 Pooja Singh
Pooja Singh Selvakumar Subbian
Selvakumar Subbian
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 30 January 2018
Sec. Antimicrobials, Resistance and Chemotherapy
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.00070
This article is part of the Research Topic Alternative Therapeutics Against Antimicrobial-Resistant Pathogens View all 27 articles
Tuberculosis (TB) remains as one of the leading killer infectious diseases of humans. At present, the standard therapeutic regimen to treat TB comprised of multiple antibiotics administered for a minimum of six months. Although these drugs are useful in controlling TB burden globally, they have not eliminated the disease. In addition, the lengthy duration of treatment with multiple drugs contributes to patient non-compliance that can result in the development of drug resistant strains (MDR and XDR) of Mycobacterium tuberculosis (Mtb), the causative agent of TB. Therefore, new and improved therapeutic strategies are urgently needed for effective control of TB worldwide. The intracellular survival of Mtb is regarded as a cumulative effect of the host immune response and the bacterial ability to resist or subvert this response. When the host innate defensive system is manipulated by Mtb for its survival and dissemination, the host develops disease conditions that are hard to overcome. The host intrinsic factors also contributes to the poor efficacy of anti-mycobacterial drugs and to the emergence of drug resistance. Hence, strengthening the immune repertoire involved in combating Mtb through host-directed therapeutics (HDT) can be one of the approaches for effective bacterial killing and clearance of infection/disease. Recently, more scientific research has been focused toward HDT strategies that empowers host cells for effective killing of Mtb, reduce the duration of treatment and/or alleviates the development of MDR/XDR, since Mtb cannot develop resistance against a drug that targets the host cell function. Autophagy is a conserved cellular process critical for maintaining cellular integrity and function. Autophagy is regulated by multiple pathways that are either dependent or independent of mTOR (mechanistic target of rapamycin; a.k.a. mammalian target of rapamycin), a master regulatory molecules that impacts several cellular functions. In this review, we summarize the role of autophagy in Mtb pathogenesis, the mTOR pathway and, modulating the mTOR pathway with inhibitors as potential adjunctive HDT, in combination with standard anti-TB antibiotics, to improve the outcome of current TB treatment.
Tuberculosis (TB) is one of the leading killer among infectious diseases of humans, accounting for about 10.4 million new cases and 1.8 million deaths in 2015 (World Health Organization, 2016). The global burden of TB has also been exacerbated by other co-morbid conditions, including diabetes and HIV-infection, and TB is a leading cause of mortality among HIV infected individuals with nearly 400,000 deaths reported in 2015. The standard therapeutic regimen recommended by the WHO for treating drug-sensitive pulmonary TB, known as DOTS (Directly Observed Treatment, Short course), is comprised of four antibiotics: isoniazid (INH), rifampicin (RIF), pyrazinamide (PZA) and ethambutol (ETH) for 2 months (initial phase) followed by INH, and RIF for 4 months (continuation phase). This multi-drug regimen is essential and necessary to ensure successful bacteriological cure in patients with TB. Although these drugs are useful in controlling the overall disease burden at the level of individual patients as well as global TB control measures, they have not eliminated the disease at both these levels (Ryan, 1992). This is in part due to the lengthy duration of treatment with multiple drugs, which promotes the fear of drug dependency and doubts of not getting cured and contributes to drug-induced tissue toxicity issues. Adverse effects, ranging from serious ones, like hepatitis and pneumonia, to minor ones, like vomiting, acne and nausea, have been reported to be associated with DOTs therapy (Michael et al., 2016). Thus, high drop-out rate of TB patients from treatment regimens (a.k.a. patient non-compliance) is a serious issue contributing directly to the development of drug resistance in Mycobacterium tuberculosis (Mtb), the causative agent of TB.
Development of drug resistance in a single bacterium has been suggested to be sufficient to create an outbreak of drug resistant bacteria (Borrell and Gagneux, 2009). In 2015, nearly 4.8 million cases of isoniazid- and rifampicin-resistant [a.k.a. multidrug-resistant TB (MDR-TB)] cases were reported. In addition to INH and RIF (the first line drugs), Mtb can develop resistance to PZA and ETH (second line drugs) and other injectable aminoglycosides, leading to extensively drug-resistant TB (XDR-TB) cases. Nearly 9.5 % of all MDR-TB cases in 2015 were estimated to be XDR-TB. A recent study aimed at predicting the future burden of TB suggests an increased prevalence of MDR and XDR cases due mainly to person-to-person transmission of drug-resistant Mtb, rather than the pathogen acquiring drug resistance within the infected host (Sharma et al., 2017). Hence, current treatment strategies demand intense patient monitoring during and after drug treatment, which poses major strategical and economical challenges for the global TB control programs conducted by various health agencies. Therefore, it is imperative that new anti-TB therapies are developed and implemented to shorten the number of antibiotics taken and/or duration of treatment, to lower the drug- induced toxicities, and to improve the drug efficacy among TB patients with co-morbid conditions, such as HIV-infection and/or patients with MDR/XDR-TB.
Development of drug resistance among infecting Mtb is also dependent on host intrinsic factors, such as genetic make-up, health, and well-being, all of which impact the immune response against the bacteria. A key component of the host innate defense system are macrophages, phagocytic cells that engulf and destroy infecting microorganisms. However, Mtb can “invade” macrophages (and other host cells), where it is able to survive, proliferate and cause infection/disease. Invasion of macrophages by Mtb brings changes to the normal phagocytosis events, such as calcium ion homeostasis, membrane protein distribution and phagosome-lysosome fusion. If/when Mtb survives, it continues to multiply intracellularly and induce a pro-inflammatory response, leading to the onset of cell mediated/adaptive immunity and granuloma formation, which is generally thought of as a region of equilibrium between the host and the bacterium. For Mtb, the granuloma serves as an environment where the bacteria can exist in a dormant, semi-and/or non-replicating state. For the host, the granulomas restrict the spread of Mtb to other tissues/organs because the diseased area is cordoned-off by the activated immune cells (Guirado et al., 2013). The host-pathogen interactions in the granuloma are highly complex, where the bacteria may get killed or able to survive and persist (Flynn and Chan, 2003). Taken together, the intracellular survival of Mtb is regarded as a cumulative effect of the host immune response and the bacterial ability to resist or subvert this response. Hence, strengthening the immune repertoire involved in combating Mtb through host-directed therapeutics (HDT) can be one of the approaches for effective bacterial killing and clearance of infection/disease.
Host directed therapy (HDT) aims at manipulating the metabolism and/or immune cell function to optimize the pro-inflammatory response or to modify the tissue physiology (Subbian et al., 2011a,b; Tobin et al., 2012). Recently, research on HDT as potential therapeutic strategy for infectious diseases has gained significant momentum due to the possibility of re-purposing drugs that have been already approved to treat chronic ailments and the advantage that pathogenic bacteria, such as Mtb, cannot develop resistance against a HDT, which targets host cell functions (Zumla et al., 2015). Autophagy is a homeostatic cellular process that removes intracellular debris derived from endo-and exo-genous sources, thus ensuring efficient functioning of cells. It is also a key innate immune response of the host cells to protect against invading pathogens. Therefore, targeting the autophagy machinery using small molecules and drugs to improve the host cell effector functions is an emerging concept in the treatment of several chronic diseases (Rubinsztein et al., 2012). Autophagy is regulated by multiple, complex networks and pathways that are either dependent or independent of mTOR (mechanistic target of rapamycin; a.k.a. mammalian target of rapamycin), a master regulatory molecule that impacts several cellular functions (Figure 1). In this review, we focus mainly on the role of autophagy in Mtb pathogenesis and modulating the mTOR pathway as potential adjunctive HDT to improve current, antibiotic-based treatment for pulmonary TB.
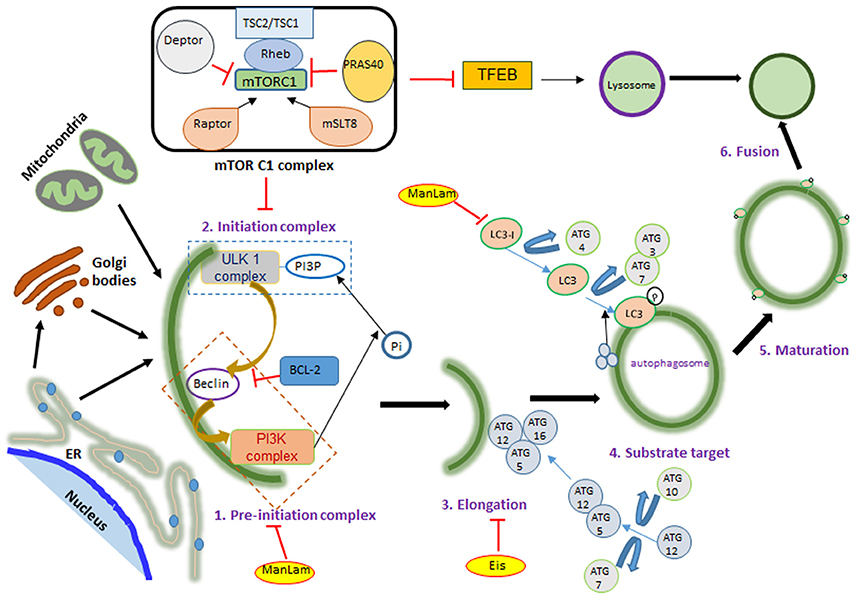
Figure 1. mTOR signaling during autophagy. mTOR has two complexes: mTORC1 and mTORC2, differentiated according to their activator proteins Raptor and Rictor. When activated, mTOR blocks ULK complex formation by phosphorylating it. ULK and PI3K complex formation marks the initiation of autophagy, followed by ATG-5, 12, and 16 binding on the phagophore membrane. This leads to LC3 translocation on the autophagosome membrane, which is required for fusion with lysosome and ultimately bacterial killing. Autophagy activating factors and pathways are denoted in black color and autophagy inhibitors are in red color letters and lines; mTOR activation leads to protein synthesis, nucleotide synthesis, cytoskeletal regulation and ion transport. These are denoted in blue color. Yellow color highlight denotes mycobacterial factors influencing autophagy.
Successful intracellular pathogens inhibit host cell antimicrobial processes involved in restricting their survival (Flynn and Chan, 2003; Kim et al., 2012). In that context, Mtb is known to inhibit killing within the phagolysosome of macrophages and other antigen presenting cells (APC) by modulating phagosome maturation and its fusion with the lysosome. In the infected APC, pathogenic Mtb inhibits actin assembly around the phagosome, thereby inhibiting host lipid molecules from interacting with phagosomal proteins necessary for further maturation and fusion with the lysosome (Vergne et al., 2003; Rohde et al., 2007; Ehrt and Schnappinger, 2009; Shui et al., 2011; Seto et al., 2012). When bacteria are phagocytosed by APC, the phagosome acquires early endosomal protein markers, such as EEA1 (early endosomal antigen 1) and Rab5, which are gradually replaced with Rab7 during the maturation of the phagosome (Chandra et al., 2015); ultimately, LAMP1 (lysosome-associated membrane protein 1) and acid hydrolases mark the late phagosome for fusion with lysosome (Huynh et al., 2007). It has been reported that phagosomes containing live Mtb do not acquire Rab5 due to the presence of tryptophan aspartate coat protein (TACO). Phagosomal association with TACO is also reported in macrophages that can engulf other pathogenic mycobacteria, which also result in the inhibition of phagosomal maturation (Pieters and Gatfield, 2002). Proper maturation of phagosomes is the key to its fusion with lysosomes, which can kill the bacteria by delivering toxic molecules. However, due to the absence of proton-ATPase molecules in Mtb-containing phagosomes, the phagosome-lysosome fusion does not take place and the bacteria survive intracellularly (Vergne et al., 2005).
Another mechanism used by Mtb to manipulate APC involves perturbation of intracellular calcium ion (Ca2+) levels (Kusner and Barton, 2001). Several studies have demonstrated fluctuations in intracellular Ca2+ levels in Mtb-infected macrophages (Vergne et al., 2003; Jayachandran et al., 2007). During phagocytosis of opsonized or heat-killed Mtb, intracellular Ca2+ concentrations increase, while macrophages infected with live pathogenic Mtb have reduced calcium ion level, which in turn significantly reduce the levels of Ca2+ associated-calmodulin and the phosphorylated Ca2+/calmodulin-dependent protein kinase II (CaMKII) (Jayachandran et al., 2007). Reduction in CaKMII level also blocks the delivery of lysosomal components to phagosome. Mtb reportedly prevents intracellular Ca2+ increase through its cell wall glycolipid, ManLAM (mannose-capped lipoarabinomannan), and by inhibiting host sphingosine kinase (SK). ManLAM also inhibits ionophore-induced increase in Ca2+ levels in macrophages. Reduced Ca2+/Calmodulin association impairs PI3K signaling, which inhibits recruitment of EEA1 to phagosomes (Rojas et al., 2000). Inhibition of SK abrogates phosphorylation of sphingosine, which is required for G-protein coupled receptor (GPCR) signaling that regulates Ca2+ homeostasis. Thus, ManLAM triggers a sequence of events leading to Ca2+ signaling disruption and phagosomal maturation arrest, which facilitate successful intracellular survival of infecting Mtb (Chan et al., 1991; Rojas et al., 2000).
Apart from its function in maintaining cellular homeostasis, autophagy is also known to sense and destroy intracellular bacteria in innate immune cells, such as macrophages. Although intracellular Mtb can efficiently modulate the bactericidal mechanisms of phagocytes, autophagy has been shown to be effective in killing Mtb (Gutierrez et al., 2004; Maiuri et al., 2007). Xenophagy, a type of autophagy whereby microorganisms can be sequestered and subject to lysosomal degradation, has been proposed to play an important role in elimination of bacteria (Gutierrez et al., 2004; Rubinsztein et al., 2012; Songane et al., 2012). In Mtb-infected host cells, the autophagosome collects ubiquitin while maturing, which then ultimately fuses with lysosome, thereby enhancing the lysosome-mediated bacterial killing. Survival of Mtb in macrophages has been reported to be dependent on the autophagosome delivery to the lysosome. However, in vivo and in vitro results have shown disparity in Mtb survival following inhibition of autophagy markers (Table 1) (Levine and Deretic, 2007; Lerena et al., 2008; Levine et al., 2011). Mutation or knockdown of autophagy associated host genes, such as Unc-51-like kinase 1 (Ulk1), Beclin1, Atg5, Atg7 or p62, has been reported to increase the survival of intracellular bacteria (Kim et al., 2011; Mizushima et al., 2011; Shang et al., 2011; Alers et al., 2012). However, although xenophagy is reported to restrict the survival of Mtb and BCG (bacille Calmette-Guerin) within macrophages, there are studies suggesting that intracellular pathogens such as Shigella flexneri, Listeria monocytogenes, Burkholderia pseudomallei, Orientia tsutsugamushi, Porphyromonas gingivalis, Staphylococcus, Brucella abortus, and Salmonella typhimurium are capable of blocking induction of autophagy by downregulating the co-localization of LC3 (microtubule-associated protein 1A/1B-light chain 3), restoring activation of mTOR, and utilizing nutrients for their growth and survival (Thurston et al., 2009; Yoshikawa et al., 2009; Zheng et al., 2009; Choy et al., 2012; Fraunholz and Sinha, 2012; Asrat et al., 2014; Yu et al., 2014).

Table 1. Major differences and similarities between mTOR complexes- mTORC1 and mTORC2.
Pathogenic Mtb also inhibits phagosome function in infected macrophages by releasing vesicle-bound lipids and glycolipids, which accumulate in lysosomes and interfere with the phagosome-lysosome fusion (Beatty et al., 2000). Taken together, pathogenic intracellular Mtb uses multiple strategies to manipulate the host defense machinery of APC for its own survival. Manipulation of APC function by Mtb impacts subsequent downstream events, including autophagy, antigen presentation, apoptosis, and activation of various signaling pathways involved in the production of cytokines, chemokines and other effector molecules that are crucial for controlling bacterial growth and replication (Briken et al., 2004; Cooper, 2009; Guenin et al., 2009; Rajaram et al., 2010).
Autophagy, a Greek word meaning “eating of self,” is a conserved cellular process critical for maintaining cellular integrity and function. This catabolic process is activated in cells due to lack of nutrient availability or cellular damage or stress, and involves degradation of damaged organelles and misfolded or abnormal proteins. During starvation, cytosolic components of cells are sequestered by autophagy to release nutrients for de novo biosynthesis of molecules (Laplante and Sabatini, 2012). Autophagy can also be activated by pathological factors, such as infections and other diseases. In these cases, normal cellular functions are facilitated by the elimination of pathogens through autophagy-dependent mechanisms, such as surface antigen presentation (Rubinsztein et al., 2012; Songane et al., 2012). Moreover, autophagy is one of the macrophage defense mechanisms against Mtb infection.
Autophagy is characterized by phagophore formation, elongation and maturation of the autophagosome, which ultimately fuse with the lysosome for the degradation of its contents. Formation of the autophagosome begins with a double membranous structure derived from the lipid bilayers of the endoplasmic reticulum (ER) or Golgi apparatus and conjugated with autophagy related (ATG) proteins (Maiuri et al., 2007; Alers et al., 2012). The three main components of autophagosome generation are: PI3KC3 (class III phosphoinositide 3-kinase complex 3), ULK1 (unc-51-like kinase 1) complex and ATG complex (Figure 1). This process is negatively regulated by mTOR kinase, which, when activated, blocks the ULK1 complex (Kim et al., 2011; Shang et al., 2011). Under stress conditions, such as nutrient deprivation or bacterial invasion, mTOR gets inactivated, enabling the ULK1 complex to recruit and activate PI3KC3 (Dibble and Cantley, 2015). This initiation complex, formed on the ER, leads to the nucleation of cell membrane, which is followed by recruitment of an ubiquitin-like molecule, LC3. In the final step, LC3 conjugates with phosphotidylethanolamine, resulting in self-fusion of the double membrane to form the autophagosome, which subsequently fuses with lysosome to degrade the engulfed contents.
Apart from mTOR signaling pathway (Lipinski et al., 2010), autophagy is also regulated by the inositol signaling pathway (Sarkar et al., 2005), Ca2+ /Calpain signaling pathway (Gordon et al., 1993) and cAMP (cyclic adenosine monophosphate) (Noda and Ohsumi, 1998). Inhibition of these mTOR-independent pathways for promoting autophagy has been studied under different disease conditions (Floto et al., 2007; Grumati et al., 2010; Hidvegi et al., 2010). Promising outcomes of autophagy induction via mTOR-independent pathway have been observed only with a combination therapy strategy, where the small molecules enhancers (SMERs) or inhibitors (SMIRs) of mTOR-independent pathway are used in combination with an mTOR inhibitor. For example, lithium, an inositol (1,4,5)-triphosphate inhibitor, when administered with rapamycin results in a stronger induction of autophagy (Sarkar et al., 2008). On the other hand, rapamycin alone can induce autophagy even at high intracellular inositol (1,4,5)-triphosphate levels, which has autophagy inhibitory effects. Since targeting ULK complex formation or ATG complex, rather than affecting the upstream pathways, seems to have a specific and stronger impact on autophagy, mTOR has been the target of interest for promoting autophagy upon infection with Mycobacteria (Gutierrez et al., 2004).
In addition to its role in autophagy, mTOR is also a master regulator of cell metabolism, growth, proliferation, translation initiation, and cytoskeletal organization. It belongs to the family of phosphoinoside 3-kinase- (PI3K-) related kinase and is a highly conserved serine/threonine protein kinase, which exists in host cells as part of two protein complexes—mTORC1 and mTORC2 (Laplante and Sabatini, 2012; Singh and Cuervo, 2012) (Figure 2). Theses complexes differ in their structure and activity, in part due to the difference in mTOR regulatory proteins such as RAPTOR (regulatory associated protein of mTOR; rapamycin sensitive) in mTORC1 and RICTOR (rapamycin-insensitive companion of mTOR) in mTORC2, as well as other accessory proteins (Laplante and Sabatini, 2009). The proteins that are common to both mTORC1 and C2 complexes are the mammalian lethal with Sec13 protein 8 (mLST8) and DEP domain containing mTOR interacting protein (DEPTOR). While mLST8 acts as a positive regulator, DEPTOR functions as a negative regulator of mTOR signaling. The mTORC1 is activated by RAPTOR, PRAS40 (proline-rich AKT substrate 40 kDa) and by phosphorylation of tuberous sclerosis protein 2 (TSC2) (Huang et al., 2008). The PI3K/AKT pathway is a positive regulator of mTOR signaling (Kim et al., 2011; Ng et al., 2011; Pan et al., 2012). Apart from PI3K/AKT, arginine, DNA damage, AMPK (AMP-activated protein kinase) and ERK1/2 (extracellular signal-regulated protein kinases 1 and 2) signaling were also reported to regulate mTORC1 activation (Kim et al., 2002; Inoki et al., 2003; Fingar and Blenis, 2004; Laplante and Sabatini, 2009). Importantly, mTORC1 activation inhibits autophagy (Jung et al., 2010). Deactivation of mTORC1 in cells under nutrition depletion or treatment with rapamycin leads to initiation of autophagy. (Seto et al., 2013). Similarly, dephosphorylating ULK1 by inactivation of mTORC1 induces autophagy (Egan et al., 2011; Kim et al., 2011; Shang et al., 2011).
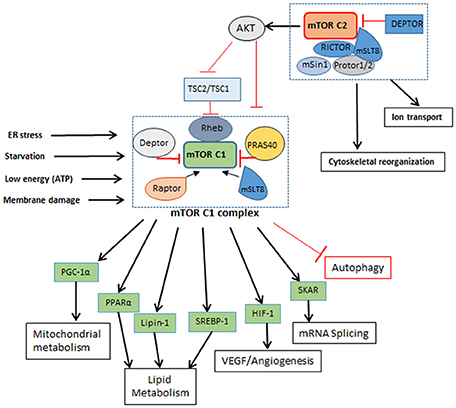
Figure 2. mTORC1 and mTORC2 complexes. mTORC1: Is a five component complex with DEPTOR and PRAS40 as negative regulators and RAPTOR and mSLT8 as positive regulators. It regulates different cellular processes like lipid metabolism and protein metabolism other than autophagy as seen in Figure 1. It is influenced by the nutrient and energy level in cell and gets shut down or inhibited when cell encounters reducing nutrient level and decrease in energy. Inhibition of mTORC1 leads to inhibition of cellular metabolic processes. mTORC2; A six component complex has DEPTOR as its negative regulator and RICTOR and mSLT8 as positive regulators. This complex influences activation of mTORC1 by phosphorylation of AKT. How nutrient level influences mTORC2 is not known yet.
The binding of mTORC2 with RICTOR facilitates the interaction of these proteins with TSC2 and mammalian stress-activated protein kinase interacting protein (mSIN-1); another protein found in association with RICTOR is PROTOR-1, which promotes activation of serum and glucorticoid-induced kinase 1 (SGK1). Interaction of all these proteins ultimately promotes mTORC2 complex formation and phosphorylation of AKT. Therefore, mTORC2 activation also regulates mTORC1 activation via AKT phosphorylation. Similarly, while mTORC1 activation is mediated by PI3K-AKT/PKB pathway in response to nutrient availability and mitogenic stimulation of the cell, phosphorylation of growth factors by autophosphorylation of their receptor tyrosine kinases activates mTORC2 complex, which also activates class I PI3K-AKT/PKB pathway (Dibble and Cantley, 2015). mTOR complexes also differ in the nature of their stimulant, for example, mTORC1 is activated by low levels of amino acids and growth factors, energy molecules and stress, while mTORC2 remains unaffected by the changing levels of these mTORC1 stimulants. However, role of mTORC2 is important for the regulation of AKT, which in turn governs mTORC1 functions. With use of TSC deficient cells importance of autophagy for cell survival was validated. In conditions like TSC (tuberous sclerosis complex), mTOR inhibition by rapamycin and pro-survival due to autophagy may have beneficial effects (Parkhitko et al., 2011).
In addition to regulating autophagy, activation of mTORC1 also promotes cellular metabolic pathways, such as glucose metabolism, protein and lipid synthesis, all of which contributes to cell growth and proliferation. S6Ks (p70 ribosomal protein S6 kinase 1/2) and 4E-BPs (eukaryotic initiation factor4 binding protein) are the two major proteins interacting with mTORC1 and play a major role in protein synthesis. mTORC1 phosphorylates 4E-BP1 thereby inhibiting its interaction with elF4E, which is then able to promote cap-dependent translation. Similarly, mTORC1 interaction with S6K1 stimulates cap-dependent translation of ribosomal proteins. Phosphorylation of S6K by mTORC1 also activates glucose transporter protein (Glut1) which activates glycolysis, lipogenesis and increases glucose uptake (Zeng et al., 2016). This increased glycolysis due to Glut1 is also reported to elevate T cell function and proliferation (Macintyre et al., 2014). Likewise, lipid synthesis is influenced by positive regulation of SREBP1 (sterol regulatory element binding protein 1) and PPARγ (peroxisome proliferator-activated receptor-γ) (Kim and Chen, 2004), which are regulated by mTORC1. The mTOR inhibitor, rapamycin, reduced phosphatidic acid phosphatase (lipin-1) phosphorylation, which is essential for glycerolipid synthesis; lipin-1 also activates PPARγ and other proteins associated with lipid synthesis (Huffman et al., 2002). Oxidative metabolism is also influenced by mTOR signaling. In a mouse model, inhibition of mTORC1 reduced the muscle mass and oxidative metabolism, leading to early death. It has been shown that PGC1-α is associated with the oxidative metabolism and that mTOR directly interacts with this regulatory protein (Laplante and Sabatini, 2012). Other proteins interacting with mTORC1 are HIF-1α (hypoxia-inducible factor 1-alpha) and STAT3 (signal transducer and activator of transcription 3), which are involved in a plethora of cellular functions, ranging from angiogenesis to inflammation and cytokine response (Laughner et al., 2001).
Since mTOR signaling pathway regulates several cellular processes, including autophagy, that are linked to the host immune response to pathogens, it is an attractive target for developing/testing small molecules to modulate host immunity for better protection against infectious agents. Moreover, the peripheral blood mononuclear cells (PBMCs) and CD4+CD25+FoxP3+Treg cells isolated from active tuberculosis patients demonstrated mTOR inhibition during infection (Zhang et al., 2017). In contrast, mTOR activation, by deletion of Tsc1 in hematopoietic stem cells, induces accumulation CDK (cyclin-dependent kinase) inhibitors p16ink4a, p19Arf, p21Cip1 leading to impaired hematopoietic system and decreased lymphopoiesis (Chen et al., 2009). These observations establish that mTOR inhibition improves cell survival and the understanding that mTOR inhibition may be promoting host cell defense mechanisms against invading pathogens (Harrison et al., 2009).
The following are some of the key mTOR inhibitors in use to treat chronic conditions in humans.
Rapamycin, specifically known for its mTOR inhibitory activity, was first isolated from Streptomyces hygroscopicus. Despite of its antifungal and antibacterial properties, Rapamycin is well-known for its immunosuppressant activity, which led to its use in organ transplant cases to reduce graft rejection. Similar to temsirolimus, rapamycin, which is also known as sirolimus, targets FKBP12 (FK506-binding protein 1A, 12 kDa) and inhibits the formation of active mTOR complex. Thus all of the currently known sirolimus derivatives target FKBP12 and inhibit mTOR complex.
In a zebrafish model of M. marinum infection, mTOR was shown to be associated with the host resistance to infection. In this model, mTOR mutants were hyper-susceptible to M. marinum at higher infection dose; however, when the inoculum size was decreased, the mTOR-deficient zebrafish cleared infection early (Pagan et al., 2016). Inhibition of mTOR in mice by rapamycin treatment at early age did not significantly affect the life expectancy or susceptibility to disease, but administration at an old age improved the survival expectancy (Harrison et al., 2009). In another study, administration of rapamycin to BCG-vaccinated mice has been shown to elicit better vaccination efficacy against Mtb infection, which is associated with induced autophagy, increased antigen presentation on dendritic cells and elevated Th1-type immune response (Gutierrez et al., 2004; Jagannath et al., 2009). Results from a low dose Mtb infection (MOI = 1) of human monocyte-derived-macrophages pre-infected with HIV, showed elevated bacterial load upon administration of rapamycin (1 μM) (Andersson et al., 2016). This study described mTOR inhibition as an advantage for the intracellular survival of Mtb; however, in an already immunocompromised cell (due to HIV infection), it is difficult to assess the impact of mTOR inhibition on Mtb growth. Although rapamycin used to be the popular drug of interest to achieve cellular mTOR inhibition, poor solubility and long intracellular half-life complicates the consideration of this molecule as potential HDT for TB therapy.
Temsirolimus, commercially known as CCI-779 or Torisel, is currently approved by the US-FDA for use in renal cell carcinoma (RCC) treatment. This prodrug can transform to sirolimus when dihydroxymethyl propionic acid ester group at C40 position is removed. Temsirolimus is metabolized by the enzyme CYP3A4 (cytochrome P450 3A4) and has a half-life of 9–27 h (MacKeigan and Krueger, 2015). Intravenous administration of temsirolimus increases its bioavailability and dose intensity (Boni et al., 2009). Mechanistically, temsirolimus targets host FKBP-12 protein. The drug-FKBP-12 interaction inhibits the formation of mTOR-FKBP-12 complex, leading to the inactivation of mTOR complex and inhibition of p70S6k and S6 phosphorylation. These effects cumulatively results in arrested cell growth, proliferation and survival in RCC patients. Nonspecific pneumonitis and gastrointestinal disorders are major side effects in RCC patients treated with this drug. In addition, metabolic diseases such as hyperglycemia, and hypercholesterolemia are associated with temsirolimus administration in these patients (Malizzia and Hsu, 2008). Importantly, temsirolimus treatment has been widely associated with reactivation of latent Mtb infection among RCC patients. Also, progression of tumor was noted in these patients when temsirolimus was administered in combination with rifampicin, a first-line anti-TB drug (Bossé et al., 2016).
Ridaforolimus (AP23573 or MK-8669) is an analog of sirolimus with improved bioavailability, solubility and half-life (30–75 h) (Rivera et al., 2011). It is administered orally or intravenously for the treatment of solid tumors of soft tissues, bone and other hematologic malignancies (Huang et al., 2015). In a phase I clinical trial with 87 ER+/high-proliferative breast cancer cases, majority of patients treated with ridaforolimus demonstrated reduced tumor activity (Di Cosimo et al., 2015). Similarly, a phase II clinical trial showed promising results for ridaforolimus to treat patients with endometrial, soft tissue and bone cancers (Palavra et al., 2017). The effect of ridaforolimus on cell metabolism and growth is largely dependent on the dose of drug used for treatment. This is due to its varying effect on mTOR inhibition with variation in dosage (Rivera et al., 2011). However, no reactivation of latent Mtb infection has been reported in these studies (Huang et al., 2015; Palavra et al., 2017).
Everolimus (40-O-(2-Hydroxy)-ethyl-rapamycin), commercially known as SDZ-RAD, RAD001, Certican and Afinitor, is a derivative of rapamycin bearing a stable 2-hydroxy ethyl chain substitution at position 40. This agent has a better solubility, oral availability, and decreased mean elimination half-life (~18–30 h), leading to early removal of drug from the body compared to the parent compound (rapamycin). Because of the better absorption, it has higher bio-availability (30–60%) and a Tmax of 1–2 h. Everolimus is an immunosuppressive, anti-inflammatory drug that inhibits host cell proliferation by arresting the progression of cell cycle from G1 to S phase; the immune suppressive function is exerted by inhibiting IL-2 and IL-15 mediated lymphocyte proliferation (Kovarik et al., 2002; Lingaraju et al., 2010; Ahya et al., 2011). In addition, everolimus promotes autophagy by inhibiting mTORC1 (Saran et al., 2015).
Inhibition of mTOR pathway by everolimus treatment has been reported to improve cellular immune response in both animal models and human studies. In a study performed with 218 healthy volunteers of >65 years of age, everolimus treatment had beneficial effects over aging-related issues (Mannick et al., 2014). Specifically, these elderly volunteers treated with a low dose of everolimus showed about 20% improvement in their protective response after influenza vaccination. This improvement was associated with reduced expression of programmed death-1 receptor, which is otherwise highly expressed in aging individuals, on CD4 and CD8 T cells, thus increasing T cell antigen processing and expression. This low dose administration (0.5 mg daily or 5 mg weekly) of eveolimus demonstrated minimum number of adverse events, (35 adverse events) compared to a higher dose administration (20 mg weekly), which resulted in 109 adverse events amongst 53 elderly individuals (Mannick et al., 2014). This study clearly highlights the importance of optimizing the dose of mTOR inhibitors, such as everolimus in this case, for better efficacy with minimal adverse effects. In contrast, case studies with organ transplant patients have mentioned a higher risk of Mtb infection and reactivation of LTBI as possible side-effects of everolimus administration (Kovarik et al., 2002; Fijałkowska-Morawska et al., 2011). Although patients in this study were treated with a higher dose of everolimus, than the influenza vaccine study mentioned above, the mechanism underlying the connection between dose of everolimus and reactivation of LTBI is not clearly understood. However, the negative consequences of high dose administration of everolimus can be overcome by co-administration with CYP3A4 enzyme inducers, such as rifampicin, which are used to treat opportunistic TB infections in organ transplant patients (Eisen et al., 2003). Thus, it is important to understand the dose-response of everolimus in the context of host cellular functions and how the drug influences phagocytosis and autophagy-mediated elimination of Mtb during infection.
All the mTOR inhibitors described above are also frequently used in the treatment of various forms of cancer, including breast cancer, renal cell carcinoma and tuberous sclerosis complex, due to their ability to inhibit host cell proliferation and growth (Pohanka, 2006; Koh et al., 2013). However, the idea of using these mTOR inhibitors as potential adjunct HDT for TB therapy needs to be substantiated through experimental evidences related to dosage, pharmacokinetics and pharmacodynamics (PK/PD) parameters and, cost vs. benefit effects on the host immunity. Such evidences need to be reinforced by series of studies on reliable and relevant pre-clinical animal models of Mtb infection. In addition, metabolic dysfunctions, such as hyperglycemia is a common side effect in cancer patients treated with mTOR inhibitors, including everolimus (Porta et al., 2011). Although the impact of such inhibitors in the context of TB treatment remains to be determined, serious side effects of HDT drugs preclude their potential use in any treatment. Moreover, as immune-suppressing agents, the application of mTOR inhibitors as a stand-alone HDT therapy for TB holds a significant risk of reactivation of latent Mtb infection, similar to the situation observed in rheumatoid arthritis patients treated with anti-TNF-α antibody (Kovarik et al., 2002). However, when used at an immune modulating-, as opposed to immune suppressing- dose, these mTOR inhibitors can be potential candidates to serve as an adjunct therapeutic molecule, along with standard anti-TB drugs, in improving the treatment outcome. Thus, fine-tuning the dose of mTOR inhibitors is an important and necessary step toward application of these HDT compounds for TB treatment. Importantly, since Mtb cannot develop resistance to a drug that targets host signaling pathway, such as mTOR or cellular processes, such as autophagy, HDT drugs has the potential to alleviate the development of MDR- and XDR-Mtb strains and their transmission in the community. Analogous to the trend in cancer treatment that have shifted from chemotherapeutic and radiologic regimens to more-host targeted treatment approaches, Mtb infection and/or disease can benefit from specific HDT drugs that targets, for example, the mTOR pathway and/or autophagy.
Pathogenic Mtb possess several virulent determinants, such as the unusually lipid-rich cell wall, that serve as permeability barriers and protects the bacteria from the harsh intracellular environment within phagocytes, and from the bactericidal activities of anti-TB drugs. Additionally, these mycobacteria-derived molecules interact with the host immune cells and modulates their function, promoting bacterial survival/persistence, causing disease within the host and enabling the development of bacterial drug resistance. Thus, when the innate defensive mechanisms of phagocytes are manipulated by the pathogen to promote its survival, the host develops active disease, which is hard to overcome. This can be one of the reasons for the inefficiency of current anti-mycobacterial drugs to eliminate TB, and for the emergence of drug-resistant Mtb strains. Perturbing host cell functions through HDT molecules has the potential to enhance the effector functions of these cells, which are the ultimate arsenals in combating bacterial infection. Moreover, these immune modulating drugs do not contribute to the emergence of drug resistance by the infecting bacteria. This criterion is crucial when considering therapy, particularly for patients with MDR, XDR-TB, as well as those patients with co-existing chronic conditions, such as diabetes or HIV infection, in which conventional antibiotics therapy has been shown to be complex, complicated, toxic and insufficient in achieving a bacteriological cure. Host cell autophagy, regulated by mTOR pathway, plays an important role in cellular homeostasis as well as in antibacterial defense mechanism. Therefore, targeting mTOR pathway with small molecules, such as everolimus, has the potential to develop novel and better combination drug therapy, along with standard anti-TB drugs to combat various forms of TB in patients with/without other co-morbid conditions. This approach can also enhance bacterial killing, reduce treatment duration, and/or improve clinical outcome. Clearly, more research and experimental evidence is warranted on these and other HDT molecules, for their efficacy, toxicity and other properties, through extensive pre-clinical studies using appropriate animal models of TB, before they are tried as therapeutic intervention for TB in human clinical trial.
SS: conceived the concept; PS and SS: wrote/edited the manuscript and agreed for submission.
This study was supported by a grant from the Bill and Melinda Gates Foundation (OPP1157210) to SS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ahya, V. N., McShane, P. J., Baz, M. A., Valentine, V. G., Arcasoy, S. M., Love, R. B., et al. (2011). Increased risk of venous thromboembolism with a sirolimus-based immunosuppression regimen in lung transplanta-tion. J. Heart Lung Transplant. 30, 175–181. doi: 10.1016/j.healun.2010.08.010
Alers, S., Löffler, A. S., Wesselborg, S., and Stork, B. (2012). Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 32, 2–11. doi: 10.1128/MCB.06159-11
Andersson, A. M., Andersson, B., Lorell, C., Raffetseder, J., Larsson, M., and Blomgran, R. (2016). Autophagy induction targeting mTORC1 enhances Mycobacterium tuberculosis replication in HIV co-infected human macrophages. Sci. Rep. 6:28171. doi: 10.1038/srep28171
Asrat, S., de Jesús, D. A., Hempstead, A. D., Ramabhadran, V., and Isberg, R. R. (2014). Bacterial pathogen manipulation of host membrane trafficking. Ann. Rev. Cell Dev. Biol. 30, 79–109. doi: 10.1146/annurev-cellbio-100913-013439
Beatty, W. L., Rhoades, E. R., Ullrich, H. J., Chatterjee, D., Heuser, J. E., and Russell, D. G. (2000). Trafficking and release of mycobacterial lipids from infected macrophages. Traffic 1, 235–247. doi: 10.1034/j.1600-0854.2000.010306.x
Boni, J. P., Hug, B., Leister, C., and Sonnichsen, D. (2009). Intravenous temsirolimus in cancer patients: clinical pharmacology and dosing considerations. Semin. Oncol. 36(Suppl. 3), S18–S25. doi: 10.1053/j.seminoncol.2009.10.009
Borrell, S., and Gagneux, S. (2009). Infectiousness, reproductive fitness and evolution of drug-resistant Mycobacterium tuberculosis. Int. J. Tuberc. Lung Dis. 13, 1456–1466.
Bossé, D., Ng, T., Ahmad, C., Alfakeeh, A., Alruzug, I., Biagi, J., et al. (2016). Eastern Canadian gastrointestinal cancer consensus conference 2016. Curr. Oncol. 23, e605–e614. doi: 10.3747/co.23.3394
Briken, V., Porcelli, S. A., Besra, G. S., and Kremer, L. (2004). Mycobacterial lipoarabinomannan and related lipoglycans: from biogenesis to modulation of the immune response. Mol. Microbiol. 53, 391–403. doi: 10.1111/j.1365-2958.2004.04183.x
Chan, J., Fan, X. D., Hunter, S. W., Brennan, P. J., and Bloom, B. R. (1991). Lipoarabinomannan, a possible virulence factor involved in persistence of Mycobacterium tuberculosis within macrophages. Infect. Immun. 59, 1755–1761.
Chandra, P., Ghanwat, S., Matta, S. K., Yadav, S. S., et al. (2015). Mycobacterium tuberculosis inhibits RAB7 recruitment to selectively modulate autophagy flux in macrophages. Sci. Rep. 5:16320. doi: 10.1038/srep16320
Chen, C., Liu, Y., Liu, Y., and Zheng, P. (2009). mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci. Signal. 2:ra75. doi: 10.1126/scisignal.2000559
Choy, A., Dancourt, J., Mugo, B., O'Connor, T. J., Isberg, R. R., Melia, T. J., et al. (2012). The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science 338, 1072–1076. doi: 10.1126/science.1227026
Cooper, A. M. (2009). Cell-mediated immune responses in tuberculosis. Annu. Rev. Immunol. 27, 393–422. doi: 10.1146/annurev.immunol.021908.132703
Di Cosimo, S., Sathyanarayanan, S., Bendell, J. C., Cervantes, A., Stein, M. N., Braña, I., et al. (2015). Combination of the mTOR inhibitor ridaforolimus and the anti-IGF1R monoclonal antibody dalotuzumab: preclinical characterization and phase I clinical trial. Clin. Cancer Res. 21, 49–59. doi: 10.1158/1078-0432.CCR-14-0940
Dibble, C. C., and Cantley, L. C. (2015). Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 25, 545–555. doi: 10.1016/j.tcb.2015.06.002
Egan, D. F., Shackelford, D. B., Mihaylova, M. M., Gelino, S., Kohnz, R. A., Mair, W., et al. (2011). Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461. doi: 10.1126/science.1196371
Ehrt, S., and Schnappinger, D. (2009). Mycobacterial survival strategies in the phagosome: defence against stresses. Cell. Microbiol. 11, 1170–1178. doi: 10.1111/j.1462-5822.2009.01335.x
Eisen, H. J., Tuzcu, E. M., Dorent, R., Kobashigawa, J., Mancini, D., Valantine-von Kaeppler, H. A., et al. (2003). Everolimus for the prevention of allograft rejection and vasculopathy in cardiac-transplant recipients. N. Engl. J. Med. 349, 847–858. doi: 10.1056/NEJMoa022171
Fijałkowska-Morawska, J. B., Jagodzinska, M., and Nowicki, M. (2011). Pulmonary embolism and reactivation of tuberculosis during everolimus therapy in a kidney transplant recipient. Ann. Transplant. 16, 107–110. doi: 10.12659/AOT.882226
Fingar, D. C., and Blenis, J. (2004). Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 23, 3151–3171. doi: 10.1038/sj.onc.1207542
Floto, R. A., Sarkar, S., Perlstein, E. O., Kampmann, B., Schreiber, S. L., and Rubinsztein, D. C. (2007). Small molecule enhancers of rapamycin-induced TOR inhibition promote autophagy, reduce toxicity in Huntington's disease models and enhance killing of mycobacteria by macrophages. Autophagy 3, 620–622. doi: 10.4161/auto.4898
Flynn, J. L., and Chan, J. (2003). Immune evasion by Mycobacterium tuberculosis: living with the enemy. Curr. Opin. Immunol. 15, 450–455. doi: 10.1016/S0952-7915(03)00075-X
Fraunholz, M., and Sinha, B. (2012). Intracellular staphylococcus aureus: Live-in and let die. Front. Cell. Inf. Microbiol. 2:43. doi: 10.3389/fcimb.2012.00043
Gordon, P. B., Holen, I., Fosse, M., Røtnes, J. S., and Seglen, P. O. (1993). Dependence of hepatocytic autophagy on intracellularly sequestered calcium. J. Biol. Chem. 268, 26107–26112.
Grumati, P., Coletto, L., Sabatelli, P., Cescon, M., Angelin, A., Bertaggia, E., et al. (2010). Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat. Med.. 16, 1313–1320. doi: 10.1038/nm.2247
Guenin-Macé, L., Siméone, R., and Demangel, C. (2009). Lipids of pathogenic Mycobacteria: contributions to virulence and host immune suppression. Trans. Emerg. Dis. 56, 255–268. doi: 10.1111/j.1865-1682.2009.01072.x
Guirado, E., Schlesinger, L. S., and Kaplan, G. (2013). Macrophages in tuberculosis: friend or foe. Semin. Immunopathol. 35, 563–583. doi: 10.1007/s00281-013-0388-2
Gutierrez, M. G., Master, S. S., Singh, S. B., Taylor, G. A., Colombo, M. I., and Deretic, V. (2004). Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119, 753–766. doi: 10.1016/j.cell.2004.11.038
Harrison, D. E., Strong, R., Sharp, Z. D., et al. (2009). Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395. doi: 10.1038/nature08221
Hidvegi, T., Ewing, M., Hale, P., Dippold, C., Beckett, C., Kemp, C., et al. (2010). An autophagy-enhancing drug promotes degradation of mutant α1-antitrypsin Z and reduces hepatic fibrosis. Science 329, 229–232. doi: 10.1126/science.1190354
Huang, J., Dibble, C. C., Matsuzaki, M., and Manning, B. D. (2008). The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol. Cell. Biol. 28, 4104–4115. doi: 10.1128/MCB.00289-08
Huang, Z., Wu, Y., Zhou, X., Qian, J., Zhu, W., Shu, Y., et al. (2015). Clinical efficacy of mTOR inhibitors in solid tumors: a systematic review. Future Oncol. 11, 1687–1699. doi: 10.2217/fon.15.70
Huffman, T. A., Mothe-Satney, I., and Lawrence, J. C. Jr. (2002). Insulin-stimulated phosphorylation of lipin mediated by the mammalian target of rapamycin. Proc. Natl. Acad. Sci. U.S.A. 99, 1047–1052. doi: 10.1073/pnas.022634399
Huynh, K. K., Eskelinen, E.-L., Scott, C. C., Malevanets, A., Saftig, P., and Grinstein, S. (2007). LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J. 26, 313–324. doi: 10.1038/sj.emboj.7601511
Inoki, K., Zhu, T., and Guan, K. L. (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. doi: 10.1016/S0092-8674(03)00929-2
Jagannath, C., Lindsey, D. R., Dhandayuthapani, S., Xu, Y., Hunter, R. L. Jr., and Eissa, N. T. (2009). Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat. Med. 15, 267–276. doi: 10.1038/nm.1928
Jayachandran, R., Sundaramurthy, V., Combaluzier, B., Mueller, P., Korf, H., Huygen, K., et al. (2007). Survival of mycobacteria in macrophages is mediated by coronin-1 dependent activation of calcineurin. Cell 130, 37–50. doi: 10.1016/j.cell.2007.04.043
Jung, C. H., Ro, S. H., Cao, J., Otto, N. M., and Kim, D. H. (2010). mTOR regulation of autophagy. FEBS Lett. 584, 1287–1295. doi: 10.1016/j.febslet.2010.01.017
Kim, D. H., Sarbassov, D. D., Ali, S. M., King, J. E., Latek, R. R., Erdjument-Bromage, H., et al. (2002). mTOR interacts with raptor to form a nutrient sensitive complex that signals to the cell growth machinery. Cell 110, 163–175. doi: 10.1016/S0092-8674(02)00808-5
Kim, J. E., and Chen, J. (2004). Regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes 53, 2748–2756. doi: 10.2337/diabetes.53.11.2748
Kim, J. J., Lee, H. M., Shin, D. M., Kim, W., Yuk, J. M., Jin, H. S., et al. (2012). Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe. 11, 457–468. doi: 10.1016/j.chom.2012.03.008
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi: 10.1038/ncb2152
Koh, Y., Lim, H. Y., Ahn, J. M., Lee, J. L., Rha, S. Y., Kim, Y. J., et al. (2013). Phase II trial of everolimus for the treatment of nonclear-cell renal cell carcinoma. Ann. Oncol. 24, 1026–1031. doi: 10.1093/annonc/mds582
Kovarik, J. M., Hartmann, S., Figueiredo, J., Rouilly, M., Port, A., and Rordorf, C. (2002). Effect of rifampin on apparent clearance of everolimus. Ann. Pharmacother. 36, 981–985. doi: 10.1345/aph.1A384
Kusner, D. J., and Barton, J. A. (2001). ATP stimulates human macrophages to kill intracellular virulent Mycobacterium tuberculosis via calcium dependent phagosome-lysosome fusion. J. Immunol. 167, 3308–3315. doi: 10.4049/jimmunol.167.6.3308
Laplante, M., and Sabatini, D. M. (2009). mTOR signaling at a glance. J. Cell. Sci. 122(Pt 20), 3589–3594. doi: 10.1242/jcs.051011
Laplante, M., and Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell 149, 274–293. doi: 10.1016/j.cell.2012.03.017
Laughner, E., Taghavi, P., Chiles, K., Mahon, P. C., and Semenza, G. L. (2001). HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1α (HIF-1α) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol. Cell. Biol. 21, 3995–4004. doi: 10.1128/MCB.21.12.3995-4004.2001
Lerena, C., Calligaris, S. D., and Colombo, M. I. (2008). Autophagy: for better or for worse, in good times or in bad times. Curr. Mol. Med. 8, 92–101. doi: 10.2174/156652408783769634
Levine, B., and Deretic, V. (2007). Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 7, 767–777. doi: 10.1038/nri2161
Levine, B., Mizushima, N., and Virgin, H. W. (2011). Autophagy in immunity and inflammation. Nature 469, 323–335. doi: 10.1038/nature09782
Lingaraju, R., Blumenthal, N. P., Mendez, J., Hadjiliadis, O., Lee, J. C., Ahya, V. N., et al. (2010). Venous thromboembolic disease after lung transplantation: special focus on sirolimus. Open. Transplant. J. 4, 1–4. doi: 10.2174/1874418401004010001
Lipinski, M. M., Hoffman, G., Ng, A., Zhou, W., Py, B. F., Hsu, E., et al. (2010). A genome-wide siRNA screen reveals multiple mTORC1 independent signaling pathways regulating autophagy under normal nutritional conditions. Dev. Cell 18, 1041–1052. doi: 10.1016/j.devcel.2010.05.005
Macintyre, A. N., Gerriets, V. A., Nichols, A. G., Michalek, R. D., Rudolph, M. C., Deoliveira, D., et al. (2014). The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 20, 61–72. doi: 10.1016/j.cmet.2014.05.004
MacKeigan, J. P., and Krueger, D. A. (2015). Differentiating the mTOR inhibitors everolimus and sirolimus in the treatment of tuberous sclerosis complex. Neuro-Oncology 17, 1550–1559. doi: 10.1093/neuonc/nov152
Maiuri, M. C., Zalckvar, E., Kimchi, A., and Kroemer, G. (2007). Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 8, 741–752. doi: 10.1038/nrm2239
Malizzia, L. J., and Hsu, A. (2008). Temsirolimus, an mTOR inhibitor for treatment of patients with advanced renal cell carcinoma. Clin. J. Oncol. Nurs. 12, 639–646. doi: 10.1188/08.CJON.639-646
Mannick, J. B., Del Giudice, D., Lattanzi, M., Valiante, N. M., Praestgaard, J., Huang, B., et al. (2014). mTOR inhibition improves immune function in the elderly. Sci. Transl. Med. 6:68ra179. doi: 10.1126/scitranslmed.3009892
Michael, O. S., Sogaolu, O. M., Fehintola, F. A., Ige, O. M., and Falade, C. O. (2016). Adverse events to first line anti-tuberculosis drugs in patients co-infected with HIV and tuberculosis. Ann. Ib. Postgrad. Med. 14, 21–29.
Mizushima, N., Yoshimori, T., and Ohsumi, Y. (2011). The role of Atg proteins in autopha-gosome formation. Annu. Rev. Cell Dev. Biol. 27, 107–132. doi: 10.1146/annurev-cellbio-092910-154005
Ng, S., Wu, Y. T., Chen, B., Zhou, J., and Shen, H. M. (2011). Impaired autophagy due to constitutive mTOR activation sensitizes TSC2-null cells to cell death under stress. Autophagy 7, 1173–1186. doi: 10.4161/auto.7.10.16681
Noda, T., and Ohsumi, Y. (1998). Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 273, 3963–3966. doi: 10.1074/jbc.273.7.3963
Pagan, A. J., Levitte, S., Berg, R. D., Hernandez, L., Zimmerman, J., Tobin, D., et al. (2016). mTOR deficiency reveals an immunological trade-off in innate resistance to mycobacterial infection in vivo. J. Immunol. 196(1 Suppl.) 200.6.
Palavra, F., Robalo, C., and Reis, F. (2017). Recent advances and challenges of mTOR inhibitors use in the treatment of patients with tuberous sclerosis complex. Oxid. Med. Cell. Longev. 2017:9820181. doi: 10.1155/2017/9820181
Pan, H., O'Brien, T. F., Zhang, P., and Zhong, X. P. (2012). The role of tuberous sclerosis complex 1 in regulating innate immunity. J. Immunol. 188, 3658–3666. doi: 10.4049/jimmunol.1102187
Parkhitko, A., Myachina, F., Morrison, T. A., Hindi, K. M., Auricchio, N., Karbowniczek, M., et al. (2011). Tumorigenesis in tuberous sclerosis complex is autophagy and p62/sequestosome 1 (SQSTM1)-dependent. Proc. Natl. Acad. Sci. U.S.A. 108, 12455–12460. doi: 10.1073/pnas.1104361108
Pieters, J., and Gatfield, J. (2002). Hijacking the host: survival of pathogenic mycobacteria inside macrophages. Trends Microbiol. 10, 142–146. doi: 10.1016/S0966-842X(02)02305-3
Pohanka, E. (2006). Conversion to everolimus in maintenance patients - current clinical strategies. Nephrol. Dial. Transplant. 21(Suppl. 3), iii24–iii29. doi: 10.1093/ndt/gfl301
Porta, C., Osanto, S., Ravaud, A., Climent, M. A., Vaishampayan, U., White, D. A., et al. (2011). Management of adverse events associated with the use of everolimus in patients with advanced renal cell carcinoma. Eur. J. Cancer 47, 1287–1298. doi: 10.1016/j.ejca.2011.02.014
Rajaram, M. V., Brooks, M. N., Morris, J. D., Torrelles, J. B., Azad, A. K., and Schlesinger, L. S. (2010). Mycobacterium tuberculosis activates human macrophage peroxisome proliferator-activated receptor gamma linking mannose receptor recognition to regulation of immune responses. J. Immunol. 185, 929–942. doi: 10.4049/jimmunol.1000866
Rivera, V. M., Miller, S. D., Berk, L., Wardwell, S. D., Ning, Y., Pollock, R., et al. (2011). Ridaforolimus (AP23573; MK-8669), a potent mTOR inhibitor, has broad antitumor activity and can be optimally administered using intermittent dosing regimens. Mol. Cancer Ther. 10, 1059–1071. doi: 10.1158/1535-7163.MCT-10-0792
Rohde, K., Yates, R. M., Purdy, G. E., and Russell, D. G. (2007). Mycobacterium tuberculosis and the environment within the phagosome. Immunol. Rev. 219, 37–54. doi: 10.1111/j.1600-065X.2007.00547.x
Rojas, M., García, L. F., Nigou, J., Puzo, G., and Olivier, M. (2000). Mannosylated lipoarabinomannan antagonizes Mycobacterium tuberculosis induced macrophage apoptosis by altering Ca2+-dependent cell signaling. J. Infect. Dis. 182, 240–251. doi: 10.1086/315676
Rubinsztein, D. C., Codogno, P., and Levine, B. (2012). Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 11, 709–730. doi: 10.1038/nrd3802
Ryan, F. (1992). The Forgotten Plague: How the Battle against Tuberculosis was Won and Lost. Boston, MA: Little Brown.
Saran, U., Foti, M., and Dufour, J. F. (2015). Cellular and molecular effects of the mTOR inhibitor everolimus. Clin. Sci. 129, 895–914. doi: 10.1042/CS20150149
Sarkar, S., Floto, R. A., Berger, Z., Imarisio, S., Cordenier, A., Pasco, M., et al. (2005). Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 170, 1101–1111. doi: 10.1083/jcb.200504035
Sarkar, S., Krishna, G., Imarisio, S., Saiki, S., O'Kane, C. J., and Rubinsztein, D. C. (2008). A rational mechanism for combination treatment of Huntington's disease using lithium and rapamycin. Hum. Mol. Genet. 17, 170–178. doi: 10.1093/hmg/ddm294
Seto, S., Tsujimura, K., Horii, T., and Koide, Y. (2013). Autophagy adaptor protein p62/SQSTM1 and autophagy-related gene Atg5 mediate autophagosome for-mation in response to Mycobacterium tuberculosis infection in dendritic cells. PLoS ONE 8:e86017. doi: 10.1371/journal.pone.0086017
Seto, S., Tsujimura, K., and Koideet, Y. (2012). Coronin-1a inhibits autophagosome formation around Mycobacterium tuberculosis-containing phago-somes and assists mycobacterial survival in macrophages. Cell. Microbiol. 14, 710–727. doi: 10.1111/j.1462-5822.2012.01754.x
Shang, L., Chen, S., Du, F., Li, S., Zhao, L., and Wang, X. (2011). Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. U.S.A. 108, 4788–4793. doi: 10.1073/pnas.1100844108
Sharma, A., Hill, A., Kurbatova, E., van der Walt, M., Kvasnovsky, C., Tupasi, T. E., et al. (2017). Estimating the future burden of multidrug-resistant and extensively drug-resistant tuberculosis in India, the Philippines, Russia, and South Africa: a mathematical modelling study. Lancet Infect. Dis. 17, 707–715. doi: 10.1016/S1473-3099(17)30247-5
Shui, W., Petzold, C. J., Redding, A., Liu, J., Pitcher, A., Sheu, L., et al. (2011). Organelle membrane proteomics reveals differential influence of mycobacterial lipoglycans on macro-phage phagosome maturation and autophagosome accumulation. J. Proteome Res. 10, 339–348. doi: 10.1021/pr100688h
Singh, R., and Cuervo, A. M. (2012). Lipophagy: connecting autophagy and lipid metabolism. Int. J. Cell Biol. 2012:282041. doi: 10.1155/2012/282041
Songane, M., Kleinnijenhuis, J., Nete, M. G., and van Crevel, R. (2012). The role of autophagy in host defense against Mycobacterium tuberculosis infection. Tuberculosis (Edinb.) 92, 388–396. doi: 10.1016/j.tube.2012.05.004
Subbian, S., Tsenova, L., O'Brien, P., Yang, G., Koo, M. S., Peixoto, B., et al. (2011a). Phosphodiesterase-4 inhibition alters gene expression and improves isoniazid-mediated clearance of Mycobacterium tuberculosis in rabbit lungs. PLoS Pathog. 7:e1002262. doi: 10.1371/journal.ppat.1002262
Subbian, S., Tsenova, L., O'Brien, P., Yang, G., Koo, M. S., Peixoto, B., et al. (2011b). Phosphodiesterase-4 inhibition combined with isoniazid treatment of rabbits with pulmonary tuberculosis reduces macrophage activation and lung pathology. Am. J. Pathol. 179, 289–301. doi: 10.1016/j.ajpath.2011.03.039
Thurston, T. L. M., Ryzhakov, G., Bloor, S., von Muhlinen, N., and Randow, F. (2009). The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 10, 1215–1221. doi: 10.1038/ni.1800
Tobin, D. M., Roca, F. J., Oh, S. F., McFarland, R., Vickery, T. W., Ray, J. P., et al. (2012). Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell 148, 434–446. doi: 10.1016/j.cell.2011.12.023
Vergne, I., Chua, J., and Deretic, V. (2003). Tuberculosis toxin blocking phagosome maturation inhibits a novel Ca2+/calmodulin–PI3K hVPS34 cascade. J. Exp. Med. 198, 653–659. doi: 10.1084/jem.20030527
Vergne, I., Chua, J., Lee, H. H., Lucas, M., Belisle, J., and Deretic, V. (2005). Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 102, 4033–4038. doi: 10.1073/pnas.0409716102
Yoshikawa, Y., Ogawa, M., Hain, T., Yoshida, M., Fukumatsu, M., Kim, M., et al. (2009). Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat. Cell Biol. 11, 1233–1240. doi: 10.1038/ncb1967
Yu, H. B., Croxen, M. A., Marchiando, A. M., Ferreira, R. B., Cadwell, K., Foster, L. J., et al. (2014). Autophagy facilitates Salmonella replication in HeLa cells. MBio 5:e00865-14. doi: 10.1128/mBio.00865-14
Zeng, H., Cohen, S., Guy, C., Shrestha, S., Neale, G., Brown, S. A., et al. (2016). mTORC1 and mTORC2 kinase signaling and glucose metabolism drive follicular helper T cell differentiation. Immunity 45, 540–554. doi: 10.1016/j.immuni.2016.08.017
Zhang, X. X., Huang, T. Y., Wu, Y., Peng, W. G., Xie, H. B., et al. (2017). Inhibition of the PI3K-Akt-mTOR signaling pathway in T lymphocytes in patients with active tuberculosis. Int. J. Infect. Dis. 59, 110–117. doi: 10.1016/j.ijid.2017.04.004
Zheng, Y. T., Shahnazari, S., Brech, A., Lamark, T., Johansen, T., and Brumell, J. H. (2009). The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J. Immunol. 183, 5909–5916. doi: 10.4049/jimmunol.0900441
Keywords: mTOR, autophagy, everolimus, host directed therapy, tuberculosis, drug resistance, adjunct therapy, phagocytosis
Citation: Singh P and Subbian S (2018) Harnessing the mTOR Pathway for Tuberculosis Treatment. Front. Microbiol. 9:70. doi: 10.3389/fmicb.2018.00070
Received: 23 October 2017; Accepted: 11 January 2018;
Published: 30 January 2018.
Edited by:
Noton Kumar Dutta, Johns Hopkins University, United StatesReviewed by:
Osmar Nascimento Silva, Universidade Católica Dom Bosco, BrazilCopyright © 2018 Singh and Subbian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Selvakumar Subbian, c3ViYmlhc2VAbmptcy5ydXRnZXJzLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.