 Margarita Gomila
Margarita Gomila Antonio Busquets
Antonio Busquets Magdalena Mulet
Magdalena Mulet Elena García-Valdés
Elena García-Valdés Jorge Lalucat
Jorge Lalucat
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 07 December 2017
Sec. Evolutionary and Genomic Microbiology
Volume 8 - 2017 | https://doi.org/10.3389/fmicb.2017.02422
This article is part of the Research Topic Microbial Taxonomy, Phylogeny and Biodiversity View all 28 articles
The Pseudomonas syringae phylogenetic group comprises 15 recognized bacterial species and more than 60 pathovars. The classification and identification of strains is relevant for practical reasons but also for understanding the epidemiology and ecology of this group of plant pathogenic bacteria. Genome-based taxonomic analyses have been introduced recently to clarify the taxonomy of the whole genus. A set of 139 draft and complete genome sequences of strains belonging to all species of the P. syringae group available in public databases were analyzed, together with the genomes of closely related species used as outgroups. Comparative genomics based on the genome sequences of the species type strains in the group allowed the delineation of phylogenomic species and demonstrated that a high proportion of strains included in the study are misclassified. Furthermore, representatives of at least 7 putative novel species were detected. It was also confirmed that P. ficuserectae, P. meliae, and P. savastanoi are later synonyms of P. amygdali and that “P. coronafaciens” should be revived as a nomenspecies.
The genus Pseudomonas is divided into two phylogenetic lineages (Pseudomonas aeruginosa and Pseudomonas fluorescens) based on inferred evolutionary relationships by using multilocus sequence analysis (MLSA) of four housekeeping genes (Mulet et al., 2010). The P. fluorescens lineage contains six phylogenetic groups, one of them represented by Pseudomonas syringae, and includes most of the phytopathogens within the genus Pseudomonas (Bull et al., 2010).
P. syringae was described by Van Hall (1902) and several closely related species have since been described. In the Approved List of Bacterial Names (Skerman et al., 1980), three other species of phytopathogenic Pseudomonas were also included: Pseudomonas cichorii (Stapp, 1928), Pseudomonas viridiflava (Burkholder, 1939), Pseudomonas caricapapayae (Robbs, 1956), and Pseudomonas amygdali (Psallidas and Panagopoulos, 1975). “Pseudomonas coronafaciens” (Schaad and Cunfer, 1979) was not included in the Approved List of Bacterial Names and is not recognized as a valid species name. Until that moment, species characterizations and proposals have been performed using physiological, biochemical, serological, and pathological traits. Later, several other species closely related to P. syringae were proposed and validated: Pseudomonas meliae (Ogimi, 1977), Pseudomonas savastanoi (Gardan et al., 1992), Pseudomonas ficuserectae (Goto, 1983), Pseudomonas avellanae (Janse et al., 1996), Pseudomonas cannabina (Gardan et al., 1999), Pseudomonas tremae (Gardan et al., 1999), Pseudomonas congelans (Behrendt et al., 2003), Pseudomonas asturiensis (González et al., 2013), Pseudomonas cerasi (Kałuzna et al., 2016), and Pseudomonas caspiana (Busquets et al., 2017). The P. syringae species complex is usually considered to include all these taxonomically closely related species.
Molecular techniques based on experimental DNA-DNA hybridizations (DDH) or on DNA sequence analysis are essential in determining actual taxonomy. DDH were used first in the P. syringae group of species by Pecnold and Grogan (1973) and when P. savastanoi was proposed (Gardan et al., 1992). Gardan and colleagues established eight genomic groups, called genomospecies, based on DDH analysis (Gardan et al., 1999) that allowed the reclassification of strains previously known as pathovars of P. syringae as the new species P. cannabina and P. tremae. A phylogenetic study based on the 16S rRNA gene sequences of species in the genus was applied first by Moore et al. (1996) to propose a phylogenetic scheme within the genus, but until the description of P. avellanae, sequence analyses were not included in new species proposals in the P. syringae species complex. Due to the limitations in sequence variation in the 16S rRNA gene, other genes have been used for species delineation, especially the rpoD gene (Yamamoto et al., 2000; Mulet et al., 2010; Parkinson et al., 2011) and the cts gene (Berge et al., 2014). These analyses have allowed the delineation of phylogenetic groups, or phylogroups, within the species complex. Multilocus sequence analyses (MLSA) based on the sequences of three or four housekeeping genes have also been very successful in clarifying the phylogeny of strains in the Pseudomonas genus (Mulet et al., 2010; Bull et al., 2011; Berge et al., 2014). More specifically, Almeida et al. (2010) have developed the Plant associated microbes database (PAMDB) that contains sequences for MLST and MLSA accessible in a useful website. As a result of the molecular techniques, many strains have been reclassified and a more stable phylogenetic classification has become possible. Determining the precise taxonomic affiliations of strains in the P. syringae species complex can be difficult when pathovars are considered (Baltrus, 2016; Vinatzer et al., 2017). Currently, the species in the P. syringae phylogenetic group are subdivided into over 60 pathovars defined by pathogenic characters, 15 genomospecies are defined by DDH, 13 phylogroups are defined by MLSA using 3 or 4 genes, and 15 validly described species are accepted in the List of Prokaryotic Names with Standing in Nomenclature (Parte, 2014; http://www.bacterio.net/). Vinatzer and Bull (2009) have published a comprehensive history of the taxonomy of plant pathogenic bacteria, the use of MLSA and the impact of genomic approaches on taxonomy of plant pathogenic bacteria.
Genome-based taxonomic analyses have been recently introduced, and several algorithms are currently used for strain comparisons, such as the average nucleotide identity based on BLAST or MUMmer algorithms (ANIb, ANIm) and genome-to-genome distance calculations (GGDC), and are substituted for experimental DDH (Konstantinidis and Tiedje, 2007). Comparative genomics provides another tool that allows core genome and pangenome analyses at different levels of classification in a phylogenomic approach, that is, phylogenetic inference by combining many genes (Jeffroy et al., 2006). Recently, it has been proposed the use of similarity-based codes, called life identification numbers (LINs) to name individual bacterial isolates in the P. syringae species complex (Vinatzer et al., 2017).
As noted by Morris et al. (2017), “delineation of pertinent phylogenetic contours of plant pathogenic bacteria and naming of strains independent of their presumed life style is one of the five challenges for understanding the ecology of plant pathogenic bacteria.” With the goal of clarifying the taxonomic delineation of species in the P. syringae phylogenetic group, 139 genomes of the 15 recognized species assigned to this group that are available in public databases have been analyzed by a phylogenomic approach. At least one member of each phylogroup described in the P. syringae phylogenetic branch by Berge et al. (2014) was included in the analyses if it was available in the public databases. “P. coronafaciens” strains and the three closely related species in the Pseudomonas lutea group (P. graminis, P. lutea, and P. abietaniphila) were also included, as well as an unclassified Pseudomonas sp. strain S25 isolated in our laboratory. MLSA and several in silico algorithms for genome comparisons (e.g., ANIb, GGDC) allowed the clustering of strains in 6 clear genomic branches. Core genome and pangenome analyses have been performed in the present study for the whole P. syringae phylogenetic group, for 5 of the 6 individual genomic branches and for 7 proposed phylogenomic species to explore their usefulness to delineate inter- and intra-species relationships. We included in our study the genome sequences of the type strains in the P. syringae group and 19 of the 56 pathotype strains recently published by Thakur et al. (2016) with the main purpose to clarify the species delineation, without considering all the pathovars. Species affiliation of the pathotypes is a prerequisite for the posterior study of the phylogeny of the pathovars.
Draft and complete genome sequences of 139 strains belonging to different species of the P. syringae group available in the NCBI database were analyzed, including the genomes of 3 P. aeruginosa strains and 2 P. stutzeri strains used as outgroup. The 139 selected strains included the genomes of 15 species type strains of the P. syringae phylogenetic group, Pseudomonas sp. S25 and representatives of “P. coronafaciens.” Genomes of strains in the P. lutea phylogenetic group (P. lutea, P. graminis, and P. abietaniphila) were used as an outgroup in the analysis because they belong to the closest phylogenetic group to the P. syringae group (Gomila et al., 2015). Six type strain genomes were analyzed in duplicate: five were representatives of the same type strain but from two different culture collections, and the sequence of P. cannabina was deposited twice by 2 different authors. The set of 139 genome sequences of Pseudomonas was retrieved from the GenBank database on 30th April 2017. The list of the 139 complete or draft genomes analyzed and additional details are provided in Supplemental Table S1.
An MLSA based on the analysis of the partial sequences of the 16S rRNA, gyrB and rpoD genes was performed. The sequences of the 16S rRNA, gyrB, and rpoD genes were extracted from each genome studied and compared with the corresponding sequences of all Pseudomonas species type strains (161) described through 2016. Sequences are available in the public National Center for Biotechnology Information (NCBI) database. A concatenated gene tree was constructed using the individual alignments in the following order: 16S rRNA (1,309 nt), gyrB (803 nt), and rpoD (791 nt) by methods previously described (Gomila et al., 2015). Genomes that did not contain the 16S rRNA, gyrB, and rpoD gene sequences were removed from the MLSA concatenated analysis.
In silico tools were used for genomic species delineation. Average nucleotide identity based on BLAST algorithm (ANIb) was calculated between all pairs of genomes, using the JSpecies software tool available at the webpage http://www.imedea.uib.es/jspecies (Konstantinidis and Tiedje, 2007; Richter and Rosselló-Móra, 2009). The recommended species cut-off was 95%. The similarity matrix obtained with all pairwise genomic comparisons was used to generate a UPGMA dendrogram using the PAST software. GGDC was performed between genome pairs on specific sets of genomes using the GGDC 2.0 update available in the web service http://ggdc.dsmz.de (Meier-Kolthoff et al., 2013).
A comparative genomic analysis was performed using the GET_HOMOLOGUES software described by Contreras-Moreira and Vinuesa (2013). All genomes were annotated with PROKKA for comparison purposes (Seemann, 2014), and the protein amino acid sequences obtained were compared using the criterion of 50% similarity over 50% of coverage alignment. Core genome and pangenome analyses were performed with three different clustering algorithms, bi-directional best-hits (BDBH), COGtriangle (COG), and OrthoMCL (OMCL). The four clusters determined from the analyses were defined as previously described (Koonin and Wolf, 2008; Kaas et al., 2012): core, soft core, shell, and cloud. Core genome and pangenome analyses were performed for all the genomes analyzed and for subsets of them.
All proteins codified by genes of the core genome that were present in monocopy were aligned, and the resulting alignments were concatenated. Elimination of poorly aligned positions and divergent regions of protein sequences were performed with Gblocks (Castresana, 2000), and the phylogenetic tree was constructed with the PhyML program (Guindon et al., 2010). Analysis of the concatenated amino acid sequences of the core proteins (core MLSA) was performed for all genomes and for the different delineated subsets.
A GET_HOMOLOGUES script was used to estimate the average amino acid identities of CDSs between individual members of specific pangenome clusters. Gower's distance matrix were determined based on the percent amino acid identities of protein coding genes in the different genome branches using a script from GET_HOMOLOGUES Those distance matrices obtained were further illustrated as heatmaps, showing similarities and differences between genomes.
Genome characteristics of the strains studied are summarized in Supplemental Table S1. The genome sequences of the 15 species type strains so far described in the P. syringae phylogenetic group were included. At least one member of 11 of the 13 phylogroups described in the P. syringae phylogenetic branch by Berge et al. (2014) was included in the analyses. Genomes of phylogroups 8 and 12 were not available in public databases. As a control, the sequences of the genomes of the P. cichorii, P. viridiflava, P. congelans, and P. meliae type strains were studied in duplicate, i.e., two type strains from two different culture collections. Two genome sequences of P. cannabina ICMP 2823T with two different accession numbers were also studied. The studied genomes included 121 genomes with a status of “contig” (the number of contigs ranged from 5 to 5,099; mean: 617 contigs) and 6 with a status of “complete genome.” The chromosome sizes ranged from 4,713,747 to 7,317,256 bp (mean: 5,976,989 bp) and the GC content in mol % ranged from 56.95 to 59.38 (mean: 58.34). Plasmids were reported in the databases for only 3 of the 6 closed genomes: P. savastanoi pv. savastanoi strain 1448A (2 plasmids, 3% of the genome content), P. cerasi strain 58T (6 plasmids, 7% of the genome), and P. syringae pv. tomato DC3000 (2 plasmids, 2% of the genome). The plasmids were not included in the comparative analyses.
The authenticity of the type strains studied was checked by analyzing their affiliation in the Pseudomonas 3-gene MLSA tree (Gomila et al., 2015). All type strains were affiliated with the previously determined gene sequences with the exception of 2 species type strains, P. tremae and P. lutea. The genome of P. tremae ICMP 9151T clustered close to those of “P. coronafaciens” strains, and the sequences were different from those published for P. tremae LMG 22121T. Therefore, the published sequences of the cts, gyrB, rpoB, rpoD, aconitase, and 16S rRNA genes of the type strains of three different culture collections (LMG 22121T, CFBP 3229T, NCPPB 3465T) were compared with the corresponding sequences of the P. tremae ICMP 9151T genome. The sequences were only 88–98% identical. We concluded that the status of the species type strain of P. tremae ICMP 9151T must be revised, and therefore it was not further considered as a type strain in the present study. Two genome sequences are available for P. lutea type strains. Surprisingly, P. lutea LMG 21974T was an outlier. The 16S rDNA gene sequence of strain LMG 21974T was 99% identical to that of Pseudomonas poae DSM 14936T in the P. fluorescens phylogenetic group. Several housekeeping genes of strain LMG 21974T were analyzed, and it was concluded that the deposited P. lutea LMG 21974T genome did not belong to the P. lutea phylogenetic group. The rest of the duplicated genome sequences were concordant.
All 139 genomes were phylogenetically analyzed using different strategies: (i) 3-gene MLSA of the partial sequences of the 16S rRNA, gyrB and rpoD genes, (ii) a phylogenomic tree based on the concatenation of all single-copy conserved protein sequences that conforms the core genome of the 139 genomes analyzed (core MLSA), and (iii) by ANIb.
(i) The 3-gene MLSA phylogenetic analysis included the 15 species type strains in the P. syringae phylogenetic group that are validly described and were used in a previous publication (Gomila et al., 2015) combined with the 139 Pseudomonas complete or draft genomes available in databases. A phylogenetic tree (Supplemental Figure S1) was generated based on the concatenated sequences with a total length of 2,796 nucleotides. One hundred and nine of the 139 strains (78% of the genomes analyzed) were affiliated with the corresponding species type strain, and their species assignments were considered correct.
(ii) One hundred and forty-nine monocopy genes were defined in the core genome of the whole set of 139 genomes. The phylogenomic tree obtained after the concatenation of the amino acid sequences of the 149 monocopy genes (core MLSA) is shown in Supplemental Figure S2. The P. aeruginosa and P. stutzeri genomes were used as outgroups. From the 33,744 positions obtained after the concatenation of the individual alignments, 93% of them were finally analyzed (31,400 positions). Bootstrap values were indicated on the nodes. In this phylogenomic tree, six main clusters or phylogenomic branches could be detected, indicated in Roman numerals from I to VI (Supplemental Figure S2).
(iii) Average nucleotide identities based on BLAST (ANIb) were calculated for the 139 genomes, obtaining a square matrix with 19,321 pairwise comparison values (Supplemental Table S2). A dendrogram was generated for this matrix to assess phylogenetic coherence (Figure 1). The ANIb dendrogram showed high topological congruence compared with the 3-gene MLSA phylogenetic tree and with the core MLSA phylogenetic tree. All duplicated type strains clustered together with ANIb values higher than 99.87%. The reference genomes of P. aeruginosa, P. stutzeri, and the species in the P. lutea group clustered outside the P. syringae group. All ANIb percentage values calculated were plotted on a graph that demonstrated a clear gap between 89 and 93% (Supplemental Figure S3). Only 206 values were observed between 93 and 96%, <2%, and corresponded to pairwise comparisons among strains in groups I and II. Six genomic branches were again delineated in the P. syringae group based on the observed tree branching in the ANIb dendrogram with an ANIb cut-off of 93%, such that members of different genomic branches could not have an ANIb of >93%. The genomic branches corresponded to the same six main phylogenetic branches detected in the 3-gene MLSA and in the core-gene phylogenetic tree. Therefore, each branch was considered a homogeneous phylogenomic branch, without taxonomic implications, to later facilitate comparative genomic analyses. The only exception detected was genomic branch II, which was divided into two clusters by ANIb value. The boundary values of the phylogenomic branches (minimal value among strains of different phylogenomic branches) were higher than 4.8%. The usually accepted cut-off for species delineation based on the ANIb lies between 95 and 96% (Richter and Rosselló-Móra, 2009). In branch I (represented by P. syringae) and branch IV (represented by P. amygdali), no clear gap at the 95–96% ANIb cut-off could be delineated; therefore, we were not able to distinguish genomic species (genospecies or genomospecies) by using only the ANIb value. Groupings of strains with intrabranch values higher than 94.3% in the other four branches were separated by clear gaps, and each group was considered a phylogenomic species. Seventeen clusters with ANIb intrabranch values higher than 94.3% could be differentiated, and their boundary values were higher than 4.4% with the exception of 9 strains in genomic branch I, which included the P. syringae type strain, as indicated in Table 1 and Supplemental Table S2. Each of the 17 clusters was considered a phylogenomic species. Genomic branches I, IV, V, and VI contain more than one validly described species.
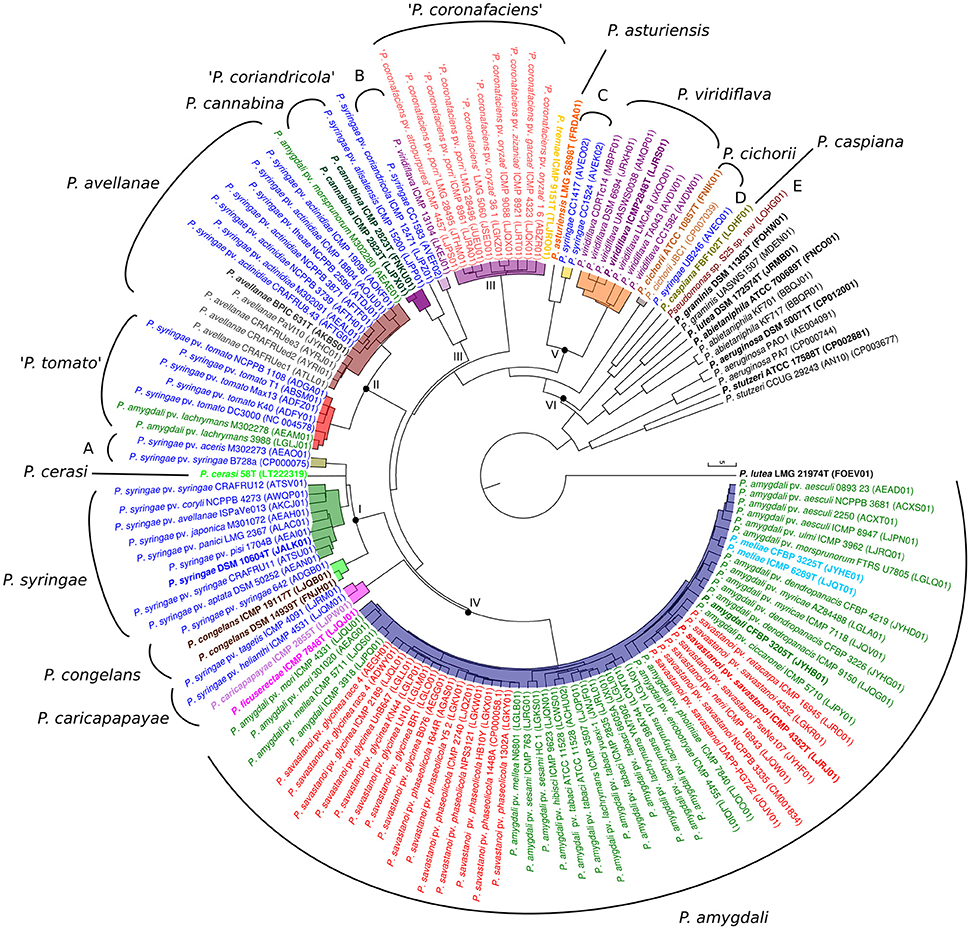
Figure 1. UPGMA dendrogram based on ANIb values of all pairwise comparisons. Each species name, as submitted in the database, is labeled with different colors. Roman numerals at the corresponding nodes indicate phylogenomic branches defined. Phylogenomic species inside each phylogenetic branch are highlighted with different colors. Species type strains are labeled in bold. Accession numbers of the corresponding genomes are given in brackets. Proposed phylogenomic species are indicated in the external circle. Putative novel species are marked in quotation marks or by capital letters (A–E).
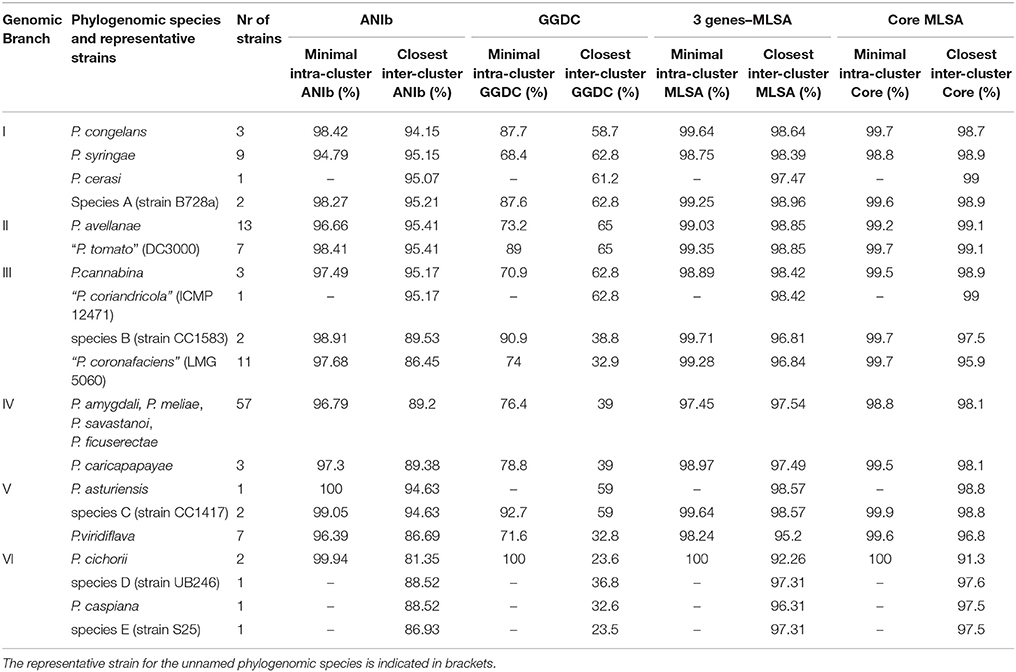
Table 1. ANIb, GGDC, 3 genes-MLSA, and core MLSA indices for the delineation of the proposed phylogenomic species in each genomic branch.
The results obtained for the three methodologies applied were compared. Strain clustering was maintained in the 3-gene MLSA at a cut-off of 97% (Table 1), which is in agreement with previous studies (Gomila et al., 2015), but some slight differences can be observed in the branching order (Supplemental Figure S1). Two strains with a 3-gene MLSA value lower than 97% cannot be assigned to the same species. Phylogenetic similarities in the analysis of the three concatenated genes were compared with the ANIb similarities calculated in the whole genome analysis and the results plotted in Supplemental Figure S4. A good correspondence between the ANIb and 3-gene MLSA indices could be observed. The six main clusters or genomic branches observed in the ANIb results were also detected in the core-gene MLSA tree, although phylogenomic branch III was divided into two closely related branches in the ANIb analysis.
GGDC similarities were also calculated for all genomes included in each ANIb/core MLSA genomic branch in order to clarify phylogenetic assignments to species. The results are shown in Supplemental Table S3 and were highly concordant with the ANIb values, accepting a species cut-off value of 70% as recommended by Meier-Kolthoff et al. (2013). It is worth mentioning that the 2 genomes available for the P. cannabina type strain were only 70.9% similar in the GGDC analysis but were almost identical in the ANIb (99.9% similar) and MLSA analyses (100% identical). This discrepancy has to be attributed to the in silico methodologies or to the quality of the genome sequences.
The combined use of the 4 indices allowed the delineation of 19 phylogenomic species, and these are described below in the context of each phylogenomic branch. ANIb values among members of different phylogenomic species were lower than 96%. Genomic branch I (intrabranch values: 93.11–98.18% for ANIb and 97.18–99.93% for 3-gene MLSA) included 15 strains divided into 4 groups, 3 of them belonging to recognized taxonomically described species: the P. syringae type strain and 8 closely related strains, P. cerasi (1 strain), and P. congelans (2 strains; the type strain is duplicated). The fourth group, designated as unnamed group A and represented by strain B728a, includes 2 strains. The boundaries at 95% ANIb were diffuse within this branch, although the 4 clusters could be clearly distinguished with GGDC values lower than the accepted cut-off of 70% (intrabranch values between the 4 groups ranged between 54.9 and 62.8%).
Genomic branch II, represented by the P. avellanae type strain and 19 other strains, presented intrabranch values of 94.30–99.99% for ANIb, 98.21–100% for 3-gene MLSA, and 59–99% for GGDC. Two homogeneous and clear sub-branches could be distinguished at a cut-off lower than 96% in ANIb (94.30–95.41%), which corresponded to 95.09% in 3-gene MLSA and values lower of 64% in GGDC and can be considered phylogenomic species. Seven strains, represented by P. syringae pv. tomato DC3000, conformed a possible phylogenomic species. They grouped with similarities higher than 98.41%. The other phylogenomic species, represented by the P. avellanae type strain, together with 12 additional strains, grouped at 96.6% of ANIb, and in both cases, GGDC values were higher than 70%. Each phylogenomic species was circumscribed by uniform intrabranch values.
Intrabranch values of genomic branch III were 85.15–99.9% ANIb, 93.90–100% 3-gene MLSA, and 30–99.9% GGDC. This genomic branch included 17 strains distributed in 4 possible phylogenomic species: one included 2 P. cannabina type strain genome sequences and another strain identified as P. syringae; P. syringae pv. coriandricola ICMP 12471 was a singleton; 11 strains (10 of them classified as “P. coronafaciens,” not taxonomically validly described, and the supposed type strain of P. tremae); and an unnamed phylogenomic species B (2 strains). The 4 sub-branches were also clearly distinguished at a threshold of 62% in GGDC.
Intrabranch values of genomic branch IV were 88.56–99.99% for ANIb, 96.13–100% for 3-gene MLSA, and 37.3–99.8% for GGDC. It included 5 validly described species type strains: the P. meliae, P. amygdali, P. savastanoi, and P. ficuserectae type strains, grouped together with 53 additional strains forming a unique phylogenomic species with clear boundaries from the rest of the strains; and P. caricapapayae (together with two additional strains). The 2 proposed phylogenomic species were clearly separated at the established cut-offs of 95% ANIb, 97% MLSA, and 70% GGDC.
Genomic branch V included two validly described species type strains with intrabranch values of 80.1–99.97% for ANIb, 91.68–100% for 3-gene MLSA, and 32.6–92.7% for GGDC. P. asturiensis was a singleton, and P. viridiflava was represented by 7 strains. Two strains formed a separate sub-branch (possible phylogenomic species C) close to P. asturiensis. The phylogenomic species were separated at cut-offs of 95% ANIb, 97% MLSA, and 70% GGDC.
Genomic branch VI was more distant and diverse. It included the type strains of P. cichorii and P. caspiana, together with other 2 strains: Pseudomonas sp. S25 and P. syringae UB246. The 4 strains were clearly separated at the accepted species cut-offs for ANIb, GGDC and 3-gene MLSA and have to be considered four different species.
As discussed later, these results suggested that a high proportion of genomes (53 of 127, 42%) were submitted in the databases with a species name affiliation different from that suggested by their affiliation in the ANIb, GGDC, 3-gene MLSA, and core MLSA dendrograms.
Sixty-two pathovars of P. syringae are listed in the “Comprehensive list of names of plant pathogenic bacteria, 1980–2007” (Bull et al., 2010). Twenty-seven strains of P. syringae assigned to 15 different pathovars were included in the present study (3 pathovars with more than 1 representative: pv. actinidiae 6 strains, pv. tomato 5 strains, and pv. syringae 4 strains) to emphasize that the correct species affiliation is a prerequisite for the posterior study of the phylogeny of the pathovars. The 6 pv. actinidiae strains clustered together with P. avellanae strains. The 5 strains of pv. tomato clustered also together in one phylogenomic species included in genomic branch II. On the contrary, the 4 strains of pv. syringae affiliated to three different phylogenomic species (P. congelans, P. syringae, and the proposed new phylogenomic species A) in branch I. The rest of pathovars represented by single strains were distributed in branches I, II, III, and IV. Only 8 of the pathovars were affiliated with P. syringae phylogenomic species (e.g., P. syringae pv. tagetis ICMP 4091 and P. syringae pv. helianthi ICMP 4531 belonged to P. caricapapayae). It is worthy of note that five of the six P. syringae pathotype strains included in this study did not affiliate with P. syringae genomic species: P. syringae pv. tagetis ICMP 4091 belongs to P. caricapapayae; P. syringae pv. actinidiae NCPPB 3739 belongs to P. avellanae; P. syringae pv. alisalensis ICMP 15200 belongs to P. cannabina; and finally P. syringae pv. coriandricola ICMP 12471 was a putative new phylogenomic species. This suggested the need to reclassify the misclassified strains.
Most of the 14 pathovars of the 33 P. amygdali strains clustered in the same phylogenomic species with the exception of P. amygdali pv. morsprunorum, that clustered with P. avellanae strains, and 2 P. amygdali pv. lachrymans that clustered with P. syringae pv. tomato strains. The 5 pathovars of the 23 P. savastanoi strains clustered together in the same phylogenomic species represented by P. amygdali. The 5 pathovars of the 9 strains of P. coronafaciens affiliated to the same phylogenomic species. Only 3 pathovars (lachrymans, morsprunorum, and syringae) were assigned to more than 1 phylogenomic species.
To facilitate later comparative genomic analyses and due to the good correspondence between the 4 methods, the 6 genomic branches were maintained for further comparative genomic analysis (Figure 1, Supplemental Figure S2 and Table 1). Similarities between the members of different branches were always lower than 90% in ANIb, lower than 97% in 3-gene MLSA and lower than 70% in GGDC analyses.
Core genome and pangenome analyses were performed for the whole set of strains selected belonging to the P. syringae group (127 genomes) and for each of the five genomic branches (I–V) delineated by the previous methodologies. Members of group VI were not included in the analyses because they were more distantly related and only 5 strains representing 4 phylogenomic species were available. Each set of genomes was analyzed with the GET_HOMOLOGUES software. Different images were produced for each pangenome analysis: (1) a Venn diagram of core genomes generated by the three algorithms BDBH, COG, and OMCL and of pangenomes generated by COG and OMCL algorithm, (2) the core genome size was estimated with the Tettelin and Willenbrock fits and the pangenome size with the Tettelin fit, and (3) the partition of the OMCL pangenomic matrix into shell, cloud, soft core, and core compartments (Figure 2, Supplemental Figures S5–S9).
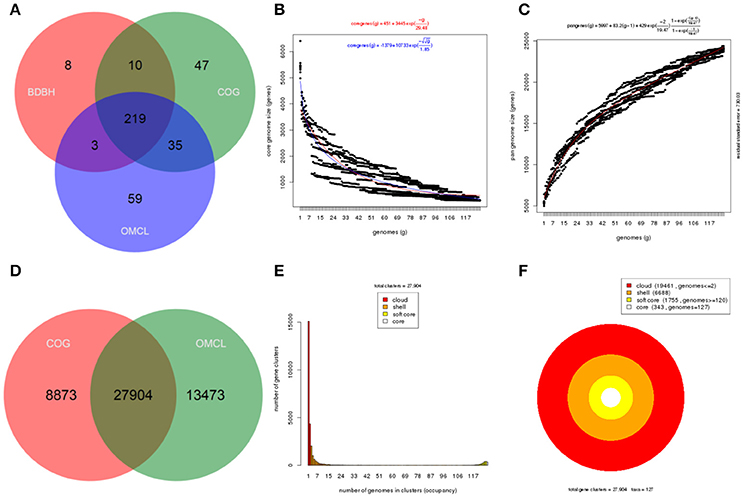
Figure 2. Core and pangenome analysis of the 127 strains in the P. syringae phylogenetic group. (A) Venn diagram of core genomes generated by the BDBH, COG, and OMCL strategies. (B) Estimate of core genome size with the Tettelin (blue) and Willenbrock (red) fits. (C) Estimate of pangenome size with the Tettelin fit. (D) Venn analysis of pangenomes generated by COG and OMCL. (E,F) Partition of the OMCL pangenomic matrix into shell, cloud, soft-core, and core compartments. These plots can be easily created with GET_HOMOLOGUES auxiliary scripts, as explained in the manual.
The core genome of the 127 strains in the P. syringae phylogenetic group contained 343 genes. Two hundred and nineteen of them were in monocopy and were concatenated and analyzed to establish their phylogenetic relationships as previously described. The clustering of strains in the 6 phylogenetic groups was identical to those obtained with the other indices (ANIb, GGDC, 3-gene MLSA, and core MLSA of 139 genomes), showing the same branching order and supported by high bootstrap values (100) (Figure 3). Genomic branch II was the only exception, being separated from groups I, III, and IV with a bootstrap value of only 12. Bull et al. (2011) also observed this result. The 5 main phylogenomic branches were also supported by a high number of shared genes, representing the core genome proteins as a minimum of 20% of the whole genome, as indicated in Table 2.
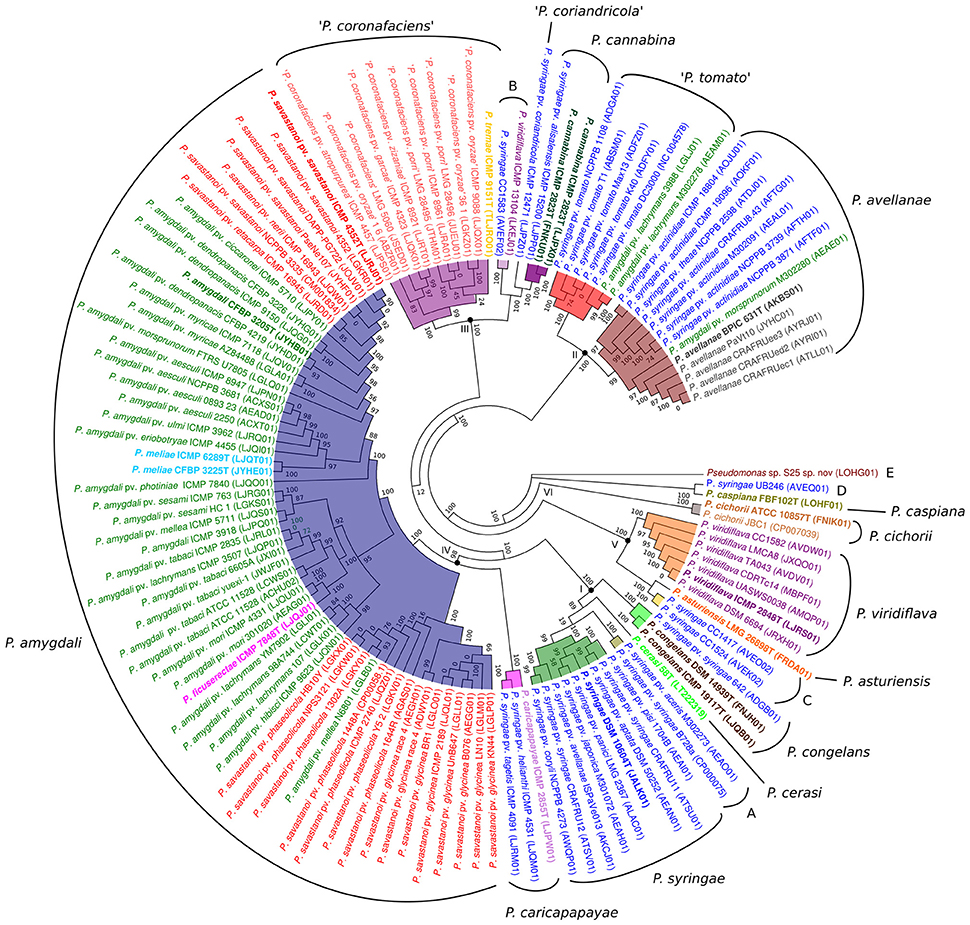
Figure 3. Phylogenetic tree of the concatenated amino acid sequences of 219 monocopy proteins of the core genome defined in the 127 genomes analyzed. Forty-three thousand one hundred thirty-three amino acid positions were used to construct the tree. Each species name, as submitted in the database, is labeled with different colors. Roman numerals at the corresponding nodes indicate phylogenomic branches defined. Phylogenomic species inside each phylogenetic branch are highlighted with different colors. Species type strains are labeled in bold. Accession numbers of the corresponding genomes are given in brackets. Proposed phylogenomic species are indicated in the external circle. Putative novel species are marked in quotation marks or by capital letters (A–E). All bootstrap values are indicated in the nodes.
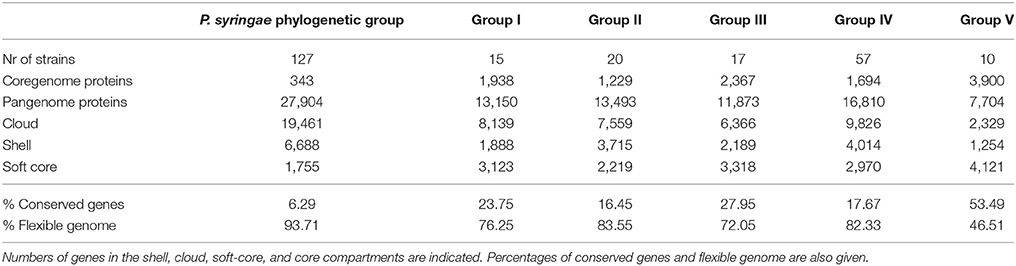
Table 2. Core and pangenome analyses of the 127 strains included in the P. syringae phylogenetic group, as well as for 5 of the individual groups defined (I–V).
Each branch was analyzed separately to assess the potential use of the shared genes for species delineation. Table 2 summarizes the number of core genome and pangenome genes calculated for all genomes and for each specific group analyzed. Table 2 shows the genes present in the “soft core” (genes present in 95% of the genome analyzed), in the “shell” cluster (genes moderately common in the pangenome, present in >10% and <89% of the genomes and in the “cloud” cluster (genes present in very few of the genomes analyzed, 2 or less). The “core” and “soft core” clusters include highly conserved genes with phylogenetic information (Bezuidt et al., 2016). The shell and the cloud clusters represent subsets of the flexible genome, which reflect the adaptation of strains to particular environments and also the evolutionary history these organisms. The 127 genomes of the P. syringae phylogenetic group show a high percentage of genes in the flexible genome, indicating that these strains are able to share genes and are highly diverse.
Average amino acid identity matrices were calculated using protein-CDSs within the 5 phylogenomic branches (Supplemental Figures S5–S9). The heatmap shows the clustering of genomes into different groups based on average similarities and differences of their CDS amino acid identities. In this case, core and flexible gene pools are combined. The clustering of the genomes in each phylogenomic branch follows the same groupings observed with the other methodologies (3-gene MLSA, core MLSA, ANIb, and GGDC). That is, in phylogenomic branch I, the four phylogenomic species detected with the previous methodologies could also be differentiated by clear boundaries.
The amino acid sequences of the core genes for each phylogenomic branch were concatenated, and the phylogenetic tree was constructed with PhyML. The phylogenetic trees are depicted in Supplemental Figures S5–S9. The clustering observed was consistent with the results obtained previously and supported the proposed phylogenomic species.
The core genome and pangenome were also analyzed for 6 delineated phylogenomic species that contained at least 7 strains each. Four of them included the type strains of the validly described species P. syringae, P. avellanae, P. amygdali, and P. viridiflava. Additionally, genomes of the proposed phylogenomic species “P. coronafaciens” and “P. tomato,” were also studied. The results are shown in Table 3 and Supplemental Figures S10–S15. The pool of conserved genes decreased with increasing genome number and represents at least 17% of the individual genomes. The percentage of the core genes in the 6 proposed phylogenomic species ranged between 17.67 and 72.60%. The low percentage of conserved genes might be directly associated with the difficulty to phylogenetically assign some strains to the correct phylogenomic species.
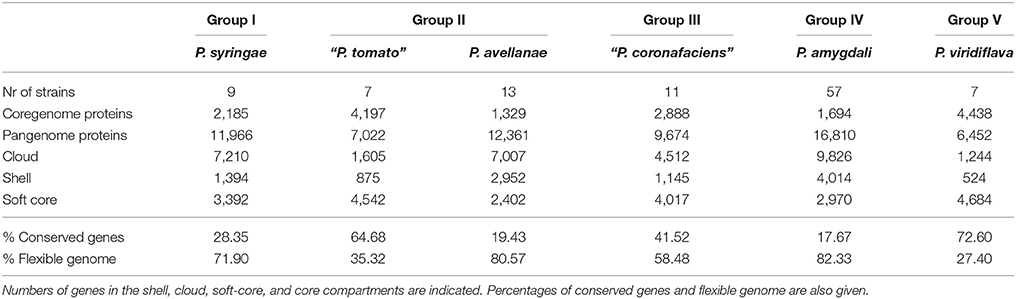
Table 3. Core and pangenome analyses of the phylogenomic species proposed with more than 6 strains in the P. syringae phylogenetic group.
This low percentage of conserved genes correlates with a higher number of cloud genes that contribute to the flexible genome.
Although the taxonomy of Pseudomonas, and more specifically of the P. syringae phylogenetic group, has been extensively analyzed, significant uncertainties remain regarding the genus boundaries and species composition of this heterogeneous taxon. Many species have been named without adequate descriptions, or their identifications have not been updated with more modern techniques. Phylogenomic insights published by Gomila et al. (2015) and more recently by Tran et al. (2017) have substantially improved the knowledge of the whole genus. Vinatzer et al. (2017) used genome similarities to study the taxonomy of plant-pathogenic bacteria and constructed core genome phylogenies for plant-pathogenic bacteria. The composition of distinct species and whether P. syringae is a cohesive unit has been debated for a long time (Janse et al., 1996; Bull et al., 2010). Several recent publications have tried to clarify this situation (Baltrus, 2016; Baltrus et al., 2017), also at the pathovar classification level (Thakur et al., 2016).
Strains were originally identified phenotypically as members of the P. syringae complex if they were fluorescent pseudomonads, positive for levan sucrase activity, negative for oxidase activity, unable to rot potato, able to produce arginine dihydrolase and able to cause a hypersensitive response on tobacco (the LOPAT group 1 strains; Lelliott et al., 1966; Sands et al., 1970). In 1975, numerous formerly distinct LOPAT group 1 plant pathogenic species were combined into the species P. syringae (Lapage et al., 1975), and the confusion increased due to subspecific pathovar names have been given to distinct pathogenic characters and host of isolation (Young, 2008). At that time a large number of nomenspecies of these bacteria were defined and became widely regarded as host-adapted pathogenic varieties (pathovars). Consequently, the Approved Lists of Bacterial Names did not list most of these nomenspecies, which thus lost standing in nomenclature. Main reason were the absence of deposited strains in culture collections, lack of adequate phenotypic descriptions and phenotypic traits that distinguished the proposed species names. The International Society of Plant Pathologists published a checklist of the earlier nomenspecies and pathovars (Dye et al., 1980) and advised that such names should be revived only for the original bacteria (Lapage et al., 1992).
Classification based only on phenotype has led to increased taxonomic confusion, as more P. syringae strains have been isolated from different environments, including non-diseased tissues and environmental sources, such as rivers, lakes, snowfields, and clouds (Morris et al., 2008). Phenotypic diversity of strains in relevant species has also been demonstrated (Demba Diallo et al., 2012; Bartoli et al., 2014) and many strains cannot be easily classified. Currently, the P. syringae species complex is subdivided into over 60 pathovars defined by pathogenic characters, nine genomospecies defined by DDH and 13 phylogenetic groups (phylogroups) defined by multilocus sequence analysis (Sarkar and Guttman, 2004; Hwang et al., 2005; Almeida et al., 2010; Bull et al., 2011; Berge et al., 2014).
The aim of this work is to try to circumscribe the P. syringae species complex and classify its strains into species according to the taxonomic rules and thresholds actually accepted in taxonomy. Considering all methods tested together, we were able to circumscribe 6 phylogenomic branches within the P. syringae phylogenetic group.
The first branch, branch I, included 15 strains divided into 4 groups corresponding to four different phylogenomic species: P. syringae, P. congelans, P. cerasi, and novel species A. The first one includes the P. syringae type strain, together with only 9 strains previously identified as P. syringae. The rest of the P. syringae strains included in the present study (23) must be reassigned to other species (see Supplemental Table S1).
A second branch, branch II, contains 20 strains: 5 P. avellanae strains, including the species type strain, together with 1 strain classified as P. amygdali pv. morsprunorum (M302280), 5 strains of P. syringae pv. tomato, 2 of P. amygdali pv. lachrymans, 7 of P. syringae pv. actinidiae, and 1 of P. syringae pv. theae. We have included in this study five strains not considered in the previous publication by Scortichini et al. (2013) on P. avellanae genomes. The 3-gene MLSA values for all strains of this group ranged from 99.46 to 100% and divided these 20 strains into two subclusters. These 2 subclusters are maintained in the analysis of 219 concatenated core genes. All strains in the branch have ANIb similarity values ranging from 94.3 to 100%, but 2 sub-branches can be delineated: one at 97.1–100%, which corresponds to the group of the P. avellanae type strain, and another at 98.4–100%, which corresponds to strains of P. syringae pv. tomato and P. amygdali pv. lachrymans. These two sub-branches can also be delineated with the GGDC values; they are in accordance with Gardan's genomospecies 3 and 8, respectively, and with the phylogroups established by Parkinson and Berge. If we apply the currently accepted species threshold, all the strains of the group might be assigned to the species P. avellanae, but attending to all indices tested and the boundaries of the two subclusters, the possibility to differentiate two species or 2 subspecies must be considered. We have distinguished 2 distinct phylogenomic species: one is P. avellanae, and we propose the provisional operative name of “Pseudomonas tomato” for the branch that includes strain DC3000, pending a deeper taxonomic analysis. To formally propose a new species clear phenotypic characteristics that differentiate the new species with its closely related species have to be found. In our experience, the whole-cell protein profiles obtained by MALDI-TOF mass spectrometry can be a method of choice to phenotypically discriminate new species in the genus Pseudomonas (Mulet et al., 2012).
Branch III includes 4 phylogenomic species: 3 strains in one cluster must be assigned to P. cannabina; 1 strain deposited as P. viridiflava and another as P. syringae must be considered representatives of new species B; P. syringae pv. coriandricola ICMP 13104 represents a phylogenomic species provisionally named “Pseudomonas coriandricola”; 10 strains initially assigned to “P. coronafaciens” cluster together in all analyses and constitute the fourth phylogenomic species. This group includes the genome of P. tremae ICMP 9151T, not considered type strain in the present study. Therefore, the genome of P. tremae LMG 22121T was sequenced and ANIb and GGDC results demonstrated that it has to be included in genomic branch IV (results not shown), that strengthen the possible misclassification of strain ICMP 9151T. The “P. coronafaciens” strains belong to phylogroup 4 of Parkinson and Berge and to genomospecies 4 of Gardan. The species “P. coronafaciens” has been proposed by Schaad and Cunfer (1979) based on phenotypic characteristics, and the indices studied clearly support the revival of “P. coronafaciens” as a nomenspecies.
Two phylogenomic species can be delineated in branch IV, which correspond to phylogroup 6 of Parkinson and Berge and genomospecies 7 of Gardan. Two strains and the P. caricapapayae type strain must be assigned to this species. The other phylogenomic species is more abundant and very homogeneous and contains 4 accepted nomenspecies. As already noted by Gardan et al. (1999), strains in this phylogenomic species have been assigned to genomospecies 3 and must be considered P. amygdali strains. The other 3 are later synonyms (P. ficuserectae, P. meliae, and P. savastanoi). As mentioned before P. tremae should be also considered in this group as a later synonym once will be assessed the correct genome.
Three well-defined phylogenomic species were distinguished in branch V. One was formed by P. asturiensis strains, another for a novel species C with 2 strains, and the other by P. viridiflava strains. Strains in this branch shared at least 53% of the genes in the pangenome. The strains P. syringae CC1417 and P. syringae CC1524 are considered non-phytopathogens, and P. asturiensis LMG 26898T is phytopathogenic. The three strains are closely related in all the indices tested and were isolated from distant geographical areas (Montana, USA; France and Spain, respectively) and ecological habitats (rocks in waterfall in pristine woods, stream water and soybeans, respectively; Morris et al., 2008; González et al., 2013; Baltrus et al., 2014). The ANIb and GGDC indices are near the borderline of the species acceptance cut-off and share at least 87% of the pangenome in the Gower analysis. Consequently, strains CC1417 and CC1524 are not members of the P. syringae phylogenomic species and might be considered strains of P. asturiensis. However, the differences in plant pathogenicity and other practical reasons suggest that the taxonomic status of both strains merit further analyses before a definitive classification and must be considered for the moment representatives of putative new species C.
In branch VI, four phylogenomic species have been defined: the P. caspiana type strain is a representative of the other 4 strains of the species (Beiki et al., 2016; Busquets et al., 2017); P. cichorii with two strains, Pseudomonas sp. S25 and P. syringae UB246 are singletons, and more closely related strains are needed for a definitive taxonomic assignment of both these strains, which are assigned to putative new species D and E.
Overall, we were able to distinguish 19 phylogenomic species in the P. syringae phylogenetic group distributed within 6 phylogenomic branches. Two strains are assigned to 2 different phylogenomic species when the following criteria are accomplished: (i) ANIb value is lower than 94.5%, (ii) GGDC values lower than 68%, and (iii) 3-gene MLSA similarity lower than 98%. ANIb values between 94.5 and 96% might be analyzed carefully with respect to other characteristics such as GGDC, the core genome and pangenome analyses. In general, very good agreement has been found between these phylogenomic species, the phylogroups of Parkinson and Berge, and the genomospecies of Gardan. In fact, Bull et al. (2011) showed also how MLSA quite accurately reflects the genomospecies described by Gardan et al. (1999) by experimental DNA-DNA hybridizations.
A strain was assigned to a given phylogenomic species when it fell into one of the 19 phylogenomic species delineated as described above. In 58% of strains, there was an agreement between strain name and genomospecies. Furthermore, 23 out of 32 (72%) strains deposited as P. syringae were not assigned to the P. syringae phylogenomic species but were scattered among 10 different phylogenomic species. This fact points out the importance of correctly assigning a genome to the right species. Thanks to NGS technologies, a remarkable increase in the number of sequenced genomes, both draft and complete, are available, but the correct assignment of the sequenced strains to the corresponding species with the accepted taxonomic tools is important before comparative analyses with other genomes can be performed.
Genomic data are very useful in the actual taxonomy to delineate phylogenomic species that merits the species status. However, it is possible that many species will be separated in several species, even when the abundance of species names can be confusing. In this sense, the use by the experts in phytopathology will consolidate or not the use of these new species names. In many practical issues it can be maintained the less precise concept of P. syringae species complex for all of them, although it is essential a proper naming of bacterial species in order to establish a truly systematic taxonomy and avoid confusions in the scientific communities.
Comparative genomics is a very useful tool for the establishment of a stable taxonomy, and we demonstrate its usefulness for the plant pathogenic bacteria studied in the present manuscript. Although further taxonomic studies are needed to support formal proposals, based on the present study of strains in the P. syringae phylogenetic group, we suggest that P. ficuserectae, P. meliae, and P. savastanoi are later synonyms of P. amygdali and, therefore, the group includes 11 recognized nomenspecies: P. amygdali, P. asturiensis, P. avellanae, P. cannabina, P. caricapapayae, P. caspiana, P. cerasi, P. cichorii, P. congelans, P. syringae, and P. viridiflava. Additionally, “P. coronafaciens” should be revived as a nomenspecies, and 27 strains representing 7 putative new species must be considered.
MG, EG-V, and JL: conceived and designed the research project, and analyzed the data; AB and MM performed the experiments; MG: performed the bioinformatic analyses of the data; MG, AB, MM, EG-V, and JL: interpreted the results and contributed to the writing of the manuscript.
Financial support was obtained from the Spanish MINECO through project CGL2015-70925-P (with FEDER cofunding). MG was supported by a postdoctoral contract from the Conselleria d'Innovació, Recerca i Turisme del Govern de les Illes Balears and the European Social Fund.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We wish to thank to Dr. Bruno Contreras-Moreira and Dr. Pablo Vinuesa for the useful help with the Get_Homologues scripts. We would like to thank the reviewers for their detailed comments and suggestions for the manuscript. Their constructive criticism and valuable comments have improved the manuscript content.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.02422/full#supplementary-material
Almeida, N. F., Yan, S., Cai, R., Clarke, C. R., Morris, C. E., Schaad, N. W., et al. (2010). PAMDB, a multilocus sequence typing and analysis database and website for plant-associated microbes. Phytopathology 100, 208–215. doi: 10.1094/PHYTO-100-3-0208
Baltrus, D. A. (2016). Divorcing strain classification from species names. Trends Microbiol. 24, 431–439. doi: 10.1016/j.tim.2016.02.004
Baltrus, D. A., McCann, H. C., and Guttman, D. S. (2017). Evolution, genomics and epidemiology of Pseudomonas syringae. Challenges in bacterial molecular pathology. Mol. Plant. Pathol. 18, 152–168. doi: 10.1111/mpp.12506
Baltrus, D. A., Yourstone, S., Lind, A., Guilbaud, C., Sands, D. C., Jones, C. D., et al. (2014). Draft genome sequences of a phylogenetically diverse suite of Pseudomonas syringae strains from multiple source populations. Genome Announc. 23:e01195-13. doi: 10.1128/genomeA.01195-13
Bartoli, C., Berge, O., Monteil, C. L., Guilbaud, C., Balestra, G. M., Varvaro, L., et al. (2014). The Pseudomonas viridiflava phylogroups in the P. syringae species complex are characterized by genetic variability and phenotypic plasticity of pathogenicity-related traits. Environ. Microbiol. 16, 2301–2315. doi: 10.1111/1462-2920.12433
Behrendt, U., Ulrich, A., and Schumann, P. (2003). Fluorescent pseudomonads associated with the phyllosphere of grasses; Pseudomonas trivialis sp. nov., Pseudomonas poae sp. nov. and Pseudomonas congelans sp. nov. Int. J. Syst. Evol. Microbiol. 53, 1461–1469. doi: 10.1099/ijs.0.02567-0
Beiki, F., Busquets, A., Gomila, M., Rahimian, H., Lalucat, J., and García-Valdés, E. (2016). New Pseudomonas spp. are pathogenic to citrus. PLoS ONE 26:e0148796. doi: 10.1371/journal.pone.0148796
Berge, O., Monteil, C. L., Bartoli, C., Chandeysson, C., Guilbaud, C., Sands, D. C., et al. (2014). A user's guide to a data base of the diversity of Pseudomonas syringae and its application to classifying strains in this phylogenetic complex. PLoS ONE 3:e105547. doi: 10.1371/journal.pone.0105547
Bezuidt, O. K., Pierneef, R., Gomri, A. M., Adesioye, F., Makhalanyane, T. P., Kharroub, K., et al. (2016). The Geobacillus pan-Genome: implications for the evolution of the genus. Front. Microbiol. 7:723. doi: 10.3389/fmicb.2016.00723
Bull, C. T., Clarke, C. R., Cai, R., Vinatzer, B. A., Jardini, T. M., and Koike, S. T. (2011). Multilocus sequence typing of Pseudomonas syringae sensu lato confirms previously described genomospecies and permits rapid identification of P. syringae pv. coriandricola and P. syringae pv. apii causing bacterial leaf spot on parsley. Phytopathology 101, 847–858. doi: 10.1094/PHYTO-11-10-0318
Bull, C. T., De Boer, S. H., Denny, T. P., Firrao, G., Fischer-Le Saux, M., Saddler, G. S., et al. (2010). Comprehensive list of names of plant pathogenic bacteria, 1980-2007. J. Plant Pathol. 92, 551–592. doi: 10.4454/jpp.v92i3.302
Burkholder, W. H. (1939). The bacterial diseases of the bean. Mem. Cornell University Agric. Exp. Stat. 127, 1–88.
Busquets, A., Gomila, M., Beiki, F., Mulet, M., Rahimian, H., García-Valdés, E., et al. (2017). Pseudomonas caspiana sp. nov., a citrus pathogen in the Pseudomonas syringae phylogenetic group. Syst. Appl. Microbiol. 40, 266–273. doi: 10.1016/j.syapm.2017.04.002
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Contreras-Moreira, B., and Vinuesa, P. (2013). GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 79, 7696–7701. doi: 10.1128/AEM.02411-13
Demba Diallo, M., Monteil, C. L., Vinatzer, B. A., Clarke, C. R., Glaux, C., Guilbaud, C., et al. (2012). Pseudomonas syringae naturally lacking the canonical type III secretion system are ubiquitous in nonagricultural habitats, are phylogenetically diverse and can be pathogenic. ISME J. 6, 1325–1335. doi: 10.1038/ismej.2011.202
Dye, D. W., Bradbury, J. F., Goto, M., Hayward, A. C., Lelliott, R. A., and Schroth, M. N. (1980). International standards for naming pathovars of phytopathogenic bacteria and a list of pathovar names and pathotype strains. Rev. Plant Pathol. 59, 153–168.
Gardan, L., Bollet, C., Abu Ghorrah, M., Grimont, F., and Grimont, P. A. D. (1992). DNA relatedness among the pathovar strains of Pseudomonas syringae subsp. savastanoi Janse (1982) and proposal of Pseudomonas savastanoi sp. nov. Int. J. Syst. Bacteriol. 42, 606–612. doi: 10.1099/00207713-42-4-606
Gardan, L., Shafik, H., Belouin, S., Broch, R., Grimont, F., and Grimont, P. A. D. (1999). DNA relatedness among the pathovars of Pseudomonas syringae and description of Pseudomonas tremae sp. nov. and Pseudomonas cannabina sp. nov. (ex Sutic and Dowson 1959). Int. J. Syst. Evol. Microbiol. 49, 469–478. doi: 10.1099/00207713-49-2-469
Gomila, M., Peña, A., Mulet, M., Lalucat, J., and García-Valdés, E. (2015). Phylogenomics and systematics in Pseudomonas. Front. Microbiol. 6:214. doi: 10.3389/fmicb.2015.00214
González, A. J., Cleenwerck, I., De Vos, P., and Fernández-Sanz, A. M. (2013). Pseudomonas asturiensis sp. nov., isolated from soybean and weeds. Syst. Appl. Microbiol. 36, 320–324. doi: 10.1016/j.syapm.2013.04.004
Goto, M. (1983). Pseudomonas ficuserectae sp. nov., the causal agent of bacterial leaf spot of Ficus erecta Thunb. Int. J. Syst. Evol. Microbiol. 33, 546–550. doi: 10.1099/00207713-33-3-546
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Hwang, M. S., Morgan, R. L., Sarkar, S. F., Wang, P. W., and Guttman, D. S. (2005). Phylogenetic characterization of virulence and resistance phenotypes of Pseudomonas syringae. Appl. Environ. Microbiol. 71, 5182–5191. doi: 10.1128/AEM.71.9.5182-5191.2005
Janse, J. D., Rossi, P., Angelucci, L., Scortichini, M., Derks, J. H. J., Akkermans, A. D. L., et al. (1996). Reclassification of Pseudomonas syringae pv. avellanae as Pseudomonas avellanae (spec. nov.), the bacterium causing canker of hazelnut (Corylus avellana L.). Syst. Appl. Microbiol. 19, 589–595. doi: 10.1016/S0723-2020(96)80030-0
Jeffroy, O., Brinkmann, H., Delsuc, F., and Philippe, H. (2006). Phylogenomics: the beginning of incongruence? Trends Genet. 22, 225–231. doi: 10.1016/j.tig.2006.02.003
Kaas, R. S., Friis, C., Ussery, D. W., and Aarestrup, F. M. (2012). Estimating variation within the genes and inferring the phylogeny of 186 sequenced diverse Escherichia coli genomes. BMC Genomics. 13:577. doi: 10.1186/1471-2164-13-577
Kałuzna, M., Willems, A., Pothier, J. F., Ruinelli, M., Sobiczewski, P., and Puławska, J. (2016). Pseudomonas cerasi sp. nov. (non Griffin, 1911) isolated from diseased tissue of cherry. Syst. Appl. Microbiol. 39, 370–377. doi: 10.1016/j.syapm.2016.05.005
Konstantinidis, K. T., and Tiedje, J. M. (2007). Prokaryotic taxonomy and phylogeny in the genomic era: advancements and challenges ahead. Curr. Opin. Microbiol. 10, 504–509. doi: 10.1016/j.mib.2007.08.006
Koonin, E. V., and Wolf, Y. I. (2008). Genomics of bacteria and Archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 36, 6688–6719. doi: 10.1093/nar/gkn668
Lapage, S. P., Sneath, P. H. A., Lessel, E. F., Skerman, V. B. D., Seeliger, H. P. R., and Clark, W. A. (1975). International Code of Nomenclature of Bacteria, 1976 Revision. Washington, DC: American Society for Microbiology.
Lapage, S. P., Sneath, P. H. A., Lessel, E. F., Skerman, V. B. D., Seeliger, H. P. R., and Clark, W. A. (eds.). (1992). “Chapter 3, Rules of nomenclature with recommendations,” in International Code of Nomenclature of Bacteria: Bacteriological Code, 1990 Revision (Washington, DC: American Society for Microbiology Press).
Lelliott, R. A., Billing, E., and Hayward, A. C. (1966). A determinative scheme for the fluorescent plant pathogenic pseudomonads. J. Appl. Bacteriol. 29, 470–489. doi: 10.1111/j.1365-2672.1966.tb03499.x
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60
Moore, E. R. B., Mau, M., Arnscheidt, A., Böttger, E. C., Hutson, R. A., Collins, M. D., et al. (1996). The determination and comparison of the 16S rRNA gene sequences of species of the genus Pseudomonas (sensu stricto) and estimation of the natural intrageneric relationships. Syst. Appl. Microbiol. 19, 478–492. doi: 10.1016/S0723-2020(96)80021-X
Morris, C. E., Barny, M. A., Berge, O., Kinkel, L. L., and Lacroix, C. (2017). Frontiers for research on the ecology of plant-pathogenic bacteria: fundamentals for sustainability. Mol. Plant Pathol. 18, 308–319. doi: 10.1111/mpp.12508
Morris, C. E., Sands, D. C., Vinatzer, B. A., Glaux, C., Guilbaud, C., Buffière, A., et al. (2008). The life history of the plant pathogen Pseudomonas syringae is linked to the water cycle. ISME J. 2, 321–334. doi: 10.1038/ismej.2007.113
Mulet, M., Lalucat, J., and García-Valdés, E. (2010). DNA sequence-based analysis of the Pseudomonas species. Environ. Microbiol. 12, 1513–1530. doi: 10.1111/j.1462-2920.2010.02181.x
Mulet, M., Gomila, M., Scotta, C., Sánchez, D., Lalucat, J., and García-Valdés, E. (2012). Concordance between whole-cell matrix-assisted laser-desorption/ionization time of flight mass spectrometry and multi locus sequence analysis approaches in the species discrimination in the genus Pseudomonas. System. Appl. Microbiol. 35, 455–464. doi: 10.1016/j.syapm.2012.08.007
Ogimi, C. (1977). Studies on bacterial gall of chinaberry (Melia Azedarach Lin.), caused by Pseudomonas meliae n. sp. Bull. Coll. Agric. Univ. Ryukyus 24, 497–556.
Parkinson, N., Bryant, R., Bew, J., and Elphinstone, J. (2011). Rapid phylogenetic identification of members of the Pseudomonas syringae species complex using the rpoD locus. Plant Pathol. 60, 338–344. doi: 10.1111/j.1365-3059.2010.02366.x
Parte, A. C. (2014). LPSN-list of prokaryotic names with standing in nomenclature. Nucleic Acids Res. 42, D613–D616. doi: 10.1093/nar/gkt1111
Pecnold, P. C., and Grogan, R. G. (1973). Deoxyribonucleic acid homology groups among phytopathogenic Pseudomonus species. Int. J. Syst. Evol. Microbiol. 23, 111–121.
Psallidas, P. G., and Panagopoulos, C. G. (1975). A new bacteriosis of almond caused by Pseudomonas amygdali sp. nov. Ann. Instit. Phytopathol. Benaki 11, 94–108.
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold stan-dard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Robbs, C. F. (1956). Uma nova doenca bacteriana do mamoeiro (Carica papaya L.). Revist. Soc. Brasil. Agron. 12, 73–76.
Sands, D. C., Schroth, M. N., and Hildebrand, D. C. (1970). Taxonomy of phytopathogenic pseudomonads. J. Bacteriol. 101, 9–23.
Sarkar, S. F., and Guttman, D. S. (2004). Evolution of the core genome of Pseudomonas syringae, a highly clonal, endemic plant pathogen. Appl. Environ. Microbiol. 70, 1999–2012. doi: 10.1128/AEM.70.4.1999-2012.2004
Schaad, N. W., and Cunfer, B. M. (1979). Synonymy of Pseudomonas coronafaciens, Pseudomonas coronafaciens pathovar zeae, Pseudomonas coronafaciens subsp. atropurpurea, and Pseudomonas striafaciens. Int. J. Syst. Bacteriol. 29, 213–221. doi: 10.1099/00207713-29-3-213
Scortichini, M., Marcelletti, S., Ferrante, P., and Firrao, G. (2013). A genomic redefinition of Pseudomonas avellanae species. PLoS ONE 25:e75794. doi: 10.1371/journal.pone.0075794
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Skerman, V. B. D., McGowan, V., and Sneath, P. H. A. (1980). Approved list of bacterial names. Int. J. Syst. Bacteriol. 30, 225–420. doi: 10.1099/00207713-30-1-225
Stapp, C. (1928). “Schizomycetes (Spaltpilze oder Bakterien)”, in Handbuch der Pflanzenkrankheiten, 5th Edn, Vol. 2, ed P. Sorauer (Berlin: Parey), 1–295.
Thakur, S., Weir, B. S., and Guttman, D. S. (2016). Phytopathogen genome announcement: draft genome sequences of 62 Pseudomonas syringae type and pathotype strains. Mol. Plant Microbe Interact. 29, 243–246. doi: 10.1094/MPMI-01-16-0013-TA
Tran, P. N., Savka, M. A., and Gan, H. M. (2017). In-silico taxonomic classification of 373 genomes reveals species misidentification and new genospecies within the genus Pseudomonas. Front. Microbiol. 8:1296. doi: 10.3389/fmicb.2017.01296
Van Hall, C. J. J. (1902). Bijdragen tot de Kennis der Bakterieele Plantenziekten. Amsterdam: Coöp. Drukk.
Vinatzer, B. A., and Bull, C. T. (2009). “The impact of genomic approaches on our understanding of diversity and taxonomy of plant pathogenic bacteria,” in Plant Pathogenic Bacteria: Genomics and Molecular Biology, ed R. W. Jackson (Caister: Academic Press), 37–61.
Vinatzer, B. A., Weisberg, A. J., Monteil, C. L., Elmarakeby, H. A., Sheppard, S. K., and Health, L. S. (2017). A proposal for a genome similarity-based taxonomy for plant-pathogenic bacteria that is sufficiently precise to reflect phylogeny, host range, and outbreak affiliation applied to Pseudomonas syringae sensu lato as a proof of concept. Phytopathology 107, 18–28. doi: 10.1094/PHYTO-07-16-0252-R
Yamamoto, S., Kasai, H., Arnold, D. L., Jackson, R. W., Vivian, A., and Harayama, S. (2000). Phylogeny of the genus Pseudomonas: intrageneric structure reconstructed from the nucleotide sequences of gyrB and rpoD genes. Microbiology 146, 2385–2394. doi: 10.1099/00221287-146-10-2385
Keywords: P. syringae, phylogenetic group, phylogenomic species, core genome, pangenome, ANIb, GGDC, MLSA
Citation: Gomila M, Busquets A, Mulet M, García-Valdés E and Lalucat J (2017) Clarification of Taxonomic Status within the Pseudomonas syringae Species Group Based on a Phylogenomic Analysis. Front. Microbiol. 8:2422. doi: 10.3389/fmicb.2017.02422
Received: 15 October 2017; Accepted: 22 November 2017;
Published: 07 December 2017.
Edited by:
Jesus L. Romalde, Universidade de Santiago de Compostela, SpainReviewed by:
Boris Alexander Vinatzer, Virginia Tech, United StatesCopyright © 2017 Gomila, Busquets, Mulet, García-Valdés and Lalucat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jorge Lalucat, amxhbHVjYXRAdWliLmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.