 Yan Xu
Yan Xu Qun Wu
Qun Wu Rubing Du
Rubing Du- State Key Laboratory of Food Science and Technology, Key Laboratory of Industrial Biotechnology, Ministry of Education, Synergetic Innovation Center of Food Safety and Nutrition, School of Biotechnology, Jiangnan University, Wuxi, China
Zygosaccharomyces bailii is a common yeast in various food fermentations. Understanding the metabolic properties and genetic mechanisms of Z. bailii is important for its industrial applications. Fermentation characteristics of Z. bailii MT15 from Chinese Maotai-flavor liquor fermentation were studied. Z. bailii MT15 produced various flavor compounds, including 19 alcohols, six acids, three esters, three ketones, and two aldehydes. Moreover, production of acids and aldehydes were increased by 110 and 41%, respectively, at 37°C (the maximum temperature in liquor fermentation) compared with that at 30°C, indicating its excellent flavor productivity. Z. bailii MT15 is a diploid with genome size of 20.19 Mb. Comparative transcriptome analysis revealed that 12 genes related to amino acid transport were significantly up-regulated (2.41- to 5.11-fold) at 37°C. Moreover, genes ARO8, ARO9, and ALDH4 involved in amino acid metabolism also showed higher expression levels (>1.71-fold) at 37°C. Increased substrate supply and a vigorous metabolism might be beneficial for the increased production of acids and aldehydes at 37°C. This work revealed the potential contribution of Z. bailii to various flavor compounds in food fermentation, and produced insights into the metabolic mechanisms of Z. bailii in flavor production.
Introduction
Zygosaccharomyces bailii is a yeast species widely present in various food fermentations, such as wine, tea, and vinegar fermentations (Teoh et al., 2004; Solieri et al., 2006; Garavaglia et al., 2015). In most food and beverage industries, Z. bailii is considered as a problematic spoilage yeast due to its high resistance to preservatives and high tolerance of various stresses (Stratford et al., 2013; Palma et al., 2015). Regardless of its association with spoilage, the potential beneficial effects of Z. bailii have also been proposed in food industries (Ciani et al., 2009; Domizio et al., 2011). In wine fermentation, owing to its high production of esters, Z. bailii in a mixed starter with Saccharomyces cerevisiae improved the production of ethyl esters (Garavaglia et al., 2015). The co-culture of Z. bailii with S. cerevisiae also increased the production of polysaccharides that improved the taste and body of wine (Domizio et al., 2011). Additionally, Z. bailii formed part of the tea fungus in Kombucha and Haipao tea fermentation that were rich in crude protein, crude fiber, and lysine (Jayabalan et al., 2010). Nevertheless, the metabolic properties and genetic mechanisms of Z. bailii in food fermentations are still unclear and need to be systematically studied.
Chinese Maotai-flavor liquor is a popular alcoholic beverage even in other parts of the world (Xu and Ji, 2012). This liquor contains over 300 influential flavor compounds including alcohols, acids, esters, ketones, and aldehydes, which greatly contribute to its unique aroma and quality (Xu and Ji, 2012). Maotai-flavor liquor is produced from grains by a spontaneous and solid-state fermentation. Yeasts play essential roles in Maotai-flavor liquor fermentation (Wu, 2013; Wu et al., 2013). Among them, S. cerevisiae is one of the most important and contributes significantly to the quantity and quality of the liquor (Wu et al., 2012; Meng et al., 2015). Z. bailii has been found to be a dominant species in Maotai-flavor liquor fermentation, with proportions close to those of S. cerevisiae (Wu, 2013). Despite its large population, the metabolic activity of Z. bailii in the liquor fermentation is still unclear.
The present work is aimed to study the fermentation characteristics of Z. bailii MT15, including the genome sequencing and comparative transcriptome analysis to unravel its metabolic mechanisms at the relatively high temperature used in liquor fermentation. The results shed new light on the function of Z. bailii and provide a guide for its efficient use in food fermentation.
Materials and Methods
Yeast Strains
Z. bailii MT15 and S. cerevisiae MT1 were previously isolated from the Maotai-flavor liquor fermentation process and were deposited in the China General Microbiological Culture Collection Center with accession number CGMCC 4745 (Xu et al., 2013), and the China Center for Type Culture Collection with accession number CCTCC M2014463 (Meng et al., 2015), respectively.
Fermentation Conditions
Sorghum extract was used as fermentation medium and was prepared according to the method below. Two kilogram of ground sorghum was added to 8 L of deionized water. Then the mixture was steamed for 2 h and subsequently saccharified by glucoamylase (5 U/L) at 60°C for 4 h. The supernatant was collected after being filtered through gauze and centrifuged at 8,000 × g for 15 min. The obtained sorghum extract was diluted with water to produce a final reducing sugar concentration of 75 ± 5 g/L before sterilization (Lu et al., 2015). Fermentation media were prepared with aliquots of 50 mL sorghum extract in 250 mL flasks, and were sterilized at 115°C for 30 min.
Z. bailii MT15 and S. cerevisiae MT1 were pre-cultured in sorghum extract at 30°C for 16 h to obtain the seed culture. Then they were inoculated into fermentation media with an initial concentration of 1 × 106 colony-forming units (CFU)/mL. For the comparison of fermentation characteristics between Z. bailii MT15 and S. cerevisiae MT1, the strains were, respectively, fermented at 30°C for 48 h, with shaking at 200 rpm. To study the effects of temperature on fermentation, Z. bailii MT15 was fermented at 30 and 37°C for 48 h, with shaking at 200 rpm. During the fermentation, 1 mL of samples were withdrawn at 8-h intervals to determine the cell numbers (Figure 1A). Each experiment was performed in triplicate.
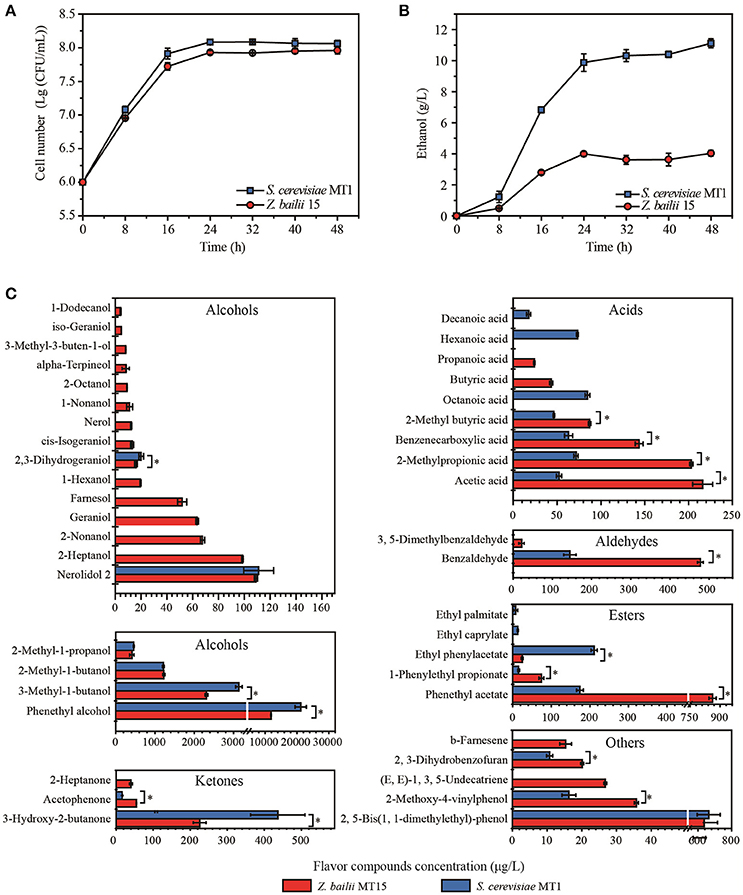
Figure 1. Biomass (A), ethanol content (B), and flavor compounds production (C) of Z. bailii MT15 and S. cerevisiae MT1. Significance level of the one-way ANOVA: *P < 0.05.
Analytical Determinations and Statistical Analysis
Yeast cells in the seed culture and during the fermentation process were obtained by centrifugation, washed three times with sterile saline solution, diluted to the applicable concentration by saline solution, and counted by hemocytometer under microscope. To analyze the contents of ethanol, flavor compounds, and amino acids, fermentation broths were centrifuged at 8,000 × g for 10 min to remove cells. Ethanol was monitored by high-performance liquid chromatography (HPLC) with a refractive index detector (Varian 355 RI) (Meng et al., 2015). Flavor compounds were analyzed by the gas chromatography-mass spectrometry method (Kong et al., 2014). Free amino acids were detected by HPLC via a pre-column derivatization method (Zhang et al., 2014).
The data are presented in terms of arithmetic averages of three replicates and the error bars indicate the standard deviations. The statistical significance of the difference between the means of samples was tested by one-way analysis of variance (ANOVA) using SPSS Statistics 22.
Genomic Sequencing and Assembly
The Z. bailii MT15 genome was sequenced using the whole-genome shotgun sequencing approach on an Illumina Miseq platform (Quail et al., 2012). Four paired-end/mate-paired sequencing libraries were constructed with insert sizes of 450, 700 bp, 3, and 8 kb. The de novo assembler Newbler and SSPACE software packages were employed to assemble raw data into contigs and scaffolds (Boetzer et al., 2011; Nederbragt, 2014). The GapCloser program was used to close gaps (Boetzer and Pirovano, 2012). The genome sequence of Z. bailii MT15 was deposited in GenBank under the Whole Genome Shotgun project number SRR5452526.
The genes of Z. bailii MT15 were predicted with the CEGMA pipeline, combining Augustus, SNAP, and Glimmer gene prediction software (Delcher et al., 1999; Stanke and Waack, 2003; Korf, 2004; Parra et al., 2007). The functional annotation of each gene was based on the eggNOG and Swissprot databases (Boeckmann et al., 2003; Powell et al., 2012). The functional classifications were performed with the eggNOG database. Genes for tRNAs and rRNAs were predicted with tRNAscan-SE and RNAmmer 1.2 Server, respectively (Schattner et al., 2005; Lagesen et al., 2007).
cDNA Preparation and Transcriptome Analysis Using RNA Sequencing
For RNA extraction, Z. bailii MT15 cells were cultivated with three parallel at 30 and 37, respectively, for 24 h. Then, the three parallel were mixed in the same volume and harvested after centrifugation at 8,000 × g for 5 min at 4°C. Subsequently, the supernatant was removed and the cell pellet was washed three times with sterile saline solution on ice. Then the washed cell pellet was immediately frozen in liquid nitrogen. Total RNA was isolated using the Trizol Reagent (Invitrogen Life Technologies, Shanghai) according to the manufacturer's instructions. Quality and integrity of total RNA were determined using a Nanodrop spectrophotometer (Thermo Scientific, USA) and Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA). Ribo-Zero rRNA Removal Kit (Illumina, San Diego, CA) was used to remove the ribosomal RNAs. The mRNA was subsequently fragmented and used as a template for oligo (dT)-primed PCR.
The cDNA libraries were prepared employing standard techniques for subsequent Illumina sequencing using the mRNA-seq Sample Prep Kit (Illumina, San Diego, CA). The cDNA libraries were sequenced on an Illumina NextSeq 500 according to the manufacturer's instructions. Sequencing raw reads were pre-processed after filtering sequencing adapters, rRNA reads, short-fragment reads, and other low-quality reads. The remaining clear reads were mapped to the reference genome of Z. bailii MT15 using Bowtie2/Tophat2 software (Langmead and Salzberg, 2012; Kim et al., 2015) based on the local alignment algorithm. Gene expression level was normalized by calculating reads per kilobase per million reads (RPKM) (Mortazavi et al., 2008). Differential expression of all of transcripts was quantified using DESeq software, and the method of FDR (False Discovery Rate) control was used to correct the results for multiple hypothesis testing (Anders and Huber, 2013). Significant DEGs were screened based on an FDR threshold of ≤0.05, and a Fold change ≥1.5. The RNA sequence data of Z. bailii MT15 at 30 and 37°C was deposited in the DNA Data Bank of Japan (DDBJ) with accession IDs DRX082892 and DRX082893, respectively.
Results and Discussion
Metabolic Properties of Z. bailii MT15
The biomass and ethanol production of Z. bailii MT15 and S. cerevisiae MT1 was determined during the fermentation process. The cell number of Z. bailii MT15 and S. cerevisiae MT1 was 9.18 × 107 and 8.18 × 107 CFU/mL at the end of fermentation (Figure 1A), respectively, which showed no significantly statistical difference (Supplementary Table 1). The highest ethanol production of Z. bailii MT15 was 4.03 g/L, which was 63.76% less than that of S. cerevisiae MT1 (Figure 1B, Supplementary Table 2). Considering the large population of Z. bailii MT15, whose proportion can reach 78% of the total yeast population (Wu, 2013), we considered that this yeast also contributes to ethanol production in Maotai-flavor liquor fermentation.
Flavor compounds play essential roles in forming the unique flavor quality of Maotai-flavor liquor (Xu and Ji, 2012). The metabolic activity of Z. bailii MT15 in flavor production was studied at the end of fermentation and compared with that of S. cerevisiae MT1 (Figure 1C). Production of alcohols by Z. bailii MT15 was 16,333.78 μg/L, which was approximately half that of S. cerevisiae MT1. Production of ketones by Z. bailii MT15 was 319.95 μg/L, which was also less than that of S. cerevisiae MT1 (452.98 μg/L). However, the production of acids, esters, and aldehydes by Z. bailii MT15 was 1.75, 2.28, and 3.45 times of that of S. cerevisiae MT1, respectively.
Additionally, Z. bailii MT15 produced 38 flavor compounds including 19 alcohols, six acids, three esters, three ketones, two aldehydes, and five other compounds (Figure 1C). Among them, 19 flavor compounds were also detected in the fermentation broth of S. cerevisiae MT1. The amounts of 10 flavor compounds, including acetic acid, benzaldehyde, phenethyl acetate, 1-phenylethyl propionate, and acetophenone, produced by Z. bailii MT15 were significantly higher than those produced by S. cerevisiae MT1 (Figure 1C). Furthermore, Z. bailii MT15 was able to produce 19 unique flavor compounds compared with S. cerevisiae MT1, including 13 alcohols, two acids, one ketone, one aldehyde, and two other flavor compounds. Some of these unique flavor compounds were influential flavor compounds and contributed significantly to the flavor of the Maotai-flavor liquor, such as 2-heptanol (fruity flavor), 2-nonanol (fruity flavor), 1-nonanol (fruity flavor), 1-hexanol (floral, green scent), geraniol (sweet, rose-like scent), 2-heptanone (fruity, spicy, cinnamon scent), propionic acid (vinegar-like scent), and butyric acid (cheesy-like scent) (Xu and Ji, 2012).
These results demonstrated that Z. bailii MT15 could generate ethanol and various flavor compounds including alcohols, acids, esters, aldehydes, and ketones during liquor fermentation, which would contribute to the flavor and quality of Maotai-flavor liquor. Moreover, previous studies showed that Z. bailii contributed to the flavor complexity of wine and could be used as a mixed starter with S. cerevisiae to improve the production of ethyl esters in wine fermentation (Ciani et al., 2009; Garavaglia et al., 2015). Therefore, except for the capacity of flavor production, Z. bailii would interact with S. cerevisiae during liquor fermentation and positively affect the flavor and quality of Maotai-flavor liquor.
Effect of Temperature on Flavor Metabolism of Z. bailii MT15
Maotai-flavor liquor is produced by a spontaneous fermentation process with a relatively high temperature of up to about 37°C (Wu et al., 2013). However, little is known about the metabolic properties and mechanisms of Z. bailii under such a relatively high temperature in food fermentation. We therefore studied the metabolic activity of Z. bailii MT15 at 37°C, and compared it with that at 30°C.
The biomass of Z. bailii MT15 decreased by 32.46% at 37°C compared with 30°C. Meanwhile, few differences were observed in the types of flavor compounds produced, but the amounts of flavor compounds per unit cell were substantially different between the two temperatures (Dataset 1). As shown in Figure 2, no significant change was observed in the production of alcohols and esters per unit cell, and the production of ketones decreased by 28.30% at the higher temperature. By contrast, the production of acids and aldehydes per unit cell at 37°C was, respectively, 110 and 41% higher than that at 30°C. Maotai-flavor liquor contains large amounts of acids and aldehydes, which have positive effects on its unique sensory characteristics (Fan et al., 2011; Xu et al., 2017). The results suggested that production of acids and aldehydes in Z. bailii MT15 was enhanced under higher temperature. This enhancement would probably contribute to the unique sensory characteristics of Maotai-flavor liquor. Nevertheless, the metabolic mechanisms for its vigorous activities under higher temperature remains unclear. Therefore, we used omics' technology including genomic and transcriptomic analysis to unravel the metabolic features and mechanisms of Z. bailii MT15.
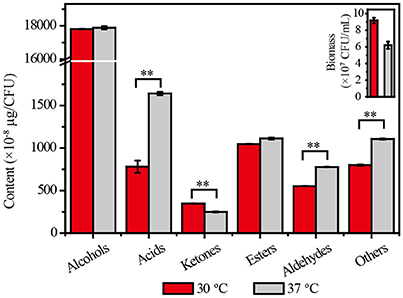
Figure 2. Cell growth and production of flavor compounds by Z. bailii MT15 at 30 and 37°C. Significance level of the one-way ANOVA: **P < 0.01.
Genomic and Transcriptomic Analysis of Z. bailii MT15
The whole genome of Z. bailii MT15 was sequenced and compared with the genome of three other Z. bailii strains (Z. bailii CLIB 213T, Z. bailii ISA1307, and Z. bailii IST302) (Galeote et al., 2013; Mira et al., 2014; Palma et al., 2017), and the results are shown in Table 1. The Z. bailii MT15 genome assembly resulted in 287 contigs (>527 bp) with an N50 value of 156,420 bp, and 95 scaffolds (>2020 bp) with an N50 value of 684,448 bp. The assembled genome was 20.19 Mb with a GC content of 42.38 mol%. The genome size of Z. bailii MT15 was 1.97-fold that of Z. bailii CLIB 213T and 1.87-fold that of Z. bailii IST302, but was similar to that of Z. bailii ISA1307. A total of 9,498 genes were predicted with an average length of 1,461 bp, occupying 68.72% of the whole genome (Table 1).
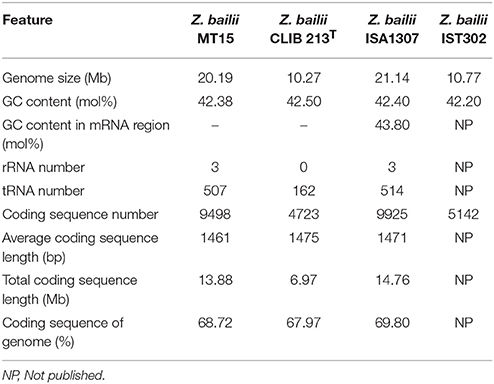
Table 1. General features of the Z. bailii MT15, Z. bailii CLIB 213T, Z. bailii ISA1307, and Z. bailii IST302 genomes.
The number of genes in the Z. bailii MT15 genome was nearly twice that of Z. bailii CLIB 213T and Z. bailii IST302, but was close to the number in Z. bailii ISA1307. It has been proven that Z. bailii ISA1307 is an interspecies hybrid strain that was generated in a stressful environment to improve strain robustness (Sipiczki, 2008; Mira et al., 2014). We analyzed the sequences of housekeeping genes including RPB1, RPB2, TBB, and EFGM in the Z. bailii MT15 genome, which are proposed to have a high capacity to discriminate Zygosaccharomyces species (Suh et al., 2013). The RPB1, RPB2, TBB, and EFGM genes were all duplicated in the Z. bailii MT15 genome, and the sequences of alleles of the four genes were different, which indicated that Z. bailii MT15 might be an interspecies hybrid strain. Furthermore, alleles of the four genes showed high sequence identity to the orthologous genes in Z. bailii CLIB 213T and Zygosaccharomyces parabailii ATCC 60483 (Supplementary Table 3). Thus, it is likely that Z. bailii MT15 is an interspecies hybrid strain generated from Z. bailii and Z. parabailii, which was beneficial for its adaptation to the relatively high temperature environment in Maotai-flavor liquor fermentation.
RNA-Seq was employed to reveal the transcriptomic features of flavor metabolism in Z. bailii MT15 under heat stress at 30and 37°C. The results showed that 257 genes (2.71% of the Z. bailii MT15 genome) were differentially expressed (≥1.5-fold, P < 0.05), including 126 up-regulated genes (Dataset 2) and 131 down-regulated genes (Dataset 3). These differentially expressed genes (DEGs) were clustered according to eggNOG functional categories (Figure 3). Among these categories, general function prediction only (8.56% of DEGs), transcription (7.39% of DEGs), amino acid transport and metabolism (7.00% of DEGs), and carbohydrate transport and metabolism (5.84% of DEGs), contained the greatest number of DEGs (Figure 3). Amino acid metabolism is important for the production of ethanol and most flavor compounds in liquor; therefore, we further analyzed the DEGs involved in amino acid transport and metabolism.
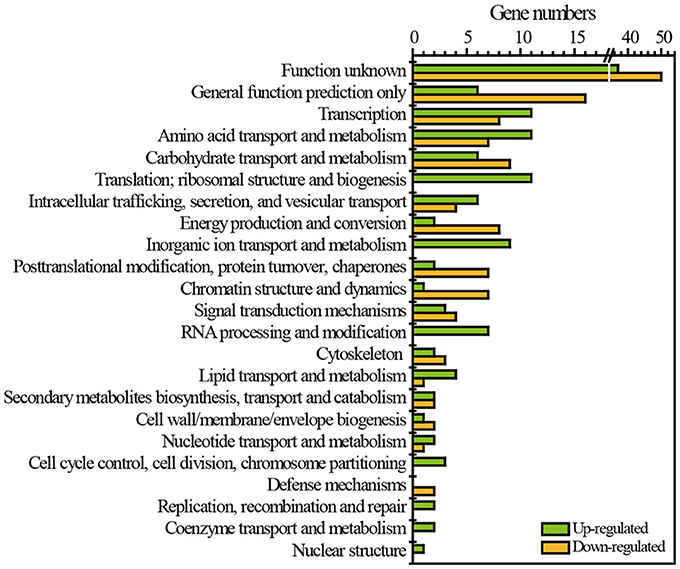
Figure 3. Functional classes of DEGs of Z. bailii MT15 at 30 and 37°C using eggNOG functional categories.
Comparative transcriptome analysis showed that 12 genes related to amino acid transport and metabolism were significantly up-regulated at 37°C (Figure 4A). Among them, eight genes encoded amino acid permeases, including one general amino acid permease and seven permeases specific for arginine, proline, lysine, and γ-aminobutyric acid. Higher transcription levels of these genes would be important for the transport of more amino acids into yeast cells (Jauniaux and Grenson, 1990; Regenberg et al., 1999). The other four genes were found to be involved in amino acids biosynthesis, including PTR2 and OPT1 associated with the transport of small peptides, and MEP21 and MEP22 encoding ammonium transporters, would also contribute to a strengthened amino acid metabolism (Meister, 1957). Thus, we speculated that these up-regulated amino acid metabolism related genes would promote the absorption and utilization of amino acid at 37°C. To validate this speculation, we detected the absorption of the most conventional amino acids by Z. bailii MT15 at 30 and 37°C after 24 h of cultivation. As shown in Figure 4B, the absorption of 14 amino acids was obviously improved by 83% at 37°C, including the uptake of glutamate acid, aspartate, proline, alanine, and arginine. Among these, the uptake of glutamate acid increased most prominently from 1.14 × 10−9 to 2.11 × 10−9 mg/CFU. By contrast, the absorption of valine and cysteine decreased, and no significant difference was found in the phenylalanine absorption. Therefore, we can conclude that Z. bailii MT15 could absorb and utilize amino acids more efficiently at 37 than at 30°C, due to the strengthened amino acid associated pathway at higher temperature. The strengthened absorption and utilization of amino acids possibly contributed to the improvement of the corresponding flavor compounds production, in particular, the production of acids and aldehydes.
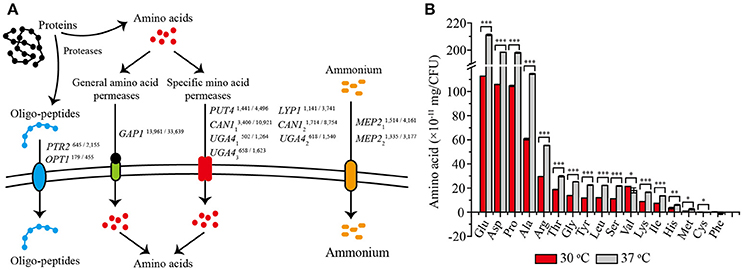
Figure 4. Differentially expressed genes related to amino acid transport (A) and the absorption of 17 conventional amino acids (B) at 30 and 37°C after 24 h of cultivation. The RPKM value of each gene is presented in the upper right corner, and the numbers on the left and right represent the RPKM values of Z. bailii MT15 at 30 and 37°C, respectively. Significance level of the one-way ANOVA: *0.05 > P > 0.01; **0.01 > P > 0.001; ***P < 0.001.
Metabolic Mechanisms of Production of Flavor Compounds in Z. bailii MT15
Z. bailii MT15 could produce various flavor compounds, especially alcohols, acids, and esters (Figure 2). During food fermentation, alcohols and acids are generated by yeasts from amino acids through the Ehrlich pathway and from sugars through the Harris pathway (Figure 5; Ehrlich, 1907; Chen, 1978). To unravel the molecular mechanisms of flavor metabolism in Z. bailii MT15, we compared its genome with that of strain S. cerevisiae MT1 and analyzed the transcription levels of genes involved in flavor metabolism at 37 and 30°C.
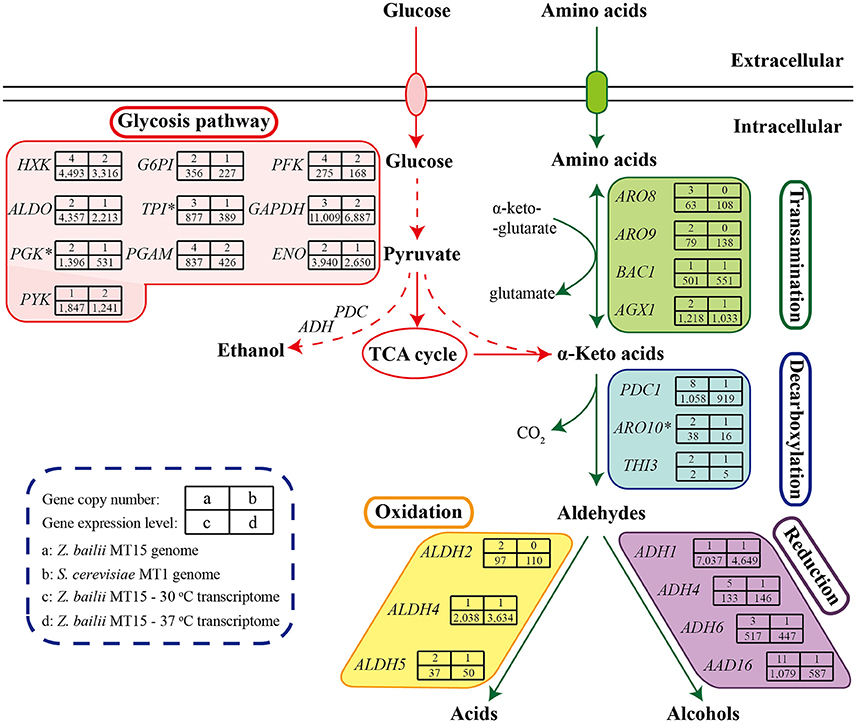
Figure 5. Genome and transcriptome analysis of Z. bailii MT15 in flavor production. Catabolism of sugars and amino acids leads to the formation of various flavor compounds. Genes encoding the key enzymes required in each step are indicated. *This gene was a DEG (≥2-fold, P < 0.05) at 37°C.
As shown in Figure 5, sugars could be converted to α-keto acids via the glycolysis and tricarboxylic acid (TCA) cycle pathways. In the Z. bailii MT15 genome, the copy number of genes involved in glycolysis and the TCA cycle was around twice those in the S. cerevisiae MT1 genome (Supplementary Table 4), which was expected as Z. bailii MT15 was considered to be an interspecies hybrid strain. In addition, α-keto acids could also be generated by various aminotransferases through the Ehrlich pathway (Hazelwood et al., 2008). In the Z. bailii MT15 genome, at least 16 different genes were annotated as aminotransferases including AGX1, BCA1, ARO9, ARO8, and YGD3. (Supplementary Table 5). Among them, ARO9 and ARO8 were uniquely found in the Z. bailii MT15 genome and might be associated with the differences in flavor metabolism compared with S. cerevisiae MT1.
The α-keto acids were further decarboxylated to the corresponding aldehydes by α-keto acid decarboxylases including PDC1, PDC5, PDC6, ARO10, and THI3 (ter Schure et al., 1998; Dickinson et al., 2003; Vuralhan et al., 2005). Among them, PDC1, ARO10, and THI3 existed in both of the two strains, while PDC5 and PDC6 were uniquely found in the S. cerevisiae MT1 genome. Pyruvate decarboxylases (PDCs) participate in alcoholic fermentation by converting pyruvate to acetaldehyde and in amino acid metabolism through the Ehrlich pathway (ter Schure et al., 1998). We found 13 genes encoding PDC enzymes in the Z. bailii MT15 genome, whereas only six were found in the S. cerevisiae MT1 genome. Since the ethanol production of Z. bailii MT15 was less than that of S. cerevisiae MT1 (Figure 1B), more PDC genes in Z. bailii MT15 did not favor its alcoholic fermentation, but might be beneficial for the conversion of amino acids and reducing sugars to aldehydes by the Ehrlich pathway.
Aldehydes could subsequently be converted to higher alcohols and acids by alcohol dehydrogenases and aldehyde dehydrogenases, respectively (Hazelwood et al., 2008). So far, at least 16 genes encoding alcohol dehydrogenases have been found to catalyze the interconversion of aldehydes and alcohols (Hazelwood et al., 2008). However, only four of these genes (ADH1, ADH4, ADH6, and AAD16) were found in the Z. bailii MT15 genome, while 11 such genes were present in the S. cerevisiae MT1 genome. Z. bailii MT15 harboring fewer alcohol dehydrogenases genes may be the reason that it produced more aldehydes and less alcohols compared with S. cerevisiae MT1. Moreover, the genome analysis showed that three genes (ALDH2, ALDH4, and ALDH5) annotated as aldehyde dehydrogenase were found in the Z. bailii MT15 genome, and ALDH2 was a specific gene compared with S. cerevisiae MT1. This would account for the higher acid productivity of Z. bailii MT15. Therefore, we can conclude that genome differences, particularly in the aspect of genes involved in amino acid metabolism between Z. bailii MT15 and S. cerevisiae MT1, would lead to the different metabolic features of the two strains (Figure 1C).
The transcriptome comparison of Z. bailii MT15 revealed that genes involved in the glycolysis pathway showed lower RPKM values at 37 than those at 30°C, which indicated that the glycolysis pathway might not be related to the increased production of acids and aldehydes at higher temperature. Moreover, in the TCA cycle, ACON2 associated with the production of α-ketoglutaric acid was significantly up-regulated at 37°C, which would promote production of α-keto acids. Meanwhile, the RPKM values of ARO8 and ARO9, which are involved in the transamination step of the Ehrlich pathway and regarded as broad-substrate-specificity aminotransferases (Iraqui et al., 1998), were higher at 37 than those at 30°C (≥1.50-fold, P ≤ 0.25). The higher expression of these genes might be beneficial for the transamination of amino acids to corresponding α-keto acids at 37°C. Furthermore, ALDH4 also showed higher RPKM values at 37°C (1.78-fold, P = 0.19), which was consistent with the increased production of acids at 37°C (Figure 2). Therefore, the higher expression of genes, including ACON2, ARO8, ARO9, and ALDH4 in the Harris pathway and Ehrlich pathway, might be beneficial for the increased production of acids and aldehydes at the relatively high fermentation temperature.
Conclusions
This study revealed the fermentation characteristics and potential function of Z. bailii MT15 that produces various flavor compounds including alcohols, acids, esters, aldehydes, and ketones. Its ability to generate acids and aldehydes is improved at the relatively high temperature used in liquor fermentation, which is beneficial for the complexity of the aroma and quality of the liquor. The genome and transcriptome analysis of Z. bailii MT15 revealed that amino acids metabolism plays important roles in flavor production. This work sheds new light on the metabolic characteristics of Z. bailii in flavor production during Maotai-flavor liquor fermentation that would be applicable to various food fermentations.
Author Contributions
YX (first author), QW, and YZ drafted the manuscript. YX (first author), QW, and RD performed the physiological studies and genome sequencing and transcriptome analysis. QW and YX (last author) participated in the design of the study. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (31371822, 31530055), the National Key R&D Program (2016YFD0400503), the Priority Academic Program Development of Jiangsu Higher Education Institutions, and the 111 Project (no. 111-2-06).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.02609/full#supplementary-material
References
Anders, S., and Huber, W. (2013). Differential Expression of RNA-Seq Data at the Gene Level—the DESeq package. Available online at: www.bioconductor.org/packages/devel/bioc/vignettes/DESeq/inst/doc/DESeq.pdf (Accessed October 17, 2013).
Boeckmann, B., Bairoch, A., Apweiler, R., Blatter, M. C., Estreicher, A., Gasteiger, E., et al. (2003). The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370. doi: 10.1093/nar/gkg095
Boetzer, M., and Pirovano, W. (2012). Toward almost closed genomes with GapFiller. Genome Biol. 13:R56. doi: 10.1186/gb-2012-13-6-r56
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D., and Pirovano, W. (2011). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. doi: 10.1093/bioinformatics/btq683
Chen, E. C. H. (1978). The relative contribution of Ehrlich and biosynthetic pathways to the formation of fusel alcohols. Am. Soc. Brew. Chem. 36, 39–43.
Ciani, M., Comitini, F., Mannazzu, I., and Domizio, P. (2009). Controlled mixed culture fermentation: a new perspective on the use of non-Saccharomyces yeasts in winemaking. FEMS Yeast Res. 10, 123–133. doi: 10.1111/j.1567-1364.2009.00579.x
Delcher, A. L., Harmon, D., Kasif, S., White, O., and Salzberg, S. L. (1999). Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 27, 4636–4641. doi: 10.1093/nar/27.23.4636
Dickinson, J. R., Salgado, L. E., and Hewlins, M. J. (2003). The catabolism of amino acids to long chain and complex alcohols in Saccharomyces cerevisiae. J. Biol. Chem. 278, 8028–8034. doi: 10.1074/jbc.M211914200
Domizio, P., Romani, C., Comitini, F., Gobbi, M., Lencioni, L., Mannazzu, I., et al. (2011). Potential spoilage non-Saccharomyces yeasts in mixed cultures with Saccharomyces cerevisiae. Ann. Microbiol. 61, 137–144. doi: 10.1007/s13213-010-0125-1
Ehrlich, F. (1907). Über die Bedingungen der Fuselölbildung und über ihren Zusammenhang mit dem Eiweissaufbau der Hefe. Ber. Dtsch. Chem. Ges. 40, 1027–1047. doi: 10.1002/cber.190704001156
Fan, W., Shen, H., and Xu, Y. (2011). Quantification of volatile compounds in Chinese soy sauce aroma type liquor by stir bar sorptive extraction and gas chromatography-mass spectrometry. J. Sci. Food Agric. 91, 1187–1198. doi: 10.1002/jsfa.4294
Galeote, V., Bigey, F., Devillers, H., Neuvéglise, C., and Dequin, S. (2013). Genome sequence of the food spoilage yeast Zygosaccharomyces bailii CLIB 213T. Genome Announc. 1, e00606–e00613. doi: 10.1128/genomeA.00606-13
Garavaglia, J., Schneider, Rde. C., Camargo Mendes, S. D., Welke, J. E., Zini, C. A., Caramão, E. B., et al. (2015). Evaluation of Zygosaccharomyces bailii BCV 08 as a co-starter in wine fermentation for the improvement of ethyl esters production. Microbiol. Res. 173, 59–65. doi: 10.1016/j.micres.2015.02.002
Hazelwood, L. A., Daran, J. M., van Maris, A. J., Pronk, J. T., and Dickinson, J. R. (2008). The Ehrlich pathway for fusel alcohol production: a century of research on Saccharomyces cerevisiae metabolism. Appl. Environ. Microbiol. 74, 3920–3920. doi: 10.1128/AEM.02625-07
Iraqui, I., Vissers, S., Cartiaux, M., and Urrestarazu, A. (1998). Characterisation of Saccharomyces cerevisiae ARO8 and ARO9 genes encoding aromatic aminotransferases I and II reveals a new aminotransferase subfamily. Mol. Gen. Genet. 257, 238–248. doi: 10.1007/s004380050644
Jauniaux, J. C., and Grenson, M. (1990). GAP1, the general amino acid permease gene of Saccharomyces cerevisiae. Nucleotide sequence, protein similarity with the other bakers yeast amino acid permeases, and nitrogen catabolite repression. Eur. J. Biochem. 190, 39–44. doi: 10.1111/j.1432-1033.1990.tb15542.x
Jayabalan, R., Malini, K., Sathishkumar, M., Swaminathan, K., and Yun, S. E. (2010). Biochemical characteristics of tea fungus produced during kombucha fermentation. Food Sci. Biotechnol. 19, 843–847. doi: 10.1007/s10068-010-0119-6
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, S. L. (2015). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14:R36. doi: 10.1186/gb-2013-14-4-r36
Kong, Y., Wu, Q., Zhang, Y., and Xu, Y. (2014). In situ analysis of metabolic characteristics reveals the key yeast in the spontaneous and solid-state fermentation process of Chinese light-style liquor. Appl. Environ. Microbiol. 80, 3667–3676. doi: 10.1128/AEM.04219-13
Korf, I. (2004). Gene finding in novel genomes. BMC Bioinformatics 5:59. doi: 10.1186/1471-2105-5-59
Lagesen, K., Hallin, P., Rødland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lu, X., Wu, Q., Zhang, Y., and Xu, Y. (2015). Genomic and transcriptomic analyses of the Chinese Maotai-flavored liquor yeast MT1 revealed its unique multi-carbon co-utilization. BMC Genomics 16:1064. doi: 10.1186/s12864-015-2263-0
Meng, X., Wu, Q., Wang, L., Wang, D., Chen, L., and Xu, Y. (2015). Improving flavor metabolism of Saccharomyces cerevisiae by mixed culture with Bacillus licheniformis for Chinese Maotai-flavor liquor making. J. Ind. Microbiol. Biotechnol. 42, 1601–1608. doi: 10.1007/s10295-015-1647-0
Mira, N. P., Münsterkötter, M., Dias-Valada, F., Santos, J., Palma, M., Roque, F. C., et al. (2014). The genome sequence of the highly acetic acid-tolerant Zygosaccharomyces bailii-derived interspecies hybrid strain ISA1307, isolated from a sparkling wine plant. DNA Res. 21, 299–313. doi: 10.1093/dnares/dst058
Mortazavi, A., Williams, B. A., Mccue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Nederbragt, A. J. (2014). On the middle ground between open source and commercial software - the case of the Newbler program. Genome Biol. 15, 1–2. doi: 10.1186/gb4173
Palma, M., Münsterkötter, M., Peça, J., Güldener, U., and Sá-Correia, I. (2017). Genome sequence of the highly weak-acid-tolerant Zygosaccharomyces bailii IST302, amenable to genetic manipulations and physiological studies. FEMS Yeast Res. 17:fox025. doi: 10.1093/femsyr/fox025
Palma, M., Roque Fde, C., Guerreiro, J. F., Mira, N. P., Queiroz, L., and Sá-Correia, I. (2015). Search for genes responsible for the remarkably high acetic acid tolerance of a Zygosaccharomyces bailii-derived interspecies hybrid strain. BMC Genomics 16:1070. doi: 10.1186/s12864-015-2278-6
Parra, G., Bradnam, K., and Korf, I. (2007). CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067. doi: 10.1093/bioinformatics/btm071
Powell, S., Szklarczyk, D., Trachana, K., Roth, A., Kuhn, M., Muller, J., et al. (2012). eggNOG v3. 0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 40, 284–289. doi: 10.1093/nar/gkr1060
Quail, M. A., Smith, M., Coupland, P., Otto, T. D., Harris, S. R., Connor, T. R., et al. (2012). A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics 13:341. doi: 10.1186/1471-2164-13-341
Regenberg, B., Düring-Olsen, L., Kielland-Brandt, M. C., and Holmberg, S. (1999). Substrate specificity and gene expression of the amino-acid permeases in Saccharomyces cerevisiae. Curr. Genet. 36, 317–328. doi: 10.1007/s002940050506
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Sipiczki, M. (2008). Interspecies hybridization and recombination in Saccharomyces wine yeasts. FEMS Yeast Res. 8, 996–1007. doi: 10.1111/j.1567-1364.2008.00369.x
Solieri, L., Landi, S., De Vero, L., and Giudici, P. (2006). Molecular assessment of indigenous yeast population from traditional balsamic vinegar. J Appl. Microbiol. 101, 63–71. doi: 10.1111/j.1365-2672.2006.02906.x
Stanke, M., and Waack, S. (2003). Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19(Suppl. 2), ii215–ii225. doi: 10.1093/bioinformatics/btg1080
Stratford, M., Steels, H., Nebe-Von-Caron, G., Novodvorska, M., Hayer, K., and Archer, D. B. (2013). Extreme resistance to weak-acid preservatives in the spoilage yeast Zygosaccharomyces bailii. Int. J. Food Microbiol. 166, 126–134. doi: 10.1016/j.ijfoodmicro.2013.06.025
Suh, S.-O., Gujjari, P., Beres, C., Beck, B., and Zhou, J. (2013). Proposal of Zygosaccharomyces parabailii sp. nov. and Zygosaccharomyces pseudobailii sp. nov., novel species closely related to Zygosaccharomyces bailii. Int. J. Syst. Evol. Microbiol. 63, 1922–1929. doi: 10.1099/ijs.0.048058-0
Teoh, A. L., Heard, G., and Cox, J. (2004). Yeast ecology of Kombucha fermentation. Int. J. Food Microbiol. 95, 119–126. doi: 10.1016/j.ijfoodmicro.2003.12.020
ter Schure, E. G., Flikweert, M. T., van Dijken, J. P., Pronk, J. T., and Verrips, C. T. (1998). Pyruvate decarboxylase catalyzes decarboxylation of branched-chain 2-oxo acids but is not essential for fusel alcohol production by Saccharomyces cerevisiae. Appl. Environ. Microbiol. 64, 1303–1307.
Vuralhan, Z., Luttik, M. A., Tai, S. L., Boer, V. M., Morais, M. A., Schipper, D., et al. (2005). Physiological characterization of the ARO10-dependent, broad-substrate-specificity 2-oxo acid decarboxylase activity of Saccharomyces cerevisiae. Appl. Environ. Microbiol. 71, 3276–3284. doi: 10.1128/AEM.71.6.3276-3284.2005
Wu, Q., Chen, L., and Xu, Y. (2013). Yeast community associated with the solid state fermentation of traditional Chinese Maotai-flavor liquor. Int. J. Food Microbiol. 166, 323–330. doi: 10.1016/j.ijfoodmicro.2013.07.003
Wu, Q., Xu, Y., and Chen, L. (2012). Diversity of yeast species during fermentative process contributing to Chinese Maotai-flavour liquor making. Lett. Appl. Microbiol. 55, 301–307. doi: 10.1111/j.1472-765X.2012.03294.x
Wu, X. (2013). Diversity and Dynamics of Yeasts and Bacteria during the solid State Fermentative Process Contributing to Chinese Maotai-Flavor Liquor Making. Master's thesis, Jiangnan University, Jiangsu.
Xu, M. L., Yu, Y., Ramaswamy, H., and Zhu, S. (2017). Characterization of Chinese liquor aroma components during aging process and liquor age discrimination using gas chromatography combined with multivariable statistics. Sci. Rep. 7:39671. doi: 10.1038/srep39671
Xu, Y., and Ji, K. (2012). 15–Moutai (Maotai): production and sensory properties. Alcoholic Beverages 25, 315–330. doi: 10.1533/9780857095176.3.315
Xu, Y., Wu, Q., and Chen, L. (2013). Screening and Application of the Yeast CGMCC 4745 with High Ethanol Production and Low Fat Fusel Oil in Chinese Maotai-Flavor Liquor Fermentation. Chinese Patent, CN102816706A.
Keywords: Zygosaccharomyces bailii, food fermentation, flavor compounds, genome, transcriptome, Chinese Maotai-flavor liquor
Citation: Xu Y, Zhi Y, Wu Q, Du R and Xu Y (2017) Zygosaccharomyces bailii Is a Potential Producer of Various Flavor Compounds in Chinese Maotai-Flavor Liquor Fermentation. Front. Microbiol. 8:2609. doi: 10.3389/fmicb.2017.02609
Received: 06 July 2017; Accepted: 14 December 2017;
Published: 22 December 2017.
Edited by:
Florence Abram, National University of Ireland Galway, IrelandReviewed by:
Jyoti Prakash Tamang, Sikkim University, IndiaValentina Bernini, Università degli Studi di Parma, Italy
Copyright © 2017 Xu, Zhi, Wu, Du and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qun Wu, d3VxQGppYW5nbmFuLmVkdS5jbg==
Yan Xu, eXh1QGppYW5nbmFuLmVkdS5jbg==