 Shicheng Chen
Shicheng Chen Jochen Blom
Jochen Blom Thomas P. Loch3
Thomas P. Loch3 Edward D. Walker
Edward D. Walker- 1Department of Microbiology and Molecular Genetics, Michigan State University, East Lansing, MI, United States
- 2Bioinformatics and Systems Biology, Justus-Liebig-University, Giessen, Germany
- 3Department of Pathobiology and Diagnostic Investigation, College of Veterinary Medicine, Michigan State University, East Lansing, MI, United States
- 4Department of Fisheries and Wildlife, College of Agriculture and Natural Resources, Michigan State University, East Lansing, MI, United States
Flavobacterium spartansii strain T16T was isolated from a disease outbreak in hatchery-reared Chinook salmon (Oncorhynchus tshawytscha) fingerlings. To gain insight into its genomic content, structure and virulence pathogenesis factors, comparative genome analyses were performed using genomes from environmental and virulent Flavobacterium strains. F. spartansii shared low average nucleotide identity (ANI) to well-known fish-pathogenic flavobacteria (e.g., F. columnare, F. psychrophilum, and F. branchiophilum), indicating that it is a new and emerging fish pathogen. The genome in T16T had a length of 5,359,952 bp, a GC-content 35.7%, and 4,422 predicted protein-coding sequences. Flavobacterium core genome analysis showed that the number of shared genes decreased with the addition of input genomes and converged at 1182 genes. At least 8 genomic islands and 5 prophages were predicted in T16T. At least 133 virulence factors associated with virulence in pathogenic bacteria were highly conserved in F. spartansii T16T. Furthermore, genes linked to virulence in other bacterial species (e.g., those encoding for a type IX secretion system, collagenase and hemolysin) were found in the genome of F. spartansii T16T and were conserved in most of the analyzed pathogenic Flavobacterium. F. spartansii was resistant to ampicillin and penicillin, consistent with the presence of multiple genes encoding diverse lactamases and the penicillin-binding protein in the genome. To allow for future investigations into F. spartansii virulence in vivo, a transposon-based random mutagenesis strategy was attempted in F. spartansii T16T using pHimarEm1. Four putative gliding motility deficient mutants were obtained and the insertion sites of pHimarEm1 in the genome of these mutants were characterized. In total, study results clarify some of the mechanisms by which emerging flavobacterial fish pathogens may cause disease and also provide direly needed tools to investigate their pathogenesis.
Introduction
Among the fish diseases that result in significant losses in farmed and wild fish populations around the world, those caused by flavobacteria (Family Flavobacteriaceae, Phylum Bacteroidetes) occupy a central position (Starliper and Schill, 2011). Although the majority of published reports on flavobacterial disease outbreaks in fish, along with the research aimed at combatting them, has focused on three Flavobacterium spp. (Flavobacterium psychrophilum, F. columnare, and F. branchiophilum) (Starliper and Schill, 2011; Loch and Faisal, 2017), multiple novel Flavobacterium spp. have recently emerged in association with fish disease outbreaks (reviewed in Loch and Faisal, 2015). Interestingly, some of these novel flavobacteria not only cause systemic disease outbreaks in fish, but also generate disease signs that mimic their better-known fish-pathogenic counterparts (Loch and Faisal, 2016), thereby complicating disease diagnosis and treatment. Unfortunately, very little is known about the disease ecology, transmission, and pathogenesis of emergent fish-associated flavobacteria.
Despite decades of research, effective means for the prevention and control of flavobacterial diseases have yet to be realized. However, with the advancement and ready availability of complete genome sequencing technologies for microbes, there have been significant efforts to elucidate the molecular mechanisms behind flavobacterial pathogenicity and thereby guide vaccine development through genome sequencing of pathogenic and environmental flavobacteria alike. Indeed, complete genomes are available for a handful of Flavobacterium spp. (McBride et al., 2009; Barbier et al., 2012; Kumru et al., 2017) and additional genome sequence data are now available for multiple strains of the fish pathogens F. columnare (Bartelme et al., 2016; Zhang et al., 2016), F. psychrophilum (Wiens et al., 2014; Wu et al., 2015), and F. branchiophilum (Touchon et al., 2011). These studies have revealed some of the processes by which flavobacterial fish pathogens are believed to colonize, attack, and survive within fish hosts (Duchaud et al., 2007; Castillo et al., 2016), as well as how some of the molecular mechanisms vary amongst strains (Wiens et al., 2014; Wu et al., 2015; Bartelme et al., 2016; Zhang et al., 2016).
Recently, we described the novel species Flavobacterium spartansii, which was originally isolated from a disease outbreak in hatchery-reared Chinook salmon (Oncorhynchus tshawytscha) fingerlings and from systemically-infected, feral, spawning Chinook salmon (Loch and Faisal, 2014a). Subsequent studies investigating the pathogenicity of two F. spartansii strains (T16T and S12) under laboratory conditions showed that they were capable of inducing morbidity and mortality in intraperitoneally injected Great Lakes Chinook salmon, although the estimated median lethal dose was relatively high and varied by strain (Loch and Faisal, 2016). Nevertheless, the induced gross and microscopic changes were severe at times, which may suggest that F. spartansii is a facultative salmonid pathogen. Therefore, a detailed examination of its possible pathogenicity is warranted. Through functional and comparative genomic analyses, the following study aimed to: (a) better understand the mechanisms of virulence for F. spartansii and investigate how its genetic repertoire compares with the better-known fish-pathogenic flavobacteria through the use of complete genome sequencing; (b) examine the antimicrobial resistance capabilities in F. spartansii; (c) further characterize the evolution of virulence among Flavobacterium species; and (d) develop additional molecular tools for genetic manipulation of flavobacteria.
Materials and Methods
Culture Conditions
Flavobacterium spartansii (T16T) was grown aerobically in CYE broth. Escherichia coli DH5α was used for cloning and plasmid propagation. E. coli S17 (λ pir) was used for conjugation. E. coli EC100D pir+ was used to recover transposons from F. spartansii. E. coli strains were routinely grown in Luria-Bertani (LB) broth. Liquid cultures were incubated with shaking (ca. 200 rpm) at either 28°C (F. spartansii) or 37°C (E. coli). For solid LB medium, Bacto agar (Difco, Detroit, MI) was added at a final concentration of 20 g/liter with kanamycin (50 μg/ml) or ampicillin (100 μg/ml) added for plasmid selection in E. coli or erythromycin (Em) added (100 μg/ml) for transposon or plasmid selection in F. spartansii. Demonstration of motility was done by culturing F. spartansii cells on PY2 agar medium at 28°C.
Molecular Manipulation
Isolation and purification of bacterial genomic DNA was performed with the Wizard Genomic DNA Purification Kit (Promega, CA, USA) according to the manufacturer's instruction. The purity, quality and quantity of purified genomic DNA were assessed using a NanoDrop 2000 UV-Vis spectrophotometer (Thermo Scientific, MA, USA) and a Qubit 2.0 fluorometer (Life Technologies, MA, USA), respectively.
Random Transposon Mutagenesis
Random transposon mutagenesis in F. spartansii T16T was performed using pHimarEm1 (Braun et al., 2005). Donor E. coli S17-1 λ pir cells with pHimarEm1 and recipient cells T16T were grown to the log phase. Cells were collected by centrifugation at 3,800 rpm for 15 min. F. spartansii T16T and E. coli cells were mixed at 1:1 ratios in CYE medium supplemented with Ca2+ and spotted onto CYE agar. After 24-h incubation at 28°C, the mixed cells were scraped from agar plates, washed once with CYE broth, and plated on the CYE agar containing erythromycin (100 μg/ml). Next, erythromycin transconjugants were transferred to PY2 agar (8%) for screening the spreading-deficient mutants. The gliding deficiency mutants were further observed for their motility on glass slides under microscopy. The pHimarEm1 insertion sites in the non-spreading mutants were identified using a published method (Braun et al., 2005). Briefly, genomic DNA was digested by XbaI overnight, purified with Qiagen PCR purification kit, and self-ligated. The ligation mixture was electroporated into E. coli Ec100D+ (Epicentre Technologies). Purified plasmid DNA from the Km-resistant colonies was sequenced using the primer Walker85 (TGGGAATCATTTGAAGGTTGG) and Walker86 (TCGGGTATCGCTCTTGAAGGG). The sequences were blasted against F. spartansii T16T genome to characterize the insertion site. Growth between the selected mutants and WT was compared by determination of the OD600nm after overnight culturing in PY2 broth (Braun et al., 2005).
Hemolysin Analysis
Hemolysin production in F. spartansii T16T was examined by inoculating bacteria on Remel Blood Agar (Thermo Scientific, KS). Hemolytic activity was evaluated following incubation at 28°C for 48 h. The controls for α- and β-hemolysin producers were Elizabethkingia meningoseptica ATCC 13253 and Staphylococcus aureus MSU001, respectively (Chen et al., 2017).
Antibiotic Susceptibility Testing
Flavobacterium spartansii isolates T16T and S12 were tested for antibiotic susceptibility using the Kirby-Bauer disk diffusion method as previously described (Loch and Faisal, 2014b). In brief, cultures grown on Hsu-Shotts medium (48 h) were re-suspended in sterile 0.85% saline and adjusted to OD600nm of 0.5 in a Biowave CO8000 Cell Density Meter (WPA Inc., Cambridge, UK). The bacterial suspension in duplicate was inoculated onto dilute Mueller-Hinton agar (Hawke and Thune, 1992) without 5% calf serum. Antibiotic-impregnated disks were stamped onto the medium and plates incubated at 22°C for 48 h, at which time the zones of inhibition were measured and means calculated. Antibiotics included polymyxin-B (PB; 300 iu), oxytetracycline (T; 30 μg), trimethoprim-sulfamethoxazole (SXT; 25 μg), erythromycin (E; 15 μg), ampicillin (AMP; 10 μg), florfenicol (FFC; 30 μg), penicillin G (P; 10 iu), and the vibriostatic agent O/129 (O129; 2,4-diamino,6,7-di-isopropyl pteridine; 10 μg). The cutoffs for sensitivity were as follows: PB < 8 mm resistant; 8–12 mm intermediate sensitivity; >12 mm sensitive; T < 15 mm resistant; 15–18 mm intermediate sensitivity; >18 mm sensitive; SXT < 11 mm resistant; 11–15 mm intermediate sensitivity; >15 mm sensitive; E < 14 mm resistant; 14–18 mm intermediate sensitivity; >18 mm sensitive; AMP < 12 mm resistant; 12–13 intermediate sensitivity; >13 mm sensitive; FFC < 16 mm resistant; 16–21 mm, intermediate sensitivity; >21 mm, sensitive; O/129, < 7 mm resistant, >7 mm sensitive (Whitman, 2004).
Genome Sequencing, Assembly, and Annotation
Next generation sequencing (NGS) libraries were prepared using the Illumina TruSeq Nano DNA Library Preparation Kit. Completed libraries were evaluated using a combination of Qubit dsDNA HS, Caliper LabChipGX HS DNA, and Kapa Illumina Library Quantification qPCR assays. Libraries were combined in a single pool for multiplexed sequencing and this pool was loaded on one standard MiSeq flow cell (v2) and sequencing was performed in a 2 × 250 bp paired end format using a v2, 500 cycle reagent cartridge. Base calling was done by Illumina Real Time Analysis v1.18.54 and the output of RTA was demultiplexed and converted to a FastQ format with Illumina Bcl2fastq v1.8.4 (Klein et al., 2013). NGS libraries were sequenced by Illumina Miseq paired-end sequencing technology at the Research Technology Support Facility (RTSF) at Michigan State University.
The reads were assembled using SPAdes (version 3.9.0. Gene annotation was carried out by NCBI Prokaryotic Genome Automatic Annotation Pipeline (PGAAP 3.3) (http://www.ncbi.nlm.nih.gov/genome/annotation_prok/). Initial prediction and annotation of open reading frames (ORF) and tRNA/rRNA gene prediction were carried out with Glimmer 3 through the Rapid Annotation using Subsystem Technology server (RAST).
Bioinformatics
The functional categorization and classification for predicted ORFs were performed by RAST server-based SEED viewer (Aziz et al., 2008). The multi-drug resistance genes were predicted by Resistance Gene Identifier (RGI) tool implemented in CARD website (McArthur et al., 2013). Determination of phage elements was conducted in PHASTER (Arndt et al., 2016). Identification of genomic islands was performed in IslandViewer3 using F. johnsoniae genome as the reference (http://www.pathogenomics.sfu.ca/islandviewer). Prophage and Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) were predicted using the CRISPRfinder (Grissa et al., 2007). Detection and identification of virulence factors were carried out using MvirDB database (Zhou et al., 2007). Some virulence factors found in Bacteroidetes were manually compared among those fish pathogens and environmental isolates (Chen et al., 2005). For genome similarity assessment, digital DNA-DNA hybridization (dDDH) values were computed using web tool GGDC 2.1 (formula 2, identities/HSP length). Further, the average nucleotide identity (ANI) values were computed by ANI calculator (http://enve-omics.ce.gatech.edu/ani/).
The pan genome, core genome, and specific genes of F. spartansii were analyzed by comparison with 13 representative Flavobacterium using EDGAR 2.0 (Blom et al., 2016). The sizes of pan genome and core genomes were approximated using the core/pan development feature.
Data Deposition
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession MIKE00000000. The version described in this paper is version MIKE00000000. The BioProject designation for this project is PRJNA341797. BioSample accession number is SAMN05728473.
Results
Genome Features and Phylogenic Inference
The general genome features of F. spartansii compared to those representative Flavobacterium, and assembly statistics, are presented in Table 1. The assembly of F. spartansii produced 5.35 Mb of sequence across 29 contigs with an N50 of 515,812 bp, the longest sequence of 1,308,521 bp and a G+C content of 36% (Table 1). The genome was predicted to have at least 4,422 protein-coding genes and 130 RNA genes. Among the protein-encoding genes, 2,864 of them could be assigned a putative function, whereas 1,772 genes were predicted to encode hypothetical proteins. At least 2,119 proteins were assigned to 25 different functional categories with 368 subsystems using SEED subsystems by RAST analysis (Figure S1). The annotated genome had 112 genes responsible for bacteriocins (2 genes), resistance to antibiotics and toxic compounds (84 genes), and invasion and intracellular resistance (26 genes; Figure S1).
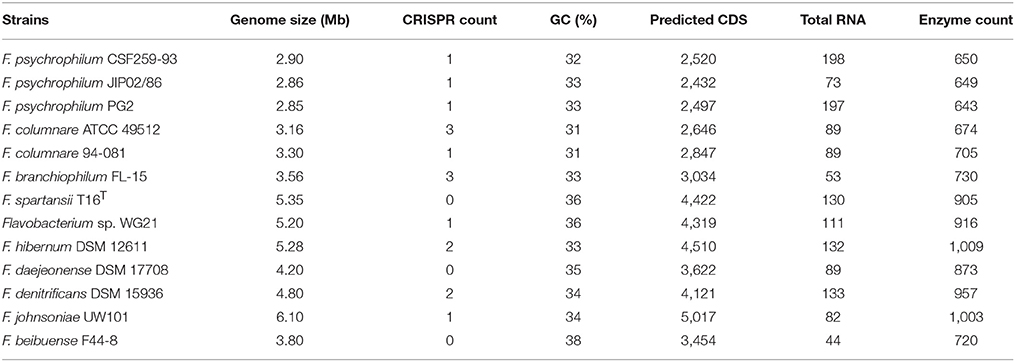
Table 1. The genome features of the selected Flavobacterium spp.
The ANI and dDDH values of representative Flavobacterium species are summarized in Table 2. F. spartansii and Flavobacterium sp. WG21 have an ANI value of 96.9% and a dDDH value of 72.7% (Table 2), suggesting that they are the same species according to the microbial taxonomy for species delineation (95 and 70% cut-off for ANI and dDDH, respectively) (Goris et al., 2007).
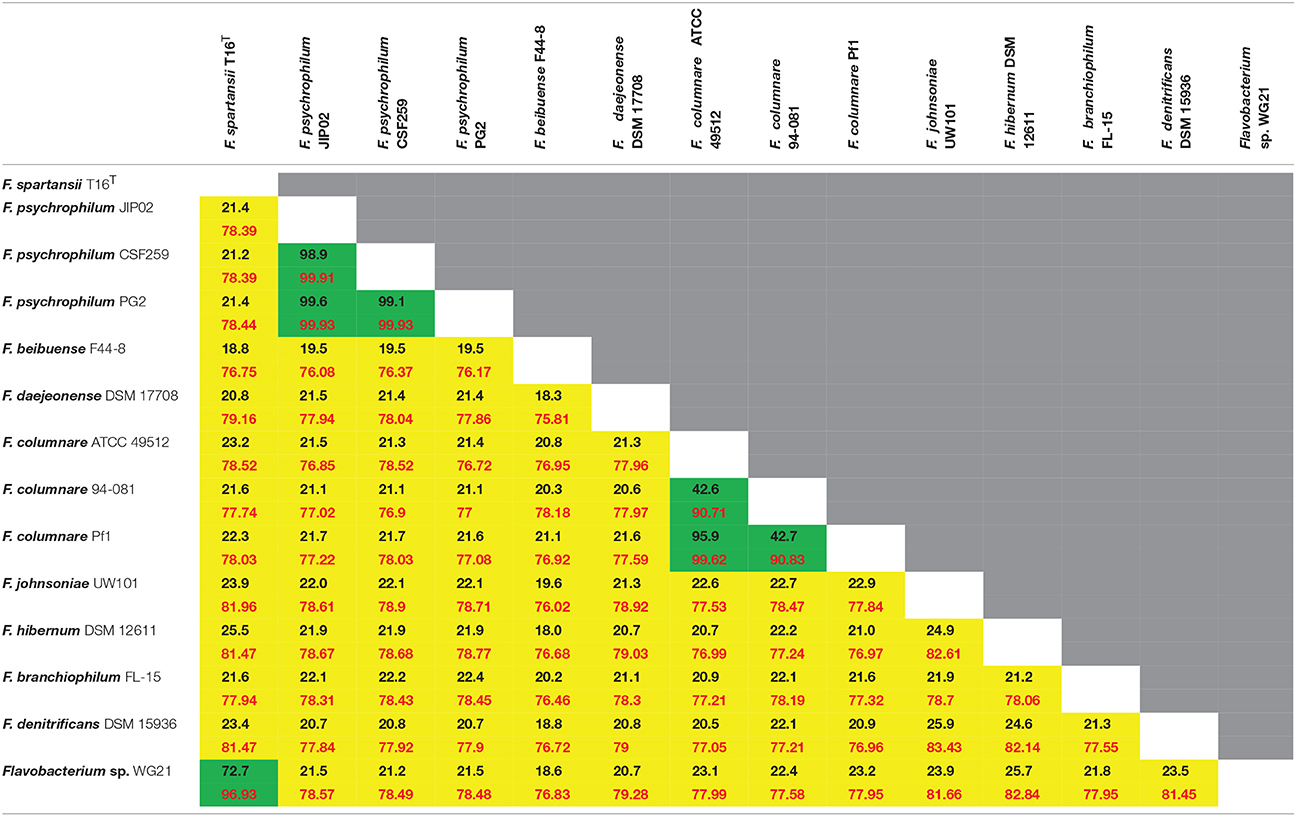
Table 2. Digital DNA-DNA Hybridization values (up, black font) and average nucleotide identity values (low, red font) amongst different Flavobacterium spp.
Gene Repertoire of Flavobacterium
The genome size for ubiquitous genes (core genome) or entire set of genome (pan-genome) amongst the selected Flavobacterium genomes was plotted against the number of genomes (Figure 1). The pan-genome plot showed that the power trend line had not reached the plateau (Figure 1), indicating that Flavobacterium possess an open pan-genome. Core genome analysis showed that the number of shared genes decreased with the addition of the input genomes and was predicted to converge against 1,182 (Figure 1). Actually, the core genome for the selected Flavobacterium in comparison was calculated to be 1,125 CDS per genome; thus, given the assumption that the prediction slightly overpredicts the core genome size, the current core genome represents the Flavobacterium genus quite well. F. spartansii shared at least 3,649, 3,201, 2,996, 2,774, 1,971, and 1,868 CDS with those environmental or opportunistic pathogenic Flavobacterium including Flavobacterium sp. WG211667 (87.2% of its total encoding genes), F. hibernum DSM 12611 (70.1%), F. denitrificans DSM 15936 (71.7%), F. beibuense F44-8 (69.6%) F. daejeonense DSM 17708 (68.2%), and F. johnsoniae UW101 (61.6%) (Figure 2). On the other hand, it shared up to 1,627, 1,651, and 1,712 CDS with the selected pathogenic Flavobacterium species such as F. columnare strains (~81%) and three F. psychrophilum strains (~75%) and F. branchiophlium (~71.3%).
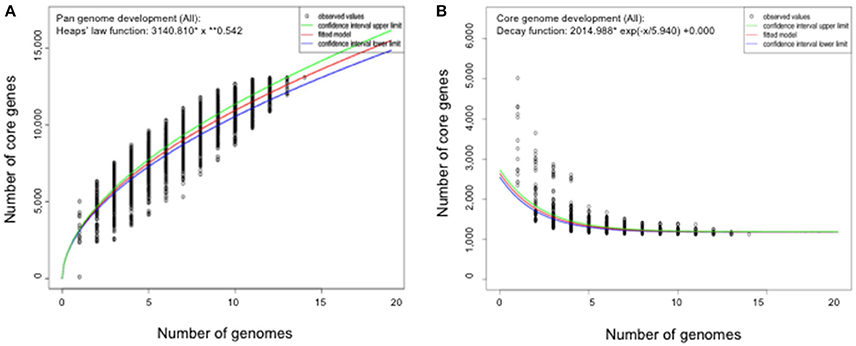
Figure 1. Pan and core genome evolution according to the number of sequenced Flavobacterium genomes. (A) Total number of genes (pan-genome) for a given number of genomes sequentially added. (B) Number of shared genes (core genome) as a function of the number of genomes sequentially added.
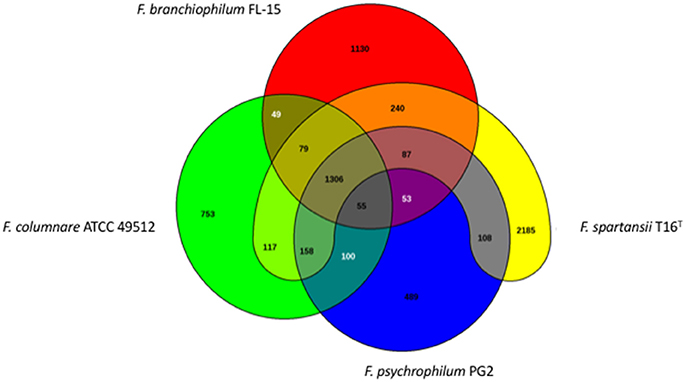
Figure 2. Venn diagram of shared and unique genes in the selected Flavobacterium. The unique and shared genome among the compared genomes were determined using the BLAST score ratio approach of EDGAR 2.0 with a cutoff of 30% (Blom et al., 2016).
Antimicrobial Resistance
Antibiotic susceptibility assays were conducted using the disk diffusion method (Table 3). Of the 8 tested antibiotics, F. spartansii T16T was intermediately sensitive to trimethoprim-sulfamethoxazole, polymyxin-B, florfenicol, erythromycin as well as oxytetracycline, while it was resistant to ampicillin, penicillin G, 2,4-diamino and 6,7-di-isopropyl pteridine (Table 3). The resistome in strain S12 was similar to that in strain T16T. However, compared to strain T16T, strain S12 was more sensitive to low concentrations of erythromycin and immediate sensitive to ampicillin. Neither of the two F. spartansii isolates (Table 3) were completely sensitive to the two antibiotics (i.e., florfenicol and oxytetracycline) that are approved to treat certain flavobacterial diseases (e.g., bacterial coldwater disease caused by F. psychrophilum, and columnaris disease caused by F. columnare) in food fish (Bowker et al., 2016).
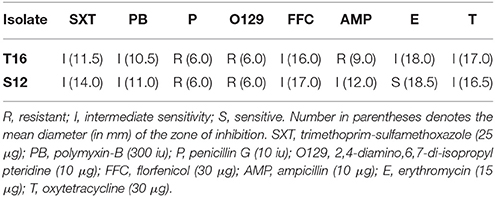
Table 3. Antibiotic susceptibility results for two F. spartansii isolates as determined via the Kirby-Bauer disk diffusion method.
Both the RAST SEED subsystem and CARD were used to identify antibiotic resistance genes within the F. spartansii genome using the default settings. The predicted genes conferring resistance to aminocoumarin, mupirocin, chloramphenicol, fluoroquinolone, tetracycline, rifampin, and β-lactam are summarized in Table S1. Interestingly, up to 10 β-lactam resistance-related genes were found in the F. spartansii T16T genome including those encoding the putative β-lactamases (8 copies), metallo-β-lactamases (1 copy) and penicillin-binding protein (1 copy). This observation is consistent with resistance to both penicillin and ampicillin in our antimicrobial tests (Table 3). Most β-lactamases in F. spartansii showed very low identity (<50%) to other fish pathogens (F. psychrophilum PG2, F. columnare and F. branchiophilum FL-15) (Table S1). However, a few lactamases have relatively high identity to those in F. johnsoniae and Flavobacterium sp. WG21. No erythromycin resistance related genes were detected in F. spartansii. More drug resistance genes and non-specific efflux pumps (5 hydrophobic/amphiphilic exporters, HAE1 family) possibly conferring antibiotic resistance are also conserved in Flavobacterium sp. WG21 and F. hibernum DSM 12611.
Prophages
At least 5 prophages were predicted in F. spartansii by PHAST (Table S2). Among them, the prophage 1 (29.4 kb) consisted of 20 CDSs that encodes 12 proteins with known functions and 8 hypothetical proteins. Of interest, prophage 1 may be complete, as it consists of a phage tail, head, portal, integrase, lysin, terminase, and other component proteins involved in phage structure and assembly. The GC content (35.9%) in prophage 1 is very close to the average of GC in the whole genome (35.5%), indicating that prophage 1 may have been integrated into F. spartansii genome long ago. Unlike prophage 1, the remaining 5 prophages seem to be incomplete, as evidenced by the absence of a portion of phage structure proteins (Table S2). However, these regions carried genes encoding some important enzymes which may contribute to flavobacterial fitness under certain conditions. For example, prophage 2 had a DNA polymerase III beta subunit, methylase, NinG recombination, ATPase and Rec T protein, indicating this region may participate in DNA replication and modification. Prophage 3 carried many genes encoding enzymes related to polysaccharide (capsule precursor) metabolism; there were at least two 1,3-N-acetylglucosaminyltransferases, four glycosyl transferases, one GDP-D-mannose 4,6-dehydratase, one alpha-1,2-fucosyltransferase, one fucose synthetase, one dTDP-d-glucose 4,6-dehydratase, one L-fucosamine transferase and one UDP-N-acetylglucosamine 2-epimerase (Table S2). Prophage 4 may be linked to RNA metabolism because ribokinase-like domain-containing protein, RNA polymerase, ribonuclease H and putative ribonuclease 3 were organized as a cluster. Prophage 5 had genes encoding arsenate reductase and metallophosphoesterase, which possibly contribute to heavy metal resistance.
Genomic Islands and Conjugative Transposon (CTnFs)
At least 8 genomic islands (GIs) were identified in F. spartansii (Figures 3A,B and Table S3) by both IslandPick and IslandPath-DIMOB methods (Nagamatsu et al., 2015). The GI size ranged from 11.3 to 63.8 Kb (Figure 3A). Genes encoding virulence factors, toxins, DNA metabolism, transposases, regulators, modification and restriction systems and resistance to antibiotics occurred in these GIs (Figure 3 and Table S3), indicating that F. spartansii T16T possibly acquired these genes, thereby forming GIs favoring adaptation to diverse environments (see below). Among the 8 GIs, GI-8 had the largest size (63.7 Kb). Notably, GI-8 has many genes encoding components of a conjugative transposon in Bacteroidetes (here, named CTnFs), e.g., traN, traM, traK, traJ, traI, and traG. Moreover, the remaining components of this large conjugative transposon CTnFs can be found in GI-7 (33.3 kb) including traD, traC, traE, traF, traG, traI, traJ, traK, traM, traI, and traQ genes (Table S3). We further examined the surrounding regions around GI-7 and found the traA and traC genes which are located upstream of traD (Table 4). The distribution spectrum of the CTnFs-like conjugative transposons were investigated by searching the conserved transfer protein gene traG (a signature gene for conjugative transposons) among the selected Flavobacterium species. No traG gene sequences (cutoff 50%) were found in F. psychrophilum, F. columnare, and F. branchiophilum FL-15. The same results were obtained when using other tra genes to search against their genomes, indicating that the conjugative transposon is absent in these fish pathogens. By contrast, the similar conjugative transposons were observed in F. johnsoniae, Flavobacterium sp. W21, F. hibernum DSM 12611, F. daejeonense DSM 17708, F. denitrificans DSM 15936, and F. beibuense F44-8 (Table 4).
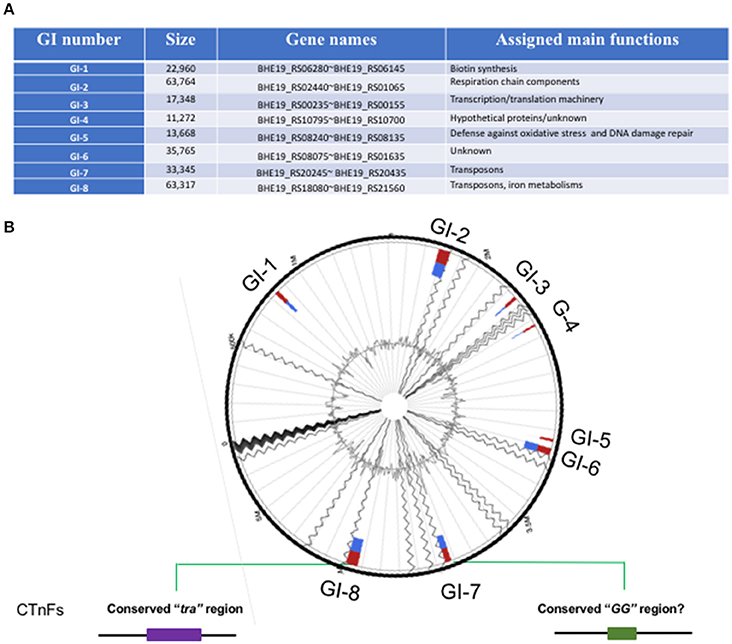
Figure 3. Genomic islands in F. spartansii T16T. (A) The 8 genomic islands were predicted by Islandviewer. (B) The schematic of GI-associated features. The relative locations of the 8 GIs were shown in the predicted genome. The conjugation protein genes belonging to “tra” and “GG” in CTnFs were centered in the GI-7 and GI-8 regions, respectively.
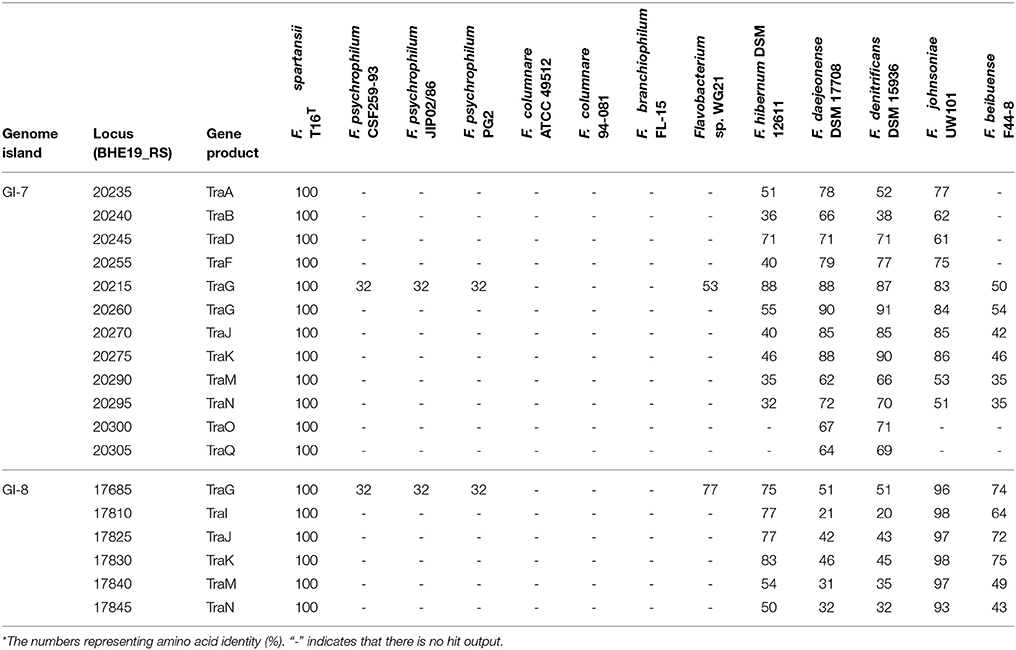
Table 4. Transfer proteins in the selected Flavobacterium spp.*.
GI-2 had many genes encoding respiratory chain components such as cytochrome C subunits, cytochrome P450, quinol:cytochrome C oxidoreductase, and several other membrane proteins (Table S3). GI-2 had the predicted virulence factor VirJ, possibly participating in the Type IV secretory pathway (Table S3). Immediately downstream of GI-2, there are genes involved in the gliding motility (RemB and SprA). Another large genomic island, GI-6, contained a gene cluster encoding DNA repair proteins and enzymes such as MsrA (preventing oxidative damage), NTP pyrophosphohydrolases, chaperone protein DnaK, deoxyribodipyrimidine photolyase (DNA repair enzyme), DNA alkylation repair enzyme, peroxiredoxin, and DNA-damage-inducible protein D (Table S3). On the same GI, there were transcriptional regulators from GntR, HxlR, and AraC families, showing that the GI-1 may be involved in the defense against oxidative stress and DNA damage repair. Compared to the above 4 GIs, the remaining ones (GI-1, GI-3, GI-4, and GI-5) were smaller (Table S3).
Prediction of Virulence Factors
When the proteome of F. spartansii T16T was used in a BLAST search against MvirDB database, 1,001 virulence factors were predicted (cutoff E-10) (Table S4). Further, 133 putative virulence proteins were found to be highly conserved in F. spartansii T16T (cutoff 50%) (Table S4). Virulence factor matches in the MvirDB database include well-known proteases, catalase/peroxidase HPI, conjugal transfer proteins (4 Tra components), molecular chaperones (GroEL, DnaJ, and DnaK), translation initiation factor IF-1, transcription regulation factors, antimicrobial resistance proteins, LPS biosynthesis protein WbpP, biopolymer transporter (ExbD and SusD/RagB), ribosomal proteins, carbon and nitrogen metabolism related proteins as well as iron-binding proteins (Table S4).
At least three hemolysin genes were present in F. spartansii (Table 5), which complements observations that strain T16T has in vitro hemolytic activity (Figure 4). When grown on sheep's blood agar plate for 48 h, colonies of F. spartansii T16T appeared greenish, suggesting α-hemolytic activity (Figure 4). Moreover, many genes encoding proteolytic enzymes were discovered in the genome of F. spartansii T16T (Table 5). Further, the amino sequence of a collagenase (BHE19_RS20565, peptidases U32) was conserved in most of the selected Flavobacterium (Table 5).
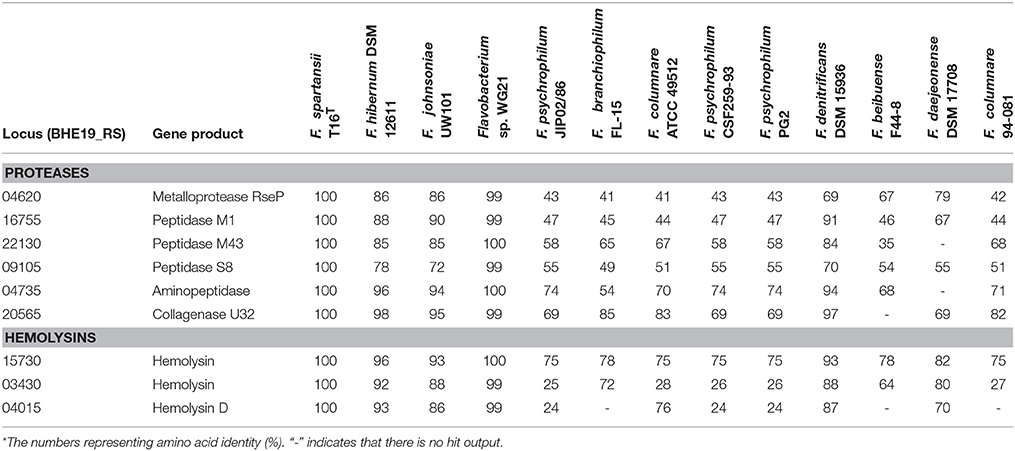
Table 5. Proteases and hemolysins in Flavobacterium spp.*.

Figure 4. Demonstration of the hemolytic activity in F. spartansii. The hemolytic activity on sheep blood agar plate was observed after 48-h incubation. (A) F. spartansii, (B) S. aureus MSU001, the β-hymolysin control, and (C) E. meningoseptica ATCC 13253, α-hemolysin control.
Gliding Machinery and Random Mutation Analysis
In F. spartansii, at least 21 genes encoding proteins associated with gliding motility were identified, including gldA, gldB, gldC, gldD, gldE, gldF, gldG, gldH, gldI, gldJ, gldK, gldL, gldM, gldN, SprA, SprB, SprC, SprD, SprE, SprF, and SprT (Table 6). The motility protein sequences were conserved among these Flavobacterium genomes except GldE, SprB, and SprC (Table 6). Genes encoding PorV and RemA also existed in F. spartansii T16T (Table 6). Therefore, F. spartansii possesses the minimal requirement of the full function of T9SSs (also important for gliding) consisting of GldK, GldL, GldM, GldN, SprA, SprE, and SprT (McBride and Nakane, 2015).
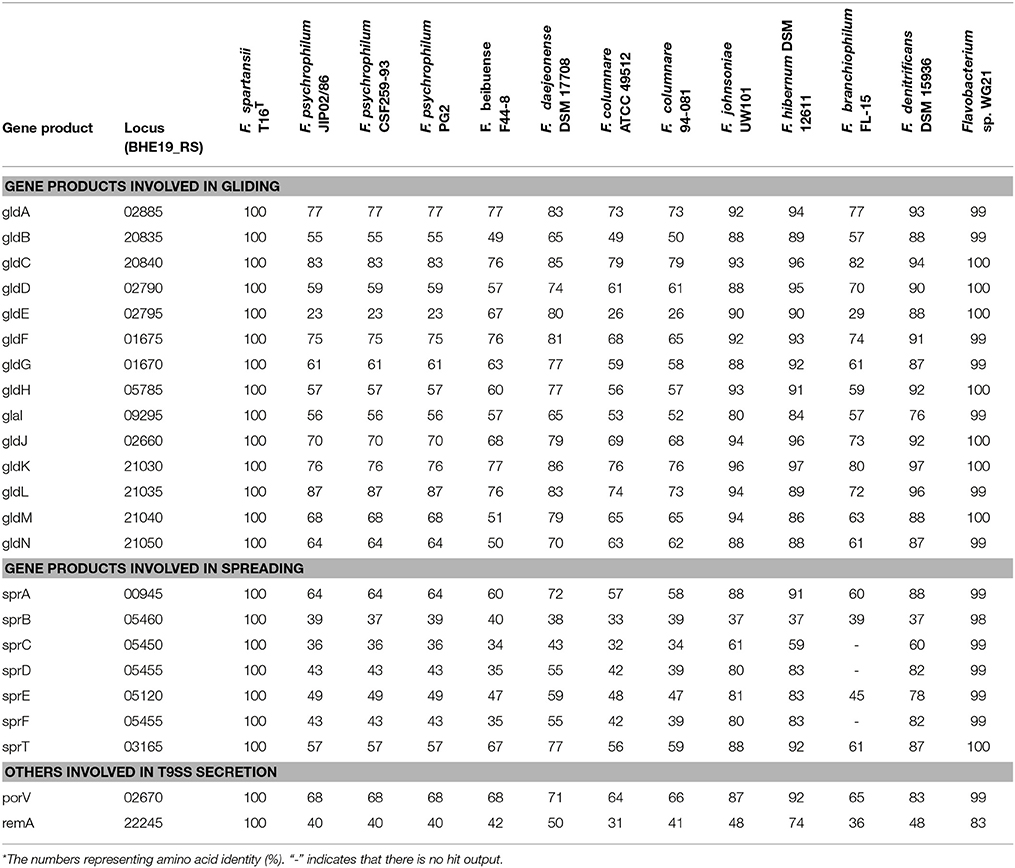
Table 6. Comparation of gliding machinery in selected Flavobacterium spp.*.
A transposon used for random mutagenesis in flavobacteria, pHimarEm1, was functional in F. spartansii T16T. Out of 2,000 conjugants, at least 4 mutants were recovered with impaired ability to spread on PY2 agar or glide on the surface of glass slides (Figure 5). For mutant 2F2-2, the transposon inserted 6-bp upstream of the ATG start codon of a gene encoding a hypothetical protein (BHE19_RS13930). For mutants 7B5-5, the insertion of pHimarEm1 transposon was 14 bp downstream of the ATG start codon of an aconitase gene (BHE19_RS00150). In mutant 10E3-2 the transposon inserted 1,380 bp downstream of the ATG start codon of the gldM gene (BHE19_RS21040). For the mutant M69, the transposon inserted at 67 bp downstream of the ATG start codon of EpsM gene (BHE19_RS15305). The difference in biomass determination was negligible between the WT and mutants, indicating that these gliding genes were not critical for cell growth in PY2 broth (Figure 5).
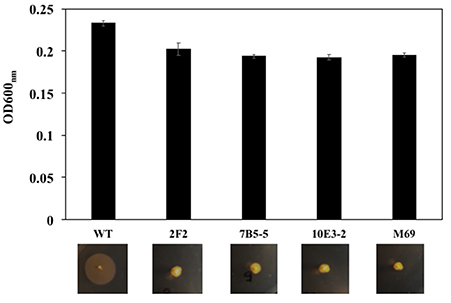
Figure 5. Comparation of the cell growth in PY2 broth and gliding motility on the PY2 agar between the WT and mutants. Transposon pHimarEm1 was introduced into F. spartansii T16T and erythromycin-resistant conjugants were sub-cultured in PY2 broth. The cell growth was compared by determining the OD600nm after overnight culture (up panel). The motility was evaluated by culture the cells on PY2 agar after 48 h (low panel).
Discussion
Our results demonstrated that strain T16T possesses some virulence factors that are similar to those previously reported in other Bacteroidetes, including metalloproteases, and hemolysins (Table 5). Moreover, the type 9 secretion system (T9SS) also occurs in T16T, which may contribute to secreting proteins that may be involved in virulence (Table 6). Nevertheless, our results also show that T16T has some virulence factors and antimicrobial genes absent in well-known fish pathogens (i.e., F. columnare, F. Branchiophilum, and F. psychrophilum; Table 5). The GC content in strain T16T (36%) was higher than in F. columnare ATCC 49512 (31%), F. branchiophilum FL-15 (33%), and F. psychrophilum JIP02/86 (33%), indicating their diverse genome evolution under differential selection (Lassalle et al., 2015). Among the selected flavobacteria, the genome size of F. spartansii T16T (5.35 Mb, next to F. johnsoniae) was remarkably larger than that in typical fish pathogens such as F. columnare ATCC 49512 (3.16 Mb), F. branchiophilum (3.56 Mb), and F. psychrophilum JIP02/86 (2.86 Mb). Genome size in opportunistic pathogens is typically larger than in virulent strains because more functional genes are required for their complicated living styles and diverse environments. Kolton et al. (2013) reported the genome size in Flavobacterium belonging to the terrestrial clade is around 40% larger than within the aquatic clade (Kolton et al., 2013).
Genomic islands often drive microbial evolution, increase fitness, and contribute to species diversity (Jackson et al., 2011). It is interesting that a large conjugative transposon (CTnFs) spanning at least two large GIs (GI-7 and GI-8) was found in F. spartansii T16T. Such conjugative transposons are widespread in Bacteroides, Prevotella, and Porphyromonas (Franco, 2004; Naito et al., 2011; Gorenc et al., 2012). Here, we extended the distribution profiles of conjugative transposons to some flavobacterial members including F. spartansii, F. johnsoniae, F. hibernum, F. denitrificans, F. beibuense, and F. daejeonense (Table 4). Gene content and organization of CTnFs-like conjugative transposons in F. spartansii mimic those reported in transposons CTn3-Bf and CTnB14 of B. fragilis YCH46 and P. bryantii B14T (Gorenc et al., 2012). One of the most intriguing features of conjugative transposon is presence of two conserved regions called “tra” and “GG” (Gorenc et al., 2012). However, CTnFs-like transposons have not been reported in F. columnare, F. branchiophilum and F. psychrophilum species based on marker gene search (traG), nor were CTnFs genes detected in Flavobacterium sp. W22 (Table 4) though its phylogeny placement is very close to F. spartansii T16T. The reason for the absence of CTnFs-like transposons in typical Flavobacterium pathogens remains unknown. The size of conjugative transposon is generally large (100–210 kb in Bacteroides) but varies considerably amongst species. CTnB14 in P. bryantii B14T is around 212 kb with a spacer (49.1 kb) where a large sugar utilization gene cluster locates (Gorenc et al., 2012). This conjugative transposon may promote the ability to scavenge the glycans in vivo in this oral cavity-adapted symbiont (Matsui et al., 2000). The accurate size, full contents and physiological roles in the CTnFs remain unclear in free-living isolates or opportunistic pathogens (see next). However, many functional gene products were predicted between/around the two regions (Gorenc et al., 2012). While the conjugative transposon is activated and mobilized, it frequently carries over its close regions to the new hosts (Juhas et al., 2009). Thus, the conjugative transposons may have important implications for spreading virulence factors, metabolism genes and some resistance genes (Burrus and Waldor, 2004; Juhas et al., 2009). Furthermore, the transfer efficiency of CTnDOT/ERL conjugative transposons in Bacteroides (also found in other Bacteroidetes members such as Prevotella), improved at least 1,000-fold when a low concentration of tetracycline was present in the culture (Jeters et al., 2009). The phenomenon indicates that antibiotics in the environment (such tetracycline) not only provide selection pressure for Bacteroidetes to acquire foreign antimicrobial genes, but also promote spreading other antibiotic genes (e.g., erythromycin gene transfer in Bacteroides) or other genetic elements (Shoemaker et al., 2001). Because the conjugative transposons present in Flavobacterium strains are very similar to CTnDOT, one can infer that horizontal transfer of flavobacterial genes involved in antibiotic resistance and/or virulence factors is very likely in their hosts and native habitats.
There are multiple antibiotics and chemotherapeutic compounds that are currently approved for use in foodfish or can be utilized under an investigational new animal drug (INAD) status (https://www.fws.gov/fisheries/aadap/aquaculture.html). Thus, generating more antibiotic sensitivity data for F. spartansii is crucial for guiding further research toward additional antibiotic approvals. F. spartansii T16T is intermediate in susceptibility to the permitted antibiotics in the USA aquaculture industry, namely oxytetracycline, sulfadimethoxine and florfenicol. One of the striking features in resistome analysis of T16T is that there are at least 10 β-lactam resistance-related genes. Putative β-lactamases (8 copies), metallo-β-lactamases (1 copy) and penicillin-binding proteins (1 copy) possibly confer resistance to penicillin and ampicillin (Table S1). Resistance toward third-generation cephalosporins (data not shown) by class C beta-lactamases seen in F. spartansii would hamper treatment of infection by Flavobacterium species. The concern is that, as a reservoir of extended-spectrum class C beta-lactamases, F. spartansii may spread these resistance gene(s) to other flavobacterial pathogens in the future. Extensive antibiotics application in the aquaculture industry may account for the multidrug resistance profile seen in resistance genes of F. spartansii (Kristiansson et al., 2011). It is interesting that two genes conferring tetracycline resistance were found in F. spartansii (as in several other Flavobacterium species) while disc analysis showed that bacterial cells were intermediate in susceptibility to tetracycline (Table 3). A tetracycline resistance (tetX) gene in transposons Tn4351 and Tn4400 did not confer resistance on anaerobically-grown Bacteroides fragilis while it functioned in aerobically-grown E. coli (Speer and Salyers, 1990; Speer et al., 1991). Gene tetX, located inside the conjugative transposon, may be acquired from other bacteria (exemplified by the GC ratio compared to the average ratio in the genome) during the transposon mobilization process. Expression of tetX may be problematic in F. spartansii because it is known that the transcriptional and translational initiation signals from Proteobacteria or the Gram-positive bacteria are not recognized well in Bacteroidetes (Chen et al., 2007a). Drug resistance mechanisms in bacteria are multifactorial, and can involve enzymatic degradation of the drugs, direct extrusion of the drug from the cells through efflux pumps, or alteration/mutation of ribosomal binding sites (Nikaido, 2009). The multidrug resistance efflux pumps predicted in F. spartansii and other flavobacteria are possibly involved in antimicrobial resistance, which does not manifest in other antibiotic degradation genes (Nikaido, 2009; Sun et al., 2014). This situation is cause for concern, and warrants more stringent surveillance in the use of antibiotics, as well as the resultant antibiotic resistance in clinically important bacterial species. Whole genome sequence of F. spartansii will be useful in future studies to determine antimicrobial resistance and virulence attributes as well as mechanisms that enhance its environmental or host fitness.
Besides the conjugative transposons and genomic islands, bacteriophages and phage-like genetic elements occupy a great amount of bacterial genome space. Bacteriophage-mediated transduction is an important contributor to spreading antimicrobial resistance and metabolism genes by the horizontal gene transfer mechanisms (Dutta and Sarkar, 2015). At least 5 prophages were predicted in F. spartansii T16T (Table S2). Only one of them (prophage 1) seems to be complete. However, the other prophages should not just be regarded as inactive because they may share machinery (e.g., structure proteins or replication enzymes) originating from prophage 1 or from the host, thus they can successfully assemble, pack and activate (Drulis-Kawa et al., 2012). Genes encoding important enzymes involved in capsule precursor biosynthesis and heavy metal resistance were found within the regions of the predicted prophage genomes, indicating that it is possible that these prophages possibly shape the bacterial genome evolution (Table S2). However, how these prophages influence the host behavior and virulence remains unexplored in flavobacteria. Indeed, prophages or phage-like genetic elements are not as prevalent in flavobacterial pathogen genomes as those in F. spartansii T16T; e.g., most of F. psychrophilum strains (such as 950106-1/1, JIP02/86, MH1, PG2, and 5) carry only one prophage (named 6H); moreover, there are only 1 and 3 incomplete phage clusters (no complete one) identified in F. columnare 94-081 and F. columnare ATCC 49512, respectively. Similarly, Touchon et al. reported that there were no incomplete prophage elements in F. branchiophilum FL-15 (Touchon et al., 2011). This may be partially explained due to the presence of multiple CRISPR loci in F. psychrophilum (1~2 loci) (Castillo et al., 2016), F. columnare (at least 3 loci) (Kayansamruaj et al., 2017), and F. branchiophilum FL-15 (at least 3 loci) (Touchon et al., 2011; Barrangou and Marraffini, 2014; van Houte et al., 2016). It is interesting that we did not detect any CRISPR elements in F. spartansii T16T, which may coincide with presence of prophages, genomic islands, and conjugative transposons in its genome. CRISPR/Cas, the prokaryotic immune system, defends the foreign DNA invasion (plasmids, phages and transposons) and may participate in regulating stress gene response and controlling bacterial virulence (Louwen et al., 2014; Laanto et al., 2017).
Prediction of virulence factors in this study contributes to our understanding of flavobacterial pathogenesis mechanisms as well as Flavobacterium/host interactions. The predicted virulence factors have good homology with those discovered in other well-known pathogenic flavobacteria (Touchon et al., 2011; Kumru et al., 2017). For example, F. spartansii T16T has the capability to digest animal erythrocytes with α-hemolytic activities, consistent with three hemolysin genes predicted in F. spartansii as virulence factors (Table 5). Hemolysins are well-documented cytolytic toxins that are important for animal pathogenesis process such as sepsis and tissue damage (Portnoy et al., 1988; Los et al., 2013). In F. columnare strain 94-081, disruption of one of the hemolysin genes (AWN65_RS11020) decreased fish mortality 15%, indicating that it contributes to virulence (Kumru et al., 2017). Moreover, this observation also showed that additional hemolysin(s) may be necessary to have full virulence against fish (Kumru et al., 2017). In addition, F. spartansii T16T has the thiol-activated cytolysin (TACYs) that possibly forms pores on the erythrocyte membrane (Morgan et al., 1996). Moreover, TACYs can also lead to the triggering signaling pathways in host cells, as is the case for listeriolysin O (LLO) (Hamon et al., 2012). LLOs induce a cytokine response by functioning as a pleiotropic pseudocytokine/chemokine, thus strongly influencing the course of infection (Baba et al., 2002). Protein sequences of a collagenase (peptidases U32) in strain T16T are conserved amongst the selected Flavobacterium strains (Table 5). Nakayama et al. (2016) found that the collagenase gene (fpcol) contributed to mortality in the Ayu (Plecoglossus altivelis) (Nakayama et al., 2016). Further, they also reported that the expression of fpcol was partially repressed by calcium or gelatin, showing that collagenase was necessary when the bacterial cells adhered to fish surface and initiated the invasion process (Nakayama et al., 2016). F. spartansii T16T has several metalloproteases (Table 5), which may facilitate bacterial dispersion and tissue damage. Conjugal transfer proteins (4 Tra components) as virulence factors probably allow these genomic elements to transfer from or to other bacteria (Vogel et al., 1998).
Fourteen gld and seven spr genes participating in the gliding motility, respectively, were found in F. spartansii T16T genome (Table 6). The amino acid sequences of the gliding and spreading proteins are highly conserved among the selected flavobacteria except GldE (Table 6). GldE (cutoff 50%) seem to be absent in the genomes of F. psychrophilum and F. columnare, indicating that they are not critical for gliding in these two Flavobacterium species. In F. johnsoniae, the gliding function of gldE can be replaced by gldB (Hunnicutt and McBride, 2001). Seven spreading protein encoding genes (SprA, SprB, SprC, SprD, SprE, SprF, and SprT) were found in F. spartansii T16T genome (Table 6). Most of the gene products (except SprB) show good homology to those in F. johnsoniae and F. hibernum DSM 12611, F. denitrificans DSM 15936 and Flavobacterium sp. WG21 though some of them (SprB and SprC) have the relative low identity to those in fish pathogens F. psychrophilum and F. columnare (Table 6).
Genetic amenability of F. spartansii T16T was exemplified by obtaining several gliding-deficient mutants using a transposon mutagenesis method. It is not surprising that we found one of the motility mutants had disruption of the gliding gene gldM. Disability of gldM gene expression in F. johnsoniae and Cellulophaga algicola caused gliding deficiency on the slide or agar surfaces, as well as failure of the delivery of SprB to the cell surface, and secretion of many cell surface and extracellular proteins (Braun et al., 2005; Zhu and McBride, 2016). Moreover, disruption of some gliding genes in flavobacteria led to attenuated virulence or biofilm formation (Álvarez et al., 2006; Sato et al., 2010). Decreasing virulence in gliding mutants may be due to the disruption of the T9SSs function (Sato et al., 2010). Diverse CTDs involved in protein secretion were recently characterized in F. johnsoniae (Chen et al., 2007b; Kulkarni et al., 2017). Proteins secreted through T9SSs possess two critical signals: the first one is the N-terminal signal peptides that are required by the Sec system to export outside the cytoplasmic membrane; and the second one is the carboxy-terminal domains (CTDs) that are needed by the T9SSs to secrete through outer membranes (McBride and Zhu, 2013; de Diego et al., 2016; Kulkarni et al., 2017). However, more research is needed to provide more evidences for virulence factors secreted through T9SSs in these flavobacterial pathogens. Furthermore, three putative gliding motility genes encoding the hypothetical protein belonging to the HCP-like family, the aniconase involved in TCA cycle for energy production, and a polysaccharide synthesis component. The complementation of these mutants is warranted to further confirm the gene function(s) involved in gliding motility.
Author Contributions
SC and EW conceived the study and participated in its design and coordination. SC performed the experiments, whole genome sequencing, annotation, and comparative analysis. JB contributed to the genome analysis. SC, TL, MF, and EW wrote the manuscript. All authors have read and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This project was funded by NIH grant R37AI21884.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.02339/full#supplementary-material
References
Álvarez, B., Secades, P., Prieto, M., McBride, M. J., and Guijarro, J. A. (2006). A mutation in Flavobacterium psychrophilum tlpB inhibits gliding motility and induces biofilm formation. Appl. Environ. Microbiol. 72, 4044–4053. doi: 10.1128/AEM.00128-06
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 9:75. doi: 10.1186/1471-2164-9-75
Baba, H., Kawamura, I., Kohda, C., Nomura, T., Ito, Y., Kimoto, T., et al. (2002). Induction of gamma interferon and nitric oxide by truncated pneumolysin that lacks pore-forming activity. Infect. Immun. 70, 107–113. doi: 10.1128/IAI.70.1.107-113.2002
Barbier, P., Houel, A., Loux, V., Poulain, J., Bernardet, J.-F., Touchon, M., et al. (2012). Complete genome sequence of Flavobacterium indicum GPSTA100-9T, isolated from warm spring water. J. Bacteriol. 194, 3024–3025. doi: 10.1128/JB.00420-12
Bartelme, R. P., Newton, R. J., Zhu, Y., Li, N., LaFrentz, B. R., and McBride, M. J. (2016). Complete genome sequence of the fish pathogen Flavobacterium columnare strain C#2. Genome Announc. 4:e00624-16. doi: 10.1128/genomeA.00624-16
Barrangou, R., and Marraffini, L. A. (2014). CRISPR-Cas systems: prokaryotes upgrade to adaptive immunity. Mol. Cell. 54, 234–244. doi: 10.1016/j.molcel.2014.03.011
Blom, J., Kreis, J., Spänig, S., Juhre, T., Bertelli, C., Ernst, C., et al. (2016). EDGAR 2.0: an enhanced software platform for comparative gene content analyses. Nucleic Acids Res. 44, W22–W28. doi: 10.1093/nar/gkw255
Bowker, J. D., Trushenski, J. T., Gaikowski, M. P., and Straus, D. L. (eds.). (2016). Guide to Using Drugs, Biologics, and Other Chemicals in Aquaculture. American Fisheries Society Fish Culture Section.
Braun, T. F., Khubbar, M. K., Saffarini, D. A., and McBride, M. J. (2005). Flavobacterium johnsoniae gliding motility genes identified by mariner mutagenesis. J. Bacteriol. 187, 6943–6952. doi: 10.1128/JB.187.20.6943-6952.2005
Burrus, V., and Waldor, M. K. (2004). Shaping bacterial genomes with integrative and conjugative elements. Res. Microbiol. 155, 376–386. doi: 10.1016/j.resmic.2004.01.012
Castillo, D., Christiansen, R. H., Dalsgaard, I., Madsen, L., Espejo, R., and Middelboe, M. (2016). Comparative genome analysis provides insights into the pathogenicity of Flavobacterium psychrophilum. PLoS ONE 11:e0152515. doi: 10.1371/journal.pone.0152515
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328. doi: 10.1093/nar/gki008
Chen, S., Bagdasarian, M., Kaufman, M., and Walker, E. (2007a). Characterization of strong promoters from an environmental Flavobacterium hibernum strain by using a green fluorescent protein-based reporter system. Appl. Environ. Microbiol. 73, 1089–1100. doi: 10.1128/AEM.01577-06
Chen, S., Bagdasarian, M., Kaufman, M., Bates, A., and Walker, E. (2007b). Mutational analysis of the ompA promoter from Flavobacterium johnsoniae. J. Bacteriol. 189, 5108–5118. doi: 10.1128/JB.00401-07
Chen, S., Blom, J., and Walker, E. D. (2017). Genomic, physiologic, and symbiotic characterization of Serratia Marcescens strains isolated from the mosquito Anopheles stephensi. Front. Microbiol. 8:1483. doi: 10.3389/fmicb.2017.01483
de Diego, I., Ksiazek, M., Mizgalska, D., Koneru, L., Golik, P., Szmigielski, B., et al. (2016). The outer-membrane export signal of Porphyromonas gingivalis type IX secretion system (T9SS) is a conserved C-terminal β-sandwich domain. Sci. Rep. 6:23123. doi: 10.1038/srep23123
Drulis-Kawa, Z., Majkowska-Skrobek, G., Maciejewska, B., Delattre, A.-S., and Lavigne, R. (2012). Learning from bacteriophages-advantages and limitations of phage and phage-encoded protein applications. Curr. Protein Pept. Sci. 13, 699–722. doi: 10.2174/138920312804871193
Duchaud, E., Boussaha, M., Loux, V., Bernardet, J., Michel, C., Kerouault, B., et al. (2007). Complete genome sequence of the fish pathogen Flavobacterium psychrophilum. Nat. Biotechnol. 25, 763–769. doi: 10.1038/nbt1313
Dutta, C., and Sarkar, M. (2015). “Horizontal gene transfer and bacterial diversity,” in Encyclopedia of Metagenomics: Genes, Genomes and Metagenomes: Basics, Methods, Databases and Tools, ed K. E. Nelson (Boston, MA: Springer), 251–257.
Franco, A. A. (2004). The Bacteroides fragilis pathogenicity island is contained in a putative novel conjugative transposon. J. Bacteriol. 186, 6077–6092. doi: 10.1128/JB.186.18.6077-6092.2004
Gorenc, K., Accetto, T., and Avguštin, G. (2012). Bioinformatic evidence and characterization of novel putative large conjugative transposons residing in genomes of genera Bacteroides and Prevotella. Folia Microbiol. (Praha) 57, 285–290. doi: 10.1007/s12223-012-0126-5
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W7. doi: 10.1093/nar/gkm360
Hamon, M. A., Ribet, D., Stavru, F., and Cossart, P. (2012). Listeriolysin O: the Swiss army knife of Listeria. Trends Microbiol. 20, 360–368. doi: 10.1016/j.tim.2012.04.006
Hawke, J. P., and Thune, R. L. (1992). Systemic isolation and antimicrobial susceptibility of Cytophaga columnaris from commercially reared channel catfish. J. Aquat. Anim. Health 4, 109–113. doi: 10.1577/1548-8667(1992)004<0109:SIAASO>2.3.CO;2
Hunnicutt, D. W., and McBride, M. J. (2001). Cloning and characterization of the Flavobacterium johnsoniae gliding motility GenesgldD and gldE. J. Bacteriol. 183, 4167–4175. doi: 10.1128/JB.183.14.4167-4175.2001
Jackson, R. W., Vinatzer, B., Arnold, D. L., Dorus, S., and Murillo, J. (2011). The influence of the accessory genome on bacterial pathogen evolution. Mob. Genet. Elements. 1, 55–65. doi: 10.4161/mge.1.1.16432
Jeters, R. T., Wang, G.-R., Moon, K., Shoemaker, N. B., and Salyers, A. A. (2009). Tetracycline-associated transcriptional regulation of transfer genes of the Bacteroides conjugative transposon CTnDOT. J. Bacteriol. 191, 6374–6382. doi: 10.1128/JB.00739-09
Juhas, M., van der Meer, J. R., Gaillard, M., Harding, R. M., Hood, D. W., and Crook, D. W. (2009). Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 33, 376–393. doi: 10.1111/j.1574-6976.2008.00136.x
Kayansamruaj, P., Dong, H. T., Hirono, I., Kondo, H., Senapin, S., and Rodkhum, C. (2017). Comparative genome analysis of fish pathogen Flavobacterium columnare reveals extensive sequence diversity within the species. Infect. Genetics Evol. 54, 7–17. doi: 10.1016/j.meegid.2017.06.012
Klein, C. C., Alves, J. M. P., Serrano, M. G., Buck, G. A., Vasconcelos, A. T. R., Sagot, M.-F., et al. (2013). Biosynthesis of vitamins and cofactors in bacterium-harbouring trypanosomatids depends on the symbiotic association as revealed by genomic analyses. PLoS ONE 8:e79786. doi: 10.1371/journal.pone.0079786
Kolton, M., Sela, N., Elad, Y., and Cytryn, E. (2013). Comparative genomic analysis indicates that niche adaptation of terrestrial flavobacteria is strongly linked to plant glycan metabolism. PLoS ONE 8:e76704. doi: 10.1371/journal.pone.0076704
Kristiansson, E., Fick, J., Janzon, A., Grabic, R., Rutgersson, C., Weijdegård, B., et al. (2011). Pyrosequencing of antibiotic-contaminated river sediments reveals high levels of resistance and gene transfer elements. PLoS ONE 6:e17038. doi: 10.1371/journal.pone.0017038
Kulkarni, S. S., Zhu, Y., Brendel, C. J., and McBride, M. J. (2017). Diverse C-Terminal sequences involved in Flavobacterium johnsoniae protein secretion. J. Bacteriol. 199:e00884-16. doi: 10.1128/JB.00884-16
Kumru, S., Tekedar, H. C., Gulsoy, N., Waldbieser, G. C., Lawrence, M. L., and Karsi, A. (2017). Comparative analysis of the Flavobacterium columnare Genomovar, I., and II Genomes. Front. Microbiol. 8:1375. doi: 10.3389/fmicb.2017.01375
Laanto, E., Hoikkala, V., Ravantti, J., and Sundberg, L.-R. (2017). Long-term genomic coevolution of host-parasite interaction in the natural environment. Nat. Commun. 8, 111. doi: 10.1038/s41467-017-00158-7
Lassalle, F., Périan, S., Bataillon, T., Nesme, X., Duret, L., and Daubin, V. (2015). GC-Content evolution in bacterial genomes: the biased gene conversion hypothesis expands. PLoS Genet. 11:e1004941. doi: 10.1371/journal.pgen.1004941
Loch, T. P., and Faisal, M. (2014a). Flavobacterium spartansii sp. nov., a pathogen of fishes, and emended descriptions of Flavobacterium aquidurense and Flavobacterium araucananum. Int. J. Syst. Evol. Microbiol. 64, 406–412. doi: 10.1099/ijs.0.051433-0
Loch, T. P., and Faisal, M. (2014b). Deciphering the biodiversity of fish-pathogenic Flavobacterium spp. recovered from the Great Lakes basin. Dis Aquat Org. 112, 45–57. doi: 10.3354/dao02791
Loch, T. P., and Faisal, M. (2015). Emerging flavobacterial infections in fish: a review. Int. J. Adv. Res. 6, 283–300. doi: 10.1016/j.jare.2014.10.009
Loch, T. P., and Faisal, M. (2016). Flavobacterium spartansii induces pathological changes and mortality in experimentally challenged Chinook salmon Oncorhynchus tshawytscha (Walbaum). J. Fish Dis. 39, 483–488. doi: 10.1111/jfd.12363
Loch, T. P., and Faisal, M. (2017). “Flavobacterium spp.: F. psychrophilum, F. columnare, and F. branchiophilum,” in Fish Viruses and Bacteria: Pathobiology and Protection, eds R. Cipriano and P. Woo (Wellingford: CABI International), 211–232.
Los, F. C. O., Randis, T. M., Aroian, R. V., and Ratner, A. J. (2013). Role of pore-forming toxins in bacterial infectious diseases. Microbiol. Mol. Biol. Rev. 77, 173–207. doi: 10.1128/MMBR.00052-12
Louwen, R., Staals, R. H. J., Endtz, H. P., van Baarlen, P., and van der Oost, J. (2014). The Role of CRISPR-Cas systems in virulence of pathogenic bacteria. Microbiol. Mol. Biol. Rev. 78, 74–88. doi: 10.1128/MMBR.00039-13
Matsui, H., Ogata, K., Tajima, K., Nakamura, M., Nagamine, T., Aminov, R. I., et al. (2000). Phenotypic characterization of polysaccharidases produced by four prevotella type strains. Curr. Microbiol. 41, 45–49. doi: 10.1007/s002840010089
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57, 3348–3357. doi: 10.1128/AAC.00419-13
McBride, M. J., and Nakane, D. (2015). Flavobacterium gliding motility and the type IX secretion system. Curr. Opin. Microbiol. 28, 72–77. doi: 10.1016/j.mib.2015.07.016
McBride, M. J., Xie, G., Martens, E. C., Lapidus, A., Henrissat, B., Rhodes, R. G., et al. (2009). Novel features of the polysaccharide-digesting gliding bacterium Flavobacterium johnsoniae as revealed by genome sequence analysis. Appl. Environ. Microbiol. 75, 6864–6875. doi: 10.1128/AEM.01495-09
McBride, M. J., and Zhu, Y. (2013). Gliding motility and por secretion system genes are widespread among members of the phylum Bacteroidetes. J. Bacteriol. 195, 270–278. doi: 10.1128/JB.01962-12
Morgan, P. J., Andrew, P. W., and Mitchell, T. J. (1996). Thiol-activated cytolysins. Rev. Med. Microbiol. 7, 221–230. doi: 10.1097/00013542-199610000-00004
Nagamatsu, K., Hannan, T. J., Guest, R. L., Kostakioti, M., Hadjifrangiskou, M., Binkley, J., et al. (2015). Dysregulation of Escherichia coli α-hemolysin expression alters the course of acute and persistent urinary tract infection. Proc. Natl. Acad. Sci. U.S.A. 112, E871–E80. doi: 10.1073/pnas.1500374112
Naito, M., Sato, K., Shoji, M., Yukitake, H., Ogura, Y., Hayashi, T., et al. (2011). Characterization of the Porphyromonas gingivalis conjugative transposon CTnPg1: determination of the integration site and the genes essential for conjugal transfer. Microbiology 157, 2022–2032. doi: 10.1099/mic.0.047803-0
Nakayama, H., Tanaka, K., Teramura, N., and Hattori, S. (2016). Expression of collagenase in Flavobacterium psychrophilum isolated from cold-water disease-affected ayu (Plecoglossus altivelis). Biosci. Biotechnol. Biochem. 80, 135–144. doi: 10.1080/09168451.2015.1079477
Nikaido, H. (2009). Multidrug resistance in bacteria. Annu. Rev. Biochem. 78, 119–146. doi: 10.1146/annurev.biochem.78.082907.145923
Portnoy, D. A., Jacks, P. S., and Hinrichs, D. J. (1988). Role of hemolysin for the intracellular growth of Listeria monocytogenes. J. Exp. Med. 167:1459. doi: 10.1084/jem.167.4.1459
Sato, K., Naito, M., Yukitake, H., Hirakawa, H., Shoji, M., McBride, M. J., et al. (2010). A protein secretion system linked to Bacteroidete gliding motility and pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 107, 276–281. doi: 10.1073/pnas.0912010107
Shoemaker, N. B., Vlamakis, H., Hayes, K., and Salyers, A. A. (2001). Evidence for extensive resistance gene transfer among Bacteroides spp. and among Bacteroides and other genera in the human colon. Appl. Environ. Microbiol. 67, 561–568. doi: 10.1128/AEM.67.2.561-568.2001
Speer, B. S., Bedzyk, L., and Salyers, A. A. (1991). Evidence that a novel tetracycline resistance gene found on two Bacteroides transposons encodes an NADP-requiring oxidoreductase. J. Bacteriol. 173, 176–183. doi: 10.1128/jb.173.1.176-183.1991
Speer, B. S., and Salyers, A. A. (1990). A tetracycline efflux gene on Bacteroides transposon Tn4400 does not contribute to tetracycline resistance. J. Bacteriol. 172, 292–298. doi: 10.1128/jb.172.1.292-298.1990
Starliper, C., and Schill, W. (2011). “Flavobacterial diseases: columnaris disease, coldwater disease, and bacterial gill disease,” in Fish Diseases and Disorders, eds P. T. K. Woo and D. B. Bruno (Oxfordshire: CAB International). 606–631.
Sun, J., Deng, Z., and Yan, A. (2014). Bacterial multidrug efflux pumps: mechanisms, physiology and pharmacological exploitations. Biochem. Biophys. Res. Commun. 453, 254–267. doi: 10.1016/j.bbrc.2014.05.090
Touchon, M., Barbier, P., Bernardet, J.-F., Loux, V., Vacherie, B., Barbe, V., et al. (2011). Complete genome sequence of the fish pathogen Flavobacterium branchiophilum. Appl. Environ. Microbiol. 77, 7656–7662. doi: 10.1128/AEM.05625-11
van Houte, S., Buckling, A., and Westra, E. R. (2016). Evolutionary ecology of prokaryotic immune mechanisms. Microbiol. Mol. Biol. Rev. 80, 745–763. doi: 10.1128/MMBR.00011-16
Vogel, J. P., Andrews, H. L., Wong, S. K., and Isberg, R. R. (1998). Conjugative transfer by the virulence system of Legionella pneumophila. Science 279:873. doi: 10.1126/science.279.5352.873
Whitman, K. A. (2004). Finfish and Shellfish Bacteriology Manual: Techniques and Procedures. Ames, IA: Iowa State Press. 258.
Wiens, G. D., LaPatra, S. E., Welch, T. J., Rexroad, C. III., Call, D. R., Cain, K. D., et al. (2014). Complete genome sequence of Flavobacterium psychrophilum strain CSF259-93, used to select rainbow trout for increased genetic resistance against bacterial cold water disease. Genome Announc. 2, e00889–e00814. doi: 10.1128/genomeA.00889-14
Wu, A. K., Kropinski, A. M., Lumsden, J. S., Dixon, B., and MacInnes, J. I. (2015). Complete genome sequence of the fish pathogen Flavobacterium psychrophilum ATCC 49418T. Stand. Genomic Sci. 10:3. doi: 10.1186/1944-3277-10-3
Zhang, Y., Nie, P., and Lin, L. (2016). Complete genome sequence of the fish pathogen Flavobacterium columnare Pf1. Genome Announc. 4:e00900-16. doi: 10.1128/genomeA.00900-16
Zhou, C. E., Smith, J., Lam, M., Zemla, A., Dyer, M. D., and Slezak, T. (2007). MvirDB—a microbial database of protein toxins, virulence factors and antibiotic resistance genes for bio-defence applications. Nucleic Acids Res. 35, D391–D394. doi: 10.1093/nar/gkl791
Keywords: Flavobacterium spartansii, genome analysis, virulence factors, mutation
Citation: Chen S, Blom J, Loch TP, Faisal M and Walker ED (2017) The Emerging Fish Pathogen Flavobacterium spartansii Isolated from Chinook Salmon: Comparative Genome Analysis and Molecular Manipulation. Front. Microbiol. 8:2339. doi: 10.3389/fmicb.2017.02339
Received: 12 September 2017; Accepted: 13 November 2017;
Published: 30 November 2017.
Edited by:
Daniela De Biase, Sapienza Università di Roma, ItalyReviewed by:
Mark J. McBride, University of Wisconsin–Milwaukee, United StatesPattanapon Kayansamruaj, Kasetsart University, Thailand
Brian Dixon, University of Waterloo, Canada
Copyright © 2017 Chen, Blom, Loch, Faisal and Walker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shicheng Chen, c2hpY2hlbmdAbXN1LmVkdQ==