 Jingshu Zhang
Jingshu Zhang Yun Lan
Yun Lan Sumana Sanyal
Sumana Sanyal
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 28 November 2017
Sec. Microbial Immunology
Volume 8 - 2017 | https://doi.org/10.3389/fmicb.2017.02286
This article is part of the Research Topic Shaping of Human Immune System and Metabolic Processes by Viruses and Microorganisms View all 64 articles
Lipid droplets (LDs) are endoplasmic reticulum (ER)-related dynamic organelles that store and regulate fatty acids and neutral lipids. They play a central role in cellular energy storage, lipid metabolism and cellular homeostasis. It has become evident that viruses have co-evolved in order to exploit host lipid metabolic pathways. This is especially characteristic of the Flaviviridae family, including hepatitis C virus (HCV) and several flaviviruses. Devoid of an appropriate lipid biosynthetic machinery of their own, these single-strand positive-sense RNA viruses can induce dramatic changes in host metabolic pathways to establish a favorable environment for viral multiplication and acquire essential components to facilitate their assembly and traffic. Here we have reviewed the current knowledge on the intracellular life cycle of those from the Flaviviridae family, with particular emphasis on HCV and dengue virus (DENV), and their association with the biosynthesis and metabolism of LDs, with the aim to identify potential antiviral targets for development of novel therapeutic interventions.
Cellular homeostasis is maintained by a constant metabolic energy flux. As one of the major energy sources, lipids are synthesized, modified and utilized through various pathways. Lipid droplets (LDs) are ubiquitous and conserved cytoplasmic compartments delineated by a phospholipid monolayer, and serve as energy reservoirs in almost all living organisms. Excess lipids are packaged, stored and distributed in LDs, an organelle which is not only important in lipid storage and metabolism, but protein quality control, pathogenesis, and immune responses (Walther and Farese, 2012).
Since viruses lack the appropriate machinery to conduct their own lipid synthesis, most have evolved mechanisms to hijack host lipid metabolic pathways (including LDs) for completing their intracellular replication cycles. Hepatitis C virus (HCV) has long been demonstrated to do so (Paul et al., 2014). Apart from the cell biology underlying infection, the interplay between viral infection and host lipid metabolic pathways is important not only to elucidate the pathogenicity of this category of viruses but also to assess how they can be targeted as a general means of combating infections.
As a consequence of development of gene editing and mass spectrometry based lipidomics and proteomics technologies, an increasing body of evidence indicates the involvement of host LDs at different steps of the intracellular life cycle of HCV and flaviviruses (Martín-Acebes et al., 2016b). Here, we have cataloged these interactions and anticipate that this knowledge will be beneficial for identification of host factors as suitable targets for antiviral interventions.
LDs are essentially the emulsion phase of insoluble oil droplets dispersed in aqueous cytoplasm. Compared to other cellular organelles with double-layered membranes, the structure of LDs is rather unique, containing a hydrophobic core and a single layer of amphipathic phospholipids. The neutral lipid core contains predominantly triacylglycerols (TAGs) and cholesterol esters (CEs) (Thiam et al., 2013). Although the composition of the phospholipid monolayer varies in different cell types, phosphatidylcholine (PC) and phosphatidylethanolamine (PE) are the two major phospholipids. The morphology and consumption of LDs are drastically altered by the composition of their phospholipid monolayer (Guo et al., 2008). The surface of the monolayer is decorated with LD-associated proteins, including lipolytic enzymes such as hormone-sensitive lipase (HSL), adipose triglyceride lipase (ATGL), and PAT-domain family (perilipin, adipophilin and TIP47) (Tauchi-Sato et al., 2002; Ohsaki et al., 2006; Wilfling et al., 2014a). Despite being present in nearly all cell types across different organisms, LDs are highly heterogeneous and dynamic with varied numbers and sizes (ranging from 100 nm to 100 mm in diameter) in otherwise identical cells. Even within the same cell, LDs expand or shrink in response to cellular signals.
In eukaryotes, LDs respond to increased cellular fatty acid levels and emerge from the accumulation of neutral lipids in the ER, which harbors enzymes necessary for neutral lipid synthesis in most cell types (Buhman et al., 2001; Pol et al., 2004). First established as an oil-in-water emulsion, the small nascent LDs undergo a series of well-organized processes and grow into larger, mature LDs. The final steps of TAG and CE synthesis are catalyzed by ER-localized diacylglycerol acyltransferases (DGATs) and acyl-CoA:cholesterol acyltranserases (ACATs), respectively. The continuous accumulation of the newly synthesized TAGs and CEs at specific sites at the ER results in separation of two phases, where globules of TAGs arise between the two leaflets of the bilayer and eventually dissociate. DGAT2, which is inserted into one leaflet of the ER membrane, is transported to LDs where it continues to catalyze synthesis of TAGs, hence promoting further growth of LDs (Kassan et al., 2013; Wilfling et al., 2013). This process is thermodynamically enabled by the unique phospholipid monolayer structure of LDs.
Long been regarded as simple and passive lipid storage compartments, LDs are currently considered highly dynamic and complex. They play a central role in lipid metabolism and are connected to diverse cellular processes like fatty acid trafficking, cellular signaling, protein storage, autophagy, immunity, and virus replication (Singh et al., 2009; Saka and Valdivia, 2012; Rambold et al., 2015; Welte, 2015; Velázquez et al., 2016).
As metabolically active organelles, LDs regulate the balance between host lipid synthesis and mobilization to maintain cellular homeostasis. Catalyzed by DGAT1 and DGAT2, cellular fatty acids together with diacylyglycerols (DAGs) are converted into TAGs and stored in LDs. TAGs can be further hydrolyzed to generate DAGs or phosphatidic acid (PA) for membrane phospholipid synthesis and free fatty acids (FFAs) for energy production (Pol et al., 2014).
Due to unique structural features and proximity to the ER, the surface of LDs can also serve as transient storage depots for proteins that are destined for degradation via the ER-associated degradation (ERAD) pathway (Gao and Goodman, 2015). Misfolded proteins in the ER are removed and degraded by the ubiquitin–proteasome system. Current evidence suggests that ubiquitinated apolipoprotein B100 (ApoB100) (Ohsaki et al., 2008) and 3-hydroxy-3-methylglutaryl CoA reductase (HMGCR) (Hartman et al., 2010) are likely degraded on the surface of LDs through proteasomal and autophagic pathways (Ohsaki et al., 2006). HMGCR is one of the rate-limiting enzymes for cholesterol synthesis in mammalian cells. Ubiquitination of HMGCR is mediated by ancient ubiquitous protein 1 (AUP1), a highly conserved monotopic membrane protein localized to both LDs and the ER membrane (Spandl et al., 2011). AUP1 recruits the ubiquitin-conjugating enzyme UBE2G2 to LDs and facilitates its binding with the ER ubiquitin ligases gp78 and Trc8, which subsequently initiates the ubiquitination/degradation of HMGCR resulting in inhibition of cholesterol synthesis (Jo et al., 2013). Apart from providing a molecular link between LDs and the ubiquitination machinery, monoubiquitinated AUP1 was reported to induce LD clustering, a widespread phenomenon observed in multiple cell types across all species (Lohmann et al., 2013). LDs may also provide sequestration platforms for protein storage (Cermelli et al., 2006), such as during the synthesis of eicosanoids, a class of signaling molecules that use LDs as distinct sites for eicosanoid generation (Bozza et al., 2011).
Depending on the cell type, starvation and/or physiological conditions, eukaryotic cells mobilize lipids stored in LDs via two major pathways termed lipolysis and lipophagy. In mammalian adipocytes, lipolysis is activated in response to changes in cellular energy and hormone levels. This allows transient docking and activation of three major lipolytic enzymes, ATGL, HSL, and monoglyceride lipase (MGL) which co-ordinate the hydrolysis of TAGs for energy production (Karlsson et al., 1997; Zimmermann et al., 2004; Dugail and Hajduch, 2007; Lass et al., 2011). Perilipins localize to LD surfaces and under basal conditions shield TAGs from cytosolic lipases. During starvation, perilipins are degraded via the chaperone-mediated autophagy (CMA) pathway to facilitate lipolysis by HSL and ATGL (Brasaemle, 2007; Sztalryd and Kimmel, 2014; Kaushik and Cuervo, 2015). Apart from LD-associated proteins, the ADP-ribosylation factor-coat protein I (ARF1-COPI) vesicular trafficking machinery is likely to play an important role in mediating lipolysis by regulating LD composition and targeting ATGL to LDs (Soni et al., 2009; Wilfling et al., 2014b).
The role of autophagy in regulating lipid metabolism has been intensively studied in recent years (Singh et al., 2009; Singh and Cuervo, 2012). Various cell types have been used to demonstrate the process of LD mobilization via the autophagy pathway, such as hypothalamic neurons (Kaushik et al., 2011), glial cells (Martinez-Vicente et al., 2010), and enterocytes (Narabayashi et al., 2015). Autophagy is a conserved cellular process that delivers cytoplasmic contents, including dysfunctional proteins, and excess or damaged organelles to lytic compartments for degradation and recycling. The process can be induced by a number of factors such as ER stress, cellular starvation, and pathogenic infection. Available data support that three distinct types of autophagy can be triggered: macro-, micro- and chaperone-mediated autophagy, amongst which, macroautophagy is the best characterized (Yoshimori, 2004; Mizushima, 2007). Upon activation, cytoplasmic components are first enclosed by a double-layered vesicular structure termed autophagosome, which fuse with lysosomes where internal cargos are degraded (Mizushima, 2007). Multiple factors such as nutrient deprivation, virus infection, and sterol (cholesterol) depletion, can trigger degradation of LDs through the autophagic machinery (Ouimet and Marcel, 2012). LC3II, a structural component of the autophagosomes, and autophagy-related proteins Atg2, Atg5, and Atg7 are recruited to the surface of LDs to form autophagosomes. LDs are engulfed for lysosomal degradation to release stored lipids, which undergo mitochondrial β-oxidation for energy production. This process is frequently manipulated by flaviviruses to promote their replication (see Usage of LD as an energy reservoir during viral life cycle) (Singh et al., 2009; Heaton and Randall, 2010; Fujimoto and Parton, 2011; Velikkakath et al., 2012). The level and distribution of cellular cholesterol is tightly regulated; excess free cholesterol stored as cholesteryl esters in LDs are hydrolyzed during sterol starvation through autophagy (Cheng et al., 2006; Ouimet and Marcel, 2012). Sterol regulatory element-binding proteins (SREBPs) are the central transcriptional regulators of cholesterol metabolism and lipogenesis. In the presence of high cholesterol content in the cytoplasm, SREBP binds to sterol regulatory element-binding protein cleavage-activating protein (SCAP) and the ER-associated protein Insig. Upon reduction of cellular cholesterol below a threshold, Insig is degraded through the ERAD pathway, the SCAP-SREBP complex is transported to the Golgi, where SREBP undergoes intramembrane proteolysis and translocates to the nucleus. This mature form of SREBP initiates transcription of a series of down-stream genes involved in the biosynthesis of cholesterol (Brown and Goldstein, 1997; Yang et al., 2002).
Viruses of the Flaviviridae family are enveloped single-strand positive-sense RNA viruses, with the nucleocapsids surrounded by two or more types of envelope glycoproteins and lipid bilayers (Lindenbach et al., 2007; Paul and Bartenschlager, 2015). It comprises several different genera including Hepacivirus (e.g., HCV), Flavivirus [e.g., Zika virus (ZIKV), dengue virus (DENV)], Pegivirus, and Pestivirus.
Persistent infection with HCV in humans can develop into serious liver diseases, including fibrosis and liver cirrhosis, which could further progress into hepatocellular carcinoma (Bartenschlager et al., 2013). Medically-relevant flaviviruses, including yellow fever virus (YFV), ZIKV, DENV, West Nile virus (WNV), and Japanese encephalitis virus (JEV), are usually arboviruses (viz., transmitted by arthropods, mainly mosquitoes and ticks) that are responsible for severe mortality in humans and animals worldwide. DENV and YFV infections are known to cause vascular leakage and hemorrhage in some infected patients (Siqueira et al., 2005; Garske et al., 2014; Thanachartwet et al., 2015). JEV and WSN infections on the other hand, tend to cause neurological diseases (Sarkari et al., 2012; Samaan et al., 2016). ZIKV infection is associated with serious birth defects—microcephaly in particular—and other neurological disorders (Petersen et al., 2016). Although there has been significant progress in therapeutic interventions for HCV and some other flaviviruses (for example YFV), there is still an urgent need for vaccines and medications against others such as DENV and ZIKV. Additionally, the ever-increasing geographical spread and number of outbreaks caused by these pathogens pose a considerable threat to public health (Gould and Solomon, 2008).
Despite significant differences in transmission, tissue tropism and pathogenesis, viruses of the Flaviviridae family employ similar intracellular replication strategies. After receptor-mediated endocytosis, the acidic environment in the endosomes triggers fusion between the virion lipid envelope and cellular membranes, followed by viral uncoating. The viral RNA is subsequently released into the cytoplasm and used directly as mRNA for translation of the viral polyprotein. Host and viral proteases cleave the newly synthesized viral polyprotein to generate the structural and non-structural (NS) proteins (Lindenbach et al., 2007). Viral replicase proteins together with other host factors induce massive rearrangements of intracellular membranes to form organelle-like membrane-delineated compartments for efficient RNA replication. At the replication sites, the positive-sense RNA is used as template to generate the negative-sense RNA intermediate, while multiple positive-sense progeny RNAs are produced to be incorporated into nascent virus particles (Paul and Bartenschlager, 2015). Progeny virions are assembled in close proximity to the ER and LDs, and appear to bud into the ER-lumen, followed by transport through the host secretory pathway where they undergo further maturation, and are eventually released from the cell surface (Lindenbach et al., 2007; Paul and Bartenschlager, 2015; Figure 1).
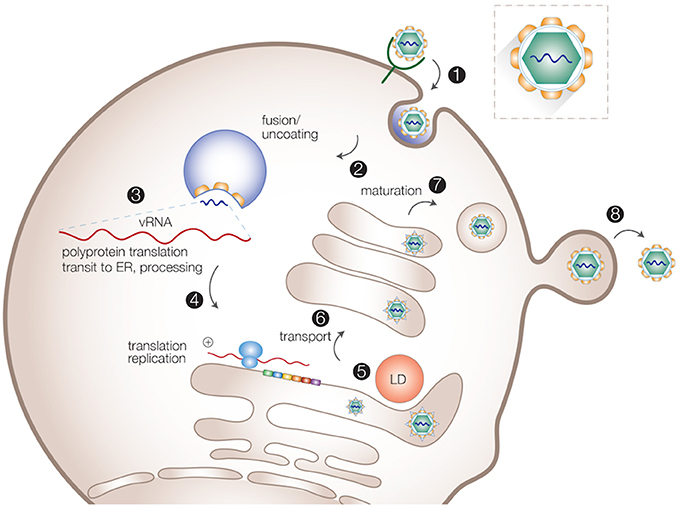
Figure 1. Intracellular life cycle of flaviviruses. Viral particles are internalized via receptor-mediated endocytosis (1). After the uncoating of viral particles (2), viral RNA is released into cytosol for translation and replication (3,4). Progeny virions are assembled in close proximity to the ER and LDs (5). Virions are transported through the host secretory pathway and undergo maturation (6,7). Mature virions are released from the cell surface (8).
HCV has historically been used for studying interactions between LD metabolism and the viral life cycle. Others from the same family, such as DENV, have recently started receiving more attention in this regard. The magnitude and complexity of these interactions underscore the significance of targeting LD metabolism to control viral infection. As a dynamic cellular lipid storage organelle, LDs participate in the viral life cycle by providing intracellular membrane surface area, lipids, energy, and proteins.
Upon infection massive intracellular membrane rearrangements are induced by perturbing lipid biosynthetic pathways to spatially segregate the replication and assembly sites (Welsch et al., 2009; Romero-Brey et al., 2012). On the one hand, the two sites need to be separated to avoid competition between the capsid protein and the viral replicase complex at the level of RNA binding. On the other hand, newly synthesized positive-sense progeny RNAs need to be transported from the replication to the assembly sites, where the capsid protein is concentrated. For maximum efficiency in virus assembly the two sites require close proximity to each other (Welsch et al., 2009; Romero-Brey et al., 2012; Figure 2).
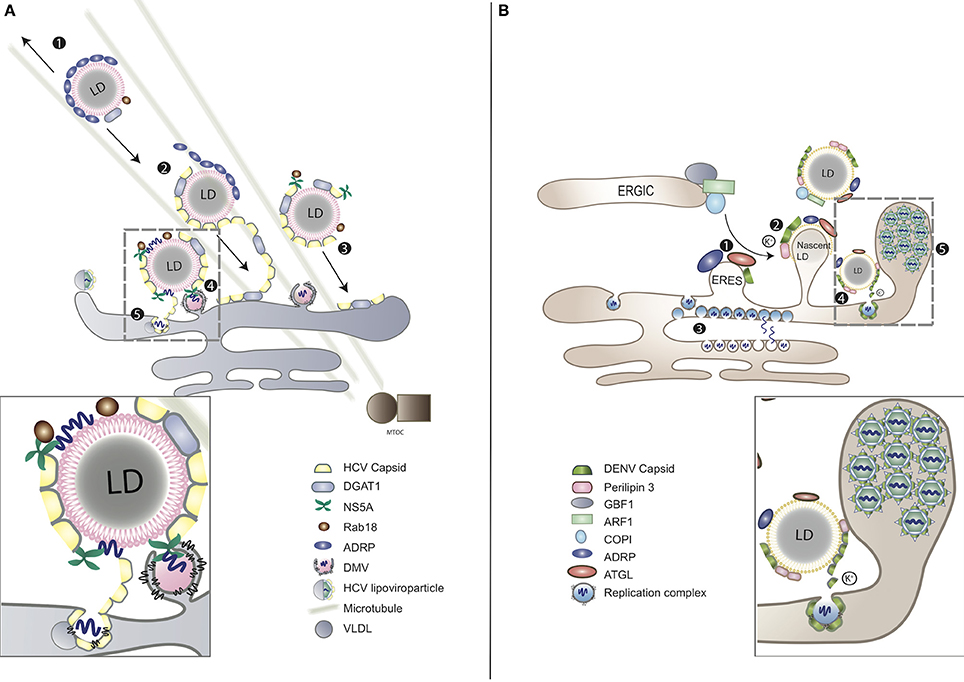
Figure 2. LDs as platforms for virion assembly in (A) HCV and (B) DENV infection. (A) (1) ADRP-coated LDs are able to interact with microtubules and move toward both plus and minus ends. (2) During HCV infection, viral capsid protein replaces ADRP from LD surface with the assistance of DGAT1. (3) As the consequence of losing ADRP, LD losses the balance of mobility, moving toward MTOC and nucleus. (4) Clustering of LDs at the peripheral of nucleus enables the contact between LDs and the replication complex of HCV. HCV RNA is delivered from ER-bound replication complexes to NS5A, obtaining access to LD surface, followed by nucleocapsid formation (gray-dashed frame and enlarged panel). (5) The nucleocapsid fuses with VLDL to form viral lipoviroparticle in ER. (B) (1) At the ER–Golgi intermediate compartment (ERGIC), ARF1 and its guanine nucleotide exchange factor (GEF) GBF1 together with COPI deliver ATGL and ADRP from ER export sites (ERES) to the surface of LD. DENV subverts this process for the transportation of capsid protein to LD surface. (2) The accumulation of DENV capsid protein on LDs associates with cellular perilipin 3 and intracellular K+ concentration. (3) Replicated DENV genomes are released through the vesicle pore and then engaged into nucleocapsids that bud through the ER membrane in close proximity. (4) Capsid protein can be released from LDs to the cytosol or other cellular compartments for subsequent viral assembly (gray-dashed frame and enlarged panel). (5) Packed virions accumulate within the lumen of the vesicle packets-containing ER network before transported to Golgi (Boulant et al., 2008; Chatel-Chaix and Bartenschlager, 2014).
LDs have been reported to associate with virus-induced membrane bound compartments believed to be replication sites. Despite belonging to the same family, HCV and DENV induce morphologically distinctive replication compartments. In the case of HCV infection, the double-membrane vesicles (DMVs) are derived from the ER (Romero-Brey et al., 2012; Figure 2A). DMVs are composed of active viral replicase proteins and double-stranded RNA (dsRNA), along with several host components including vesicle-associated membrane protein-associated protein A (VAP-A) and VAP-B that are crucial for viral RNA replication (Evans et al., 2004; Gao et al., 2004). The highly hydrophobic NS4B of HCV, together with NS5A, are the major viral factors that contribute to DMV formation (Lundin et al., 2006). These virus-induced compartments use cholesterol as a structural component and can be visualized in close proximity to LDs (Romero-Brey et al., 2012; Paul et al., 2013). While DMVs are considered as replication factories of HCV, their association to LDs is still unclear. The interferon-induced antiviral protein viperin, which inhibits HCV RNA replication, localizes to LDs using a similar mechanism as HCV NS5A, indicating the importance of LD-NS5A association during HCV replication (Jiang et al., 2008; Hinson and Cresswell, 2009). LDs release free cholesterol from the esterified form for membrane biogenesis as per the host cellular requirements (Maxfield and Tabas, 2005) and, therefore, may serve as reservoirs for lipids required for expanding the intracellular membrane surface to form DMVs (see Association of LDs to Viral Replication Compartments). Besides, HCV replication triggers the activation of the cellular SREBP pathway for de novo synthesis of membrane lipids, which, in turn, regulate biogenesis of LDs (see Manipulation of LD Reserves during Viral life Cycle) (Li et al., 2013). Another possibility is that LDs themselves provide a platform for virus assembly and, therefore, require close proximity to the replication sites for efficient recruitment of newly synthesized viral proteins and subsequent virion packaging (see LDs as a Platform for Virion Assembly) (Miyanari et al., 2007).
Unlike HCV, DENV infection induces formation of single-membrane in-folding into the ER lumen and unstructured convoluted membranes (Welsch et al., 2009; Figure 2B). These DENV-induced vesicle-like structures contain viral replicase and dsRNA. Pore-like openings on these structures enable release of newly synthesized viral RNA, facilitating replication and efficient encapsidation (Welsch et al., 2009). Other flaviviruses, such as WNV and tick-borne encephalitis virus (TBEV) share similar features of intracellular membrane rearrangements (Gillespie et al., 2010; Miorin et al., 2013). DENV replication activates the autophagy pathway to mobilize FFAs from LDs and co-opts FA synthase (FASN). FFAs released from LDs are consumed by oxidation in mitochondria to generate ATP, which is required for viral RNA replication (see Usage of LD as an Energy Reservoir during Viral life Cycle) (Heaton and Randall, 2010). Moreover, DENV NS3 recruits FASN to virus replication sites during membrane remodeling in a Rab18-dependent fashion, engaging both LDs and the viral replication complexes in the process (Heaton et al., 2010; Tang et al., 2014). Regardless of the distinct membrane compartmentalization strategies of HCV and DENV both require close juxtaposition of LDs for energy supply and subsequent virion assembly, as reviewed below.
In the case of HCV infection, after being generated at the ER, the capsid protein localizes to LDs via its domain 2 in a time-dependent manner. They accumulate on discrete regions of LDs before fully covering the surface of LDs (Boulant et al., 2007; Shavinskaya et al., 2007). Host DGAT1 that synthesizes triglycerides stored within LDs, binds to the HCV capsid protein, which in turn acquires access to DGAT1-generated LDs. Viral RNA replication complexes are subsequently recruited to appropriate sites of virus assembly. LD-localized capsid protein provides stability to these structures via interfering with TAG turnover and inducing aggregation of LDs (Boulant et al., 2008; Herker et al., 2010; Harris et al., 2011). Additionally, by replacing LD-localized ADRP, the capsid protein induces imbalance between the minus-end-directed and the plus-end-directed motors, causing movement of LDs on microtubules toward the nucleus so as to enhance interactions between sites of HCV RNA replication and virion assembly (Boulant et al., 2008). The capsid protein recruits viral NS5A, while the N-terminal of NS5A engages viral replication complexes to LD-associated membranes (Boulant et al., 2007; Appel et al., 2008). HCV NS5A also associates with Rab18, a member of the Rab GTPase family that plays an essential role in membrane traffic (Salloum et al., 2013). Rab18 localizes directly to the monolayer surface of LDs in response to lipolytic stimulation (Martin et al., 2005), and facilitates association of NS5A and other replicase components with LDs (Salloum et al., 2013; Figure 2A). HCV infection increases the expression of apolipoprotein J, which further stabilizes LD-associated capsid protein and NS5A, thereby facilitating virion assembly (Lin et al., 2014). Cellular CD2 associated protein (CD2AP) also regulates HCV assembly by interacting with HCV NS5A while modulating LD biogenesis at the same time (Li, 2017). Dissociation of HCV capsid protein from LDs has no effect on viral RNA replication but decreases production of infectious virions, indicating that LDs either directly provide a platform for HCV assembly or facilitate transport of the capsid protein from RNA translation/replication to the assembly sites (Boulant et al., 2007, 2008; Miyanari et al., 2007). Additionally, during chronic HCV infection, LDs in liver tissues increase in number and size, causing pathological accumulation of liver lipids, also known as hepatic steatosis. The interaction between the HCV capsid protein and LDs is critical for this development. An LD membrane protein, perilipin 3, regulates the capsid-induced steatosis, indicating host LD-associated proteins as an effective preventive measure of HCV-induced pathology (Ferguson et al., 2017).
The DENV capsid protein also interacts with LDs but in a mechanistically distinct manner as compared to HCV. DENV capsid protein accumulates on the surface of LDs via its center domain and the N-terminal disordered region (Samsa et al., 2009; Martins et al., 2012). Additionally, the binding between DENV capsid protein and LDs may also be attributed to the association between capsid protein and LD membrane protein perilipin 3 in a potassium ion-dependent fashion. Changing the concentration of potassium ion concentration regulates the binding and release of capsid protein from LDs. This phenomenon indicates that DENV may manipulate specific intracellular ion concentrations to favor viral replication (Carvalho et al., 2012). HCV may use the same potassium ion-dependent strategy to interact with LDs via its p7 and NS5A proteins (Carvalho et al., 2012). Contrary to DGAT1-dependent trafficking to LDs, the DENV capsid protein utilizes host Golgi-specific brefeldin A-resistance guanine nucleotide exchange factor 1 (GBF1)-ARF-COPI pathway to localize to the surface of LDs (Iglesias et al., 2015; Figure 2B). Similar to HCV infection, inhibiting the association between DENV capsid protein and LDs results in attenuated infectious virion production but not viral RNA replication, underscoring the function of LDs as a scaffold for DENV assembly through exposure of the protein cationic surface toward the aqueous environment (Carvalho et al., 2012).
DENV and HCV capsid proteins use distinct mechanisms for LD association. The process by which LDs gain or release viral capsid proteins remain unknown. However, current evidence on the involvement of LDs provides several possible targets for developing antiviral approaches (Table 1) (section Targeting LD Metabolism as Antiviral Strategies).
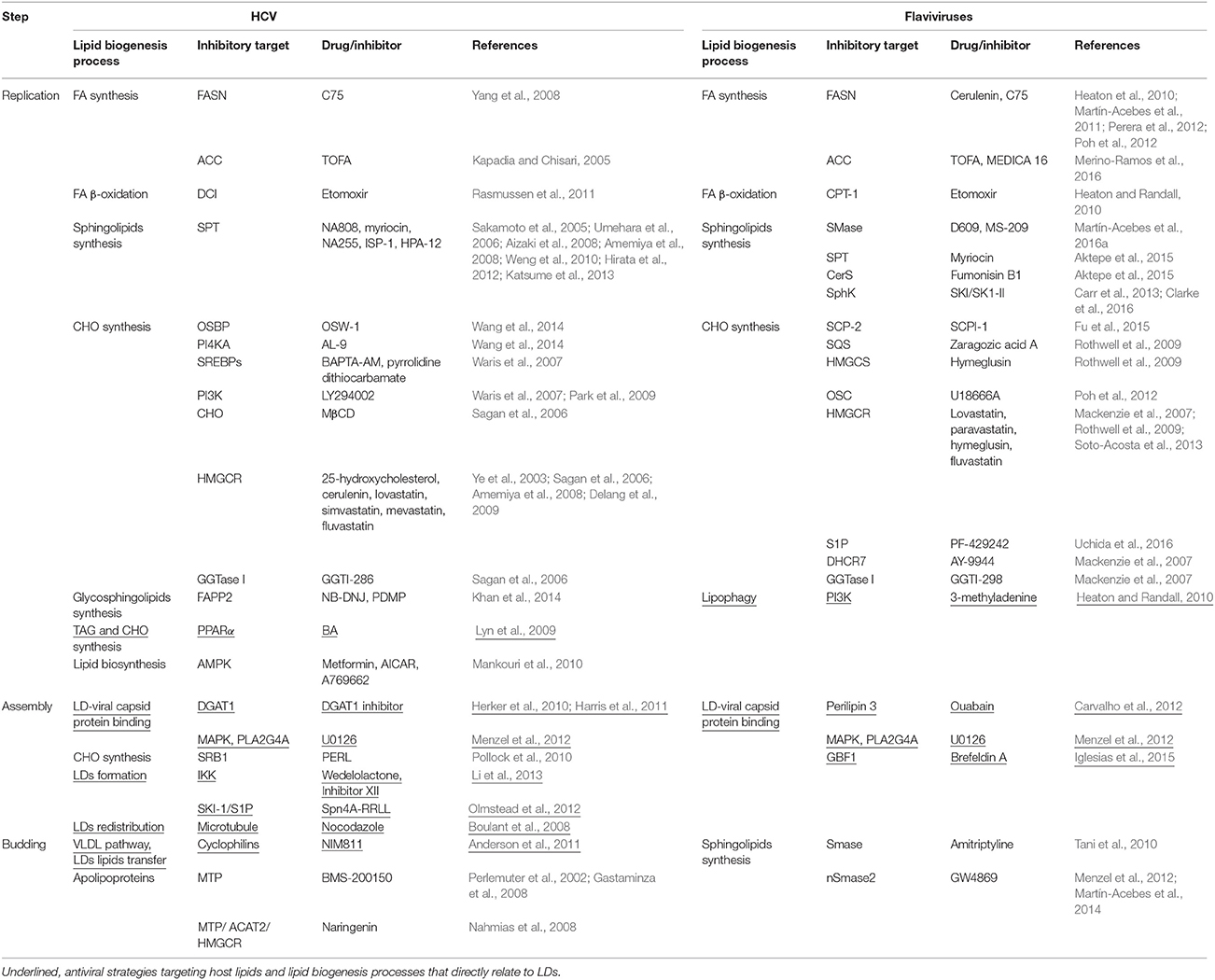
Table 1. Examples antiviral strategies against HCV and flaviviruses interfering with lipid metabolism-related processes.
Replication of the viral genome is an energy-consuming process. In HCV infected cells, cytoplasmic ATP levels decrease dramatically, as a result of active energy consumption. Meanwhile, elevated ATP levels at replication compartments within infected cells have also been reported (Ando et al., 2012). This would involve either incorporation of ATP-generating machinery into the membrane-associated replication site, or transport of ATP though membrane-to-membrane communication between mitochondria and replication compartments (Ando et al., 2012). The C terminus of Flaviviridae NS3 encodes a DExH/D-box RNA helicase that functions to unwind dsRNA molecules through ATP-hydrolysis (Tai et al., 1996; Dumont et al., 2006). Many of the cellular signaling events activated during viral infection are also regulated by ATP levels (Hardie, 2011). Given the highly reduced and hydrophobic lipids at the core, LDs serve as an efficient storage for energy (Walther and Farese, 2012). FA hydrolysis releases 2.5 times more ATPs per gram compared to glucose, which provides a tremendous reservoir for supplying energy during viral replication. Not surprisingly, many other pathogens also manipulate LD metabolism to acquire fuel for replication.
Energy stored in LDs is released through lipolysis. Mobilization of TAG stores from LDs by lipases produces significant amounts of FFAs that can be used in β-oxidation, generating ATP and other intermediates for the cell. In addition to lipolysis, an alternative route through autophagy, commonly referred to as lipophagy, can also take up and deliver LDs to lytic compartments for lipid hydrolysis (see Mobilization of Lipids from LDs) (Wang, 2016).
A model proposed by Randall and Heaton suggested that DENV infection triggers lipophagy to deplete LDs, releasing FFAs. DENV also induces cellular β-oxidation to consume the FFAs released from lipophagy for energy production. Exogenously supplemented FAs can replace the need for lipophagy during DENV replication, suggesting that flaviviruses manipulate cellular lipid metabolism to create an environment that favors virus replication (Heaton and Randall, 2010). Our own data support this model. AUP1, a monotopic membrane protein localized to both LDs and ER membranes, was identified as a key component in DENV biogenesis. Expression of AUP1 was up-regulated during DENV infection and was found to be necessary for virus-triggered lipophagy to proceed (Zhang et al., 2016). The requirement of lipophagy during other flavivirus infections is still to be investigated.
Virus-induced lipophagy for energy production remains unclear in the context of HCV infection. HCV uses membranes of autophagic vacuoles for viral RNA replication. The induction of autophagosomes is nutrient starvation-independent. An impaired autophagy pathway results in attenuated virion production (Dreux et al., 2009; Sir et al., 2012). Proteomic and lipidomic studies showed an up-regulation of lipogenic enzymes and proteins related to β-oxidation, such as 3,2-trans-enoyl-CoA isomerase (DCI) (Diamond et al., 2010). In line with this study, DCI was reported to be essential for productive HCV infection through regulation of mitochondrial FA oxidation (Rasmussen et al., 2011). Another microarray analysis revealed a down-regulation of genes involved in degradation and oxidation of FAs, and an elevation of genes that control metabolism and transport of FAs (Blackham et al., 2010). Although a direct experimental evidence of lipophagy induced by HCV is still missing, data from several indirect sources strongly suggest the utilization of cellular pathways for β-oxidation of FFAs.
Apart from providing FFAs for β-oxidation during Flaviviridae infection, LDs also function as a reservoir for lipids that are essential for viral replication.
Flaviviridae replication organelles consist of FAs, specific phospholipids, sphingolipids, and cholesterol (Heaton et al., 2010; Perera et al., 2012; Paul et al., 2013; Martín-Acebes et al., 2016a). While DENV obtains FAs by breakdown of LDs via lipophagy (Heaton and Randall, 2010), HCV controls the transcriptional induction of lipid biosynthetic and related genes through SREBP signaling (Olmstead et al., 2012). HCV infection activates the SREBP precursor that localizes to the ER, and triggers its trafficking to the Golgi. Thereafter, the SREBP precursor is proteolytically processed by site 1 protease (S1P) and S2P at Golgi, releasing its N-terminal fragment that is transported into the nucleus and initiates transcription of lipogenic factors such as FASN and 3-hydroxy-3-methylglutaryl CoA (HMGCoA). The 3′ untranslated region of the HCV RNA genome with DEAD box polypeptide 3 X-linked (DDX3X) further activates IκB kinase (IKK)-α, which translocates to the nucleus and stimulates SREBP transcriptional activity, thus modulating LD biogenesis (Olmstead et al., 2012; Li et al., 2013).
HCV replication organelles use cholesterol as a structural component (Romero-Brey et al., 2012; Paul et al., 2013). Cellular oxysterol-binding protein (OSBP) and phosphatidylinositol 4-kinases (PI4KA) facilitate trafficking of cholesterol to the HCV-rearranged membrane-like structures during replication, highlighting the need for both factors in supporting HCV replication (Wang et al., 2014). OSBPs are speculated to be sterol carriers and might function to transport sterols out of the ER and incorporate them into LDs in a phosphatidylinositol 4-phosphate (PI(4)P)-dependent manner. Sterols and cholesterol are exchanged by OSBP at the ER-Golgi interface (Mesmin et al., 2013). OSBP-related protein 2 that resides on the surface of LDs may also participate in the process of lipid exchange (Hynynen et al., 2009). Notwithstanding its cellular function, the activity of OSBP appears to be dispensable for DENV replication (Hynynen et al., 2009). DENV replication is regulated by endogenous cholesterol production that is controlled by mevalonate (diphospho) decarboxylase (MVD) and exogenous cholesterol uptake (Rothwell et al., 2009). Similarly, WNV also hijacks cellular cholesterol and redistributes it to viral RNA replication compartments (Mackenzie et al., 2007).
Besides cholesterol, sphingomyelin is another essential membrane component of HCV replication organelles. An active role for sphingolipids in HCV RNA replication has been reported. Sphingomyelin enhances binding of the RNA dependent RNA polymerase NS5B to the template RNA and is therefore important for HCV replication (Weng et al., 2010; Hirata et al., 2012). Expression of genes that encode sphingomyelin synthases 1 and 2 is up-regulated upon HCV infection, resulting in enhanced synthesis of sphingomyelin (Hirata et al., 2012). Dynamic pools of sphingomyelin were observed in LDs, with the high affinity sphingomyelin-binding protein ADRP on the surface of LDs (McIntosh et al., 2010). It is likely that LDs participate in the biogenesis of sphingolipids necessary for HCV replication.
In addition to consumption of lipids that are stored in LDs, HCV can also obstruct the turnover of LDs to establish a microenvironment that is more favorable to viral infection. Release of infectious HCV particles relies on secretion of hepatic very low-density lipoprotein (VLDL)—a TAG-rich lipoprotein. For hijacking VLDL secretion, HCV inhibits the function of the putative TAG lipase, arylacetamide deacetylase (AADAC), thus, further impairing TAG lipolysis (Nourbakhsh et al., 2013). Moreover, HCV capsid protein that localizes to LDs through the activity of DGAT1 (Harris et al., 2011), restrains lipolysis of TAG by interacting with ATGL and its activator comparative gene identification-58 (CGI-58) (Camus et al., 2014).
As with LD association, distinct strategies are employed by HCV and DENV for mobilizing lipids within LDs, hence providing insights into LD catabolism and cellular factors as possible targets (Table 1).
Although viruses of the Flaviviridae family cause severe human diseases, there are currently no clinically approved drugs available for treatment against them, other than for HCV. Historically, the development of antiviral therapy has largely focused on directly targeting viral components involved in multiple stages of the virus life cycle.
Entry of flaviviruses is mediated by fusion of the viral envelope (E) protein with the host membrane. Blocking virus entry via targeting the viral E protein offers a means to suppress the onset of infection. A few heterocyclic compounds, such as compound 6, NITD-448 and P02, have been identified to directly bind to the hydrophobic pocket of viral E protein and block its conformational change, which is essential for virus-host fusion (Modis et al., 2003, 2004; Zhou et al., 2008; Poh et al., 2009; Wang et al., 2009). Due to the multifunctional nature of the E protein, its inhibitors may potentially block multiple steps in the viral life cycle, including entry and virion assembly/maturation. More importantly, these inhibitors can exert their effect through direct binding to virions without the need to cross the hydrophobic membrane bilayer and be delivered into infected cells. However, due to the complexity and high variability of flaviviral E protein, it is challenging to develop pan-serotype inhibitors (Wang and Shi, 2015).
During replication, the viral genome is translated into a single polyprotein which is cleaved into individual proteins by a viral protease complex. Since polyprotein processing is a prerequisite for viral replication and assembly, these virally encoded proteases are one of the most attractive antiviral targets (Chambers et al., 1990, 1993; Luo et al., 2015). Two HCV NS3/4A serine protease inhibitors, boceprevir and telaprevir, have been approved in combination with PEG-interferon plus ribavirin for treatment of chronic HCV genotype 1 (Ghany et al., 2011). Recent study by Shiryaev and colleagues have identified a group of small molecule antiviral inhibitors that interfering with the productive fold of the NS2B cofactor in the two-component protease, inhibit its cleavage activity and therefore suppress ZIKV infection. The most potent inhibitor NSC157058 was shown to inhibit ZIKV infection in both cultured hfNPCs and mice without significant toxicity (Shiryaev et al., 2017). Despite these advances, resistance to protease inhibitors can occur rapidly, especially for chronic infections such as HCV due to the genetic variability of the virus and high mutation rate (Rong et al., 2010; Wu et al., 2013). Another concern in developing protease-based antiviral therapy is toxicity. Similarities in viral and host cellular serine proteases would presumably create problems in specificity while targeting the virus.
The flaviviral NS3 RNA helicase is located adjacent to the C terminal of the NS3 protease (Luo et al., 2008). The RNA helicase is believed to be required for several different functions such as initiation of RNA synthesis, separating dsRNA structures formed during viral RNA synthesis and as a translocase that eliminates proteins bound to the viral RNA (Sampath and Padmanabhan, 2009). Viruses with a mutated NS3 helicase are unable to replicate properly (Matusan et al., 2001). Several RNA helicase inhibitors have been identified. The antiparasitic drug ivermectin was shown to inhibit WNV, YFV, and DENV at submicromolar levels, and a small molecule inhibitor ST-610 was found to potently and selectively inhibit all four serotypes of DENV in vivo (Mastrangelo et al., 2012; Lim et al., 2013). However, due to a lack of specific binding pockets for RNA and NTPs, molecules that target the RNA helicase via these binding sites might also non-selectively bind to other cellular proteins with helicase/NTPase activities, resulting in significant toxicity (Luo et al., 2015).
The NS5 RNA-dependent RNA polymerase (RdRp) is the most conserved amongst the flavivirus proteins, and is essential for viral RNA synthesis. Since host cells lack these enzymes, the specificity makes them one of the most promising and intensively studied classes of antiviral targets. RdRp can be targeted by non-nucleoside inhibitors (NNIs) and nucleoside/nucleotide analog inhibitors (NIs) (Malet et al., 2008). NNIs directly target the binding pocket of the polymerase and block its conformational change from its inactive to active form (Biswal et al., 2005). Although a number of NNI candidates for HCV are under clinical development, there hasn't been any FDA approved NNIs for flaviviruses yet. The major challenge in the use of NNIs in antiviral therapy is the structural variability of the binding pockets across different serotypes or genotypes as well as the resistant mutation in or near the binding pocket which results in resistance to the NNIs (Sofia et al., 2012). NIs have been widely used in clinics for treatment of hepatitis, HIV and herpesvirus infections (Jordheim et al., 2013; Menéndez-Arias et al., 2014). Compared to other classes of inhibitors, NIs have a higher threshold for developing resistance, and a relatively broad-antiviral spectrum due to the relatively conserved polymerase structure (Delang et al., 2011; Lim et al., 2013). Unlike NNIs which directly bind to RNA polymerase, NIs have to convert into its triphosphate form inside cells by host kinases before exerting their antiviral effects (Stein and Moore, 2001). However, the kinase activity varies significantly in different cell types/hosts, causing variable efficacy of the same NI. Another major issue associated with NIs is the unpredictable toxicity in vitro. Although the toxicity of NIs is often associated with the inhibition of mitochondrial polymerases (Arnold et al., 2012), other mitochondrial perturbations may also attribute to toxicity (Selvaraj et al., 2014).
The N-terminal domain of NS5 contains one methyltransferase (MTase) that catalyzes guanine N-7 and ribose 2′-O-methylations using S-adenosylmethionine (SAM) as a methyl donor during viral cap formation (Zhou et al., 2007). Non-selective competitive inhibitors, such as S-adenosylhomocysteine and sinefungin bind to SAM binding sites and inhibit its function (Boldescu et al., 2017). Using virtual screening, a group of small compound molecules have been identified with broad-spectrum activity against the MTase proteins of multiple flaviviruses, including DENV2, DENV3, and YFV (Brecher et al., 2015). Apart from the most important antiviral targets such as E protein, NS3 protease and NS5 polymerase, other viral targets such as capsid protein, NS1 and NS4 proteins are also under evaluation. The details of different viral targets have been reviewed elsewhere (Boldescu et al., 2017; García et al., 2017).
Due to extensive dependence of viruses (replication, assembly, and budding) on host LDs, the interface of virus-host interactions with LDs and/or LD metabolism provides a rich source for potential antiviral interventions (Table 1). First, targeting host factors may produce potential broad-spectrum activity against multiple viral infections due to similar intracellular pathways employed by viruses within the same genus or family. Second, given the high replication and mutation rates of viruses, long-term antiviral therapy against chronic infections inevitably selects for the resistant variants which alter the drug target and therefore are less susceptible to the inhibitory effects of the treatment. The resistant mutants eventually become the dominant species and lead to treatment failure and persistent infection. Development of drug-resistance has become a major challenge with direct-acting antivirals when treating chronic infections (Rong et al., 2010). Unlike viral elements, host cellular factors are much less prone to mutation; thus targeting host lipid metabolism provides an attractive approach for long-term treatment of diseases caused by viral infection. However, since LDs play a role in lipid metabolism in-vivo, manipulating a major metabolic pathway may have a more pleiotropic impact on cellular homeostasis (Georgel et al., 2010). Such consequences need to be carefully assessed to hit the right balance between causing host toxicity while preventing viral pathogenesis. Third, several inhibitors targeting host lipid metabolic pathways are well characterized, which can greatly accelerate the process of drug development. Moreover, targeting specific steps of LD biosynthesis, distribution, trafficking, and metabolism which viruses routinely exploit, allows us to design antiviral strategies with an enhanced therapeutic window. For example, triglyceride-synthesizing enzyme DGAT1 has been identified as an important host factor which is required for trafficking of viral capsid protein to LDs, facilitating early steps of viral assembly. Of note, RNAi-mediated silencing of DGAT1 resulted in impaired viral particle production without affecting LD composition (Herker et al., 2010). Currently, novel classes of pharmacological inhibitors targeting DGAT1 have been developed for clinical applications (DeVita and Pinto, 2013). In addition, regulating enzymes in the FA synthesis pathway has been shown to inhibit production of different viruses. C75, a FA synthase inhibitor, displayed a strong inhibitory effect on HCV replication (Yang et al., 2008), DENV production (Samsa et al., 2009), as well as WNV and YFV replication (Martín-Acebes et al., 2011) without causing significant toxicity to host cells. A series of chemical probes (ML-206, ML-219 and ML-220) has been shown to reduce the biogenesis and consumption of LDs without toxicity to mammalian cells (Boxer et al., 2013). These probes may prove to be beneficial in inhibiting virus production. A noteworthy and indirect strategy to interrupt the association between virus and LDs during viral replication and assembly is to target involved viral proteins. During the biosynthesis of the HCV polyproteins, an internal signal sequence between the capsid protein and envelope protein E1 can be preceded by cellular signal peptide peptidase (SPP). This process releases the capsid protein from the ER, followed by its transport to LDs. SPP inhibitor (Z-LL)2-ketone abolishes the cleavage of capsid protein by SPP and thereby inhibits production of infectious HCV (McLauchlan et al., 2002).
Ideally, antiviral treatments should exert their effects as early as possible after infection. This is particularly true for acute flaviviral infections such as DENV. Targeting intracellular host factors, however, is perhaps less effective in preventing the onset of an infection compared to other inhibitors, which block viral entry. The advantages and disadvantages of antiviral strategies against HCV and flaviviruses by targeting viral components and host factors including those involved in LD metabolism are summarized in Table 2.
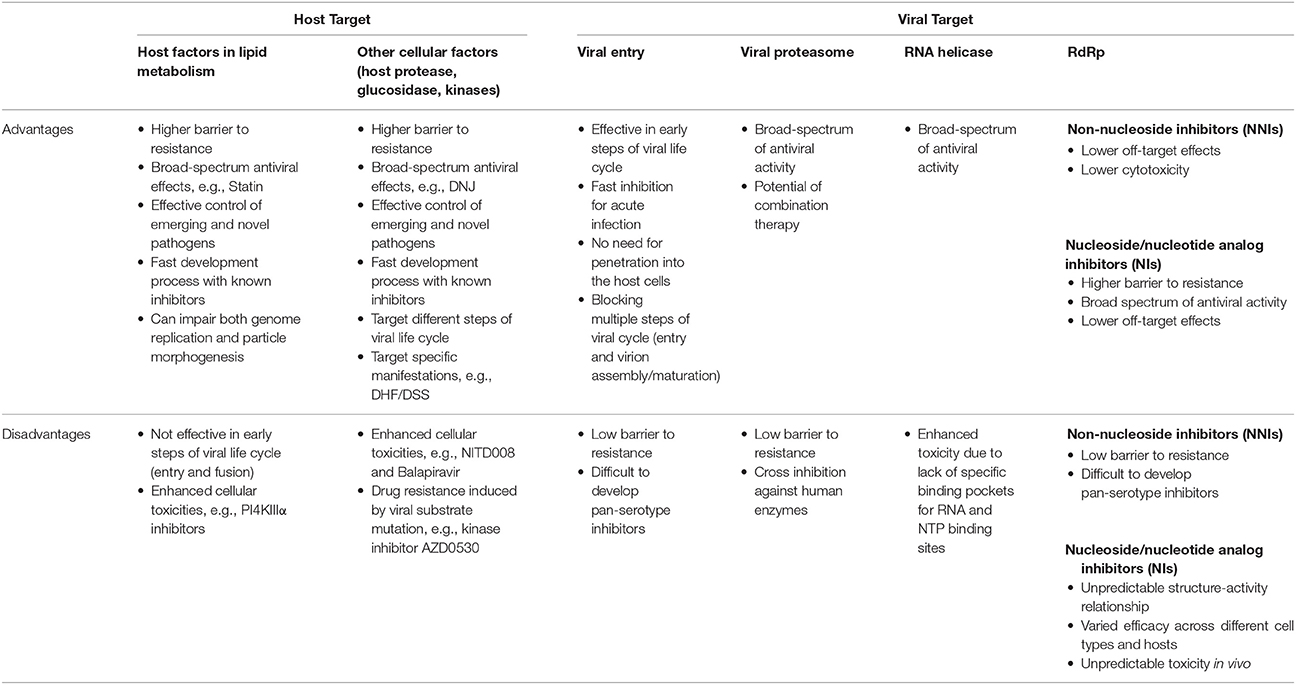
Table 2. Comparison of advantages and disadvantages of different antiviral strategies against HCV and flaviviruses.
Despite being an immense global health problem, there are no affordable and efficient prophylactic or therapeutic treatments for some pathogenic flaviviruses. It is imperative to have alternative therapeutic strategies of inhibiting specific steps in the intracellular virus life cycle to combat infection. Viruses from the Flaviviridae family often cause perturbations in cellular energy and lipid homeostasis during infection. This has been reported for DENV, WNV, and HCV infection. Therefore, targeting cellular LDs offers possibilities for such interventions, including inhibition of lipid metabolism and disruption of interactions with viral components. Although knowledge on the participation of LDs during infection of HCV and flaviviruses has significantly progressed, comparative studies that aim to determine the shared or specific requirements of LD components for these pathogens are still lacking. In addition, much of the information available is from in-vitro studies, while the in-vivo relevance remains unexplored. Therefore, a more comprehensive understanding of the molecular biology of viruses and their dependence on host LD metabolism is of utmost priority for development of broad-spectrum and specific anti-flaviviral strategies.
JZ and YL drafted the manuscript and contributed equally to this work. SS supervised, evaluated, and edited the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was funded by Research Grants Council (GRF grants 17117914 and 17113915), and partially supported by Health and Medical Research Funds (16150592), theme based research grant from the Research Grants Council (Project No. T11-705/14N) and research funds from Institut Pasteur (PTR 546). SS is supported by the Croucher Foundation.
769662, 6,7-Dihydro-4-hydroxy-3-(2′-hydroxy[1,1′-biphenyl]-4-yl)-6-oxo-thieno[2,3-b]pyridine-5-carbonitrile; AADAC, arylacetamide deacetylase; ACAT, acyl-CoA, cholesterol acyltranserases; ACC, acetyl coenzyme a carboxylase; ADRP, adipose differentiation-related protein; AICAR, aminoimidazole carboxamide ribonucleotide; AMPK, 5′ AMP-activated protein kinase; ApoB100, apolipoprotein B100; ARF, ADP-ribosylation factor; ARF1-COP I, ADP-ribosylation factor-coat protein I; ATGL, adipose triglyceride lipase; AUP1, ancient ubiquitous protein 1; AY9944, trans-1,4-bis(2-Chlorobenzylaminoethyl) cyclohexane dihydrochloride; BA, 2-chloro-5-nitro-N-(pyridyl)benzamide; BAPTA-AM, 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester); BMS-200150, 2-[1-(3,3-diphenylpropyl)-4-piperidinyl]-2,3-dihydro-1H-isoindol-1-one; C75, 4-Methylene-2-octyl-5-oxotetrahydrofuran-3-carboxylic acid; CD2AP, CD2 associated protein; CE, cholesterol ester; CerS, ceramide synthase; CGI58, comparative gene identification-58; CHO, cholesterol; CMA, chaperone-mediated autophagy; COP, coat protein; CPT-1, carnitine palmitoyltransferase 1; D609, tricyclodecan-9-yl-xanthogenate; DAG, diacylyglycerol; DCI, 3,2-trans-enoyl-CoA iosmerase; DDX3X, DEAD box polypeptide 3 X-linked; DENV, dengue virus; DGAT, diacylglycerol acyltransferases; DHCR, dehydrocholesterol reductase; DHF, dengue hemorrhagic fever; DMV, double-membrane vesicle; DNJ, 1-deoxynojirimycin; dsRNA, double-stranded RNA; DSS, dengue shock syndrome; E protein, envelope protein; ER, endoplasmic reticulum; ERAD, ER-associated degradation; ERGIC, ER–Golgi intermediate compartment; FAPP2, Golgi-associated four-phosphate adaptor protein 2; FASN, fatty acid synthase; GBF1, golgi-specific brefeldin A-resistance guanine nucleotide exchange factor 1; GGTase I, geranylgeranyltransferase; GGTI-286, N-4-[2(R)-Amino-3-mercaptopropyl]amino-2-phenylbenzoyl-(L)-leucine methyl ester; GGTI-298, N-4-[2(R)-Amino-3-mercaptopropyl]amino-2-naphthylbenzoyl-(L)-leucine methyl ester trifluoroacetate salt; GW4869, N,N′-Bis[4-(4,5-dihydro-1H-imidazol-2-yl)phenyl]-3,3′-p-phenylene-bis-acrylamide dihydrochloride; HCV, hepatitis C virus; HMGCoA, 3-hydroxy-3-methylglutaryl CoA; HMGCR, 3-hydroxy-methyglutaryl-Coenzyme A reductase; HMGCS, hydroxymethylglutaryl-CoA synthase; HSL, hormone-sensitive lipase; IKK, IkB kinase; JEV, Japanese encephalitis virus; LD, lipid droplet; LY294002, 2-(4-Morpholinyl)-8-phenyl-4H-1-benzopyran-4-one; MAPK, mitogen-activated protein kinases; MEDICA 16:3,3,14,14-Tetramethylhexadecanedioic acid; MGL, monoglyceride lipase; MS-209, dofequidar fumarate; MTase, methyltransferase; MTOC, microtubule-organizing center; MTP, microsomal triglyceride transfer protein large subunit; MVD, mevalonate (diphospho) decarboxylase; MβCD, methyl-β-cyclodextrin; NIM811, N-methyl-4-isoleucine cyclosporine; NB-DNJ, N-Butyldeoxynojirimycin; NIs, nucleoside/nucleotide analog inhibitors; NNIs, non-nucleoside inhibitors; NS, non-structural; nSMase2, neutral sphingomyelinase 2; OSBP, oxysterol-binding protein; OSC, oxidosqualene cyclase; OSW-1, 3β,16β,17α-trihydroxycholest-5-en-22-one 16-O-(2-O-4-methoxybenzoyl-β-D-xylopyranosyl)-(1 → 3)-(2-O-acetyl-α-l-arabinopyranoside); PC, phosphatidylcholine; PDMP, D,L-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol; PE, phosphatidylethanolamine; PERL, polyunsaturated ER liposomes; PF-429242, 4-[(Diethylamino)methyl]-N-[2-(2-methoxyphenyl)ethyl]-N-(3R)-3-pyrrolidinyl-benzamide; PI3K, phosphatidylinositide 3-kinases; PI4KA, phosphatidylinositol 4-kinases; PI4P, phosphatidylinositol 4-phosphate; PLA2G4A, cytosolic phospholipase A2; PNPLA5, patatin-like phospholipase domain-containing protein 5; PPARα, peroxisome proliferator-activated receptor α; RdRp, RNA-dependent RNA polymerase; S1P, site 1 protease; SAM, S-adenosylmethionine; SCAP, Sterol regulatory element-binding protein cleavage-activating protein; SCPI-1, [N-(4-{[4-(3,4-dichlorophenyl)-1,3-thiazol-2-yl]amino}-phenyl)acetamidehydrobromide]; SCP-2, sterol carrier protein 2; SKI-1, subtilisin/kexin-isozyme-1; Smase, sphingomyelinase; SphK, sphingosine kinase; SPP, signal peptide peptidase; SPT, serine palmitoyltransferase; SQS, squalene synthetase; SRB1, scavenger receptor class B member 1; SREBP, sterol regulatory element–binding protein; TAG, triacylglycerol; TBEV, tick-borne encephalitis virus; TOFA,5-tetradecyl-oxy-2-furoic acid; U0126, 1,4-Diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene; U18666A, 3β-(2-Diethylaminoethoxy)androst-5-en-17-one; VAP, vesicle-associated membrane protein-associated protein; VLDL, very low-density lipoprotein; WNV, West Nile virus; YFV, yellow fever virus; ZIKV, Zika virus.
Aizaki, H., Morikawa, K., Fukasawa, M., Hara, H., Inoue, Y., Tani, H., et al. (2008). Critical role of virion-associated cholesterol and sphingolipid in hepatitis C virus infection. J. Virol. 82, 5715–5724. doi: 10.1128/JVI.02530-07
Aktepe, T. E., Pham, H., and Mackenzie, J. M. (2015). Differential utilisation of ceramide during replication of the flaviviruses West Nile and dengue virus. Virology 484, 241–250. doi: 10.1016/j.virol.2015.06.015
Amemiya, F., Maekawa, S., Itakura, Y., Kanayama, A., Matsui, A., Takano, S., et al. (2008). Targeting lipid metabolism in the treatment of hepatitis C virus infection. J. Infect. Dis. 197, 361–370. doi: 10.1086/525287
Anderson, L. J., Lin, K., Compton, T., and Wiedmann, B. (2011). Inhibition of cyclophilins alters lipid trafficking and blocks hepatitis C virus secretion. Virol. J. 8:329. doi: 10.1186/1743-422X-8-329
Ando, T., Imamura, H., Suzuki, R., Aizaki, H., Watanabe, T., Wakita, T., et al. (2012). Visualization and measurement of ATP levels in living cells replicating hepatitis C virus genome RNA. PLoS Pathog. 8:e1002561. doi: 10.1371/journal.ppat.1002561
Appel, N., Zayas, M., Miller, S., Krijnse-Locker, J., Schaller, T., Friebe, P., et al. (2008). Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 4:e1000035. doi: 10.1371/journal.ppat.1000035
Arnold, J. J., Sharma, S. D., Feng, J. Y., Ray, A. S., Smidansky, E. D., Kireeva, M. L., et al. (2012). Sensitivity of mitochondrial transcription and resistance of RNA polymerase II dependent nuclear transcription to antiviral ribonucleosides. PLoS Pathog. 8:e1003030. doi: 10.1371/journal.ppat.1003030
Bartenschlager, R., Lohmann, V., and Penin, F. (2013). The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat. Rev. Microbiol. 11, 482–496. doi: 10.1038/nrmicro3046
Biswal, B. K., Cherney, M. M., Wang, M., Chan, L., Yannopoulos, C. G., Bilimoria, D., et al. (2005). Crystal structures of the RNA-dependent RNA polymerase genotype 2a of hepatitis C virus reveal two conformations and suggest mechanisms of inhibition by non-nucleoside inhibitors. J. Biol. Chem. 280, 18202–18210. doi: 10.1074/jbc.M413410200
Blackham, S., Baillie, A., Al-Hababi, F., Remlinger, K., You, S., Hamatake, R., et al. (2010). Gene expression profiling indicates the roles of host oxidative stress, apoptosis, lipid metabolism, and intracellular transport genes in the replication of hepatitis C virus. J. Virol. 84, 5404–5414. doi: 10.1128/JVI.02529-09
Boldescu, V., Behnam, M. A. M., Vasilakis, N., and Klein, C. D. (2017). Broad-spectrum agents for flaviviral infections: dengue, Zika and beyond. Nat. Rev. Drug Discov. 16, 565–586. doi: 10.1038/nrd.2017.33
Boulant, S., Douglas, M. W., Moody, L., Budkowska, A., Targett-Adams, P., and McLauchlan, J. (2008). Hepatitis C virus core protein induces lipid droplet redistribution in a microtubule- and dynein-dependent manner. Traffic 9, 1268–1282. doi: 10.1111/j.1600-0854.2008.00767.x
Boulant, S., Targett-Adams, P., and McLauchlan, J. (2007). Disrupting the association of hepatitis C virus core protein with lipid droplets correlates with a loss in production of infectious virus. J. Gen. Virol. 88(Pt 8), 2204–2213. doi: 10.1099/vir.0.82898-0
Boxer, M. B., Shen, M., Zhang, Y. Q., Liu, L., Auld, D. S., and Beller, M. (2013). Modulators of Lipid Storage. Probe Reports from the NIH Molecular Libraries Program. Bethesda, MD: National Center for Biotechnology Information (US).
Bozza, P. T., Bakker-Abreu, I., Navarro-Xavier, R. A., and Bandeira-Melo, C. (2011). Lipid body function in eicosanoid synthesis: an update. Prostaglandins Leukot. Essent. Fatty Acids 85, 205–213. doi: 10.1016/j.plefa.2011.04.020
Brasaemle, D. L. (2007). Thematic review series: adipocyte biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J. Lipid Res. 48, 2547–2559. doi: 10.1194/jlr.R700014-JLR200
Brecher, M., Chen, H., Liu, B., Banavali, N. K., Jones, S. A., Zhang, J., et al. (2015). Novel broad spectrum inhibitors targeting the flavivirus methyltransferase. PLoS ONE 10:e0130062. doi: 10.1371/journal.pone.0130062
Brown, M. S., and Goldstein, J. L. (1997). The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89, 331–340. doi: 10.1016/S0092-8674(00)80213-5
Buhman, K. K., Chen, H. C., and Farese, R. V. (2001). The enzymes of neutral lipid synthesis. J. Biol. Chem. 276, 40369–40372. doi: 10.1074/jbc.R100050200
Camus, G., Schweiger, M., Herker, E., Harris, C., Kondratowicz, A. S., Tsou, C. L., et al. (2014). The hepatitis C virus core protein inhibits adipose triglyceride lipase (ATGL)-mediated lipid mobilization and enhances the ATGL interaction with comparative gene identification 58 (CGI-58) and lipid droplets. J. Biol. Chem. 289, 35770–35780. doi: 10.1074/jbc.M114.587816
Carr, J. M., Kua, T., Clarke, J. N., Calvert, J. K., Zebol, J. R., Beard, M. R., et al. (2013). Reduced sphingosine kinase 1 activity in dengue virus type-2 infected cells can be mediated by the 3′ untranslated region of dengue virus type-2 RNA. J. Gen. Virol. 94, 2437–2448. doi: 10.1099/vir.0.055616-0
Carvalho, F. A., Carneiro, F. A., Martins, I. C., Assunção-Miranda, I., Faustino, A. F., Pereira, R. M., et al. (2012). Dengue virus capsid protein binding to hepatic lipid droplets (LD) is potassium ion dependent and is mediated by ld surface proteins. J. Virol. 86, 2096–2108. doi: 10.1128/JVI.06796-11
Cermelli, S., Guo, Y., Gross, S. P., and Welte, M. A. (2006). The lipid-droplet proteome reveals that droplets are a protein-storage depot. Curr. Biol. 16, 1783–1795. doi: 10.1016/j.cub.2006.07.062
Chambers, T. J., Nestorowicz, A., Amberg, S. M., and Rice, C. M. (1993). Mutagenesis of the yellow fever virus NS2B protein: effects on proteolytic processing, NS2B-NS3 complex formation, and viral replication. J. Virol. 67, 6797–6807.
Chambers, T. J., Weir, R. C., Grakoui, A., McCourt, D. W., Bazan, J. F., Fletterick, R. J., et al. (1990). Evidence that the N-terminal domain of nonstructural protein NS3 from yellow fever virus is a serine protease responsible for site-specific cleavages in the viral polyprotein. Proc. Natl. Acad. Sci. U.S.A. 87, 8898–8902. doi: 10.1073/pnas.87.22.8898
Chatel-Chaix, L., and Bartenschlager, R. (2014). Dengue virus- and hepatitis C virus-induced replication and assembly compartments: the enemy inside–caught in the web. J. Virol. 88, 5907–5911. doi: 10.1128/JVI.03404-13
Cheng, J., Ohsaki, Y., Tauchi-Sato, K., Fujita, A., and Fujimoto, T. (2006). Cholesterol depletion induces autophagy. Biochem. Biophys. Res. Commun. 351, 246–252. doi: 10.1016/j.bbrc.2006.10.042
Clarke, J. N., Davies, L. K., Calvert, J. K., Gliddon, B. L., Al Shujari, W. H., Aloia, A. L., et al. (2016). Reduction in sphingosine kinase 1 influences the susceptibility to dengue virus infection by altering antiviral responses. J. Gen. Virol. 97, 95–109. doi: 10.1099/jgv.0.000334
Delang, L., Paeshuyse, J., Vliegen, I., Leyssen, P., Obeid, S., Durantel, D., et al. (2009). Statins potentiate the in vitro anti-hepatitis C virus activity of selective hepatitis C virus inhibitors and delay or prevent resistance development. Hepatology 50, 6–16. doi: 10.1002/hep.22916
Delang, L., Vliegen, I., Froeyen, M., and Neyts, J. (2011). Comparative study of the genetic barriers and pathways towards resistance of selective inhibitors of hepatitis C virus replication. Antimicrob. Agents Chemother. 55, 4103–4113. doi: 10.1128/AAC.00294-11
DeVita, R. J., and Pinto, S. (2013). Current status of the research and development of diacylglycerol O-acyltransferase 1 (DGAT1) inhibitors: miniperspective. J. Med. Chem. 56, 9820–9825. doi: 10.1021/jm4007033
Diamond, D. L., Syder, A. J., Jacobs, J. M., Sorensen, C. M., Walters, K. A., Proll, S. C., et al. (2010). Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 6:e1000719. doi: 10.1371/journal.ppat.1000719
Dreux, M., Gastaminza, P., Wieland, S. F., and Chisari, F. V. (2009). The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. U.S.A. 106, 14046–14051. doi: 10.1073/pnas.0907344106
Dugail, I., and Hajduch, E. (2007). A new look at adipocyte lipid droplets: towards a role in the sensing of triacylglycerol stores. Cell. Mol. Life Sci. 64, 2452–2458. doi: 10.1007/s00018-007-7277-6
Dumont, S., Cheng, W., Serebrov, V., Beran, R. K., Tinoco, I. Jr., Pyle, A. M., et al. (2006). RNA translocation and unwinding mechanism of HCV NS3 helicase and its coordination by ATP. Nature 439, 105–108. doi: 10.1038/nature04331
Evans, M. J., Rice, C. M., and Goff, S. P. (2004). Phosphorylation of hepatitis C virus nonstructural protein 5A modulates its protein interactions and viral RNA replication. Proc. Natl. Acad. Sci. U.S.A. 101, 13038–13043. doi: 10.1073/pnas.0405152101
Ferguson, D., Zhang, J., Davis, M. A., Helsley, R. N., Vedin, L. L., Lee, R. G., et al. (2017). The lipid droplet-associated protein perilipin 3 facilitates hepatitis C virus-driven hepatic steatosis. J. Lipid Res. 58, 420–432. doi: 10.1194/jlr.M073734
Fu, Q., Inankur, B., Yin, J., Striker, R., and Lan, Q. (2015). Sterol carrier protein 2, a critical host factor for dengue virus infection, alters the cholesterol distribution in mosquito Aag2 cells. J. Med. Entomol. 52, 1124–1134. doi: 10.1093/jme/tjv101
Fujimoto, T., and Parton, R. G. (2011). Not just fat: the structure and function of the lipid droplet. Cold Spring Harb. Perspect. Biol. 3:a004838. doi: 10.1101/cshperspect.a004838
Gao, L., Aizaki, H., He, J. W., and Lai, M. M. (2004). Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J. Virol. 78, 3480–3488. doi: 10.1128/JVI.78.7.3480-3488.2004
Gao, Q., and Goodman, J. M. (2015). The lipid droplet—a well-connected organelle. Front. Cell Dev. Biol. 3:49. doi: 10.3389/fcell.2015.00049
García, L. L., Padilla, L., and Castano, J. C. (2017). Inhibitors compounds of the flavivirus replication process. Virol. J. 14:95. doi: 10.1186/s12985-017-0761-1
Garske, T., Van Kerkhove, M. D., Yactayo, S., Ronveaux, O., Lewis, R. F., Staples, J. E., et al. (2014). Yellow Fever in Africa: estimating the burden of disease and impact of mass vaccination from outbreak and serological data. PLoS Med. 11:e1001638. doi: 10.1371/journal.pmed.1001638
Gastaminza, P., Cheng, G., Wieland, S., Zhong, J., Liao, W., and Chisari, F. V. (2008). Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 82, 2120–2129. doi: 10.1128/JVI.02053-07
Georgel, P., Schuster, C., Zeisel, M. B., Stoll-Keller, F., Berg, T., Bahram, S., et al. (2010). Virus–host interactions in hepatitis C virus infection: implications for molecular pathogenesis and antiviral strategies. Trends Mol. Med. 16, 277–286. doi: 10.1016/j.molmed.2010.04.003
Ghany, M. G., Nelson, D. R., Strader, D. B., Thomas, D. L., and Seeff, L. B. (2011). An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 54, 1433–1444. doi: 10.1002/hep.24641
Gillespie, L. K., Hoenen, A., Morgan, G., and Mackenzie, J. M. (2010). The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J. Virol. 84, 10438–10447. doi: 10.1128/JVI.00986-10
Gould, E. A., and Solomon, T. (2008). Pathogenic flaviviruses. Lancet 371, 500–509. doi: 10.1016/S0140-6736(08)60238-X
Guo, Y., Walther, T. C., Rao, M., Stuurman, N., Goshima, G., Terayama, K., et al. (2008). Functional genomic screen reveals genes involved in lipid-droplet formation and utilization. Nature 453, 657–661. doi: 10.1038/nature06928
Hardie, D. G. (2011). AMP-activated protein kinase-an energy sensor that regulates all aspects of cell function. Genes Dev. 25, 1895–1908. doi: 10.1101/gad.17420111
Harris, C., Herker, E., Farese, R. V. Jr., and Ott, M. (2011). Hepatitis C virus core protein decreases lipid droplet turnover: a mechanism for core-induced steatosis. J. Biol. Chem. 286, 42615–42625. doi: 10.1074/jbc.M111.285148
Hartman, I. Z., Liu, P., Zehmer, J. K., Luby-Phelps, K., Jo, Y., Anderson, R. G., et al. (2010). Sterol-induced dislocation of HMG CoA reductase from ER membranes into the cytosol through a subcellular compartment resembling lipid droplets. J Biol Chem. 110:134213. doi: 10.1074/jbc.M110.134213
Heaton, N. S., Perera, R., Berger, K. L., Khadka, S., Lacount, D. J., Kuhn, R. J., et al. (2010). Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc. Natl. Acad. Sci. U.S.A. 107, 17345–17350. doi: 10.1073/pnas.1010811107
Heaton, N. S., and Randall, G. (2010). Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 8, 422–432. doi: 10.1016/j.chom.2010.10.006
Herker, E., Harris, C., Hernandez, C., Carpentier, A., Kaehlcke, K., Rosenberg, A. R., et al. (2010). Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat. Med. 16, 1295–1298. doi: 10.1038/nm.2238
Hinson, E. R., and Cresswell, P. (2009). The antiviral protein, viperin, localizes to lipid droplets via its N-terminal amphipathic alpha-helix. Proc. Natl. Acad. Sci. U.S.A. 106, 20452–20457. doi: 10.1073/pnas.0911679106
Hirata, Y., Ikeda, K., Sudoh, M., Tokunaga, Y., Suzuki, A., Weng, L., et al. (2012). Self-enhancement of hepatitis C virus replication by promotion of specific sphingolipid biosynthesis. PLoS Pathog. 8:e1002860. doi: 10.1371/journal.ppat.1002860
Hynynen, R., Suchanek, M., Spandl, J., Bäck, N., Thiele, C., and Olkkonen, V. M. (2009). OSBP-related protein 2 is a sterol receptor on lipid droplets that regulates the metabolism of neutral lipids. J. Lipid Res. 50, 1305–1315. doi: 10.1194/jlr.M800661-JLR200
Iglesias, N. G., Mondotte, J. A., Byk, L. A., De Maio, F. A., Samsa, M. M., Alvarez, C., et al. (2015). dengue virus uses a non-canonical function of the host GBF1-Arf-COPI system for capsid protein accumulation on lipid droplets. Traffic 16, 962–977. doi: 10.1111/tra.12305
Jiang, D., Guo, H., Xu, C., Chang, J., Gu, B., Wang, L., et al. (2008). Identification of three interferon-inducible cellular enzymes that inhibit the replication of hepatitis C virus. J. Virol. 82, 1665–1678. doi: 10.1128/JVI.02113-07
Jo, Y., Hartman, I. Z., and DeBose-Boyd, R. A. (2013). Ancient ubiquitous protein-1 mediates sterol-induced ubiquitination of 3-hydroxy-3-methylglutaryl CoA reductase in lipid droplet–associated endoplasmic reticulum membranes. Mol. Biol. Cell 24, 169–183. doi: 10.1091/mbc.E12-07-0564
Jordheim, L. P., Durantel, D., Zoulim, F., and Dumontet, C. (2013). Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 12, 447–464. doi: 10.1038/nrd4010
Kapadia, S. B., and Chisari, F. V. (2005). Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc. Natl. Acad. Sci. U.S.A. 102, 2561–2566. doi: 10.1073/pnas.0409834102
Karlsson, M., Contreras, J. A., Hellman, U., Tornqvist, H., and Holm, C. (1997). cDNA cloning, tissue distribution, and identification of the catalytic triad of monoglyceride lipase evolutionary relationship to esterases, lysophospholipases, and haloperoxidases. J. Biol. Chem. 272, 27218–27223. doi: 10.1074/jbc.272.43.27218
Kassan, A., Herms, A., Fernández-Vidal, A., Bosch, M., Schieber, N. L., Reddy, B. J., et al. (2013). Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in ER microdomains. J. Cell Biol. 203, 985–1001. doi: 10.1083/jcb.201305142
Katsume, A., Tokunaga, Y., Hirata, Y., Munakata, T., Saito, M., Hayashi, H., et al. (2013). A serine palmitoyltransferase inhibitor blocks hepatitis C virus replication in human hepatocytes. Gastroenterology 145, 865–873. doi: 10.1053/j.gastro.2013.06.012
Kaushik, S., and Cuervo, A. M. (2015). Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol. 17, 759–770. doi: 10.1038/ncb3166
Kaushik, S., Rodriguez-Navarro, J. A., Arias, E., Kiffin, R., Sahu, S., Schwartz, G. J., et al. (2011). Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab. 14, 173–183. doi: 10.1016/j.cmet.2011.06.008
Khan, I., Katikaneni, D. S., Han, Q., Sanchez-Felipe, L., Hanada, K., Ambrose, R. L., et al. (2014). Modulation of hepatitis C virus genome replication by glycosphingolipids and four-phosphate adaptor protein 2. J. Virol. 88, 12276–12295. doi: 10.1128/JVI.00970-14
Lass, A., Zimmermann, R., Oberer, M., and Zechner, R. (2011). Lipolysis–a highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog. Lipid Res. 50, 14–27. doi: 10.1016/j.plipres.2010.10.004
Li, C. Y. (2017). CD2AP facilitates HCV production by targeting NS5A to lipid droplets and regulating lipid droplet biogenesis. FASEB J. 31(1 Suppl. 184.181). doi: 10.1096/fj.1530-6860
Li, Q., Pène, V., Krishnamurthy, S., Cha, H., and Liang, T. J. (2013). Hepatitis C virus infection activates an innate pathway involving IKK-alpha in lipogenesis and viral assembly. Nat. Med. 19, 722–729. doi: 10.1038/nm.3190
Lim, S. P., Wang, Q. Y., Noble, C. G., Chen, Y. L., Dong, H., Zou, B., et al. (2013). Ten years of dengue drug discovery: progress and prospects. Antiviral Res. 100, 500–519. doi: 10.1016/j.antiviral.2013.09.013
Lin, C. C., Tsai, P., Sun, H. Y., Hsu, M. C., Lee, J. C., Wu, I. C., et al. (2014). Apolipoprotein J, a glucose-upregulated molecular chaperone, stabilizes core and NS5A to promote infectious hepatitis C virus virion production. J. Hepatol. 61, 984–993. doi: 10.1016/j.jhep.2014.06.026
Lindenbach, B. D., Thiel, H.-J., and Rice, C. M. (2007). “Flaviviridae: the viruses and their replication,” in Fields Virology, 5th Edn., eds D. M. Knipe and P. M. Howley (Philadelphia, PA: Lippincott-Raven Publishers), 1101–1152.
Lohmann, D., Spandl, J., Stevanovic, A., Schoene, M., Philippou-Massier, J., and Thiele, C. (2013). Monoubiquitination of ancient ubiquitous protein 1 promotes lipid droplet clustering. PLoS ONE 8:e72453. doi: 10.1371/journal.pone.0072453
Lundin, M., Lindström, H., Grönwall, C., and Persson, M. A. (2006). Dual topology of the processed hepatitis C virus protein NS4B is influenced by the NS5A protein. J. Gen. Virol. 87(Pt 11), 3263–3272. doi: 10.1099/vir.0.82211-0
Luo, D., Vasudevan, S. G., and Lescar, J. (2015). The flavivirus NS2B-NS3 protease-helicase as a target for antiviral drug development. Antiviral Res. 118, 148–158. doi: 10.1016/j.antiviral.2015.03.014
Luo, D., Xu, T., Hunke, C., Grüber, G., Vasudevan, S. G., and Lescar, J. (2008). Crystal structure of the NS3 protease-helicase from dengue virus. J. Virol. 82, 173–183. doi: 10.1128/JVI.01788-07
Lyn, R. K., Kennedy, D. C., Sagan, S. M., Blais, D. R., Rouleau, Y., Pegoraro, A. F., et al. (2009). Direct imaging of the disruption of hepatitis C virus replication complexes by inhibitors of lipid metabolism. Virology 394, 130–142. doi: 10.1016/j.virol.2009.08.022
Mackenzie, J. M., Khromykh, A. A., and Parton, R. G. (2007). Cholesterol manipulation by west nile virus perturbs the cellular immune response. Cell Host Microbe 2, 229–239. doi: 10.1016/j.chom.2007.09.003
Malet, H., Massé, N., Selisko, B., Romette, J. L., Alvarez, K., Guillemot, J. C., et al. (2008). The flavivirus polymerase as a target for drug discovery. Antiviral Res. 80, 23–35. doi: 10.1016/j.antiviral.2008.06.007
Mankouri, J., Tedbury, P. R., Gretton, S., Hughes, M. E., Griffin, S. D., Dallas, M. L., et al. (2010). Enhanced hepatitis C virus genome replication and lipid accumulation mediated by inhibition of AMP-activated protein kinase. Proc. Natl. Acad. Sci. U.S.A. 107, 11549–11554. doi: 10.1073/pnas.0912426107
Martin, S., Driessen, K., Nixon, S. J., Zerial, M., and Parton, R. G. (2005). Regulated localization of Rab18 to lipid droplets: effects of lipolytic stimulation and inhibition of lipid droplet catabolism. J. Biol. Chem. 280, 42325–42335. doi: 10.1074/jbc.M506651200
Martín-Acebes, M. A., Blázquez, A. B., Jiménez de Oya, N., Escribano-Romero, E., and Saiz, J. C. (2011). West Nile virus replication requires fatty acid synthesis but is independent on phosphatidylinositol-4-phosphate lipids. PLoS ONE 6:e24970. doi: 10.1371/journal.pone.0024970
Martín-Acebes, M. A., Gabandé-Rodríguez, E., García-Cabrero, A. M., Sánchez, M. P., Ledesma, M. D., Sobrino, F., et al. (2016a). Host sphingomyelin increases West Nile virus infection in vivo. J. Lipid Res. 57, 422–432. doi: 10.1194/jlr.M064212
Martín-Acebes, M. A., Merino-Ramos, T., Blázquez, A. B., Casas, J., Escribano-Romero, E., Sobrino, F., et al. (2014). The composition of West Nile virus lipid envelope unveils a role of sphingolipid metabolism in flavivirus biogenesis. J. Virol. 88, 12041–12054. doi: 10.1128/JVI.02061-14
Martín-Acebes, M. A., Vázquez-Calvo, Á., and Saiz, J. C. (2016b). Lipids and flaviviruses, present and future perspectives for the control of dengue, Zika, and West Nile viruses. Prog. Lipid Res. 64, 123–137. doi: 10.1016/j.plipres.2016.09.005
Martinez-Vicente, M., Talloczy, Z., Wong, E., Tang, G., Koga, H., Kaushik, S., et al. (2010). Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat. Neurosci. 13, 567–576. doi: 10.1038/nn.2528
Martins, I. C., Gomes-Neto, F., Faustino, A. F., Carvalho, F. A., Carneiro, F. A., Bozza, P. T., et al. (2012). The disordered N-terminal region of dengue virus capsid protein contains a lipid-droplet-binding motif. Biochem. J. 444, 405–415. doi: 10.1042/BJ20112219
Mastrangelo, E., Pezzullo, M., De Burghgraeve, T., Kaptein, S., Pastorino, B., Dallmeier, K., et al. (2012). Ivermectin is a potent inhibitor of flavivirus replication specifically targeting NS3 helicase activity: new prospects for an old drug. J. Antimicrob. Chemother. 67, 1884–1894. doi: 10.1093/jac/dks147
Matusan, A. E., Pryor, M. J., Davidson, A. D., and Wright, P. J. (2001). Mutagenesis of the Dengue virus type 2 NS3 protein within and outside helicase motifs: effects on enzyme activity and virus replication. J. Virol. 75, 9633–9643. doi: 10.1128/JVI.75.20.9633-9643.2001
Maxfield, F. R., and Tabas, I. (2005). Role of cholesterol and lipid organization in disease. Nature 438, 612–621. doi: 10.1038/nature04399
McIntosh, A. L., Storey, S. M., and Atshaves, B. P. (2010). Intracellular lipid droplets contain dynamic pools of sphingomyelin: ADRP binds phospholipids with high affinity. Lipids 45, 465–477. doi: 10.1007/s11745-010-3424-1
McLauchlan, J., Lemberg, M. K., Hope, G., and Martoglio, B. (2002). Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. Embo J. 21, 3980–3988. doi: 10.1093/emboj/cdf414
Menéndez-Arias, L., Álvarez, M., and Pacheco, B. (2014). Nucleoside/nucleotide analog inhibitors of hepatitis B virus polymerase: mechanism of action and resistance. Curr. Opin. Virol. 8, 1–9. doi: 10.1016/j.coviro.2014.04.005
Menzel, N., Fischl, W., Hueging, K., Bankwitz, D., Frentzen, A., Haid, S., et al. (2012). MAP-kinase regulated cytosolic phospholipase A2 activity is essential for production of infectious hepatitis C virus particles. PLoS Pathog. 8:e1002829. doi: 10.1371/journal.ppat.1002829
Merino-Ramos, T., Vázquez-Calvo, Á., Casas, J., Sobrino, F., Saiz, J. C., and Martín-Acebes, M. A. (2016). Modification of the Host cell lipid metabolism induced by hypolipidemic drugs targeting the acetyl coenzyme A carboxylase impairs west nile virus replication. Antimicrob. Agents Chemother. 60, 307–315. doi: 10.1128/AAC.01578-15
Mesmin, B., Bigay, J., Moser von Filseck, J., Lacas-Gervais, S., Drin, G., and Antonny, B. (2013). A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell 155, 830–843. doi: 10.1016/j.cell.2013.09.056
Miorin, L., Romero-Brey, I., Maiuri, P., Hoppe, S., Krijnse-Locker, J., Bartenschlager, R., et al. (2013). Three-dimensional architecture of tick-borne encephalitis virus replication sites and trafficking of the replicated RNA. J. Virol. 87, 6469–6481. doi: 10.1128/JVI.03456-12
Miyanari, Y., Atsuzawa, K., Usuda, N., Watashi, K., Hishiki, T., Zayas, M., et al. (2007). The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9, 1089–1097. doi: 10.1038/ncb1631
Mizushima, N. (2007). Autophagy: process and function. Genes Dev. 21, 2861–2873. doi: 10.1101/gad.1599207
Modis, Y., Ogata, S., Clements, D., and Harrison, S. C. (2003). A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 100, 6986–6991. doi: 10.1073/pnas.0832193100
Modis, Y., Ogata, S., Clements, D., and Harrison, S. C. (2004). Structure of the dengue virus envelope protein after membrane fusion. Nature 427, 313–319. doi: 10.1038/nature02165
Nahmias, Y., Goldwasser, J., Casali, M., van Poll, D., Wakita, T., Chung, R. T., et al. (2008). Apolipoprotein B-dependent hepatitis C virus secretion is inhibited by the grapefruit flavonoid naringenin. Hepatology 47, 1437–1445. doi: 10.1002/hep.22197
Narabayashi, K., Ito, Y., Eid, N., Maemura, K., Inoue, T., Takeuchi, T., et al. (2015). Indomethacin suppresses LAMP-2 expression and induces lipophagy and lipoapoptosis in rat enterocytes via the ER stress pathway. J. Gastroenterol. 50, 541–554. doi: 10.1007/s00535-014-0995-2
Nourbakhsh, M., Douglas, D. N., Pu, C. H., Lewis, J. T., Kawahara, T., Lisboa, L. F., et al. (2013). Arylacetamide deacetylase: a novel host factor with important roles in the lipolysis of cellular triacylglycerol stores, VLDL assembly and HCV production. J. Hepatol. 59, 336–343. doi: 10.1016/j.jhep.2013.03.022
Ohsaki, Y., Cheng, J., Fujita, A., Tokumoto, T., and Fujimoto, T. (2006). Cytoplasmic lipid droplets are sites of convergence of proteasomal and autophagic degradation of apolipoprotein B. Mol. Biol. Cell 17, 2674–2683. doi: 10.1091/mbc.E05-07-0659
Ohsaki, Y., Cheng, J., Suzuki, M., Fujita, A., and Fujimoto, T. (2008). Lipid droplets are arrested in the ER membrane by tight binding of lipidated apolipoprotein B-100. J. Cell Sci. 121, 2415–2422. doi: 10.1242/jcs.025452
Olmstead, A. D., Knecht, W., Lazarov, I., Dixit, S. B., and Jean, F. (2012). Human subtilase SKI-1/S1P is a master regulator of the hcv lifecycle and a potential host cell target for developing indirect-acting antiviral agents. PLoS Pathog. 8:e1002468. doi: 10.1371/journal.ppat.1002468
Ouimet, M., and Marcel, Y. L. (2012). Regulation of lipid droplet cholesterol efflux from macrophage foam cells. Arterioscler. Thromb. Vasc. Biol. 32, 575–581. doi: 10.1161/ATVBAHA.111.240705
Park, C. Y., Jun, H. J., Wakita, T., Cheong, J. H., and Hwang, S. B. (2009). Hepatitis C virus nonstructural 4B protein modulates sterol regulatory element-binding protein signaling via the AKT pathway. J. Biol. Chem. 284, 9237–9246. doi: 10.1074/jbc.M808773200
Paul, D., and Bartenschlager, R. (2015). Flaviviridae replication organelles: oh, what a tangled web we weave. Annu. Rev. Virol. 2, 289–310. doi: 10.1146/annurev-virology-100114-055007
Paul, D., Hoppe, S., Saher, G., Krijnse-Locker, J., and Bartenschlager, R. (2013). Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J. Virol. 87, 10612–10627. doi: 10.1128/JVI.01370-13
Paul, D., Madan, V., and Bartenschlager, R. (2014). Hepatitis C virus RNA replication and assembly: living on the fat of the land. Cell Host Microbe 16, 569–579. doi: 10.1016/j.chom.2014.10.008
Perera, R., Riley, C., Isaac, G., Hopf-Jannasch, A. S., Moore, R. J., Weitz, K. W., et al. (2012). Dengue virus infection perturbs lipid homeostasis in infected mosquito cells. PLoS Pathog. 8:e1002584. doi: 10.1371/journal.ppat.1002584
Perlemuter, G., Sabile, A., Letteron, P., Vona, G., Topilco, A., Chrétien, Y., et al. (2002). Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: a model of viral-related steatosis. FASEB J. 16, 185–194. doi: 10.1096/fj.01-0396com
Petersen, L. R., Jamieson, D. J., and Honein, M. A. (2016). Zika virus. N. Engl. J. Med. 375, 294–295. doi: 10.1056/NEJMra1602113
Poh, M. K., Shui, G., Xie, X., Shi, P. Y., Wenk, M. R., and Gu, F. (2012). U18666A, an intra-cellular cholesterol transport inhibitor, inhibits dengue virus entry and replication. Antiviral Res. 93, 191–198. doi: 10.1016/j.antiviral.2011.11.014
Poh, M. K., Yip, A., Zhang, S., Priestle, J. P., Ma, N. L., Smit, J. M., et al. (2009). A small molecule fusion inhibitor of dengue virus. Antiviral Res. 84, 260–266. doi: 10.1016/j.antiviral.2009.09.011
Pol, A., Gross, S. P., and Parton, R. G. (2014). Biogenesis of the multifunctional lipid droplet: lipids, proteins, and sites. J. Cell Biol. 204, 635–646. doi: 10.1083/jcb.201311051
Pol, A., Martin, S., Fernandez, M. A., Ferguson, C., Carozzi, A., Luetterforst, R., et al. (2004). Dynamic and regulated association of caveolin with lipid bodies: modulation of lipid body motility and function by a dominant negative mutant. Mol. Biol. Cell 15, 99–110. doi: 10.1091/mbc.E03-06-0368
Pollock, S., Nichita, N. B., Böhmer, A., Radulescu, C., Dwek, R. A., and Zitzmann, N. (2010). Polyunsaturated liposomes are antiviral against hepatitis B and C viruses and HIV by decreasing cholesterol levels in infected cells. Proc. Natl. Acad. Sci. U.S.A. 107, 17176–17181. doi: 10.1073/pnas.1009445107
Rambold, A. S., Cohen, S., and Lippincott-Schwartz, J. (2015). Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 32, 678–692. doi: 10.1016/j.devcel.2015.01.029
Rasmussen, A. L., Diamond, D. L., McDermott, J. E., Gao, X., Metz, T. O., Matzke, M. M., et al. (2011). Systems virology identifies a mitochondrial fatty acid oxidation enzyme, dodecenoyl coenzyme A delta isomerase, required for hepatitis C virus replication and likely pathogenesis. J. Virol. 85, 11646–11654. doi: 10.1128/JVI.05605-11
Romero-Brey, I., Merz, A., Chiramel, A., Lee, J. Y., Chlanda, P., Haselman, U., et al. (2012). Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 8:e1003056. doi: 10.1371/journal.ppat.1003056
Rong, L., Dahari, H., Ribeiro, R. M., and Perelson, A. S. (2010). Rapid emergence of protease inhibitor resistance in hepatitis C virus. Sci. Transl. Med. 2:30ra32. doi: 10.1126/scitranslmed.3000544
Rothwell, C., Lebreton, A., Young Ng, C., Lim, J. Y., Liu, W., Vasudevan, S., et al. (2009). Cholesterol biosynthesis modulation regulates dengue viral replication. Virology 389, 8–19. doi: 10.1016/j.virol.2009.03.025
Sagan, S. M., Rouleau, Y., Leggiadro, C., Supekova, L., Schultz, P. G., Su, A. I., et al. (2006). The influence of cholesterol and lipid metabolism on host cell structure and hepatitis C virus replication. Biochem. Cell Biol. 84, 67–79. doi: 10.1139/o05-149
Saka, H. A., and Valdivia, R. (2012). Emerging roles for lipid droplets in immunity and host-pathogen interactions. Annu. Rev. Cell Dev. Biol. 28, 411–437. doi: 10.1146/annurev-cellbio-092910-153958
Sakamoto, H., Okamoto, K., Aoki, M., Kato, H., Katsume, A., Ohta, A., et al. (2005). Host sphingolipid biosynthesis as a target for hepatitis C virus therapy. Nat. Chem. Biol. 1, 333–337. doi: 10.1038/nchembio742
Salloum, S., Wang, H., Ferguson, C., Parton, R. G., and Tai, A. W. (2013). Rab18 binds to hepatitis C virus NS5A and promotes interaction between sites of viral replication and lipid droplets. PLoS Pathog. 9:e1003513. doi: 10.1371/journal.ppat.1003513
Samaan, Z., McDermid Vaz, S., Bawor, M., Potter, T. H., Eskandarian, S., and Loeb, M. (2016). Neuropsychological impact of West Nile virus infection: an extensive neuropsychiatric assessment of 49 cases in Canada. PLoS ONE 11:e0158364. doi: 10.1371/journal.pone.0158364
Sampath, A., and Padmanabhan, R. (2009). Molecular targets for flavivirus drug discovery. Antiviral Res. 81, 6–15. doi: 10.1016/j.antiviral.2008.08.004
Samsa, M. M., Mondotte, J. A., Iglesias, N. G., Assunçã, I., Barbosa-Lima, G., Da Poian, A. T., et al. (2009). Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 5:e1000632. doi: 10.1371/journal.ppat.1000632
Sarkari, N. B., Thacker, A. K., Barthwal, S. P., Mishra, V. K., Prapann, S., Srivastava, D., et al. (2012). Japanese encephalitis (JE). Part I: clinical profile of 1,282 adult acute cases of four epidemics. J. Neurol. 259, 47–57. doi: 10.1007/s00415-011-6118-6
Selvaraj, S., Ghebremichael, M., Li, M., Foli, Y., Langs-Barlow, A., Ogbuagu, A., et al. (2014). Antiretroviral therapy-induced mitochondrial toxicity: potential mechanisms beyond polymerase-gamma inhibition. Clin. Pharmacol. Ther. 96, 110–120. doi: 10.1038/clpt.2014.64
Shavinskaya, A., Boulant, S., Penin, F., McLauchlan, J., and Bartenschlager, R. (2007). The lipid droplet binding domain of hepatitis C virus core protein is a major determinant for efficient virus assembly. J. Biol. Chem. 282, 37158–37169. doi: 10.1074/jbc.M707329200
Shiryaev, S. A., Farhy, C., Pinto, A., Huang, C. T., Simonetti, N., Ngono, A. E., et al. (2017). Characterization of the Zika virus two-component NS2B-NS3 protease and structure-assisted identification of allosteric small-molecule antagonists. Antiviral Res. 143, 218–229. doi: 10.1016/j.antiviral.2017.04.015
Singh, R., and Cuervo, A. M. (2012). Lipophagy: connecting autophagy and lipid metabolism. Int. J. Cell Biol. 2012:282041. doi: 10.1155/2012/282041
Singh, R., Kaushik, S., Wang, Y., Xiang, Y., Novak, I., Komatsu, M., et al. (2009). Autophagy regulates lipid metabolism. Nature 458, 1131–1135. doi: 10.1038/nature07976
Siqueira, J. B. Jr., Martelli, C. M., Coelho, G. E., Simplicio, A. C., and Hatch, D. L. (2005). Dengue and dengue hemorrhagic fever, Brazil, 1981-2002. Emerg. Infect. Dis. 11, 48–53. doi: 10.3201/eid1101.031091
Sir, D., Kuo, C. F., Tian, Y., Liu, H. M., Huang, E. J., Jung, J. U., et al. (2012). Replication of hepatitis C virus RNA on autophagosomal membranes. J. Biol. Chem. 287, 18036–18043. doi: 10.1074/jbc.M111.320085
Sofia, M. J., Chang, W., Furman, P. A., Mosley, R. T., and Ross, B. S. (2012). Nucleoside, nucleotide, and non-nucleoside inhibitors of hepatitis C virus NS5B RNA-dependent RNA-polymerase. J. Med. Chem. 55, 2481–2531. doi: 10.1021/jm201384j
Soni, K. G., Mardones, G. A., Sougrat, R., Smirnova, E., Jackson, C. L., and Bonifacino, J. S. (2009). Coatomer-dependent protein delivery to lipid droplets. J. Cell Sci. 122, 1834–1841. doi: 10.1242/jcs.045849
Soto-Acosta, R., Mosso, C., Cervantes-Salazar, M., Puerta-Guardo, H., Medina, F., Favari, L., et al. (2013). The increase in cholesterol levels at early stages after dengue virus infection. correlates with an augment in LDL particle uptake and HMG-CoA reductase activity. Virology 442, 132–147. doi: 10.1016/j.virol.2013.04.003
Spandl, J., Lohmann, D., Kuerschner, L., Moessinger, C., and Thiele, C. (2011). Ancient ubiquitous protein 1 (AUP1) localizes to lipid droplets and binds the E2 ubiquitin conjugase G2 (Ube2g2) via its G2 binding region. J. Biol. Chem. 286, 5599–5606. doi: 10.1074/jbc.M110.190785
Stein, D. S., and Moore, K. H. (2001). Phosphorylation of nucleoside analog antiretrovirals: a review for clinicians. Pharmacotherapy 21, 11–34. doi: 10.1592/phco.21.1.11.34439
Sztalryd, C., and Kimmel, A. R. (2014). Perilipins: lipid droplet coat proteins adapted for tissue-specific energy storage and utilization, and lipid cytoprotection. Biochimie 96, 96–101. doi: 10.1016/j.biochi.2013.08.026
Tai, C. L., Chi, W. K., Chen, D. S., and Hwang, L. H. (1996). The helicase activity associated with hepatitis C virus nonstructural protein 3 (NS3). J. Virol. 70, 8477–8484.
Tang, W. C., Lin, R. J., Liao, C. L., and Lin, Y. L. (2014). Rab18 facilitates dengue virus infection by targeting fatty acid synthase to sites of viral replication. J. Virol. 88, 6793–6804. doi: 10.1128/JVI.00045-14
Tani, H., Shiokawa, M., Kaname, Y., Kambara, H., Mori, Y., Abe, T., et al. (2010). Involvement of ceramide in the propagation of japanese encephalitis virus. J. Virol. 84, 2798–2807. doi: 10.1128/JVI.02499-09
Tauchi-Sato, K., Ozeki, S., Houjou, T., Taguchi, R., and Fujimoto, T. (2002). The surface of lipid droplets is a phospholipid monolayer with a unique fatty acid composition. J. Biol. Chem. 277, 44507–44512. doi: 10.1074/jbc.M207712200
Thanachartwet, V., Oer-Areemitr, N., Chamnanchanunt, S., Sahassananda, D., Jittmittraphap, A., Suwannakudt, P., et al. (2015). Identification of clinical factors associated with severe dengue among Thai adults: a prospective study. BMC Infect. Dis. 15:420. doi: 10.1186/s12879-015-1150-2
Thiam, A. R., Farese, R. V. Jr., and Walther, T. C. (2013). The biophysics and cell biology of lipid droplets. Nat. Rev. Mol. Cell Biol. 14, 775–786. doi: 10.1038/nrm3699
Uchida, L., Urata, S., Ulanday, G. E., Takamatsu, Y., Yasuda, J., Morita, K., et al. (2016). Suppressive effects of the Site 1 Protease (S1P) inhibitor, PF-429242, on dengue virus propagation. Viruses 8:e46. doi: 10.3390/v8020046
Umehara, T., Sudoh, M., Yasui, F., Matsuda, C., Hayashi, Y., Chayama, K., et al. (2006). Serine palmitoyltransferase inhibitor suppresses HCV replication in a mouse model. Biochem. Biophys. Res. Commun. 346, 67–73. doi: 10.1016/j.bbrc.2006.05.085
Velázquez, A. P., Tatsuta, T., Ghillebert, R., Drescher, I., and Graef, M. (2016). Lipid droplet-mediated ER homeostasis regulates autophagy and cell survival during starvation. J. Cell Biol. 212, 621–631. doi: 10.1083/jcb.201508102
Velikkakath, A. K., Nishimura, T., Oita, E., Ishihara, N., and Mizushima, N. (2012). Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol. Biol. Cell 23, 896–909. doi: 10.1091/mbc.E11-09-0785
Walther, T. C., and Farese, R. V. Jr. (2012). Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 81, 687–714. doi: 10.1146/annurev-biochem-061009-102430
Wang, C. W. (2016). Lipid droplets, lipophagy, and beyond. Biochim. Biophys. Acta 1861(8 Pt B), 793–805. doi: 10.1016/j.bbalip.2015.12.010
Wang, H., Perry, J. W., Lauring, A. S., Neddermann, P., De Francesco, R., and Tai, A. W. (2014). Oxysterol-binding protein is a phosphatidylinositol 4-kinase effector required for HCV replication membrane integrity and cholesterol trafficking. Gastroenterology 146, 1373–1385. doi: 10.1053/j.gastro.2014.02.002
Wang, Q. Y., Patel, S. J., Vangrevelinghe, E., Xu, H. Y., Rao, R., Jaber, D., et al. (2009). A small-molecule dengue virus entry inhibitor. Antimicrob. Agents Chemother. 53, 1823–1831. doi: 10.1128/AAC.01148-08
Wang, Q. Y., and Shi, P. Y. (2015). Flavivirus entry inhibitors. ACS Infect. Dis. 1, 428–434. doi: 10.1021/acsinfecdis.5b00066
Waris, G., Felmlee, D. J., Negro, F., and Siddiqui, A. (2007). Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J. Virol. 81, 8122–8130. doi: 10.1128/JVI.00125-07
Welsch, S., Miller, S., Romero-Brey, I., Merz, A., Bleck, C. K., Walther, P., et al. (2009). Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 5, 365–375. doi: 10.1016/j.chom.2009.03.007
Welte, M. A. (2015). Expanding roles for lipid droplets. Curr. Biol. 25, R470–R481. doi: 10.1016/j.cub.2015.04.004
Weng, L. Y., Hirata, Y., Arai, M., Kohara, M., Wakita, T., Watashi, K., et al. (2010). Sphingomyelin activates hepatitis C virus RNA polymerase in a genotype-specific manner. J. Virol. 84, 11761–11770. doi: 10.1128/JVI.00638-10
Wilfling, F., Haas, J. T., Walther, T. C., and Farese, R. V. Jr. (2014a). Lipid droplet biogenesis. Curr. Opin. Cell Biol. 29, 39–45. doi: 10.1016/j.ceb.2014.03.008
Wilfling, F., Thiam, A. R., Olarte, M. J., Wang, J., Beck, R., Gould, T. J., et al. (2014b). Arf1/COPI machinery acts directly on lipid droplets and enables their connection to the ER for protein targeting. Elife 3:e01607. doi: 10.7554/eLife.01607
Wilfling, F., Wang, H., Haas, J. T., Krahmer, N., Gould, T. J., Uchida, A., et al. (2013). Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev. Cell 24, 384–399. doi: 10.1016/j.devcel.2013.01.013
Wu, S., Kanda, T., Nakamoto, S., Imazeki, F., and Yokosuka, O. (2013). Hepatitis C virus protease inhibitor-resistance mutations: our experience and review. World J. Gastroenterol. 19, 8940–8948. doi: 10.3748/wjg.v19.i47.8940
Yang, T., Espenshade, P. J., Wright, M. E., Yabe, D., Gong, Y., Aebersold, R., et al. (2002). Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 110, 489–500. doi: 10.1016/S0092-8674(02)00872-3
Yang, W., Hood, B. L., Chadwick, S. L., Liu, S., Watkins, S. C., Luo, G., et al. (2008). Fatty acid synthase is up-regulated during hepatitis C virus infection and regulates hepatitis C virus entry and production. Hepatology 48, 1396–1403. doi: 10.1002/hep.22508
Ye, J., Wang, C., Sumpter, R., Brown, M. S., Goldstein, J. L., and Gale, M. (2003). Disruption of hepatitis C virus RNA replication through inhibition of host protein geranylgeranylation. Proc. Natl. Acad. Sci. U.S.A. 100, 15865–15870. doi: 10.1073/pnas.2237238100
Yoshimori, T. (2004). Autophagy: a regulated bulk degradation process inside cells. Biochem. Biophys. Res. Commun. 313, 453–458. doi: 10.1016/j.bbrc.2003.07.023
Zhang, J. S., Lamers, M. M., Li, M. Y., Angyal, D., Ashour, J., and Sanyal, S. (2016). “Lipid droplet associated membrane protein Aup1 facilitates dengue virus release through induction of lipophagy,” in 12th GERLI Lipidomics meeting “Microbe and host lipids” (Toulouse).
Zhou, Y., Ray, D., Zhao, Y., Dong, H., Ren, S., Li, Z., et al. (2007). Structure and function of flavivirus NS5 methyltransferase. J. Virol. 81, 3891–3903. doi: 10.1128/JVI.02704-06
Zhou, Z., Khaliq, M., Suk, J. E., Patkar, C., Li, L., Kuhn, R. J., et al. (2008). Antiviral compounds discovered by virtual screening of small-molecule libraries against dengue virus E protein. ACS Chem. Biol. 3, 765–775. doi: 10.1021/cb800176t
Keywords: lipid droplet, lipid metabolism, HCV, flavivirus, dengue
Citation: Zhang J, Lan Y and Sanyal S (2017) Modulation of Lipid Droplet Metabolism—A Potential Target for Therapeutic Intervention in Flaviviridae Infections. Front. Microbiol. 8:2286. doi: 10.3389/fmicb.2017.02286
Received: 28 September 2017; Accepted: 06 November 2017;
Published: 28 November 2017.
Edited by:
Wesley H. Brooks, University of South Florida, United StatesReviewed by:
Abdulsamie Hanano, Atomic Energy Commission of Syria, SyriaCopyright © 2017 Zhang, Lan and Sanyal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sumana Sanyal, c2FueWFsQGhrdS5oaw==
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.