 Ye Sun1
Ye Sun1 Yu-Qin Zhang
Yu-Qin Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 23 May 2018
Sec. Extreme Microbiology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.01068
This article is part of the Research Topic Actinobacteria in Special and Extreme Habitats: Diversity, Function Roles and Environmental Adaptations, Second Edition View all 17 articles
To assess the diversity of actinobacterial taxa in desert sands and obtain the novel microbial resources, 79 and 50 samples were collected from the Badain Jaran (BJD) and Tengger Deserts (TGD) of China, respectively. High-throughput sequencing (HTS) of environmental 16S rRNA genes within these samples was conducted on an Illumina Miseq platform, using universal bacterial primers targeting the V3–V4 hypervariable region. Based on the HTS analyses, cultivation-dependent (CULD) techniques were optimized to identify the cultivable Actinobacteria members. A total of 346,766 16S rRNA gene reads comprising 3,365 operational taxonomic units (OTUs) were obtained from the BJD sands using HTS, while 170,583 reads comprising 1,250 OTUs were detected in the TGD sands. Taxonomic classification indicated that Actinobacteria was the predominant phylum, comprising 35.0 and 29.4% of the communities in BJD and TGD sands, respectively. Among the Actinobacteria, members of the Geodermatophilaceae were considerably abundant in both deserts, indicating that they represent ubiquitous populations within the deserts. At the genus level, Arthrobacter spp. and Kocuria spp. were dominant, and corresponded to 21.2 and 5.3% of the actinobacterial communities in BJD and TGD deserts, respectively. A total of 786 and 376 actinobacterial strains were isolated and identified from BJD and TGD samples, respectively. The isolates comprised 73 genera of 30 families within the phylum Actinobacteria. In addition to the Geodermatophilaceae, Streptomyces spp. were a prominent component of the isolates, comprising 25% of the isolates from BJD and 17.5% of those from TGD. Comparison of the actinobacterial community structure in other ecosystems indicated that Geodermatophilaceae was the main actinobacterial group in desert sands, which is consistent with our results. Additionally, in these desert habits, Geodermatophilaceae and some other core groups may promote or inhabit the subsequent members' occurrence or prosper to shape the bacteria community structure. However, it should be noted that a number of other low-abundance bacteria appear to be specific to desert sands, which are worth further investigation. In antimicrobial activity assays, 10.36 % of the tested isolates showed antimicrobial activities in one or more screens. Importantly, 37 of the newly isolated strains reported here represent novel taxa that could be valuable resources for further research of novel secondary metabolites and their ecological significance in deserts.
Extremophiles havex generated significant interest in the biological sciences in recent years, due to their unique genotypes, physiological functions, and secondary metabolites that hold great scientific and industrial value (Ciaramella, 1995). Deserts are extreme environments that are characterized by extreme aridity, intense solar UV radiation, and extreme shifts of temperature in day-night cycles. Consequently, deserts harbor numerous extremophiles (Subramani and Aalbersberg, 2013; Júlia et al., 2016). In addition to the above conditions, deserts feature chronic oligotrophy. Thus, sporadic vascular vegetation and microbiological crusts play critical roles in the primary production of this ecosystem, in addition to keeping soils from undergoing further desertification (Hawkes and Flechtner, 2002). Moreover, vegetation and microbiological crusts contribute greatly in shaping soil micro-ecosystem bacterial community structure in desert environments (Evans and Johansen, 1999; Belnap and Lange, 2003; Sun et al., 2015).
Deserts are typical of extreme, harsh ecosystems, where the availability of water is the cardinal parameter affecting organisms. Consequently, xerophilous microorganisms that are adapted to relatively high temperatures and radiation levels are likely to be the dominant populations in these ecosystems, including desiccation and radiation resistant phyla, such as Actinobacteria, Proteobacteria, and Bacteroidetes (Vikram et al., 2016).
The Badain Jaran Desert (BJD) is located in the northwest region of the Alxa Plateau of the Inner Mongolia Autonomous Region (39°20′–41°30′ N, 100°–104° E), and comprises an area of 49,000 km2, making it the third largest desert in China (Yan et al., 2001). This region is known for its extreme continental climate and experiences the East Asian monsoon in its southwestern quadrant. The East Asian monsoon provides most of the precipitation for this area, accounting for 70% of the annualrainfall, which occurs within 10–35 days between July and September. The average annual rainfall is 150 mm while evaporation can reach as much as 1,500 mm. The Tengger Desert (TGD) is located in the southwest region of the Alashan Left Banner in the Inner Mongolia Autonomous Region and along the central border of Gansu Province (34°30′–39°00′ N, 103°–106°E). The TGD comprises a total area of about 43,000 km2, and is the fourth largest desert in China. A typical continental climate dominates this region, with an annual average temperature of 7–9°C, and annual rainfall of 116–148 mm that primarily occurs in July and August. The annual evaporation of this region is 3,000–3,600 mm, and the average annual wind speed is 3–4 m/s. With increasing grassland degeneration and increases in desertification, the two deserts are almost contiguous with each other in some places (Figure 1).
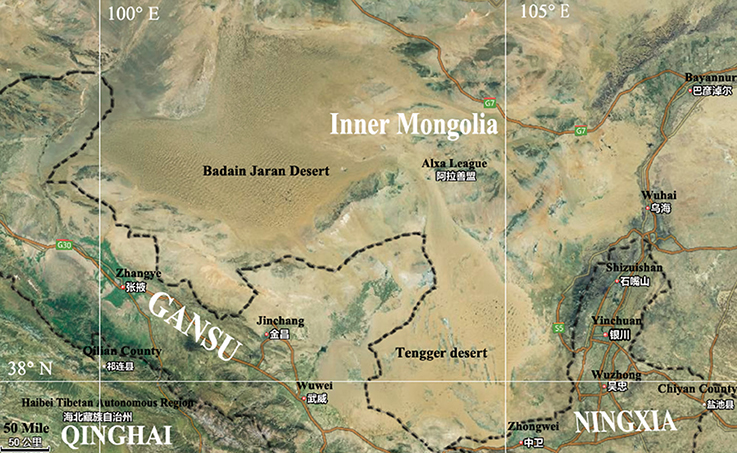
Figure 1. Map showing Badain Jaran desert location and Tengger desert location.
Actinobacteria are a group of bacteria with high G+C content in the genomic DNA, and well-known for producing abundant secondary active metabolites. Since Waksman discovered streptomycin from Streptomyces sp. in 1943, actinobacterial members have been considered as an important source of new antibiotic-producing bacteria. Currently, about 70% antibiotics in clinical use are produced by various actinomyces (Doull and Vining, 1990; Jose and Jebakumar, 2013). However, decades later, people found it is increasingly difficult to obtain novel compounds from the actinobacterial strains from normal environments. Therefore, more and more researchers proposed to find potential functional strains from various extreme environments, in which the external stress factors endow the microorganisms the unique defense mechanisms and metabolic systems, which may be more likely to produce novel antibiotics. Consequently, detecting various actinobacteria from extreme environments has become an important strategy for the discovery of new antibiotics (Phoebe et al., 2001; Wilson and Brimble, 2009). Till now, no systematic studies on actinobacteria regarding their potential abilities to produce bio-active substances and their ecological function in deserts have been reported.
The primary goal of the present research was to obtain a systematic understanding of the bacterial community structure and diversity of actinobacteria inhabiting the two immense BJD and TGD deserts of China. Further, we attempted to better characterize the distribution characteristics of actinobacterial population in these environments. Based on the above findings, we intended to optimize the isolation and cultivation strategies to explore the specific actinobacterial strains and to assess their bio-characteristics, potential functions and applications in depth.
A total of 79 samples were collected from the BJD (38°20′–41°30′N, 100°–104°E). Sporadic vascular vegetation and small salt lakes characterize this desert. We thus assigned samples to the following three types based on vegetation cover and salt lake presence: sands with sporadic plants (SP), sands without any vegetation (NV), and sands around salt lakes (SL). Detailed information of samples including types, and the specific collection locations are provided in Table S1. Fifty samples were collected from the TGD, where the microbial crusts are generally categorized into the following three types: Cyanobacteria-dominated crusts (CC), moss-dominated crusts (MC), and lichen-dominated crusts (LC). Samples from the TGD also included bare sands (BS) (Sun et al., 2015), and sample information is provided in Table S2.
All samples were placed in sterilized envelopes following collection and then taken to the laboratory within 3 days of collection. The samples were immediately processed for cultivation assays and total DNA extraction after arriving at the laboratory.
The following seven media were prepared to isolate microbial strains:
M1: 1/5 strength R2A (Difco).
M2: 5 g/L yeast extract, 2 g/L cellobiose, 2 g/L CaCO3, 0.5 g/L MgSO4•7H2O, 1 g/L K2HPO4, and 15 g/L agar.
M3: 0.1 g/L NH 4NO3, 2 g/L sodium propionate, 0.05 g/L MgSO4·7H2O, 0.1 g/L KCl, and 15 g/L agar.
M4: 1 g/L humic acid, 1 g/L asparagine, 0.01 g/L FeSO4, 0.5 g/L Na2HPO4, 1.7 g/L KCl, 0.02 g/L CaCO3, and 15 g/L agar.
M5: 2 g/L trehalose, 5 g/L yeast extract, 2 g/L CaCO3, 0.5 g/L MgSO4·7H2O, 1 g/L K2HPO4, and 15 g/L agar.
M6: 0.5 g/L K2HPO4, 0.25 g/L yeast extract, and 15 g/L agar.
M7: 5 g/L yeast extract, 3 g/L peptone, 10 g/L glycerol, 1.25 g/L sodium pyruvate, 1.25 g/L glycine betaine, and 15 g/L agar.
Media were adjusted to pH 7.2–7.5 using 1M HCl and/or 1M NaOH. In addition, betaine (0.125% w/v), sodium pyruvate (0.125% w/v), compound trace salts solution (0.1% v/v), and compound vitamins (0.1% w/v) were added to the media as described in Sun et al. (2015). Nystatin (25 mg/L) and potassium dichromate (50 mg/L) were added to the media to inhibit the growth of fungi and Gram negative bacteria.
Based the obsevationof the colony diversity growth in the isolation media, the PYG medium was selected to cultivate isolates after initial isolation and consisted of 3 g/L peptone, 10 g/L glycerol, 5 g/L yeast extract, 1.25 g/L sodium pyruvate, 1.25 g/L glycine betaine, and 15 g/L agar, adjusted to pH 7.5. PYG medium was supplemented with compound trace salts solution (0.1% v/v), and compound vitamins (0.1% w/v).
The 79 sand samples from BJD were pooled into seven composite samples, and the 50 sand samples from TGD were pooled into eight composite samples, according to the environments where the sands were collected. Detailed information of the 15 samples is provided in Table S3. Total genomic DNA from each of the 15 pooled samples was extracted with a PowerSoil DNA isolation kit (MoBio, USA) according to the manufacturer's protocols. Total DNA was then used as template for PCR amplification of 16S rRNA genes. The V3 to V4 hypervariable regions of 16S rRNA genes were PCR amplified using the universal bacterial primers 5′-ACTCCTACGGGAGGCAGCAG-3′ (338F) and 5′-GGACTACHVGGGTWTCTAAT-3′ (806R). PCR amplifications were performed using high fidelity TransStart Fastpfu DNA Polymerase (Transgen, China) in 20 μL reaction mixtures containing 4 μL of 5× FastPfu Buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer (5 μM), 0.4 μL of FastPfu Polymerase, and 10 ng of template DNA. The reaction cycling conditions consisted of the following steps: 5 min of an initial denaturation at 94°C followed by 35 cycles of denaturation at 94°C for 30 s, 45 s of primer annealing at 55°C, 40 s of elongation at 72°C, and then a final 10 min elongation at 72°C.
An AxyPrep DNA Gel Extraction Kit (Axygen, USA) and QuantiFluorTM-ST system (Promega, USA) were used to purify and quantify amplicons, respectively. Purified amplicons were pooled in equimolar concentrations and sequenced on an Illumina MiSeq platform with paired-end sequencing (2 × 250 bp). The QIIME software package (version 1.18; White et al., 2009; Caporaso et al., 2010) was used to demultiplex and quality-filter raw reads by removing reads meeting the following criteria: (a) 300 bp reads exhibiting any 50-bp sliding window with an average quality score < 20, and discarding truncated reads that were shorter than 50 bp; (b) any barcode mismatch, greater than two nucleotide mismatches in primers, and reads containing ambiguous characters (c). Lastly, paired sequences were assembled based on their overlapping sequences that were longer than 10 bp. Unassembled reads were discarded.
Chimeras were identified and removed using the UCHIME program (Robert et al., 2001). The Unite database (Release 7.0, http://unite.ut.ee/index.php; Kõljalg et al., 2013) was used as the taxonomic reference database to assess the taxonomic affiliation of each 16S rRNA gene sequence using a confidence threshold of 70% and the RDP Classifier (Wang et al., 2007). Sequences were clustered into operational taxonomic units (OTUs) using a 97% similarity threshold in the UPARSE program (version 7.1, http://drive5.com/uparse/). Alpha diversity and beta diversity metrics were calculated based on OTU abundances within and among samples. Specifically, alpha diversity was measured using the Chao1 richness estimator, and Shannon's diversity index, while measuring the coverage of richness using Good's coverage. Principal co-ordinates analysis (PCoA) was used to assess differences in OTU composition among samples. An analysis of similarities (ANOSIM) test was conducted to determine if sample types contained significantly different bacterial communities. Metastats and LEfSe (LDA effect size) analysis were employed to identify the bacterial groups that were significantly differentiated among sample types. Lastly, co-occurrence networks were constructed to visualize the OTU-based similarities among different communities (White et al., 2009; Caporaso et al., 2010).
The dilution plating method was used to isolate microbial strains from the sand samples. Approximately 0.3 mL of a 10−4 dilution of a sand sample suspension was spread on each isolation plate. Every kind of media was parallelly spread with the suspension on two same plates and then incubated at 28°C and 45°C for 21 days, respectively. Single colonies were transferred to freshly prepared PYG plates that were supplemented with compound trace salts solution and compound vitamins, followed by subsequent purification of isolates. Purified isolates were maintained on PYG slants at 4°C and also in glycerol suspensions (20%, v/v) at −80°C (Yue et al., 2006). Genomic DNA was extracted from pure cultures and PCR amplification of 16S rRNA genes was conducted as described in Xu et al. (2003). The sequences of the isolates were compared with available 16S rRNA gene sequences from GenBank using the BLAST program and the EzBioCloud (https://www.ezbiocloud.net/) (Yoon et al., 2017).
Antimicrobial activities of the isolates were investigated by using media containing Escherichia coli ATCC 25922, Pseudomonas aeruginosa ATCC 27853, Enterococcus faecalis ATCC 29212, Klebsiella pneumonia subsp. pneumoniae ATCC 700603, and Candida albicans ATCC 10231, with a concentration of 108 colony forming units (CFU) per mL. These assays were performed using Kirby Bauer Method with a culture broth concentration of 1% (v/v).
The SRA accession number in DDBJ/EMBL/GenBank for the sequences data is SRP134260.
A total of 349,374 reads with an average of 23,291 reads per sample remained after quality filtering, and were clustered into 4,298 OTUs at the 97% sequence similarity level. The alpha diversity of each sample estimated by the Chao1 estimator, and Shannon's index, in addition to the coverage estimated by Good's coverage, are provided in Table 1.
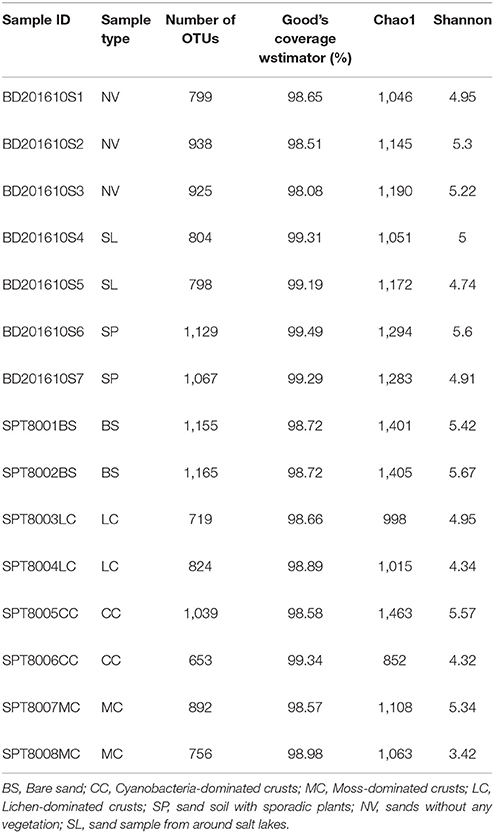
Table 1. Summary of high-throughput sequencing data from the 15 samples.
Rarefaction analyses using the Shannon index as a diversity metric indicated that our sequencing efforts covered nearly all of the diversity that would be expected to be found in these samples (Figure S1). On average, 923 (95% CI: 799–1,029) and 900 (95% CI: 653–1,165) OTUs were found in each of the BJD and TGD samples, respectively. Samples from the desert sands exhibited almost identical Chao1 and Shannon index values, as indicated by ANOVA tests, suggesting no significant difference in the richness or diversity of bacteria between the two deserts (Figure 2).
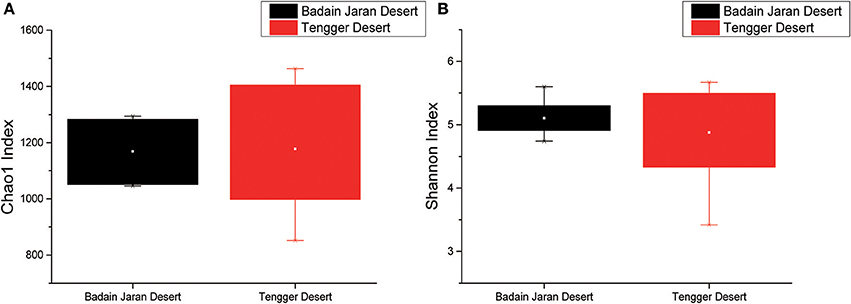
Figure 2. Statistical comparisons of Chao1 and Shannon indices between two desert sand samples. (A) Values of Chao1 index; (B) values of Shannon index.
Among the BJD samples, SP samples had the highest Chao1 values, whereas samples from NV, SL and SP did not significantly differ. Among the TGD samples, bacterial richness and diversity was slightly higher in BS samples compared to the others, which was reflected in higher Chao1 and Shannon index values (Figure 3).
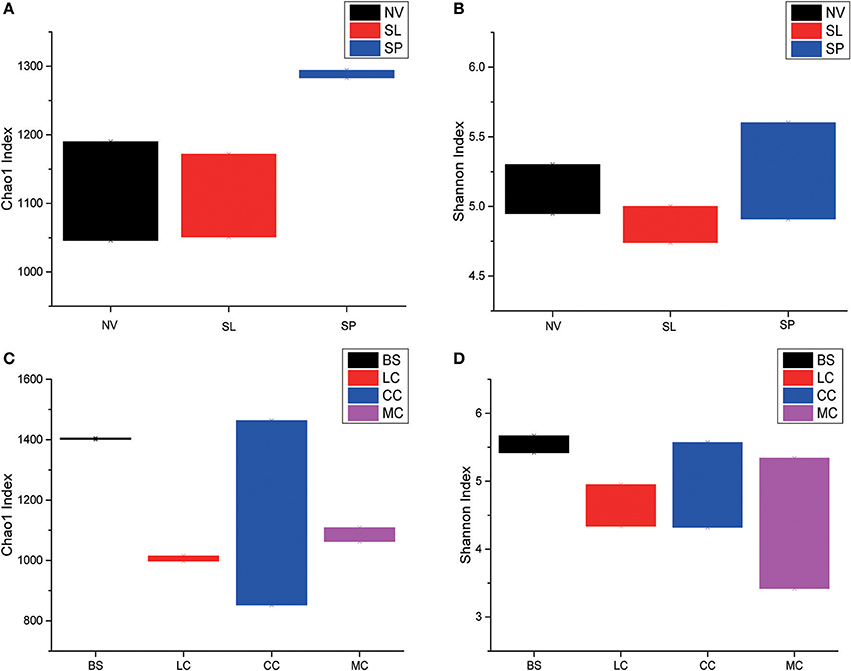
Figure 3. Statistical comparisons of Chao1 and Shannon indices among three types of Badain Jaran desert sand samples (NV, SP, and SL) and four types of Tengger desert sand samples (BS, LC, MC, and CC). (A) Values of Chao1 in of BJD; (B) values of Shannon index of BJD; (C) values of Chao1 of TGD; (D) values of Shannon index of TGD.
A total of 32 phyla, 97 orders, and 470 genera were identified in BJD samples, while 28 phyla, 73 orders, and 302 genera were identified in TGD samples. Actinobacteria dominated the bacterial communities of both deserts (35 and 29% relative abundance in BJD and TGD, respectively), followed by Proteobacteria, Bacteroidetes, and Chloroflexi (Figure 4). Frankiales, Micrococcales, Micromonosporales, and Acidimicrobiales were the most abundant orders within the 15 sand samples. Among these, the families Geodermatophilaceae, Micrococcaceae, Micromonosporaceae, and Rubrobacteriaceae were the predominant actinobacterial populations in both deserts. Lastly, the most abundant genera were Arthrobacter and Rubrobacter in the actinobacterial community, followed by Blastococcus, Modestobacter, and Geodermatophilus, which are members of the Geodermatophilaceae (Figure 5). In BJD, Micrococcaceae was much more prevalent than Geodermatophilaceae, while the reverse was observed in TGD samples. Arthrobacter was the most abundant genus in BJD samples and the second most abundant genus in TGD samples. Kocuria was a dominant genus in BJD samples, but only a few Kocuria OTUs were detected in TGD samples. Conversely, Actinoplanes were more abundant in TGD samples than in BJD samples. In addition to these dominant genera, a large number of rare bacterial genera were also detected (we defined rare bacterial groups as less than 0.1% abundance of communities), and included the genera Conexibacter, Longispora, Dactylosporangium, Umezawaea, Demequina, Janibacter, and Motilibacter in BJD and genera Ornithinimicrobium, Angustibacter, Nakamurella, Aquiluna, Williamsia, Amycolatopsis, and Kineosporia in TGD (Figure 5).
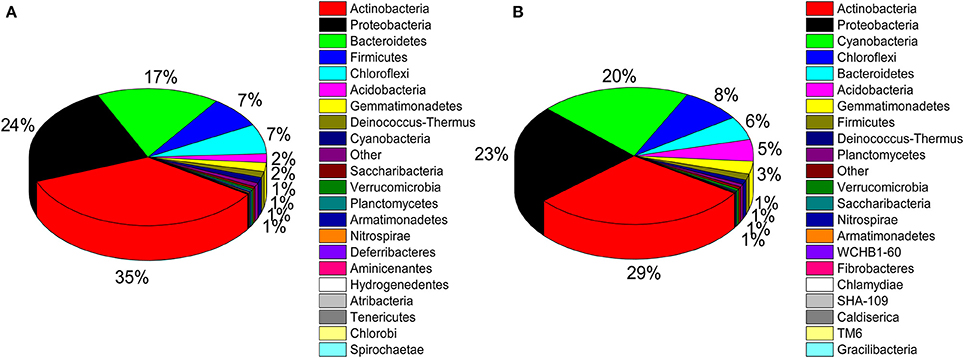
Figure 4. Pie chart showing the percentage of relative abundance of bacterial communities from two desert sand samples in HTS method. (A) Badain Jaran desert sands; (B) Tengger Desert sands.
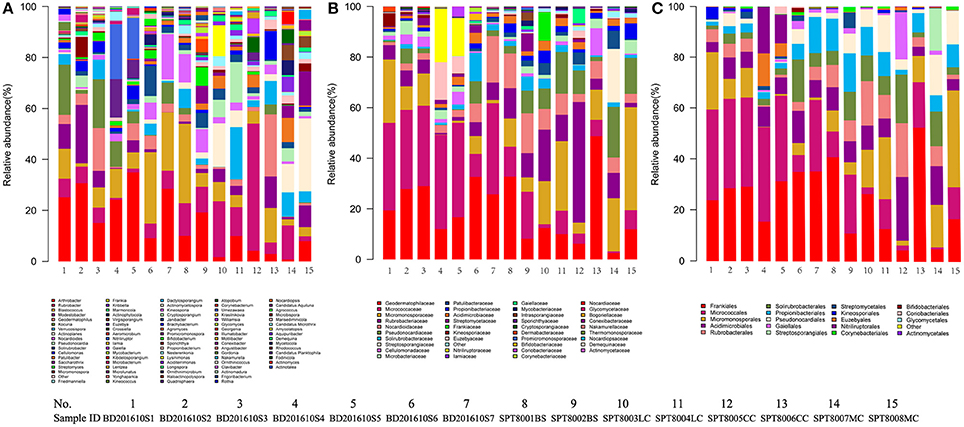
Figure 5. The relative abundances of different actinobacterial taxon level of two desert sand samples in HTS method. (A) Genus taxon level; (B) family taxon level; (C) order taxon level.
Metastats analysis indicated that the composition of actinobacterial genera in the two deserts were not significantly different (P = 1) (Table S4). However, different sand types represented distinct ecosystems, and generally, bacterial community composition was more strongly correlated with sampling site rather than micro-ecosystem type (Figure 6).
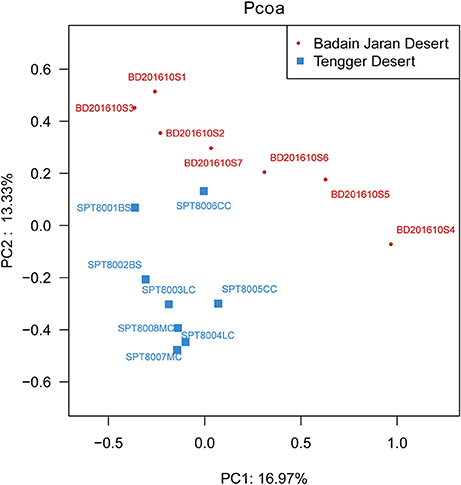
Figure 6. PCoA diagram showing the significant difference in different desert samples.
In particular, ANOSIM (R = 0.328, p = 0.001) indicated that community structures were not different between samples taken from sampling sites that were close together. LEfSe analyses indicated that there were some significant differences in community composition among the three sample types from BJD. For example, the abundances of the genera Virgisorangium, Nocardiopsis, Brachybacterium, Kineococcus, Georgenia, and Streptosporangium were highly differential among sample types within BJD (Figure 7). However, no significant differences in the abundances of these genera were observed for samples from the TGD. The Geodermatophilaceae and Micrococcaceae were the dominant families in NV, SL, SP, and BS samples, whereas Micromonosporaceae were more abundant in the LC, CC, and MC samples. Further, Pseudonocardiaceae were more abundant in NV, SP, BS, LC, CC, and MC samples, but not in SL samples. Arthrobacter was the predominant genus in NV, SL, SP, and BS samples, but not in LC, CC, or MC samples. Lastly Rubrobacter were abundant in LC, CC and MC samples, but not NV, SL, SP, or BS samples (Figure S2).
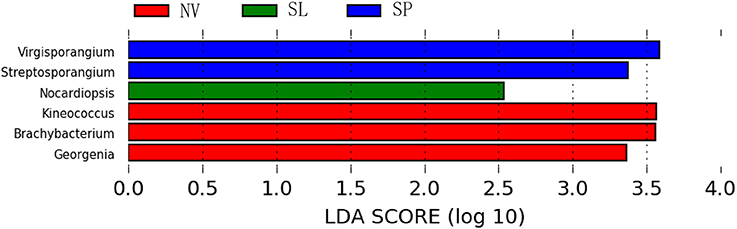
Figure 7. The LEfSe analysis indicating the significant difference among three types of Badain Jaran desert sand samples.
We used co-occurrence network analysis to identify correlations between different genera among samples. A total of 27 nodes and 84 correlation edges were used to represent the dominant genera (>1% relative abundance). Correlation analysis indicated 77 positive and seven negative correlations. The genera Solirubrobacter and Actinoplanes exhibited the most correlations (12 edges for each), followed by the Pseudonocardia, Marmoricola, Saccharothrix, and Friedmanniella which each comprised 10 edges. These results indicated that the six genera were likely to be the key actinobacterial members in the sand communities of the two deserts. Among the family Geodermatophilaceae, three genera appeared to also be important with Geodermatophilus exhibiting six edges, Modestobacter with five, and Blastococcus with two (Figure 8).
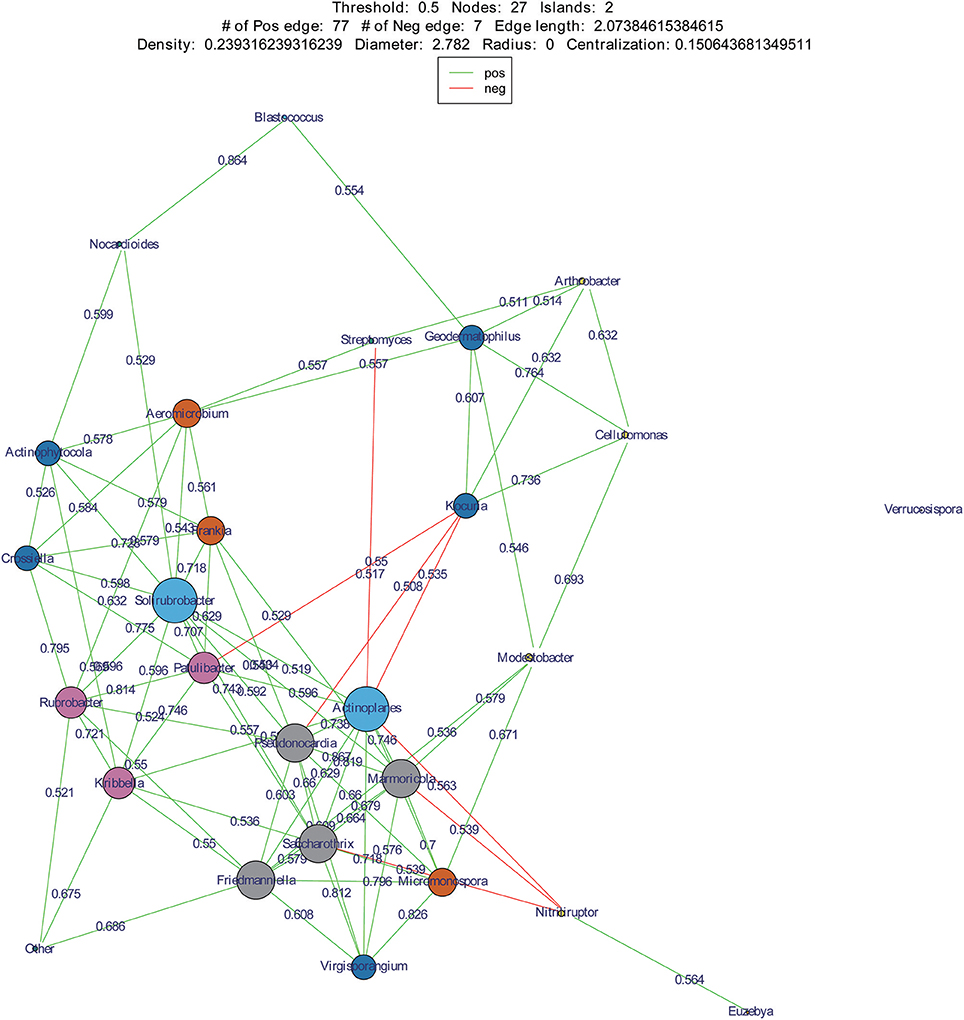
Figure 8. Co-occurrence networks of dominant genera in two deserts. Each node means a dominant genus, each edge means two correlated genera with an absolute Spearman's correlation above 0.5 at a 0.05 significance level; the edge length is based on Bray-curtis distance and the node size means the number of its connection with other genera.
A total of 786 actinobacterial strains were isolated and identified from the BJD samples. 16S rRNA gene sequencing indicated that these 786 strains belonged to 30 families and 73 genera (Figure S3A). In addition, 376 actinobacterial strains were isolated from TGD samples, comprising 18 families and 29 genera (Figure S3B). 16S rRNA gene sequencing indicated that members of Geodermatophilaceae were the major cultivated actinobacterial taxa in both of these deserts, which is consistent with the cultivation-independent analyses. Moreover, the taxonomic distributions of isolates were highly similar to those of the cultivation-independent analyses. Arthrobacter spp. and Kocuria spp. were the primary cultivated genera, and comprised 13.4 and 4.1% of the actinobacterial community isolates in BJD, respectively (Figure S4; Figure 9). Streptomyces spp. were also dominant among the isolated genera, comprising 25% of BJD isolates and 17.5% of TGD isolates. In addition, some low abundance taxa that were detected using cultivation-independent analysis including Deinococcus spp., Skermanella spp., Actinophytocola spp., Patulibacter spp., Rufibacter spp., and Geminicoccus spp. were also isolated.
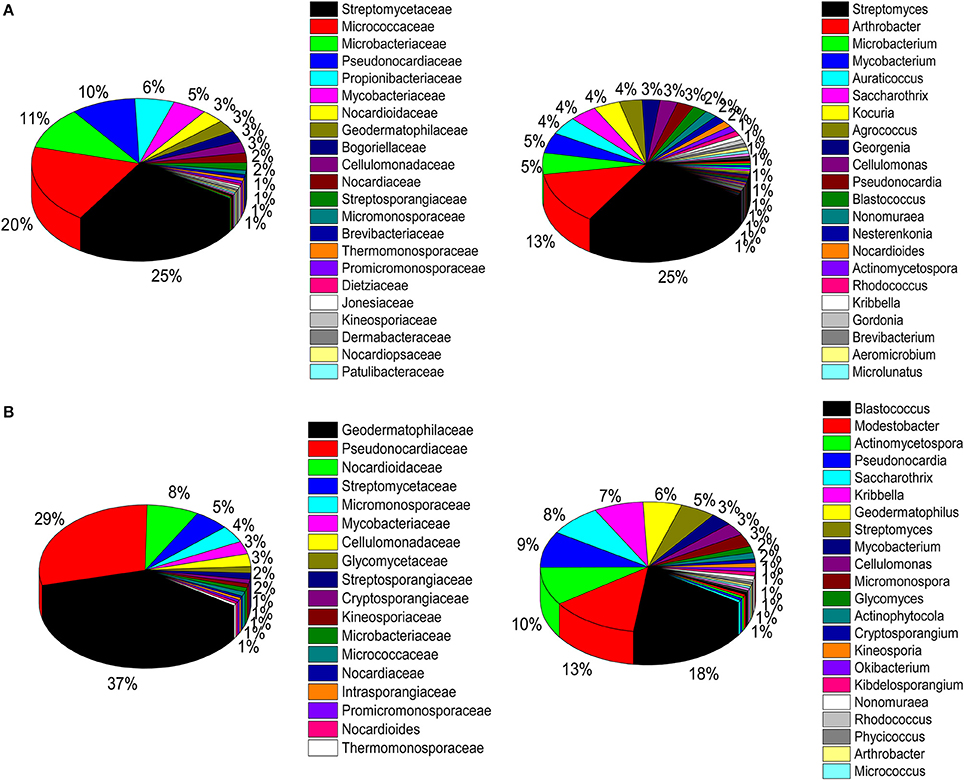
Figure 9. Pie chart showing the percentage of relative abundance of bacterial communities from two desert sand samples in CULD method. (A) Badain Jaran desert sands; (B) Tengger desert sands. The sequences of the bacteria were assigned to family (1/3) and genus level (2/4).
Of the dominant genera (>1%), 4.3 and 32.2% were detected using the two methodologies in BJD and TGD samples, respectively (Figure 10). In addition, 14.0 and 17.1% of the rare genera (<0.1%) were identified by both methodologies in BJD and TGD, respectively. However, there were some genera that were only detected by either cultivation-dependent (CULD) or high-throughput sequencing (HTS) analysis (Figure 10). These results indicate that the composition of the native microbial communities are not only driven by the dominant groups, but also rare taxa that are active and integral members of community structure.
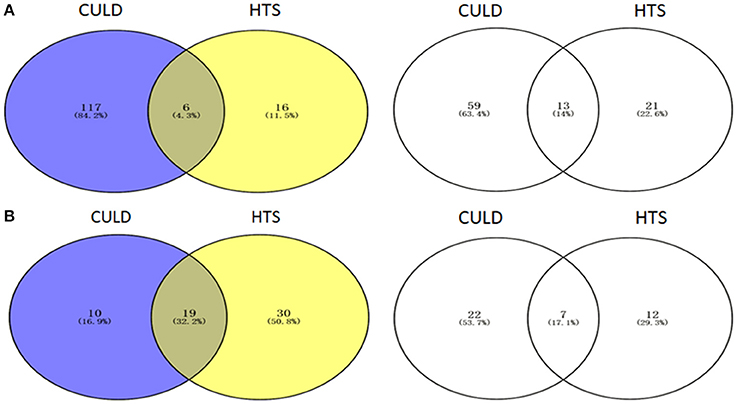
Figure 10. Venn diagrams illustrating the number of genera between High-throughput sequencing analysis and Cultivation-dependent method. (A) Badain Jaran desert sands; (B) Tengger desert sands. The Venn diagrams were compared by dominant genera (1/3) and rare genera (2/4).
Geodermatophilaceae populations were the dominant group in both the BJD and TGD deserts, comprising 27.4 and 18.2% of the Actinobacteria according to cultivation-independent analyses. The genera Blastococcus, Modestobacter, and Geodermatophilus exhibited nearly equivalent diversity in the two deserts (Figure S4). However, Blastococcus was more ubiquitous and abundant in all samples, compared to the other two genera. Modestobacter was more abundant in NV, MC, and CC samples, while OTUs associated with this genus were rare in SL, SP, and BS samples. Geodermatophilus was abundant in NV, SP, and CC samples, but not in LC or MC samples. Generally, NV, SP, BS, and CC samples harbored the most abundant Geodermatophilaceae communities (Figure 11). Futher, more Geodermatophilaceae isolates were obtained form the NV and BS samples, which could be regarded as more oligotrophic environments. In total, 52 Geodermatophilaceae strains were obtained from BJD, including 36 Blastococcus spp., six Geodermatophilus spp. and 10 Modestobacter spp. In addition, 34 Blastococcus spp., 11 Geodermatophilus spp., and 25 Modestobacter spp. were obtained from TGD. Importantly, 21 actinobacterial isolates from BJD representing novel species within the following 15 genera were obtained: Actinokineospora, Actinomycetospora, Auraticoccus, Blastococcus, Cellulomonas, Kibdelosporangium, Mariniluteicoccus, Microbacterium, Nesterenkonia, Nocardia, Nocardioides, Pseudonocardia, and Solirubrobacter. Likewise, 16 novel taxa were isolated from TGD that were distributed among 11 genera (Figure 12). These novel actinobacterial taxa are worthy of further bioprospecting studies.
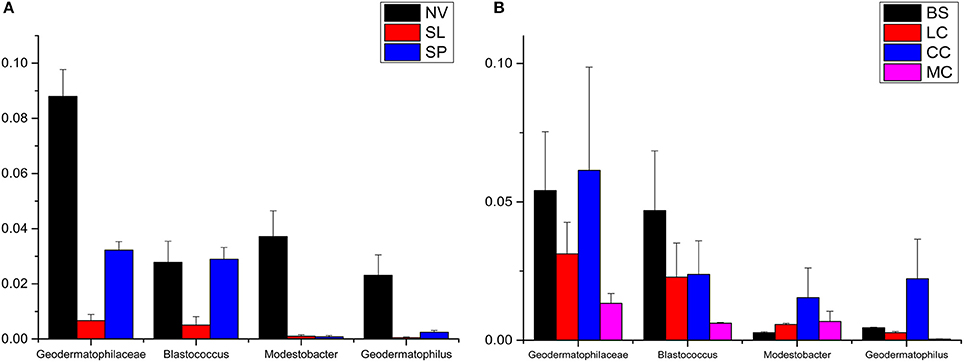
Figure 11. The relative abundances of Geodermatophilaceae in different types of the two deserts. (A) Badain Jaran Desert sand samples; (B) Tengger Desert sand samples.
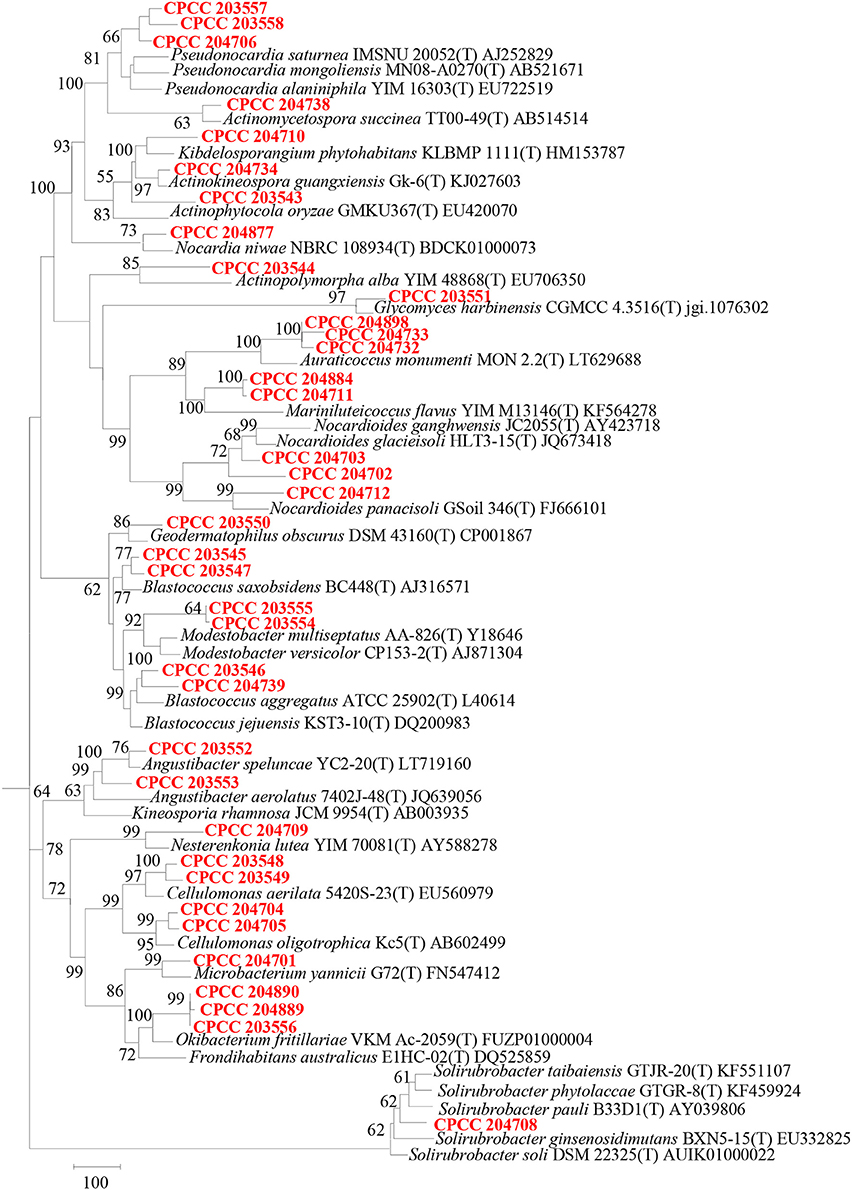
Figure 12. Dendrogram based on 16S rRNA gene sequences analysis of 37 novel species and some relevant strains in the two deserts. Bar, 100 substitutions per nucleotide position.
Five-hundred actinobacterial isolates from BJD were screened for antimicrobial activities. Of these, 1.6% of the isolates exhibited antimicrobial activity against Escherichia coli ATCC 25922, 3.5% against Enterococcus faecalis ATCC 29212, 1.7% against Klebsiella pneumonia subsp. pneumoniae ATCC 700603, 1.2% against Pseudomonas aeruginosa ATCC 27853, and 2.2% against Candida albicans ATCC 10231. Overall, 10.36% of the tested isolates from BJD exhibited antimicrobial activity in one or more screens.
With the rapid technological innovation in molecular biology in recent years, various new techniques, and in particular high-throughput cultivation-independent approaches, have been extensively applied in microbial ecology research. These new methodologies have greatly expanded our knowledge of bacterial community composition. Consequently, numerous studies have shown that microbial diversity is far higher and more complex than previously thought. 16S rRNA gene cloning and denaturing gradient gel electrophoresis (DGGE) have been traditionally used to identify uncultured microbial community structures in the past. However, these methods are particularly susceptible to 16S rRNA gene copy numbers, and the methods are both complicated and costly. High-throughput sequencing precludes the need to build clone libraries, and the efficiency associated with HTS sequencing approaches and the widespread adoption of the Illumina Miseq sequencing platform has accelerated analytical capabilities. Consequently, HTS analysis on the Illumina Miseq platform has provided comprehensive and efficient analyses of environmental microbial communities. (Navarro-Noya et al., 2013; Poulsen et al., 2013; Sul et al., 2013; Yu et al., 2013; Wu et al., 2014). Claesson et al. (2010) conducted a comprehensive analysis of 9 variable regions of the 16S rRNA gene. The results showed that V1 and V9 had the worst effects, followed by V7 and V8 regions, while V3, V4, and V5 regions were relatively better. In addition, by comparing the regions of V1/V2, V2/V3, V3/V4, V5/V6, and V7/V8 in series, the accuracy of the tandem region V3/V4 and V4/V5 were the highest. Considering the classification efficiency (CE), the V3 / V4 region had significant amplification preference relative to other regions (Claesson et al., 2010). Here, we analyzed the bacterial communities of two Chinese deserts using HTS, and accordingly, modified the media and temperature for isolation and cultivation the targeted actinobacterial strains. Our results provide evidence for a number of actinobacterial strains that are specific for these desert sands and were isolated and identified via cultivation-dependent and -independent analyses. Importantly, these results describe the diversity and composition of actinobacterial communities in different sand types, which has not been systematically investigated in these two deserts (Sun et al., 2015; Li et al., 2016).
Comparative results from the two deserts and the different mico-ecosystems indicated that geographical barrier contributed much more in shaping the bacterial communities than micro-ecological types. On the other side, even in a certain same terrain, the indigenous microorganisms' ecological service function resulted in discriminating micro-ecosystem.
Deserts are typical of extreme, harsh ecosystems, where the availability of water is the cardinal parameter affecting organisms. Consequently, xerophilous microorganisms that are adapted to relatively high temperatures and radiation levels are likely to be the dominant populations in these ecosystems. Actinobacteria are a ubiquitously distributed bacterial phylum inhabiting diverse terrestrial ecosystems. Based on this observation, we approved that Actinobacteria is the predominant microorganisms in desert sand microbial communities using HTS and CULD data. Generally, Actinobacteria, characterized by high genomic GC content, complex peptidoglycan layer containing in the thick cell wall, motility with spore-bearing cells and pigmentation in colonies, may have evolved into some genetic and metabolic functions to help them inhabit in the deserts with drought and high ultraviolet radiation (Mohammadipanah and Wink, 2012).
A total of 4,298 OTUs were obtained using cultivation-independent HTS, indicating a high level of bacterial diversity in these desert environments. Importantly, the cultivation-independent and -dependent analyses provide evidence for valuable microbial resources that warrant further research. In particular, some specific, novel Actinobacteria members and rare microbial groups were identified that could serve as highly valuable microbial resources. OTUs affiliated with the Geodermatophilaceae were frequently detected among the 15 sample types analyzed here, which is consistent with our previous supposition that Geodermatophilaceae are ubiquitous within arid deserts (Sun et al., 2015). Geodermatophilaceae have been detected from various relatively extreme environments, including stone habitats (Salazar et al., 2006; Chouaia et al., 2012; Gtari et al., 2012; Normand et al., 2012), dry hot valleys (Nie et al., 2012), and arid sand from deserts (Montero-Calasanz et al., 2012; Montero-Calasanz C. et al., 2013; Montero-Calasanz M. C. et al., 2013; Saul-Tcherkas et al., 2013), among others. Thus, Geodermatophilaceae strains may be pioneer microorganisms that play a key role in shaping the bacterial community structure in these harsh environments. Furthermore, to some extent, in these desert habits, the microbial community structures may always be doomed from some pioneer population's colonization, where the core groups may promote or inhabit the subsequent members' occurrence and prosper.
Actinoplanes and Kocuria were abundant members of their communities and were also two key genera of the desert sand communities based on co-occurrence network analyses. Interestingly, Actinoplanes and Kocuria were negatively correlated in the co-occurrence network. A large number of Kocuria spp. were isolated from our cultivation assays (21 strains) from sand samples, yet only a few Actinoplanes strains were obtained. In addition, Streptomyces was an abundant component of both the HTS and cultivation assays. However, Streptomyces and Actinoplanes were negatively correlated in the co-occurrence network analysis. These results point to a potentially intriguing negative interaction between Streptomyces, Kocuria and Actinoplanes that may explain the paucity of Actinoplanes isolates that were obtained. Future research should investigate the potential for supplements that inhibit the growth of Streptomyces and Kocuria in order to promote the isolation of Actinoplanes. Regardless, our results suggest the presence of valuable microbial strains, particularly rare community members that are present within these desert sands, and which may hold great significance in understanding the ecology of these systems or in novel microbiological drug research. The analyses here, particularly the co-occurrence networks, provide a framework from which to design isolation media that may promote the growth of these potentially valuable taxa.
YS carried out the experiments and prepared the manuscript. Y-LS helped prepare some experiments. TZ, L-YY, and HW contributed in sampling from deserts. HS helped correct the writing. Y-QZ is responsible for designing the research and preparing the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This research was supported by the National Infrastructure of Microbial Resources (NIMR-2016-3), the CAMS Innovation Fund for Medical Sciences (CIFMS) (2016-I2M-2-002), and the National Natural Science Foundation of China (NSFC 31670010).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01068/full#supplementary-material
Belnap, J., and Lange, O. L. (2003). Biological soil crusts: structure, function, and management. Biol. Conserv. 108, 129–130. doi: 10.1007/978-3-642-56475-8
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chouaia, B., Crotti, E., Brusetti, L., Daffonchio, D., Essoussi, I., Nouioui, I., et al. (2012). Genome sequence of Blastococcus saxobsidens DD2, a stone-inhabiting bacterium. J. Bacteriol. 194, 2752–2753. doi: 10.1128/JB.00320-12
Ciaramella, M. (1995). Molecular biology of extremophiles. World J. Microb. Biot. 11, 71–84. doi: 10.1007/BF00339137
Claesson, M. J., Wang, Q., O'Sullivian, O., Greene-Diniz, R., Cole, J. R., Ross, R. P., et al. (2010). Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res. 38, 985–989. doi: 10.1093/nar/gkq873
Doull, J. L., and Vining, L. C. (1990). Physiology of antibiotic production in actinomycetes and some underlying control mechanisms. Biotechnol. Adv. 8, 141–158. doi: 10.1016/0734-9750(90)90010-9
Evans, R. D., and Johansen, J. R. (1999). Microbiotic crusts and ecosystem processes. Crit. Rev. Plant Sci. 18, 183–225. doi: 10.1080/07352689991309199
Gtari, M., Essoussi, I., Maaoui, R., Sghaier, H., Boujmil, R., Gury, J., et al. (2012). Contrasted resistance of stone-dwelling Geodermatophilaceae species to stresses known to give rise to reactive oxygen species. FEMS Microbiol. Ecol. 80, 566–577. doi: 10.1111/j.1574-6941.2012.01320.x
Hawkes, C. V., and Flechtner, V. R. (2002). Biological soil crustsin a xeric Florida shrubland: composition, abundance, and spatial heterogeneity of crusts with different disturbancehistories. Microb. Ecol. 43, 1–12. doi: 10.1007/s00248-001-1017-5
Jose, P. A., and Jebakumar, S. R. (2013). Non-streptomycete actinomycetes nourish the current microbial antibiotic drug discovery. Front. Microbiol. 4:240. doi: 10.3389/fmicb.2013.00240
Júlia, M. A., Gergely, K., Dóra, A., Károly, M., Balázs, N., and Andrea, K. B. (2016). Diversity of extremophilic bacteria in the sediment of high-altitude lakes located in the mountain desert of Ojos del Salado volcano, Dry-Andes. Extremophiles 20, 603–620. doi: 10.1007/s00792-016-0849-3
Kõljalg, U., Nilsson, R. H., Abarenkov, K., Tedersoo, L., Taylor, A. F., Bahram, M., et al. (2013). Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 22, 5271–5277. doi: 10.1111/mec.12481
Li, J. Y., Zhang, X., Sun, M., and Zhang, Y. L. (2016). Analysis of soil microbial diversity in shapotou area of tengger desert. J. Eco. Rural Environ. 32, 780–787. doi: 10.11934/j.issn.1673-4831.2016.05.014
Mohammadipanah, F., and Wink, J. (2012). Actinobacteria from arid and desert habitats: diversity and biological activity. Front. Microbiol. 6:1541. doi: 10.3389/fmicb.2015.01541
Montero-Calasanz, M. C., Göker, M., Pötter, G., Rohde, M., Spröer, C., Schumann, P., et al. (2012). Geodermatophilus arenarius sp. nov., a xerophilic actinomycete isolated from Saharan desert sand in Chad. Extremophiles 16, 903–909. doi: 10.1007/s00792-012-0486-4
Montero-Calasanz Mdel, C., Göker, M., Pötter, G., Rohde, M., Spröer, C., Schumann, P., et al. (2013). Geodermatophilus africanus sp. nov., a halotolerant actinomycete isolated from Saharan desert sand. Antonie Van Leeuwenhoek 104, 207–216. doi: 10.1007/s10482-013-9939-8
Montero-Calasanz, M. C., Göker, M., Pötter, G., Rohde, M., Spröer, C., Schumann, P., et al. (2013). Geodermatophilus saharensis sp. nov., isolated from sand of the Saharan desert in Chad. Arch. Microbiol. 195, 153–159. doi: 10.1007/s00203-012-0860-8
Navarro-Noya, Y. E., Gómez-Acata, S., Montoya-Ciriaco, N., Rojas-Valdez, A., Suarez-Arriaga, C., Valenzuela-Encinas, M. C., et al. (2013). Relative impacts of tillage, residue management and croprotation on soil bacterial communities in a semiarid agroeco system. Soil Biol. Biochem. 65, 86–95. doi: 10.1016/j.soilbio.2013.05.009
Nie, G. X., Ming, H., Li, S., Zhou, E. M., Cheng, J., Yu, T. T., et al. (2012). Geodermatophilus nigrescens sp. nov., isolated from a dry-hot valley. Antonie Van Leeuwenhoek 101, 811–817. doi: 10.1007/s10482-012-9696-0
Normand, P., Gury, J., Pujic, P., Chouaia, B., Crotti, E., Brusetti, L., et al. (2012). Genome sequence of radio-resistant Modestobacter marinus strain BC501, a representative actinobacterium thriving on calcareous stone surfaces. J. Bacteriol. 194, 4773–4774. doi: 10.1128/JB.01029-12
Phoebe, J. C. H., Combie, J., Albert, F. G., Van, T. K., Cabrera, J., Correira, H. J., et al. (2001). Extremophilic organisms as an unexplored source of antifungal compounds. J. Antibiot. 54, 56–65. doi: 10.7164/antibiotics.54.56
Poulsen, P. H. B., Al-Soud, W. A., Bergmark, L., Magid, J., and Hansen, L. H. (2013). Effects of fertilization with urban and agricultural organic wastes in a field trial – prokaryotic diversity investigated by pyrosequencing. Soil Biol. Biochem. 57, 784–793. doi: 10.1016/j.soilbio.2011.12.023
Robert, C. E., Brain, J. H., Jose, C. C., Christopher, Q., and Rob, K. (2001). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Salazar, O., Valverde, A., and Genilloud, O. (2006). Real-time PCR for the detection and quantification of Geodermatophilaceae from stone samples and identification of new members of the genus Blastococcus. Appl. Environ. Microbiol. 72, 346–352. doi: 10.1128/AEM.72.1.346-352.2006
Saul-Tcherkas, V., Unc, A., and Steinberger, Y. (2013). Soil microbial diversity in the vicinity of desert shrubs. Microbial. Ecol. 65, 689–699. doi: 10.1007/s00248-012-0141-8
Subramani, R., and Aalbersberg, W. (2013). Culturable rare actinomycetes: diversity, isolation and marine natural product discovery. Appl. Microbiol. Biotechnol. 97, 9291–9321. doi: 10.1007/s00253-013-5229-7
Sul, W. J., Asuming-Brempong, S., Wang, Q., Tourlousse, D. M., Penton, R. C., Deng, Y., et al. (2013). Tropical agricultural land management influences on soil microbial communities through its effect on soil organic carbon. Soil Biol. Biochem. 65, 33–38. doi: 10.1016/j.soilbio.2013.05.007
Sun, H. M., Zhang, T., Yu, L. Y., Keya, S., and Zhang, Y. Q. (2015). Ubiquity, diversity and physiological characteristics of Geodermatophilaceae in Shapotou National Desert Ecological Reserve. Front. Microbiol. 6:1059. doi: 10.3389/fmicb.2015.01059
Vikram, S., Guerrero, L. D., Makhalanyane, T. P., Le, P. T., Seely, M., and Cowan, D. A. (2016). Metagenomic analysis provides insights into functional capacity in a hyperarid desert soil niche community. Environ. Microbiol. 18, 1875–1888. doi: 10.1111/1462-2920.13088
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
White, J. R., Nagarajan, N., and Pop, M. (2009). Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 5:e1000352. doi: 10.1371/journal.pcbi.1000352
Wilson, Z. E., and Brimble, M. A. (2009). Molecules derived from the extremes of life. Nat. Prod. Rep. 26, 44–71. doi: 10.1039/B800164M
Wu, R. N., Yu, X. L., Meng, L. S., Xu, X., and Yue, Q. X. Wu, J. R. (2014). PCR-DGGE analysis of the microbial diversity in naturally fermented suan-cai from Northeast China. Mod. Food Sci. Technol. 30, 8–12. doi: 10.13982/j.mfst.1673-9078.2014.10.002
Xu, P., Li, W. J., Xu, L. H., and Jiang, C. L. (2003). Amicrowave–based method for genomic DNA extraction from actinomycetes. Microbiology 30, 82–84. doi: 10.3969/j.issn.0253-2654.2003.04.020
Yan, M. C., Wang, G. Q., Li, B. S., and Dong, G. R. (2001). Formation and growth of high megadunes in Badain Jaran Desert. J. Geogr. Sci. 56, 83–91. doi: 10.3724/SP.J.1148.2008.00304
Yoon, S. H., Ha, S. M., Kwon, S., Lim, J., Kim, Y., Seo, H., et al. (2017). Introducing EzBioCloud: a taxonomically united database of 16S rRNA and whole genome assemblies. Int. J. Syst. Evol. Microbiol. 67, 1613–1617. doi: 10.1099/ijsem.0.001755
Yu, L., Nicolaisen, M., Larsen, J., and Ravnskov, S. (2013). Organic fertilization alters the community composition of root associated fungi in Pisum sativum. Soil Biol. Biochem. 58, 36–41. doi: 10.1016/j.soilbio.2012.11.004
Keywords: Actinobacteria, diversity, microbial community, desert sands, 16S rRNA
Citation: Sun Y, Shi Y-L, Wang H, Zhang T, Yu L-Y, Sun H and Zhang Y-Q (2018) Diversity of Bacteria and the Characteristics of Actinobacteria Community Structure in Badain Jaran Desert and Tengger Desert of China. Front. Microbiol. 9:1068. doi: 10.3389/fmicb.2018.01068
Received: 07 December 2017; Accepted: 04 May 2018;
Published: 23 May 2018.
Edited by:
Sheng Qin, Jiangsu Normal University, ChinaReviewed by:
Kimberley Warren-Rhodes, Ames Research Center, United StatesCopyright © 2018 Sun, Shi, Wang, Zhang, Yu, Sun and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu-Qin Zhang, emh5dXFpbkAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.