 Cristina Romero-López
Cristina Romero-López Alfredo Berzal-Herranz
Alfredo Berzal-Herranz
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 31 October 2017
Sec. Virology
Volume 8 - 2017 | https://doi.org/10.3389/fmicb.2017.02093
This article is part of the Research Topic Structural Motifs and Biological Functions of RNA Viruses View all 23 articles
Viral genomes are complexly folded entities that carry all the information required for the infective cycle. The nucleotide sequence of the RNA virus genome encodes proteins and functional information contained in discrete, highly conserved structural units. These so-called functional RNA domains play essential roles in the progression of infection, which requires their preservation from one generation to the next. Numerous functional RNA domains exist in the genome of the hepatitis C virus (HCV). Among them, the 5BSL3.2 domain in the cis-acting replication element (CRE) at the 3′ end of the viral open reading frame has become of particular interest given its role in HCV RNA replication and as a regulator of viral protein synthesis. These functionalities are achieved via the establishment of a complex network of long-distance RNA–RNA contacts involving (at least as known to date) the highly conserved 3′X tail, the apical loop of domain IIId in the internal ribosome entry site, and/or the so-called Alt region upstream of the CRE. Changing contacts promotes the execution of different stages of the viral cycle. The 5BSL3.2 domain thus operates at the core of a system that governs the progression of HCV infection. This review summarizes our knowledge of the long-range RNA–RNA interaction network in the HCV genome, with special attention paid to the structural and functional consequences derived from the establishment of different contacts. The potential implications of such interactions in switching between the different stages of the viral cycle are discussed.
Predicting how RNA virus populations might evolve – a major public health goal – is a challenging task. RNA viruses, such as hepatitis C virus (HCV), replicate by virtue of a relatively low fidelity viral genome-encoded RNA-dependent RNA polymerase. It has been calculated that for each round of replication the mutation rate in HCV reaches a maximum of 1.3 nucleotides (nt) (Bartenschlager and Lohmann, 2000; Cuevas et al., 2009; Ribeiro et al., 2012). The variation introduced generates a dynamic genome pool composed of different but closely related sequences, referred to as a quasispecies (Martell et al., 1992). Quasispecies dynamics involve the constant sampling and selection of mutations that improve viral fitness. Such mutations are critical for escaping the host immune response, for drug resistance, the infection of new host species, and disease emergence. The acquisition of new mutations by viral genomes therefore provides a background for the action of natural selection. The magnitude of the effect of the acquired mutations is dependent on the environment: changes can be favorable for the infection in a given host but may have important fitness costs in a different environment, even within the same infected individual. This phenomenon creates a complex picture for the prediction of the disease severity and impedes the development of novel therapies (Manrubia and Lazaro, 2016; Perales and Domingo, 2016).
The balance between sequence promiscuity and functional conservation in viral genomes is made possible via the use of highly structured genomic regions that can absorb nucleotide variations – as long as these do not alter their active conformation. These are organized as discrete, in cis-functional RNA domains that operate in an interconnected manner to perform functions essential to the execution of the viral cycle (Romero-López and Berzal-Herranz, 2013). Such domains are considered information-carrying units beyond the nucleotide sequence. The study of the cis-acting functional elements, including their localization, sequence and structural conservation, is key to understanding viral infections at the molecular level. In addition, the essential role of genomic functional RNA domains in viral propagation and persistence makes them potential therapeutic targets.
Hepatitis C virus infection, which has a global prevalence of 2.8% (more than 185 million people are infected worldwide; Mohd Hanafiah et al., 2013), causes severe chronic disease that in many patients may eventually make liver transplantation necessary (Hoofnagle, 2002). Currently, the standard of care (SOC) involves the use of pegylated α-interferon combined with the modified nucleosides ribavirin and sofosbuvir (Kowdley et al., 2013; Lawitz et al., 2013). Unfortunately, interferon is not always well tolerated; in addition, the variability of the viral genome prevents a sustained therapeutic response. As mutants resistant to the current SOC arise, new targets and therapeutic agents must be ready. In this context, recent advances in the development of direct antiviral agents (DAAs) have prompted the progress of a new combined therapy consisting in the use of sofosbuvir plus the NS5A inhibitor velpatasvir and the NS3-NS4A inhibitor voxilaprevir (see below) (Bourliere et al., 2017). The two major benefits of this new strategy rely, on one hand, in the lack of α-interferon in this drug cocktail, which significantly reduces the secondary effects of the treatment; on the other, both velpatasvir and voxilaprevir operate as pangenotypic inhibitors, thus simplifying the customization of the therapy.
Hepatitis C virus is a member of the family Flaviviridae and the genus Hepacivirus. Based on the phylogenetic analysis of genomic sequences, up to seven genotypes have been described for HCV showing more than 30% divergence at the nucleotide level. Closely related sets of subtypes and isolates have been identified as well (Smith et al., 2014). The viral genome is a positive RNA molecule of ∼9.6 kb that encodes a single ORF flanked by highly conserved untranslated regions (5′ and 3′UTR; Figure 1) (Choo et al., 1989). Viral proteins are produced as a single polypeptide that is co- and post-translationally processed by viral and cellular proteases to yield structural (core, E1, and E2) and non-structural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B; Figure 1) (Grakoui et al., 1993). It is noteworthy that genetic variation does not occur evenly over the viral genome. On average, the complete sequence genome differs by about 31–33% per position (Smith et al., 2014), but only ∼10% of the entire viral genome is susceptible to positive selection (Patino-Galindo and Gonzalez-Candelas, 2017). The affected regions are intimately related to genotype prevalence (Messina et al., 2015). The 5′ end of the HCV genome and the viral capsid-encoding sequence are the most conserved regions, with around 80–90% sequence identity. The hypervariable region and the envelope protein-encoding sequence show the least sequence identity among isolates (Figure 1) (Le Guillou-Guillemette et al., 2007). These observations agree with the existence of highly conserved structural units throughout the HCV genome (Fricke et al., 2015; Mauger et al., 2015; Pirakitikulr et al., 2016). Some of these operate as essential functional domains – key targets for the development of new therapeutics and diagnostic agents. This review provides a brief overview of the cis-acting signals critical for HCV infection, with special emphasis on the core partner, i.e., the so-called 5BSL3.2 domain at the 3′ end of the viral ORF. It also focuses on the sequences and structural units of the viral genome essential to the preservation of the roles of 5BSL3.2 during the infective cycle.
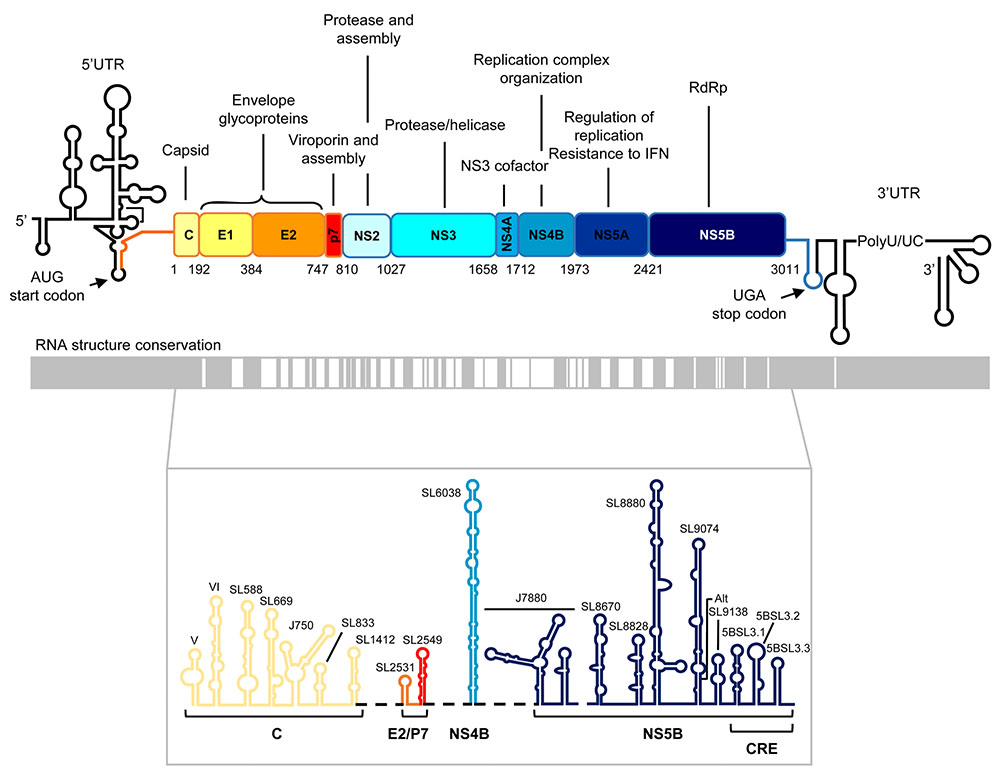
FIGURE 1. Genetic organization and functional RNA domains in the hepatitis C virus (HCV) genome. Upper panel: Diagram showing the genetic organization of the viral genome with the highly structured 5′ and 3′ untranslated regions flanking the single open reading frame (ORF). Viral structural and non-structural (NS) proteins and their functions are indicated. The translation start and stop codons are marked by arrows. Numbering corresponds to codon positions in the ORF according to the HCV Con1 isolate, genotype 1b. Middle panel: Structural conservation map of HCV RNA. Gray boxes denote those structurally conserved regions among different viral isolates. Lower panel: Conserved secondary structural elements in the ORF of the viral RNA genome. Stem-loop structures names are shown. CRE, cis-acting replication element, located at the 3′ end of the ORF. It consists of three highly conserved stem-loops, 5BSL3.1, 5BSL3.2, and 5BSL3.3. Color code and labels at the bottom indicate the position where each stem-loop is located.
The HCV 5′UTR occupies the first 341 nucleotides of the HCV genome (Figure 2). This is one of the most conserved genomic regions, with ∼85% of sequence identity preserved across viral isolates (Bukh et al., 1992; Smith et al., 1995). Importantly, HCV protein synthesis and replication are governed by functional domains in the 5′UTR.
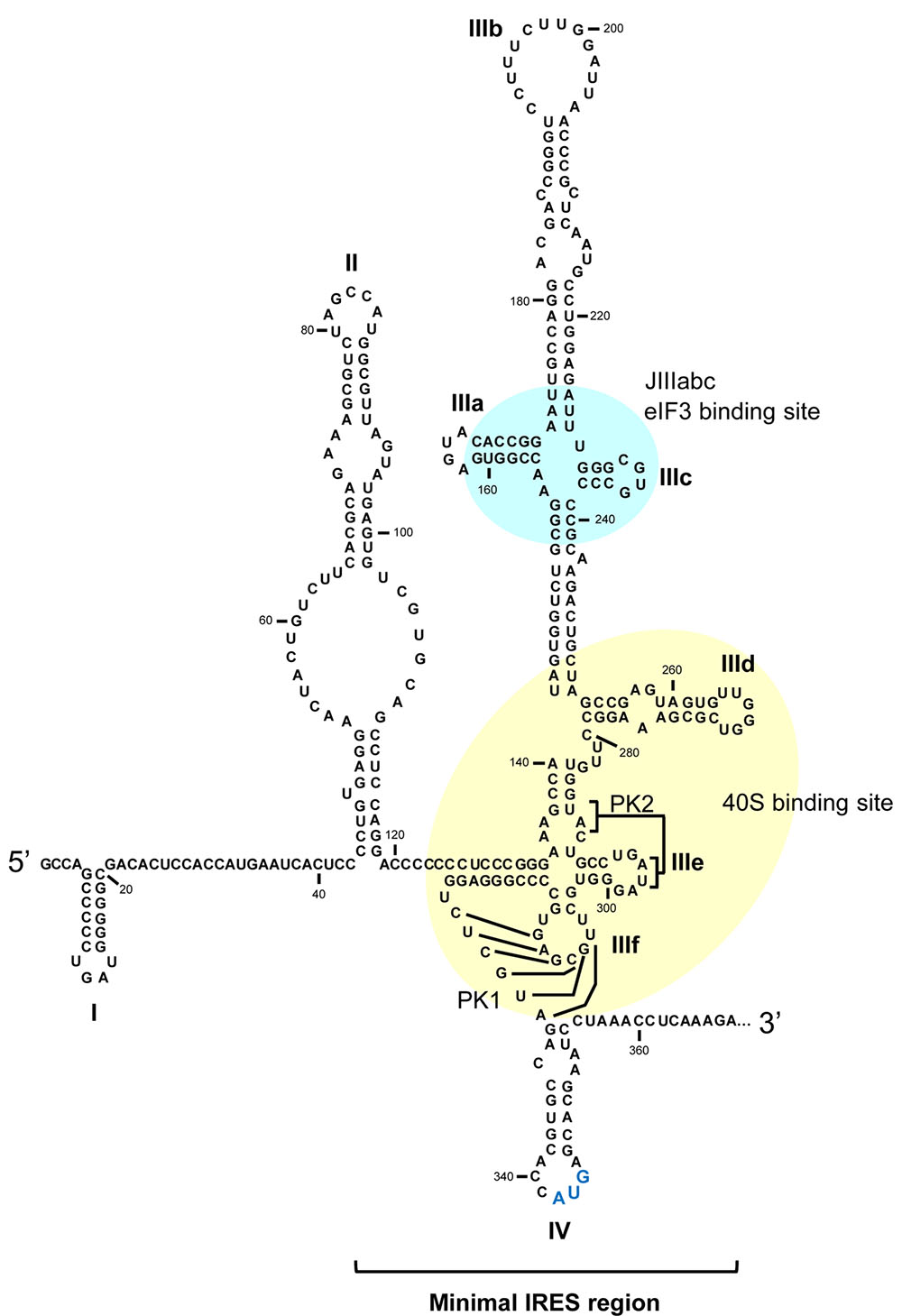
FIGURE 2. The HCV IRES region. Secondary structure of the 5′UTR in the HCV genome including the minimal internal ribosome entry site (IRES). Domains involved in the interaction with eIF3 and the 40S ribosomal subunit are marked in cyan and yellow, respectively. The translation start codon is shown in enlarged blue lettering. PK, pseudoknot. Numbering corresponds to nucleotide positions of HCV Con1 isolate, genotype 1b.
Viral translation is initiated by a mechanism different to the canonical cap-dependent pathway that dictates most cellular protein production. In HCV, an internal ribosome entry site (IRES), in the absence of any other factor, can direct the recruitment of the 40S ribosomal subunit (Tsukiyama-Kohara et al., 1992; Wang et al., 1993; Pestova et al., 1998; Lytle et al., 2002). The 48S particle is then constituted via interaction with the eIF3 and eIF2/GTP/Met-tRNA ternary complex (Sizova et al., 1998; Otto et al., 2002; Berry et al., 2011; Sun et al., 2013). The three-dimensional structure of the 48S complex allows for the proper positioning of the translation start codon in the P site (Berry et al., 2010, 2011; Quade et al., 2015; Angulo et al., 2016). Viral protein synthesis initiates after the binding of the 60S particle, plus the eIF5 and eIF5B factors, to the 48S complex, thus forming the productive 80S ribosomal complex (Yamamoto et al., 2014). This mechanism can be simplified by bypassing the recruitment of eIF3 and eIF2. In fact, eIF3-IRES binding is not essential for the initiation of viral translation per se. A recent publication has shown that this binding displaces the eIF3 from the canonical 43S pre-initiation translation complex, thus releasing the IRES binding site in the 40S ribosomal subunit and promoting the initiation of protein synthesis (Hashem et al., 2013). In addition, under certain stress conditions during which eIF2 is inactivated by phosphorylation, the HCV IRES can circumvent the eIF2-dependent delivery of the tRNA-Met by the GTPase-activating protein eIF5 (Terenin et al., 2008), or perhaps eIF2A (Kim et al., 2011) or 2D (Dmitriev et al., 2010). This strategy imitates the assembly of the translation machinery seen in prokaryotic cells.
The different proposed mechanisms provide important insights in the translation initiation pathway mediated by the HCV IRES (and other related viral and cellular IRESs). However, several questions remained unclear during past years. In a recent work (Jaafar et al., 2016), new roles for eIF1A were discovered, which may help to illustrate and complete the real stepwise pathway during HCV IRES-dependent translation initiation. The findings include the stabilization of the Met-tRNA binding to the IRES-40S pre-initiation complex, the help in the discrimination of improper AUG start codons and the stimulation of the eIF5B-dependent GTP hydrolysis. The authors proposed a revisited IRES-driven translation initiation model by which the HCV IRES would bind to a pre-assembled translation initiation complex, which is a remainder from the translation termination and ribosomal recycling events. This complex encompasses the 40S subunit, eIF3 and eIF1A. In the absence of the ternary complex eIF2/GTP/Met-tRNA, the IRES can easily dock to occupy the decoding groove, in good agreement with previous reports (Quade et al., 2015). In this step, the IRES can displace the eIF3 from its position in the 40S subunit (Hashem et al., 2013). The conformation of this new complex favors the recruitment of the Met-tRNA, in a process that should not necessarily require eIF2. This would be followed by GTP hydrolysis and the release of eIF2. Then, eIF1A and eIF5 could work together to confirm the use of the functional AUG codon, with the subsequent 60S ribosomal subunit binding. Both the model proposed by Jaafar et al. (2016) and the previous simplified version are supported by experimental data and can overlap to provide a wide view of the potential of the IRES to accomplish the complex task of translation initiation under adverse cellular conditions.
The extreme simplification of the initiation translation mechanism shown by the HCV IRES, compared to the canonical mechanism, is achieved through its complex, high-order structure, in which the canonical protein factors are substituted by functional RNA domains present in the viral genome. The minimal IRES region encompasses most of the 5′UTR and spans to nucleotide 372 within the coding sequence (Figure 2) (Reynolds et al., 1995; Honda et al., 1996b). Under physiological magnesium conditions, the IRES folds autonomously into three major domains – II, III, and IV – defined by simple or branched stem-loops (Figure 2) (Kieft et al., 1999; Perard et al., 2013). Far from being a rigid entity, the HCV IRES is an articulated region in which different domains move collectively to achieve the initiation of translation (Perard et al., 2013). Domains II and III appear aligned at both sides of a double pseudoknot motif (PK1 and PK2; Figure 2) (Berry et al., 2010), which guides the correct positioning of the translation start codon (domain IV) into the P site (Berry et al., 2011). Domain II is mostly involved in the constitution of the pre-initiation 48S complex and in inducing changes in the conformation of the 40S ribosomal subunit to promote the first round of ribosomal translocation (Spahn et al., 2001; Filbin and Kieft, 2011; Filbin et al., 2013; Yamamoto et al., 2015).
The highly branched domain III bears critical partners for the execution of viral protein synthesis. A collection of three- and four-way junctions organize different stem-loops (designated subdomains IIIa to IIIf; Figure 2), which operate as recruiting platforms for the binding of eIF3 (in the JIIIabc junction) (Lukavsky et al., 2000) or the 40S ribosomal subunit (mainly anchored in the critical subdomain IIId; Figure 2) (Jubin et al., 2000; Kolupaeva et al., 2000; Babaylova et al., 2009).
In addition to the well-known IRES-protein interactions, three GGG residues within the essential apical loop of subdomain IIId specifically recognize (via canonical Watson-Crick base-pairing) the highly conserved CCC triplet in structural element helix 26 of the rRNA 18S (Malygin et al., 2013a; Matsuda and Mauro, 2014). This interaction favors efficient and stable HCV IRES-40S binding, and seems to be specific for HCV RNA. Certainly, mutations in the rRNA 18S disrupting the IRES-40S contact do not affect cap-dependent translation (Matsuda and Mauro, 2014). Further, changes in the GGG triplet also block IRES translational activity (Jubin et al., 2000; Matsuda and Mauro, 2014; Angulo et al., 2016). A direct consequence of the interaction between IIId and 18S rRNA is a conformational rearrangement in the region surrounding the universally conserved nucleotide G1639 in rRNA, which unleashes the tRNA discrimination mechanism and the subsequent initiation of translation (Malygin et al., 2013a).
The molecular and functional organization of the HCV 5′UTR provides clear evidence of how information contained in genomic RNA domains governs the progress of the infective cycle.
Further proof exists of the essential nature of genomic functional domains in HCV RNA. The HCV 3′UTR is a ∼200–250 nt-long sequence that contains essential functional domains required for viral replication and translational control (Kolykhalov et al., 1996; Fricke et al., 2015). Three well-defined regions are (Figure 3):
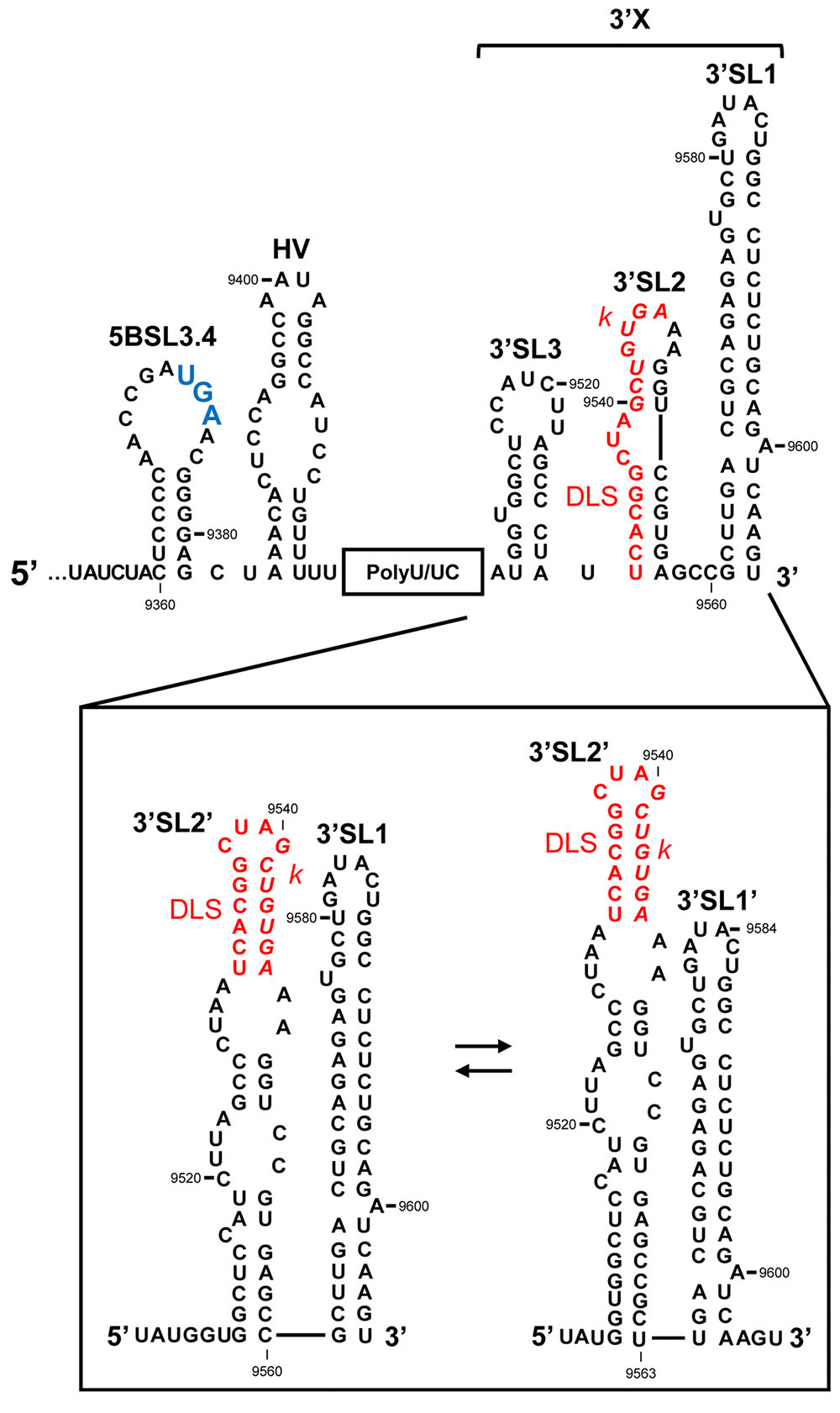
FIGURE 3. The 3′UTR of the HCV RNA genome. Secondary structure model proposed for the 3′UTR, showing the theoretical alternative conformations acquired in the 3′X tail. The palindromic motif involved in viral genome dimerization (DLS, dimer linkage sequence) is shown in red. The k sequence required for the interaction with the apical loop of the 5BSL3.2 domain is shown in italics. The translation stop codon is marked in enlarged blue text. Nucleotide numbering is as in Figure 2.
(i) The hypervariable region (HV); this is around 40 nts long and shows low-level sequence conservation among HCV isolates (Tanaka et al., 1995, 1996; Yamada et al., 1996). However, its secondary structure is preserved as a single stem-loop plus a 5′ region that partially overlaps the 5BSL3.4 domain (Figure 3) (Tanaka et al., 1996; Yamada et al., 1996; Ito and Lai, 1997). Though the hypervariable region seems to be dispensable for virus viability, its complete deletion leads to a significant reduction in HCV replication efficiency in cell culture (Kolykhalov et al., 2000; Friebe and Bartenschlager, 2002; Yi and Lemon, 2003).
(ii) The polyU/UC tract. This varies in length from one viral isolate to another, ranging from 30 to 80 nts (Figure 3; Kolykhalov et al., 1996). It consists of a homopolyuridine stretch interrupted by cytosine residues. Shortening the poly(U) tract below 27 nts, or interruption by CC dinucleotides, leads to the instability of the viral genome (Friebe and Bartenschlager, 2002; You and Rice, 2008). This suggests the recruitment to the polyU/UC tract of protein factors with special preferences for uridine over other pyrimidines. Interestingly, the HCV NS3 helicase, NS5A and NS5B (Figure 1) have been shown to preferentially bind poly(U) sequences in vitro (Gwack et al., 1996; Lohmann et al., 1997; Huang et al., 2005), which suggests a role for the poly(U/UC) tract in HCV replication.
(iii) The 3′X tail, located at the very 3′ end of the HCV RNA genome, is one of the most sequence- and structurally-conserved regions of the entire genome (Figure 3) (Kolykhalov et al., 1996; Blight and Rice, 1997). This 98 nt-long sequence is of major structural interest given its dynamic folding, which endorses the idea that the 3′X tail has an important regulatory function. Two mutually exclusive conformations (Figure 3) have been identified by chemical modification assays (Cristofari et al., 2004; Ivanyi-Nagy et al., 2006) and NMR (Cantero-Camacho and Gallego, 2015): a three stem-loop (3′SL3, 3′SL2, and 3′SL1) conformer (Figure 3, upper panel), which exposes the highly conserved k sequence motif complementary to the apical loop of the upstream 5BSL3.2 domain (see below) (Friebe et al., 2005), and a two stem-loop (3′SL2′ and 3′SL1/3′SL1′) conformer (Figure 3, lower panel). Nevertheless, recent NMR and SAXS studies have led to the proposal of the existence of a predominant, relatively rigid conformation defined by two stem-loops exposing the palindromic sequence motif DLS (dimer linkage sequence; Figure 3) (Ivanyi-Nagy et al., 2006; Shetty et al., 2010; Cantero-Camacho and Gallego, 2015; Cantero-Camacho et al., 2017). The DLS motif is involved in the formation of homodimeric genomes. In the two stem-loop conformation, a fine-tuned balance between two further isoforms is achieved, mediated by long-distance RNA–RNA interactions (see below). This conformational switch works via a slight gliding of 3 nts at the base of 3′SL1 (Figure 3) (Cantero-Camacho and Gallego, 2015; Cantero-Camacho et al., 2017) allowing the formation of 3′SL1′ (Figure 3). This conformation favors the initiation of primer-independent RNA synthesis via the viral RNA-dependent RNA polymerase NS5B protein (Kao et al., 2000). All this is consistent with the fact that a homodimeric genome operates as a preferential template for HCV polymerase (Masante et al., 2015), suggesting that the acquisition of the two stem-loop conformation is a pre-requisite for the progression of the infective cycle (Romero-López et al., 2014; Masante et al., 2015). Dimer genomic formation might also promote the generation of new recombinant variants during RNA synthesis (Morel et al., 2011; Scheel et al., 2013; Galli and Bukh, 2014), helping to improve viral fitness.
As well as its essential role in RNA replication, the 3′UTR functions as a translation enhancer (Mccaffrey et al., 2002; Bradrick et al., 2006; Song et al., 2006; Bung et al., 2010). This is mediated by the acquisition of a viral genome’s closed-loop topology, which resembles that adopted by mRNAs that are translated in a cap-dependent manner (Song et al., 2006; Weinlich et al., 2009; Bai et al., 2013). This conformation depends on the establishment of distant, direct long-range RNA–RNA contacts (Romero-López and Berzal-Herranz, 2009, 2012; Romero-López et al., 2012, 2014; Shetty et al., 2013). The interaction of the translational machinery with both ends of the viral genome is also critical in the viral genome achieving a circular isoform (Bai et al., 2013), which helps to retain the 40S ribosomal subunit during the translation termination step and favors ribosome recycling for the next round of protein synthesis (Bai et al., 2013).
The HCV 3′UTR thus plays various roles during the viral cycle, controlling the progress of infection via a collection of functional elements that can switch between different structural states.
In addition to RNA domains being located in the untranslated regions, numerous cis-acting signals have been identified in the ORFs of RNA viruses (for a review, see Romero-López and Berzal-Herranz, 2013). The search for unique, highly conserved structural and functional elements encoded in the HCV ORF has been going on for 15 years. The use of bioinformatic tools and secondary structure mapping, combined with genetic strategies, has finally provided a good overview of the global and local folding of HCV RNA (Tuplin et al., 2002, 2004; Mcmullan et al., 2007; Davis et al., 2008; Diviney et al., 2008; Chu et al., 2013; Fricke et al., 2015; Mauger et al., 2015; Pirakitikulr et al., 2016). The HCV genome is a highly compact molecule, with extensive base-pairing, helping to preserve viral RNA from cellular endonuclease-mediated degradation. Two of these degradation systems, both of which are involved in the innate immune response, include RNase L, which is specific for single-stranded regions, and the double-stranded-specific interference pathway (Li and Lemon, 2013). Importantly, by constraining the length of the helix segments, the viral genome has reached a perfect balance between single- and double-stranded regions with the aim of minimizing the effect of RNase L activity without unleashing the interference mechanism. This is consistent with the presence of alternate, extensive regions of compact folding throughout viral RNA genomes – the so-called GORS (genome-scale ordered RNA structure) elements. The existence of these elements correlates positively with host persistence and cell-to-cell movement (Simmonds et al., 2004; Davis et al., 2008; Witteveldt et al., 2014).
Up to 20 conserved RNA structural elements are scattered throughout the HCV ORF (Figure 1), expanding the functional repertoire of the viral genome via their participation in viral translation, replication, and infectivity (Tuplin et al., 2002, 2004; Mcmullan et al., 2007; Diviney et al., 2008; Chu et al., 2013; Fricke et al., 2015; Mauger et al., 2015; Pirakitikulr et al., 2016). Interestingly, the NS5B coding sequence is specifically enriched in functional RNA domains, with up to 10 distinct conserved stem-loop structural units able to drive HCV RNA and protein synthesis (Figure 1) (Smith and Simmonds, 1997; Walewski et al., 2001; Tuplin et al., 2002, 2004; Hofacker, 2004; Lee et al., 2004; You et al., 2004; Chu et al., 2013; Mauger et al., 2015; Pirakitikulr et al., 2016). One of the best-characterized functional RNA domains in the NS5B coding sequence is the so-called 5BSL3.2 domain (also known as SL9266 following the new standardized nomenclature system according to the genomic nucleotide position of the first 5′ paired residue of the stem-loop). The following sections focus on the molecular and functional features of this domain and its relationships with distant RNA elements in the HCV genome.
Using a combination of phylogenetic comparisons and thermodynamic prediction methods, Tuplin et al. (2002), defined the precise and conserved boundaries within the NS5B coding sequence that mark the limits of a set of genotypically well-conserved secondary structure elements. Five of these had already been proposed by other groups, either in whole or in part (Smith and Simmonds, 1997; Hofacker et al., 1998; Walewski et al., 2001; Tuplin et al., 2002). Nevertheless, it was in 2004 when three independent groups described the existence of a preserved sequence and structural region composed of three stem-loops in the 3′ terminus of the coding sequence. These were named 5BSL3.1, 5BSL3.2, and 5BSL3.3 (or SL9217, SL9266, and SL9324, respectively; Figures 1, 4; Lee et al., 2004; You et al., 2004; Friebe et al., 2005). Reverse genetic analyses in hepatocytes bearing subgenomic replicon constructs, and in full-length viral replication models, showed these stem-loops to operate as a cis-acting replication elements (CRE). The central domain 5BSL3.2 was shown indispensable for HCV propagation (Lee et al., 2004; You et al., 2004; Friebe et al., 2005; Masante et al., 2015; Tuplin et al., 2015).
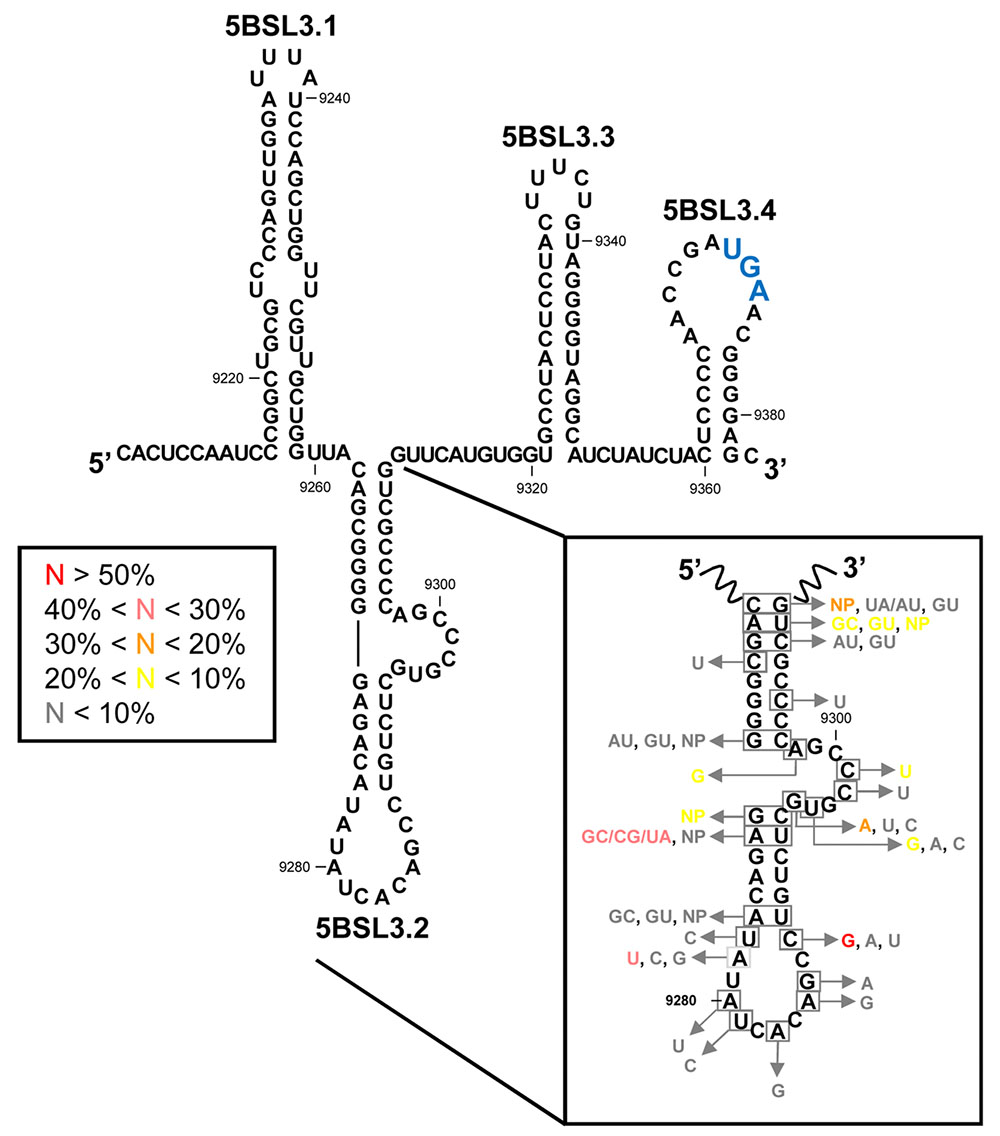
FIGURE 4. The CRE region. Sequence and secondary structure of the HCV CRE region, including the functional domains 5BSL3.1, 5BSL3.2, and 5BSL3.3 plus the 5BSL3.4 stem-loop. Conserved residues and covariant base pairs in the essential 5BSL3.2 domain, as described by You et al. (2004), are indicated in boxes. Colors denote the frequency for each nucleotide variation, as indicated. NP (non-paired) represents nucleotide variations impeding the formation of canonical base pairs. The translation stop codon is indicated in enlarged blue letters. Position numbering is as in Figure 2.
Bioinformatic prediction, biochemical structural mapping and NMR studies have all been undertaken to try to decipher the three-dimensional folding of 5BSL3.2 (Lee et al., 2004; You et al., 2004; Friebe et al., 2005; Tuplin et al., 2012), which was found to be a 48 nt-long imperfect hairpin with a 12 nt-long apical loop (Figure 4). The stem is interrupted at its 3′ end by an 8 nt-long bulge. Interestingly, both unpaired regions are phylogenetically conserved across different genotypes and show low synonymous site sequence variation (Figure 4) (You et al., 2004) that cannot be explained only by the need to preserve the NS5B coding sequence. Such a high degree of conservation undoubtedly points to a role in the functional control of the infective cycle, mediated by the apical loop and the bulge of the 5BSL3.2 domain (Lee et al., 2004; You et al., 2004; Friebe et al., 2005; Tuplin et al., 2012, 2015). Interestingly, the 5′-CACAGC-3′ sequence motif in the apical loop is found in other conserved elements of distantly related flaviviruses, such as Kunjin virus, West Nile virus or Dengue virus, where it operates as a single CRE (Markoff, 2003; Friebe et al., 2005). This observation points to the existence of common molecular mechanisms for viral RNA synthesis across different members of the family Flaviviridae.
Co-variation data confirm the existence in the basal part of the stem-loop of three out of the eight base pairs. In the upper stem, three of the six base pairs are invariable and two of the other remaining three can be predicted through compensatory base pair changes (Figure 4) (You et al., 2004). These phylogenetic data provide a convincing clue about the existence of the 5BSL3.2 hairpin in vivo.
From a structural point of view, the molecular context surrounding the 5BSL3.2 domain is striking. It is embedded between two other stem-loops, 5BSL3.1 and 5BSL3.3, to yield a high-order structure that can be depicted as a cruciform element (Figure 5) (Lee et al., 2004; You et al., 2004; Friebe et al., 2005). Domains 5BSL3.1 and 5BSL3.3 were initially identified as evolutionarily conserved elements (Smith and Simmonds, 1997; Tuplin et al., 2004) that fold into stable stem-loop structures (Figure 4). RNase and chemical mapping have confirmed this secondary structure and support the proposed large cruciform structure (You et al., 2004). However, neither mutagenesis nor biophysical methods have yet confirmed the existence of this high-order structure in vivo. This might be due to the dynamic and relatively unstable nature of long-distance RNA–RNA contacts, which can promote the switch between different metastable structural states in the RNA molecule, depending on the presence of specific ligands or external stimuli. Thus, such contacts suggest a regulatory operation mode that might rely on the versatility and efficiency of the HCV CRE region.
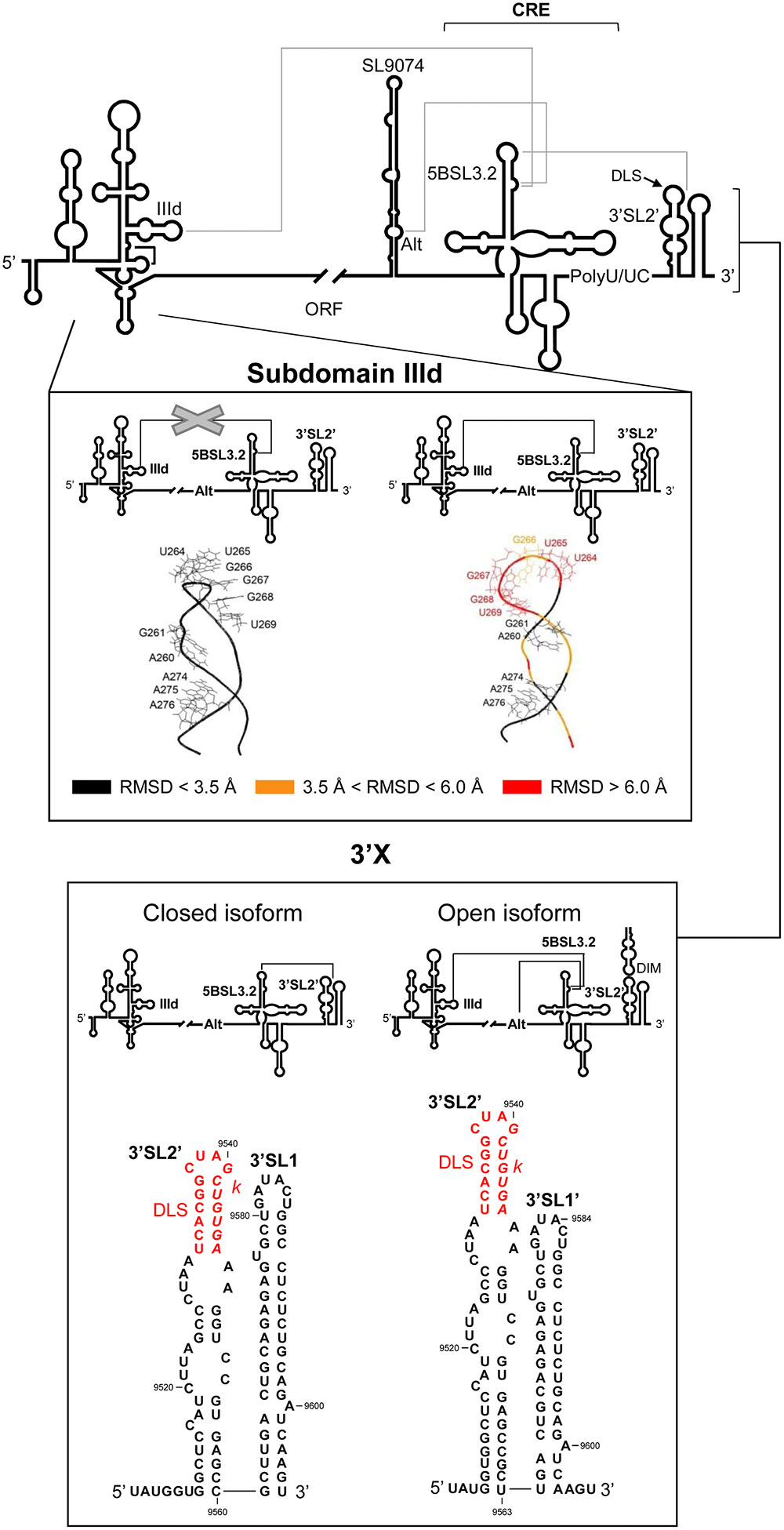
FIGURE 5. Long-range RNA–RNA interactions in the HCV genome promote conformational rearrangements in essential functional domains. Upper panel: Diagram of the secondary structure of the 5′ and 3′ ends of the HCV genome, including the functional domains involved in long-distance RNA–RNA contacts (solid gray lines). Middle panel: It shows the prediction for the structure of subdomain IIId (Romero-López and Berzal-Herranz, 2015). Root mean-square deviation (RMSD) values reflect differences in the conformation of subdomain IIId in the presence of the IIId-5BSL3.2 interaction (right) compared to its absence (left). Color code: Black, residues with an RMSD < 3.5 Å; orange, nucleotides with an RMSD ranging from 3.5 to 6.0 Å; red, residues with a RMSD of >6.0 Å. Lower panel: Structural reorganization of the 3′X tail depending on the establishment of different RNA–RNA contacts. The interaction of the 5BSL3.2 domain with the k motif in the 3′X region renders a two-stem-loop conformation in which the residues located at the 3′ end of the viral genome appear completely base-paired (left, closed isoform). However, contacts between 5BSL3.2 and upstream domains, such as the Alt sequence or subdomain IIId, release a three-nucleotide 3′ overhang in the 3′X tail (right, open isoform). DLS and k motifs are indicated according to Figure 3. Nucleotide numbering is as mentioned in Figure 2.
The remarkable structural features of the 5BSL3.2 domain, as well as its strong conservation, point to its critical involvement in the HCV infective cycle. As mentioned above, the observation that both the apical loop and the bulge of the 5BSL3.2 domain are highly preserved among different genotypes and viral isolates suggests their participation in interactions with other viral RNA elements. Several reports have provided substantial proof of the existence of long-distance RNA–RNA contacts, together forming a complex network of interactions that appears to be governed by 5BSL3.2 (Friebe et al., 2005; Romero-López and Berzal-Herranz, 2009; Romero-López et al., 2012, 2014, 2017; Tuplin et al., 2012, 2015; Shetty et al., 2013; Fricke et al., 2015). Such a network would organize the three-dimensional folding of the viral genome into a compact conformation that correlates with virulence and the persistence of infection (Davis et al., 2008). To date, two conformational rearrangements of the HCV genome are known to be mediated by the 5BSL3.2 domain: viral genome circularization and the structural tuning of the 3′ end of the HCV genome.
Genome circularization is a crucial step in the initiation of viral protein and RNA synthesis in many positive-stranded RNA viruses (Villordo and Gamarnik, 2009). It depends on either direct, long-distance RNA–RNA interactions, or protein bridges that bring the ends of the viral RNA together. In most cases, a combination of both mechanisms is used. While no conclusive evidence exists for the acquisition of a closed-loop topology by the HCV genome, indirect data suggests it does indeed occur and depends on long-range RNA–RNA contacts (Romero-López and Berzal-Herranz, 2009; Romero-López et al., 2012, 2014; Shetty et al., 2013; Fricke et al., 2015). Using bioinformatic, biochemical, and biophysical methods, several independent groups have shown the establishment of a direct interaction involving the bulge of the 5BSL3.2 domain and the apical loop of subdomain IIId in the IRES element (Figure 5) (Romero-López and Berzal-Herranz, 2009; Shetty et al., 2013; Fricke et al., 2015). Importantly, this interaction occurs in the absence of protein factors. According to the proposed model, the essential nucleotide G263, located at the base of the apical loop of the subdomain IIId, would establish the initial contact with C9301 on the 3′ side of the bulge in the 5BSL3.2 domain (Romero-López and Berzal-Herranz, 2009). This contact theoretically extends along the unpaired regions of the participating domains. Nucleotide complementarity runs through the flanking stems up to A288 for the 5′ end, and to A9275 for the 3′ end (Romero-López and Berzal-Herranz, 2009). However, no such extended complex has ever been detected.
Conformational consequences derived from the interaction 5BSL3.2-IIId have been mapped by chemical probing coupled to three-dimensional structure prediction (Figure 5) (Romero-López et al., 2012; Romero-López and Berzal-Herranz, 2015). The aromatic rings of the residues located in the apical loop of subdomain IIId have been reported to change their orientation with respect to the solvent because of their interaction with 5BSL3.2 (Figure 5). Such conformational rearrangements would affect IRES function due to their interference with the efficient recruitment of the 40S ribosomal subunit mediated by the subdomain IIId (Malygin et al., 2013a,b). This is in good agreement with the idea that the 5BSL3.2 domain operates as a specific and efficient negative regulator during HCV IRES-dependent translation (Romero-López and Berzal-Herranz, 2012). The 5BSL3.2 is therefore considered a versatile multifunctional genomic element that takes part in different steps of the infective cycle and controls transitions between them.
The apical loop of the 5BSL3.2 domain establishes a long-range interaction with a complementary sequence, k, located in the apical loop of the downstream 3′SL2 element within the 3′X tail (Figures 3, 5) (Friebe et al., 2005; Cantero-Camacho et al., 2017). The biochemical and structural properties of this interaction have been studied in depth following different biophysical and biochemical strategies (Friebe et al., 2005; You and Rice, 2008; Tuplin et al., 2012; Palau et al., 2013; Shetty et al., 2013; Cantero-Camacho et al., 2017). These analyses showed that the contact 5BSL3.2-3′X occurs in the absence of RNA chaperone proteins and is preferred over the formation of homodimeric viral genomes (Cantero-Camacho et al., 2017). Recent studies have determined that even for the dimerizable conformation of the 3′X region, which partially occludes k, the interaction with the 5BSL3.2 domain is made possible by the induction of the structural disruption of the two stem-loop conformations of the 3′X tail, thus unfolding the motif k (Cantero-Camacho et al., 2017). Hence, 5BSL3.2 may act as a structural cofactor promoting the acquisition of a new, functionally active folding of the HCV genome.
Conversely, the 5BSL3.2 bulge element may interact with the complementary sequence motif centered on position 9110 – the so-called Alt sequence (Figure 5) (Diviney et al., 2008; Tuplin et al., 2012; Shetty et al., 2013). This contact overlaps with that involving subdomain IIId. Both interactions show dissociation constant values in the same range, and seem equally likely to occur (Shetty et al., 2013). Choosing between them is affected by additional structural constraints and the presence of different cofactors (see below).
A significant feature of the interactions mediated by 5BSL3.2 at the 3′ end of the viral RNA is that they can be established in an independent and simultaneous manner (Tuplin et al., 2012; Palau et al., 2013; Shetty et al., 2013). This suggests that the binding sites are structurally independent. The connections established at the 3′ end render the formation of an intricate tertiary structure in which the 5BSL3.2 forms the core of an extended and dynamic pseudoknot (Tuplin et al., 2012). Reverse genetic and biochemical structural data have revealed this pseudoknot able to promote two alternative conformations in the 3′X region (Tuplin et al., 2012; Cantero-Camacho and Gallego, 2015; Kranawetter et al., 2017). In the open form, contact with the Alt sequence or with subdomain IIId would favor the establishment of the preferred two stem-loop conformation bearing three overhang nucleotides at the 3′ end of the HCV RNA (Figures 3, 5). As mentioned above, this is a favored folding state for virus replication (Kao et al., 2000) and for genome dimerization (Palau et al., 2013; Romero-López et al., 2014; Cantero-Camacho et al., 2017). Alternatively, with the closed isoform, the interaction of 5BSL3.2 with the k motif in the 3′X tail would induce the local melting of the upper stem of 3′SL2′ and induce conformational rearrangements in the base of the 3′SL1′, leading to the formation of the extended stem-loop 3′SL1 (Figures 3, 5) (Tuplin et al., 2012; Cantero-Camacho and Gallego, 2015). Interestingly, the thermodynamic equilibrium between these two isoforms seems to be dependent on the viral genotype, with the open conformation being favored by genotype 1b and the closed form by genotype 2a (Tuplin et al., 2012). These structural data undoubtedly suggest clear correlations between HCV RNA conformation, genotype-dependent virulence and even the viral sustained response to the treatment shown by the infected patient (Irshad et al., 2010; Carter et al., 2017).
The creation of this complex network of RNA–RNA interactions promotes the structural remodeling of the viral genome, not only in the directly involved domains, but also in distant regions. This phenomenon achieves complex and interconnected genomic-RNA-folding-dependent regulatory pathways. Indeed, the conformational rearrangement mediated by the 5BSL3.2 domain at the 3′ end is regulated by the IRES region, most likely as a consequence of the interaction IIId-5BSL3.2 (Romero-López and Berzal-Herranz, 2009; Romero-López et al., 2014). Using chemical structural mapping coupled to secondary structure prediction, it has been shown that the presence of both the IRES and the CRE regions favors the acquisition of the dimerizable two stem-loop conformation in the 3′X tail, exposing the DLS motif (Romero-López et al., 2014). This led to the assumption that the contact IIId-5BSL3.2 improves the dimerization of the viral genome. However, it has been recently reported that while the two stem-loop isoform is a requisite, it is not the only determinant affecting genomic dimer formation (Cantero-Camacho et al., 2017; Romero-López et al., 2017). In summary, the IRES and the CRE regions might be considered both cofactors and chaperoning agents that finely tune the three-dimensional folding of the 3′ end of the HCV RNA in order to control the different stages of the infective cycle, and the transitions between them.
The folding of the IRES region is also influenced by the presence of the 3′ end of the HCV genome. The 3′UTR and the CRE region have been shown to effect remarkable changes in subdomains IIIe and IIIf (Romero-López et al., 2012) – essential elements involved in the proper positioning of the translation start codon in the P site of the 40S ribosomal subunit (Berry et al., 2011). In addition, the three-dimensional organization of the eIF3 binding platform within the IRES can be modified by the interaction IIId-5BSL3.2, turning the typical S-turn conformation (Collier et al., 2002) into a more rigid form with altered functionality (Romero-López et al., 2012). Domain IV appears less stable in the presence of the 3′ end of the HCV RNA, which might contribute toward the proper positioning of the translation start codon (Honda et al., 1996a; Romero-López et al., 2012). All these findings are in good agreement with the acquisition of a circular topology by the HCV genomic RNA.
The 5BSL3.2 domain thus participates in the establishment of different long-range RNA–RNA interactions in the absence of proteins. This helps create a complex network of contacts, the careful regulation of which allows the HCV genome to acquire different functional conformational states, facilitating proper switching between the different stages of the viral cycle.
The recruitment of both host and viral components is important in the regulatory activities of the CRE region (Lourenco et al., 2008). The apical loop of the 5BSL3.2 domain binds to the viral polymerase NS5B protein (Zhang et al., 2005), favoring the positioning of the polymerase in the 3′X tail and thus the initiation of replication. The interaction of the NS5B protein with the apical loop of 5BSL3.2 might compete with the contact 5BSL3.2-3′X. Swapping from NS5B recruitment to 3′X interaction could therefore be used by the 5BSL3.2 domain as a regulatory mechanism for promoting transitions between steps during the infective cycle.
In recent years, exhaustive lists of cellular proteins susceptible to recruitment by the CRE region have been produced (Oakland et al., 2013; Ríos-Marco et al., 2016). Interestingly, some have been shown to influence viral translation, replication or both (Oakland et al., 2013; Ríos-Marco et al., 2016). Oakland et al. (2013) showed Ewing’s Sarcoma binding protein 1 (EWSR1) to interact with the CRE region; EWSR1 is a nuclear factor that regulates RNA synthesis and processing as well as the transport of pre-mRNAs involved in cell cycle progression and the response to DNA damage (Zinszner et al., 1994; Paronetto et al., 2011). It also binds other factors related to splicing, such as those belonging to the heterogeneous ribonuclear protein family (hnRNPs). Its regulatory role during mitosis has also been reported (Wang et al., 2016). The involvement of ESWR1 in tumorigenesis has attracted much attention since HCV infection can lead to the development of hepatocellular carcinoma. Though EWSR1 is preferentially located in the nucleus, it can be translocated to the cytosol of HCV-infected hepatocytes (Oakland et al., 2013). Here it binds to the viral genome in a manner dependent on the structure resulting from the interaction 5BSL3.2-3′X, promoting efficient HCV RNA replication, at least in cell culture (Oakland et al., 2013).
Subsequent proteomic analyses have identified a collection of RNA-binding proteins with highly conserved RNA recognition motifs able to bind to the CRE region (Ríos-Marco et al., 2016). hnRNPA1 is a very abundant nuclear and cytosolic protein involved in the packaging of pre-mRNAs into spliceosomal particles, and in the translocation of processed poly(A) mRNAs from the nucleus to the cytoplasm (Dreyfuss et al., 2002). During HCV infection, hnRNPA1 operates as an IRES-dependent translation initiation enhancer via its interaction with the IRES region (Lu et al., 2004), and as a negative regulatory partner of viral RNA synthesis, most likely by competing with NS5B for a common interacting site in the 5BSL3.2 domain (Ríos-Marco et al., 2016). Additionally, hnRNPA1 plays an important role as a splicing factor in the maturation of IFR3 (interferon regulatory factor 3), which is involved in interferon-mediated immunity (Guo et al., 2013). Sequestering hnRNPA1 by 5BSL3.2 would, therefore, influence interferon production, helping HCV escape the cellular immune response. HMGB1 (high mobility group box 1 protein) also interferes with HCV replication by binding to the CRE (Jung et al., 2011; Ríos-Marco et al., 2016), and is considered an antiviral factor (Jung et al., 2011). Finally, host proteins with helicase activity, such as DDX3, DDX5, and DDX17, might recognize the 5BSL3.2 domain and promote HCV replication and/or translation (Ríos-Marco et al., 2016). This last observation confirms the role of the CRE as a regulator of viral protein and RNA synthesis (Ariumi et al., 2007; Randall et al., 2007; Oakland et al., 2013; Ríos-Marco et al., 2016).
MicroRNAs (miRNAs) produced by the host cell have been identified as agents that regulate viral infection (Bruscella et al., 2017). Via a little-understood mechanism, miRNAs influence HCV infection at different stages. For example, miR-122, a highly abundant miRNA in hepatocytes, promotes viral translation and replication via its interaction with the IRES region at different target sites (Jopling et al., 2005; Jangra et al., 2010; Roberts et al., 2011), while miR-199a∗ represses RNA replication (Murakami et al., 2009). Let-7b also acts as a negative regulatory agent of HCV replication via its direct interaction with the region connecting domains 5BSL3.2 and 5BSL3.3 (Cheng et al., 2012). Let-7b is involved in cell differentiation and has been intimately associated with the development of cancer (Takamizawa et al., 2004; Yu et al., 2007). This, along with the observation that cell cycle regulatory proteins bind to the CRE, provides additional evidence of the potential role of the CRE region in HCV-associated hepatocellular carcinoma.
The HCV cycle is extensively compartmentalized, both spatially and temporally: viral protein synthesis, replication, and encapsidation occur in different cellular localizations and do not overlap in time (Shulla and Randall, 2015). The switch between one stage and the next must therefore be carefully controlled. The machinery required for this is not well-understood, but it is widely assumed that functional genomic domains, along with host and viral factors, together control the progression of the infective cycle.
During early infection, HCV virions are endocytosed and their genomes released into the cytosol by a little-understood mechanism (Figure 6; for a review see Scheel and Rice, 2013). Endoplasmic reticulum (ER)-associated translation is then initiated by the IRES region. During this stage, subdomain IIId of the IRES is preferentially occupied by the translational machinery, thus favoring the contacts 5BSL3.2-Alt and 5BSL3.2-3′X (Figure 6). The emerging HCV polyprotein is co- and post-translationally cleaved by cellular proteases and viral NS2-NS3 and NS3-NS4A proteases to release 10 HCV proteins (Grakoui et al., 1993). The accumulation of non-structural viral proteins promotes significant rearrangements in the ER membrane to create optimized microenvironments for RNA replication (as seen for other positive-strand RNA viruses) (Egger et al., 2002; Moradpour et al., 2003; Meyers et al., 2016; Falcon et al., 2017). The binding of the replicase complex to the 3′X tail of the viral genome, and the concomitant recruitment of cellular components at the CRE region (Oakland et al., 2013; Ríos-Marco et al., 2016), releases the 5BSL3.2 domain for new interactions with distant genomic RNA elements such as subdomain IIId within the IRES (Figure 6). This contact promotes the viral genome’s acquisition of a circular form. In addition, the interaction IIId-5BSL3.2 blocks the recruitment of 40S ribosomal particles by the IRES, contributing to the creation of a translationally repressed-state(Romero-López and Berzal-Herranz, 2012). 5BSL3.2 is thus considered a specific inhibitor of HCV IRES function (Romero-López and Berzal-Herranz, 2012). The IIId-5BSL3.2 interaction also controls the structural switch at the 3′X tail and partially interferes with the formation of dimeric genomes (Romero-López et al., 2017). From a thermodynamic point of view, the interaction IIId-5BSL3.2 can swap with the contact Alt-5BSL3.2 (Shetty et al., 2013) to create a favorable replicative environment (Figure 6) (Diviney et al., 2008; Tuplin et al., 2012). Viral RNA synthesis is then initiated using the positive genome strand as a template, yielding the complementary, negative strand, which is used for the generation of new progeny RNA genomes (for a review, see Kim and Chang, 2013). In a later stage of the cycle, viral genomes accumulate in the cytosol, where they serve as mRNAs for the production of HCV proteins, thus initiating new rounds of translation-replication (Figure 6). At this time, the IRES region is occluded by the translational machinery, favoring the interaction Alt-5BSL3.2. Because of the high concentration of viral RNAs, genomic dimerization is thermodynamically favored (Figure 6). This results in viral RNA storage, but HCV dimeric genomes are also excellent templates for the initiation of replication (Cantero-Camacho and Gallego, 2015; Masante et al., 2015; Cantero-Camacho et al., 2017). According to this model, HCV genomic dimerization may play a replication enhancing role during late infection. Finally, a fraction of the newly synthesized RNA molecules is shuttled for encapsidation and released to the extracellular medium (Figure 6).
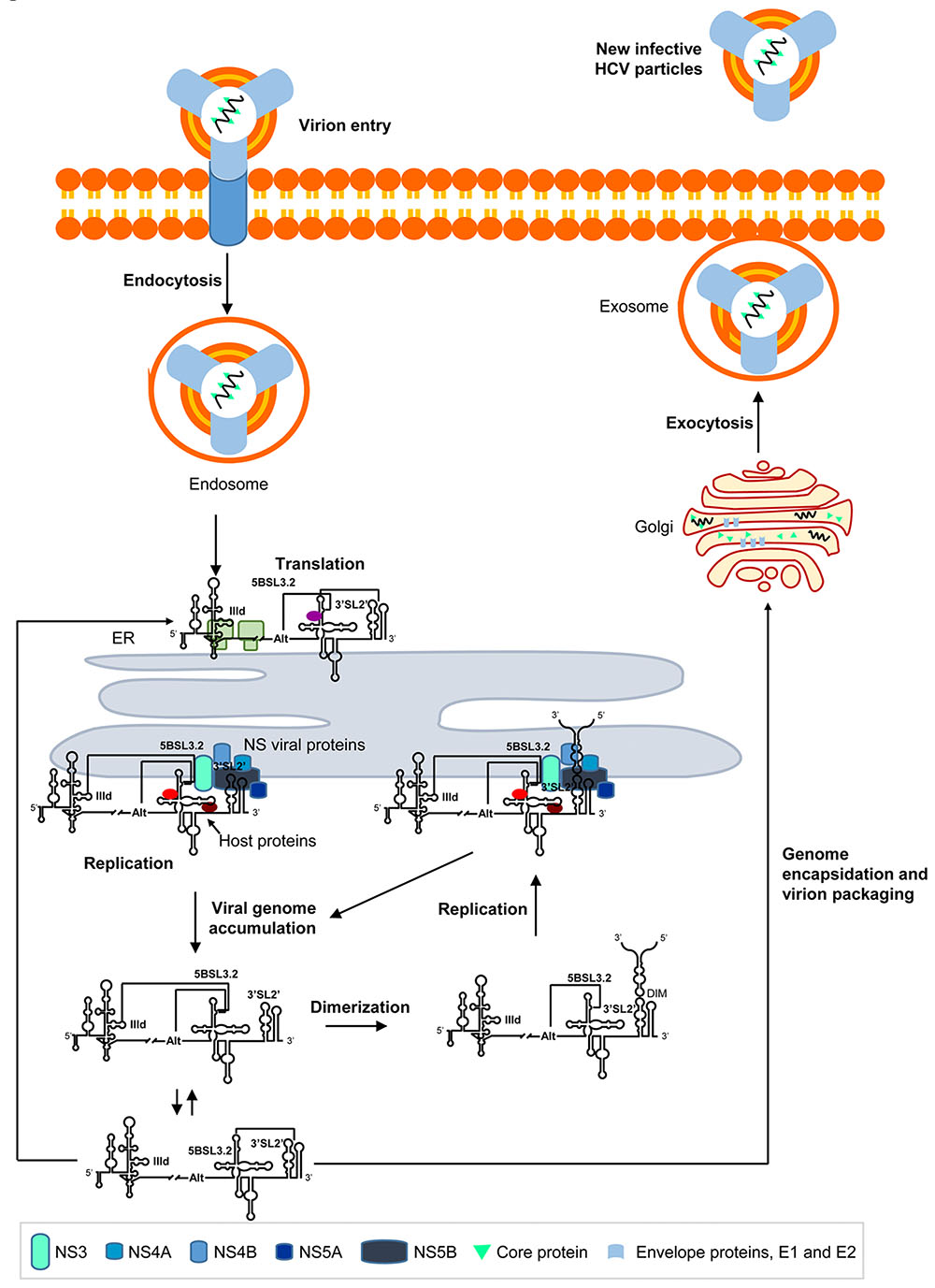
FIGURE 6. Long-range RNA–RNA interactions regulate the different steps in the HCV infective cycle. The figure shows a working model demonstrating the long-range RNA–RNA interactions in the HCV genome described to date, and their role in the progression of the infective cycle. Briefly, during early infection, the viral genome is released to the cytosol and viral translation initiates in an IRES-dependent manner on the surface of the endoplasmic reticulum (ER). Subdomain IIId is then occupied by the 40S ribosomal subunit, which impedes the interaction IIId-5BSL3.2 and enhances the conformational rearrangement at the 3′ end mediated by the 5BSL3.2 domain. The accumulation of NS viral proteins induces the formation of replication complexes, which preferentially recruit viral genomes showing the interaction IIId-5BSL3.2 or Alt-5BSL3.2. This contact favors a translationally repressed-state and enhanced replication dependent on the interaction Alt-5BSL3.2. It also interferes with the formation of dimeric genomic particles. The accumulation of newly synthesized HCV RNA genomes induces the initiation of new rounds of translation. The dimerization process is thermodynamically favored under these conditions, and the dimeric genomes produced offer optimal templates for viral replication. Alternatively, a fraction of the HCV RNA molecules is encapsidated and released to the extracellular medium by exocytosis.
The model proposed above recapitulates many of the findings made regarding the molecular biology of HCV. More importantly, it provides an overview of the critical role of long-distance RNA–RNA contacts in the progression of the infective cycle, and points to 5BSL3.2 as a multifunctional component encoded in the viral genome.
The contrast between the high structural conservation and sequence variability of the HCV RNA genome reflects an efficient mechanism for retaining essential functionalities in certain structural elements while favoring the acquisition of novel capabilities. Whereas many genomic domains govern essential steps in infection, the control exerted by 5BSL3.2 at multiple levels reflects the functional versatility and fine regulation that can be achieved by a single stem-loop. This domain is thus an interesting target for novel therapeutic agents. Exploring the structural dynamics of functional RNA domains offers a powerful, informative methodology for future drug design. Extending the knowledge acquired in HCV to related viruses, such as flaviviruses, will allow conformational maps of common structural domains to be produced, help reveal similar viral mechanisms involved in the infective cycle, and perhaps contribute to the development of novel therapeutic and diagnostic strategies.
CR-L and AB-H designed and wrote the paper.
This work was supported by the Spanish Ministerio de Economía y Competitividad [BFU2015-64359-P] to AB-H. Work at our laboratory is partially supported by FEDER funds from the EU.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Angulo, J., Ulryck, N., Deforges, J., Chamond, N., Lopez-Lastra, M., Masquida, B., et al. (2016). LOOP IIId of the HCV IRES is essential for the structural rearrangement of the 40S-HCV IRES complex. Nucleic Acids Res. 44, 1309–1325. doi: 10.1093/nar/gkv1325
Ariumi, Y., Kuroki, M., Abe, K., Dansako, H., Ikeda, M., Wakita, T., et al. (2007). DDX3 DEAD-box RNA helicase is required for hepatitis C virus RNA replication. J. Virol. 81, 13922–13926. doi: 10.1128/JVI.01517-07
Babaylova, E., Graifer, D., Malygin, A., Stahl, J., Shatsky, I., and Karpova, G. (2009). Positioning of subdomain IIId and apical loop of domain II of the hepatitis C IRES on the human 40S ribosome. Nucleic Acids Res. 37, 1141–1151. doi: 10.1093/nar/gkn1026
Bai, Y., Zhou, K., and Doudna, J. A. (2013). Hepatitis C virus 3′UTR regulates viral translation through direct interactions with the host translation machinery. Nucleic Acids Res. 41, 7861–7874. doi: 10.1093/nar/gkt543
Bartenschlager, R., and Lohmann, V. (2000). Replication of hepatitis C virus. J. Gen. Virol. 81, 1631–1648. doi: 10.1099/0022-1317-81-7-1631
Berry, K. E., Waghray, S., and Doudna, J. A. (2010). The HCV IRES pseudoknot positions the initiation codon on the 40S ribosomal subunit. RNA 16, 1559–1569. doi: 10.1261/rna.2197210
Berry, K. E., Waghray, S., Mortimer, S. A., Bai, Y., and Doudna, J. A. (2011). Crystal structure of the HCV IRES central domain reveals strategy for start-codon positioning. Structure 19, 1456–1466. doi: 10.1016/j.str.2011.08.002
Blight, K. J., and Rice, C. M. (1997). Secondary structure determination of the conserved 98-base sequence at the 3′ terminus of hepatitis C virus genome RNA. J. Virol. 71, 7345–7352.
Bourliere, M., Gordon, S. C., Flamm, S. L., Cooper, C. L., Ramji, A., Tong, M., et al. (2017). Sofosbuvir, Velpatasvir, and Voxilaprevir for previously treated HCV infection. N. Engl. J. Med. 376, 2134–2146. doi: 10.1056/NEJMoa1613512
Bradrick, S. S., Walters, R. W., and Gromeier, M. (2006). The hepatitis C virus 3′-untranslated region or a poly(A) tract promote efficient translation subsequent to the initiation phase. Nucleic Acids Res. 34, 1293–1303. doi: 10.1093/nar/gkl019
Bruscella, P., Bottini, S., Baudesson, C., Pawlotsky, J. M., Feray, C., and Trabucchi, M. (2017). Viruses and miRNAs: more friends than foes. Front. Microbiol. 8:824. doi: 10.3389/fmicb.2017.00824
Bukh, J., Purcell, R. H., and Miller, R. H. (1992). Sequence analysis of the 5′ noncoding region of hepatitis C virus. Proc. Natl. Acad. Sci. U.S.A. 89, 4942–4946. doi: 10.1073/pnas.89.11.4942
Bung, C., Bochkaeva, Z., Terenin, I., Zinovkin, R., Shatsky, I. N., and Niepmann, M. (2010). Influence of the hepatitis C virus 3′-untranslated region on IRES-dependent and cap-dependent translation initiation. FEBS Lett. 584, 837–842. doi: 10.1016/j.febslet.2010.01.015
Cantero-Camacho, A., Fan, L., Wang, Y. X., and Gallego, J. (2017). Three-dimensional structure of the 3′X-tail of hepatitis C virus RNA in monomeric and dimeric states. RNA 23, 1465–1476. doi: 10.1261/rna.060632.117
Cantero-Camacho, A., and Gallego, J. (2015). The conserved 3′X terminal domain of hepatitis C virus genomic RNA forms a two-stem structure that promotes viral RNA dimerization. Nucleic Acids Res. 43, 8529–8539. doi: 10.1093/nar/gkv786
Carter, W., Connelly, S., and Struble, K. (2017). Reinventing HCV treatment: past and future perspectives. J. Clin. Pharmacol. 57, 287–296. doi: 10.1002/jcph.830
Cheng, J. C., Yeh, Y. J., Tseng, C. P., Hsu, S. D., Chang, Y. L., Sakamoto, N., et al. (2012). Let-7b is a novel regulator of hepatitis C virus replication. Cell Mol. Life Sci. 69, 2621–2633. doi: 10.1007/s00018-012-0940-6
Choo, Q. L., Kuo, G., Weiner, A. J., Overby, L. R., Bradley, D. W., and Houghton, M. (1989). Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244, 359–362. doi: 10.1126/science.2523562
Chu, D., Ren, S., Hu, S., Wang, W. G., Subramanian, A., Contreras, D., et al. (2013). Systematic analysis of enhancer and critical cis-acting RNA elements in the protein-encoding region of the hepatitis C virus genome. J. Virol. 87, 5678–5696. doi: 10.1128/JVI.00840-12
Collier, A. J., Gallego, J., Klinck, R., Cole, P. T., Harris, S. J., Harrison, G. P., et al. (2002). A conserved RNA structure within the HCV IRES eIF3-binding site. Nat. Struct. Biol. 9, 375–380. doi: 10.1038/nsb785
Cristofari, G., Ivanyi-Nagy, R., Gabus, C., Boulant, S., Lavergne, J. P., Penin, F., et al. (2004). The hepatitis C virus core protein is a potent nucleic acid chaperone that directs dimerization of the viral (+) strand RNA in vitro. Nucleic Acids Res. 32, 2623–2631. doi: 10.1093/nar/gkh579
Cuevas, J. M., Gonzalez-Candelas, F., Moya, A., and Sanjuan, R. (2009). Effect of ribavirin on the mutation rate and spectrum of hepatitis C virus in vivo. J. Virol. 83, 5760–5764. doi: 10.1128/JVI.00201-09
Davis, M., Sagan, S. M., Pezacki, J. P., Evans, D. J., and Simmonds, P. (2008). Bioinformatic and physical characterizations of genome-scale ordered RNA structure in mammalian RNA viruses. J. Virol. 82, 11824–11836. doi: 10.1128/JVI.01078-08
Diviney, S., Tuplin, A., Struthers, M., Armstrong, V., Elliott, R. M., Simmonds, P., et al. (2008). A hepatitis C virus cis-acting replication element forms a long-range RNA-RNA interaction with upstream RNA sequences in NS5B. J. Virol. 82, 9008–9022. doi: 10.1128/JVI.02326-07
Dmitriev, S. E., Terenin, I. M., Andreev, D. E., Ivanov, P. A., Dunaevsky, J. E., Merrick, W. C., et al. (2010). GTP-independent tRNA delivery to the ribosomal P-site by a novel eukaryotic translation factor. J. Biol. Chem. 285, 26779–26787. doi: 10.1074/jbc.M110.119693
Dreyfuss, G., Kim, V. N., and Kataoka, N. (2002). Messenger-RNA-binding proteins and the messages they carry. Nat. Rev. Mol. Cell Biol. 3, 195–205. doi: 10.1038/nrm760
Egger, D., Wolk, B., Gosert, R., Bianchi, L., Blum, H. E., Moradpour, D., et al. (2002). Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 76, 5974–5984. doi: 10.1128/JVI.76.12.5974-5984.2002
Falcon, V., Acosta-Rivero, N., Gonzalez, S., Duenas-Carrera, S., Martinez-Donato, G., Menendez, I., et al. (2017). Ultrastructural and biochemical basis for hepatitis C virus morphogenesis. Virus Genes 53, 151–164. doi: 10.1007/s11262-017-1426-2
Filbin, M. E., and Kieft, J. S. (2011). HCV IRES domain IIb affects the configuration of coding RNA in the 40S subunit’s decoding groove. RNA 17, 1258–1273. doi: 10.1261/rna.2594011
Filbin, M. E., Vollmar, B. S., Shi, D., Gonen, T., and Kieft, J. S. (2013). HCV IRES manipulates the ribosome to promote the switch from translation initiation to elongation. Nat. Struct. Mol. Biol. 20, 150–158. doi: 10.1038/nsmb.2465
Fricke, M., Dunnes, N., Zayas, M., Bartenschlager, R., Niepmann, M., and Marz, M. (2015). Conserved RNA secondary structures and long-range interactions in hepatitis C viruses. RNA 21, 1219–1232. doi: 10.1261/rna.049338.114
Friebe, P., and Bartenschlager, R. (2002). Genetic analysis of sequences in the 3′ nontranslated region of hepatitis C virus that are important for RNA replication. J. Virol. 76, 5326–5338. doi: 10.1128/JVI.76.11.5326-5338.2002
Friebe, P., Boudet, J., Simorre, J. P., and Bartenschlager, R. (2005). Kissing-loop interaction in the 3′ end of the hepatitis C virus genome essential for RNA replication. J. Virol. 79, 380–392. doi: 10.1128/JVI.79.1.380-392.2005
Galli, A., and Bukh, J. (2014). Comparative analysis of the molecular mechanisms of recombination in hepatitis C virus. Trends Microbiol. 22, 354–364. doi: 10.1016/j.tim.2014.02.005
Grakoui, A., Wychowski, C., Lin, C., Feinstone, S. M., and Rice, C. M. (1993). Expression and identification of hepatitis C virus polyprotein cleavage products. J. Virol. 67, 1385–1395.
Guo, R., Li, Y., Ning, J., Sun, D., Lin, L., and Liu, X. (2013). HnRNP A1/A2 and SF2/ASF regulate alternative splicing of interferon regulatory factor-3 and affect immunomodulatory functions in human non-small cell lung cancer cells. PLOS ONE 8:e62729. doi: 10.1371/journal.pone.0062729
Gwack, Y., Kim, D. W., Han, J. H., and Choe, J. (1996). Characterization of RNA binding activity and RNA helicase activity of the hepatitis C virus NS3 protein. Biochem. Biophys. Res. Commun. 225, 654–659. doi: 10.1006/bbrc.1996.1225
Hashem, Y., Des Georges, A., Dhote, V., Langlois, R., Liao, H. Y., Grassucci, R. A., et al. (2013). Hepatitis-C-virus-like internal ribosome entry sites displace eIF3 to gain access to the 40S subunit. Nature 503, 539–543. doi: 10.1038/nature12658
Hofacker, I. L. (2004). RNA secondary structure analysis using the Vienna RNA package. Curr. Protoc. Bioinformatics 26:12.2.1–12.2.16. doi: 10.1002/0471250953.bi1202s04
Hofacker, I. L., Fekete, M., Flamm, C., Huynen, M. A., Rauscher, S., Stolorz, P. E., et al. (1998). Automatic detection of conserved RNA structure elements in complete RNA virus genomes. Nucleic Acids Res. 26, 3825–3836. doi: 10.1093/nar/26.16.3825
Honda, M., Brown, E. A., and Lemon, S. M. (1996a). Stability of a stem-loop involving the initiator AUG controls the efficiency of internal initiation of translation on hepatitis C virus RNA. RNA 2, 955–968.
Honda, M., Ping, L. H., Rijnbrand, R. C., Amphlett, E., Clarke, B., Rowlands, D., et al. (1996b). Structural requirements for initiation of translation by internal ribosome entry within genome-length hepatitis C virus RNA. Virology 222, 31–42.
Hoofnagle, J. H. (2002). Course and outcome of hepatitis C. Hepatology 36, S21–S29. doi: 10.1053/jhep.2002.36227
Huang, L., Hwang, J., Sharma, S. D., Hargittai, M. R., Chen, Y., Arnold, J. J., et al. (2005). Hepatitis C virus non-structural protein 5A (NS5A) is a RNA-binding protein. J. Biol. Chem. 280, 36417–36428. doi: 10.1074/jbc.M508175200
Irshad, M., Ansari, M. A., Singh, A., Nag, P., Raghvendra, L., Singh, S., et al. (2010). HCV-genotypes: a review on their origin, global status, assay system, pathogenecity and response to treatment. Hepatogastroenterology 57, 1529–1538.
Ito, T., and Lai, M. M. (1997). Determination of the secondary structure of and cellular protein binding to the 3′-untranslated region of the hepatitis C virus RNA genome. J. Virol. 71, 8698–8706.
Ivanyi-Nagy, R., Kanevsky, I., Gabus, C., Lavergne, J. P., Ficheux, D., Penin, F., et al. (2006). Analysis of hepatitis C virus RNA dimerization and core-RNA interactions. Nucleic Acids Res. 34, 2618–2633. doi: 10.1093/nar/gkl240
Jaafar, Z. A., Oguro, A., Nakamura, Y., and Kieft, J. S. (2016). Translation initiation by the hepatitis C virus IRES requires eIF1A and ribosomal complex remodeling. Elife 5:e21198. doi: 10.7554/eLife.21198
Jangra, R. K., Yi, M., and Lemon, S. M. (2010). Regulation of hepatitis C virus translation and infectious virus production by the microRNA miR-122. J. Virol. 84, 6615–6625. doi: 10.1128/JVI.00417-10
Jopling, C. L., Yi, M., Lancaster, A. M., Lemon, S. M., and Sarnow, P. (2005). Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 309, 1577–1581. doi: 10.1126/science.1113329
Jubin, R., Vantuno, N. E., Kieft, J. S., Murray, M. G., Doudna, J. A., Lau, J. Y., et al. (2000). Hepatitis C virus internal ribosome entry site (IRES) stem loop IIId contains a phylogenetically conserved GGG triplet essential for translation and IRES folding. J. Virol. 74, 10430–10437. doi: 10.1128/JVI.74.22.10430-10437.2000
Jung, J. H., Park, J. H., Jee, M. H., Keum, S. J., Cho, M. S., Yoon, S. K., et al. (2011). Hepatitis C virus infection is blocked by HMGB1 released from virus-infected cells. J. Virol. 85, 9359–9368. doi: 10.1128/JVI.00682-11
Kao, C. C., Yang, X., Kline, A., Wang, Q. M., Barket, D., and Heinz, B. A. (2000). Template requirements for RNA synthesis by a recombinant hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 74, 11121–11128. doi: 10.1128/JVI.74.23.11121-11128.2000
Kieft, J. S., Zhou, K., Jubin, R., Murray, M. G., Lau, J. Y., and Doudna, J. A. (1999). The hepatitis C virus internal ribosome entry site adopts an ion-dependent tertiary fold. J. Mol. Biol. 292, 513–529. doi: 10.1006/jmbi.1999.3095
Kim, C. W., and Chang, K. M. (2013). Hepatitis C virus: virology and life cycle. Clin. Mol. Hepatol. 19, 17–25. doi: 10.3350/cmh.2013.19.1.17
Kim, J. H., Park, S. M., Park, J. H., Keum, S. J., and Jang, S. K. (2011). eIF2A mediates translation of hepatitis C viral mRNA under stress conditions. EMBO J. 30, 2454–2464. doi: 10.1038/emboj.2011.146
Kolupaeva, V. G., Pestova, T. V., and Hellen, C. U. (2000). An enzymatic footprinting analysis of the interaction of 40S ribosomal subunits with the internal ribosomal entry site of hepatitis C virus. J. Virol. 74, 6242–6250. doi: 10.1128/JVI.74.14.6242-6250.2000
Kolykhalov, A. A., Feinstone, S. M., and Rice, C. M. (1996). Identification of a highly conserved sequence element at the 3′ terminus of hepatitis C virus genome RNA. J. Virol. 70, 3363–3371.
Kolykhalov, A. A., Mihalik, K., Feinstone, S. M., and Rice, C. M. (2000). Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3′ nontranslated region are essential for virus replication in vivo. J. Virol. 74, 2046–2051. doi: 10.1128/JVI.74.4.2046-2051.2000
Kowdley, K. V., Lawitz, E., Crespo, I., Hassanein, T., Davis, M. N., Demicco, M., et al. (2013). Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatment-naive patients with hepatitis C genotype-1 infection (ATOMIC): an open-label, randomised, multicentre phase 2 trial. Lancet 381, 2100–2107. doi: 10.1016/S0140-6736(13)60247-0
Kranawetter, C., Brady, S., Sun, L., Schroeder, M., Chen, S. J., and Heng, X. (2017). Nuclear magnetic resonance study of RNA structures at the 3′-end of the hepatitis C virus genome. Biochemistry 56, 4972–4984. doi: 10.1021/acs.biochem.7b00573
Lawitz, E., Lalezari, J. P., Hassanein, T., Kowdley, K. V., Poordad, F. F., Sheikh, A. M., et al. (2013). Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naive patients with genotypes 1, 2, and 3 hepatitis C infection: a randomised, double-blind, phase 2 trial. Lancet Infect. Dis. 13, 401–408. doi: 10.1016/S1473-3099(13)70033-1
Le Guillou-Guillemette, H., Vallet, S., Gaudy-Graffin, C., Payan, C., Pivert, A., Goudeau, A., et al. (2007). Genetic diversity of the hepatitis C virus: impact and issues in the antiviral therapy. World J. Gastroenterol. 13, 2416–2426. doi: 10.3748/wjg.v13.i17.2416
Lee, H., Shin, H., Wimmer, E., and Paul, A. V. (2004). cis-acting RNA signals in the NS5B C-terminal coding sequence of the hepatitis C virus genome. J. Virol. 78, 10865–10877. doi: 10.1128/JVI.78.20.10865-10877.2004
Li, K., and Lemon, S. M. (2013). Innate immune responses in hepatitis C virus infection. Semin. Immunopathol. 35, 53–72. doi: 10.1007/s00281-012-0332-x
Lohmann, V., Korner, F., Herian, U., and Bartenschlager, R. (1997). Biochemical properties of hepatitis C virus NS5B RNA-dependent RNA polymerase and identification of amino acid sequence motifs essential for enzymatic activity. J. Virol. 71, 8416–8428.
Lourenco, S., Costa, F., Debarges, B., Andrieu, T., and Cahour, A. (2008). Hepatitis C virus internal ribosome entry site-mediated translation is stimulated by cis-acting RNA elements and trans-acting viral factors. FEBS J. 275, 4179–4197. doi: 10.1111/j.1742-4658.2008.06566.x
Lu, H., Li, W., Noble, W. S., Payan, D., and Anderson, D. C. (2004). Riboproteomics of the hepatitis C virus internal ribosomal entry site. J. Proteome Res. 3, 949–957. doi: 10.1021/pr0499592
Lukavsky, P. J., Otto, G. A., Lancaster, A. M., Sarnow, P., and Puglisi, J. D. (2000). Structures of two RNA domains essential for hepatitis C virus internal ribosome entry site function. Nat. Struct. Biol. 7, 1105–1110. doi: 10.1038/81951
Lytle, J. R., Wu, L., and Robertson, H. D. (2002). Domains on the hepatitis C virus internal ribosome entry site for 40s subunit binding. RNA 8, 1045–1055. doi: 10.1017/S1355838202029965
Malygin, A. A., Kossinova, O. A., Shatsky, I. N., and Karpova, G. G. (2013a). HCV IRES interacts with the 18S rRNA to activate the 40S ribosome for subsequent steps of translation initiation. Nucleic Acids Res. 41, 8706–8714. doi: 10.1093/nar/gkt632
Malygin, A. A., Shatsky, I. N., and Karpova, G. G. (2013b). Proteins of the human 40S ribosomal subunit involved in hepatitis C IRES binding as revealed from fluorescent labeling. Biochemistry 78, 53–59. doi: 10.1134/S0006297913010069
Manrubia, S., and Lazaro, E. (2016). Getting to know viral evolutionary strategies: towards the next generation of quasispecies models. Curr. Top. Microbiol. Immunol. 392, 201–217. doi: 10.1007/82_2015_457
Markoff, L. (2003). 5′- and 3′-noncoding regions in flavivirus RNA. Adv. Virus Res. 59, 177–228. doi: 10.1016/S0065-3527(03)59006-6
Martell, M., Esteban, J. I., Quer, J., Genesca, J., Weiner, A., Esteban, R., et al. (1992). Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: quasispecies nature of HCV genome distribution. J. Virol. 66, 3225–3229.
Masante, C., Jaubert, C., Palau, W., Plissonneau, J., Besnard, L., Ventura, M., et al. (2015). Mutations of the SL2 dimerization sequence of the hepatitis C genome abrogate viral replication. Cell Mol. Life Sci. 72, 3375–3385. doi: 10.1007/s00018-015-1893-3
Matsuda, D., and Mauro, V. P. (2014). Base pairing between hepatitis C virus RNA and 18S rRNA is required for IRES-dependent translation initiation in vivo. Proc. Natl. Acad. Sci. U.S.A. 111, 15385–15389. doi: 10.1073/pnas.1413472111
Mauger, D. M., Golden, M., Yamane, D., Williford, S., Lemon, S. M., Martin, D. P., et al. (2015). Functionally conserved architecture of hepatitis C virus RNA genomes. Proc. Natl. Acad. Sci. U.S.A. 112, 3692–3697. doi: 10.1073/pnas.1416266112
Mccaffrey, A. P., Ohashi, K., Meuse, L., Shen, S., Lancaster, A. M., Lukavsky, P. J., et al. (2002). Determinants of hepatitis C translational initiation in vitro, in cultured cells and mice. Mol. Ther. 5, 676–684. doi: 10.1006/mthe.2002.0600
Mcmullan, L. K., Grakoui, A., Evans, M. J., Mihalik, K., Puig, M., Branch, A. D., et al. (2007). Evidence for a functional RNA element in the hepatitis C virus core gene. Proc. Natl. Acad. Sci. U.S.A. 104, 2879–2884. doi: 10.1073/pnas.0611267104
Messina, J. P., Humphreys, I., Flaxman, A., Brown, A., Cooke, G. S., Pybus, O. G., et al. (2015). Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 61, 77–87. doi: 10.1002/hep.27259
Meyers, N. L., Fontaine, K. A., Kumar, G. R., and Ott, M. (2016). Entangled in a membranous web: ER and lipid droplet reorganization during hepatitis C virus infection. Curr. Opin. Cell Biol. 41, 117–124. doi: 10.1016/j.ceb.2016.05.003
Mohd Hanafiah, K., Groeger, J., Flaxman, A. D., and Wiersma, S. T. (2013). Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology 57, 1333–1342. doi: 10.1002/hep.26141
Moradpour, D., Gosert, R., Egger, D., Penin, F., Blum, H. E., and Bienz, K. (2003). Membrane association of hepatitis C virus nonstructural proteins and identification of the membrane alteration that harbors the viral replication complex. Antiviral Res. 60, 103–109. doi: 10.1016/j.antiviral.2003.08.017
Morel, V., Fournier, C., Francois, C., Brochot, E., Helle, F., Duverlie, G., et al. (2011). Genetic recombination of the hepatitis C virus: clinical implications. J. Viral. Hepat. 18, 77–83. doi: 10.1111/j.1365-2893.2010.01367.x
Murakami, Y., Aly, H. H., Tajima, A., Inoue, I., and Shimotohno, K. (2009). Regulation of the hepatitis C virus genome replication by miR-199a. J. Hepatol. 50, 453–460. doi: 10.1016/j.jhep.2008.06.010
Oakland, T. E., Haselton, K. J., and Randall, G. (2013). EWSR1 binds the hepatitis C virus cis-acting replication element and is required for efficient viral replication. J. Virol. 87, 6625–6634. doi: 10.1128/JVI.01006-12
Otto, G. A., Lukavsky, P. J., Lancaster, A. M., Sarnow, P., and Puglisi, J. D. (2002). Ribosomal proteins mediate the hepatitis C virus IRES-HeLa 40S interaction. RNA 8, 913–923. doi: 10.1017/S1355838202022057
Palau, W., Masante, C., Ventura, M., and Di Primo, C. (2013). Direct evidence for RNA-RNA interactions at the 3′ end of the Hepatitis C virus genome using surface plasmon resonance. RNA 19, 982–991. doi: 10.1261/rna.037606.112
Paronetto, M. P., Minana, B., and Valcarcel, J. (2011). The Ewing sarcoma protein regulates DNA damage-induced alternative splicing. Mol. Cell 43, 353–368. doi: 10.1016/j.molcel.2011.05.035
Patino-Galindo, J. A., and Gonzalez-Candelas, F. (2017). Comparative analysis of variation and selection in the HCV genome. Infect. Genet. Evol. 49, 104–110. doi: 10.1016/j.meegid.2017.01.010
Perales, C., and Domingo, E. (2016). Antiviral strategies based on lethal mutagenesis and error threshold. Curr. Top. Microbiol. Immunol. 392, 323–339. doi: 10.1007/82_2015_459
Perard, J., Leyrat, C., Baudin, F., Drouet, E., and Jamin, M. (2013). Structure of the full-length HCV IRES in solution. Nat. Commun. 4:1612. doi: 10.1038/ncomms2611
Pestova, T. V., Shatsky, I. N., Fletcher, S. P., Jackson, R. J., and Hellen, C. U. (1998). A prokaryotic-like mode of cytoplasmic eukaryotic ribosome binding to the initiation codon during internal translation initiation of hepatitis C and classical swine fever virus RNAs. Genes Dev. 12, 67–83. doi: 10.1101/gad.12.1.67
Pirakitikulr, N., Kohlway, A., Lindenbach, B. D., and Pyle, A. M. (2016). The coding region of the HCV genome contains a network of regulatory RNA structures. Mol. Cell 62, 111–120. doi: 10.1016/j.molcel.2016.01.024
Quade, N., Boehringer, D., Leibundgut, M., Van Den Heuvel, J., and Ban, N. (2015). Cryo-EM structure of hepatitis C virus IRES bound to the human ribosome at 3.9-A resolution. Nat. Commun. 6:7646. doi: 10.1038/ncomms8646
Randall, G., Panis, M., Cooper, J. D., Tellinghuisen, T. L., Sukhodolets, K. E., Pfeffer, S., et al. (2007). Cellular cofactors affecting hepatitis C virus infection and replication. Proc. Natl. Acad. Sci. U.S.A. 104, 12884–12889. doi: 10.1073/pnas.0704894104
Reynolds, J. E., Kaminski, A., Kettinen, H. J., Grace, K., Clarke, B. E., Carroll, A. R., et al. (1995). Unique features of internal initiation of hepatitis C virus RNA translation. EMBO J. 14, 6010–6020.
Ribeiro, R. M., Li, H., Wang, S., Stoddard, M. B., Learn, G. H., Korber, B. T., et al. (2012). Quantifying the diversification of hepatitis C virus (HCV) during primary infection: estimates of the in vivo mutation rate. PLOS Pathog. 8:e1002881. doi: 10.1371/journal.ppat.1002881
Ríos-Marco, P., Romero-López, C., and Berzal-Herranz, A. (2016). The cis-acting replication element of the hepatitis C virus genome recruits host factors that influence viral replication and translation. Sci. Rep. 6:25729. doi: 10.1038/srep25729
Roberts, A. P., Lewis, A. P., and Jopling, C. L. (2011). miR-122 activates hepatitis C virus translation by a specialized mechanism requiring particular RNA components. Nucleic Acids Res. 39, 7716–7729. doi: 10.1093/nar/gkr426
Romero-López, C., Barroso-delJesus, A., and Berzal-Herranz, A. (2017). The chaperone-like activity of the hepatitis C virus IRES and CRE elements regulates genome dimerization. Sci. Rep. 7:43415. doi: 10.1038/srep43415
Romero-López, C., Barroso-delJesus, A., García-Sacristán, A., Briones, C., and Berzal-Herranz, A. (2012). The folding of the hepatitis C virus internal ribosome entry site depends on the 3′-end of the viral genome. Nucleic Acids Res. 40, 11697–11713. doi: 10.1093/nar/gks927
Romero-López, C., Barroso-delJesus, A., García-Sacristán, A., Briones, C., and Berzal-Herranz, A. (2014). End-to-end crosstalk within the hepatitis C virus genome mediates the conformational switch of the 3′X-tail region. Nucleic Acids Res. 42, 567–582. doi: 10.1093/nar/gkt841
Romero-López, C., and Berzal-Herranz, A. (2009). A long-range RNA-RNA interaction between the 5′ and 3′ ends of the HCV genome. RNA 15, 1740–1752. doi: 10.1261/rna.1680809
Romero-López, C., and Berzal-Herranz, A. (2012). The functional RNA domain 5BSL3.2 within the NS5B coding sequence influences hepatitis C virus IRES-mediated translation. Cell Mol. Life Sci. 69, 103–113. doi: 10.1007/s00018-011-0729-z
Romero-López, C., and Berzal-Herranz, A. (2013). Unmasking the information encoded as structural motifs of viral RNA genomes: a potential antiviral target. Rev. Med. Virol. 23, 340–354. doi: 10.1002/rmv.1756
Romero-López, C., and Berzal-Herranz, A. (2015). Current and emerging themes in the structural analysis of viral RNA genomes: applications for the development of novel therapeutic drugs. Genom. Comp. Biol. 1:e15. doi: 10.18547/gcb.2015.vol1.iss1.e15
Scheel, T. K., Galli, A., Li, Y. P., Mikkelsen, L. S., Gottwein, J. M., and Bukh, J. (2013). Productive homologous and non-homologous recombination of hepatitis C virus in cell culture. PLOS Pathog. 9:e1003228. doi: 10.1371/journal.ppat.1003228
Scheel, T. K., and Rice, C. M. (2013). Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat. Med. 19, 837–849. doi: 10.1038/nm.3248
Shetty, S., Kim, S., Shimakami, T., Lemon, S. M., and Mihailescu, M. R. (2010). Hepatitis C virus genomic RNA dimerization is mediated via a kissing complex intermediate. RNA 16, 913–925. doi: 10.1261/rna.1960410
Shetty, S., Stefanovic, S., and Mihailescu, M. R. (2013). Hepatitis C virus RNA: molecular switches mediated by long-range RNA-RNA interactions? Nucleic Acids Res. 41, 2526–2540. doi: 10.1093/nar/gks1318
Shulla, A., and Randall, G. (2015). Spatiotemporal analysis of hepatitis C virus infection. PLOS Pathog. 11:e1004758. doi: 10.1371/journal.ppat.1004758
Simmonds, P., Tuplin, A., and Evans, D. J. (2004). Detection of genome-scale ordered RNA structure (GORS) in genomes of positive-stranded RNA viruses: implications for virus evolution and host persistence. RNA 10, 1337–1351. doi: 10.1261/rna.7640104
Sizova, D. V., Kolupaeva, V. G., Pestova, T. V., Shatsky, I. N., and Hellen, C. U. (1998). Specific interaction of eukaryotic translation initiation factor 3 with the 5′ nontranslated regions of hepatitis C virus and classical swine fever virus RNAs. J. Virol. 72, 4775–4782.
Smith, D. B., Bukh, J., Kuiken, C., Muerhoff, A. S., Rice, C. M., Stapleton, J. T., et al. (2014). Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology 59, 318–327. doi: 10.1002/hep.26744
Smith, D. B., Mellor, J., Jarvis, L. M., Davidson, F., Kolberg, J., Urdea, M., et al. (1995). Variation of the hepatitis C virus 5′ non-coding region: implications for secondary structure, virus detection and typing. The International HCV collaborative study group. J. Gen. Virol. 76 (Pt 7), 1749–1761. doi: 10.1099/0022-1317-76-7-1749
Smith, D. B., and Simmonds, P. (1997). Characteristics of nucleotide substitution in the hepatitis C virus genome: constraints on sequence change in coding regions at both ends of the genome. J. Mol. Evol. 45, 238–246. doi: 10.1007/PL00006226
Song, Y., Friebe, P., Tzima, E., Junemann, C., Bartenschlager, R., and Niepmann, M. (2006). The hepatitis C virus RNA 3′-untranslated region strongly enhances translation directed by the internal ribosome entry site. J. Virol. 80, 11579–11588. doi: 10.1128/JVI.00675-06
Spahn, C. M., Kieft, J. S., Grassucci, R. A., Penczek, P. A., Zhou, K., Doudna, J. A., et al. (2001). Hepatitis C virus IRES RNA-induced changes in the conformation of the 40S ribosomal subunit. Science 291, 1959–1962. doi: 10.1126/science.1058409
Sun, C., Querol-Audi, J., Mortimer, S. A., Arias-Palomo, E., Doudna, J. A., Nogales, E., et al. (2013). Two RNA-binding motifs in eIF3 direct HCV IRES-dependent translation. Nucleic Acids Res. 41, 7512–7521. doi: 10.1093/nar/gkt510
Takamizawa, J., Konishi, H., Yanagisawa, K., Tomida, S., Osada, H., Endoh, H., et al. (2004). Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 64, 3753–3756. doi: 10.1158/0008-5472.CAN-04-0637
Tanaka, T., Kato, N., Cho, M. J., and Shimotohno, K. (1995). A novel sequence found at the 3′ terminus of hepatitis C virus genome. Biochem. Biophys. Res. Commun. 215, 744–749. doi: 10.1006/bbrc.1995.2526
Tanaka, T., Kato, N., Cho, M. J., Sugiyama, K., and Shimotohno, K. (1996). Structure of the 3′ terminus of the hepatitis C virus genome. J. Virol. 70, 3307–3312.
Terenin, I. M., Dmitriev, S. E., Andreev, D. E., and Shatsky, I. N. (2008). Eukaryotic translation initiation machinery can operate in a bacterial-like mode without eIF2. Nat. Struct. Mol. Biol. 15, 836–841. doi: 10.1038/nsmb.1445
Tsukiyama-Kohara, K., Iizuka, N., Kohara, M., and Nomoto, A. (1992). Internal ribosome entry site within hepatitis C virus RNA. J. Virol. 66, 1476–1483.
Tuplin, A., Evans, D. J., and Simmonds, P. (2004). Detailed mapping of RNA secondary structures in core and NS5B-encoding region sequences of hepatitis C virus by RNase cleavage and novel bioinformatic prediction methods. J. Gen. Virol. 85, 3037–3047. doi: 10.1099/vir.0.80141-0
Tuplin, A., Struthers, M., Cook, J., Bentley, K., and Evans, D. J. (2015). Inhibition of HCV translation by disrupting the structure and interactions of the viral CRE and 3′ X-tail. Nucleic Acids Res. 43, 2914–2926. doi: 10.1093/nar/gkv142
Tuplin, A., Struthers, M., Simmonds, P., and Evans, D. J. (2012). A twist in the tail: SHAPE mapping of long-range interactions and structural rearrangements of RNA elements involved in HCV replication. Nucleic Acids Res. 40, 6908–6921. doi: 10.1093/nar/gks370
Tuplin, A., Wood, J., Evans, D. J., Patel, A. H., and Simmonds, P. (2002). Thermodynamic and phylogenetic prediction of RNA secondary structures in the coding region of hepatitis C virus. RNA 8, 824–841. doi: 10.1017/S1355838202554066
Villordo, S. M., and Gamarnik, A. V. (2009). Genome cyclization as strategy for flavivirus RNA replication. Virus Res. 139, 230–239. doi: 10.1016/j.virusres.2008.07.016
Walewski, J. L., Keller, T. R., Stump, D. D., and Branch, A. D. (2001). Evidence for a new hepatitis C virus antigen encoded in an overlapping reading frame. RNA 7, 710–721. doi: 10.1017/S1355838201010111
Wang, C., Sarnow, P., and Siddiqui, A. (1993). Translation of human hepatitis C virus RNA in cultured cells is mediated by an internal ribosome-binding mechanism. J. Virol. 67, 3338–3344.
Wang, Y. L., Chen, H., Zhan, Y. Q., Yin, R. H., Li, C. Y., Ge, C. H., et al. (2016). EWSR1 regulates mitosis by dynamically influencing microtubule acetylation. Cell Cycle 15, 2202–2215. doi: 10.1080/15384101.2016.1200774
Weinlich, S., Huttelmaier, S., Schierhorn, A., Behrens, S. E., Ostareck-Lederer, A., and Ostareck, D. H. (2009). IGF2BP1 enhances HCV IRES-mediated translation initiation via the 3′UTR. RNA 15, 1528–1542. doi: 10.1261/rna.1578409
Witteveldt, J., Blundell, R., Maarleveld, J. J., Mcfadden, N., Evans, D. J., and Simmonds, P. (2014). The influence of viral RNA secondary structure on interactions with innate host cell defences. Nucleic Acids Res. 42, 3314–3329. doi: 10.1093/nar/gkt1291
Yamada, N., Tanihara, K., Takada, A., Yorihuzi, T., Tsutsumi, M., Shimomura, H., et al. (1996). Genetic organization and diversity of the 3′ noncoding region of the hepatitis C virus genome. Virology 223, 255–261. doi: 10.1006/viro.1996.0476
Yamamoto, H., Collier, M., Loerke, J., Ismer, J., Schmidt, A., Hilal, T., et al. (2015). Molecular architecture of the ribosome-bound Hepatitis C Virus internal ribosomal entry site RNA. EMBO J. 34, 3042–3058. doi: 10.15252/embj.201592469
Yamamoto, H., Unbehaun, A., Loerke, J., Behrmann, E., Collier, M., Burger, J., et al. (2014). Structure of the mammalian 80S initiation complex with initiation factor 5B on HCV-IRES RNA. Nat. Struct. Mol. Biol. 21, 721–727. doi: 10.1038/nsmb.2859
Yi, M., and Lemon, S. M. (2003). 3′ nontranslated RNA signals required for replication of hepatitis C virus RNA. J. Virol. 77, 3557–3568. doi: 10.1128/JVI.77.6.3557-3568.2003
You, S., and Rice, C. M. (2008). 3′ RNA elements in hepatitis C virus replication: kissing partners and long poly(U). J. Virol. 82, 184–195. doi: 10.1128/JVI.01796-07
You, S., Stump, D. D., Branch, A. D., and Rice, C. M. (2004). A cis-acting replication element in the sequence encoding the NS5B RNA-dependent RNA polymerase is required for hepatitis C virus RNA replication. J. Virol. 78, 1352–1366. doi: 10.1128/JVI.78.3.1352-1366.2004
Yu, F., Yao, H., Zhu, P., Zhang, X., Pan, Q., Gong, C., et al. (2007). let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 131, 1109–1123. doi: 10.1016/j.cell.2007.10.054
Zhang, J., Yamada, O., Sakamoto, T., Yoshida, H., Araki, H., Murata, T., et al. (2005). Inhibition of hepatitis C virus replication by pol III-directed overexpression of RNA decoys corresponding to stem-loop structures in the NS5B coding region. Virology 342, 276–285. doi: 10.1016/j.virol.2005.08.003
Keywords: HCV, functional RNA domains, 5BSL3.2, HCV IRES, HCV CRE, long-distant RNA–RNA interactions
Citation: Romero-López C and Berzal-Herranz A (2017) The 5BSL3.2 Functional RNA Domain Connects Distant Regions in the Hepatitis C Virus Genome. Front. Microbiol. 8:2093. doi: 10.3389/fmicb.2017.02093
Received: 11 September 2017; Accepted: 12 October 2017;
Published: 31 October 2017.
Edited by:
Ricardo Flores, Instituto de Biología Molecular y Celular de Plantas (CSIC), SpainReviewed by:
Antonio Mas, Universidad de Castilla – La Mancha, SpainCopyright © 2017 Romero-López and Berzal-Herranz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Romero-López, Y3Jpc3RpbmFfcm9tZXJvQGlwYi5jc2ljLmVz Alfredo Berzal-Herranz, YWJlcnphbGhAaXBiLmNzaWMuZXM=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.