-A3, and OTU-A4 grouped with SCA1145. OTU-A4 was especially close to Nitrocosmicus spp., suggesting that this OTU is probably the closest to the species prevalently distributed worldwide. Also, when the substitution changes for the branches with known species were compared, the phylogenetic tree suggests that there are probably several new species of Thaumarchaeota to be described in the Canadian Prairies.
OTU-F9 belonging to O. brassicae was the only fungal OTU classified as core for canola, with abundances that varied significantly between treatments (up to 83%) in roots but not in the rhizosphere (Table S5). OTU-F9 was also detected in the microbiomes of wheat and pea roots, but in much lower abundances than in canola roots (Table S5), while it was not detected in the rhizosphere of these reference crops. In comparison with the canola fungal core microbiomes, both reference crops harbored a higher fungal diversity (Table 5). Three OTUs were shared between wheat and pea roots and were absent from canola roots. Among them, OTU-F16 was first classified as O. brassicae using the UNITE database (Table 5), but since OTU-F16 differed from OTU-F9 (from canola roots) (lengths of 102 vs. 132 bp; identity of 86% within 78% coverage), they were furthered compared. A BLASTn search in the NCBI database found them to be 100% identical with 100% coverage to the sequences AB205208 and AB205213, respectively classified as O. virulentus and O. brassicae in a taxonomic study of Olpidium spp. (Sasaya and Koganezawa, 2006) that sought to differentiate the non-virus-carrying and virus-carrying species within this genus. Our classification results were therefore updated accordingly. The OTU belonging to O. virulentus, which is the virus-carrying Olpidium species, then appeared as part of the microbiome of wheat and pea roots but was not detected in canola. Among the 15 OTUs retained as part of the fungal core microbiomes of the canola, wheat, and pea rhizospheres (Table 5), 2 OTUs were found associated with all crops, 5 OTUs were shared between canola and pea, 2 OTUs were shared between canola and wheat, 7 OTUs were shared between wheat and pea, and 5 OTUs were associated with the rhizosphere of one crop only. The Sørensen index showed that the shared fungal core and eco microbiomes were more similar between pea and canola than between wheat and canola (Table 5), either in roots or in rhizospheres.
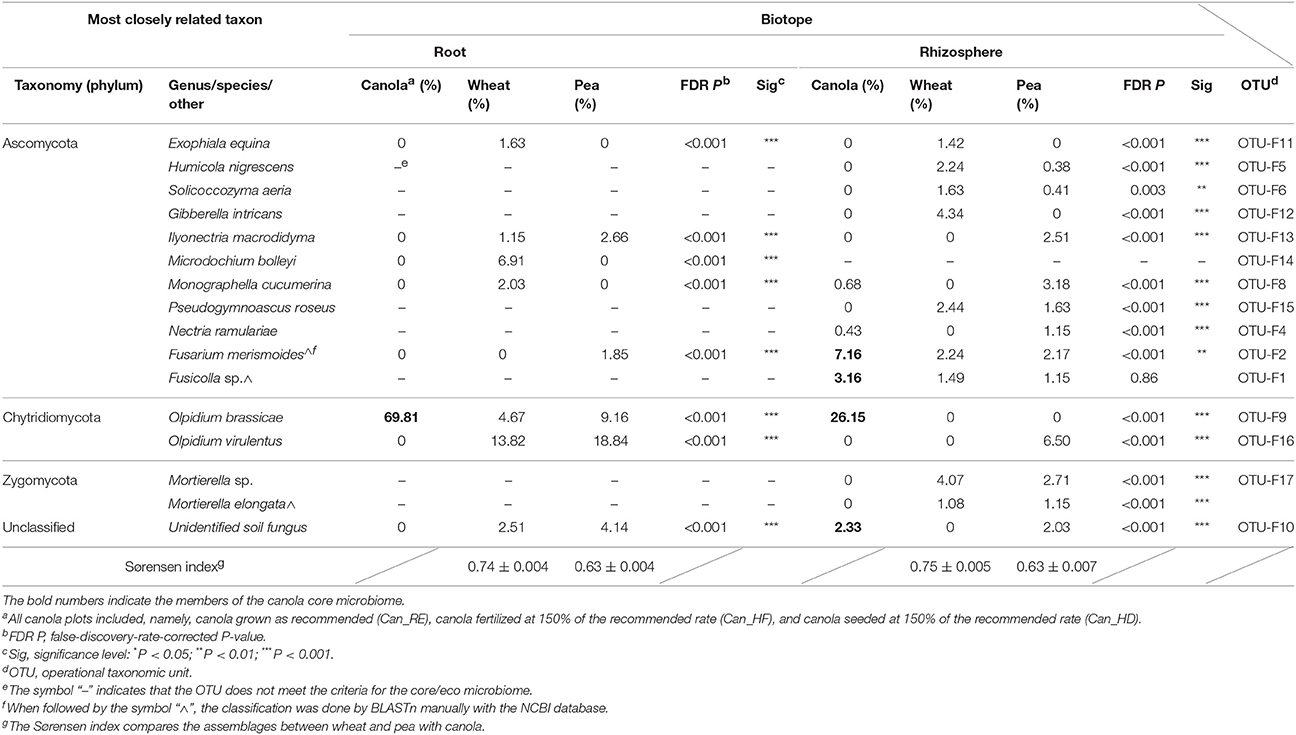
Table 5. Canola core microbiomes of fungi compared with wheat and pea core microbiomes.
Within each canola treatment, we examined the relationships and interactions of the bacterial and fungal core members using CoIA based on the PCA matrices, except for canola roots from the Can_HF and Can_HD treatments, where the fungal core microbiome contained only O. brassicae, which was not suitable to be analyzed by co-inertia analysis. This approach showed putative interactions between bacteria and fungi under different treatments and biotopes. Five main relationships were significant (P < 0.05), and most of them were from rhizospheres, namely, the rhizospheres of canola from the Can_RE, Can_HF, and Can_HD treatments, the rhizosphere of pea, and from the roots of wheat (Table S6, Figure 3 and Figure S6). The overall analyses showed that the bacterial and fungal core and eco microbiomes were more strongly correlated in the rhizospheres than in the roots (Table S6). The first two axes of the corresponding CoIA hyperspace (Figure 3) represented 92.58%, 96.49%, and 95.88% of the total variation for the Can_RE, Can_HF, and Can_HD treatments, respectively, in the rhizospheres. The values of the RV coefficient (a multivariate generalization of the squared Pearson correlation coefficient) for these three treatments in the rhizospheres were higher in the Can_RE treatment (0.605) than in the Can_HF (0.456) and Can_HD (0.491) treatments, indicating that the relationships between bacterial and fungal core and eco microbiomes were higher in the canola grown as recommended (Can_RE) than in the other two canola treatments. It suggests that increases in fertilization and seeding density might lower down the interactions of these two communities of microorganisms although we could not evaluate if this was positive or negative to the crops. The most obvious grouping was related to the locations. According to the projection of the fungal and bacterial OTUs on the co-inertia plane of each figure, O. brassicae was positively correlated with Kaistobacter sp. in all the canola treatments. Correlations between O. brassicae and Blastococcus sp. were stronger in the Can_HF and Can_HD treatments than in Can_RE (Figure 3A). In the Can_RE treatment, Fusicolla sp. and Fusarium merismoides were positively correlated with Stenotrophomonas sp. and Serratia proteamaculans. However, in the Can_HF plots (Figure 3B), S. proteamaculans was the only OTU showing a positive correlation with these two fungi. Further, in the Can_HD plots (Figure 3C), these two fungal OTUs showed a positive correlation with Stenotrophomonas sp. and Arthrobacter sp. The RV coefficient values for the pea rhizosphere and wheat root biotopes were 0.492 and 0.510, respectively. The two first axes represented 95.21 and 89.41% of the co-inertia variance (Figure S6).
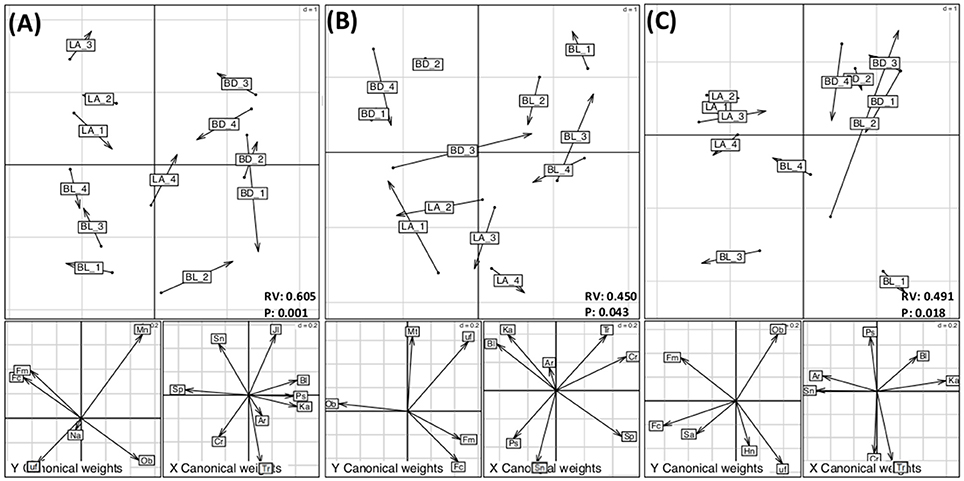
Figure 3. Co-inertia analysis showing the relationship between rhizosphere bacterial and fungal core and eco microbiomes in (A) canola grown as recommended (Can_RE), (B) canola fertilized at 150% of the recommended rate (Can_HF), and (C) canola seeded at 150% of the recommended rate (Can_HD). For each treatment, the top graph is the projection of both bacterial and fungal operational taxonomic units (OTUs) onto the co-inertia plane. The cumulative projective inertias for the first two axes were (A) 92.58%, (B) 96.49%, and (C) 95.88% of the total variation. The label abbreviations are as follows: BL, Beaverlodge; BD, Brandon; LA, Lacombe. Arrow length is proportional to the difference between the ordinations of the two data sets: longer arrows denote less concordance between the two assemblage data sets. The two lower panels are the projection of fungi (left panel) and bacteria (right panel) onto the co-inertia plane for each treatment. The abbreviations for bacterial and fungal species are as follows: Ar, Arthrobacter sp.; Bl, Blastococcus sp.; Cr, Cryococcus sp.; Jl, Janthinobacterium lividum; Ka, Kaistobacter sp.; Ps, Pseudomonas sp.; Sn, Stenotrophomonas sp.; Sp, Serratia proteamaculans; Tr, Terracoccus sp.; Fc, Fusicolla sp.; Fm, Fusarium merismoides; Hn, Humicola nigrescens; Ob, Olpidium brassicae; Mn, Monographella cucumerina; Mt, Mortierella sp.; Na, Nectria ramulariae; uf, unclassified fungus. The RV coefficients are the values of the multivariate generalization of the squared Pearson correlation coefficient. P indicates the P-values of each analysis.
We used RDA and Spearman's correlation to assess the relationships between the bacterial and fungal core and eco microbiomes of canola on one hand and canola yield, canola emergence, and post-spray weed count (Figure S7) on the other hand. Overall, location was the only factor that was found to significantly affect all members of the bacteria and fungi core and eco microbiomes as well as canola yield, according to the RDA model. However, neither post-spray weed counts nor emergence counts were significantly correlated with the core and eco microbiome members. Spearman's correlation showed that individual members of the core and eco microbiomes (Table 6) that were significantly correlated with yield were mostly bacteria and were mostly members of the core microbiomes, either of the roots or of the rhizosphere. Three fungal members of the rhizosphere microbiome were found to be significantly correlated with yield: Monographella cucumerina (negatively correlated), Fusicolla sp. (positively correlated), and F. merismoides (positively correlated). In roots, five bacteria were positively correlated with yield: Amycolatopsis sp., Stenotrophomonas sp., S. proteamaculans, Arthrobacter sp., and Pedobacter sp. The remaining bacteria were negatively correlated with yield. Amycolatopsis sp. and Stenotrophomonas sp. were members of the eco microbiome associated with higher fertilization (Can_HF treatment). In the rhizosphere, S. proteamaculans and Stenotrophomonas sp. were also positively correlated with yield. S. proteamaculans was also a member of the eco microbiome associated with higher fertilization. Interestingly, in the rhizosphere, Fusicolla sp. and F. merismoides, which can be considered potential pathogens (Gräfenhan et al., 2011), showed positive correlations with yield. On the other hand, the very abundant O. brassicae was not significantly correlated with canola yield. The canola archaeal core OTUs did not show significant correlation with canola yield.
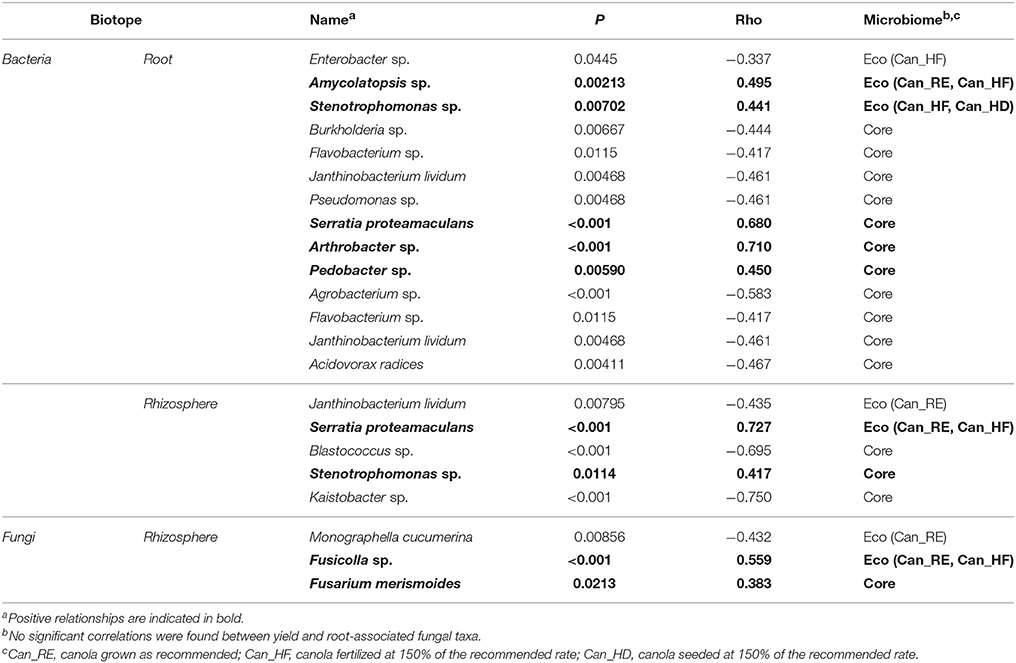
Table 6. Bacterial and fungal taxa from the core and eco microbiomes of canola showing a significant Spearman's correlation between their relative abundance and canola yield.
In this study, we compared the canola root-associated microbiome with those of wheat and pea grown alongside canola in the same fields, and the effect of selected agronomic treatments on the canola microbiome. We assessed the effect of crop and treatment on the α- and β-diversities of the bacterial, fungal, and archaeal assemblages associated with the roots and rhizosphere soil. The results show that canola has a core microbiome distinct from those of wheat and pea and that the root and rhizosphere microbiomes significantly responded to the agronomic treatments. We also found treatment-specific changes in the relationship between bacterial and fungal microbiome members.
The canola microbiome was significantly different from those of the reference crops in all three microbial domains, both in the roots and in the rhizosphere soil, according to PERMANOVA and core microbiome analyses. Different plant species have often been reported to select for different root-associated microorganisms (e.g., Hallmann et al., 1997; Tkacz et al., 2015). Previous work has been done on the canola-root-associated bacterial community (Germida et al., 1998; Macrae et al., 2000; Alström, 2001; Misko and Germida, 2002; Farina et al., 2012; Croes et al., 2013; de Campos et al., 2013). Table S7 provides a list of the bacterial taxa forming the core microbiomes of canola roots and rhizosphere from the present study in comparison with previous research and the PGPR references (Kloepper et al., 1988; Belimov and Dietz, 2000; Alström, 2001; Poly et al., 2001; Gray and Smith, 2005; Cruz et al., 2008; Kumar et al., 2011; Pliego et al., 2011). Although the studied areas were quite diverse, there were a large number of common species found in each study. In our study, the core microbiome encompasses 12 different genera that were also detected in previous research. In particular, Pseudomonas spp. and Stenotrophomonas sp. were detected in five previous studies (Table S7). According to Table 4, these two bacterial genera were present at very low abundance or were absent from the wheat and pea core microbiomes. This observation indicates that these taxa are quite unique to the canola microbiome. Interestingly, the members of the canola core microbiome that we identified were not that similar to those in the other two Canadian studies by Misko and Germida (2002) and Germida et al. (1998). The difference was probably due to different sampling times and locations since the microbiome was shown to change significantly between different growth stages (Farina et al., 2012) and soil types (Garbeva et al., 2004).
The three agronomic treatments applied to the canola plots consisted of the recommended practice as well as a higher fertilization rate and a higher seeding density. Higher fertilization rate and seeding density were shown to increase canola yields (as detailed in the introduction). In previous studies, higher fertilization of wheat fields also increase soil microbial biomass and activities, as determined by enzyme assays (Goyal et al., 1999; Raiesi, 2004). Our results show that the abundances of Amycolatopsis sp., Enterobacter sp., Stenotrophomonas sp., Janthinobacterium lividum, S. proteamaculans, and the archaeal OTU-A2 and OTU-A3 were significantly affected by the treatments in canola roots or rhizosphere. The other OTUs remained quite stable between the different treatments. Except for Enterobacter sp., J. lividum, and the archaeal OTUs, these microbial taxa were all positively correlated with canola yield. In addition, two members of the core microbiome that were not affected by the treatments, Arthrobacter sp. and Pedobacter sp., were also positively correlated with yield.
Even with a relatively low Good's coverage (0.62) for fungi, due to subsampling to 123 reads per sample, the analysis showed that the situation was quite different with the canola fungal core microbiome. In previous studies (Bennett et al., 2014; Tkacz et al., 2015; Gkarmiri et al., 2017), O. brassicae, a fungal parasite, was found to be the dominant and most active fungal species in the canola rhizosphere and root environments. Our results confirmed these findings; moreover, in our study, it was the only fungus forming the core microbiome of canola roots. However, the abundance of this taxon was highly reduced in wheat and pea roots, along with a concurrent increase in the abundance of O. virulentus, which was the dominant fungus in the roots of those crops. In canola roots, the abundance of O. brassicae was also highly reduced in densely seeded plots in comparison with the other agronomic treatments, perhaps because of the higher soil-drying potential of a dense plant stand, or due to the higher amount of fungicide brought to the same volume of soil since the commercial seeds were coated with pesticides. Nevertheless, in our study, the abundance of O. brassicae was not negatively correlated with canola yield. A previous study (Hilton et al., 2013) showed that infection with a high number of O. brassicae zoospores reduced the above-ground growth and root growth of canola seedlings and also affected pod and seed production. The absence of a negative correlation with yield in the present study suggests that in our experiment, either the minimum threshold of Olpidium zoospores at the sensitive growth stage was not met or the environmental conditions that are required for disease expression were not present. Further research would therefore be needed to determine if increasing the seeding density of canola would reduce the detrimental effect of O. brassicae when the conditions required for disease development are present. We also observed a positive statistical interaction between the OTUs of O. brassicae and those of Kaistobacter, based on Co-intertia analysis. These two taxa were both recently found among the most active microbes in the rhizosphere of B. napus (Gkarmiri et al., 2017). While the role of Kaistobacter in canola rhizosphere is unknown, more investigation would be required to determine if the biological interaction between these species is direct or indirect.
Many microorganisms promote plant growth (Gray and Smith, 2005), but evidence shows that some species of microorganisms acting as plant-growth-promoting microorganisms in most conditions can also reduce crop yield under certain circumstances (Bennett et al., 2012). Thus, while identifying PGPRs, one needs to consider the conditions where the PGPR were observed. In this study, we assessed the members of the core and eco microbiomes of canola using Spearman's correlation analysis to identify the microorganisms related to canola yield, in order to highlight their potentially beneficial or detrimental effect on canola productivity. It is well known that plant growth promoting characteristics are strain-dependent so these correlations are only indicative of potential PGPR microorganisms. Sixteen bacterial OTUs and three fungal OTUs, mainly from the root microbiomes, were significantly correlated with canola yield. Among them, OTUs related to Amycolatopsis sp., S. proteamaculans, Pedobacter sp., Arthrobacter sp., Stenotrophomonas sp., F. merismoides, and Fusicolla sp. were positively related to yield (Table 6), while others, including some supposed PGPR such as Flavobacterium sp. and Pseudomonas sp., were negatively correlated with canola yield.
Serratia proteamaculans is well known as a PGPR that facilitates nodulation and nitrogen fixation in soybeans and lentils (Chanway et al., 1989; Dashti et al., 1998). This taxon was also shown to promote growth and resistance to fungal pathogens in rapeseed (Alström, 2001; Neupane et al., 2013b). As a member of the canola root core microbiome in our work, this species had high specificity to canola, but in contrast, pea and wheat did not appear to recruit the OTU related to S. proteamaculans as a member of their core microbiomes. In our study, the OTU related to S. proteamaculans is also a potentially beneficial rhizosphere and root microorganism that was highly correlated with canola yield.
Arthrobacter spp. has been studied in fields for a long time and was shown in a previous study to increase canola yield significantly when applied as a bacterial suspension to seeds (Kloepper et al., 1988). Arthrobacter was also recognized as fast-growing bacteria in the rhizosphere of B. rapa canola (Tkacz et al., 2015). In our study, one OTU related to Arthrobacter was a member of the canola root core microbiome, with significantly lower abundances in pea and wheat. The fact that Arthrobacter had the highest correlation with canola yield among the members of the root core microbiome suggests an important beneficial influence of this bacterium on canola.
The OTU related to an unknown Stenotrophomonas sp. present in canola roots and rhizosphere was significantly correlated with yield. The related bacterium Stenotrophomonas rhizophila, which was originally identified from oilseed rape and potato roots and was reported to have antagonistic activity against fungal plant pathogens (Wolf et al., 2002), was also present in the core microbiome of canola. Stenotrophomonas sp. was previously shown to inhibit the growth of Verticillium dahliae in oilseed rape plants (Alström, 2001). The OTU related to unknown Stenotrophomonas was more abundant in the canola plots in the high-seeding-rate and high-fertilization-rate treatments, while S. rhizophila seemingly benefits from the use of a high fertilization rate. Therefore, it is unclear whether those species are beneficial to canola or whether they instead benefit from large, well-fertilized canola crops.
Endophytism was reported in several Amycolatopsis spp. (Miao et al., 2011; Xing et al., 2013; Klykleung et al., 2015; Axenov-Gribanov et al., 2016), and accordingly, an OTU related to Amycolatopsis sp. was detected only in the roots of canola in our study. Some Amycolatopsis spp. have antimicrobial and antifungal activities (Saito et al., 2009; Axenov-Gribanov et al., 2016), which may explain the positive correlation of this bacterium with canola yield in our experiment. Another OTU related to a commonly observed endophyte, Pedobacter sp., was detected in the roots of canola and wheat in our experiment. This OTU was positively correlated with canola yield, which may be explained by the bacterium's ability to produce indole acetic acid (IAA) (Yuan et al., 2011). Pedobacter sp. was reported in a previous study to be more abundant at the flowering stage of canola (de Campos et al., 2013). The production of IAA by this bacterium may trigger the flowering of canola, which could explain the presence of a positive correlation with yield.
Fusarium spp. and Fusicolla spp. are closely related (Gräfenhan et al., 2011), and many species from these two genera are plant pathogens (van der lee et al., 2015). Two OTU detected in the canola rhizosphere showed best match to sequences from these genera and were positively related to canola yield. However, it should be noted that the short ITS barcode sequences used in this study are known to not be particularly suitable for Fusarium species identification. Thus, these identifications should be interpreted with caution and further work will be required to confirm their identity more accurately. The Fusarium OTU that we detected in the rhizosphere was related to F. merismoides, a species considered to cause tomato stem rot (Fletcher and Lord, 1985). Non-pathogenic Fusarium spp. were sometimes detected in healthy roots from Fusarium-suppressive soils, and these non-pathogenic species or strains may antagonize pathogenic Fusarium spp. through competition. However, the cause of the suppression may also be the presence of other microorganisms in the soils (Weller et al., 2002), such as certain Actinomycetes and Gammaproteobacteria, including some species of pseudomonads (Haas and Défago, 2005). Another study showed that indigenous bacterial species, including Stenotrophomonas spp. and Serratia spp., could parasitize pathogenic Fusarium, reducing the production of aerial hyphae and microconidia (Minerdi et al., 2008). The colonization of Fusarium by beneficial bacteria may explain the positive correlations found between Fusarium spp. and Fusicolla spp. on one hand and canola yield on the other hand. The CoIA clearly showed a correlation between the two potentially pathogenic fungal species, F. merismoides and Fusicolla sp., and two potentially beneficial bacteria, Stenotrophomonas sp. and S. proteamaculans. Previous studies reported that the incidence of Fusarium disease and Fusarium populations was related to changes in soil microbial community structure in asparagus (Hamel et al., 2005; Yergeau et al., 2010). It was also shown that there is no relationship between the abundance of Fusarium in soil and Fusarium disease, and even no clear relationship between the presence of Fusarium in plant roots and Fusarium disease (Vujanovic et al., 2006; Yergeau et al., 2006).
Although some members of Pseudomonas and Flavobacterium are generally reported to be beneficial bacteria in most studies (Poly et al., 2001; Gray and Smith, 2005; Farina et al., 2012; de Campos et al., 2013), some records indicate that Pseudomonas spp. and Flavobacterium spp. may produce chemicals inhibiting plant growth (Bennett et al., 2012). Even though Pseudomonas is a broad genus, the negative relationships of the OTUs related to this genus with plant yield that we found concur with these reports.
Archaea have traditionally been considered to be extremophiles, but mesophilic archaea were found in the soil of soybean and paddy fields (Ueda et al., 1995; Kudo et al., 1997). Archaea were also observed on the rhizoplane and older root hairs of tomato by means of in situ staining and microscopy (Simon et al., 2000). The richness of archaeal assemblages was found to be higher in plant rhizospheres than in bulk soils of 76 different plant samples from diverse locations but plant identity had no influence (Sliwinski and Goodman, 2004). Archaea related to the genus Nitrososphaera were recently detected in sorghum and sunflower rhizospheres (Oberholster et al., 2018). Mesophilic archaea are also well-known as ubiquitous ammonia oxidizers (Kim et al., 2011). In our study, most archaeal OTUs belonged to Thaumarchaeota, and a similar non-specific prevalence happened only with OTU-A4, which was similarly abundant in all crops and under all agronomic treatments. However, PERMANOVA tests indicated that the relative abundance in the overall archaeal community and the archaeal OTUs forming the core microbiome significantly changed between crops. Contrary to the active components of B. napus roots/rhizosphere archaea that were reported to be mostly related to Nitrososphaera (Gkarmiri et al., 2017), all of the core archaeal OTUs we found here were closer to Nitrocosmicus spp. The abundance of OTU-A2 and OTU-A3 in canola roots and rhizosphere soil also shifted significantly with treatments, especially with the addition of fertilizers. Although we did not detect any correlation between the members of the canola archaeal core microbiome and canola yield, the significant differences in abundance of OTU-A2 and OTU-A3 in the high-fertilization-rate treatment suggest that the members of Thaumarchaeota may benefit from the organic matter released from plant roots or utilize ammonia in the root environment. However, the way in which canola plants may influence and manipulate Archaea is still unknown.
In this work, we described canola microbiomes for three communities of microorganisms, namely, bacteria, fungi, and archaea. We found that canola microbiomes were distinguished between the two biotopes (roots and rhizosphere) and were significantly different from those of the reference crops (wheat and pea). We highlighted the potential PGPR among those microorganisms by correlating the core microbiome members in the Canadian Prairies with canola yield. Taxa related to Amycolatopsis sp., S. proteamaculans, Pedobacter sp., Arthrobacter sp., Stenotrophomonas sp., F. merismoides, and Fusicolla sp. are potentially beneficial to canola due to their status as members of the core or eco microbiome and their positive correlation with canola yield. Fertilization and seeding rates seem to influence certain taxa forming the core and eco microbiomes of canola based on the relative abundances profiles, notably the parasite O. brassicae which was less abundant at the higher seeding rate. Certain archaeal taxa showed some specificity to crops and treatments. Furthermore, the putative interactions between the members of bacterial and fungal core microbiomes were weaker with higher fertilization and seeding than the recommended treatments in canola rhizospheres. Our study provides information about the canola root microbiome that is fundamental for the design of microbiome management strategies for improving canola yield and health.
KH, TB, CH, and MS-A conceived and designed the experiments. KH, RM, and C-YL performed the experiments. C-YL analyzed the data. MS-A, CG, and EY contributed reagents, materials, analysis tools. C-YL, MS-A, CH, TB, and EY wrote the paper.
This research was supported by Agri-Food Canada and the Canola Council of Canada, but these partners have in no way influenced or modified this manuscript or the analysis of the results presented.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Keith Hanson from the Swift Current Research and Development Centre, Agriculture, and Agri-Food Canada (AAFC), Saskatchewan, for organizing and collecting the samples from different AAFC fields, Dr. Sébastien Renaut for the consulting on bioinformatic processes, Dr. Vitor Pylro for the useful discussions regarding the Brazilian Microbiome Project pipelines, Dr. Saraswoti Neupane and Dr. Sally Hilton for the discussions regarding plant-growth-promoting rhizobacteria and Olpidium brassicae, and Dr. Jacynthe Masse and Stéphane Daigle for the statistical consulting. We also appreciate the kind sponsorship from AAFC, the NSERC (STPGP 494702), and the Canola Council of Canada in support of the research.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01188/full#supplementary-material
Figure S1. Maximum likelihood phylogenetic tree based on 175 bases of the archaeal 16S ribosomal RNA (rRNA) fragments. The branch length is proportional to the number of substitutions per site.
Figure S2. Rarefaction curves based on Good's coverage per sample of (A) bacterial 16S rRNA, (B) fungal internal transcribed spacer (ITS), and (C) archaeal 16S rRNA libraries.
Figure S3. Biodiversity indices [Chao1, Simpson's reciprocal (Simpson R), and evenness] of bacterial assemblages based on the operational taxonomic units (OTUs) of 16S rRNA gene fragments. The figure shows the comparisons between crops (left) and canola treatments (right). The canola treatments were canola grown as recommended (Can_RE), canola fertilized at 150% of the recommended rate (Can_HF), and canola seeded at 150% of the recommended rate (Can_HD). The black and white boxes represent the median to the upper quartile and the median to the lower quartile, respectively.
Figure S4. Biodiversity indices [Chao1, Simpson's reciprocal (Simpson R), and evenness] of fungal assemblages based on the operational taxonomic units (OTUs) of internal transcribed spacer (ITS) fragments. The figure shows the comparisons between crops (left) and canola treatments (right). The canola treatments were canola grown as recommended (Can_RE), canola fertilized at 150% of the recommended rate (Can_HF), and canola seeded at 150% of the recommended rate (Can_HD). The black and white boxes represent the median to the upper quartile and the median to the lower quartile, respectively.
Figure S5. Biodiversity indices [Chao1, Simpson's reciprocal (Simpson R), and evenness] of archaeal assemblages based on the operational taxonomic units (OTUs) of 16S rRNA fragments. The figure shows the comparisons between crops (left) and canola treatments (right). The canola treatments were canola grown as recommended (Can_RE), canola fertilized at 150% of the recommended rate (Can_HF), and canola seeded at 150% of the recommended rate (Can_HD). The black and white boxes represent the median to the upper quartile and the median to the lower quartile, respectively.
Figure S6. Co-inertia analysis showing the relationship between bacterial and fungal core and eco microbiomes in (A) the rhizosphere of pea and (B) the roots of wheat. For each crop, the top graph is the projection of both bacterial and fungal operational taxonomic units (OTUs) onto the co-inertia plane. The cumulative projective inertias for the first two axes were (A) 95.21% and (B) 89.41% of the total variation. The label abbreviations are as follows: BL, Beaverlodge; BD, Brandon; LA, Lacombe. Arrow length is proportional to the difference between the ordinations of the two data sets: longer arrows denote less concordance between the two assemblage data sets. The two sub-panels are the projection of fungi (left sub-panel) and bacteria (right sub-panel) onto the co-inertia plane for each crop. The abbreviations for bacterial and fungal species are as follows: Ac, Acidovorax radius; Ar, Arthrobacter sp.; Ag, Agrobacterium sp.; Bl, Blastococcus sp.; Bk, Burkholderia sp.; Cr, Cryococcus sp.; Fl, Flavobacterium sp.; Jl, Janthinobacterium lividum; Ka, Kaistobacter sp.; Pe, Pedobacter sp.; Pm, Promicromonospora sp.; Ps, Pseudomonas sp.; Rl, Rhizobium leguminosarum; Sg, Sphingomonas sp.; Sl, Salinibacterium sp.; Sn, Stenotrophomonas sp.; Sp, Serratia proteamaculans; St, Streptomyces sp.; Tm, TM7-3; Tr, Terracoccus sp.; Ex, Exophiala equine; Hn, Humicola nigrescens; Im, Ilyonectria macrodidyma; Ob, Olpidium brassicae; Ov, Olpidium virulentus; Mb, Microdochium bolleyi; Mn, Monographella cucumerina; Mt, Mortierella sp.; Na, Nectria ramulariae; Pr, Pseudogymnoascus roseus; uf, unclassified fungus. The RV coefficients are the values of the multivariate generalization of the squared Pearson correlation coefficient. P indicates the P-values of each analysis.
Figure S7. Agronomic variables of (A) canola emergence counts, (B) post-spray weed count, and (C) canola yield. The canola treatments were canola grown as recommended (Can_RE), canola fertilized at 150% of the recommended rate (Can_HF), and canola seeded at 150% of the recommended rate (Can_HD).
Table S1. Soil properties at each location.
Table S2. The raw sequencing counts of each sample for bacteria, fungi and archaea by MiSeq Sequencing.
Table S3. Effects of crop identity and canola treatments on the Chao1, Simpson's reciprocal (Simpson R), and evenness indices, as determined by analysis of variance (ANOVA). Significant effects greater than P < 0.05 are indicated in bold.
Table S4. Canola bacterial and archaeal core and eco microbiomes among different treatments.
Table S5. Canola fungal core and eco microbiomes among different treatments.
Table S6. Significance of the relationships between the bacterial and fungal core/eco microbiomes, as determined by co-inertia analysis.
Table S7. Comparison of the bacterial core microbiome of canola from this study with bacterial taxa considered important in other studies.
Alström, S. (2001). Characteristics of bacteria from oilseed rape in relation to their biocontrol activity against Verticillium dahliae. J. Phytopathol. 149, 57–64. doi: 10.1046/j.1439-0434.2001.00585.x
Axenov-Gribanov, D. V., Voytsekhovskaya, I. V., Rebets, Y. V., Tokovenko, B. T., Penzina, T. A., Gornostay, T. G., et al. (2016). Actinobacteria possessing antimicrobial and antioxidant activities isolated from the pollen of scots pine (Pinus sylvestris) grown on the Baikal shore. Antonie Leeuwenhoek 109, 1307–1322. doi: 10.1007/s10482-016-0730-5
Baker, G. C., Smith, J. J., and Cowan, D. A. (2003). Review and re-analysis of domain-specific 16S primers. J Microbiol. Methods 55, 541–555. doi: 10.1016/j.mimet.2003.08.009
Belimov, A. A., and Dietz, K. J. (2000). Effect of associative bacteria on element composition of barley seedlings grown in solution culture at toxic cadmium concentrations. Microbiol. Res. 155, 113–121. doi: 10.1016/S0944-5013(00)80046-4
Bengtsson-Palme, J., Ryberg, M., Hartmann, M., Branco, S., Wang, Z., Godhe, A., et al. (2013). Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol. Evol. 4, 914–919. doi: 10.1111/2041-210X.12073
Bennett, A. J., Bending, G. D., Chandler, D., Hilton, S., and Mills, P. (2012). Meeting the demand for crop production: the challenge of yield decline in crops grown in short rotations. Biol. Rev. 87, 52–71. doi: 10.1111/j.1469-185X.2011.00184.x
Bennett, A. J., Hilton, S., Bending, G. D., Chandler, D., and Mills, P. (2014). Impact of fresh root material and mature crop residues of oilseed rape (Brassica napus) on microbial communities associated with subsequent oilseed rape. Biol. Fertil. Soils 50, 1267–1279. doi: 10.1007/s00374-014-0934-7
Berendsen, R. L., Pieterse, C. M. J., and Bakker, P. A. H. M. (2012). The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478–486. doi: 10.1016/j.tplants.2012.04.001
Bintrim, S. B., Donohue, T. J., Handelsman, J., Roberts, G. P., and Goodman, R. M. (1997). Molecular phylogeny of archaea from soil. Proc. Natl. Acad. Sci. U.S.A. 94, 277–282. doi: 10.1073/pnas.94.1.277
Burggraf, S., Huber, H., and Stetter, K. O. (1997). Reclassification of the crenarchaeal orders and families in accordance with 16S rRNA sequence data. Int. J. Syst. Bacteriol. 47, 657–660. doi: 10.1099/00207713-47-3-657
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chanway, C. P., Hynes, R. K., and Nelson, L. M. (1989). Plant growth-promoting rhizobacteria - effects on growth and nitrogen-fixation of lentil (Lens esculenta Moench) and Pea (Pisum sativum L). Soil Biol. Biochem. 21, 511–517. doi: 10.1016/0038-0717(89)90123-5
Chaparro, J. M., Sheflin, A. M., Manter, D. K., and Vivanco, J. M. (2012). Manipulating the soil microbiome to increase soil health and plant fertility. Biol. Fertil. Soils 48, 489–499. doi: 10.1007/s00374-012-0691-4
Croes, S., Weyens, N., Janssen, J., Vercampt, H., Colpaert, J. V., Carleer, R., et al. (2013). Bacterial communities associated with Brassica napus L. grown on trace element-contaminated and non-contaminated fields: a genotypic and phenotypic comparison. Microb. Biotechnol. 6, 371–384. doi: 10.1111/1751-7915.12057
Cruz, A. F., Horii, S., Ochiai, S., Yasuda, A., and Ishii, T. (2008). Isolation and analysis of bacteria associated with spores of Gigaspora margarita. J. Appl. Microbiol. 104, 1711–1717. doi: 10.1111/j.1365-2672.2007.03695.x
Dashti, N., Zhang, F., Hynes, R., and Smith, D. L. (1998). Plant growth promoting rhizobacteria accelerate nodulation and increase nitrogen fixation activity by field grown soybean [Glycine max (L.) Merr.] under short season conditions. Plant Soil 200, 205–213. doi: 10.1023/A:1004358100856
de Campos, S. B., Youn, J. W., Farina, R., Jaenicke, S., Junemann, S., Szczepanowski, R., et al. (2013). Changes in root bacterial communities associated to two different development stages of canola (Brassica napus L. var oleifera) evaluated through next-generation sequencing technology. Microb. Ecol. 65, 593–601. doi: 10.1007/s00248-012-0132-9
Dereeper, A., Guignon, V., Blanc, G., Audic, S., Buffet, S., Chevenet, F., et al. (2008). Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36, W465–W469. doi: 10.1093/nar/gkn180
Dodd, I. C., Zinovkina, N. Y., Safronova, V. I., and Belimov, A. A. (2010). Rhizobacterial mediation of plant hormone status. Ann. Appl. Biol. 157, 361–379. doi: 10.1111/j.1744-7348.2010.00439.x
Dunfield, K. E., and Germida, J. J. (2003). Seasonal changes in the rhizosphere microbial communities associated with field-grown genetically modified canola (Brassica napus). Appl. Environ. Microbiol. 69, 7310–7318. doi: 10.1128/AEM.69.12.7310-7318.2003
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Farina, R., Beneduzi, A., Ambrosini, A., De Camposa, S. B., Lisboa, B. B., Wendisch, V., et al. (2012). Diversity of plant growth-promoting rhizobacteria communities associated with the stages of canola growth. Agric. Ecosyst. Environ. Appl. Soil Ecol. 55, 44–52. doi: 10.1016/j.apsoil.2011.12.011
Fletcher, J. T., and Lord, K. L. (1985). A stem rot of tomato caused by Fusarium merismoides. Plant Pathol. 34, 443–445. doi: 10.1111/j.1365-3059.1985.tb01387.x
Garbeva, P., van Veen, J. A., and van Elsas, J. D. (2004). Microbial diversity in soil: selection of microbial populations by plant and soil type and implications for disease suppressiveness. Annu. Rev. Phytopathol. 42, 243–270. doi: 10.1146/annurev.phyto.42.012604.135455
Germida, J. J., Siciliano, S. D., De Freitas, J. R., and Seib, A. M. (1998). Diversity of root-associated bacteria associated with field-grown canola (Brassica napus L.) and wheat (Triticum aestivum L.). FEMS Microbiol. Ecol. 26, 43–50. doi: 10.1111/j.1574-6941.1998.tb01560.x
Gkarmiri, K., Mahmood, S., Ekblad, A., Alström, S., Högberg, N., and Finlay, R. (2017). Identifying the active microbiome associated with roots and rhizosphere soil of oilseed rape. Appl. Environ. Microbiol. 83, e01938–e01917. doi: 10.1128/AEM.01938-17
Goeman, J. J., and Solari, A. (2014). Multiple hypothesis testing in genomics. Stat. Med. 33, 1946–1978. doi: 10.1002/sim.6082
Gomiero, T., Pimentel, D., and Paoletti, M. G. (2011). Environmental impact of different agricultural management practices: conventional vs. organic agriculture. Crit. Rev. Plant Sci. 30, 95–124. doi: 10.1080/07352689.2011.554355
Goyal, S., Chander, K., Mundra, M. C., and Kapoor, K. K. (1999). Influence of inorganic fertilizers and organic amendments on soil organic matter and soil microbial properties under tropical conditions. Biol. Fertil. Soils 29, 196–200. doi: 10.1007/s003740050544
Gräfenhan, T., Schroers, H. J., Nirenberg, H. I., and Seifert, K. A. (2011). An overview of the taxonomy, phylogeny, and typification of nectriaceous fungi in Cosmospora, Acremonium, Fusarium, Stilbella, and Volutella. Stud. Mycol. 68, 79–113. doi: 10.3114/sim.2011.68.04
Grant, C. A., Wu, R., Selles, F., Harker, K. N., Clayton, G. W., Bittman, S., et al. (2012). Crop yield and nitrogen concentration with controlled release urea and split applications of nitrogen as compared to non-coated urea applied at seeding. Field Crop Res. 127, 170–180. doi: 10.1016/j.fcr.2011.11.002
Grant, C., and Wu, R. (2008). Enhanced-efficiency fertilizers for use on the Canadian Prairies. Crop Manage. 7. doi: 10.1094/CM-2008-0730-01-RV
Gray, E. J., and Smith, D. L. (2005). Intracellular and extracellular PGPR: commonalities and distinctions in the plant-bacterium signaling processes. Soil Biol. Biochem. 37, 395–412. doi: 10.1016/j.soilbio.2004.08.030
Guo, X. W., Fernando, W. G. D., and Entz, M. (2005). Effects of crop rotation and tillage on blackleg disease of canola. Can. J. Plant Pathol. 27, 53–57. doi: 10.1080/07060660509507193
Haas, D., and Défago, G. (2005). Biological control of soil-borne pathogens by fluorescent pseudomonads. Nat. Rev. Microbiol. 3, 307–319. doi: 10.1038/nrmicro1129
Hallmann, J., Quadt-Hallmann, A., Mahaffee, W. F., and Kloepper, J. W. (1997). Bacterial endophytes in agricultural crops. Can. J. Microbiol. 43, 895–914. doi: 10.1139/m97-131
Hamel, C., Vujanovic, V., Jeannotte, R., Nakano-Hylander, A., and St-Arnaud, M. (2005). Negative feedback on a perennial crop: Fusarium crown and root rot of asparagus is related to changes in soil microbial community structure. Plant Soil 268, 75–87. doi: 10.1007/s11104-004-0228-1
Harker, K. N., Clayton, G. W., Blackshaw, R. E., O'Donovan, J. T., and Stevenson, F. C. (2003). Seeding rate, herbicide timing and competitive hybrids contribute to integrated weed management in canola (Brassica napus). Can. J. Plant Sci. 83, 433–440. doi: 10.4141/P02-073
Harker, K. N., O'Donovan, J. T., Smith, E. G., Johnson, E. N., Peng, G., Willenborg, C. J., et al. (2015a). Seed size and seeding rate effects on canola emergence, development, yield and seed weight. Can. J. Plant Sci. 95, 1–8. doi: 10.4141/cjps-2014-222
Harker, K. N., O'Donovan, J. T., Turkington, T. K., Blackshaw, R. E., Lupwayi, N. Z., Smith, E. G., et al. (2015b). Canola cultivar mixtures and rotations do not mitigate the negative impacts of continuous canola. Can. J. Plant Sci. 95, 1085–1099. doi: 10.4141/cjps-2015-126
Harker, K. N., O'Donovan, J. T., Turkington, T. K., Blackshaw, R. E., Lupwayi, N. Z., McLaren, D. G., et al. (2012). High-yield no-till canola production on the Canadian prairies. Can. J. Plant Sci. 92, 221–233. doi: 10.4141/cjps2011-125
Hartwright, L. M., Hunter, P. J., and Walsh, J. A. (2010). A comparison of Olpidium isolates from a range of host plants using internal transcribed spacer sequence analysis and host range studies. Fungal Biol. 114, 26–33. doi: 10.1016/j.mycres.2009.09.008
Hilton, S., Bennett, A. J., Keane, G., Bending, G. D., Chandler, D., Stobart, R., et al. (2013). Impact of shortened crop rotation of oilseed rape on soil and rhizosphere microbial diversity in relation to yield decline. PLoS ONE 8:e59859. doi: 10.1371/journal.pone.0059859
Hwang, S. F., Ahmed, H. U., Gossen, B. D., Kutcher, H. R., Brandt, S. A., Strelkov, S. E., et al. (2009). Effect of crop rotation on the soil pathogen population dynamics and canola seedling establishment. Plant Pathol. J. 8, 106–112. doi: 10.3923/ppj.2009.106.112
Kim, B. K., Jung, M. Y., Yu, D. S., Park, S. J., Oh, T. K., Rhee, S. K., et al. (2011). Genome sequence of an ammonia-oxidizing Soil archaeon, “Candidatus Nitrosoarchaeum koreensis” MY1. J. Bacteriol. 193, 5539–5540. doi: 10.1128/JB.05717-11
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41:e1. doi: 10.1093/nar/gks808
Kloepper, J. W., Hume, D. J., Scher, F. M., Singleton, C., Tipping, B., Laliberte, M., et al. (1988). Plant growth-promoting rhizobacteria on canola (rapeseed). Plant Dis. 72, 42–46. doi: 10.1094/PD-72-0042
Klykleung, N., Tanasupawat, S., Pittayakhajonwut, P., Ohkuma, M., and Kudo, T. (2015). Amycolatopsis stemonae sp. nov., isolated from Thai medicinal plant. Int. J. Syst. Evol. Microbiol. 65, 3894–3899. doi: 10.1099/ijsem.0.000509
Kõljalg, U., Nilsson, R. H., Abarenkov, K., Tedersoo, L., Taylor, A. F. S., Bahram, M., et al. (2013). Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 22, 5271–5277. doi: 10.1111/mec.12481
Kudo, Y., Shibata, S., Miyaki, T., Aono, T., and Oyaizu, H. (1997). Peculiar archaea found in Japanese paddy soils. Biosci. Biotechnol. Biochem. 61, 917–920. doi: 10.1271/bbb.61.917
Kumar, A., Prakash, A., and Johri, B. N. (2011). “Bacillus as PGPR in crop ecosystem,” in Bacteria in Agrobiology: Crop Ecosystems, ed D. K. Maheshwari (Berlin; Heidelberg: Springer), 37–59.
Macrae, A., Rimmer, D. L., and O'donnell, A. G. (2000). Novel bacterial diversity recovered from the rhizosphere of oilseed rape (Brassica napus) determined by the analysis of 16S ribosomal DNA. Antonie Leeuwenhoek 78, 13–21. doi: 10.1023/A:1002745312030
Manter, D. K., and Vivanco, J. M. (2007). Use of the ITS primers, ITS1F and ITS4, to characterize fungal abundance and diversity in mixed-template samples by qPCR and length heterogeneity analysis. J. Microbiol. Methods 71, 7–14. doi: 10.1016/j.mimet.2007.06.016
Martin, K. J., and Rygiewicz, P. T. (2005). Fungal-specific PCR primers developed for analysis of the ITS region of environmental DNA extracts. BMC Microbiol. 5:28. doi: 10.1186/1471-2180-5-28
Miao, Q., Qin, S., Bian, G. K., Yuan, B., Xing, K., Zhang, Y. J., et al. (2011). Amycolatopsis endophytica sp. nov., a novel endophytic actinomycete isolated from oil-seed plant Jatropha curcas L. Antonie Leeuwenhoek 100, 333–339. doi: 10.1007/s10482-011-9588-8
Minerdi, D., Moretti, M., Gilardi, G., Barberio, C., Gullino, M. L., and Garibaldi, A. (2008). Bacterial ectosymbionts and virulence silencing in a Fusarium oxysporum strain. Environ. Microbiol. 10, 1725–1741. doi: 10.1111/j.1462-2920.2008.01594.x
Misko, A. L., and Germida, J. J. (2002). Taxonomic and functional diversity of pseudomonads isolated from the roots of field-grown canola. FEMS Microbiol. Ecol. 42, 399–407. doi: 10.1111/j.1574-6941.2002.tb01029.x
Neupane, S., Andersson, B., Högberg, N., Ihrmark, K., and Alström, S. (2013a). Fungal communities associated with field grown oilseed rape (Brassica napus L.) - their possible role in early crop establishment. Acta Agric. Scand. 63, 241–252. doi: 10.1080/09064710.2012.756117
Neupane, S., Goodwin, L. A., Hogberg, N., Kyrpides, N. C., Alstrom, S., Bruce, D., et al. (2013b). Non-contiguous finished genome sequence of plant-growth promoting Serratia proteamaculans S4. Stand. Genomic Sci. 8, 441–449. doi: 10.4056/sigs.4027757
Oberholster, T., Vikram, S., Cowan, D., and Valverde, A. (2018). Key microbial taxa in the rhizosphere of sorghum and sunflower grown in crop rotation. Sci. Total Environ. 15, 530–539. doi: 10.1016/j.scitotenv.2017.12.170
Pliego, C., Kamilova, F., and Lugtenberg, B. (2011). “Plant growth-promoting bacteria: fundamentals and exploitation,” in Bacteria in Agrobiology: Crop Ecosystems, ed D. K. Maheshwari (Berlin; Heidelberg: Springer), 295–343.
Poly, F., Monrozier, L. J., and Bally, R. (2001). Improvement in the RFLP procedure for studying the diversity of nifH genes in communities of nitrogen fixers in soil. Res. Microbiol. 152, 95–103. doi: 10.1016/S0923-2508(00)01172-4
Pylro, V. S., Roesch, L. F. W., Morais, D. K., Clark, I. M., Hirsch, P. R., and Totola, M. R. (2014a). Data analysis for 16S microbial profiling from different benchtop sequencing platforms. J. Microbiol. Methods 107, 30–37. doi: 10.1016/j.mimet.2014.08.018
Pylro, V. S., Roesch, L. F. W., Ortega, J. M., Do Amaral, A. M., Totola, M. R., Hirsch, P. R., et al. (2014b). Brazilian microbiome project: revealing the unexplored microbial diversity-challenges and prospects. Microb. Ecol. 67, 237–241. doi: 10.1007/s00248-013-0302-4
Raiesi, F. (2004). Soil properties and N application effects on microbial activities in two winter wheat cropping systems. Biol. Fertil. Soils 40, 88–92. doi: 10.1007/s00374-004-0741-7
Saito, A., Ooya, T., Miyatsuchi, D., Fuchigami, H., Terakado, K., Nakayama, S. Y., et al. (2009). Molecular characterization and antifungal activity of a family 46 chitosanase from Amycolatopsis sp. CsO-2. FEMS Microbiol. Lett. 293, 79–84. doi: 10.1111/j.1574-6968.2009.01507.x
Sasaya, T., and Koganezawa, H. (2006). Molecular analysis and virus transmission tests place Olpidium virulentus, a vector of Mirafiori lettuce big-vein virus and tobacco stunt virus, as a distinct species rather than a strain of Olpidium brassicae. J. Gen. Plant Pathol. 72, 20–25. doi: 10.1007/s10327-005-0239-7
Sauder, L. A., Albertsen, M., Engel, K., Schwarz, J., Nielsen, P. H., Wagner, M., et al. (2017). Cultivation and characterization of Candidatus Nitrosocosmicus exaquare, an ammonia-oxidizing archaeon from a municipal wastewater treatment system. ISME J. 11, 1142–1157. doi: 10.1038/ismej.2016.192
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Simon, H. M., Dodsworth, J. A., and Goodman, R. M. (2000). Crenarchaeota colonize terrestrial plant roots. Environ. Microbiol. 2, 495–505. doi: 10.1046/j.1462-2920.2000.00131.x
Sliwinski, M. K., and Goodman, R. M. (2004). Comparison of crenarchaeal consortia inhabiting the rhizosphere of diverse terrestrial plants with those in bulk soil in native environments. Appl. Environ. Microbiol. 70, 1821–1826. doi: 10.1128/AEM.70.3.1821-1826.2004
Smalla, K., Wieland, G., Buchner, A., Zock, A., Parzy, J., Kaiser, S., et al. (2001). Bulk and rhizosphere soil bacterial communities studied by denaturing gradient gel electrophoresis: plant-dependent enrichment and seasonal shifts revealed. Appl. Environ. Microbiol. 67, 4742–4751. doi: 10.1128/AEM.67.10.4742-4751.2001
Tkacz, A., Cheema, J., Chandra, G., Grant, A., and Poole, P. S. (2015). Stability and succession of the rhizosphere microbiota depends upon plant type and soil composition. ISME J.. doi: 10.1038/ismej.2015.41
Ueda, T., Suga, Y., and Matsuguchi, T. (1995). Molecular phylogenetic analysis of a soil microbial community in a soybean field. Eur. J. Soil Sci. 46, 415–421. doi: 10.1111/j.1365-2389.1995.tb01337.x
USDA (2017). Production, Supply and Distribution. Market and Trade Data. Available online at: https://apps.fas.usda.gov/psdonline/app/index.html#/app/topCountriesByCommodity (January 9, 2017).
van der lee, T., Zhang, H., Van Diepeningen, A., and Waalwijk, C. (2015). Biogeography of Fusarium graminearum species complex and chemotypes: a review. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 32, 453–460. doi: 10.1080/19440049.2014.984244
Vandenkoornhuyse, P., Quaiser, A., Duhamel, M., Le Van, A., and Dufresne, A. (2015). The importance of the microbiome of the plant holobiont. New Phytol. 206, 1196–1206. doi: 10.1111/nph.13312
Vujanovic, V., Hamel, C., Yergeau, É., and St-Arnaud, M. (2006). Biodiversity and biogeography of Fusarium species from northeastern North American asparagus fields based on microbiological and molecular approaches. Microb. Ecol. 51, 242–255. doi: 10.1007/s00248-005-0046-x
Weber, E., and Bleiholder, H. (1990). Explanations of the BBCH decimal codes for the growth stages of maize, rape, faba beans, sunflowers and peas - with illustrations. Gesunde Pflanzen 42, 308–321.
Weller, D. M., Raaijmakers, J. M., Gardener, B. B., and Thomashow, L. S. (2002). Microbial populations responsible for specific soil suppressiveness to plant pathogens. Annu. Rev. Phytopathol. 40, 309–348. doi: 10.1146/annurev.phyto.40.030402.110010
Wolf, A., Fritze, A., Hagemann, M., and Berg, G. (2002). Stenotrophomonas rhizophila sp. nov., a novel plant-associated bacterium with antifungal properties. Int. J. Syst. Evol. Microbiol. 52, 1937–1944. doi: 10.1099/00207713-52-6-1937
Xing, K., Liu, W., Zhang, Y. J., Bian, G. K., Zhang, W. D., Tamura, T., et al. (2013). Amycolatopsis jiangsuensis sp. nov., a novel endophytic actinomycete isolated from a coastal plant in Jiangsu, China. Antonie Leeuwenhoek 103, 433–439. doi: 10.1007/s10482-012-9823-y
Yang, J., Kloepper, J. W., and Ryu, C. M. (2009). Rhizosphere bacteria help plants tolerate abiotic stress. Trends Plant Sci. 14, 1–4. doi: 10.1016/j.tplants.2008.10.004
Yergeau, É., Labour, K., Hamel, C., Vujanovic, V., Nakano-Hylander, A., Jeannotte, R., et al. (2010). Patterns of Fusarium community structure and abundance in relation to spatial, abiotic and biotic factors in soil. FEMS Microbiol. Ecol. 71, 34–42. doi: 10.1111/j.1574-6941.2009.00777.x
Yergeau, É., Sommerville, D. W., Maheux, E., Vujanovic, V., Hamel, C., Whalen, J. K., et al. (2006). Relationships between Fusarium population structure, soil nutrient status and disease incidence in field-grown asparagus. FEMS Microbiol. Ecol. 58, 394–403. doi: 10.1111/j.1574-6941.2006.00161.x
Keywords: rhizosphere, canola, root, Olpidium, PGPR, rotation, seeding density, oilseed rape
Citation: Lay C-Y, Bell TH, Hamel C, Harker KN, Mohr R, Greer CW, Yergeau É and St-Arnaud M (2018) Canola Root–Associated Microbiomes in the Canadian Prairies. Front. Microbiol. 9:1188. doi: 10.3389/fmicb.2018.01188
Received: 18 December 2017; Accepted: 16 May 2018;
Published: 08 June 2018.
Edited by:
Malin Bomberg, VTT Technical Research Centre of Finland Ltd, FinlandReviewed by:
Matthew G. Bakker, Agricultural Research Service (USDA), United StatesCopyright © 2018 Lay, Bell, Hamel, Harker, Mohr, Greer, Yergeau and St-Arnaud. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marc St-Arnaud, bWFyYy5zdC1hcm5hdWRAdW1vbnRyZWFsLmNhLg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.