 Sergio Antoraz1
Sergio Antoraz1 Sergio Rico1
Sergio Rico1 Héctor Rodríguez1,2
Héctor Rodríguez1,2 Laura Sevillano1
Laura Sevillano1 Juan F. Alzate3
Juan F. Alzate3 Ramón I. Santamaría1*
Ramón I. Santamaría1* Margarita Díaz1*
Margarita Díaz1*- 1Departamento de Microbiología y Genética, Instituto de Biología Funcional y Genómica, Consejo Superior de Investigaciones Científicas, Universidad de Salamanca, Salamanca, Spain
- 2Cic bioGUNE, Derio, Spain
- 3Departamento de Microbiología y Parasitología, Facultad de Medicina, Centro Nacional de Secuenciación Genómica, Sede de Investigación Universitaria, Universidad de Antioquia, Medellín, Colombia
Streptomyces coelicolor, the best-known biological antibiotic producer, encodes 29 predicted orphan response regulators (RR) with a putative role in the response to environmental stimuli. However, their implication in relation to secondary metabolite production is mostly unexplored. Here, we show how the deletion of the orphan RR Aor1 (SCO2281) provoked a drastic decrease in the production of the three main antibiotics produced by S. coelicolor and a delay in morphological differentiation. With the aim to better understand the transcriptional events underpinning these phenotypes, and the global role of Aor1 in Streptomyces, a transcriptional fingerprint of the Δaor1 mutant was compared to a wild-type strain. RNA-Seq analysis revealed that the deletion of this orphan regulator affects a strikingly high number of genes, such as the genes involved in secondary metabolism, which matches the antibiotic production profiles observed. Of particular note, the sigma factor SigB and all of the genes comprising its regulon were up regulated in the mutant. Our results show that this event links osmotic stress to secondary metabolite production in S. coelicolor and indicates that the RR encoded by aor1 could be a key regulator in both of these processes.
Introduction
The ubiquity of the genus Streptomyces, comprised of Gram-positive filamentous bacteria, has greatly facilitated its presence as a normal inhabitant of soil, marine niches, and also as a symbiotic partner of sponges, plants, and a plethora of other types of organisms (Chankhamhaengdecha et al., 2013; Braña et al., 2015). All species of this genus harbor the genetic information to produce around 20–30 different secondary metabolites, such as antibiotics (Chaudhary et al., 2013), which in nature may be used as signals in the intra- and interspecies communication that establishes relationships among the organisms within a community (symbiotic, antagonistic, etc.) (Vetsigian et al., 2011; Diminic et al., 2014). The production of secondary metabolites takes place as a response to the different physical (pH, temperature, pressure, etc.) and chemical (nutrients, elicitors, etc.) signals present in the environment through complex regulatory networks (Liu et al., 2013). The wide range of environmental and physiological responses to stimuli generated by bacteria can be appreciated within the database of the Prokaryotic 2-Component Systems (P2CS1). Two-component systems (TCSs) are able to recognize change, and integrate this information to elaborate a proper response, which includes the production of antibiotics.
A model of a typical TCS consists of a membrane-bound Histidine Kinase (HK) that acts as a sensor and is auto-phosphorylated at a specific histidine residue upon receiving a stimulus. Later on, the phosphoryl group is transferred to an aspartic acid of the cytoplasmic Response Regulator (RR), which triggers its activation through a conformational change. Finally, this protein completes the response by mainly regulating the expression of target genes. Both HK and RR genes usually lie in close proximity to one another on a chromosome and form an operon. However, there are also a considerable number of orphans, genes that lack a counterpart in the genome, which ultimately broadens the response capacity of cells (Schaller et al., 2011).
Three main mechanisms ensure partner specificity in bacteria: molecular recognition, phosphatase activity, and substrate competition (Podgornaia and Laub, 2013). A small number of residues are responsible for maintaining the specific molecular recognition in protein–protein interaction interfaces between HKs and RRs (Capra et al., 2010). Furthermore, the HK may act as both a kinase and a phosphatase in these systems. The phosphatase reaction serves, in part, to modulate the level of pathway output and to inhibit the pathway after an activating signal has subsided (Podgornaia and Laub, 2013). Crosstalk between different pathways, leading to the activation of a RR by a non-cognate HK or by promiscuous phosphodonors, such as acetyl-phosphate, can also be avoided by the phosphatase activity of the cognate HK (Salazar and Laub, 2015). Nevertheless, the existence of different inner checkpoints for maintaining fidelity does not imply that such TCSs systems cannot play a role in the activity of more than one interacting partner. However, although adding crosstalk in TCSs may have disastrous consequences for cell fitness, it is important for the evolution of new TCS pathways after the duplication of an existing pathway being immediately unavoidable the crosstalk (Capra et al., 2012; Rowland and Deeds, 2014).
The number of TCSs present in an organism is directly correlated to its habitat (Schaller et al., 2011) and can be variable: zero in Mycoplasma genitalium; 30 HKs and 32 RRs in Escherichia coli; 36 HKs and 34 RRs in Bacillus subtilis; up to 131 HKs and 80 RRs in Anabaena sp.; and 132 HKs and 119 RRs in Myxococcus xanthus (Jung et al., 2012). This evolutionary advantage may allow an organism to respond quickly to all possible situations.
Many Streptomyces genomes have been sequenced to date, and more than 50 TCSs and approximately 20 orphan HKs and 20 orphan RRs (P2CS database) have been identified in all of them. This fact reinforces the idea that Streptomyces requires a high number of these TCSs for surviving in a changeable and hostile environment and to cope with a complex lifestyle. The present study has been carried out using Streptomyces coelicolor, whose genome contains 100 HKs, 87 RRs, 20 orphan HKs and 29 RRs (P2CS database) (Bentley et al., 2002). However, only 17 TCSs have so far been studied, and most of these systems are, at least partially, involved in antibiotic biosynthesis (Rodríguez et al., 2013).
One of the TCS previously described by our group, AbrC (SCO4596/97/98) (Yepes et al., 2011; Rico et al., 2014a; Rodríguez et al., 2015), positively regulates the production of antibiotics. Wang et al. reported that AbrC3 (SCO4596) has a high amino acid sequence identity (59%) with the orphan RR SCO2281, where the conserved identity among most RRs is around 30% (Wang et al., 2009). These authors propose that such highly conserved RRs could be regulated by the same HK, which may suggest possible crosstalk between HK AbrC2 (SCO4597) and the RR SCO2281 (Wang et al., 2009). Moreover, the pleiotropic phenotypes of the triple mutant of the AbrC system and the single ΔabrC3 mutant also suggest crosstalk between AbrC HKs and other RRs (Rico et al., 2014a).
In this work we describe for the first time, the involvement of the orphan RR SCO2281 in the regulation of antibiotic production. This gene has been named aor1 from antibiotic orphan regulator, which we will use from now on. Here, we show how the deletion of this gene is characterized by a drastic decrease in the production of the three main antibiotics produced by S. coelicolor (actinorhodin – ACT, undecylprodiginine – RED, and calcium dependent antibiotic – CDA), and also by a delay in morphological differentiation, suggesting that Aor1, like AbrC3, acts as a positive regulator of both processes. Our RNA-Seq results show that Aor1 plays a crucial role in a gene regulation cascade of S. coelicolor probably integrating a response to different signals, and establishing a connection between secondary metabolism (through several regulators) and the response to osmotic stress (mediated partially by SigB) within a high level of the regulatory network.
Materials and Methods
Strains, Media and Growth Conditions
Escherichia coli strain BW25113 (pIJ790) (containing the λRed system) (Datsenko and Wanner, 2000) and non-methylating ET12567 (pUZ8002) (harboring the tra genes in the non-transmissible RP4-derivative plasmid pUZ8002) (MacNeil et al., 1992) were used for PCR-targeted mutagenesis of S. coelicolor M145. E. coli DH5α and ET12567 (a dam- strain) were used to obtain the DNA to transform S. coelicolor. For CDA bioassays, a parent strain of Bacillus subtilis (CECT 4522) was grown as an overlay on NA medium (Hopwood et al., 1985). S. coelicolor M145 (prototroph; SCP1-, SCP2-) and the Δaor1 mutant strain were grown on R2YE, MSA, YEPD, NMMP, PGA, R5 and LB solid media for transformation, sporulation, spore quantification, and phenotypic assays (Fernández et al., 1998; Kieser et al., 2000). LB medium (Sambrook et al., 1989) was used for RNA-Seq experiment. When necessary, the medium was supplemented with antibiotics: E. coli media - ampicillin (100 μg mL-1), apramycin (50 μg mL-1), kanamycin (50 μg mL-1), chloramphenicol (25 μg mL-1), or nalidixic acid (25 μg mL-1) and S. coelicolor media – neomycin (20 μg mL-1) or thiostrepton (10 μg mL-1).
DNA Manipulation
Plasmid isolation, restriction enzyme digestion, ligation, and transformation of E. coli and S. coelicolor were carried out using the methods of Green and Sambrook (2012), and Kieser et al. (2000), respectively. The plasmids and cosmids used are listed in Supplementary Table S1.
Mutant Construction
REDIRECT PCR-targeting technology was used to replace the coding region of the SCO2281 gene with an apramycin resistance cassette [aac(3)IV gene] in cosmid SCC30 (Gust et al., 2003). The primers used to amplify the mutagenesis cassette (SRG-28 and SRG-30) with pIJ773 as the template are listed in Supplementary Table S2. The mutated cosmid SCC30 ΔSCO2281::acc(3)IV (ΔSCC30-1) obtained in E. coli BW25113(pIJ790) were demethylated in the ET12567(pUZ8002) strain and transferred by conjugation to S. coelicolor M145. The desired double recombinants carrying apramycin resistance and sensitive to kanamycin (the selection marker for the vector sequences) were selected. Southern blotting and PCR assays confirmed the deletion of the SCO2281 gene in S. coelicolor M145.
Antibiotic Production Analysis
ACT and RED antibiotic production was assayed on solid LB medium plates inoculated with 105 spores added in a 5 μL drop at 30°C. RED production was detected after 2 days as the red color of colonies. ACT production was observed after 3 days of growth as a blue halo around the colonies. For CDA, after 2 days of growth on NA, the plates were overlaid with 5 mL of soft agar plus Ca(NO3)2 (70 mM) inoculated with B. subtilis as the test microorganism (0.2 mL, 0.25 DO) and incubated at 30°C for 24 h. A replica plate without calcium was used as a negative control. These experiments were all performed in triplicate.
Antibiotic production was assayed from cultures in liquid LB medium inoculated with 106 spores mL-1 at 30°C. ACT and RED antibiotic production were quantified using the spectrophotometric method described in Yepes et al. (2011). For CDA, plates of NA with and without Ca(NO3)2 (70 mM) were inoculated on the surface with 200 μL of B. subtilis (DO600 0.25) using sterile glass balls. Afterward, a sterile cork borer was used on these plates to make wells with a diameter 0.8 cm. 150 μL of the supernatant of a 48 h Streptomyces culture were added into the wells and incubated at 30°C for 24 h.
Plasmid Construction
The integrative plasmid pSETaor1 (Supplementary Table S1) was constructed by cloning the aor1 gene and its promoter region in the shuttle Streptomyces integrative plasmid pSET152t. First, the aor1 sequence was cloned in the bifunctional E. coli–Streptomyces plasmid pXHis1 in the NdeI and XhoI sites, remaining under the control of the xysA promoter and yielding the intermediate plasmid pXHisaor1. Second, the PCR-amplified aor1 promoter, cloned with the oligonucleotides SRG-37 and SRG-38 and the NdeI and EcoRI sites, replaced the xysA promoter, obtaining the pHisaor1 vector. This promoter corresponds to the 545 bp upstream region of SCO2282 that forms an operon with aor1 gene. Finally, the BglII–BglII fragment of pHisaor1, containing the aor1 gene plus its promoter, was cloned into the previously de-phosphorylated BamHI site in pSET152t, obtaining the integrative plasmid pSETaor1.
The multicopy plasmid pNXaor1 was obtained by cloning the NdeI/HindIII fragment from the plasmid pXHisaor1 into the same sites of pNX24 (pN702GEM3 derivative). To construct the multicopy plasmid pNaor1, its own promoter (upstream region of SCO2282) previously amplified by PCR was cloned as an EcoRI/NdeI fragment into the same sites of pNXaor1 (Supplementary Table S1).
qRT-PCR
Specific primers for the genes tested were designed using the Primer3 web-based tool2 (Supplementary Table S2). Five microgram of the RNA samples were treated with RNAase-free DNaseI (Promega) according to the manufacturer’s instructions. One microgram of the resulting RNA was used as template for cDNA synthesis using iScript Reverse Transcription Supermix for RT-qPCR (Bio-Rad) in 20 μL reaction volumes. The samples were diluted 1:1 with distilled water and 2 μL was used in the quantitative PCR reaction with 10 pmol of forward and reverse primer and 5 μL of SsoAdvanced SYBR Green Supermix (Bio-rad).
Each assay was performed in duplicate using the CFX96 TouchTM Real-Time PCR Detection System (Bio-Rad). Control PCRs were included to check that there was neither DNA (without RT, enzyme negative control) nor environmental contamination. Absolute quantification was performed using decimal serial dilutions of S. coelicolor genomic DNA with a known number of copies as standard.
Comparative Transcriptome Analysis
Streptomyces coelicolor M145 and S. coelicolor Δaor1 were grown in LB medium inoculated with 106 sp mL-1 for 36 h at 28°C. The cells from three biological replicates were collected and treated with RNA protect, and afterward, RNA was extracted with RNeasy Mini Kit (Qiagen) following the company instructions. RNA samples quantity and quality were performed by Nanodrop and Bioanalyzer (Agilent). All samples passed the quality threshold of RIN > 7.
Macrogen Inc. (South Korea) provided the RNA-Seq data. The rRNA was removed with Ribo-Zero rRNA Removal Kit (Bacteria) and one library per sample was made using the Illumina TruSeq Stranded Total RNA kits. An independent library was constructed and sequenced for each triplicate. Sequencing was carried out in a HiSeq 2500 (Illumina) instrument with 100 bases PE reads. Reads were quality trimmed at Q35 with the script prinseq-lite, and only those that exceeded 50 bases in length were kept. Singletons were also excluded from the clean read set. For each library, the number of reads pairs obtained ranged between 21.2 and 24.4 millions, and around 99% passed the quality filter.
Mapping was carried out with the program Bowtiew2 using default settings and the Streptomyces coelicolor A3 (2) reference genome (GenBank NC_003888). SAM file was converted to BAM format using samtools, then sorted and indexed. Between 95 and 98% of the cleaned-read set was successfully mapped to the reference genome.
Reads for rRNA origin were detected counting those that mapped to the genome coordinates of the genes 16S, 23S and 5S. In all libraries the proportion of ribosomal reads was below 8%.
For the differential expression analysis, strand specific reads were counted for each CDS feature annotated in the genome reference using HTSEQ v0.6.1p1. A table containing the raw read counts for each CDS feature was generated and used for the subsequent comparative analysis using the BIOCONDUCTOR package EDGER v3. The analysis was performed using the protocol described for RNA-Seq experiments in the user’s guide revised in April 20th 2016.
The RNA-Seq data analyzed here has been deposited in the Sequence Read Archives (SRA, Accession code: PRJNA380047).
Results
Aor1 Acts as a Positive Regulator of Antibiotic Production and Differentiation
The gene aor1 (SCO2281) from S. coelicolor has 687 nucleotides and 73.36% GC content and encodes a putative NarL orphan RR containing two domains: a REC domain (position 9–128) containing the phosphoaceptor Asp from the HK (CheY-homologous receiver) and an HTH-LuxR DNA binding domain (position 156–218) (SMART database3). The encoded protein contains 228 amino acids, with an isoelectric point of 5.47 and a molecular weight of 24.68 kDa. It is highly conserved in all of the Streptomyces spp. sequenced to date, with identities ranging from 99 to 75%. Since Aor1 is an orphan RR, there is no HK codifying gene within its genomic context (Figure 1A). The phylogenetic circular dendrogram of S. coelicolor RRs (P2CS database) shows that aor1 (SCO2281) and abrC3 (SCO4596) are closely related and share 59% protein sequence identity (Figure 1B). This fact points to the possibility of crosstalk between the HKs AbrC1 and/or AbrC2 of the AbrC system and Aor1. In addition, the other closely related RR gene SCO2358 shares 55.7% protein sequence identity with Aor1, and forms part of an operon with the HK SCO2359. Remarkably, the P2CS database also considered the three genes comprising the AbrC TCS as orphans because the intergenic space between the genes is large enough to contain a promoter region. Moreover, the Prediction of Interaction Specificity in Two-Component Systems website of Biozentrum4 showed that there were 20 orphan kinases and 29 orphan regulators in S. coelicolor, including the AbrC genes. The existence of a common RNA has been shown for the two HKs AbrC1 and AbrC2, however, no common RNA has been obtained for the HK AbrC2 and the RR AbrC3 (Rodríguez et al., 2015). Using this prediction tool, the HKs AbrC1 (SCO4598) and AbrC2 (SCO4597) were in forth and fifth place as being probable partners with Aor1 (after SCO3750, SCO6424, and SCO0211).
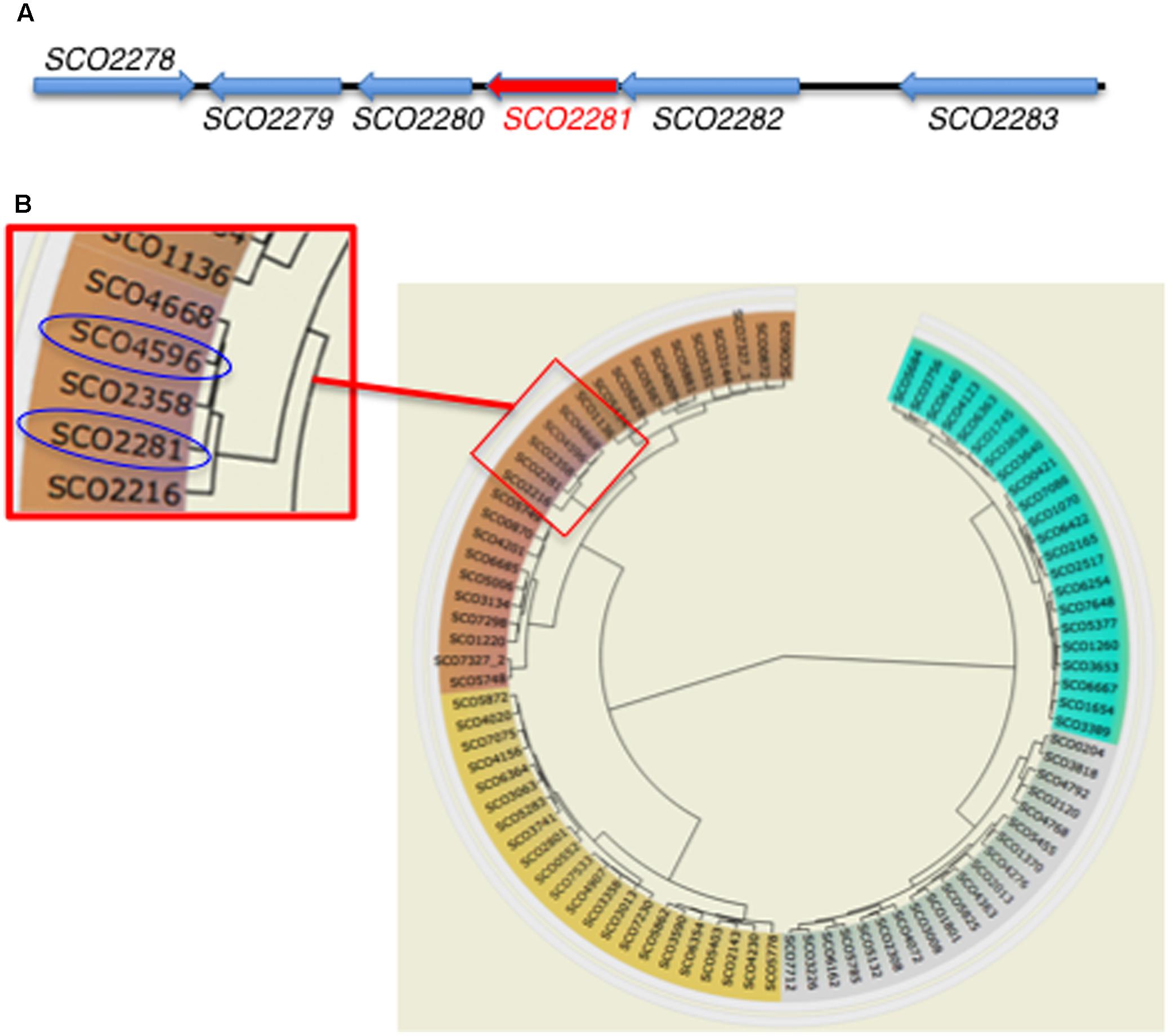
FIGURE 1. Genomic information of orphan RR aor1 (SCO2281) gene. (A) Genomic context (SCO2278: Hypothetical protein (HP), SCO2279: HP, SCO2280: putative TetR regulator, SCO2282: HP, SCO2283: putative secreted esterase) (source: StrepDB database). (B) Circular dendrogram from S. coelicolor RRs. Detailed red box: phylogenetic relationship between SCO2281 and abrC3 (SCO4596) RRs (source: P2CS database).
The functional role of Aor1 was analyzed replacing the gene by an apramycin resistance cassette, using the REDIRECT technology (Material and Methods). This mutant strain exhibited a phenotype of an hypo-production of the antibiotics ACT, RED, and CDA, and a delayed morphological development in different media (Supplementary Figure S1), with more extreme phenotypes than those reported for the ΔabrC mutant strain (Rico et al., 2014a). Due to the drastic phenotype (Figure 2A), and highly reproducibility found in LB medium, complementation assays and RNA-seq analysis (see below) were performed in that medium. These results suggest that Aor1, as previously shown for AbrC3, acts as a positive RR of antibiotic production and differentiation. The Δaor1 mutant phenotypes were complemented by the single copy integrated plasmid pSETaor1 (Supplementary Table S1), which discarded the possibility of polarity effects caused by the mutation as the origin of the phenotypes (Figure 2B).
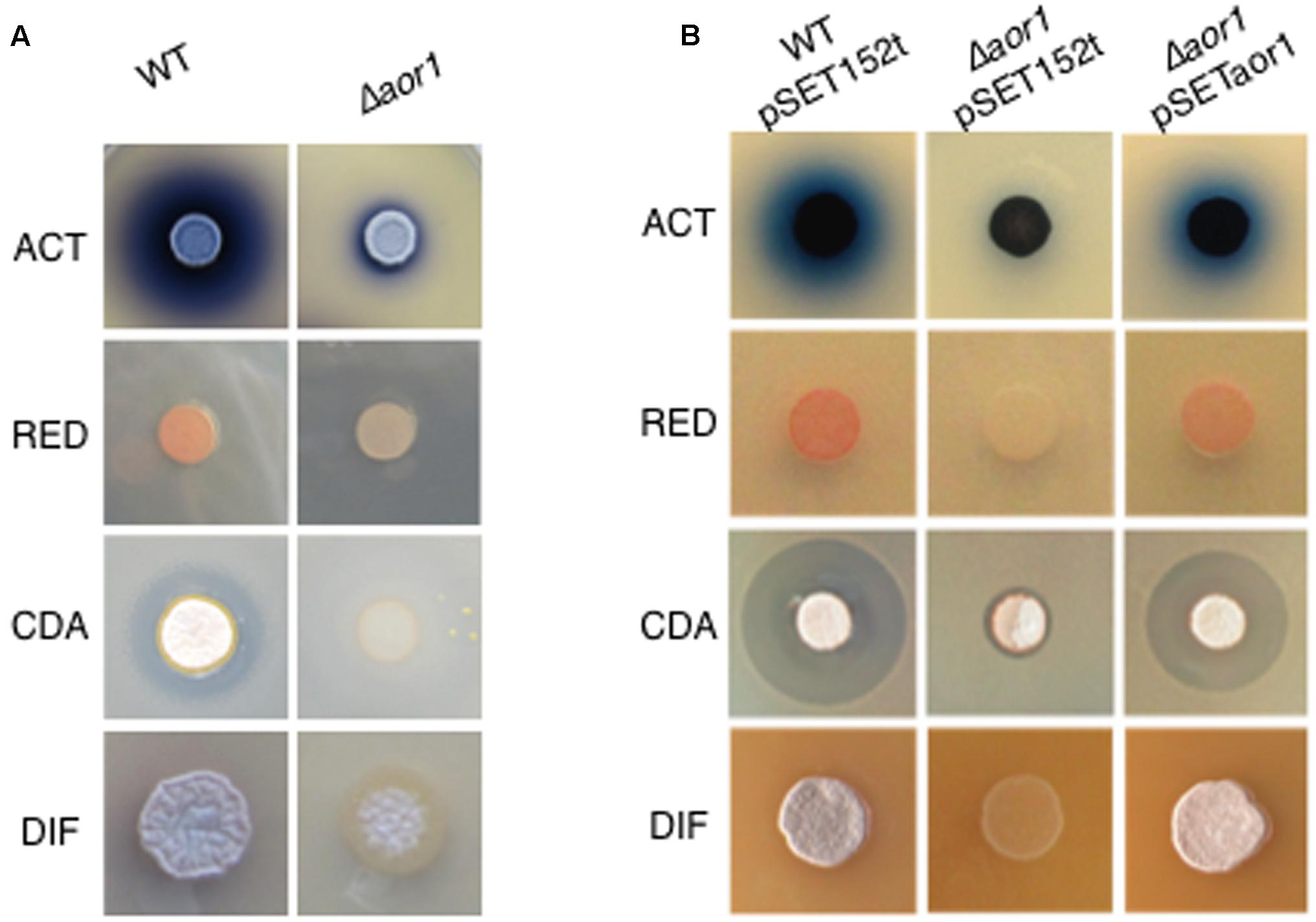
FIGURE 2. Phenotypes of mutant Δaor1. (A) Comparison of phenotypes of Δaor1 with wild-type strain. (B) Complementation of the Δaor1 phenotypes by the integrative plasmid pSETaor1 (pSET152t derivative). ACT, actinorhodin; RED, undecylprodiginine; CDA, calcium dependent antibiotic; DIF, morphological differentiation.
The Gene aor1 Is Part of a Four-Gene Operon
As shown in Figure 1A, aor1 is located in the vicinity of three genes; two of them (SCO2279 and SCO2282) have been annotated as hypothetical proteins, while SCO2280 has been annotated as a TetR family transcriptional regulator.
In order to determine if all four genes were co-transcribed and participants of a common regulation, qRT-PCR experiments were carried out to detect transcripts within the intergenic regions between the genes. It was found that the intergenic regions were not large enough to contain promoters regions, except for the region between SCO2279 and SCO2280 and SCO2280 and SCO2281. qRT-PCR experiments using primers designed to these intergenic regions (Supplementary Table S2) were positive and showed the amplification of a transcript in all cases (Figure 3), revealing the existence of a transcript that included all of the genomic region from SCO2279 to SCO2282. This result suggested the existence of an operon with the promoter region located upstream of SCO2282.
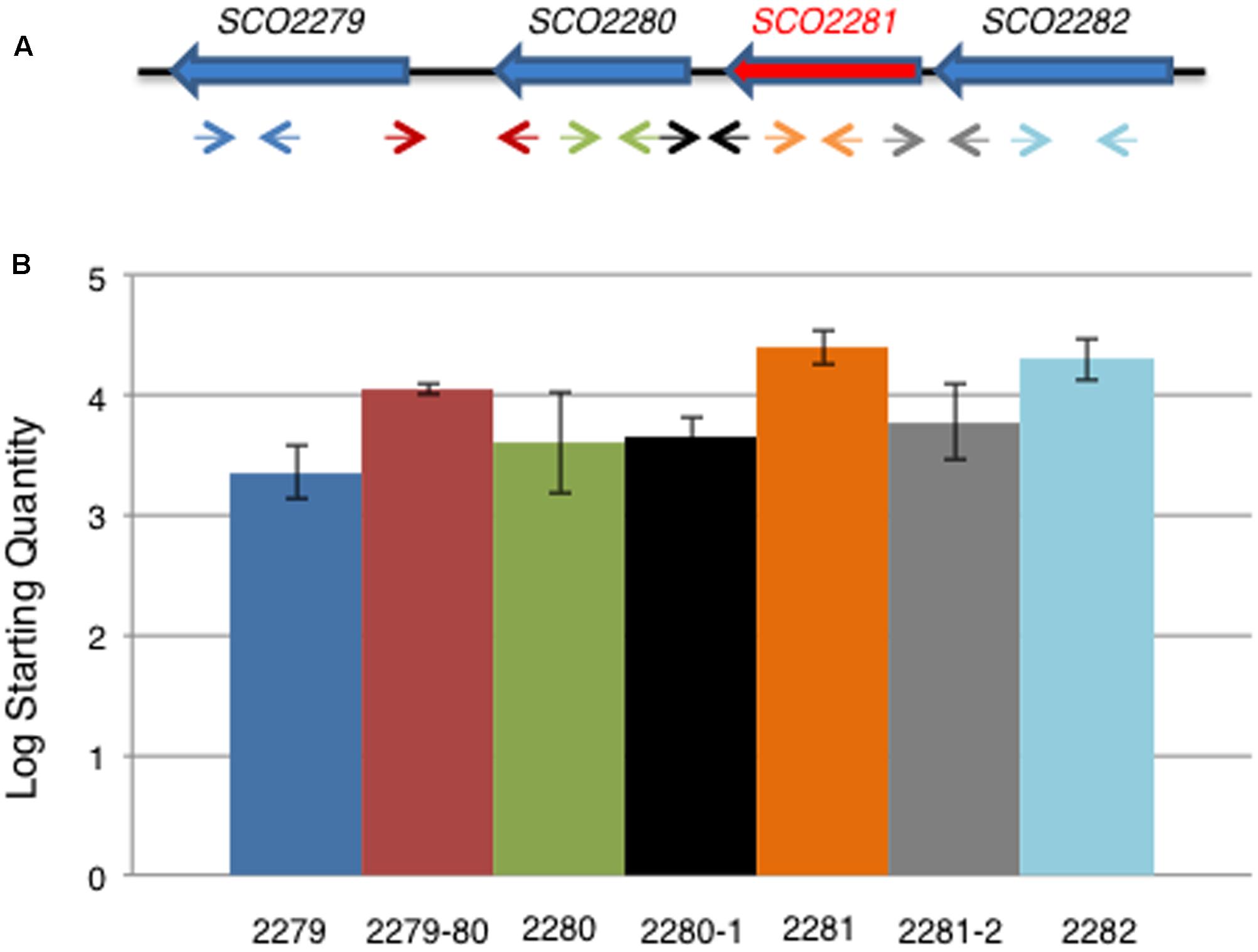
FIGURE 3. qRT-PCR of aor1 operon genes. (A) Scheme of the gene structure of the putative aor1 operon and location of the oligonucleotides used. (B) Expression profile found by qRT-PCR of the four genes of the aor1 putative operon and the corresponding intergenic regions between the different genes in NB medium at 48 h. Data represent the Log Starting Quantity of each sample.
Overexpression of aor1 Does Not Provoke Increased Antibiotic Production
Overexpression of the aor1 gene was performed under the control of the SCO2282 upstream region, which corresponded to the promoter region of the aor1 operon. The plasmid pNaor1 was obtained (Material and Methods) and introduced into the M145 wild-type strain. Antibiotic production was compared to the strain transformed with the empty plasmid used as control on LB agar plates and in liquid LB medium. The high copy number plasmid used to overexpress of the aor1 gene did not provoke an increase in the production of the antibiotic ACT, as would be expected if it were a positive regulator. This result suggests that Aor1 could possibly be exerting negative control over other negative regulator/s, a situation that will be addressed in the “Discussion” section of this paper (Supplementary Figure S2).
The Δaor1 Mutant Transcriptomic Profile Compared to Wild Type
To study the role of the Aor1 RR in a gene regulatory cascade in S. coelicolor, we compared the transcriptome of S. coelicolor M145 with S. coelicolor Δaor1. Stranded RNA-Seq was performed using RNA from samples of each strain, in triplicate, at 36 h (Material and Methods). The principal component analysis (PCA) and the sample distance matrix showed a distinct gene expression signature between M145 and the Δaor1 mutant strains (Supplementary Figures S3, S4). According to the PCA, the first principal component accounted for 82.5% of the variance, and clearly separated both groups. Moreover, the global analysis of the data showed that the absence of the aor1 gene provoked a huge impact on the transcriptome (Figure 4 and Supplementary Figures S3, S4).
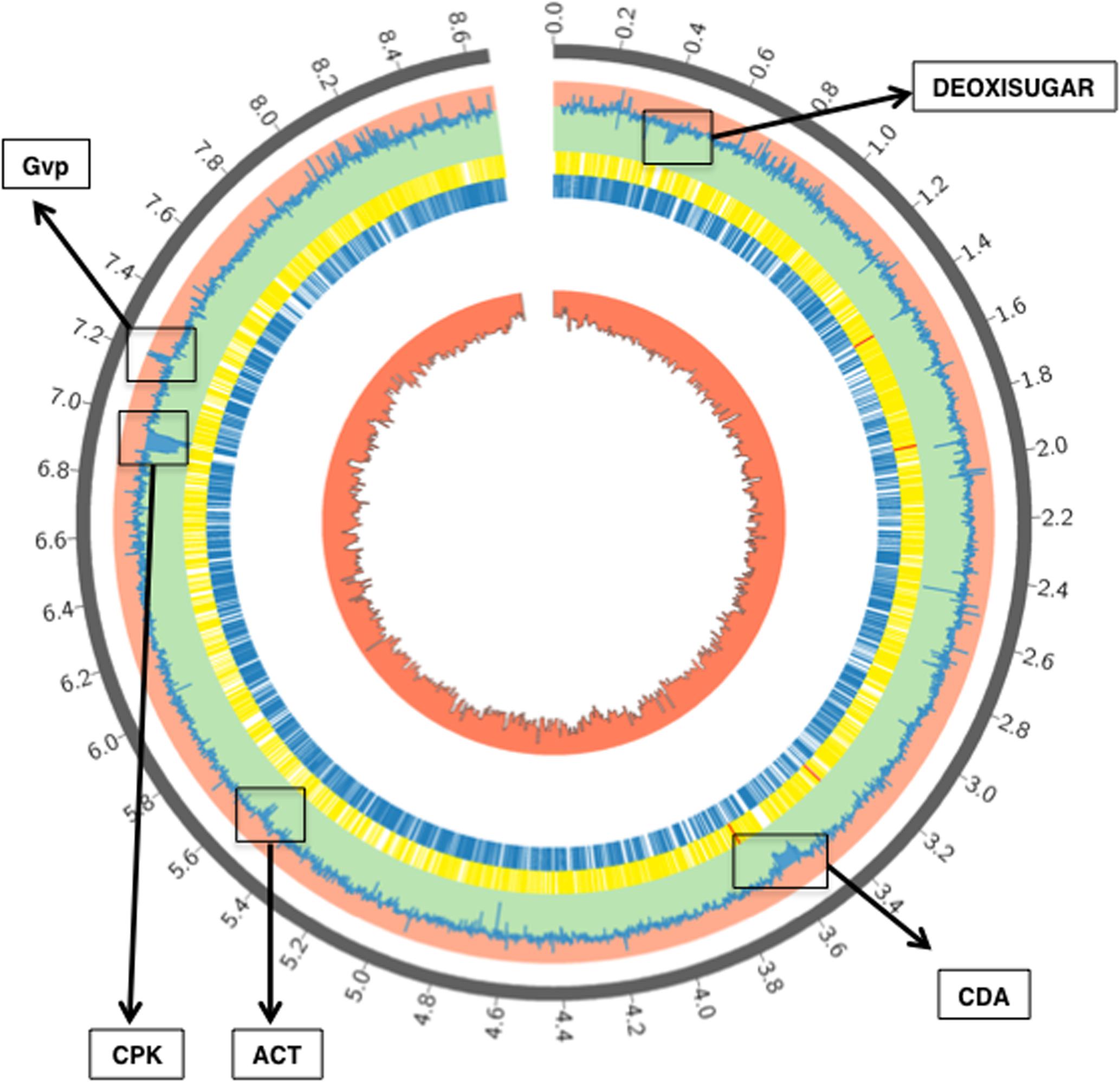
FIGURE 4. RNA-Seq gene coverage results of S. coelicolor M145 vs.Δaor1. Description from the outer circle to the inner one: gray: chromosome scale (Mb); pale blue: logFC M145 vs. Δaor1 strain (toward rose zone genes up-regulated in Δaor1 and toward green zone genes down-regulated in Δaor1; yellow: CDS in minus strand; blue: CDS in plus strand; red: GC percentage. Some of the most significant clusters differentially expressed are depicted: deoxisugar, CDA, ACT, CPK and Gvp (Gas vesicle proteins).
To facilitate the analysis of the RNA-Seq data, an additional analysis was performed but only on those transcripts whose levels changed at least three-fold in the Δaor1 mutant compared with the levels in M145. Five hundred and four genes showed high differential expression (287 down-regulated and 217 up-regulated) between both strains in the conditions studied, with a significant p-value ≤ 0.002 and FDR ≤ 0.01. A complete list of the genes corresponding to these transcripts can be found in Supplementary Tables S3, S4. These selected differentially expressed genes were categorized into seven general clusters of functional categories, using David Gene Ontology analysis terms as the reference (Huang da et al., 2009a,b), and are listed in Table 1. The relevance of the differentially expressed genes and their correlation with the phenotypic changes observed in the Δaor1 mutant were analyzed (see below). Several genes were chosen to perform q-RT-PCR and validate the RNA-Seq results (Supplementary Table S5).
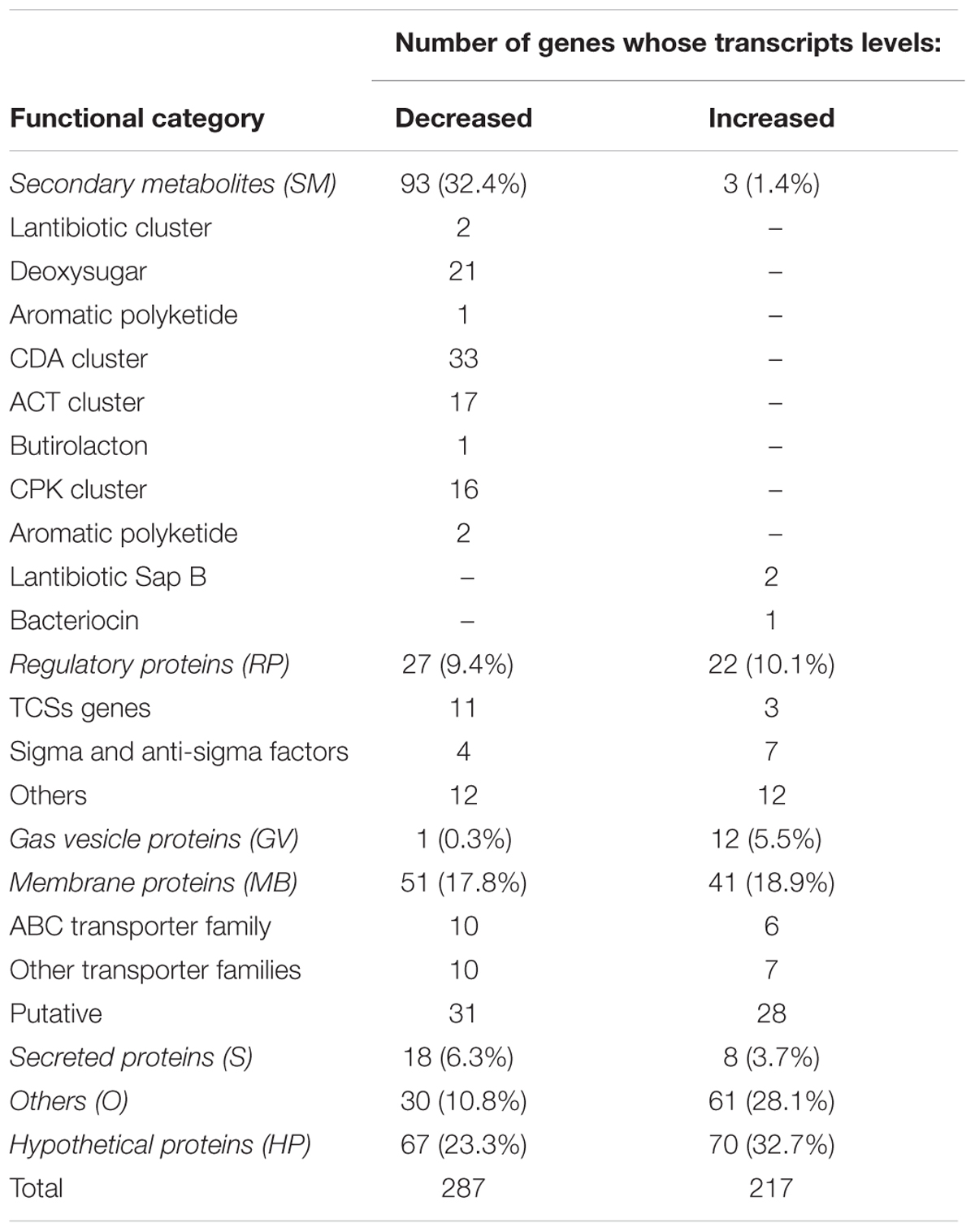
TABLE 1. Differentially expressed genes (RNA-Seq): Functional categories of transcripts whose levels changed at least 3-fold in S. coelicolor Δaor1 compared with those of M145 (p-value ≤ 0.002 and FDR ≤ 0.01).
In addition, samples were collected in parallel at 36, 48, 62, and 134 hours (h) to quantify antibiotic production in the tested growth conditions. ACT production was significantly detected at 48 h in the wild-type strain M145, and this production increased exponentially at 62 and 134 h. However, the Δaor1 mutant strain presented very small production even at 62 h, and had a delayed trigger in ACT production at 134 h, yielding slightly lower levels than that of the M145 strain at this time (Figure 5A). Regarding RED production, a high level of this antibiotic was produced by M145 at the first time point of 36 h, with a maximum of production at 48 h. RED production in the mutant strain was reduced in all of the times analyzed (Figure 5B). Bioassays against Bacillus subtilis were used to test the CDA production using the supernatants of 48 h cultures (Material and Methods). There was a small inhibition halo in the M145 strain without calcium that could be due to the production of the other antibiotics (RED and ACT). This halo was much more considerable with plates containing calcium, corresponding to CDA production. In the mutant strain, no inhibition halo was observed in the two conditions used (+Ca and –Ca), indicating that the mutant strain produced less of this antibiotic (Figure 5C).
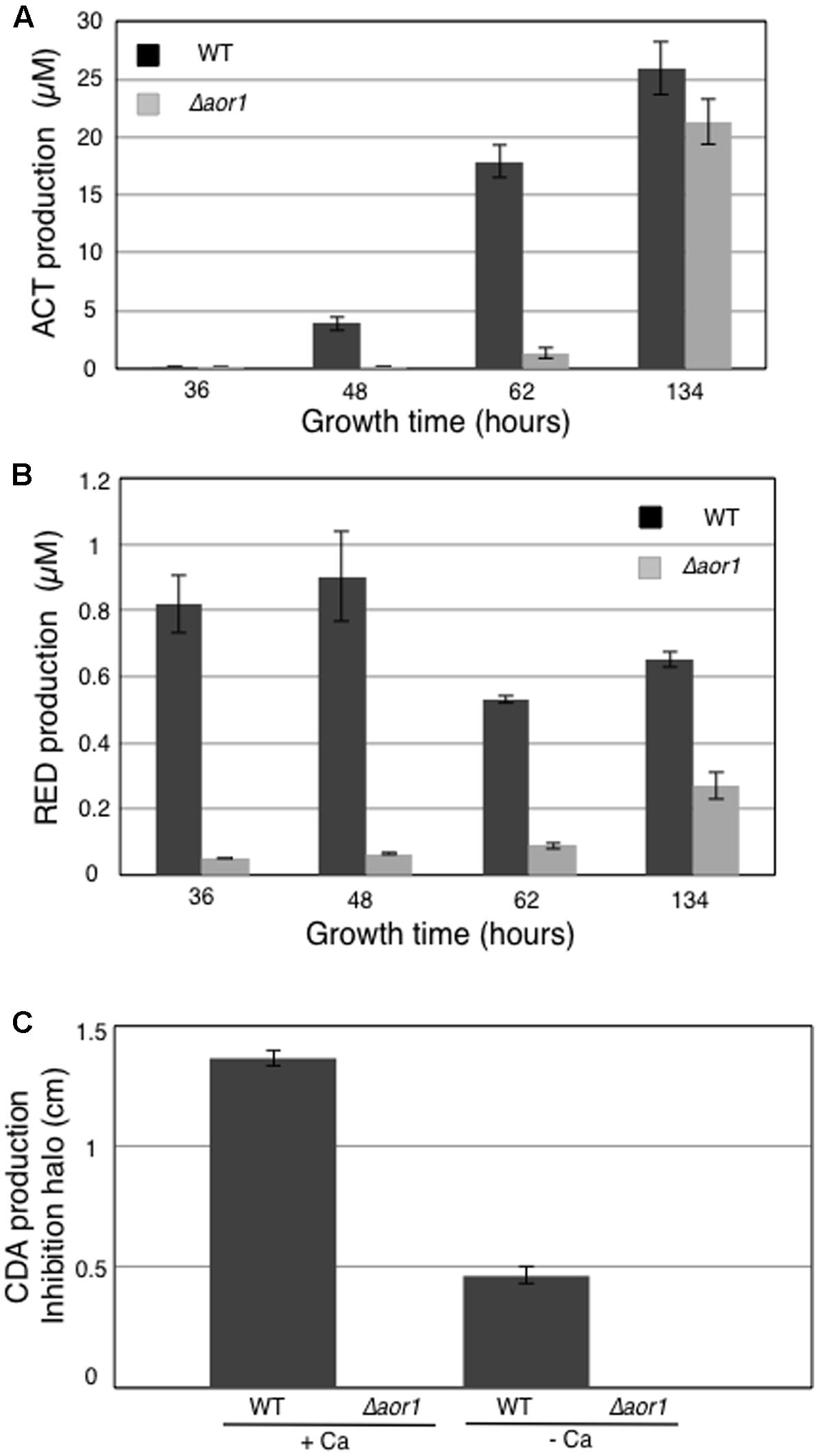
FIGURE 5. Antibiotic production of S. coelicolor M145 (WT) and S. coelicolor Δaor1 in LB medium. (A) Actinorhodin production (ACT). (B) Undecylprodiginine production (RED). (C) CDA production with and without Ca(NO3)2 (70 mM) measured in a bioassay against B. subtilis using 48 h culture supernatant. Error bar correspond to triplicate measurements.
Genes Implied in Secondary Metabolism Affected
The expression of 93 out of the 287 down-regulated genes found in the S. coelicolor Δaor1 mutant strain corresponded to the genes of many clusters encoding secondary metabolites (SM) (32.4%) (Nett et al., 2009). Among these genes, most of the ACT genes clustered together (SCOs5071-5092; Figure 4, coordinates 5514323–5534546; Supplementary Figure S5) and CDA (SCOs3210-3249; Figure 4, coordinates 3519449–3602320; Supplementary Figure S5) were found; a result that is in agreement with the phenotype observed in Δaor1 strain, including the cluster situated regulators (CSR) genes actII-ORF4 (SCO5085) and cdaR (SCO3217), although this latter with a FC of 1,9 (Supplementary Table S3). Also, almost all of the genes within several clusters, whose products were not assayed in this study, were found to be down-regulated such as a deoxysugar (SCOs0381-0401; Figure 4, coordinates 395939–423596), a CPK (SCOs6273-6288; Figure 4, coordinates 6900898–6907356) and its CSR cpkO (SCO6280), some genes within the lantibiotic cluster (SCOs0267-0270), two aromatic polyketides (SCO1266 and SCOs7669-7671) and the butirolactone (SCOs6265-66; Figure 4, coordinates 8493549–8496340). Nevertheless, only 3 genes out of 217 (1.4%) in this functional category of secondary metabolites were found to be up-regulated in the Δaor1 strain, which were a gene from a bacteriocine cluster (SCO0753) and two genes of the lantibiotic cluster containing the SapB gene (SCOs6682-85; Figure 4, coordinates 7422494–7427004).
Genes Encoding Regulatory Proteins Are Affected
Another group of genes with a high percentage of differential expression between the wild-type and the Δaor1 mutant strain corresponded to the genes encoding regulatory proteins. As mentioned above, CSRs (SARP proteins coding genes regulating antibiotic production) were down regulated, such as actII-ORF4, cdaR, cpkO, and also two CPK regulatory proteins (SCO6285 and SCO6288). This was also the case of ScbR (SCO6265), AtrA (SCO4118), Aba-like (SCO4214), 2 sigma factors including SigU, (SCO2954) and 2 putative sigma factors, 6 genes from different regulatory families (LysR, LacI, GntR and, TetR) and 11 TCS genes (including one orphan RR and, two orphan HKs) (Table 1). A total of 27 genes of regulatory proteins had a significantly reduced level of expression in the mutant strain (more than 3-fold), corresponding to 19.4% of the down-regulated genes.
On the other hand, 22 genes of this functional category were up-regulated (10.1%), and of these 7 were sigma or anti-sigma factors. These up-regulated genes included the SigB encoding gene (SCO0600), its reported regulatory network that is triggered by osmotic or heat shock (SigL, SigM, SigH, SigK), and the anti-anti-SigB RsbV (SCO7325) (Supplementary Figure S5 and Supplementary Table S4) (Lee et al., 2004a; Facey et al., 2011), whose relevance in the Δaor mutant will be discussed below.
Genes Related to Osmotic/Heat Protection Differentially Expressed
Moreover, in addition to the regulatory genes mentioned above, many other genes regulated by SigB (Lee et al., 2004b) were also over-expressed in the Δaor1 mutant compared to the wild-type. These included: catB (SCO0666) (Cho et al., 2000), coding for a catalase that was 11.4-fold up-regulated; trxC, coding for a thioredoxin C (4.7-fold); ssgC (SCO7289) implied in differentiation (8.4-fold); 12 genes of an operon for gas vesicles proteins (GV) (SCOs6496-6508; Figure 4, coordinates 7189075-7199188), whose promoter is regulated by SigB and were more than 5-fold up-regulated (Supplementary Table S4); and another 15 genes, some of them with an unknown function.
Other genes not dependent on SigB and related to osmotic protection were also up-regulated, such as a putative osmoprotectant transporter (SCO1225), the bacterioferritine gene (SCO2113), and SCO2641 a putative resistance protein implied in detoxification5).
Membrane and Transport Proteins Differentially Expressed
Those genes that encoded membrane proteins among the differentially expressed genes (92 genes, 51 down- and 41 up-regulated, 17.8 and 18.9 % respectively) were also overrepresented. These genes included 33 members of different superfamilies of transporter proteins (20 down- and 13 up-regulated), such as the ABC (ATP-binding Cassette), MFS (Mayor Facilitator Superfamily), and RND (Resistance-Nodulation-Cell Division) protein superfamilies, among others. Four of the down-regulated putative membrane proteins formed part of the regulon reported for SigU (also down-regulated as mentioned previously), as was the case for 3 putative secreted proteins found to be down-regulated (Gordon et al., 2008). In fact, 24 genes of the secreted proteins were also found to be differentially expressed; 18 genes were down-regulated and 8 up-regulated (6.3 and 3.7 %, respectively).
Discussion
Due to the increasing number of antibiotic resistant bacteria it is important to revitalize the discovery of new antibiotics (Demain, 2014). In addition, the study of the regulatory networks that control their biosynthesis is also a key goal. In this work, we report the importance of the orphan RR Aor1, which could be considered a positive regulator of antibiotic production and morphological differentiation based on the observed phenotypes of the null mutant (Figures 2, 5 and Supplementary Figure S1), although part of its action seems to be done through the inactivation of some inhibitors, as discussed below.
The RNA-Seq data have provided a revealing look at the differences between the transcriptomes of S. coelicolor M145 and Δaor1, showing that Aor1 is a global regulator that acts in a very high level of the complex regulatory network. In the S. coelicolorΔaor1 strain, deletion of the aor1 gene led to significant changes in the global transcriptional landscape, with a three-fold differential expression of 504 genes (287 down-regulated and 217 up-regulated) as compared to the wild-type strain.
In the RNA-Seq data analysis it was found that Aor1 was necessary for triggering the expression of many of the genes encoding the 30 putative secondary metabolites of S. coelicolor (Nett et al., 2009). In fact, in addition to the 93 genes down-regulated (32.4%) in Δaor1, as described in the Results section, many other genes in this functional category were down-regulated in the Δaor1 mutant, with a significant FDR and p-value. However, the differences with respect to the wild-type were less than three-fold (as in the case of cdaR). For example, 6 out of 22 genes of the RED cluster (SCOs5877-5898) and 4 genes of the eicosapentanoic acid cluster (SCOs0124-0127) were found with a FC > 1.7. In contrast, the genes included in the 5-hydroxiectoin cluster (SCOs1864-1867) were up-regulated (FC > 1.69) (Supplementary Tables S3, S4). As shown in Figure 5, RED production was triggered before 36 h, indicating that the biosynthetic genes were produced before this time. Additionally, this result helps to explain why the differential expression of these genes observed at 36 h was not as significant as the one observed for ACT or CDA.
The onset of some of these secondary metabolites by Aor1 seems to be mediated by the activation of other regulatory proteins that have been previously reported to control antibiotic production (all of them down regulated in the Δaor1) such as the antibiotic CSRs, ActII-ORF4, CdaR and, CpkO, the γ-butirolactone (ScbA) and its regulator (ScbR), AbaA-like and, AtrA (Liu et al., 2013). Other regulatory genes were also down- or up-regulated, including global regulators like TCSs that have not been characterized yet. It is also worth mentioning that in the CDA cluster (SCOs3210-3279), only the RR gene AbsA2 (SCO3226) was up-regulated (with a FC = 1.8) (Supplementary Table S4). Therefore, the regulation of some of the CSRs by Aor1 might be partly due to a negative regulation of AbsA2, which has been reported to be a negative global regulator (Sheeler et al., 2005). This could explain why deletion of the aor1 gene had such strong phenotypic effects and that a drastic increment in antibiotic production was not observed with a high copy number plasmid expression harboring aor1.
On the other hand, the regulatory network and targets that respond to osmotic stress and heat shock mediated by the master sigma factor SigB, such as SigL, SigM, SigH, SigK, CatB, TrxC, SsgC and a whole gas vesicle (GV) operon (Lee et al., 2004b, 2005; Facey et al., 2011), were found up-regulated in the S. coelicolor Δaor1 mutant strain. The genes encoding WhiB (SCO3034) (indirect target of SigB) and DpsA (SCO0596) (direct target of SigH and SigB) were also differentially expressed and were up-regulated by 2.68-fold and 2.6-fold, respectively.
One hypothesis, that needs to be addressed in the future, is if Aor1 may be modulating the osmotic stress response and antibiotic production of the cell, integrating different external signals through HKs (Figure 6). The aor1 deletion triggered the cellular responses mediated by SigB regulon in order to protect the cell against osmotic stress as reported for the GV operon (van Keulen et al., 2005), trxC and catB. Thus, Aor1 could be acting as a negative regulator of sigB. Several osmotic stress related responses, not yet described as being dependent on SigB, have also been found to be up-regulated, such as the already mentioned putative osmoprotectant transporter of proline/betaine from the MFS (SCO1225)6 and 5-hydroxiectoin compatible solute’ overexpression (with a FC > 1.7) (Eilert et al., 2013; Sadeghi et al., 2014a,b). At the same time, a general decrease in secondary metabolite production in the Δaor1 mutant could take place, as explained above. These results are in accordance with the inverse correlation between osmotic stress response and antibiotic production reported previously in Streptomyces avermitilis (Godinez et al., 2015). A precocious overproduction of ACT has also been reported in several S. coelicolor sigB mutant strains, reinforcing this correlation (Cho et al., 2001; Fernández Martínez et al., 2009). Moreover, as part of the stress response provoked by the absence of Aor1, the transport functions of the mutant cells seemed to be altered, owing to the high number of transporter proteins that were found affected, as the mentioned above SCO1225. In addition, when mutant cells sense osmotic stress they need to control their secretion in order to maintain high intracellular osmolarity. Similarly, it has also been reported that the extracytoplasmic function sigma factor (ECF) SigU elicits secretion and increases protease activity. In the Δaor1 mutant, the secreted and membrane proteins whose expression is triggered by SigU are down-regulated, and the protease inhibitor Sti1 (SCO0762) is 2.5-fold up-regulated. These results are in line with this working model (Supplementary Table S4) (Gordon et al., 2008).
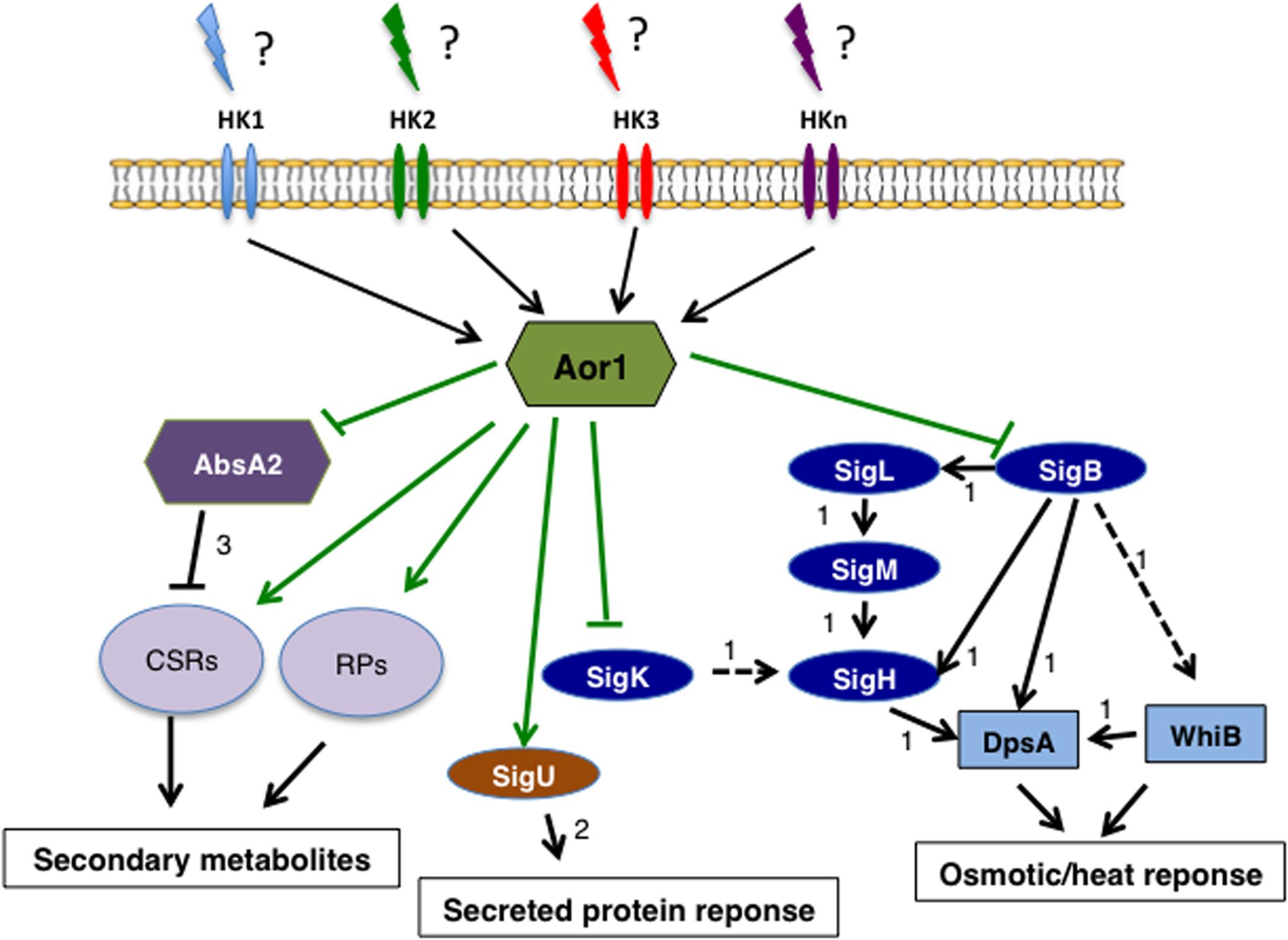
FIGURE 6. Scheme of the Aor1 network of S. coelicolor. The schematic model shows the putative gene network governed by Aor1 and its pleiotropic regulatory roles that connect secondary metabolism, secreted protein response mediated by SigU, and osmotic/heat response mediated by Sig B. Arrow: positive control; blunt line: negative control; solid black arrow: direct control; dashed black arrow: indirect control. Green lines: this work (to determine the direct or indirect control). The data for arrows 1 are from the work of Facey et al. (2011); arrow 2 from Gordon et al. (2008); CSR, Cluster Situated Regulators; RPs, Regulatory proteins.
Since Aor1 is an orphan RR, this opens up the possibility for it to be phosphorylated by different HKs, (Whitworth and Cock, 2009) allowing Aor1 to respond to many environmental cues that might exert a strict control on the osmotic stress response mediated by SigB and correct secondary metabolite production. Clarifying the function of Aor1 and situating it in a concrete point of the S. coelicolor complex regulatory network is a difficult task, as is demonstrated by the few regulators with known stimuli found in Streptomyces (Rodríguez et al., 2013). This task acquires an additional difficulty level in the case of orphan regulators. Relation between an orphan regulator and an HK has rarely been demonstrated previously, but combination of BLASTP search and trans-phosphorylation may serve to elucidate the role of Aor1 in the future (Wang et al., 2009). Several HKs have been reported as probable partners of Aor1 such as the above-mentioned AbrC1, AbrC2 (Rodríguez et al., 2015), SCO3750, SCO6424, and SCO0211. The phenotypes observed in Δaor1 were more drastic than those reported for ΔabrC, reinforcing the idea that more than one HK might be controlling the activity of this orphan regulator. As mentioned before, Streptomyces has a high number of TCSs due to its complex lifestyle and genome size, and several TCSs have been found down-regulated in Δaor1, which also suggests a transcriptional cross-regulation exerted by Aor1 on these signal transduction systems. As Zhou et al. stated, key features of Streptomyces, such as gene regulation, stress response, secondary metabolism, and morphological differentiation, are not stand-alone properties but are related to each other (Zhou et al., 2011). Aor1 may be establishing a connection that integrates some of these pathways in a high level of the regulatory network. Other TCSs as OsdK/R (SCO0203/0204), (Urem et al., 2016) and OsaA/B (SCOs5748/5749) have also been reported to respond to oxygen and osmotic stress responses, respectively (Godinez et al., 2015).
Conclusion
It has been demonstrated using transcriptome profiling that the aor1 deletion results in a highly altered transcription pattern compared to that of the parental strain. Thus, these results highlight that Aor1 plays a role in the regulation of genes related to osmotic stress and secondary metabolite production in S. coelicolor. Nevertheless, these data include changes that are both direct and indirect results of the effects of the orphan RR Aor1 on gene expression. Further experiments, such as a ChIP-Seq, must be carried out in the future to determine the direct targets of this RR. Another challenge is finding the corresponding cognate HKs, which to date has proven to be a difficult task.
Author Contributions
SA, SR, HR, LS, and RS conducted the experiment(s). SA, JA, and MD analyzed the results. SA, RS, and MD conceived the experiment(s) and wrote the manuscript. All authors read and approved the final manuscript.
Funding
Our laboratory is funded by the Spanish Ministry of Economy, Industry and Competitiveness (MINECO) [PCIN-2014-067 and BIO2015-66958-R]. SA has been supported by a Junta de Castilla y León fellowship (grant CSI099A12-1).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Ana María Martínez-Carrasco and Itziar Martín Ruíz for their technical skills and to Emma Keck for her help with the English language editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.02444/full#supplementary-material
Footnotes
- ^ http://www.p2cs.org/
- ^ https://sourceforge.net/projects/primer3/
- ^ http://smart.embl-heidelberg.de/
- ^ http://www.swissregulon.unibas.ch/cgi-bin/TCS.pl
- ^ http://dbkgroup.org/s_coeli/referencegel/proteinDetails.php?proteinId=SCO2641
- ^ http://www.membranetransport.org/other_type.php?fType=Secondary+Transporter&oOID=scoe1
References
Bentley, S. D., Chater, K. F., Cerdeño-Tárraga, A. M., Challis, G. L., Thomson, N. R., James, K. D., et al. (2002). Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417, 141–147. doi: 10.1038/417141a
Braña, A. F., Fiedler, H. P., Nava, H., González, V., Sarmiento-Vizcaíno, A., Molina, A., et al. (2015). Two streptomyces species producing antibiotic, antitumor, and anti-inflammatory compounds are widespread among intertidal macroalgae and deep-sea coral reef invertebrates from the central cantabrian sea. Microb. Ecol. 69, 512–524. doi: 10.1007/s00248-014-0508-0
Capra, E. J., Perchuk, B. S., Ashenberg, O., Seid, C. A., Snow, H. R., Skerker, J. M., et al. (2012). Spatial tethering of kinases to their substrates relaxes evolutionary constraints on specificity. Mol. Microbiol. 86, 1393–1403. doi: 10.1111/mmi.12064
Capra, E. J., Perchuk, B. S., Lubin, E. A., Ashenberg, O., Skerker, J. M., and Laub, M. T. (2010). Systematic dissection and trajectory-scanning mutagenesis of the molecular interface that ensures specificity of two-component signaling pathways. PLOS Genet. 6:e1001220. doi: 10.1371/journal.pgen.1001220
Chankhamhaengdecha, S., Hongvijit, S., Srichaisupakit, A., Charnchai, P., and Panbangred, W. (2013). Endophytic actinomycetes: a novel source of potential acyl homoserine lactone degrading enzymes. Biomed. Res. Int. 2013:782847. doi: 10.1155/2013/782847
Chaudhary, A. K., Dhakal, D., and Sohng, J. K. (2013). An insight into the ”-omics” based engineering of Streptomycetes for secondary metabolite overproduction. Biomed. Res. Int. 2013:968518. doi: 10.1155/2013/968518
Cho, Y. H., Lee, E. J., Ahn, B. E., and Roe, J. H. (2001). SigB, an RNA polymerase sigma factor required for osmoprotection and proper differentiation of Streptomyces coelicolor. Mol. Microbiol. 42, 205–214. doi: 10.1046/j.1365-2958.2001.02622.x
Cho, Y. H., Lee, E. J., and Roe, J. H. (2000). A developmentally regulated catalase required for proper differentiation and osmoprotection of Streptomyces coelicolor. Mol. Microbiol. 35, 150–160. doi: 10.1046/j.1365-2958.2000.01685.x
Datsenko, K. A., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645. doi: 10.1073/pnas.120163297
Demain, A. L. (2014). Importance of microbial natural products and the need to revitalize their discovery. J. Ind. Microbiol. Biotechnol. 41, 185–201. doi: 10.1007/s10295-013-1325-z
Diminic, J., Starcevic, A., Lisfi, M., Baranasic, D., Gacesa, R., Hranueli, D., et al. (2014). Evolutionary concepts in natural products discovery: what actinomycetes have taught us. J. Ind. Microbiol. Biotechnol. 41, 211–217. doi: 10.1007/s10295-013-1337-8
Eilert, E., Kranz, A., Hollenberg, C. P., Piontek, M., and Suckow, M. (2013). Synthesis and release of the bacterial compatible solute 5-hydroxyectoine in Hansenula polymorpha. J. Biotechnol. 167, 85–93. doi: 10.1016/j.jbiotec.2013.02.005
Facey, P. D., Sevcikova, B., Novakova, R., Hitchings, M. D., Crack, J. C., Kormanec, J., et al. (2011). The dpsA gene of Streptomyces coelicolor: induction of expression from a single promoter in response to environmental stress or during development. PLOS ONE 6:e25593. doi: 10.1371/journal.pone.0025593
Fernández, E., Weissbach, U., Sánchez Reillo, C., Braña, A. F., Méndez, C., Rohr, J., et al. (1998). Identification of two genes from Streptomyces argillaceus encoding glycosyltransferases involved in transfer of a disaccharide during biosynthesis of the antitumor drug mithramycin. J. Bacteriol. 180, 4929–4937.
Fernández Martínez, L., Bishop, A., Parkes, L., Del Sol, R., Salerno, P., Sevcikova, B., et al. (2009). Osmoregulation in Streptomyces coelicolor: modulation of SigB activity by OsaC. Mol. Microbiol. 71, 1250–1262. doi: 10.1111/j.1365-2958.2009.06599.x
Godinez, O., Dyson, P., del Sol, R., Barrios-Gonzalez, J., Millán-Pacheco, C., and Mejía, A. (2015). Targeting the osmotic stress response for strain improvement of an industrial producer of secondary metabolites. J. Microbiol. Biotechnol. 25, 1787–1795. doi: 10.4014/jmb.1503.03042
Gordon, N. D., Ottaviano, G. L., Connell, S. E., Tobkin, G. V., Son, C. H., Shterental, S., et al. (2008). Secreted-protein response to sigmaU activity in Streptomyces coelicolor. J. Bacteriol. 190, 894–904. doi: 10.1128/JB.01759-07
Green, M. R., and Sambrook, J. (2012). Molecular Cloning: A Laboratory Manual, Fourth Edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Gust, B., Challis, G. L., Fowler, K., Kieser, T., and Chater, K. F. (2003). PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. U.S.A. 100, 1541–1546. doi: 10.1073/pnas.0337542100
Hopwood, D. A., Bibb, J. M., Chater, K. F., Kieser, T., Bruton, C. J., Kieser, H. M., et al. (1985). Genetic Manipulation of Streptomyces: A Laboratory Manual. Norwich: John Innes Foundation.
Huang da, W., Sherman, B. T., and Lempicki, R. A. (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. doi: 10.1093/nar/gkn923
Huang da, W., Sherman, B. T., and Lempicki, R. A. (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. doi: 10.1038/nprot.2008.211
Jung, K., Fried, L., Behr, S., and Heermann, R. (2012). Histidine kinases and response regulators in networks. Curr. Opin. Microbiol. 15, 118–124. doi: 10.1016/j.mib.2011.11.009
Kieser, T., Hopwood, D. A., Bibb, J. M., Chater, K. F., and Buttner, M. J. (2000). Practical Streptomyces Genetics. Norwich: John Innes Foundation.
Lee, E. J., Cho, Y. H., Kim, H. S., Ahn, B. E., and Roe, J. H. (2004a). Regulation of sigmaB by an anti- and an anti-anti-sigma factor in Streptomyces coelicolor in response to osmotic stress. J. Bacteriol. 186, 8490–8498. doi: 10.1128/JB.186.24.8490-8498.2004
Lee, E. J., Cho, Y. H., Kim, H. S., and Roe, J. H. (2004b). Identification of sigmaB-dependent promoters using consensus-directed search of Streptomyces coelicolor genome. J. Microbiol. 42, 147–151.
Lee, E. J., Karoonuthaisiri, N., Kim, H. S., Park, J. H., Cha, C. J., Kao, C. M., et al. (2005). A master regulator sigmaB governs osmotic and oxidative response as well as differentiation via a network of sigma factors in Streptomyces coelicolor. Mol. Microbiol. 57, 1252–1264. doi: 10.1111/j.1365-2958.2005.04761.x
Liu, G., Chater, K. F., Chandra, G., Niu, G., and Tan, H. (2013). Molecular regulation of antibiotic biosynthesis in Streptomyces. Microbiol. Mol. Biol. Rev. 77, 112–143. doi: 10.1128/MMBR.00054-12
MacNeil, D. J., Gewain, K. M., Ruby, C. L., Dezeny, G., Gibbons, P. H., and MacNeil, T. (1992). Analysis of Streptomyces avermitilis genes required for avermectin biosynthesis utilizing a novel integration vector. Gene 111, 61–68. doi: 10.1016/0378-1119(92)90603-M
Nett, M., Ikeda, H., and Moore, B. S. (2009). Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat. Prod. Rep. 26, 1362–1384. doi: 10.1039/b817069j
Podgornaia, A. I., and Laub, M. T. (2013). Determinants of specificity in two-component signal transduction. Curr. Opin. Microbiol. 16, 156–162. doi: 10.1016/j.mib.2013.01.004
Rico, S., Santamaría, R. I., Yepes, A., Rodríguez, H., Laing, E., Bucca, G., et al. (2014a). Deciphering the regulon of Streptomyces coelicolor AbrC3, a positive response regulator of antibiotic production. Appl. Environ. Microbiol. 80, 2417–2428. doi: 10.1128/AEM.03378-13
Rodríguez, H., Rico, S., Díaz, M., and Santamaría, R. I. (2013). Two-component systems in Streptomyces: key regulators of antibiotic complex pathways. Microb. Cell Fact. 12:127. doi: 10.1186/1475-2859-12-127
Rodríguez, H., Rico, S., Yepes, A., Franco-Echevarría, E., Antoraz, S., Santamaría, R. I., et al. (2015). The two kinases, AbrC1 and AbrC2, of the atypical two-component system AbrC are needed to regulate antibiotic production and differentiation in Streptomyces coelicolor. Front. Microbiol. 6:450. doi: 10.3389/fmicb.2015.00450
Rowland, M. A., and Deeds, E. J. (2014). Crosstalk and the evolution of specificity in two-component signaling. Proc. Natl. Acad. Sci. U.S.A. 111, 5550–5555. doi: 10.1073/pnas.1317178111
Sadeghi, A., Soltani, B. M., Jouzani, G. S., Karimi, E., Nekouei, M. K., and Sadeghizadeh, M. (2014a). Taxonomic study of a salt tolerant Streptomyces sp. strain C-2012 and the effect of salt and ectoine on lon expression level. Microbiol. Res. 169, 232–238. doi: 10.1016/j.micres.2013.06.010
Sadeghi, A., Soltani, B. M., Nekouei, M. K., Jouzani, G. S., Mirzaei, H. H., and Sadeghizadeh, M. (2014b). Diversity of the ectoines biosynthesis genes in the salt tolerant Streptomyces and evidence for inductive effect of ectoines on their accumulation. Microbiol. Res. 169, 699–708. doi: 10.1016/j.micres.2014.02.005
Salazar, M. E., and Laub, M. T. (2015). Temporal and evolutionary dynamics of two-component signaling pathways. Curr. Opin. Microbiol. 24, 7–14. doi: 10.1016/j.mib.2014.12.003
Sambrook, J., Fritsch, E., and Maniatis, T. (1989). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
Schaller, G. E., Shiu, S. H., and Armitage, J. P. (2011). Two-component systems and their co-option for eukaryotic signal transduction. Curr. Biol. 21, R320–R330. doi: 10.1016/j.cub.2011.02.045
Sheeler, N. L., MacMillan, S. V., and Nodwell, J. R. (2005). Biochemical activities of the absA two-component system of Streptomyces coelicolor. J. Bacteriol. 187, 687–696. doi: 10.1128/JB.187.2.687-696.2005
Urem, M., van Rossum, T., Bucca, G., Moolenaar, G. F., Laing, E., Swiatek-Polatynska, M. A., et al. (2016). OsdR of Streptomyces coelicolor and the dormancy regulator DevR of Mycobacterium tuberculosis control overlapping regulons. mSystems 1:e00014–16. doi: 10.1128/mSystems.00014-16
van Keulen, G., Hopwood, D. A., Dijkhuizen, L., and Sawers, R. G. (2005). Gas vesicles in actinomycetes: old buoys in novel habitats? Trends Microbiol. 13, 350–354. doi: 10.1016/j.tim.2005.06.006
Vetsigian, K., Jajoo, R., and Kishony, R. (2011). Structure and evolution of Streptomyces interaction networks in soil and in silico. PLOS Biol. 9:e1001184. doi: 10.1371/journal.pbio.1001184
Wang, W., Shu, D., Chen, L., Jiang, W., and Lu, Y. (2009). Cross-talk between an orphan response regulator and a noncognate histidine kinase in Streptomyces coelicolor. FEMS Microbiol. Lett. 294, 150–156. doi: 10.1111/j.1574-6968.2009.01563.x
Whitworth, D. E., and Cock, P. J. (2009). Evolution of prokaryotic two-component systems: insights from comparative genomics. Amino Acids 37, 459–466. doi: 10.1007/s00726-009-0259-2
Yepes, A., Rico, S., Rodríguez-García, A., Santamaría, R. I., and Díaz, M. (2011). Novel two-component systems implied in antibiotic production in Streptomyces coelicolor. PLOS ONE 6:e19980. doi: 10.1371/journal.pone.0019980
Keywords: Streptomyces, two-component systems, antibiotic production, orphan response regulator, crosstalk, stress response
Citation: Antoraz S, Rico S, Rodríguez H, Sevillano L, Alzate JF, Santamaría RI and Díaz M (2017) The Orphan Response Regulator Aor1 Is a New Relevant Piece in the Complex Puzzle of Streptomyces coelicolor Antibiotic Regulatory Network. Front. Microbiol. 8:2444. doi: 10.3389/fmicb.2017.02444
Received: 05 September 2017; Accepted: 24 November 2017;
Published: 12 December 2017.
Edited by:
Bart Devreese, Ghent University, BelgiumReviewed by:
Yinhua Lu, Shanghai Institutes for Biological Sciences (CAS), ChinaSébastien Rigali, University of Liège, Belgium
Copyright © 2017 Antoraz, Rico, Rodríguez, Sevillano, Alzate, Santamaría and Díaz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margarita Díaz, bWFyZGlAdXNhbC5lcw== Ramón I. Santamaría, c2FudGFAdXNhbC5lcw==