
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 20 December 2017
Sec. Systems Microbiology
Volume 8 - 2017 | https://doi.org/10.3389/fmicb.2017.02504
This article is part of the Research Topic The Potential of Gut Microbiomes for Biotechnological Applications View all 6 articles
 Mabel T. Wong1
Mabel T. Wong1 Weijun Wang1Marie Couturier2,3
Weijun Wang1Marie Couturier2,3 Fakhria M. Razeq1
Fakhria M. Razeq1 Vincent Lombard4,5
Vincent Lombard4,5 Pascal Lapebie4
Pascal Lapebie4 Elizabeth A. Edwards1
Elizabeth A. Edwards1 Nicolas Terrapon4
Nicolas Terrapon4 Bernard Henrissat5,6
Bernard Henrissat5,6 Emma R. Master1,7*
Emma R. Master1,7*To identify carbohydrate-active enzymes (CAZymes) that might be particularly relevant for wood fiber processing, we performed a comparative metagenomic analysis of digestive systems from Canadian beaver (Castor canadensis) and North American moose (Alces americanus) following 3 years of enrichment on either microcrystalline cellulose or poplar hydrolysate. In total, 9,386 genes encoding CAZymes and carbohydrate-binding modules (CBMs) were identified, with up to half predicted to originate from Firmicutes, Bacteroidetes, Chloroflexi, and Proteobacteria phyla, and up to 17% from unknown phyla. Both PCA and hierarchical cluster analysis distinguished the annotated glycoside hydrolase (GH) distributions identified herein, from those previously reported for grass-feeding mammals and herbivorous foragers. The CAZyme profile of moose rumen enrichments also differed from a recently reported moose rumen metagenome, most notably by the absence of GH13-appended dockerins. Consistent with substrate-driven convergence, CAZyme profiles from both poplar hydrolysate-fed cultures differed from cellulose-fed cultures, most notably by increased numbers of unique sequences belonging to families GH3, GH5, GH43, GH53, and CE1. Moreover, pairwise comparisons of moose rumen enrichments further revealed higher counts of GH127 and CE15 families in cultures fed with poplar hydrolysate. To expand our scope to lesser known carbohydrate-active proteins, we identified and compared multi-domain proteins comprising both a CBM and domain of unknown function (DUF) as well as proteins with unknown function within the 416 predicted polysaccharide utilization loci (PULs). Interestingly, DUF362, identified in iron–sulfur proteins, was consistently appended to CBM9; on the other hand, proteins with unknown function from PULs shared little identity unless from identical PULs. Overall, this study sheds new light on the lignocellulose degrading capabilities of microbes originating from digestive systems of mammals known for fiber-rich diets, and highlights the value of enrichment to select new CAZymes from metagenome sequences for future biochemical characterization.
Lignocellulose comprises the non-edible fraction of plant biomass and as such is a recognized resource for the production of renewable energy, chemicals, and materials. The main components of lignocellulose are cellulose, hemicellulose, pectin, and lignin, the proportions and particular chemistries of which depend largely on the plant type and fraction (Kumar et al., 2008). For instance, glucuronoarabinoxylan is a typical hemicellulose found in agricultural crops, whereas glucuronoxylan is the predominant hemicellulose in wood tissue of deciduous trees, including poplar (Peng et al., 2012). Bioconversion pathways that convert various lignocellulose sources to targeted end products require the concerted action of CAZymes, which include glycosyl hydrolases (GHs), carbohydrate esterases (CEs), polysaccharide lyases (PLs), auxiliary activities (AAs), and of carbohydrate binding modules (CBMs) that are classified into families according to amino acid sequence similarity in the CAZy database1 (Lombard et al., 2014). Various pretreatment methods, including steam explosion, have been developed to maximize enzymatic conversion of lignocellulosic resources (Excoffier et al., 1991).
Metagenomic approaches to identify CAZymes relevant to the conversion of a given biomass feedstock have considered environmental samples persistently subjected to the targeted feedstock. For example, metagenomic analyses aimed at identifying CAZymes most relevant to bioconversion of non-woody biomass have sampled digestive systems of animals that graze on straw, grasses, and lichens (Pope et al., 2010). Parallel metagenomic analyses to identify CAZymes contributing to bioconversion of woody biomass have included samples ranging from forest soils (Damon et al., 2012; Pold et al., 2016), to insects (Warnecke et al., 2007; He et al., 2013; Rossmassler et al., 2015), and wood-feeding mollusks (O’Connor et al., 2014). Alternatively, enrichment of environmental samples on specific biomass feedstocks prior to metagenome sequencing can improve sequence assembly (DeAngelis et al., 2010), while facilitating the identification of most pertinent CAZymes (van der Lelie et al., 2012). In a few cases, direct comparison of CAZyme profiles has also been performed for enrichment cultures originating from the same source. Examples include soil-derived microbial communities enriched with wheat straw, switchgrass, and corn stover (Jimenéz et al., 2016), and those digesting mixed lignocellulosic substrates in stationary versus submerged and agitated conditions (Heiss-Blanquet et al., 2016; Wang et al., 2016). In addition to the influence of enrichment condition, such metagenomic studies highlight the increase in number of CAZyme sequences from families associated with hydrolysis of oligosaccharides and side groups of hemicelluloses and/or pectins (e.g., GH3, GH43).
Aside from identifying CAZyme families most pertinent to conversion of specific biomass feedstocks, metagenomic analysis of biomass-degrading communities has uncovered a diverse array of encoded polysaccharide utilization loci (PULs) and multi-modular proteins. Briefly, PULs comprise physically linked genes that encode CAZymes and other proteins that work in concert to degrade specific glycans. Accordingly, PULs have emerged as especially fruitful regions within metagenome sequences for enzyme discovery (Larsbrink et al., 2016; Patrascu et al., 2017). For instance, in the past year alone, detailed biochemical characterization of PULs with different selectivity has uncovered novel activities that contribute to the degradation of pectin (Ndeh et al., 2017), xylan (Wang et al., 2016), and galactomannan (Bagenholm et al., 2017), as well as fungal cell wall components including chitin (Larsbrink et al., 2016) and β-glucans (Temple et al., 2017). Likewise, multi-modular proteins and cellulosomal subunits identified from metagenome sequences and bacterial isolates constitute an additional reservoir for CAZyme discovery (Zhang et al., 2014). Most recently, CAZyme-linked dockerins were reported in PULs in a moose rumen microbiome (Svartström et al., 2017).
With the aim to identify CAZymes and novel proteins that target woody biomass, here we applied a comparative metagenomics approach to identify microbial enzymes encoded by the gut digestive microflora of wood-feeding Canadian beaver (Castor canadensis) and North American moose (Alces americanus) that are likely to promote the conversion of pretreated wood chips. Briefly, in winter months especially, beavers apply an obligate woody diet consisting of twigs, bark, and tree trunks; such seasonal confinement to a wood-based diet is also common to moose, ungulates that consume twigs, shrubs, and bark during the winter (Chaney, 2003; Hood and Bayley, 2009). In an earlier study, we confirmed the existence of biomass-degrading microorganisms in gut digestive microflora of beaver and moose, and their ability to transform lignocellulosic substrates under anaerobic conditions (Wong et al., 2016). Herein, we report the metagenomes of corresponding gut digestive microflora enriched for over 3 years on either microcrystalline cellulose or pretreated poplar wood chips. In particular, metagenome sequences were compared to reveal CAZyme families that are consistently enriched following growth on poplar hydrolysate compared to growth on cellulose. The four metagenomes were also mined in an effort to identify novel candidate enzymes for future characterization. Two approaches were devised to facilitate this analysis: (1) prediction of bacterial PULs and analysis of encoded proteins with unknown function within PULs, and (2) prediction of multi-modular proteins that comprise both modules recognized to contribute to polysaccharide conversion (e.g., a carbohydrate-binding domain) and domains with unknown function.
An ethics approval from an Animal Care and Use Committee was not required by the Office of Research Ethics of the University of Toronto, as the moose rumen sample was collected from a dead moose that was hunted in the wild for meat by a registered hunter with a license authorized by the Ministry of Natural Resources and Forestry under Government of Ontario, Canada.
As previously described (Wong et al., 2016), lignocellulose-degrading microorganisms from the digestive systems of Canadian beaver (Castor canadensis) and North American moose (Alces americanus) were sampled and enriched under anaerobic conditions at 36°C. Briefly, approximately 15 ml of the beaver dropping and moose rumen inocula were transferred to separate 160 mM Wheaton glass serum bottles, which were amended with 45 mL of sulfide-reduced mineral medium and 36 mg COD equivalents of microcrystalline cellulose (Avicel PH101; purchased from Sigma–Aldrich) or steam exploded poplar hydrolysate (provided by SunOpta Inc., Canada) (Wong et al., 2016). Biogas production by resulting cultures was carefully monitored to track metabolic activity.
Following 3 years of cultivation and 10 enrichment phases, 10 ml of each enrichment culture was harvested at early stationary phase of biogas production. Samples were centrifuged at 15,000 × g for 15 min at 4°C, and total community DNA was extracted using the QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany) (Supplementary Table S1). The concentration and quality of the extracted metagenomic DNA were assessed by measuring the 260/280 absorbance ratio using a Nanodrop 2000 Spectrophotometer (Thermo Scientific, MA, United States), and then stored at -80°C. A TruSeq library was constructed for each DNA sample. Illumina paired-end sequencing was chosen and performed with Illumina HiSeq 2000 (Illumina Inc., San Diego, CA, United States) at Génome Québec Innovation Centre.
The output reads were processed by Trimmomatic 0.32 for the removal of adapters and quality filtering (Bolger et al., 2014). The quality-trimmed metagenomic reads were assembled using ABySS with minimum coverage of 20, and minimum kmer length of 96 nucleotides (Simpson et al., 2009). Using an in house Perl script, NKD of the assembled contigs were calculated using the formula NKD = n/(L-k+1), where n is the total number of kmers assembled to the contig, L is the contig length, and k is the kmer length (96) used in the assembly. Contig distributions were then visualized by plotting the calculated NKD against contig length.
Assembled contigs were subjected to ORF prediction using Prodigal (Hyatt et al., 2010); predicted proteins were then assigned to CAZyme families using a combination of BLAST and HMM searches against CAZy reference sequences and families as already described (Al-Masaudi et al., 2017). Counts of CAZyme sequences were normalized to compare the diversity of CAZyme sequences identified within each enrichment culture. Specifically, since the number of predicted ORFs was highest for the metagenome of beaver droppings enriched on poplar hydrolysate (BD-PH, Table 1) then:
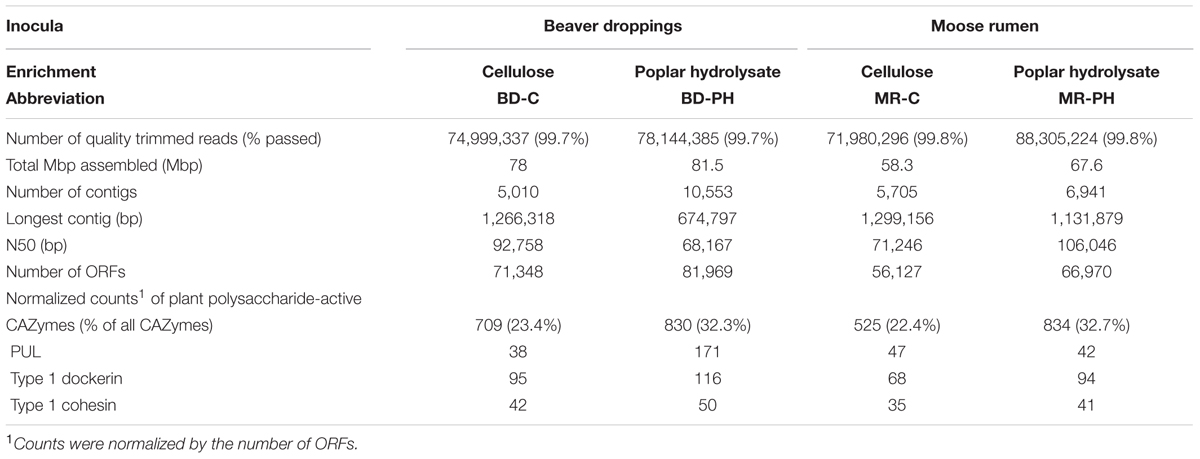
TABLE 1. Statistics of the sequencing and assembly of the metagenomes of cellulose (C) and poplar hydrolysate (PH)-fed enrichment cultures from beaver dropping (BD) and moose rumen (MR).
Relative abundance for a given CAZyme family in a metagenome was calculated by
Public metagenomes from the digestive systems of cow (Hess et al., 2011), moose (Svartström et al., 2017), panda (Zhu et al., 2011), reindeer (Pope et al., 2012), Saudi sheep (Al-Masaudi et al., 2017), termite (Warnecke et al., 2007), and wallaby (Pope et al., 2010) were also reannotated based on the latest version of the CAZy database and included in this calculation. Relative abundances of predicted plant (poly)saccharide-active CAZyme families (Figure 4) were then extracted for hierarchical clustering (correlation clustering and average linkage) and PCA using R statistics in ClustVis (Metsalu and Vilo, 2015). Taxonomic assignment of predicted CAZymes from each metagenome was determined using protein sequences belonging to archaea, bacteria, fungi, other microbial eukaryotes, and viruses reported in the NCBI-NR database (downloaded 16 May 2017) using Kaiju in greedy mode with default settings (Menzel et al., 2016). The phylogenetic distributions of the top 10 identified organisms were visualized at phylum and class levels in a chord diagram using Circos (Krzywinski et al., 2009).
Cellulosomal modules (dockerin and cohesin domains) as well as S-layer homology domains were identified using reference sequences and models built from the literature (Mesnage et al., 2000; Venditto et al., 2016; Artzi et al., 2017). Proteins with CBMs appended to >200 amino acids not covered by any CAZy module were subjected to Pfam domain annotation (Finn et al., 2016) using InterProScan (Jones et al., 2014) to identify conserved DUF (Goodacre et al., 2014). PULs were predicted around susCD-like genes, and boundaries were extended based on intergenic distances, the presence of CAZymes and of regulatory genes (e.g., hybrid two-component system protein, extracytoplasmic function (ECF) σ/anti-σ factors, etc.) following the automatic method used in PULDB (Terrapon et al., 2015). The proteins with unknown functions from the PULs reported here and in PULDB2 were pooled and submitted to CD-HIT web server (Huang et al., 2010) to identify proteins that meet a similarity threshold, which was defined by being ≥70% identical to the representative sequence, and having ≥70% alignment coverage. The sequence reads were submitted to NCBI under the Bioproject ID SUB1022597.
Each metagenomic DNA sample (Supplementary Table S1) yielded 71–88 Mb high-quality reads (150 bp long each), which were assembled into 5,010 to 10,553 contigs per metagenome (Table 1). The N50 (i.e., the length at which 50% of the assembled contigs were equal to or longer than) ranged between 68 and 105 kbp, and longest contigs were between 674 kbp and 1.13 Mbp. As shown in the summary of contig profiles (Supplementary Figure S1), longer contigs were generally present at lower NKD.
In total, 9,386 genes encoding CAZymes and CBMs were predicted from the four metagenomes (Supplementary Table S2). These sequences were assigned to 100 distinct families of GHs, 13 families of CEs, 15 families of PLs, and 39 families of GTs, as well as 43 families of associated CBMs. As observed for other anaerobic microbial communities (Al-Masaudi et al., 2017; Svartström et al., 2017), no auxiliary redox enzymes were identified.
On average, 13% (up to 17% in cellulose-fed moose rumen culture) of the annotated CAZymes were taxonomically unassigned or assigned to unknown species. The phylogenetic origins of the remaining varied among the metagenomes, with most of them derived from Firmicutes, Bacteroidetes, Chloroflexi, and Proteobacteria phyla (Figure 1). Up to 14% of predicted CAZyme sequences from cellulose-fed enrichments were assigned to class Anaerolineae, whereas CAZyme assignments to class Gammaproteobacteria were unique to cultures fed on poplar hydrolysate (Figure 1 and Supplementary Figure S2). By contrast, members from Clostridia and Bacteroidia classes contributed to 23–52% of the annotated CAZymes across the metagenomes, where representation by these classes was between two and five times higher in poplar hydrolysate-fed cultures than in cellulose-fed ones. Moreover, CAZyme families that comprise plant polysaccharide-active enzymes (i.e., families GH2, GH3, GH5, GH9, GH43, GH51, and CE1) were most frequently assigned to either Clostridia or Bacteroidia (Supplementary Figure S2). In particular, GH5 sequences were most frequently assigned to Clostridia in poplar hydrolysate-fed cultures, whereas GH2, GH3, and GH43 were most frequently assigned to class Bacteroidia in poplar hydrolysate-fed cultures.
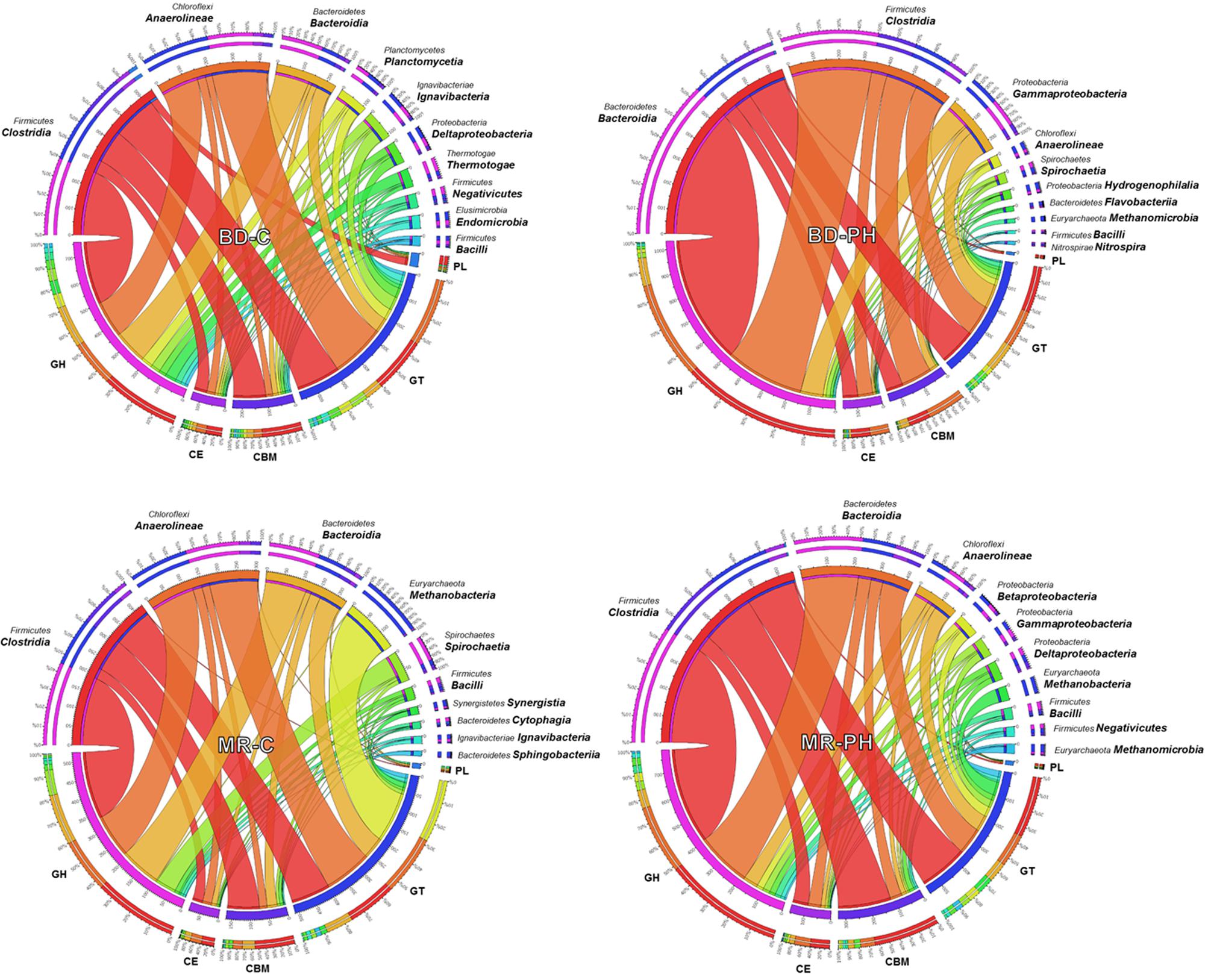
FIGURE 1. Phylogenetic distribution of CAZyme sequences assigned to the top 10 identified classes.
Considering all plant polysaccharides-degrading CAZymes predicted in each metagenome, more than half were less than 60% identical to the CAZyme amino acid sequences reported in the CAZy database (Figure 2). While percent identities varied depending on CAZyme family (Supplementary Figure S3), the most divergent sequences belong to families GH113 (on average 34% identical to their closest blast hits), PL22 (35%), GH5 (40%), GH74 (40%), and PL9 (43%).
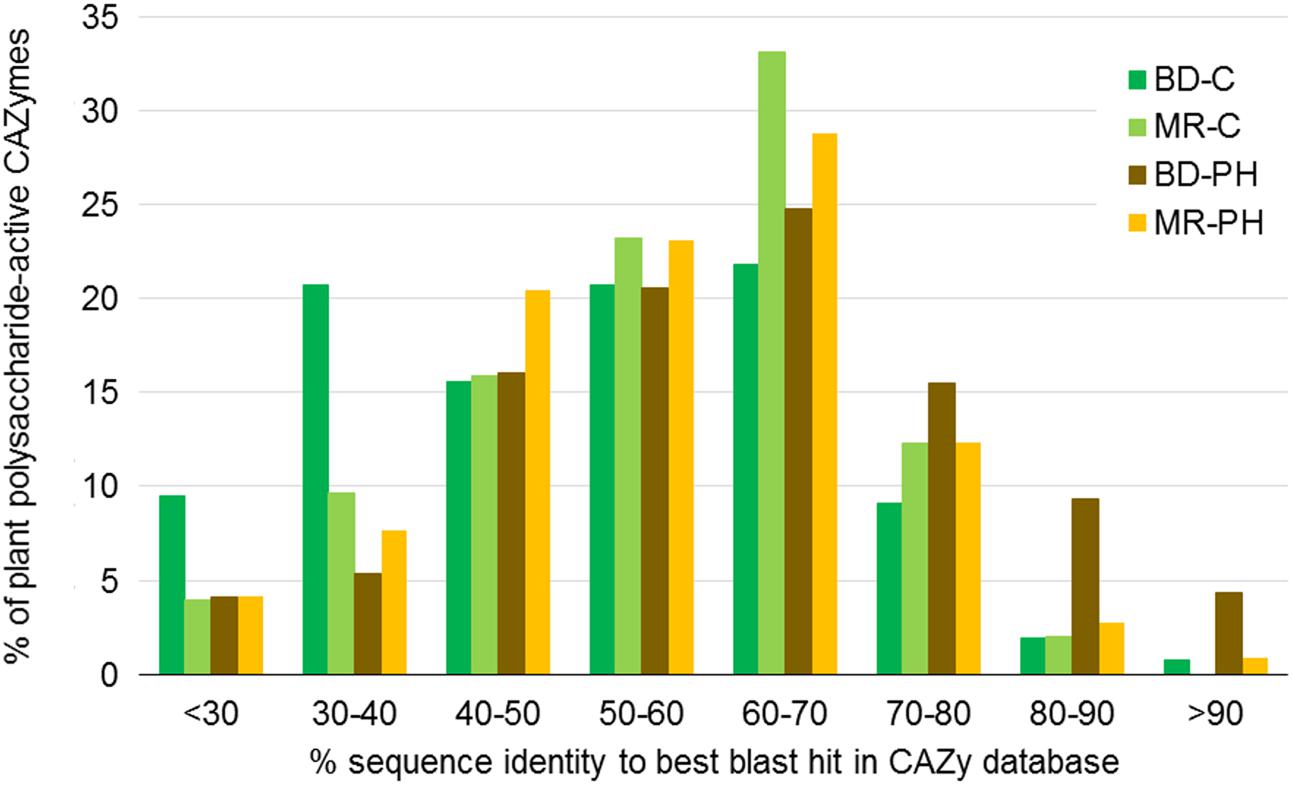
FIGURE 2. Percent identity between amino acid sequences in the CAZy database and CAZyme sequences predicted in beaver dropping (BD) and moose rumen (MR) microcosms enriched with cellulose (C) and poplar hydrolysate (PH). Percent identities correspond to best blast hits in the CAZy database, and were obtained for CAZyme sequences belonging to CAZyme families known to contain enzymes that act on plant cell wall carbohydrates (Figure 4A).
Hierarchical clustering analysis of plant polysaccharide-active CAZyme families distinguished those reported in this study from those previously predicted from grass-feeding mammals or mixed plants foragers (Figure 3A). In particular, the distribution of the CAZyme families predicted in moose rumen enrichments differed from that recently reported for the moose rumen metagenome (Svartström et al., 2017), where highest contributing factors were attributed to relatively high abundance of CE4, GH94, and GH78 families, and low abundance of GH2, GH43 in the moose rumen enrichments (Supplementary Table S3). Consistent with substrate-induced convergence (Wong et al., 2016), long-term ex situ enrichment prior to metagenome sequencing also led to higher similarity of CAZyme distributions for cultures fed with the same carbon source (i.e., poplar hydrolysate or cellulose) as opposed to originating from the same environmental source (i.e., moose rumen or beaver droppings) (Figure 3A). The observed substrate-driven convergence of metagenomes was mostly attributed to higher relative abundances of GH2, GH3, GH43, CE1, CE4 in cultures enriched on poplar hydrolysate (Supplementary Table S3). At the same time, a greater overlap of unique CAZyme sequences was observed between cultures fed with the same substrate than those with the same inoculum (Figure 3B). It is also worthwhile to note that the plant polysaccharide-active CAZyme families from termite gut, albeit wood-feeding, do not cluster closely with those from the poplar hydrolysate enrichments due to the latter’s lower relative abundances of GH5, GH10, and GH94 (Supplementary Table S3). This likely reflects differences in the wood substrates consumed, as well as intrinsic differences in the gut microbiome of mammals and insects. Nonetheless, along PC2 where these metagenomes diverge the most, the PCA plot depicted a closer resemblance of CAZyme profiles microbiomes from termite gut and moose rumen samples enriched on poplar hydrolysate, than that between the former and the non-enriched moose rumen.
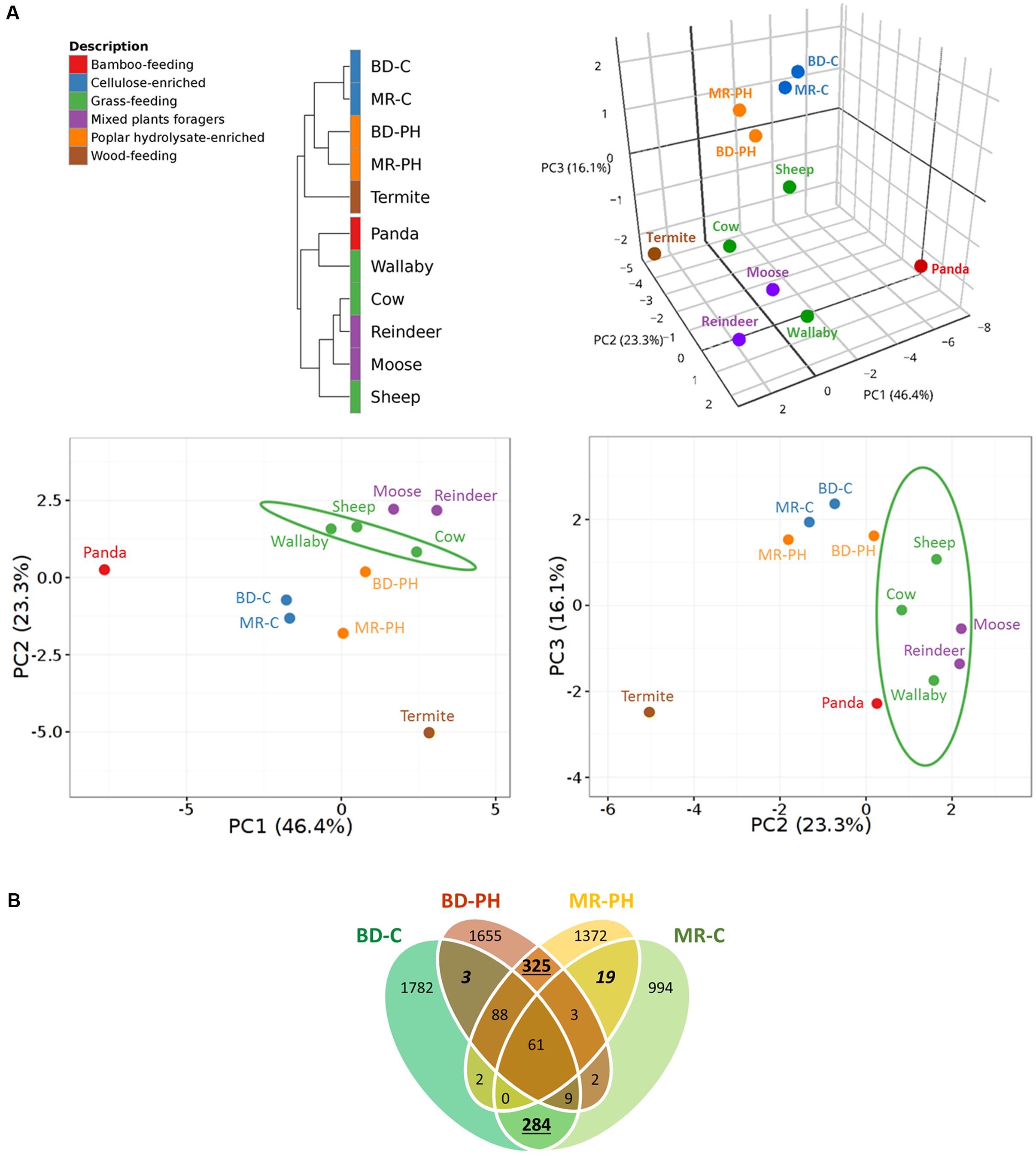
FIGURE 3. (A) Correlation clustering and PCA plots of CAZyme profiles encoded by metagenomes from lignocellulose degrading microbial communities. CAZyme families known to contain enzymes that act on plant cell wall carbohydrates were considered in the analysis. Public datasets included cow (Hess et al., 2011), moose (Svartström et al., 2017), panda (Zhu et al., 2011), reindeer (Pope et al., 2012), Saudi sheep (Al-Masaudi et al., 2017), termite (Warnecke et al., 2007), and wallaby (Pope et al., 2010). A 3D PCA plot is shown on the top right corner with the corresponding 2D PCA plots shown at the bottom; confidence intervals (95%) are indicated by the ellipses. (B) Venn diagram showing a greater overlap of unique CAZyme sequences in cultures fed with the same substrates (numbers underlined) than those that originate from the same inocula (numbers in italics).
Metagenomes of cultures enriched on poplar hydrolysate yielded a higher proportion of predicted plant polysaccharides-active CAZymes (∼33%) compared to metagenomes of cultures enriched on cellulose (∼23%) (Table 1 and Figure 4A). In particular, poplar hydrolysate-degrading communities were enriched in sequence counts from families GH3, GH5, GH43, CE1, and GH53 (Figure 4A). Additional substrate-induced differences were noted when considering moose rumen and beaver dropping enrichments separately. Beaver dropping samples enriched on poplar hydrolysate encoded higher counts of GH2 (1.5 times higher) and GH106 (4.6 times) than corresponding samples enriched on cellulose (Figure 4B). Meanwhile, moose rumen samples enriched on poplar hydrolysate encoded higher counts of GH9 (2 times higher), CE4 (1.8 times), GH127 (9 times), and CE15 (11 times) compared to corresponding samples enriched on cellulose (Figure 4B).
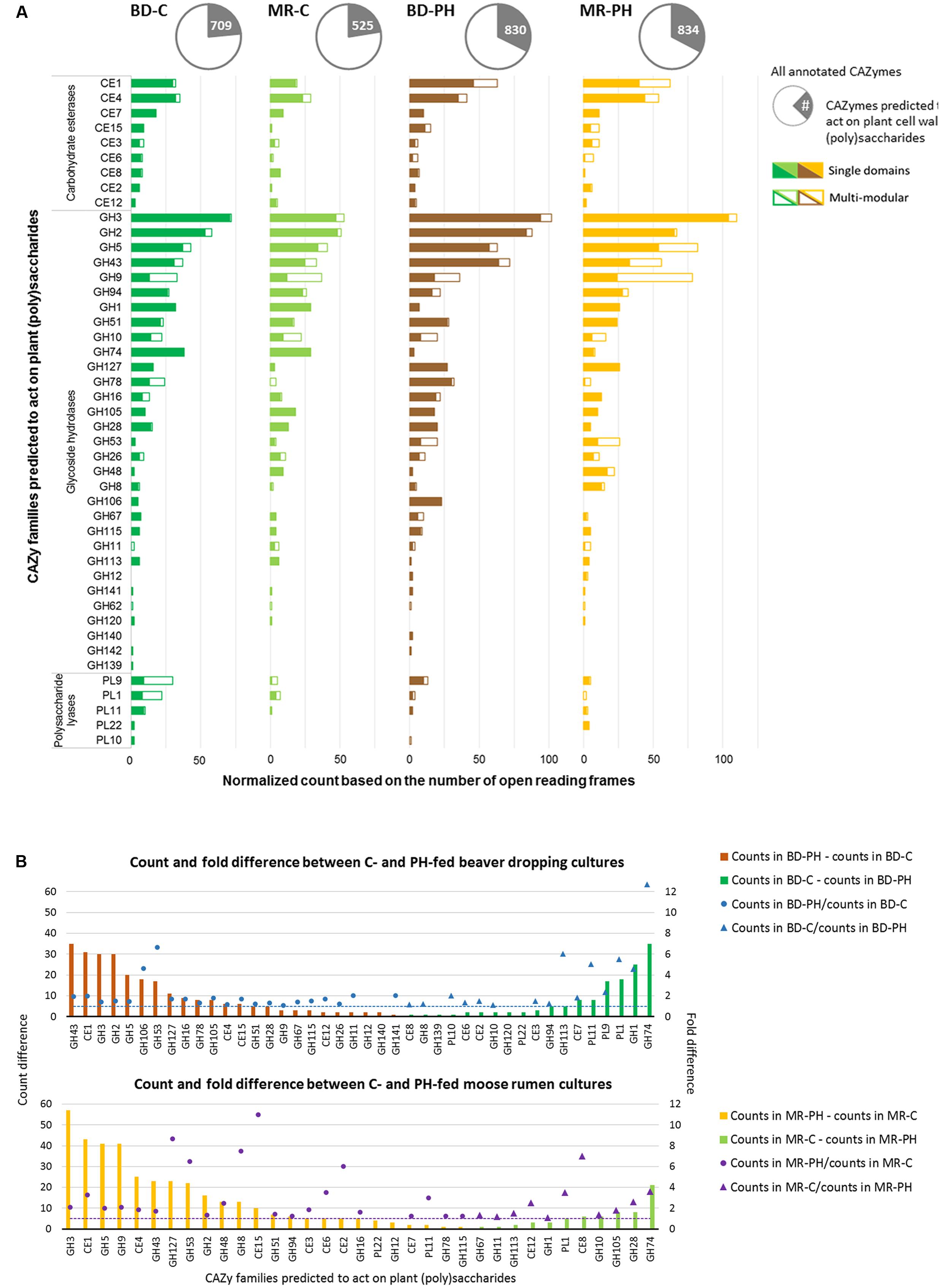
FIGURE 4. (A) Distribution of plant (poly)saccharide degrading-CAZyme families as single and multi-modular domains. (B) Normalized count and fold difference of CAZyme families predicted to act on plant polysaccharides between poplar hydrolysate (PH)- and cellulose (C)-fed beaver dropping (BD) and moose rumen (MR) cultures. Fold difference was only calculated for non-zero counts.
Carbohydrate-active enzymes families that were enriched through growth on poplar hydrolysate included those that comprise enzymes involved in plant polysaccharide deconstruction. For example, family GH43 includes enzymes that target arabinoxylan (Borsenberger et al., 2014; Mewis et al., 2016), family CE1 members were shown to deacetylate polymeric xylans (Neumuller et al., 2015; Mai-Gisondi et al., 2017), and family GH5 members include endoxylanases that targets xylans with or without methyl-glucuronic acid side chain (Gallardo et al., 2010), as well as enzymes that target cellulose and mannans (Aspeborg et al., 2012). Notably, enzymes belonging to families GH2 and GH3 were also abundant in the moose rumen microbiome, and predicted to participate in plant cell wall deconstruction (Svartström et al., 2017). Characterized CE15 members display 4-O-methyl-glucuronoyl methylesterase activity, which are thought to hydrolyze ester linkages that may form between hydroxyl groups in lignin and 4-O-methyl-D-glucuronic acid residues in glucuronoxylans that dominate in hardwood fiber (Biely et al., 2015; Biely, 2016). Recently, a marine bacterial CE15 enzyme predicted to act on alginates was also reported, suggesting a broader substrate range for this CE family (De Santi et al., 2016; Agger et al., 2017). On the other hand, GH127 enzymes typically contain β-L-arabinofuranosidases that have been shown to target plant cell wall glycoproteins, such as extensin (Fujita et al., 2011). By contrast, between 3 and 13 times more GH74 sequences were identified in cellulose-fed enrichments compared to corresponding cultures enriched on poplar hydrolysate, fitting with the endoglucanase activity reported for this CAZyme family (Song et al., 2017). Similarly, 4.6 times more GH1 sequences were identified in the beaver dropping culture enriched on cellulose than that on poplar hydrolysate; characterized bacteria members from this family large act as β-glucosidases that hydrolyze cellobiose and soluble cellodextrins to glucose (Singhania et al., 2013). Interestingly, families capable of pectin degradation (PL1, PL9) were also found at higher abundances in the cellulose-fed beaver dropping culture than that fed with poplar hydrolysate.
Carbohydrate-binding modules can impact enzyme performance through targeting catalytic modules to polysaccharide substrates, and in some cases promote non-hydrolytic fiber disruption (Boraston et al., 2004; Gourlay et al., 2012); accordingly, CAZymes with cognate CBMs were also predicted from each metagenome sequence. About 20% of the sequences predicted to encode plant polysaccharides degrading enzymes (i.e., 669 sequences) were predicted to form multi-domain proteins (Supplementary Table S4). Most frequent domain organizations included CBMs, such as CBM48-GH13_9 (7–8% in cellulose enrichments), GH9-CBM3-CBM3 (∼6% in moose rumen samples enriched on poplar hydrolysate), and CBM50-CBM50-GH18 (∼6% in beaver dropping and moose rumen samples enriched on poplar hydrolysate and cellulose, respectively). While CBM48-GH13 is a documented architecture for starch-degrading enzymes (Machovic and Janecek, 2008), the modular architecture GH9-CBM3-CBM3 was previously only reported as a non-cellulosomal enzyme encoded by Clostridium thermocellum (Anitori, 2012). CBM50-CBM50-GH18, like other GH18 chitinases with multiple CBM50 domains, was predicted to bind peptidoglycan-like and chitin-derived oligosaccharides (Bateman and Bycroft, 2000). Contrary to findings recently reported for the moose rumen microbiome (Svartström et al., 2017), the multi-modular enzymes comprising CBM50 and GH23 or GH73 were identified at low abundance (<3.5%) in the enriched moose rumen metagenomes reported herein.
In addition to multi-modular CAZymes comprising CBMs, those comprising potential cellulosomal subunits were also predicted. Cellulosomes are cell-associated multi-enzyme complexes that are produced by certain anaerobic bacteria to promote polysaccharide degradation (White et al., 2014; Artzi et al., 2017; Smith et al., 2017). When expressed as cell-bound cellulosomes (opposed to cell-free cellulosomes), the primary scaffoldins are connected through type II dockerin-cohesin interactions to specialized anchoring scaffoldins, which contain peptidoglycan-binding S-layer homology modules that anchor to the cell surface. Type I interactions, on the other hand, occur between the dockerin-containing enzymatic subunits and the cohesins on the primary scaffoldins.
For all enrichments, approximately twice the number of dockerins compared to cohesins were predicted (Table 1), and 56% of dockerins were appended to CAZyme sequences (Supplementary Table S5). The most frequently occurring CAZyme module was family GH9 (∼29%), followed by GH5 (∼11%), CE3 (∼8%), GH43 (∼7%), and GH3 (∼6%) (Figure 5 and Supplementary Table S4). And unlike the recent study of the moose rumen metagenome, the recurrent GH13 appended-dockerins (Svartström et al., 2017) were not identified in the current moose rumen metagenomes, likely due to their long enrichment on cellulosic carbon sources. Other common components of cellulosome systems (Artzi et al., 2017), such as GH10 (∼4%), GH11 (∼4%), and GH48 (∼4%) were also identified in the metagenomes of both moose rumen and beaver dropping enrichments, albeit at a lower abundance. Notably, the identification of few sequences containing a GH48 module and the approximately nine-fold higher number of those containing a GH9 module is consistent with the earlier analyses of cellulose-degrading anaerobic bacteria that generate high levels of a single GH48 and diverse GH9 enzymes with potential synergistic action (Morag et al., 1991; Ravachol et al., 2014; Artzi et al., 2015).
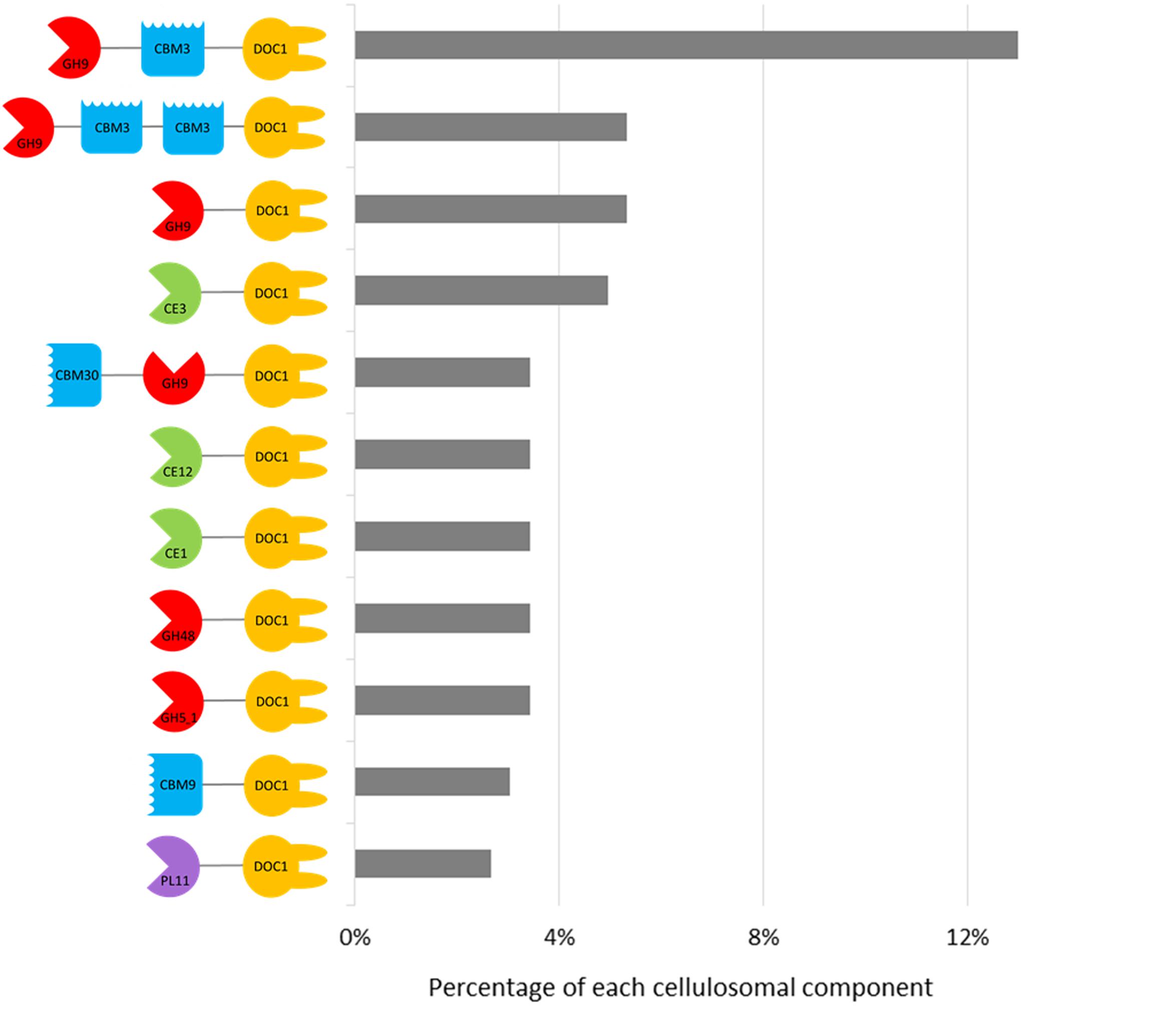
FIGURE 5. Catalog of domain architectures of top 11 abundant CAZy-dockerins in the cellulose- and poplar hydrolysate-fed microbial enrichments from beaver droppings and moose rumen. CBM, carbohydrate-binding module; CE, carbohydrate esterase; DOC1, type 1 dockerin; GH, glycoside hydrolases; PL, polysaccharide lyase.
As summarized above, PULs have emerged as especially fruitful regions within genomic sequences for enzyme discovery (Larsbrink et al., 2016). Herein, 416 PULs were predicted (Supplementary Figure S4), where the normalized number predicted from poplar hydrolysate-fed microcosms of beaver droppings was 4.5 times that predicted from cellulose-fed microcosms. Consistent with the overall distribution of predicted CAZyme sequences, those belonging to families CE1, GH3, and GH43, were most frequently identified in the predicted PULs (Figure 6A and Supplementary Table S6). Moreover, PULs comprising members of families GH127 and GH9 were exclusively identified in metagenomes from cultures enriched on poplar hydrolysate. Based on the family composition of a given PUL, substrate category of the PUL-encoded enzymes can be inferred. For example, Figure 6B illustrates PULs that potentially target xylan and pectin based on the established activities of the CAZyme families. Sequences annotated as unknown may include novel enzyme functions, for instance as shown recently in the case of the type II rhamnogalacturonan PUL of B. thetaiotaomicron (Ndeh et al., 2017).
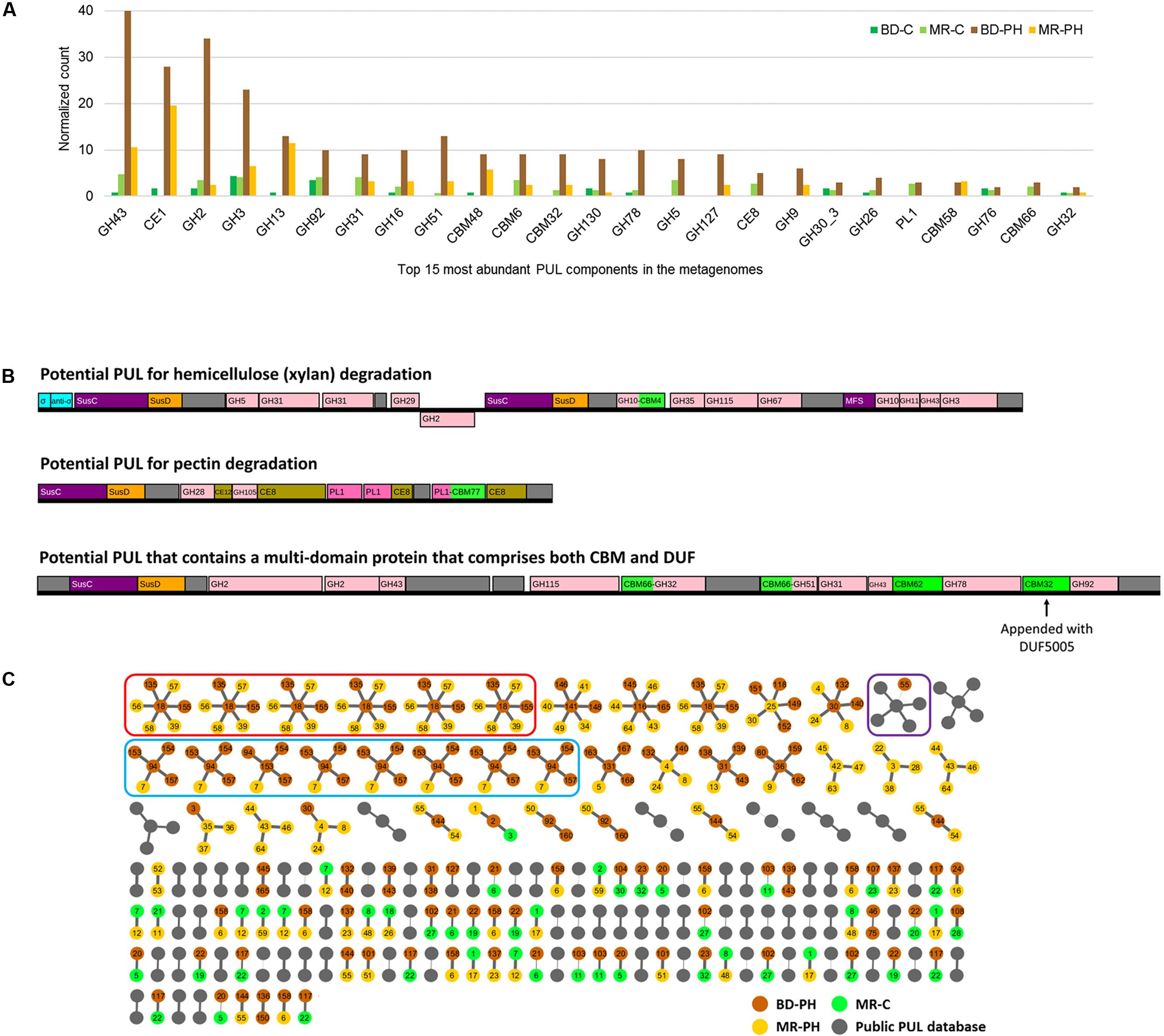
FIGURE 6. (A) Top 15 most abundant CAZyme families identified in predicted PULs from cellulose (C)- and poplar hydrolysate (PH)-fed microbial enrichments of beaver droppings and moose rumen. (B) Examples of predicted PULs from beaver droppings enriched on poplar hydrolysate (BD-PH). (C) Similarity-based clustering (≥70%) of proteins with unknown function positioned in PULs identified herein and listed in the public PUL database (http://www.cazy.org/PULDB/). Each cluster contains a central node that denotes the representative protein with unknown function (defined by the longest length) and connected nodes that represent a protein with unknown function that is ≥70% identical to the representative sequence (see the section “Materials and Methods”). PUL identifiers are shown on each node; the thickness of the edges correlates to percent identity between sequences. Circled in red and blue are proteins with unknown functions that are ≥95% identical to one another; the architecture of PULs circled in red is identical, whereas those circled in blue share a common central architecture but differ at flanking regions. Circled in purple is the only cluster that comprises proteins with unknown function from both PULs predicted herein and those from the public PUL database.
Of note, 620 sequences annotated as proteins with unknown function (with lengths ranging from 32 to 1,320 amino acids) were identified in all candidate PULs (Supplementary Figure S5A). Among these, eight sequences annotated as proteins with unknown function were found to comprise a CBM from family CBM32, CBM35, CBM51, or CBM66. In an effort to prioritize additional sequences for future characterization, a clustering network diagram was generated to uncover protein sequences with unknown function that reoccurred in the predicted PULs. However, little similarity was revealed between such sequences from PULs predicted herein and those reported in the public PULDB (Supplementary Figure S5B). In fact, only one such sequence from beaver droppings enriched on poplar hydrolysate was ≥70% identical to those annotated in the PULDB, and the few sequences with unknown function that did cluster typically originated from PULs with similar architecture (Figure 6C and Supplementary Figure S4).
A second approach to assist the discovery of potentially new CAZyme families considered multi-modular proteins predicted to comprise a DUF, or a sequence of unknown function, appended to a CBM or dockerins (cellulosomal subunit).
Considering all four metagenome sequences reported herein, 62 DUFs were identified that co-occurred with a CBM (Figure 7 and Supplementary Figure S6). The most frequent organizations were: DUF3794-CBM50 (10 identified), DUF362-CBM9-DOC1 (4 identified), DUF4366-CBM16 (4 identified), which were identified in all four metagenomes; five DUF3459-CBM48-GH13_10 sequences were also identified in the metagenome of beaver droppings enriched on poplar hydrolysate (Supplementary Figure S6). As shown herein and also described in the Pfam database (Finn et al., 2016), DUF3794 was often found in association with CBM50. On the other hand, DUF362 is often present in proteins with domains that bind to iron–sulfur clusters, and its coexistence with CBM9_1 in an uncharacterized protein from soil bacteria Sorangium cellulosum was previously observed (UniProt entry S4XJL8). The structure of DUF3459 has been determined (UniProt entries B2IUW9, Q9RX51, Q8ZPF0, Q8P5I2, Q2PS28, M1E1F6, M1E1F3, H3K096), and as observed here, was previously shown to be part of multi-modular proteins comprising GH13 and CBM48 domains (UniProt entries W6LS46, R4KHQ4, C7RTS8). Although not frequently observed, one PUL identified in the poplar hydrolysate-fed beaver droppings culture contained DUF5005 appended to a predicted CBM32 (Figure 6B).
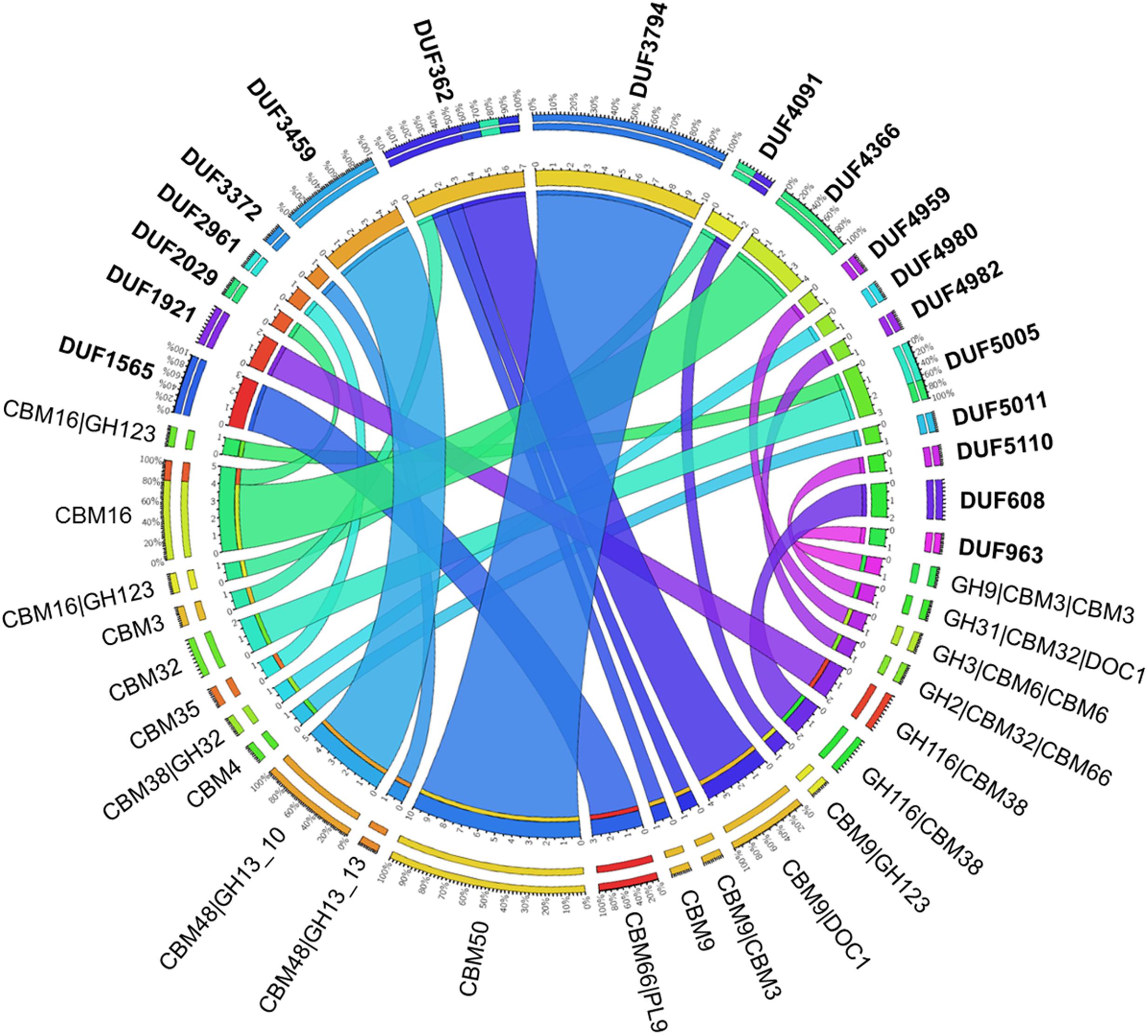
FIGURE 7. Carbohydrate-active proteins with domains of unknown functions (DUFs) identified in the metagenomes. In bold are the DUFs that are identified with various CBMs and CAZymes as shown via the connections color-coded in accordance to the DUFs. The number and corresponding percentage of each combination of modules are shown in the outer scale.
Of the predicted dockerin sequences, 44% lacked known appended CAZyme modules. Similar to previous reports (Finn et al., 2016), a few dockerin sequences were predicted to have appended DUF (i.e., DUF362, DUF1533, and DUF3237); however, the majority were not annotated as containing modules or domains functionally attributed to cellulosomes. In many cases, this could reflect sequence gaps due to incomplete metagenome assembly; however, it is also conceivable that dockerin–cohesin proteins might in fact participate in other biological functions, as suggested by a phylogenetically distinct group of cohesins discovered in the cow rumen metagenome (Bensoussan et al., 2017).
Given their natural dietary habits, the Canadian beaver and North American moose have likely evolved digestive microbiomes with the ability to degrade diverse wood polysaccharides. To identify enzymes that may be most relevant to wood fiber bioprocessing, corresponding microbial communities were enriched for approximately 3 years on comparatively complex (poplar hydrolysate) and defined (microcrystalline cellulose) carbon sources. Enrichment led to substrate-induced convergence of CAZyme profiles, which significantly narrowed the number of CAZyme families and corresponding members that could be targeted and tested for improved enzymatic conversion of wood fiber. For example, in addition to families GH2, GH3, GH5, and GH43 which previous reports have also identified, GH127 and CE15 may be especially relevant for the anaerobic conversion of pretreated wood fiber. Protein sequences containing both a CBM and a DUF, as well as proteins with unknown function yet having signals for secretion and position within PULs, were also identified and may facilitate the discovery of new CAZyme activities. Proteomic analysis of secretomes from the enrichment cultures prepared herein will provide an additional filter for protein selection and characterization. In the interim, however, enrichment followed by comparative metagenomics sufficiently narrowed protein lists of primary interest, enabling direct recombinant production and characterization.
MW performed the sequence analyses, data interpretation, and compiled the manuscript. WW maintained the enrichment cultures, prepared DNA samples for sequencing, and contributed to data interpretation. MC and FR contributed to data interpretation. BH, NT, VL, and PL contributed to the search and annotation of CAZyme modules and PULs, as well as data interpretation. EM and EE conceived and coordinated the study. All authors contributed to the revision of manuscript and approved the final version.
This work was funded by the Government of Ontario for the project “Forest FAB: Applied Genomics for Functionalized Fibre and Biochemicals” (ORF-RE-05-005), the Natural Sciences and Engineering Research Council of Canada for the Strategic Network Grant “Industrial Biocatalysis Network,” the European Research Council (ERC) Grant Agreement 322820 to BH and an ERC Consolidator Grant to EM (BHIVE – 648925). MW was funded by a University of Toronto Fellowship and Professor William F. Graydon Memorial Graduate Fellowship. MC was funded by a Marie Curie International Outgoing Fellowship within the 7th European Community Framework Program.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank C. Washer and O. Molenda for training on using the Trimmomatic and AbySS assembly programs, and also W. Gao and D. Robson for IT support.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.02504/full#supplementary-material
AA, auxiliary activity; ABySS, Assembly By Short Sequences; CAZyme, carbohydrate-active enzyme; CBM, carbohydrate-binding module; CE, carbohydrate esterase; COD, chemical oxygen demand; DUF, domain of unknown function; ECF, extracytoplasmic function; GH, glycoside hydrolase; GT, glycosyltransferase; NCBI-NR, National Center for Biotechnology Information Non-redundant; NKD, normalized kmer depths; ORF, open-reading frame; PCA, principal component analysis; PUL, polysaccharide utilization locus; RPKM, reads per kilobase per million mapped reads.
Agger, J. W., Busk, P. K., Pilgaard, B., Meyer, A. S., and Lange, L. (2017). A new functional classification of glucuronoyl esterases by peptide pattern recognition. Front. Microbiol. 8:309. doi: 10.3389/fmicb.2017.00309
Al-Masaudi, S., El Kaoutari, A., Drula, E., Al-Mehdar, H., Redwan, E. M., Lombard, V., et al. (2017). A metagenomics investigation of carbohydrate-active enzymes along the gastrointestinal tract of Saudi sheep. Front. Microbiol. 8:666. doi: 10.3389/fmicb.2017.00666
Anitori, R. P. (2012). Extremophiles: Microbiology and Biotechnology. Poole: Caister Academic Press.
Artzi, L., Bayer, E. A., and Morais, S. (2017). Cellulosomes: bacterial nanomachines for dismantling plant polysaccharides. Nat. Rev. Microbiol. 15, 83–95. doi: 10.1038/nrmicro.2016.164
Artzi, L., Morag, E., Barak, Y., Lamed, R., and Bayer, E. A. (2015). Clostridium clariflavum: key cellulosome players are revealed by proteomic analysis. mBio 6:e00411-15. doi: 10.1128/mBio.00411-15
Aspeborg, H., Coutinho, P. M., Wang, Y., Brumer, H., and Henrissat, B. (2012). Evolution, substrate specificity and subfamily classification of glycoside hydrolase family 5 (GH5). BMC Evol. Biol. 12:186. doi: 10.1186/1471-2148-12-186
Bagenholm, V., Reddy, S. K., Bouraoui, H., Morrill, J., Kulcinskaja, E., Bahr, C. M., et al. (2017). Galactomannan catabolism conferred by a polysaccharide utilization locus of Bacteroides ovatus- enzyme synergy and crystal structure of a beta-mannanase. J. Biol. Chem. 292, 229–243. doi: 10.1074/jbc.M116.746438
Bateman, A., and Bycroft, M. (2000). The structure of a LysM domain from E. coli membrane-bound lytic murein transglycosylase D (MltD). J. Mol. Biol. 299, 1113–1119. doi: 10.1006/jmbi.2000.3778
Bensoussan, L., Morais, S., Dassa, B., Friedman, N., Henrissat, B., Lombard, V., et al. (2017). Broad phylogeny and functionality of cellulosomal components in the bovine rumen microbiome. Environ. Microbiol. 19, 185–197. doi: 10.1111/1462-2920.13561
Biely, P. (2016). Microbial glucuronoyl esterases: 10 years after discovery. Appl. Environ. Microbiol. 82, 7014–7018. doi: 10.1128/AEM.02396-16
Biely, P., Malovikova, A., Uhliarikova, I., Li, X. L., and Wong, D. W. S. (2015). Glucuronoyl esterases are active on the polymeric substrate methyl esterified glucuronoxylan. FEBS Lett. 589, 2334–2339. doi: 10.1016/j.febslet.2015.07.019
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Boraston, A. B., Bolam, D. N., Gilbert, H. J., and Davies, G. J. (2004). Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem. J. 382, 769–781. doi: 10.1042/BJ20040892
Borsenberger, V., Dornez, E., Desrousseaux, M. L., Massou, S., Tenkanen, M., Courtin, C. M., et al. (2014). A H-1 NMR study of the specificity of alpha-L-arabinofuranosidases on natural and unnatural substrates. Biochim. Biophys. Acta 1840, 3106–3114. doi: 10.1016/j.bbagen.2014.07.001
Chaney, W. R. (2003). Why Do Animals Eat the Bark and Wood of Trees and Shrubs? Available at: https://www.extension.purdue.edu/extmedia/fnr/fnr_203.pdf [accessed October 8, 2017].
Damon, C., Lehembre, F., Oger-Desfeux, C., Luis, P., Ranger, J., Fraissinet-Tachet, L., et al. (2012). Metatranscriptomics reveals the diversity of genes expressed by eukaryotes in forest soils. PLOS ONE 7:e28967. doi: 10.1371/journal.pone.0028967
De Santi, C., Willassen, N. P., and Williamson, A. (2016). Biochemical characterization of a family 15 carbohydrate esterase from a bacterial marine arctic metagenome. PLOS ONE 11:e0159345. doi: 10.1371/journal.pone.0159345
DeAngelis, K. M., Silver, W. L., Thompson, A. W., and Firestone, M. K. (2010). Microbial communities acclimate to recurring changes in soil redox potential status. Environ. Microbiol. 12, 3137–3149. doi: 10.1111/j.1462-2920.2010.02286.x
Excoffier, G., Toussaint, B., and Vignon, M. R. (1991). Saccharification of steam-exploded poplar wood. Biotechnol. Bioeng. 38, 1308–1317. doi: 10.1002/bit.260381108
Finn, R. D., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Mistry, J., Mitchell, A. L., et al. (2016). The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285. doi: 10.1093/nar/gkv1344
Fujita, K., Sakamoto, S., Ono, Y., Wakao, M., Suda, Y., Kitahara, K., et al. (2011). Molecular cloning and characterization of a beta-l-arabinobiosidase in Bifidobacterium longum that belongs to a novel glycoside hydrolase family. J. Biol. Chem. 286, 5143–5150. doi: 10.1074/jbc.M110.190512
Gallardo, O., Fernandez-Fernandez, M., Valls, C., Valenzuela, S. V., Roncero, M. B., Vidal, T., et al. (2010). Characterization of a family GH5 xylanase with activity on neutral oligosaccharides and evaluation as a pulp bleaching aid. Appl. Environ. Microbiol. 76, 6290–6294. doi: 10.1128/AEM.00871-10
Gourlay, K., Arantes, V., and Saddler, J. N. (2012). Use of substructure-specific carbohydrate binding modules to track changes in cellulose accessibility and surface morphology during the amorphogenesis step of enzymatic hydrolysis. Biotechnol. Biofuels 5:51. doi: 10.1186/1754-6834-5-51
He, S. M., Ivanova, N., Kirton, E., Allgaier, M., Bergin, C., Scheffrahn, R. H., et al. (2013). Comparative metagenomic and metatranscriptomic analysis of hindgut paunch microbiota in wood- and dung-feeding higher termites. PLOS ONE 8:e61126. doi: 10.1371/journal.pone.0061126
Heiss-Blanquet, S., Fayolle-Guichard, F., Lombard, V., Hebert, A., Coutinho, P. M., Groppi, A., et al. (2016). Composting-like conditions are more efficient for enrichment and diversity of organisms containing cellulase-encoding genes than submerged cultures. PLOS ONE 11:e0167216. doi: 10.1371/journal.pone.0167216
Hess, M., Sczyrba, A., Egan, R., Kim, T. W., Chokhawala, H., Schroth, G., et al. (2011). Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331, 463–467. doi: 10.1126/science.1200387
Hood, G. A., and Bayley, S. E. (2009). A comparison of riparian plant community response to herbivory by beavers (Castor canadensis) and ungulates in Canada’s boreal mixed-wood forest. For. Ecol. Manage. 258, 1979–1989. doi: 10.1016/j.foreco.2009.07.052
Huang, Y., Niu, B. F., Gao, Y., Fu, L. M., and Li, W. Z. (2010). CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinformatics 26, 680–682. doi: 10.1093/bioinformatics/btq003
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Jimenéz, D. J., Brossi, M. J. D., Schuckel, J., Kracun, S. K., Willats, W. G. T., and Van Elsas, J. D. (2016). Characterization of three plant biomass-degrading microbial consortia by metagenomics- and metasecretomics-based approaches. Appl. Microbiol. Biotechnol. 100, 10463–10477. doi: 10.1007/s00253-016-7713-3
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W. Z., Mcanulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kumar, R., Singh, S., and Singh, O. V. (2008). Bioconversion of lignocellulosic biomass: biochemical and molecular perspectives. J. Ind. Microbiol. Biotechnol. 35, 377–391. doi: 10.1007/s10295-008-0327-8
Larsbrink, J., Zhu, Y., Kharade, S. S., Kwiatkowski, K. J., Eijsink, V. G. H., Koropatkin, N. M., et al. (2016). A polysaccharide utilization locus from Flavobacterium johnsoniae enables conversion of recalcitrant chitin. Biotechnol. Biofuels 9:260. doi: 10.1186/s13068-016-0674-z
Lombard, V., Ramulu, H. G., Drula, E., Coutinho, P. M., and Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495. doi: 10.1093/nar/gkt1178
Machovic, M., and Janecek, S. (2008). Domain evolution in the GH13 pullulanase subfamily with focus on the carbohydrate-binding module family 48. Biologia 63, 1057–1068. doi: 10.2478/s11756-008-0162-4
Mai-Gisondi, G., Maaheimo, H., Chong, S. L., Hinz, S., Tenkanen, M., and Master, E. (2017). Functional comparison of versatile carbohydrate esterases from families CE1, CE6 and CE16 on acetyl-4-O-methylglucuronoxylan and acetyl-galactoglucomannan. Biochim. Biophys. Acta 1861, 2398–2405. doi: 10.1016/j.bbagen.2017.06.002
Menzel, P., Ng, K. L., and Krogh, A. (2016). Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 7:11257. doi: 10.1038/ncomms11257
Mesnage, S., Fontaine, T., Mignot, T., Delepierre, M., Mock, M., and Fouet, A. (2000). Bacterial SLH domain proteins are non-covalently anchored to the cell surface via a conserved mechanism involving wall polysaccharide pyruvylation. EMBO J. 19, 4473–4484. doi: 10.1093/emboj/19.17.4473
Metsalu, T., and Vilo, J. (2015). ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 43, W566–W570. doi: 10.1093/nar/gkv468
Mewis, K., Lenfant, N., Lombard, V., and Henrissat, B. (2016). Dividing the large glycoside hydrolase family 43 into subfamilies: a motivation for detailed enzyme characterization. Appl. Environ. Microbiol. 82, 1686–1692. doi: 10.1128/AEM.03453-15
Morag, E., Halevy, I., Bayer, E. A., and Lamed, R. (1991). Isolation and properties of a major cellobiohydrolase from the cellulosome of Clostridium thermocellum. J. Bacteriol. 173, 4155–4162. doi: 10.1128/jb.173.13.4155-4162.1991
Ndeh, D., Rogowski, A., Cartmell, A., Luis, A. S., Basle, A., Gray, J., et al. (2017). Complex pectin metabolism by gut bacteria reveals novel catalytic functions. Nature 544, 65–70. doi: 10.1038/nature21725
Neumuller, K. G., De Souza, A. C., Van Rijn, J. H. J., Streekstra, H., Gruppen, H., and Schols, H. A. (2015). Positional preferences of acetyl esterases from different CE families towards acetylated 4-O-methyl glucuronic acid-substituted xylo-oligosaccharides. Biotechnol. Biofuels 8:7. doi: 10.1186/s13068-014-0187-6
O’Connor, R. M., Fung, J. M., Sharp, K. H., Benner, J. S., Mcclung, C., Cushing, S., et al. (2014). Gill bacteria enable a novel digestive strategy in a wood-feeding mollusk. Proc. Natl. Acad. Sci. U.S.A. 111, E5096–E5104. doi: 10.1073/pnas.1413110111
Patrascu, O., Beguet-Crespel, F., Marinelli, L., Le Chatelier, E., Abraham, A. L., Leclerc, M., et al. (2017). A fibrolytic potential in the human ileum mucosal microbiota revealed by functional metagenomic. Sci. Rep. 7:40248. doi: 10.1038/srep40248
Peng, F., Peng, P., Xu, F., and Sun, R. C. (2012). Fractional purification and bioconversion of hemicelluloses. Biotechnol. Adv. 30, 879–903. doi: 10.1016/j.biotechadv.2012.01.018
Pold, G., Billings, A. F., Blanchard, J. L., Burkhardt, D. B., Frey, S. D., Melillo, J. M., et al. (2016). Long-term warming alters carbohydrate degradation potential in temperate forest soils. Appl. Environ. Microbiol. 82, 6518–6530. doi: 10.1128/AEM.02012-16
Pope, P. B., Denman, S. E., Jones, M., Tringe, S. G., Barry, K., Malfatti, S. A., et al. (2010). Adaptation to herbivory by the Tammar wallaby includes bacterial and glycoside hydrolase profiles different from other herbivores. Proc. Natl. Acad. Sci. U.S.A. 107, 14793–14798. doi: 10.1073/pnas.1005297107
Pope, P. B., Mackenzie, A. K., Gregor, I., Smith, W., Sundset, M. A., Mchardy, A. C., et al. (2012). Metagenomics of the Svalbard reindeer rumen microbiome reveals abundance of polysaccharide utilization loci. PLOS ONE 7:e38571. doi: 10.1371/journal.pone.0038571
Ravachol, J., Borne, R., Tardif, C., De Philip, P., and Fierobe, H. P. (2014). Characterization of all family-9 glycoside hydrolases synthesized by the cellulosome-producing bacterium Clostridium cellulolyticum. J. Biol. Chem. 289, 7335–7348. doi: 10.1074/jbc.M113.545046
Rossmassler, K., Dietrich, C., Thompson, C., Mikaelyan, A., Nonoh, J. O., Scheffrahn, R. H., et al. (2015). Metagenomic analysis of the microbiota in the highly compartmented hindguts of six wood- or soil-feeding higher termites. Microbiome 3:56. doi: 10.1186/s40168-015-0118-1
Simpson, J. T., Wong, K., Jackman, S. D., Schein, J. E., Jones, S. J., and Birol, I. (2009). ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123. doi: 10.1101/gr.089532.108
Singhania, R. R., Patel, A. K., Sukumaran, R. K., Larroche, C., and Pandey, A. (2013). Role and significance of beta-glucosidases in the hydrolysis of cellulose for bioethanol production. Bioresour. Technol. 127, 500–507. doi: 10.1016/j.biortech.2012.09.012
Smith, S. P., Bayer, E. A., and Czjzek, M. (2017). Continually emerging mechanistic complexity of the multi-enzyme cellulosome complex. Curr. Opin. Struct. Biol. 44, 151–160. doi: 10.1016/j.sbi.2017.03.009
Song, Y. H., Lee, K. T., Baek, J. Y., Kim, M. J., Kwon, M. R., Kim, Y. J., et al. (2017). Isolation and characterization of a novel glycosyl hydrolase family 74 (GH74) cellulase from the black goat rumen metagenomic library. Folia Microbiol. 62, 175–181. doi: 10.1007/s12223-016-0486-3
Svartström, O., Alneberg, J., Terrapon, N., Lombard, V., De Bruijn, I., Malmsten, J., et al. (2017). Ninety-nine de novo assembled genomes from the moose (Alces alces) rumen microbiome provide new insights into microbial plant biomass degradation. ISME J. 11, 2538–2551. doi: 10.1038/ismej.2017.108
Temple, M. J., Cuskin, F., Baslé, A., Hickey, N., Speciale, G., Williams, S. J., et al. (2017). A Bacteroidetes locus dedicated to fungal 1,6-β-glucan degradation: unique substrate conformation drives specificity of the key endo-1,6-β-glucanase. J. Biol. Chem. 292, 10639–10650. doi: 10.1074/jbc.M117.787606
Terrapon, N., Lombard, V., Gilbert, H. J., and Henrissat, B. (2015). Automatic prediction of polysaccharide utilization loci in Bacteroidetes species. Bioinformatics 31, 647–655. doi: 10.1093/bioinformatics/btu716
van der Lelie, D., Taghavi, S., Mccorkle, S. M., Li, L. L., Malfatti, S. A., Monteleone, D., et al. (2012). The metagenome of an anaerobic microbial community decomposing poplar wood chips. PLOS ONE 7:e36740. doi: 10.1371/journal.pone.0036740
Venditto, I., Luis, A. S., Rydahl, M., Schuckel, J., Fernandes, V. O., Vidal-Melgosa, S., et al. (2016). Complexity of the Ruminococcus flavefaciens cellulosome reflects an expansion in glycan recognition. Proc. Natl. Acad. Sci. U.S.A. 113, 7136–7141. doi: 10.1073/pnas.1601558113
Wang, K., Pereira, G. V., Cavalcante, J. J. V., Zhang, M. L., Mackie, R., and Cann, I. (2016). Bacteroides intestinalis DSM 17393, a member of the human colonic microbiome, upregulates multiple endoxylanases during growth on xylan. Sci. Rep. 6:34360. doi: 10.1038/srep34360
Warnecke, F., Luginbuhl, P., Ivanova, N., Ghassemian, M., Richardson, T. H., Stege, J. T., et al. (2007). Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 450, 560–565. doi: 10.1038/nature06269
White, B. A., Lamed, R., Bayer, E. A., and Flint, H. J. (2014). Biomass utilization by gut microbiomes. Annu. Rev. Microbiol. 68, 279–296. doi: 10.1146/annurev-micro-092412-155618
Wong, M. T., Wang, W. J., Lacourt, M., Couturier, M., Edwards, E. A., and Master, E. R. (2016). Substrate-driven convergence of the microbial community in lignocellulose-amended enrichments of gut microflora from the Canadian beaver (Castor canadensis) and North American moose (Alces americanus). Front. Microbiol. 7:961. doi: 10.3389/fmicb.2016.00961
Zhang, M. L., Chekan, J. R., Dodd, D., Hong, P. Y., Radlinski, L., Revindran, V., et al. (2014). Xylan utilization in human gut commensal bacteria is orchestrated by unique modular organization of polysaccharide-degrading enzymes. Proc. Natl. Acad. Sci. U.S.A. 111, E3708–E3717. doi: 10.1073/pnas.1406156111
Keywords: comparative metagenomics, lignocellulose degradation, carbohydrate-active enzymes (CAZymes), polysaccharide utilization loci (PULs), microbial enrichment, digestive microbiome, beaver, moose
Citation: Wong MT, Wang W, Couturier M, Razeq FM, Lombard V, Lapebie P, Edwards EA, Terrapon N, Henrissat B and Master ER (2017) Comparative Metagenomics of Cellulose- and Poplar Hydrolysate-Degrading Microcosms from Gut Microflora of the Canadian Beaver (Castor canadensis) and North American Moose (Alces americanus) after Long-Term Enrichment. Front. Microbiol. 8:2504. doi: 10.3389/fmicb.2017.02504
Received: 12 October 2017; Accepted: 01 December 2017;
Published: 20 December 2017.
Edited by:
Guillermina Hernandez-Raquet, Institut National de la Recherche Agronomique (INRA), FranceReviewed by:
Xiuzhu Dong, Institute of Microbiology (CAS), ChinaCopyright © 2017 Wong, Wang, Couturier, Razeq, Lombard, Lapebie, Edwards, Terrapon, Henrissat and Master. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emma R. Master, ZW1tYS5tYXN0ZXJAdXRvcm9udG8uY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.