 Graciela M. Dias1,2
Graciela M. Dias1,2 Adeline Bidault1
Adeline Bidault1 Patrick Le Chevalier3Gwenaëlle Choquet1
Patrick Le Chevalier3Gwenaëlle Choquet1 Clio Der Sarkissian4Ludovic Orlando4,5Claudine Medigue6
Clio Der Sarkissian4Ludovic Orlando4,5Claudine Medigue6 Valerie Barbe6Sophie Mangenot7
Valerie Barbe6Sophie Mangenot7 Cristiane C. Thompson2
Cristiane C. Thompson2 Fabiano L. Thompson2
Fabiano L. Thompson2 Annick Jacq8
Annick Jacq8 Vianney Pichereau1
Vianney Pichereau1 Christine Paillard1*
Christine Paillard1*- 1Laboratoire des Sciences de l'Environnement Marin, Université de Bretagne Occidentale, UMR 6539 UBO/Centre National de la Recherche Scientifique/IRD/Ifremer, Institut Universitaire Européen de la Mer, Plouzané, France
- 2Laboratório de Microbiologia, Instituto de Biologia, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
- 3Laboratoire de Biotechnologie et Chimie Marine, Université de Bretagne Occidentale, Quimper, France
- 4Centre for GeoGenetics, Natural History Museum of Denmark, University of Copenhagen, Copenhagen, Denmark
- 5Laboratoire d'Anthropobiologie Moléculaire et d'Imagerie de Synthèse, Centre National de la Recherche Scientifique UMR 5288, Université de Toulouse, Université Paul Sabatier, Toulouse, France
- 6CEA, Genoscope, Laboratoire d'Analyses Bioinformatiques pour la Génomique et le Métabolisme, Université d'Evry, Centre National de la Recherche Scientifique-UMR 8030, Evry, France
- 7CEA, Institut de biologie François-Jacob, Genoscope, Laboratoire de Biologie Moléculaire pour l'Etude des Génomes, Evry, France
- 8Institute for Integrative Biology of the Cell, CEA, Centre National de la Recherche Scientifique, Univ. Paris-Sud, Université Paris-Saclay, Gif-sur-Yvette, France
The Brown Ring Disease (BRD) caused high mortality rates since 1986 in the Manila clam Venerupis philippinarum introduced and cultured in Western Europe from the 1970s. The causative agent of BRD is a Gram-Negative bacterium, Vibrio tapetis, which is also pathogenic to fish. Here we report the first assembly of the complete genome of V. tapetis CECT4600T, together with the genome sequences of 16 additional strains isolated across a broad host and geographic range. Our extensive genome dataset allowed us to describe the pathogen pan- and core genomes and to identify putative virulence factors. The V. tapetis core genome consists of 3,352 genes, including multiple potential virulence factors represented by haemolysins, transcriptional regulators, Type I restriction modification system, GGDEF domain proteins, several conjugative plasmids, and a Type IV secretion system. Future research on the coevolutionary arms race between V. tapetis virulence factors and host resistance mechanisms will improve our understanding of how pathogenicity develops in this emerging pathogen.
Introduction
Almost 16 million metric tons of molluscs are cultured each year on a global scale (FishStatJ 2014). In Europe, mollusc aquaculture is a traditional, well-established industry, essential to the socio-economic development of most coastal regions. A number of diseases, however, represent an important threat to this industry (Paillard et al., 2004; Travers et al., 2015). This is the case of the Brown Ring Disease (BRD), which has plagued French aquaculture since 1986, especially the production of Venerupis philippinarum, a species native of Japan and introduced in the 1970s (Flassch and Leborgne, 1992).
BRD develops following infection by the bacterium Vibrio tapetis and is characterized by the formation of a conchiolin deposit between the edge of the mollusc shell and the pallial line (Paillard et al., 1989; Paillard and Maes, 1994). A number of pathogenic strains have been isolated from diseased clams in France, Spain, the United Kingdom, and Norway (Paillard and Maes, 1990; Paillard et al., 1994, 2008; Novoa et al., 1998; Allam et al., 2000b), as well as from other marine animals, in particular fish such as Symphodus melops and Hippoglossus hippoglossus in Norway and Scotland (Jensen et al., 2003; Reid et al., 2003). Additional V. tapetis strains isolated from the fish Dicologoglossa cuneata, Solea solea, Genypterus chilensis, and Paralichthys adspersus have been recently proposed as potential causative agents of emergent diseases in Belgium, Spain, England, and Chile (López et al., 2011; Declercq et al., 2015; Levican et al., 2016).
Several V. tapetis strains have been studied to characterize the mechanisms underlying the bacterial pathogenicity, including virulence factors, but also the host immune response (Allam et al., 2000a, 2001). Toxins, such as haemolysin and cytotoxins, represent the main virulence factors hitherto identified (Borrego et al., 1996) but other proteins related to virulence have also been identified. For example, null mutants of djlA, which encodes an inner membrane chaperone involved in Legionella sp. pathogenesis, display a loss of cytotoxic activity against V. philippinarum hemocytes in vitro (Lakhal et al., 2008). Additionally, VirB4, a component of the Type IV secretion system (Juhas et al., 2008; Christie et al., 2014), is systematically present in the genome of pathogenic V. tapetis strains but absent from non-pathogenic strains (ie. not able to reproduce BRD significantly after in vivo pallial challenge in V. philippinarum) (Bidault et al., 2015).
The complete repertoire of genes associated with V. tapetis pathogenicity remains, however, largely unknown, despite the availability of the full sequence of the CECT4600T pVT1 plasmid (Erauso et al., 2011), the identification of molecules involved in pathogen recognition, biomineralization, and cytoskeleton disruption in the transcriptome of the Manila clam (Brulle et al., 2012; Jeffroy et al., 2013; Allam et al., 2014), and the identification of extracellular enzymes, e.g., endochitinases, lipases, and proteases, in the secretome of V. tapetis (Madec et al., 2014).
Comparative genomics has become a powerful tool to better understand the pathogenic potential of marine microbes (Medini et al., 2008; Haft, 2015), revealing a fraction of the virulence repertoire in some Vibrio genomes (Goudenège et al., 2013; Cordero and Polz, 2014). In the present study, we sequenced and analyzed the first high quality complete genome of a V. tapetis pathogenic strain, i.e., CECT4600, and compared it to 16 additional draft genomes of strains isolated from a broad a range of hosts, geographic locations and sampling times (from 1988 to 2008). Two of these strains, namely HH6087 and RP2-3, were recently released as part of a study exploring the long-term persistence of ancient DNA in mollusc shells (Der Sarkissian et al., 2017). Combining comparative genomic analysis and functional information assessing strain virulence using in vivo and in vitro infection experiments allowed us to further our understanding of the genomic structure and genetic variations underlying V. tapetis pathogenicity.
Materials and Methods
Strains Isolation and Infection Challenge Procedures
The V. tapetis strains used for genomic analysis are described in Table 1. These were mostly isolated from bivalves showing signs of BRD (V. philippinarum, Venerupis decussatus, Polititapes aureus, Polititapes rhomboides, Dosinia exoleta, all belonging to the Veneridae family), or from asymptomatic bivalves (Cerastoderma edule) but living close to V. philippinarum culture areas affected by BRD. All the strains were isolated following the procedures described in Maes and Paillard (1992). Briefly, molluscs were washed with 70% ethanol, dried, and opened aseptically by cutting adductor muscles. The pallial and extrapallial fluids were collected to inoculate Zobell agar plates (4 g/L of peptone, 1 g/L of yeast extract, 0.1 g/L of ferric phosphate, 30 g/L of sea salts, 15 g/L of agar). Additionally, two strains, LP2 and HH6087, were isolated respectively from the posterior kidney of the fish S. melops (Jensen et al., 2003) and from the head kidney samples of H. hippoglossus (Reid et al., 2003). These strains were kindly provided by Ø. Bergh and H. Birkbeck, respectively.
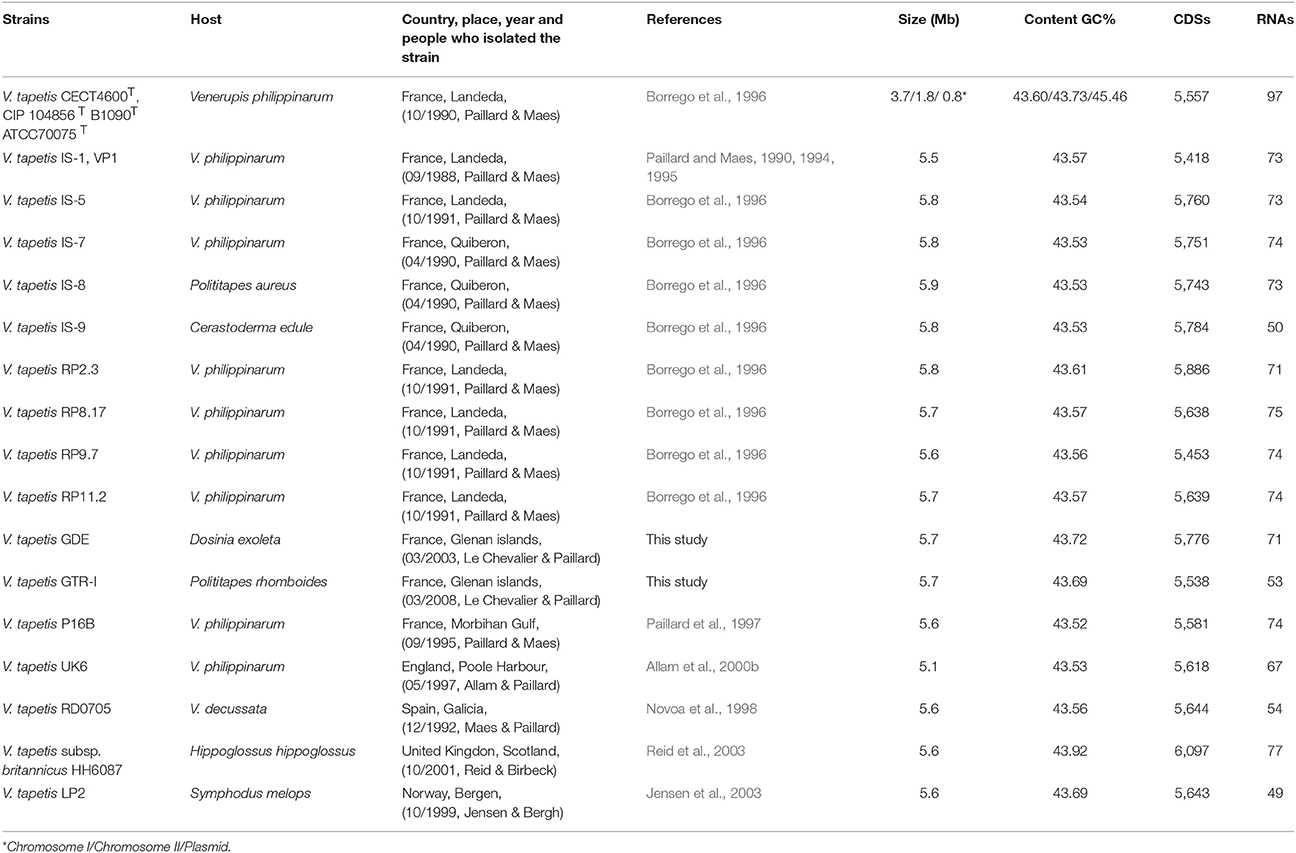
Table 1. General features of Vibrio tapetis group.
Virulence Assays
In vivo pathogenicity tests of V. tapetis strains were carried out using two different inoculation procedures in V. philippinarum. The first procedure was tailored to test the ability of V. tapetis strains to reproduce BRD symptoms. Inoculation was performed following the standardized protocol described by Paillard and Maes (1994). Briefly, a total of 107 bacteria were injected in the pallial cavity of juvenile clams (15–20 mm) that were maintained for 4 weeks, at 14°C, without feeding and water renewal. Each experimental batch consisted of 100 or 50 clams in duplicate. BRD prevalence has been measured 30 days after each V. tapetis strains challenge. In vitro hemocyte cytotoxicity assays were performed as described by Choquet et al. (2003). This test was performed with all the 17 V. tapetis strains at the same time and with the same pool of hemocytes. The non-adherent hemocyte ratio corresponds to the number of non-adherent hemocytes incubated with bacteria divided by the number of hemocytes incubated with filter-sterilized seawater (FSW). A non-adherent cell ratio above 1 represents a cytotoxic effect of the tested bacteria (Choquet et al., 2003). For each of the two pathogenicity tests, V. splendidus (ATCC 25914) and FSW were used in the same conditions (negative controls).
Genome Sequencing and Assembly
For the 17 V. tapetis strains, total DNA was isolated from cultures by lysis of the cells with sodium dodecyl sulfate, EDTA and lysozyme, and extraction was done with a phenol/chloroform solution (Sambrook et al., 1989). The complete genome sequence of V. tapetis CECT4600T was obtained using two sequencing technologies: (1) a Sanger library sequencing leading to a 4-fold coverage; (2) a 454-single read library sequencing leading to a 16-fold coverage (in the framework of the Vibrioscope project, Genoscope, Resps. D. Mazel and F. Le Roux). The draft genome of strain LP2, was also performed at Genoscope using 454 GS FLX Titanium paired end sequencing (in the framework of a partnership project of LEMAR). Each of the 15 remaining V. tapetis strains was processed in the post-PCR facilities of the Center for GeoGenetics, University of Copenhagen, Denmark. A total of 1 μg of DNA was sheared using a Diagenode Bioruptor ultrasonicator (four cycles of: 15 s at high intensity; 90 s off), and built into a blunt-ended DNA library for Illumina shotgun sequencing as in Orlando et al. (2013). We used the NEBNext Quick DNA Library Prep Master Mix Set (New England BioLabs) with the following conditions: 12°C for 20 min then 37°C for 15 min for end-repair, 20°C for 20 min for ligation (0.5 μM Illumina adapter final concentration), 37°C for 20 min then 80°C for 20 min for fill-in. A volume of 12.5 mL library was amplified by PCR in a 50 μL reaction mix containing: 5 units AmpliTaq Gold, 1X Gold Buffer, 4 mM MgCl2 (ThermoFisher), 1 mg/mL BSA, 0.25 mM each dNTP, 0.5 μM Primer inPE1.0 (5′-AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT CTT TCC CTA CAC GAC GCT CTT CCG ATC T-3′), and 0.5 μM Illumina 6 bp-indexed (“I”) primer (5′-CAA GCA GAA GAC GGC ATA CGA GAT III III GTG ACT GGA GTT CAG ACG TGT GCT CTT CCG-3′). Thermo-cycling conditions were: activation at 92°C for 10 min; 7–9 cycles of: denaturation at 92°C for 30 s, annealing at 60°C for 30 s, elongation at 72°C for 30 s; final elongation at 72°C for 7 min. The indexed DNA libraries were pooled and sequenced in single-end mode (94 cycles) on an Illumina HiSeq2500 platform at the Danish National High-Throughput DNA Sequencing Center, with depths of coverage ranging from 50 to 200X. Sequence reads were trimmed by using AdapterRemoval v2.1.5 (Schubert et al., 2016). De novo assembly was performed using the SPAdes assembler (v3.7.0, Bankevich et al., 2012) and four k-mer sizes (21, 33, 55, and 77), allowing mismatch correction. QC was performed using QUAST. Downstream analyses were limited to contigs showing a minimum size of 300 bp.
Genome Annotation, Comparative Genomics, and Core Genome Phylogeny
Expert annotation of the reference strain CECT4600T was done using the MaGe tools of the MicroScope platform (Vallenet et al., 2017). The other 16 draft genomes were then aligned on the complete genome sequence of V. tapetis CECT4600T using ABACAS (Assefa et al., 2009) and annotations were collected from MicroScope (Vallenet et al., 2017). Orthologous clusters were computed using GET_HOMOLOGUES (Contreras-Moreira and Vinuesa, 2013), a software relying on three clustering algorithms. We defined the core genome as the consensus of these three algorithms, using a maximal e-value of 1e-05 and a minimum coverage of 75% in pairwise alignments. The pangenome was defined using the sole OrthoMCl clustering algorithm instead of the three implemented in GET_HOMOLOGUES (Contreras-Moreira and Vinuesa, 2013). Circular comparison of the genomes was carried out to identify the genome architecture and the highly conserved and divergent regions using V. tapetis CECT4600T as reference and the BRIG software with results plotted as series of lanes, all colored according to BLAST alignment of coding sequences (Alikhan et al., 2011). To perform phylogenetic analyses, we retrieved the single-only copy of core genes following alignment of each individual orthologous gene cluster with Muscle v6.1 (Edgar, 2004). The concatenated alignment was used to construct a core genome maximum-likelihood (ML) phylogenetic tree using the LG amino acid evolutionary model with an estimated gamma distribution with four categories using PhyML (Guindon et al., 2010). Robustness of branching was estimated with 1000 bootstrap pseudoreplicates. The resulting dendrogram was visualized using Figtree v. 1.4.2 (http://tree.bio.ed.ac.uk/).
Defining Pan and Core Genome Size
The size of the core genome and the pangenome were estimated following Tettelin et al. (2005), by fitting an exponential decay function on the distribution of shared genes identified after sequential addition of each new genome sequence. The core genome was fitted by the functions Fc = Ω + kcexp[−n/τs] and Fs = tg(θ) + ksexp[−n/ τs], where Ω is the extrapolated core genome size and tg(θ) represents the extrapolated rate of growth of the pangenome size. We adopted the pangenome concept proposed by Koonin and Wolf (2008). In this definition, the distribution of the prokaryotic genes is not limited to two groups only (core and accessory genes), but includes conserved gene core (genomes ≥ 16), the shell of moderately common genes (2 > genomes < 17), and the cloud of genes shared by a small number of organisms (≤2 genomes).
Identifying Type IV Secretion System and Secondary Metabolites
The Type IV secretion system was predicted by CONJscan-T4SSscan (Guglielmini et al., 2011), a tool which scans a set of protein sequences associated and co-localized with VirB4 using HMM profiles. The secondary metabolites biosynthesis gene clusters were identified and annotated using Antismash (Weber et al., 2015).
Genome Accession Numbers
The complete genome of V. tapetis CECT4600T and the draft genomes of all other V. tapetis strains are available from the European Nucleotide Archive (project PRJEB22962).
Results and Discussion
General Features of V. tapetis Genomes
In this study, we generated the complete de novo genome assembly of the V. tapetis strain CECT4600T, and delivered de novo genome draft assemblies for 16 additional V. tapetis strains (genomic data from two of them were previously released in Der Sarkissian et al., 2017), this represents a total of 12 strains isolated from molluscs belonging to the Venerupis genus, three from other molluscs of the Veneridae family (Cerastoderma edule, P. rhomboides, D. exoleta), and two from fish (H. hippoglossus and S. melops). The complete genome of V. tapetis CECT4600T used as the reference consists of two circular chromosomes of 3.7 Mb (chromosome I, %GC = 43.60) and 1.8 Mb (chromosome II, %GC = 43.73), for which a total number of 3,664 and 1,833 open reading frames (ORFs) were predicted, respectively. Additionally, the CECT4600T strain carries the 88 Kb (%GC = 45%) pVT1 plasmid, which presents 88 ORFs (Erauso et al., 2011). All the other 16 V. tapetis strains harbors the pVT1 plasmid. Similar genome sizes (5.5–5.9 Mb) and numbers of predicted genes (5,418–6,097 protein coding DNA sequences, CDS) were observed in the draft genomes of all other strains (Table 1). On average, the genomes of V. tapetis encode 5,678 proteins, and show moderate genetic conservation of ~68%. Orthologous gene clustering revealed that CECT4600T and the strains isolated from clams and cockles (Venerupis, Cerastoderma, and Polititapes genus) share more genes [on average 4,768 out of 5,678 (~83%)] than strains isolated from fish [on average 4,142 out of 5,678 (~72%)]. To obtain a global comparison of the genomes, we constructed a circular blast atlas. V. tapetis genomes were compared by blastn alignment using V. tapetis CECT4600T as the reference. The blast atlas showed the highest conservation of genes between species isolated from clams before year 2000, in comparison to strains isolated from clams after year 2000 and those isolated from fish (Figure 1, red and blue lanes). Interestingly, genome alignments for these strains showed multiple gaps, illustrating that genetic variations are not evenly distributed along the genome, underscoring the importance of genomic rearrangements such as deletion and horizontal gene transfers in evolution (Figure 1).
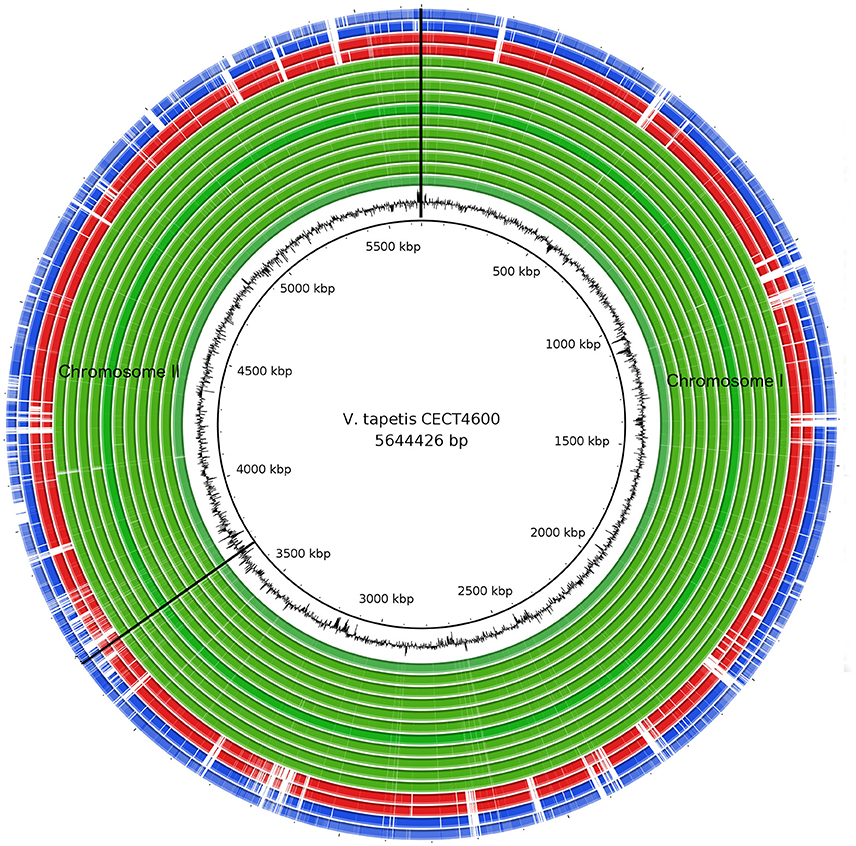
Figure 1. Comparison of V. tapetis genomes by blastn analysis. The inner most circle represents the reference genome V. tapetis CECT4600T. The second internal black circle represents the GC content. The green lanes represent genomes isolated from clams and native cockles before the year 1999 (IS1, IS5, IS7, IS8, IS9, P16B, RD0705, RP2.3, RP8.17, RP11.2, and UK6); red lanes, GDE and GTR-I strains isolated after 2000 from native clams Dosinia exoleta and Polititapes rhomboides, respectively; blue lanes, strains isolated after 1999 (HH6087, LP2) from fish (Hypoglossus hypoglossus, Symphodus melops). Genomic regions corresponding to the absence of genes in a given strain are in white.
Phylogenetic Analysis Based on the Core Genome
We performed a phylogenetic analysis based on the 2,577 single-copy genes present in the V. tapetis core genome (see the pan-/core-genome paragraph below). This showed that the reference strain CECT4600T clustered together with 12 additional strains, namely IS1, IS5, IS7, IS8, IS9, UK6, RD0705, RP2.3, RP8.17, RP9.7, RP11.2, and P16B. This cluster showed limited genetic variation (Figure 2; Figure S1), suggesting a recent common origin for those pathogenic strains that infected clams and cockles prior to 2000 in France, the UK, and in Spain. This is consistent with the phylogenetic tree based on MLSA (Figure S2). A second cluster included 3 more divergent strains (GDE, GTR-I, and LP2), isolated in 1999 and 2000 from a range of hosts. Finally, the HH6087 strain appeared divergent from all other strains. The taxonomic position of this strain has been debated since it was first isolated in 2001 (Reid et al., 2003). In spite of an early assignment as a V. tapetis member based on almost perfect 16S rRNA sequence identity with the CECT4600T strain and additional common biochemical features (Reid et al., 2003), another study on the basis of a 9-gene MLSA showed that both strains were more divergent (Balboa and Romalde, 2013), consistent with our results. DNA-DNA hybridization experiments revealed reassociation values of 69.5–74.8% (Balboa and Romalde, 2013), thus close to the 70% threshold commonly used for species determination (Wayne et al., 1987; Tindall et al., 2010), which supported its description as a new subspecies named V. tapetis subsp. britannicus (Balboa and Romalde, 2013). Additional members of the HH6087 cluster are needed before the taxonomic status of this group as a subspecies or as a new species of its own can be determined.
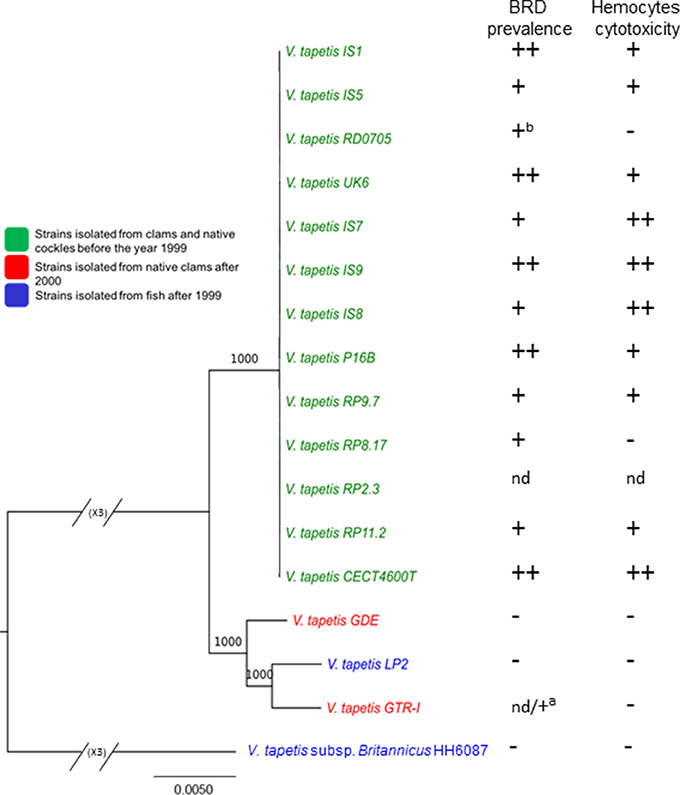
Figure 2. Maximum-likelihood tree of V. tapetis genomes based on single-copy genes of the V. tapetis core genome and pathogenicity assays. V. tapetis strains can be divided in three groups. The first group includes the virulent strains (green), while the two other groups comprise the less virulent strains (red and blue). Note that the branching pattern for the first group cannot be visible on this figure, due to the huge difference with the HH6087 strain; a specific tree for these strains is given as additional data (Figure S1). The numbers at the nodes indicate the levels of bootstrap support based on 1,000 replicates. Hosts and years of isolates are indicated in the legend. On the right of the figure, BRD prevalence in clams (in vivo) after pallial inoculation (average obtained from 1988 to 2007) and hemocyte cytotoxic assay (in vitro) performed in 2014 are given. nd: not determined, aPathogenicity of GTR-I strain after pallial cavity inoculation in Polititapes rhomboides. bObtained from both this study and Novoa et al. (1998). BRD prevalence; (−): average lesser than 20%, (+): 20%<average< 65%, (++): average greater or equal to 65%. Hemocyte cytotoxic assay; (−): ratio ≤1, (+): 1<ratio<1,5, (++): ratio>1,5.
This phylogenetic analyses showing closer ancestry between CECT4600T and clam/cockle strains than fish strains is in agreement with previous results on global genomic comparison of V. tapetis genomes (Figure 1).
Virulence Assays
In order to test the potential virulence of the strains, we used the BRD-induction challenge that appears as the most discriminant in vivo pathogenicity test between the V. tapetis strains. It mimics the natural transmission of bacteria into pallial fluids through the siphons, the adhesion and proliferation of virulent V. tapetis within the shell, and induces secretions linked to BRD development (Paillard, 2016). Using this in vivo BRD induction challenge, all strains isolated between 1988 and 1998 were able to induce BRD development in V. philippinarum, with prevalence rates ranging from 39 to 84% (Figure 2, Figure S3). Strains CECT4600T, IS1, IS9, UK6, and P16B showed the highest BRD prevalence rates (above 70%). The strains isolated from another clam species, D. exoleta (GDE), and from fish (HH6087 and LP2) showed limited prevalence rates (about 15%). It is noteworthy that approximately ~11,500 clams were inoculated for all these assays and the mortality rate was always limited to 3%. Using the in vitro cytotoxic bioassays based on the reduction of adhesion properties of Ruditapes philippinarum hemocytes after V. tapetis contact, the fours trains LP2, HH6087, GDE, and GTR-I showed also low toxicity compared to the other V. tapetis strains (Figure 2). These results are consistent with those already obtained for some strains of V. tapetis and in particular for LP2 (Choquet et al., 2003). In conclusion, virulence assays allowed to show that the four V. tapetis strains LP2, HH6087, GDE, and GTR-I display a weak virulence against the manila clams.
Vibrio tapetis Pangenome and Core Genome Size
We estimated the V. tapetis pangenome, that comprises both the core genome, i.e., the set of genes found across all strains, the variable (flexible) genome consisting of genes absent in one or more strains, and genes unique to some strains. The pangenome and core genome sizes were calculated using the fitting regression model of by Tettelin et al. (2008). As a rule, the number of genes in the core genome depends of the number of samples included in the analysis and the organisms. For example, the core genome of Vibrio vulnificus, calculated from the analysis of 17 genomes, contains 3,068 genes, a number similar to the one we found in V. tapetis (Koton et al., 2015). By contrast, a study based on 42 V. cholerae genomes identified a core genome containing 2,089 genes (Orata et al., 2015). As expected, our core genome size tended to decrease when more genomes were considered in the analysis, with a concomitant increasing importance of the variable genome, the core final size approaching 3,352 genes (Figure 3A). According to the Tettelin model, for a given species, the pangenome can be classified as “open” or “closed,” whether or not its size increases with the addition of genomes to the analysis (Medini et al., 2005; Tettelin et al., 2008). For instance, previous studies showed that the Streptococcus agalactiae pangenome increases on average by 33 genes per genome added to the analysis, and that of Pseudomonas aeruginosa increases by 29 (Tettelin et al., 2008; Mosquera-Rendón et al., 2016). We found that the V. tapetis pangenome increased by an average of 140 genes following the inclusion of one single genome in the analysis (Figure 3B). This indicates that the V. tapetis pangenome is more open than that characterized for important human pathogens, probably pertaining to differences in the spectrum of their respective habitats and environmental exposures. Future studies providing additional V. tapetis genomes are likely to lead to the discovery of additional pangenome genes.
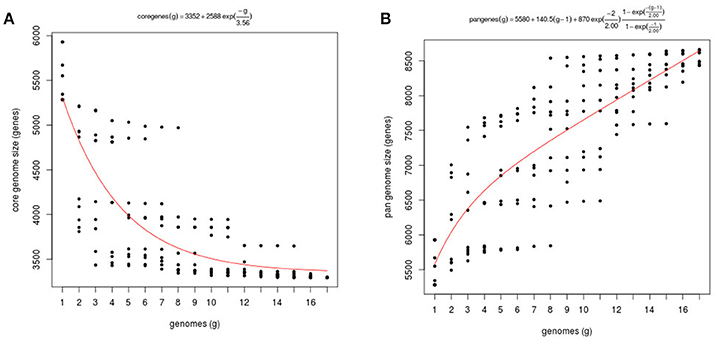
Figure 3. Core and pangenome of V. tapetis. (A) Plot of the exponential decay model of Tettelin et al. (2008) fitted to the core genome data using the OMCL algorithm. (B) Estimation of pangenome size of 17 taxa through function proposed by Tettelin et al. (2005).
Functional Classification of the Core, Cloud, and Shell Genomes
We next carried out a functional analysis of the core, shell, and cloud genomes in order to identify the diversity and composition of the global gene repertoire of V. tapetis strains (see Material and Methods). A total number of 11,213 genes were identified in the V. tapetis pangenome. The core genome contained 3,881 genes, while shell and cloud genomes contained 2,378 and 4,954 genes, respectively (Figure 4). In total, 82% of the genes included in the core genome, as well as 47% of the shell and 30% of the cloud genomes, could be assigned to a COG category. Some COG categories were over-represented in the core genome, such as “Amino acid transport and metabolism” (E), “Coenzyme transport and metabolism” (H), “Energy production and conversion” (C), and “Inorganic ion transport and metabolism” (P). The categories that were over-represented in the shell genome were “Cell Motility” (N), “Intracellular trafficking, secretion and vesicular transport” (U), “Lipid transport and metabolism” (I) and “Transcription” (K), while those pertaining to the cloud genome were “Cell wall/membrane/envelope biogenesis” (M), “Replication/recombination and repair” (L), and “Defense mechanisms” (V). Exploring the genes of each category, we found several genes of the core genome involved in sulfate and phosphate metabolism. We speculate that these could be involved in scavenging sulfate and phosphate ions from the host, thus providing sulfur, phosphate and/or carbon sources, which could facilitate survival within the host. The shell genome included several genes involved in the conjugative transfer of F-plasmid homologs (traALEKBV), several proteins containing GGDEF/EAL domains, proteins involved in the utilization of sialic acid, O-antigen genes, toxin-antitoxin system, and genes involved in biofilm formation (tad locus). Finally, the cloud genome consists mainly of genes related to integrative and conjugative elements, prophages and phage-like elements, transposons, insertion sequences, genes encoding acriflavin resistance and genes involved in Type I restriction-modification system (Tables S2, S3).
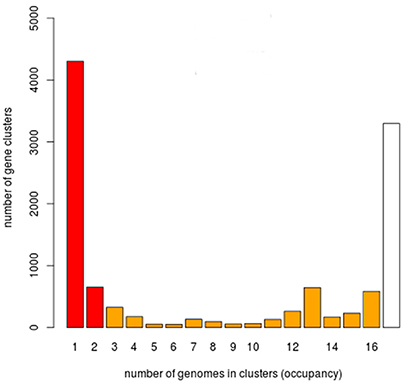
Figure 4. Histogram for the prevalence of genes of the V. tapetis pangenome. 11,213 genes of the V. tapetis pangenome were distributed according to their frequencies across all the strains analyzed. White, orange, and red bars represent the core, shell and cloud genomes, respectively.
Accessing the Differences between the V. tapetis Genomes
Our experimental assays showed that the V. tapetis strains belonging to the first monophyletic cluster (Figure 1) are able to induce the development of BRD with a 75% prevalence on average. By contrast, the average prevalence of BRD induction for GDE, LP2, and HH6087 was more limited (<20%, Figure S3). This supports the hypothesis of two independent origins for the strains, either time of isolation (before and after year 2000), or host species specificity. In order to identify the set of genes common to the most virulent strains, we compared the genomes of the 13 virulent strains (CECT4600T, IS1/5/7/8/9, RP2.2, RP8.17, RP9.7, RP11.2, UK6, RD0705, and P16B) to those of the 4 strains showing lower virulence (GDE, GTR-I, LP2, and HH6087). A total of 444 protein coding genes (24% of which are annotated as hypothetical) were present in the virulent strains and absent in less virulent strains. These included CTX phage elements, Type IV secretion system (T4SS) components (see below), signal transduction proteins such as GGDEF/EAL family proteins, and proteins encoded by regions similar to the pVT1 plasmid. Importantly, V. tapetis strains showing maximal BRD induction levels (Figure S3) display unique putative virulence genes (Table 2), such as genes encoding a Type IV secretion system, genes for toxins (RTX and esterase LpqC), and genes involved in polysaccharide metabolism. The potential contribution of these genes to virulence is discussed in more details below.
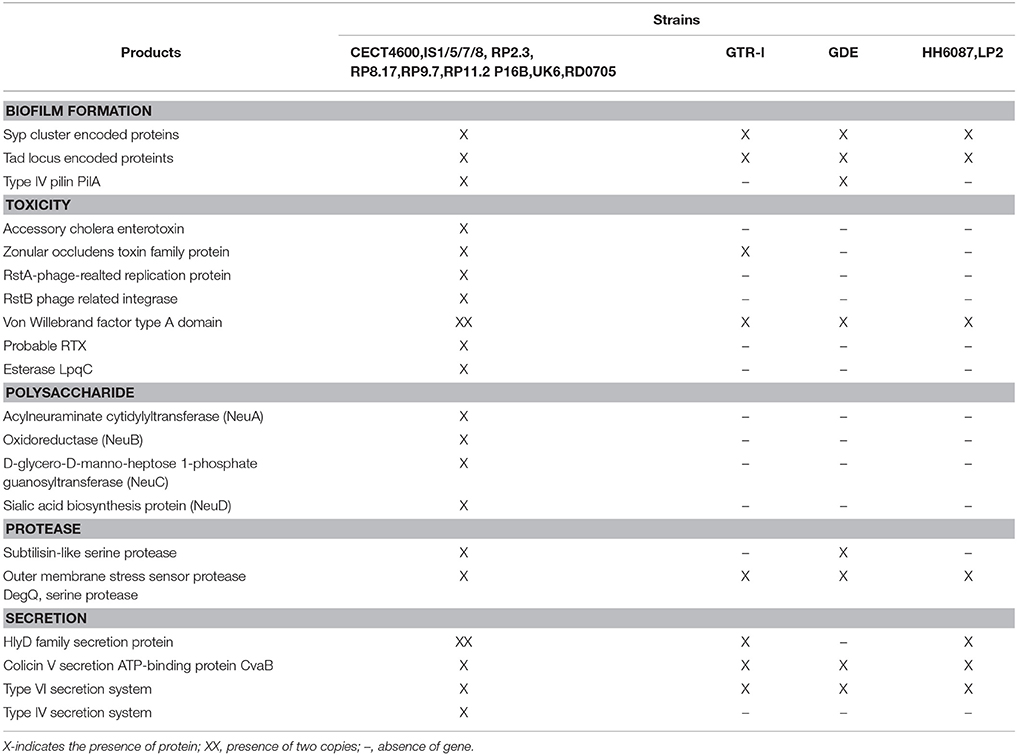
Table 2. Gene products putatively involved in virulence in V. tapetis.
Host Immunity Resistance Genes
Genes related to catabolism and transport of sialic acid were found in several pathogenic bacteria including V. cholerae and V. vulnificus (Almagro-Moreno et al., 2009; Lubin et al., 2012; Kim et al., 2013). These were shown to contribute to provide resistance to components of the host innate immune response and the ability to use such components as a nutrient source (Severi et al., 2007). For all of the 17 V. tapetis strains included in our study, except HH6087 and LP2 strains, we identified putative orthologs to genes involved in the catabolism and transport of sialic acid, in particular nanA, nanK, nanE, and TRAP transporters. We could also identify homologs of the nanH gene, which encodes the sialidase enzyme involved in the release of free sialic acid. At the sequence level, V. tapetis strains, V. cholerae, and V. vulnificus show >80% identity in their sialidase genes. This suggests that most of the analyzed V. tapetis strains have the capacity to degrade and/or capture sialic acids from their hosts in order to escape recognition by the host immune system. This is in agreement with previous studies showing that the transcripts of sialic acid binding lectins are up-regulated during BRD in clams (Allam et al., 2014).
Secondary Metabolites
All V. tapetis strains shared six secondary metabolites biosynthetic gene clusters, except the HH6087 strain, which shows only three clusters. The compounds identified through the antiSMASH prediction tool showed that these clusters should be involved in the synthesis of siderophores, polyketide synthesis (PKS), non-ribosomal peptides synthesis (NRPS), the osmoprotectant ectoine, and polyunsaturated fatty acids (PUFA). The three clusters present in the HH6087 genome contained only clusters related to PUFA, bacteriocin, and siderophore biosynthesis. Finally, one additional cluster related to aryl polyene biosynthesis could be detected only in the IS5, IS7, IS8, and IS9 strains.
Siderophores
The NRPS and NRPS-independent clusters that are responsible for the biosynthesis of vanchrobactin and achromobactin-like siderophores were found in all V. tapetis strains. By contrast, the HH6087 genome did not contain any gene related to vanchrobactin siderophore biosynthesis. The non-ribosomal biosynthetic pathway of vanchrobactin was previously identified in V. anguillarum and V. campbellii species (Balado et al., 2006; Dias et al., 2012). The putative NRPS-independent gene cluster showed similarity (>70%) to the achromobactin biosynthesis cluster of Pseudomonas syringae pv. Syringae (i.e., dimethyl menaquinone methyltransferase, AcsA, AcsB, AcsC, and AscE proteins, Table S1), a pathogen of both monocot and dicot plants (Berti and Thomas, 2009; Ravindran et al., 2015). In P. syringae pv. Syringae mutants defective for the synthesis of achromobactin siderophores exhibit reduced growth in low-iron conditions (Berti and Thomas, 2009). The exact role of siderophores is still unclear in bacteria-bivalve interactions. However, iron is known as one essential minor element in the bivalve shell (Swinehart and Smith, 1979; Zhang et al., 2003). Presumably the ability of most V. tapetis strains to produce and secrete siderophores could induce a competition for iron between bacteria and their hosts. It follows that the presence of siderophores in V. tapetis may confer an advantage in host colonization, which remains to be tested through functional experimental assays.
Polyketide Synthases (PKS)
The presence of PKS genes in both marine animals and microbiota presents a high potential for the production of polyketide secondary metabolites in marine communities/organisms. These metabolites are of paramount importance for human health and the industry, and they have important ecological roles as antibiotics and toxins, and providing chemical defenses against pathogens (Ziemert et al., 2014; Amoutzias et al., 2016). We found a Type-I PKS cluster in all the genomes analyzed, except for the HH6087 strain. This cluster has an average size of 65 Kb, and includes one copy of polyketide synthase (PKs), a non-ribosomal peptide synthetase, transporters, transcriptional regulators, the multidrug toxin extrusion (MATE) family efflux pump YdhE/NorM and a number of thioesterases. We also identified tRNAs at both ends of the cluster, suggesting the possible cluster acquisition through horizontal gene transfer.
Arylpolyene
We found an aryl polyene (APEs) cluster in IS5, IS7, IS8, IS9, RP2.3, RP8.17, RP11.2, and RD0705 strains. The aryl polyene products are responsible for the pigmentation of Gram-negative bacteria where they are widely distributed, including in commensals or pathogens of eukaryotic hosts, and could be involved in the protection against exogenous oxidative stress (Cimermancic et al., 2014). APEs are related to the antioxidative carotenoid group (Schöner et al., 2016). The V. tapetis APE cluster displays 80% similarity to most of the proteins from the V. fischeri APE cluster which confers a yellow pigmentation to wild-type V. fischeri ES114 (Cimermancic et al., 2014). Sequence analysis revealed that the APE cluster is likely to have been introduced into the V. fischeri ES114 genome as a result of a recent horizontal gene transfer. The presence of this cluster in V. tapetis may protect the bacterium from the oxidative stress induced by immune cells during the colonization or infection.
Ectoine Biosynthesis
We found genes related to the ectoine biosynthesis cluster in all strains, except in the HH6087 genome. Ectoine, a cyclic tetrahydropyrimidine (1,4,5,6-tetrahydro-2-methyl-4-pyrimidinecarboxylic acid) can be considered as a marker for halotolerant bacteria. Intracellular osmolytes accumulation in hyperosmotic environments is a growth support and survival strategy in most living organisms, and acts by controlling the osmotic equilibrium in the cells; compatible solutes can be either neosynthesized or imported by various bacteria (Pichereau et al., 2000; Roberts, 2005). Ectoine accumulation confers a competitive advantage in the growth and survival of V. parahaemolyticus and V. cholerae in high osmolarity environments (Pflughoeft et al., 2003; Ongagna-Yhombi and Boyd, 2013), therefore the ability of V. tapetis strains to accumulate ectoine may pertain to an ecological advantage both in the marine environment and during host colonization.
Characterization of the Lipopolysaccharide O-Antigen of V. tapetis
The O-antigen biosynthesis gene cluster was identified in all strains investigated and ranged in size from 38 Kb (GDE) to 76 Kb (CECT4600T). All V. tapetis strains isolated from the Venerupis genus displayed the same genetic organization as those isolated from P. aureus (IS8) and C. edule (IS9), with two main regions separated by a 7.6 Kb region containing ~15 genes encoding hypothetical proteins, a number of mobile elements, and one toxin-antitoxin system. Strikingly, the O-antigen cluster of the remaining V. tapetis strains, namely those forming the second phylogenetic lineage (GDE, GTR-I, and LP2), as well as the more divergent strain HH6087, displayed a different organization (Figure 5). The first and second regions of the O-antigen biosynthesis gene cluster are flanked by the gpm and gmhd genes, and include respectively 32 and 34 genes involved in the production of nucleotide sugars used as building blocks in polysaccharide biosynthesis, glucose, rhamnose, and the unusual legionaminic acid synthesis pathways. The legionaminic acid is a component of lipopolysaccharide in several bacteria and is best known for its presence in the O-antigen of the causative agent of Legionnaires' disease, Legionella pneumophila (Glaze et al., 2008). The legionaminic acid belongs to the NulOs family that includes other acids, such as neuraminic and pseudaminic acids. Biochemical and computational analyses revealed that the production of NulOs is functional in several Vibrio isolates. V. vulnificus clinical isolates expressed higher levels of NulOs than environmental isolates. It was found that role of NulOS in motility, biofilm formation, and pathogenicity to aquatic or terrestrial animals (Lewis et al., 2011). The exact role of the V. tapetis O-antigen is still unknown, but V. fischeri waal deficient mutants show motility defect, resulting in colonization delays (Post et al., 2012). The O-antigen and the core components of lipopolysaccharides are in direct physical interaction with the surrounding substrates and are thus subject to environmental selective pressures. The structural and gene content variation observed within the V. tapetis O-antigen cluster suggests a coevolutionary diversification process in relation to interactions with different hosts.
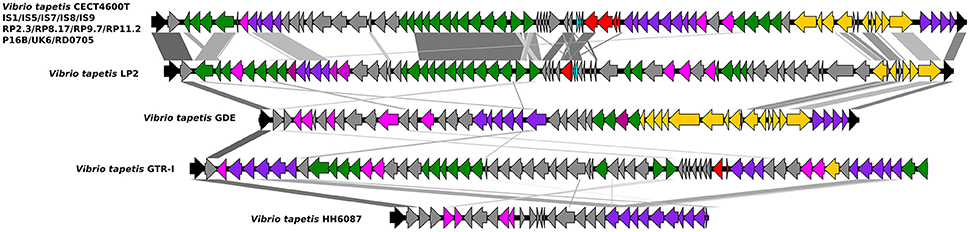
Figure 5. Genetic organization of the O-antigen locus in V. tapetis strains. The region is flanked by the gmhD and gpm genes (black arrows). Positions and orientations of CDSs are indicated with different color, gray arrows: hypothetical proteins and other; acqua arrows: toxin/antitoxin system; red arrows: mobile element proteins; green arrows: genes involved to glycosylation protein and legionaminic acid biosynthesis; yellow arrows: genes involved to capsular polysaccharides biosynthesis and assembly; purple arrows: genes involved to dTDP-rhamnose synthesis and mannose metabolism; fuschia arrows: glycosyltransferases.
Type IV Secretion System (T4SS)
We found a conserved gene cluster that potentially encodes a Type IV secretion system in 13 of the 17 genomes (Table S4) and was absent in the non-virulent strains GDE, GTRI, LP2, and HH6087 (Figure 6). This finding is in line with recent real time PCR experiments targeting T4SS elements in these strains (Bidault et al., 2015). The T4SS cluster of V. tapetis strains consists of the vir operon (virB2, virB3, virB4, virB6, virB8, virb9, virB10, virB11) plus genes of unknown functions. VirB1 is a lytic transglycosylase that degrades the peptidoglycan cell wall at the site of T4SS assembly. VirB2 and VirB5 are pili components, VirB3 and VirB7 pili associated proteins, VirB4 and VirB11 are nucleoside triphosphatases that provide energy for transfer and VirB6, VirB7, VirB8, VirB9, and VirB10 are the protein components of the transfer channel (Juhas et al., 2008). This system is closely related to the well-characterized phytopathogen A. tumefaciens VirB system which mediates the transfer of oncogenic T-DNA into plant cells, inducing tumorous growth of infected plant tissues (Escobar and Dandekar, 2003). However, T4SS can transport a wide variety of compounds from DNA during conjugation to effector proteins, in case of pathogenic bacteria. T4SS have been shown to be essential for virulence in several pathogenic species such as H. pylori, L. pneumophila, or Bordetella pertussis (Voth et al., 2012). The presence of T4SS gene cluster in V. tapetis strains that present a high degree of virulence, and the absence of these genes in strains less virulent, suggest that T4SS have a crucial role in host adaptation and virulence through the delivery of effector proteins in the bivalves.

Figure 6. Organization of the Type IV secretion system locus of V. tapetis strains isolated from different bivalves. The arrow indicates each CDS of the cluster according to function. Cyan arrows: hypothetical proteins; purple arrows: virB4 and virB11; dark pink: virB3, blue: virB2; green: virB6, virB8, virB9, and virB10.
Conclusions
Brown Ring Disease has decimated clam cultures in several European countries since the late 1980s. Previous studies focused on the ecological, physiological, and epidemiological characterization of the disease. This paper provides the first study of the genome organization and sequence of the BRD etiological agent, V. tapetis. We sequenced and analyzed 17 genomes from V. tapetis strains isolated from a broad range of hosts and geographical origins and at different times. We found that the pan-genome of V. tapetis consists of 11,253 genes, and that the core, shell, and cloud genomes contain a diversity of putative virulence factor and secondary metabolite pathway encoding genes. Importantly, V. tapetis strains showing maximal BRD induction levels display unique putative virulence genes such as a Type IV secretion system gene cluster, genes for toxins (RTX) and proteases. Phylogenetic analysis has shown that the strains isolated from clams before year 2000 cluster together within a monophyletic group despite deriving from a wide range of locations. This analysis also identified two additional groups, encompassing strains isolated from clams after the year 2000 and from fish, respectively, with the strain HH6087 being the most divergent, both at the sequence and structural levels. These two groups displayed limited BRD prevalence rates. In addition, a T4SS gene cluster has been identified only in the genomes of V. tapetis strains virulent to V. philippinarum, strongly suggesting a role for this T4SS system in pathogenicity. To the best of our knowledge, this study is the first to describe a complete T4SS gene cluster in a vibrio pathogenic to mollusc.
Author Contributions
CP conceived and coordinated the study. AB performed molecular analyses. CM, VB, and SM performed genome sequencing and assembly of V. tapetis CECT4600 and LP2 strains at Genoscope (Evry, France). AJ performed the expert annotation of V. tapetis CECT4600 reference strain on Mage. CDS and LO performed molecular experiments related to the sequencing of 15 V. tapetis genomes at the Center for GeoGenetics (Copenhagen, Denmark). VP performed the genome assembly of 15 V. tapetis strains. GD performed computational analyses with input from VP, LO, FT, CT, and CP. GC, PLC, and CP performed virulence assays. GD, VP, and CP wrote the article, with great inputs from LO and AJ.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This project received grants from the Region Bretagne, from the CNRS, IFREMER, and MENSR (Biological Resource Center, CRB 2002) “collection of pathogenic bacteria of marine aquaculture species”, the FP7 European project Bivalife (grant agreement N°266157), GIS EUROPOLE MER “VIVREM”, CNRS INEE APEGE “PaleoCOQ”, CAPES-COFECUB “MICROBIV” (Sv 818-14), PVE Brésil (N°03/2014), the cluster of excellence LabexMER (ANR-10-LABX-19), and ArcheoSHELL (Danish Council for Independent Research, Natural Sciences (FNU, 4002-00152B), and H2020 VIVALDI project (grant agreement N°678589). Acknowledgement to the LABGeM and the National Infrastructure “France Genomique” Special thanks to Philippe Maes, who isolated, together with CP, all bivalve V. tapetis strains from 1988 to 1992 in Brittany and Galicia and Jean Louis Nicolas for his inestimable collaboration in microbial ecology. We thank Christelle Ho-Fiat-Hee, Ciptaning Weargo Jati, Marie Filippi, Nolwenn Trinkler, Vanessa Thomann, Fatma Lakhal for the help with in vivo pathogenicity tests; Mathieu Paillard, for preliminary CECT4600T and LP2 bioinformatic analysis; Nelly le Goïc, for CRB collection maintenance; Didier Mazel and Frederique Le Roux for the coordination of the Vibrioscope project, Marcel Koken for his help for DNA extraction of V. tapetis CECT4600 and LP2 Jean-François Auvray (Satmar) for supplying standard clams, and; Morgan Smits for proof reading the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00227/full#supplementary-material
References
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Allam, B., Ashton-Alcox, K. A., and Ford, S. E. (2001). Haemocyte parameters associated with resistance to brown ring disease in Ruditapes spp. clams. Dev. Comp. Immunol. 25, 365–375. doi: 10.1016/S0145-305X(00)00072-0
Allam, B., Paillard, C., and Auffret, M. (2000a). Alterations in hemolymph and extrapallial fluid parameters in the Manila Clam, Ruditapes philippinarum, challenged with the pathogen Vibrio tapetis. J. Invertebr. Pathol. 76, 63–69. doi: 10.1006/jipa.2000.4940
Allam, B., Paillard, C., Howard, A., and Le Pennec, M. (2000b). Isolation of the pathogen Vibrio tapetis and defense parameters in brown ring diseased Manila clams Ruditapes philippinarum cultivated in England. Dis. Aquat. Organ. 41, 105–113. doi: 10.3354/dao041105
Allam, B., Pales Espinosa, E., Tanguy, A., Jeffroy, F., Le Bris, C., and Paillard, C. (2014). Transcriptional changes in Manila clam (Ruditapes philippinarum) in response to Brown Ring Disease. Fish Shellfish Immunol. 41, 2–11. doi: 10.1016/j.fsi.2014.05.022
Almagro-Moreno, S., Boyd, E. F., Vimr, E., Kalivoda, K., Deszo, E., Steenbergen, S., et al. (2009). Insights into the evolution of sialic acid catabolism among bacteria. BMC Evol. Biol. 9:118. doi: 10.1186/1471-2148-9-118
Amoutzias, G., Chaliotis, A., and Mossialos, D. (2016). Discovery strategies of bioactive compounds synthesized by nonribosomal peptide synthetases and type-I polyketide synthases derived from marine microbiomes. Mar. Drugs 14:80. doi: 10.3390/md14040080
Assefa, S., Keane, T. M., Otto, T. D., Newbold, C., and Berriman, M. (2009). ABACAS: algorithm-based automatic contiguation of assembled sequences. Bioinformatics 25, 1968–1969. doi: 10.1093/bioinformatics/btp347
Balado, M., Osorio, C. R., and Lemos, M. L. (2006). A gene cluster involved in the biosynthesis of vanchrobactin, a chromosome-encoded siderophore produced by Vibrio anguillarum. Microbiology 152, 3517–3528. doi: 10.1099/mic.0.29298-0
Balboa, S., and Romalde, J. (2013). Multilocus sequence analysis of Vibrio tapetis, the causative agent of Brown Ring Disease: description of Vibrio tapetis subsp. britannicus subsp. nov. Syst. Appl. Microbiol. 36, 183–187. doi: 10.1016/j.syapm.2012.12.004
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Berti, A. D., and Thomas, M. G. (2009). Analysis of achromobactin biosynthesis by Pseudomonas syringae pv. syringae B728a. J. Bacteriol. 191, 4594–4604. doi: 10.1128/JB.00457-09
Bidault, A., Richard, G. G., Le Bris, C., and Paillard, C. (2015). Development of a Taqman real-time PCR assay for rapid detection and quantification of Vibrio tapetis in extrapallial fluids of clams. Peer J. 3:e1484. doi: 10.7717/peerj.1484
Borrego, J. J., Luque, A., Castro, D., Santamaría, J. A., and Martínez-Manzanares, E. (1996). Virulence factors of Vibrio P1, the causative agent of brown ring disease in the Manila clam, Ruditapes philippinarum. Aquat. Living Resour. 9, 125–136. doi: 10.1051/alr:1996016
Brulle, F., Jeffroy, F., Madec, S., Nicolas, J.-L., and Paillard, C. (2012). Transcriptomic analysis of Ruditapes philippinarum hemocytes reveals cytoskeleton disruption after in vitro Vibrio tapetis challenge. Dev. Comp. Immunol. 38, 368–376. doi: 10.1016/j.dci.2012.03.003
Choquet, G., Soudant, P., Lambert, C., Nicolas, J. L., and Paillard, C. (2003). Reduction of adhesion properties on Ruditapes philippinarum hemocytes exposed to Vibrio tapetis. Dis. Aquat. Org. 57, 109–116. doi: 10.3354/dao057109
Christie, P. J., Whitaker, N., and González-Rivera, C. (2014). Mechanism and structure of the bacterial type IV secretion systems. Biochim. Biophys. Acta. 1843, 1578–1591. doi: 10.1016/j.bbamcr.2013.12.019
Cimermancic, P., Medema, M. H., Claesen, J., Kurita, K., Wieland Brown, L. C., Mavrommatis, K., et al. (2014). Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell 158, 412–421. doi: 10.1016/j.cell.2014.06.034
Contreras-Moreira, B., and Vinuesa, P. (2013). GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 79, 7696–7701. doi: 10.1128/AEM.02411-13
Cordero, O. X., and Polz, M. F. (2014). Explaining microbial genomic diversity in light of evolutionary ecology. Nat. Rev. Microbiol. 12, 263–273. doi: 10.1038/nrmicro3218
Declercq, A., Chiers, K., Soetaert, M., Lasa, A., Romalde, J., Polet, H., et al. (2015). Vibrio tapetis isolated from vesicular skin lesions in Dover sole Solea solea. Dis. Aquat. Organ. 115, 81–86. doi: 10.3354/dao02880
Der Sarkissian, C., Pichereau, V., Dupont, C. T., Schiøtte, P. C., Ilsøe, M., Perrigault, P., et al. (2017). Ancient DNA analysis identifies marine mollusc shells as new metagenomic archives of the past. Mol. Ecol. Resour. 17, 835–853. doi: 10.1111/1755-0998.12679.
Dias, G. M., Thompson, C. C., Fishman, B., Naka, H., Haygood, M. G., Crosa, J. H., et al. (2012). Genome sequence of the marine bacterium Vibrio campbellii DS40M4, isolated from open ocean water. J. Bacteriol. 194:904. doi: 10.1128/JB.06583-11
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Erauso, G., Lakhal, F., Bidault-Toffin, A., Le Chevalier, P., Bouloc, P., Paillard, C., et al. (2011). Evidence for the role of horizontal transfer in generating pVT1, a large mosaic conjugative plasmid from the clam pathogen, Vibrio tapetis. PLoS ONE 6:e16759. doi: 10.1371/journal.pone.0016759
Escobar, M. A., and Dandekar, A. M. (2003). Agrobacterium tumefaciens as an agent of disease. Trends Plant Sci. 8, 380–386. doi: 10.1016/S1360-1385(03)00162-6
Flassch, J. P. and Leborgne, Y. (1992). Introduction in Europe, from 1972 to 1980; of the Japanese manila clam (Tapes philippinarum) and effects on aquaculture production and natural settlement. ICES Mar. Sci. Symp. 194, 92–96.
Glaze, P. A., Watson, D. C., Young, N. M., and Tanner, M. E. (2008). Biosynthesis of CMP- N, N ′-diacetyllegionaminic acid from UDP- N, N ′-diacetylbacillosamine in Legionella pneumophila. Biochemistry 47, 3272–3282. doi: 10.1021/bi702364s
Goudenège, D., Labreuche, Y., Krin, E., Ansquer, D., Mangenot, S., Calteau, A., et al. (2013). Comparative genomics of pathogenic lineages of Vibrio nigripulchritudo identifies virulence-associated traits. ISME J. 7, 1985–1996. doi: 10.1038/ismej.2013.90
Guglielmini, J., Quintais, L., Garcillán-Barcia, M. P., de la Cruz, F., and Rocha, E. P. C. (2011). The repertoire of ICE in prokaryotes underscores the unity, diversity, and ubiquity of conjugation. PLoS Genet. 7:e1002222. doi: 10.1371/journal.pgen.1002222
Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Haft, D. H. (2015). Using comparative genomics to drive new discoveries in microbiology. Curr. Opin. Microbiol. 23, 189–196. doi: 10.1016/j.mib.2014.11.017
Jeffroy, F., Brulle, F., and Paillard, C. (2013). Differential expression of genes involved in immunity and biomineralization during Brown Ring Disease development and shell repair in the Manila clam, Ruditapes philippinarum. J. Invertebr. Pathol. 113, 129–136. doi: 10.1016/j.jip.2013.03.001
Jensen, S., Samuelsen, O., Andersen, K., Torkildsen, L., Lambert, C., Choquet, G., et al. (2003). Characterization of strains of Vibrio splendidus and V. tapetis isolated from corkwing wrasse Symphodus melops suffering vibriosis. Dis. Aquat. Organ. 53, 25–31. doi: 10.3354/dao053025
Juhas, M., Crook, D. W., and Hood, D. W. (2008). Type IV secretion systems: tools of bacterial horizontal gene transfer and virulence. Cell Microbiol. 10, 2377–2386. doi: 10.1111/j.1462-5822.2008.01187.x
Kim, I. H., Son, J., Wen, Y., Jeong, S., Min, G., Park, N., et al. (2013). Transcriptomic analysis of genes modulated by cyclo((L-Phenylalanine- L-Proline) in Vibrio vulnificus. J. Microbiol. Biotechnol. 23, 1791–1801. doi: 10.4014/jmb.1308.08068
Koonin, E. V., and Wolf, Y. I. (2008). Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 36, 6688–6719. doi: 10.1093/nar/gkn668
Koton, Y., Gordon, M., Chalifa-Caspi, V., and Bisharat, N. (2015). Comparative genomic analysis of clinical and environmental Vibrio vulnificus isolates revealed biotype 3 evolutionary relationships. Front. Microbiol. 5:803. doi: 10.3389/fmicb.2014.00803
Lakhal, F., Bury-Moné, S., Nomane, Y., Le Goïc, N., Paillard, C., and Jacq, A. (2008). DjlA, a membrane-anchored DnaJ-like protein, is required for cytotoxicity of clam pathogen Vibrio tapetis to hemocytes. Appl. Environ. Microbiol. 74, 5750–5758. doi: 10.1128/AEM.01043-08
Levican, A., Lasa, A., Irgang, R., Romalde, J. L., Poblete-Morales, M., and Avenda-o-Herrera, R. (2016). Isolation of Vibrio tapetis from two native fish species (Genypterus chilensis and Paralichthys adspersus) reared in Chile and description of Vibrio tapetis subsp. quintayensis subsp. nov. Int. J. Syst. Evol. Microbiol. 67, 716–723. doi: 10.1099/ijsem.0.001705
Lewis, A. L., Lubin, J.-B., Argade, S., Naidu, N., Choudhury, B., and Boyd, E. F. (2011). Genomic and metabolic profiling of nonulosonic acids in Vibrionaceae reveal biochemical phenotypes of allelic divergence in Vibrio vulnificus. Appl. Environ. Microbiol. 77, 5782–5793. doi: 10.1128/AEM.00712-11
López, J. R., Balboa, S., Núñez, S., de la Roca, E., de la Herran, R., Navas, J. I., et al. (2011). Characterization of Vibrio tapetis strains isolated from diseased cultured Wedge sole (Dicologoglossa cuneata Moreau). Res. Vet. Sci. 90, 189–195. doi: 10.1016/j.rvsc.2010.05.030
Lubin, J.-B., Kingston, J. J., Chowdhury, N., and Boyd, E. F. (2012). Sialic acid catabolism and transport gene clusters are lineage specific in Vibrio vulnificus. Appl. Environ. Microbiol. 78, 3407–3415. doi: 10.1128/AEM.07395-11
Madec, S., Pichereau, V., Jacq, A., Paillard, M., Boisset, C., Guérard, F., et al. (2014). Characterization of the secretomes of two vibrios pathogenic to mollusks. PLoS ONE 9:e113097. doi: 10.1371/journal.pone.0113097
Maes, P., and Paillard, C. (1992). “Effect du Vibrio P1, pathogene de Ruditapes philippinarum, sur d'autres especes de bivalves,” in Les Mollusques Marins: Biologie et Aquaculture (Brest).
Medini, D., Donati, C., Tettelin, H., Masignani, V., and Rappuoli, R. (2005). The microbial pan-genome. Curr. Opin. Genet. Dev. 15, 589–594. doi: 10.1016/j.gde.2005.09.006
Medini, D., Serruto, D., Parkhill, J., Relman, D. A., Donati, C., Moxon, R., et al. (2008). Microbiology in the post-genomic era. Nat. Rev. Microbiol. 6, 419–430. doi: 10.1038/nrmicro1901
Mosquera-Rendón, J., Rada-Bravo, A. M., Cárdenas-Brito, S., Corredor, M., Restrepo-Pineda, E., and Benítez-Páez, A. (2016). Pangenome-wide and molecular evolution analyses of the Pseudomonas aeruginosa species. BMC Genomics 17:45. doi: 10.1186/s12864-016-2364-4
Novoa, B., Luque, A., Castro, D., Borrego, J., and Figueras, A. (1998). Characterization and infectivity of four bacterial strains isolated from brown ring disease-affected clams. J. Invertebr. Pathol. 71, 34–41. doi: 10.1006/jipa.1997.4704
Ongagna-Yhombi, S. Y., and Boyd, E. F. (2013). Biosynthesis of the osmoprotectant ectoine, but not glycine betaine, is critical for survival of osmotically stressed Vibrio parahaemolyticus cells. Appl. Environ. Microbiol. 79, 5038–5049. doi: 10.1128/AEM.01008-13
Orata, F. D., Kirchberger, P. C., Méheust, R., Barlow, E. J., Tarr, C. L., and Boucher, Y. (2015). The dynamics of genetic interactions between Vibrio metoecus and Vibrio cholerae, two close relatives co-occurring in the environment. Genome Biol. Evol. 7, 2941–2954. doi: 10.1093/gbe/evv193
Orlando, L., Ginolhac, A., Zhang, G., Froese, D., Albrechtsen, A., Stiller, M., et al. (2013). Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature 499, 74–78. doi: 10.1038/nature12323
Paillard, C. (2016). “An ecological approach to understanding host-pathogen-environment interactions: the case of Brown Ring Disease in clams,” in Oysters and Clams: Cultivation, Habitat Threats, and Ecological Impact, ed J. L. Romalde (Hauppauge, NY: Nova Science Publishers), 97–112.
Paillard, C., Korsnes, K., Le Chevalier, P., Le Boulay, C., Harkestad, L., Eriksen, A., et al. (2008). Vibrio tapetis-like strain isolated from introduced Manila clams Ruditapes philippinarum showing symptoms of brown ring disease in Norway. Dis. Aquat. Organ. 81, 153–161. doi: 10.3354/dao01950
Paillard, C., Le Roux, F., and Borrego, J. J. (2004). Bacterial disease in marine bivalves, a review of recent studies: trends and evolution. Aquat. Living Resour. 17, 477–498. doi: 10.1051/alr:2004054
Paillard, C., and Maes, P. (1990). Etiologie de la maladie l'anneau brun chez Tapes philippinarum: pathogenicite d'un Vibrio sp. Comptes rendus l'Académie des Sci. 310, 15–20.
Paillard, C., and Maes, P. (1994). Brown ring disease in the Manila clam Ruditapes philippinarum: establishment of a classification svstem. Dis. Aquat. Organ. 19, 137–146. doi: 10.3354/dao019137
Paillard, C., and Maes, P. (1995). The brown ring disease in the manila clam, Ruditapes philippinarum. Part 2. Microscopic study of the brown ring symptom. J. Invertebr. Pathol. 65, 101–110. doi: 10.1006/jipa.1995.1016
Paillard, C., Maes, P., Mazurie, J., Claude, S., Marhic, A., and Le Pennec, M. (1997). “Epidemiological survey of the brown ring disease in clams of Atlantic coast: role of temperature in variations of prevalence,” in Proccedings of VIIIe Symposium of the International Society for Veterinary Epidemiology and Economics (Paris: AEEMA publications), 14.03.1–14.03.3.
Paillard, C., Maes, P., and Oubella, R. (1994). Brown ring disease in clams. Annu. Rev. Fish Dis. 4, 219–240. doi: 10.1016/0959-8030(94)90030-2
Paillard, C., Perceley, L., Le Penec, M., and Le Picard, D. (1989). Origine pathogène de l'anneau brun chez Tapes philippinarum (Mollusque, bivalve). Comptes rendus l'Académie des Sci. Série 3, Sci. 309, 235–241.
Pflughoeft, K. J., Kierek, K., and Watnick, P. I. (2003). Role of ectoine in Vibrio cholerae osmoadaptation. Appl. Environ. Microbiol. 69, 5919–5927. doi: 10.1128/AEM.69.10.5919-5927.2003
Pichereau, V., Hartke, A., and Auffray, Y. (2000). Starvation and osmotic stress induced multiresistances. Int. J. Food Microbiol. 55, 19–25. doi: 10.1016/S0168-1605(00)00208-7
Post, D. M. B., Yu, L., Krasity, B. C., Choudhury, B., Mandel, M. J., Brennan, C. A., et al. (2012). O-antigen and core carbohydrate of Vibrio fischeri lipopolysaccharide: composition and analysis of their role in Euprymna scolopes light organ colonization. J. Biol. Chem. 287, 8515–8530. doi: 10.1074/jbc.M111.324012
Ravindran, A., Jalan, N., Yuan, J. S., Wang, N., and Gross, D. C. (2015). Comparative genomics of Pseudomonas syringae pv. syringae strains B301D and HS191 and insights into intrapathovar traits associated with plant pathogenesis. Microbiologyopen 4, 553–573. doi: 10.1002/mbo3.261
Reid, H. I., Duncan, H. L., Laidler, L. A., Hunter, D., and Birkbeck, T. H. (2003). Isolation of Vibrio tapetis from cultivated Atlantic halibut (Hippoglossus hippoglossus L.). Aquaculture 221, 65–74. doi: 10.1016/S0044-8486(03)00060-7
Roberts, M. F. (2005). Organic compatible solutes of halotolerant and halophilic microorganisms. Saline Syst. 1:5. doi: 10.1186/1746-1448-1-5
Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). Molecular Cloning: A Laboratory Manual, 2nd Edn. New York, NY: old Spring Harbor Laboratory; Cold Spring Harbor.
Schubert, M., Lindgreen, S., and Orlando, L. (2016). AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes 9:88. doi: 10.1186/s13104-016-1900-2
Schöner, T. A., Gassel, S., Osawa, A., Tobias, N. J., Okuno, Y., Sakakibara, Y., et al. (2016). Aryl polyenes, a highly abundant class of bacterial natural products, are functionally related to antioxidative carotenoids. ChemBioChem. 17, 247–253. doi: 10.1002/cbic.201500474
Severi, E., Hood, D. W., and Thomas, G. H. (2007). Sialic acid utilization by bacterial pathogens. Microbiology 153, 2817–2822. doi: 10.1099/mic.0.2007/009480-0
Swinehart, J. H., and Smith, K. W. (1979). Iron and Managanese deposition in the periostraca of several bivale molluscs. Biol. Bull. 156, 369–381. doi: 10.2307/1540924
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955. doi: 10.1073/pnas.0506758102
Tettelin, H., Riley, D., Cattuto, C., and Medini, D. (2008). Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 11, 472–477. doi: 10.1016/j.mib.2008.09.006
Tindall, B. J., Rossello-Mora, R., Busse, H. J., Ludwig, W., Kampfer, P., et al. (2010). Notes on the characterization of prokaryote strains for taxonomic. Int. J. Syst. Evol. Microbiol. 60, 249–266. doi: 10.1099/ijs.0.016949-0
Travers, M.-A., Boettcher Miller, K., Roque, A., and Friedman, C. S. (2015). Bacterial diseases in marine bivalves. J. Invertebr. Pathol. 131, 11–31. doi: 10.1016/j.jip.2015.07.010
Vallenet, D., Calteau, A., Cruveiller, S., Gachet, M., Lajus, A., Josso, A., et al. (2017). MicroScope in 2017: an expanding and evolving integrated resource for community expertise of microbial genomes. Nucleic Acids Res. 45, D517–D528. doi: 10.1093/nar/gkw1101
Voth, D. E., Broederdorf, L. J., and Graham, J. G. (2012). Bacterial type IV secretion systems: versatile virulence machines. Future Microbiol. 7, 241–257. doi: 10.2217/fmb.11.150
Wayne, G., Brenner, R., Colwell, R., Grimont, P., Kandler, O., Krichevsky, M. I., et al. (1987). Report of the Ad Hoc committee on reconciliation of approaches to bacterial systematics. Inte. J. Syst. Bacteriol. 37, 463–464. doi: 10.1099/00207713-37-4-463
Weber, T., Blin, K., Duddela, S., Krug, D., Kim, H. U., Bruccoleri, R., et al. (2015). antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 43, W237–W243. doi: 10.1093/nar/gkv437
Zhang, Y., Meng, Q., Jiang, T., Wang, H., Xie, L., and Zhang, R. (2003). A novel ferritin subunit involved in shell formation from the pearl oyster (Pinctada fucata). Comp. Biochem. Physiol. B Biochem. Mol. Biol. 135, 43–54. doi: 10.1016/S1096-4959(03)00050-2
Keywords: Vibrio tapetis, Venerupis philippinarum, comparative genomics, pathogenicity, T4SS, pangenome, core genome
Citation: Dias GM, Bidault A, Le Chevalier P, Choquet G, Der Sarkissian C, Orlando L, Medigue C, Barbe V, Mangenot S, Thompson CC, Thompson FL, Jacq A, Pichereau V and Paillard C (2018) Vibrio tapetis Displays an Original Type IV Secretion System in Strains Pathogenic for Bivalve Molluscs. Front. Microbiol. 9:227. doi: 10.3389/fmicb.2018.00227
Received: 21 October 2017; Accepted: 30 January 2018;
Published: 19 February 2018.
Edited by:
Feng Gao, Tianjin University, ChinaReviewed by:
Stephen Lloyd Michell, University of Exeter, United KingdomChih-Horng Kuo, Academia Sinica, Taiwan
Copyright © 2018 Dias, Bidault, Le Chevalier, Choquet, Der Sarkissian, Orlando, Medigue, Barbe, Mangenot, Thompson, Thompson, Jacq, Pichereau and Paillard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christine Paillard, cGFpbGxhcmRAdW5pdi1icmVzdC5mcg==