 Filomena Perrone1
Filomena Perrone1 Barbara De Siena1
Barbara De Siena1 Lidia Muscariello1
Lidia Muscariello1 Sharon L. Kendall2
Sharon L. Kendall2 Simon J. Waddell3
Simon J. Waddell3 Margherita Sacco1*
Margherita Sacco1*- 1Dipartimento di Scienze e Tecnologie Ambientali Biologiche e Farmaceutiche, Università della Campania “Luigi Vanvitelli”, Caserta, Italy
- 2Department of Pathobiology and Population Science, Royal Veterinary College, London, United Kingdom
- 3Wellcome Trust Brighton and Sussex Centre for Global Health Research, Brighton and Sussex Medical School, University of Sussex, Brighton, United Kingdom
Mycobacterium tuberculosis has the ability to survive inside macrophages under acid-nitrosative stress. M. tuberculosis Rv1685c and its ortholog in M. smegmatis, MSMEG_3765, are induced on exposure to acid-nitrosative stress. Both genes are annotated as TetR transcriptional regulators, a family of proteins that regulate a wide range of cellular activities, including multidrug resistance, carbon catabolism and virulence. Here, we demonstrate that MSMEG_3765 is co-transcribed with the upstream genes MSMEG_3762 and MSMEG_3763, encoding efflux pump components. RTq-PCR and GFP-reporter assays showed that the MSMEG_3762/63/65 gene cluster, and the orthologous region in M. tuberculosis (Rv1687c/86c/85c), was up-regulated in a MSMEG_3765 null mutant, suggesting that MSMEG_3765 acts as a repressor, typical of this family of regulators. We further defined the MSMEG_3765 regulon using genome-wide transcriptional profiling and used reporter assays to confirm that the MSMEG_3762/63/65 promoter was induced under acid-nitrosative stress. A putative 36 bp regulatory motif was identified upstream of the gene clusters in both M. smegmatis and M. tuberculosis and purified recombinant MSMEG_3765 protein was found to bind to DNA fragments containing this motif from both M. smegmatis and M. tuberculosis upstream regulatory regions. These results suggest that the TetR repressor MSMEG_3765/Rv1685c controls expression of an efflux pump with an, as yet, undefined role in the mycobacterial response to acid-nitrosative stress.
Introduction
Tuberculosis (TB) is still endemic in many low and middle-income countries and the high incidence of Mycobacterium tuberculosis multi-drug resistant strains continues to plague the control of TB worldwide (Ayabina et al., 2016; Malone and Gordon, 2016; Dheda et al., 2017). Indeed, M. tuberculosis, the causative agent of tuberculosis, is one of the most successful bacterial pathogens with approximately 10 million new infections and 2 million deaths per year; furthermore, it is estimated that one third of the world’s populations are latently infected. The success of M. tuberculosis is partly due to its ability to manipulate the intracellular environment of macrophages. Indeed, it is able to survive within macrophages by preventing phagosome maturation, antigen processing and apoptosis, and by creating a niche where bacteria remain metabolically active and capable of replication (Gan et al., 2008; Garton and O’Hare, 2013). Bacilli are able to grow in intra- and extra-cellular environments in spite of the presence of anti-microbial effectors such as reactive oxygen and nitric oxide intermediates, acidification or metal accumulation (Butler et al., 2010; Awuh and Flo, 2017). This is due to the low permeability of the mycobacterial cell envelope for many toxic molecules, to the ability of mycobacteria to detoxify reactive oxygen and reactive nitrogen molecules, and to the maintenance of a neutral intra-bacterial pH within acidic environments. Many of these mechanisms are still poorly understood (Ehrt and Schnappinger, 2009; Evangelopoulos et al., 2015). Model systems, including the fast-growing non-pathogenic M. smegmatis, have been widely used to highlight relevant aspects of M. tuberculosis physiology, exemplified by the modulation of gene expression by the DosR and Lsr2 regulators involved in dormancy and adaptation to hypoxic stress (Colangeli et al., 2009; Trauner et al., 2012; Agrawal et al., 2015).
In recent work, Cossu et al. (2013) profiled the responses of M. tuberculosis and M. smegmatis to acid-nitrosative multi-stress, simulating a macrophage-like environment. In these conditions, Rv1685c in M. tuberculosis and its ortholog MSMEG_3765 in M. smegmatis were found to be up-regulated. Both genes are annotated as transcriptional regulators of the TetR family, sharing a high percentage of identity between their deduced amino acid sequences. Members of the TetR family of transcriptional regulators are widespread among bacteria (Cuthbertson and Nodwell, 2013). They control a wide range of cellular activities, like pathogenicity, osmotic stress responses and drug efflux (Balhana et al., 2015). The members of this family usually act as repressors, with an N-terminal DNA binding domain and a larger C-terminal domain that interacts with one or more ligands modulating the regulator’s ability to bind DNA. A diversity of ligands have been described, including antibiotics, bile acids and other toxic molecules, cell–cell signaling molecules, carbon sources, proteins and metal ions, underscoring a central role of the TetR-regulators in the adaptation of cellular processes to the changing environment (Cuthbertson and Nodwell, 2013).
Here, we characterize the TetR regulator MSMEG_3765 (Rv1685c) using a combination of mutagenesis, local and global gene expression analyses, and DNA binding studies to show that it regulates the MSMEG_3762/63/65 (and Rv1687c/86c/85c) operon encoding an efflux pump. This system is conserved in M. tuberculosis and our experiments suggest that this TetR regulator plays a novel role in the mycobacterial response to the intracellular environment.
Materials and Methods
Bacterial Strains and Culture Conditions
Escherichia coli TOP10 and DH5α were used as strains for cloning and E. coli BL21 (DE3) was used as a host for protein expression. M. smegmatis mc2155 and M. tuberculosis H37Rv were used throughout this work. The E. coli strains were grown in Luria-Bertani (LB) broth, while the mycobacterial strains were cultured in Middlebrook 7H9 broth (Difco) containing 10% oleic acid-albumin-dextrose-catalase supplement (Becton Dickinson) and 0.05% Tween 80. All strains were grown at 37°C with shaking. Hygromycin (200 μg ml-1 for E. coli and 100 μg ml-1 for M. smegmatis), kanamycin (50 μg ml-1 for E. coli and 25 μg ml-1 for M. smegmatis), 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (Xgal 50 μg ml-1) and sucrose (2% w/v) were used for selection or screening as appropriate. Acid-nitrosative multi-stress was induced in 7H9 buffered medium at pH 5.3 by the addition of NaNO2 up to a final concentration of 5 mM for 5 h. Plasmids used throughout this work are listed in Supplementary Table S1. All primer sequences used in this study are listed in Supplementary Table S2.
Bioinformatic Analysis
The bioinformatic analysis of the MSMEG_3765 locus was conducted using BLAST and Clustal Omega. The analysis to identify putative TetR binding sites was conducted using MEME1 (Bailey et al., 2009). MEME was set to find palindromic motifs with a minimum width of 6 bp and a maximum of 50 bp. MEME was set to return a maximum of three motifs.
Construction of the M. smegmatis ΔMSMEG_3765 Mutant Strain
The M. smegmatis ΔMSMEG_3765 mutant strain was isolated using a two-step homologous recombination strategy (Parish et al., 1999). A 804 bp fragment (up), containing the upstream flanking regions of MSMEG_3765 (from 3829208 to 3829941), was PCR-amplified from M. smegmatis mc2155 genomic DNA using the forward upMS3765f and reverse upMS3765r primers, with HindIII-BamHI sites respectively, to clone the up fragment into p2NIL, yielding the pFP2 plasmid. A 906 bp fragment (dw), containing the downstream flanking regions of MSMEG_3765 (from 3830395 to 3831300), was PCR-amplified from M. smegmatis mc2155 genomic DNA using the forward dwMS3765f and reverse dwMS3765r primers, with BamHI-PacI sites respectively, to clone the dw fragment into pFP2, yielding the pFP3 plasmid. To obtain the suicide delivery vector (pFP4), the PacI cassette from pGOAL19 was cloned into pFP3. pFP4 was electroporated into M. smegmatis mc2155 and single crossovers were selected using kanamycin, hygromycin and Xgal. A single blue kanamycin and hygromycin-resistant colony was streaked onto fresh media without selection, and incubated at 37°C for 3–5 days to allow for second recombination events, before selection on plates containing sucrose and Xgal. The white sucrose-resistant colonies were screened for kanamycin and hygromycin sensitivity, then analyzed by PCR to confirm the deletion in MSMEG_3765. The deletion event in M. smegmatis ΔMSMEG_3765 was verified by sequencing.
Construction of the M. smegmatis ΔMSMEG_3765 Complemented Strain
To complement M. smegmatis ΔMSMEG_3765, a DNA fragment containing the 636 bp coding sequence of MSMEG_3765 (including start and stop codons) was amplified from M. smegmatis mc2155 with forward cMS3765f and reverse cMS3765r primers. The forward primer included an optimized Shine–Dalgarno sequence (Andreu et al., 2010). The 636 bp fragment was cloned into the EcoRI-XhoI sites of pMV306hsp (Addgene#26155) to obtain pFP6, which was electroporated into M. smegmatis ΔMSMEG_3765. Recombinant clones were selected using kanamycin, and the insertion confirmed by PCR.
RNA Extraction and Reverse Transcription Reactions
RNA for RT-PCR and RTq-PCR analyses were extracted from wild-type mc2155, knockout mutant and complemented strains from log-phase cultures under standard growth conditions. The bacteria were harvested, resuspended in 4 ml of RLT buffer (RNeasy kit – Qiagen), and vortexed using glass beads. Cell lysates were recovered by centrifugation and RNAs was extracted using RNeasy kit (Qiagen), according to the manufacturer’s instructions. RNA samples were treated with RQ1 DNase (Promega) for 30 min at 37°C, followed by heat inactivation. Finally, the quality and quantity of RNA was assessed using NanoDrop spectrophotometer analysis (Nanodrop, Thermo Scientific) and gel electrophoresis. Reverse transcription was performed for 15 min at 42°C in a total volume of 20 μl containing 1 μg total RNA (QuantiTect Reverse Transcription kit-Qiagen). As negative controls, samples without the reverse transcription step were used as template.
RT-PCR and RT-qPCR Analyses
RT-PCR was performed to evaluate the transcriptional organization of the MSMEG_3765 locus. Oligonucleotides were designed to span the intergenic regions from MSMEG_3760 to MSMEG_3765 (Supplementary Figure S1). As positive controls, each region was amplified using M. smegmatis mc2155 chromosomal DNA as template. Analysis by Real time PCR was performed using SYBR green technology on a 7500 RT-qPCR system instrument (Applied Biosystems). The sigA gene was used as an internal standard for expression analysis. The PCR conditions included an initial denaturation at 95°C for 10 min, followed by 40 cycles of amplification of 15 s at 95°C, 1 min at 60°C, and 30 s at 72°C. RT-qPCR analysis was performed in triplicate, and each assay included standard curves for both internal control and target genes, obtained by amplifying serial dilutions (ratio 1:10) of the samples. Relative expression levels were normalized using sigA and calculated using the 2-ΔΔCt method (Livak and Schmittgen, 2001).
Microarray Hybridization, and Data Analysis
Total RNA was extracted from mid-log phase M. smegmatis wt, ΔMSMEG_3765 and complemented strains grown in 7H9/ADC at 37°C with shaking using the GTC/Trizol method (Waddell and Butcher, 2010). Samples were DNase-treated and purified using RNeasy columns (Qiagen), and the quality assessed by Agilent 2100 Bioanalyzer. Mycobacterial RNA (1 μg) was directly labeled with Cy3 fluorophore using the Universal Linkage System (ULS, Kreatech Diagnostics). Microarray hybridizations from three biological replicates (two for the complemented strain) were conducted as previously described (Tailleux et al., 2008) using an M. smegmatis microarray (Agilent Technologies) designed by the Bacterial Microarray Group at St. George’s (ArrayExpress accession number A-BUGS-39). To define the ΔMSMEG_3765 regulon, significantly differentially expressed genes were identified comparing ΔMSMEG_3765 to both wt and complemented strains using a moderated t-test (p-value < 0.05 with Benjamini and Hochberg multiple testing correction) and a >2.5-fold change. Fully annotated microarray data have been deposited in ArrayExpress (accession number E-MTAB-5869).
GFP-Reporter Assay
To identify the region containing putative MSMEG_3765-responding operators, we cloned: (1) a 277 bp DNA fragment containing 43 bp coding sequence and 234 bp upstream from the ATG codon of MSMEG_3760, using MS13f and MS13r primers; (2) a 218 bp DNA fragment containing 106 bp of MSMEG_3761 coding sequence, 36 bp of MSMEG_3762 coding sequence and 76 bp of intergenic non-coding region, using MS14f and MS14r primers. The PCR products, obtained with iProof high-fidelity Taq (Bio-Rad), were cloned into the BamHI and ApaI sites of the E. coli–mycobacteria shuttle vector pFPV27 (Valdivia et al., 1996), yielding pFP13 and pFP14 plasmids, respectively. To analyze the promoter region of the M. tuberculosis H37Rv Rv1687c gene, we cloned a 200 bp DNA fragment, containing 40 bp coding sequence and 160 bp upstream from the ATG codon of Rv1687c into the pFPV27 plasmid, using Rv10f and Rv10r primers, yielding the pFP10 plasmid.
The resulting recombinant plasmids, together with the pFPV27 negative control plasmid (empty vector) and the pFPV27hsp positive control plasmid (carrying the mycobacterial hsp60 promoter) (Dhandayuthapani et al., 1995), were electroporated separately into M. smegmatis wt and M. smegmatis ΔMSMEG_3765 cells. GFP fluorescence was measured as described previously (Carroll and James, 2009), with excitation at 490 nm and emission at 510 nm. To evaluate promoter activity of MSMEG_3762 under acid-nitrosative multi-stress, M. smegmatis (pFP14) log-phase cells were incubated for 5 h in 7H9 buffered medium at pH 5.3 and supplemented with 5 mM NaNO2 before measuring fluorescence.
Expression and Purification of Recombinant MSMEG_3765
The MSMEG_3765 coding region (630 bp) was PCR-amplified from M. smegmatis mc2155 genomic DNA using eMS3765f and eMS3765r primers, and the product was cloned into the NdeI-XhoI sites of the pET-22b(+) expression vector. The recombinant plasmid, pFP5, was sequence verified and used to express and purify the C-terminally His-tagged TetR3765 protein. For expression, E. coli BL21 (DE3) cultures containing pFP5 were grown at 37°C to mid-exponential phase. Cultures were induced with 1 mM IPTG for 2 h at 37°C and harvested by centrifugation. The recombinant protein was purified using Profinity IMAC resins (Bio-Rad) following manufacturer’s instructions and concentrated using Centricon ultra-filtration spin columns (cut-off 10 kDa; Millipore) in 20 mM Tris HCl pH 8, 500 mM NaCl. The purified protein was correctly folded, as evaluated by circular dichroism spectroscopy (data not shown).
Electrophoretic Mobility Shift Assays
Binding assays were performed incubating increasing concentrations of purified recombinant MSMEG_3765 (from 1.5 to 10 pmol) in HEPES 40 mM (pH 8.0), NaCl 150 mM, MgCl2 20 mM, 10% glycerol with 0.5 pmol of MSMEG_3762 (218 bp) or Rv1687c (200 bp) upstream fragments (used in the GFP assays). The binding reaction was performed at 37°C for 20 min. The resulting DNA/protein complexes were resolved on native PAGE gels (8% acrylamide:bisacrylamide 30:1) and stained with SYBR Safe DNA Gel Stain (Invitrogen). To probe the specificity of MSMEG_3765 binding activity to MSMEG_3762 and Rv1687c upstream regions, reactions were incubated in the presence of 0.5 pmol MSMEG_3760 upstream fragment (127 bp, primers mot3760f1/MS13r) as non-specific competitor DNA. The MSMEG_3762 upstream fragment of 218 bp was refined into four shorter fragments for further EMSA analysis: (a) 133 bp containing 21 bp of the MSMEG_3761 coding sequence, 36 bp of MSMEG_3762 coding sequence and 76 bp of intergenic non-coding region (primers mot3762f1/MS14r); (b) 182 bp containing 106 bp of the MSMEG_3761 coding sequence and 76 bp of intergenic non-coding region (primers MS14f/mot3762r1); (c) 97 bp containing 21 bp of the MSMEG_3761 coding sequence and 76 bp of intergenic non-coding region (primers mot3762f1/mot3762r1); (d) 36 bp putative TetR binding motif.
Results
MSMEG_3765 and the Surrounding Region Is Conserved in M. tuberculosis and Other Pathogenic Mycobacteria
Mycobacterium tuberculosis H37Rv Rv1685c and M. smegmatis MSMEG_3765 TetR transcriptional regulators share 71% amino acid identity. In order to identify their distribution among other mycobacteria, we searched for orthologs in other species. The regulator is conserved in mycobacteria with amino acid identities ranging from 62 to 73% (Figure 1A). In all species, the regulator is preceded by two genes in the same transcriptional orientation, annotated as an ABC transporter ATP-binding protein and an ABC transporter, that also share high percentage of identity between species (68–79%).
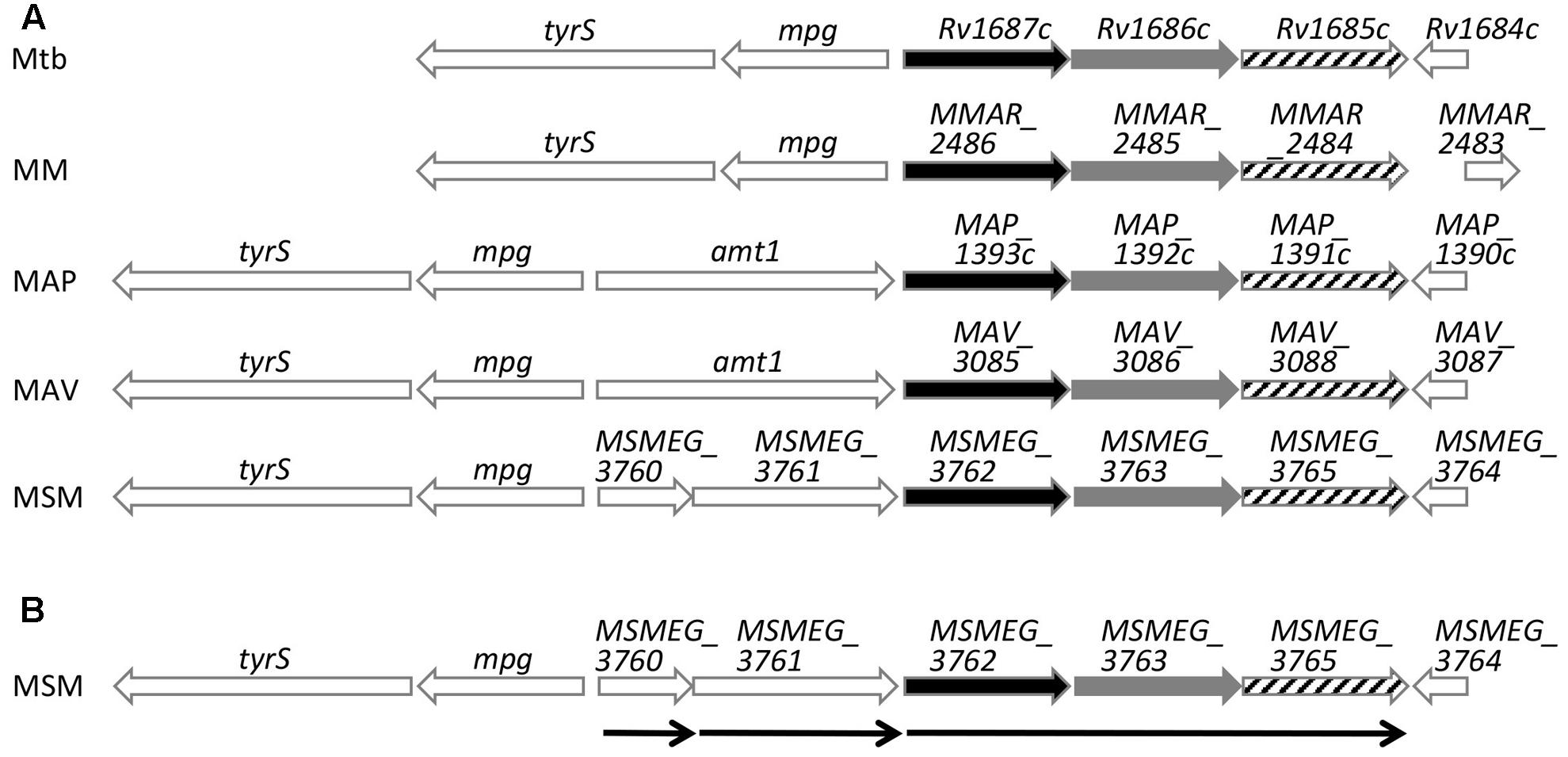
FIGURE 1. Schematic representation of Rv1685c TetR loci sequence alignments. (A) Alignment of M. tuberculosis H37Rv (Mtb), M. marinum (MM), M. avium subsp. paratuberculosis str. k10 (MAP), M. avium (MAV), and M. smegmatis (MSM); black bars indicate ABC transporter ATP-binding proteins, gray bars indicate ABC transporters and stripped bars indicate TetR transcriptional regulators. (B) Schematic representation of transcriptional units of the M. smegmatis MSMEG_3765 tetR locus determined by RT-PCR. Thin arrows indicate transcriptional units and direction of transcription.
MSMEG_3765 Is Co-transcribed with the Upstream Genes MSMEG_3763 and MSMEG_3762
In order to define the transcriptional unit(s) in the MSMEG_3765 locus, an RT-PCR analysis was performed on total RNA extracted from log phase M. smegmatis mc2155 using oligonucleotide pairs designed to detect transcripts of individual or co-transcribed genes. MSMEG_3762, MSMEG_3763 and MSMEG_3765 were found to be co-transcribed defining the MSMEG_3762/63/65 operon (Figure 1B and Supplementary Figure S1). The analysis indicated that MSMEG_3760 and MSMEG_3761, were not part of the 3 gene operon but were transcribed as monocistronic units (Supplementary Figure S1).
MSMEG_3765 Acts as a Transcriptional Repressor of the MSMEG_3762/63/65 Operon
Many TetR-like proteins have been shown to regulate adjacent genes (Balhana et al., 2015). RTq-PCR analysis was used to determine the effect of deletion of MSMEG_3765 on the expression of the surrounding genes. A M. smegmatis mutant strain carrying a 495 bp deletion in MSMEG_3765 was generated, ΔMSMEG_3765. The deletion was also complemented using the integrative recombinant pFP6 plasmid, harboring the MSMEG_3765 coding sequence under the control of the hsp60 promoter. Deletion or complementation of MSMEG_3765 did not affect growth in standard conditions, with a doubling time of ∼3 h for wt, mutant and complemented strains. Transcription of the MSMEG_3762/63/65 operon (as assessed by measuring the first gene in the operon MSMEG_3762) was upregulated by 55-fold in the mutant strain versus the wt (Figure 2A), whereas the expression of MSMEG_3760 and MSMEG_3761 was unchanged. The expression of the MSMEG_3762/63/65 operon was restored to wt levels in the complemented strain (Figure 2B). These data strongly suggest that MSMEG_3765 acts as a repressor of its own operon but does not control the expression of MSMEG_3760 or MSMEG_3761.
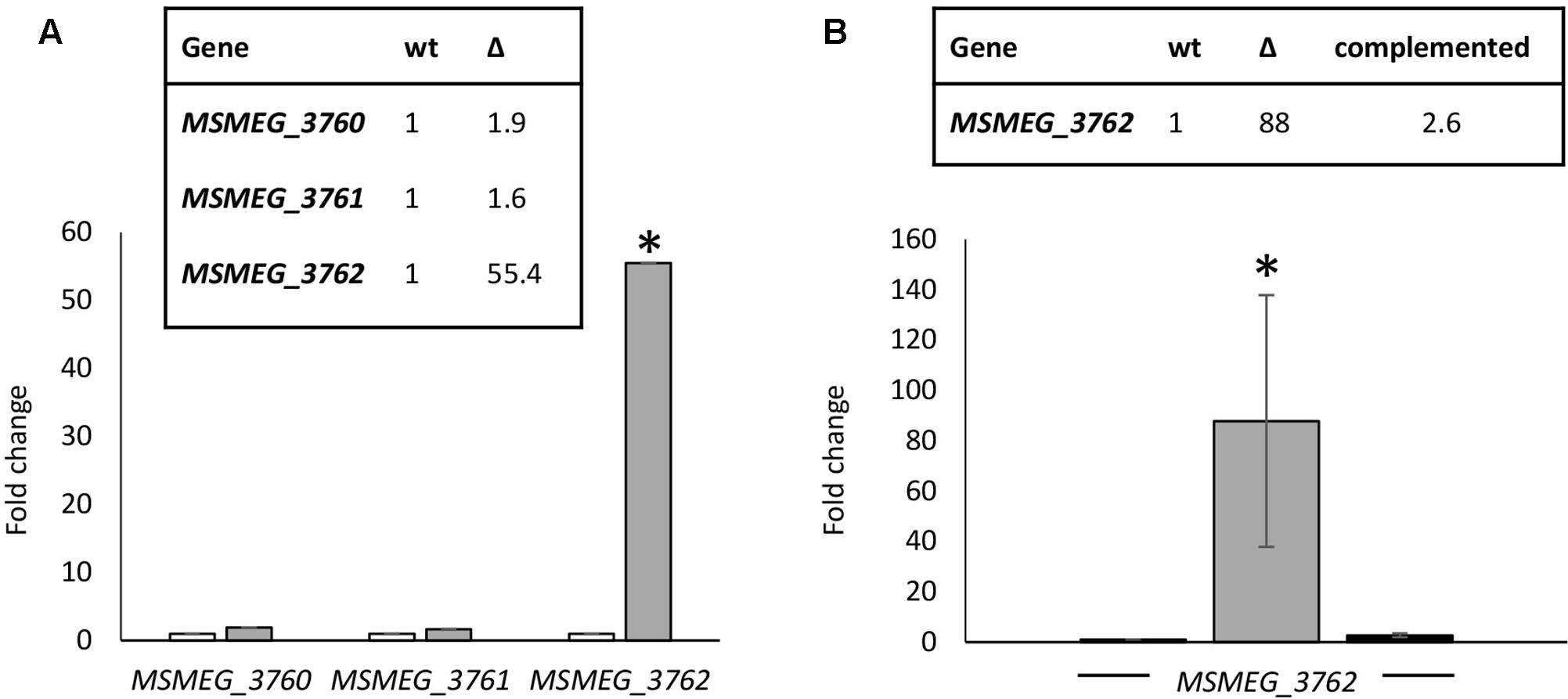
FIGURE 2. Comparative transcriptional analysis of the M. smegmatis TetR locus. (A) Expression levels of MSMEG_3760, MSMEG_3761, and MSMEG_3762-63-65 were evaluated by RT-qPCR in wt (white) and ΔMSMEG_3765 (gray); (B) RT-qPCR analysis of the MSMEG_3762-63-65 operon in wt (white), ΔMSMEG_3765 (gray) and complemented ΔMSMEG_3765 pFP6 (black) strains. Expression of each gene in the wild-type was arbitrarily assigned as 1. Error bars represent standard deviation of the mean values of three independent experiments and each RT-qPCR was carried out in triplicate (mean values are also shown in the boxes). sigA was used as reference gene and relative expression levels were calculated with the 2-ΔΔCt method; ∗p < 0.001.
Genome-wide transcriptional profiling was applied as an unbiased approach to further describe the MSMEG_3765 regulon. The MSMEG_3765 regulon was defined as genes significantly differentially expressed in M. smegmatis ΔMSMEG_3765 compared to both wt and complemented strains to control for the possibility of polar effects of gene manipulation. Microarray analysis confirmed that MSMEG_3765 acts as a repressor of MSMEG_3762 and MSMEG_3763 with both genes induced in the ΔMSMEG_3765 genetic background (Table 1).

TABLE 1. The TetR-MSMEG_3765 regulon determined by genome-wide transcriptional profiling.
Identification of a Regulatory Motif Upstream of the MSMEG_3762/63/65 Operon
TetR proteins often bind palindromic regulatory sequences, therefore we searched for palindromic motifs upstream of the MSMEG_3762/63/65 operon. A conserved 36 bp region, containing a 34 bp palindrome, at the 5′ intergenic region extending into the coding sequence of M. tuberculosis Rv1687c, M. smegmatis MSMEG_3762, M. marinum MMAR_2486, M. avium subsp. paratuberculosis MAP_1393c, M. avium MAV_3085 genes was found (Figure 3A). This motif overlaps with a putative TATA box found 10 bp upstream of the translation start site for each gene.

FIGURE 3. MEME motif analysis for putative TetR binding sites. (A) Palindromic motif (36 bp) identified in the intergenic region of M. tuberculosis Rv1687c, M. marinum MMAR_2486, M. avium subsp. paratuberculosis MAP_1393c, M. avium MAV_3085 and M. smegmatis MSMEG_3762 genes, extending into the predicted coding regions (e-value = 3.1 × 10-27). (B) Palindromic motifs identified in the MSMEG_3760 upstream region, located –47 bp (e-value = 7.53 × 10-10) and –19 bp (e-value = 2.06 × 10-9) upstream of the ATG start codon. Asterisks indicate the predicted start codon; motifs identified by MEME are underlined.
TetR proteins often control divergently oriented genes, therefore we searched for additional motifs in the intergenic region upstream of mpg/amt1 in M. avium paratuberculosis and M. avium and mpg/MSMEG_3760 in M. smegmatis. Two copies of a 16 bp motif containing a 6 bp palindrome were found in this region in the M. smegmatis genome but not in the other genomes (Figure 3B). The RTq-PCR data suggested that this motif was not recognized by MSMEG_3765, however, reporter assays were performed to determine the functionality of both motifs.
Characterization of MSMEG_3765 Binding Sites Using GFP-Promoter Probes
Several reporter strains were made by cloning the promoter regions (including the motif regions) from MSMEG_3760, MSMEG_3762/63/65, and Rv1687c/85c/85c upstream of GFP to assay promoter activity. Promoterless GFP (empty vector) and GFP under the control of the hsp60 promoter were used as negative and positive controls, respectively. Expression of GFP from the MSMEG_3760 promoter resulted in a small but significant increase in fluorescence compared to empty vector (p < 0.01), but no significant difference was observed between wt and ΔMSMEG_3765 backgrounds (Figure 4A). Conversely, expression of GFP from the MSMEG_3762/63/65 promoter showed a significant increase (3.2-fold, p < 0.0001) in the ΔMSMEG_3765 compared to wt (Figure 4B). These results suggest that the promoter region upstream of MSMEG_3760 is active but not controlled by MSMEG_3765, while the MSMEG_3762/63/65 promoter is active and negatively regulated by MSMEG_3765. Since the motif identified in this region was also present in the M. tuberculosis Rv1687c/86c/85c gene cluster, a 200 bp fragment, containing the Rv1687c upstream sequence was assayed in M. smegmatis wt and ΔMSMEG_3765 strains. Promoter activity was detected in both genetic backgrounds with a fivefold increase (p < 0.0001) in expression in the mutant strain (Figure 4C), suggesting that the regulation of this gene cluster is conserved in M. tuberculosis.
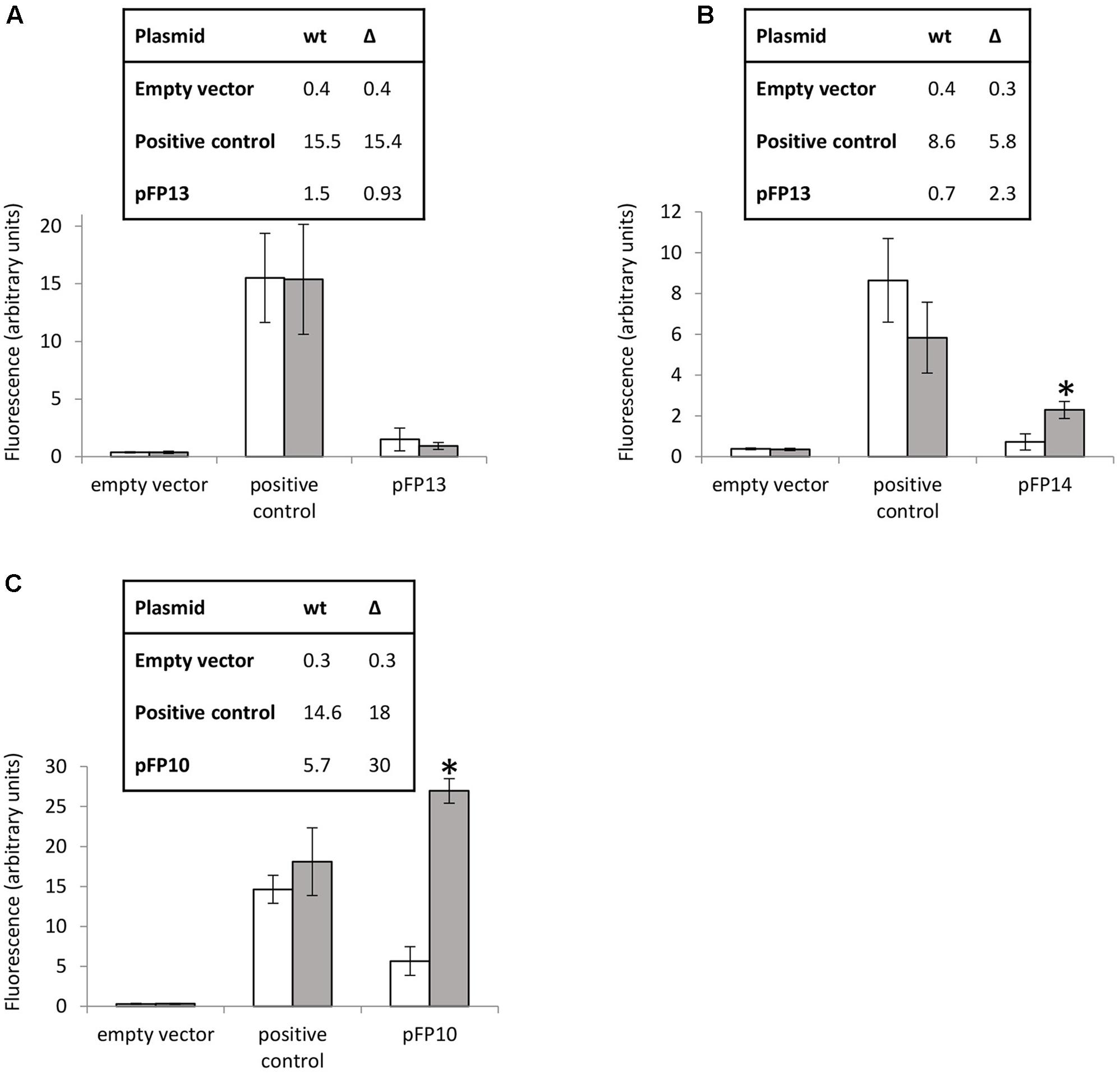
FIGURE 4. MSMEG_3765 binding upstream of MSMEG_3762 and Rv1685c established by promoter activity. Promoter probe assays of (A) MSMEG_3760 upstream region (pFP13); (B) MSMEG_3762 upstream region (pFP14); and (C) Rv1687c upstream region (pFP10). Empty vector (pFPV27) acted as the negative control; pFPV27hsp, carrying the mycobacterial hsp60 promoter, acted as the positive control. The analysis was performed in M. smegmatis wt (white) and ΔMSMEG_3765 (gray) genetic backgrounds. All the experiments were performed three times in triplicate. Error bars represent standard deviation of the mean (mean values are also shown in the boxes); ∗p < 0.0001.
Previous reports indicated that the Rv1687c/86c/85c gene cluster is induced by acid-nitrosative stress (Cossu et al., 2013). Therefore, we measured the promoter activity of the MSMEG_3762/63/65 upstream region under the same conditions. A small but significant increase in promoter activity was detected after exposure to 5 mM NaNO2 at pH 5.3 (Figure 5), further evidence that this TetR-regulator is involved in the response to acid-nitrosative stress in M. smegmatis and M. tuberculosis.
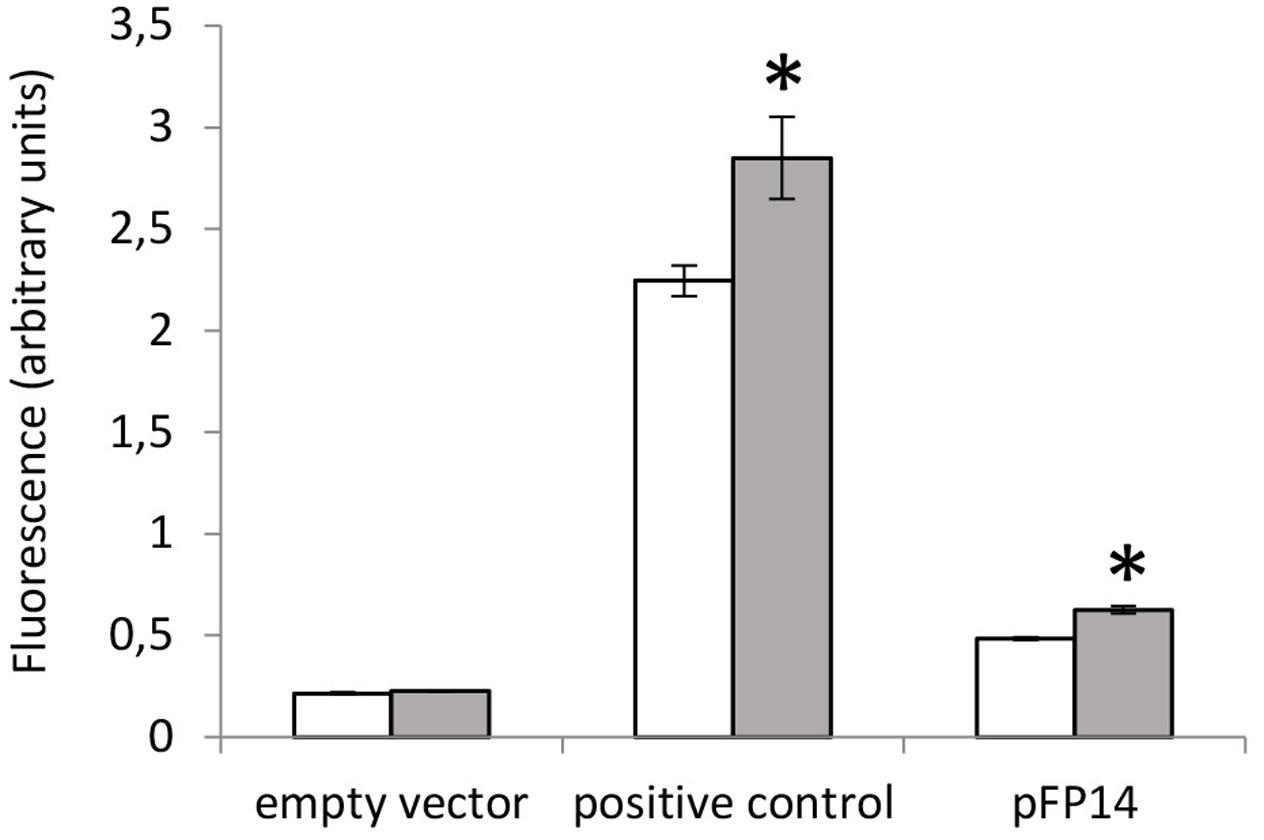
FIGURE 5. Acid-nitrosative stress induced-activity at the MSMEG_3762 upstream region by promoter probe assay. GFP assay of the MSMEG_3762 upstream region (pFP14) in wt M. smegmatis in standard conditions (white) and after acid-nitrosative stress (gray). Empty vector (pFPV27) acted as the negative control; pFPV27hsp, carrying the mycobacterial hsp60 promoter, acted as the positive control. The experiment was performed twice in triplicate. Error bars represent standard deviation of the mean; ∗p < 0.01.
Confirmation of MSMEG_3765 DNA Binding and Target Sequence by EMSA
The promoter studies suggested that MSMEG_3765 binds to the motif upstream of the MSMEG_3762/63/65 cluster but not to the motif identified upstream of MSMEG_3760. To verify the binding activity of MSMEG_3765, we conducted an electrophoretic mobility shift assay (EMSA) with the purified recombinant protein. A preliminary experiment using the three DNA fragments upstream of the MSMEG_3760, MSMEG_3762/63/65, and Rv1687c86c/85c genes (from the GFP assays) was performed. The upstream MSMEG_3760 fragment did not show a shift of electrophoretic mobility in the presence of the MSMEG_3765 recombinant protein, while the other two DNA fragments were shifted in the presence of the protein (Supplementary Figure S2).
Electrophoretic mobility shift assay experiments were then performed using the 127 bp upstream MSMEG_3760 fragment as a negative control in co-migration experiments either with MSMEG_3762 or Rv1687c upstream sequences. Increasing input of recombinant MSMEG_3765 protein resulted in enhanced shift of the MSMEG_3762 upstream fragment (218 bp) up to saturation (Figure 6A, lanes 2–4), while no shift was detected for the negative control fragment. In addition, a MSMEG_3765 concentration-dependent shift in electrophoretic mobility was observed for the Rv1687c upstream fragment (200 bp), also up to saturation with the highest concentration of MSMEG_3765 tested (Figure 6B, lanes 2–4). The EMSA analysis shows specific binding activity of MSMEG_3765 at the upstream regulatory regions of the M. smegmatis MSMEG_3762/63/65 and homologous M. tuberculosis Rv1687c/86c/85c operons.
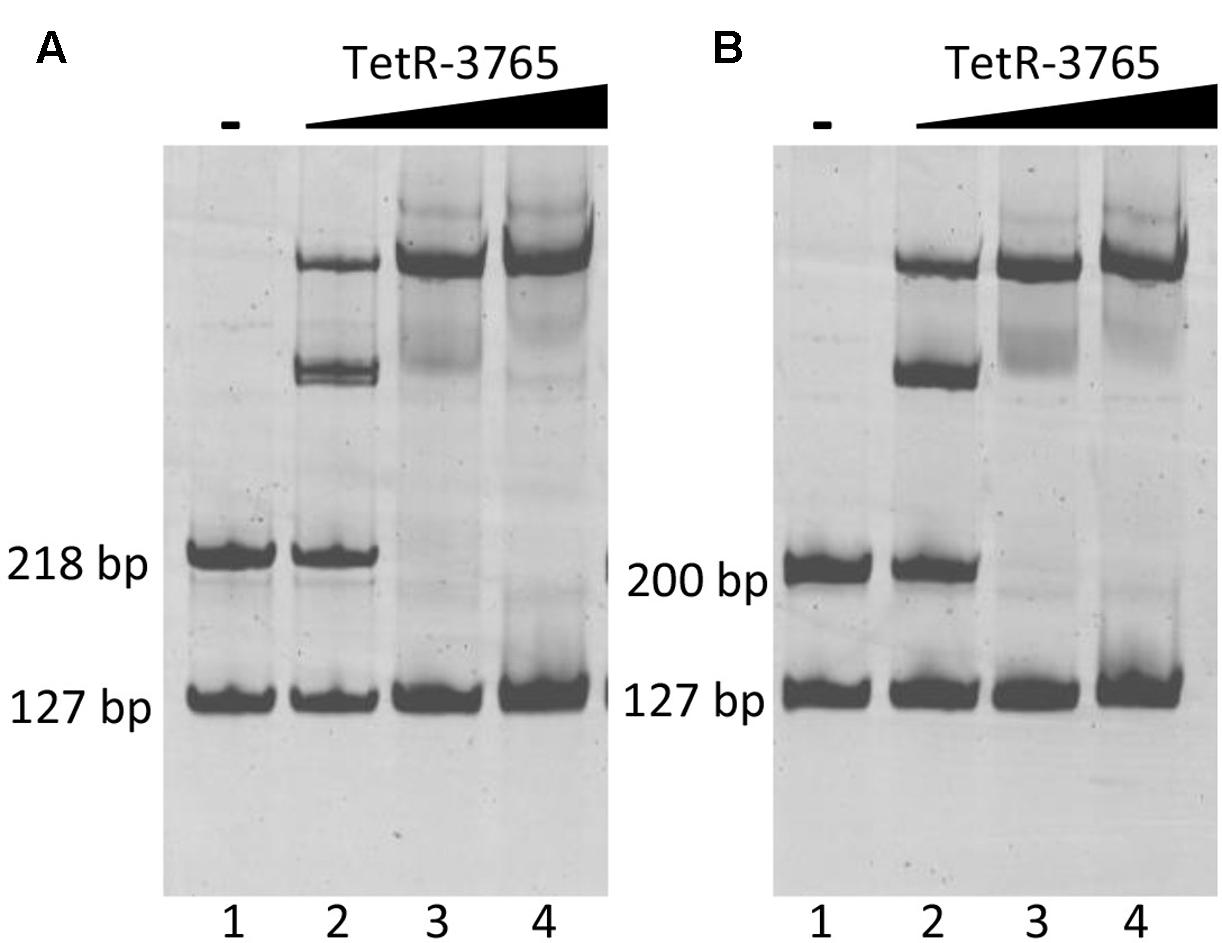
FIGURE 6. Binding of TetR-MSMEG_3765 to MSMEG_3762 and Rv1687c promoters verified by EMSA. (A) M. smegmatis MSMEG_3762 upstream fragment (218 bp); (B) M. tuberculosis Rv1687c upstream fragment (200 bp). Lane 1: negative control in which no protein was added. Lanes 2–4: binding reactions in the presence of 1.5, 5, 10 pmol of recombinant MSMEG_3765 showing a concentration-dependent gel shift in (A,B). All reactions were carried out in the presence of the MSMEG_3760 upstream fragment (127 bp) as the negative control fragment.
The 218 bp MSMEG_3762 upstream fragment was truncated for further EMSA. Three DNA fragments were analyzed: (a) a 133 bp DNA fragment depleted of 85 bp MSMEG_3761 coding sequence (truncated at the 5′ end); (b) a 182 bp DNA fragment depleted of 36 bp MSMEG_3762 coding sequence (truncated at the 3′ end); (c) a 97 bp DNA fragment depleted of 85 bp MSMEG_3761 coding sequence and of 36 bp MSMEG_3762 coding sequence (truncated at the 5′ and 3′ ends) (Figure 7A). Only the 133 bp fragment, extending up to the 12th codon into the MSMEG_3762 coding region, retained binding activity for the MSMEG_3765 protein (Figure 7B), showing that truncation of the motif at the 3′ end destroyed binding.
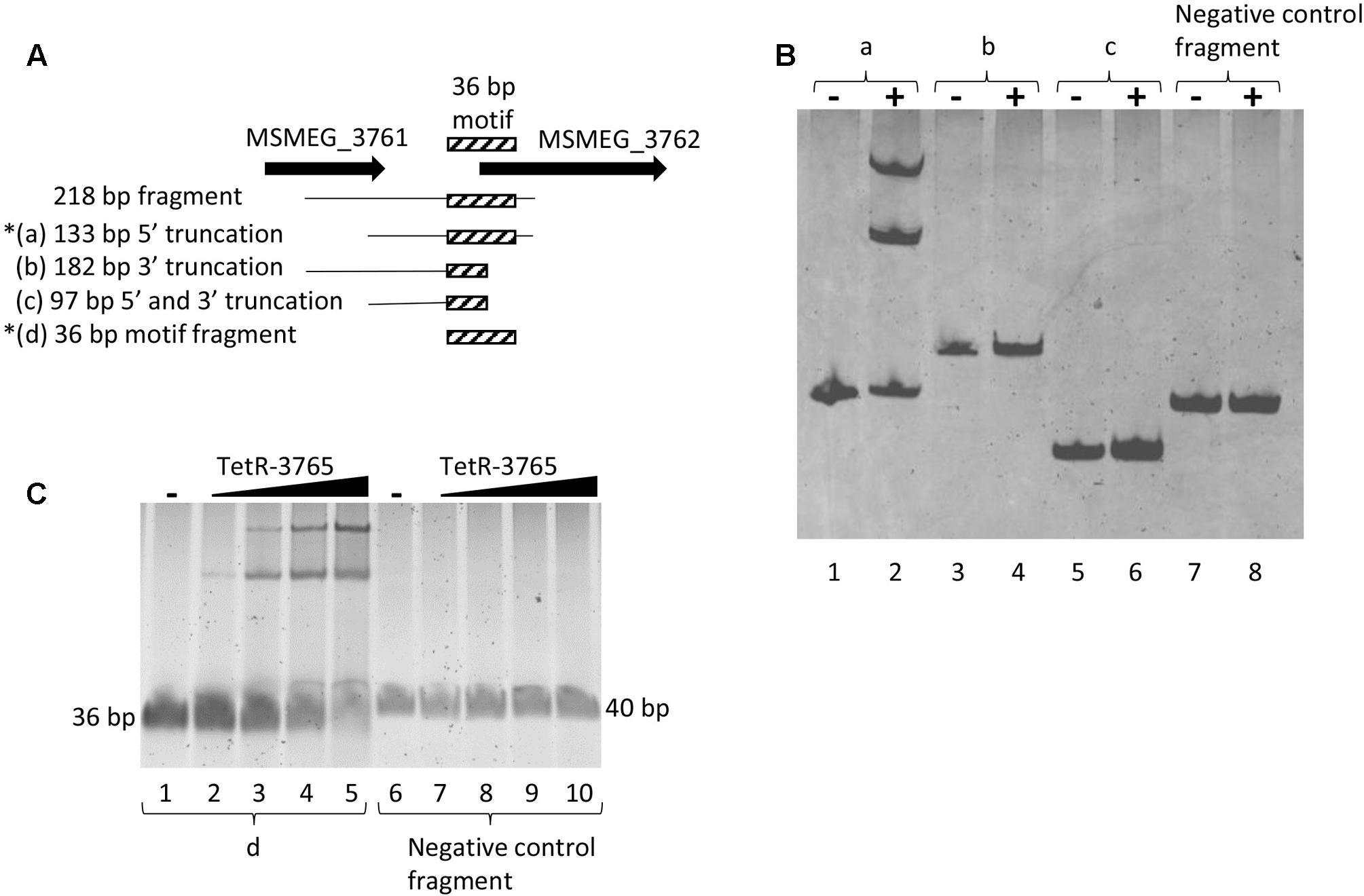
FIGURE 7. Refinement of the MSMEG_3765 binding site to 133 bp MSMEG_3762 upstream sequence. (A) Schematic representation of DNA fragments used in the EMSA assay; the hatched box indicates the position of the conserved 36 bp TetR-binding motif (Figure 3A) truncated in (b) and in (c). Asterisks mark MSMEG_3765-binding by EMSA. (B) EMSA assay using the (a), (b) and (c) fragments and a 127 bp fragment containing the MSMEG_3760 promoter as negative control (Negative control fragment). Five pmol of the recombinant MSMEG_3765 protein was used in the binding reactions (+), while no protein was added in the controls (–). (C) EMSA assay using the d fragment (lanes 1–5) and a 40 bp MSMEG_3760 upstream fragment (Negative control fragment, lanes 6–10) containing the palindromic motif shown in Figure 3B. Lanes 1 and 6: no protein was added (–). Lanes 2–5 and 7–10: binding reactions in the presence of 1.5, 5, 10, and 20 pmol recombinant MSMEG_3765.
To further characterize the DNA motif for MSMEG_3765 binding, the 36 bp MSMEG_3762 upstream fragment (Figure 3A) and a 40 bp MSMEG_3760 upstream fragment, containing the palindromic motif shown in Figure 3B, were tested by EMSA. The former fragment showed a MSMEG_3765 concentration-dependent shift in electrophoretic mobility, while the latter fragment was not affected by the presence of the protein (Figure 7C). These data correlate with the MEME analysis (Figure 3A) and demonstrate that MSMEG_3765, acting as transcriptional repressor, binds to a 36 bp TetR-like motif extending into the coding sequence of MSMEG_3762 and M. tuberculosis ortholog Rv1687c operons, controlling expression.
Discussion
Mycobacterium tuberculosis is a well-adapted intracellular pathogen, employing multiple strategies to survive within macrophages. Many of the mechanisms that enable M. tuberculosis to survive stresses encountered in macrophages are still poorly understood. Experimental strategies involving in vitro-simulated phagosomal environments have been widely used to highlight differential gene expression of M. tuberculosis to the changing microenvironment (Kendall et al., 2004; Bacon and Marsh, 2007; Voskuil et al., 2011; Green et al., 2014; Salina et al., 2014; Namouchi et al., 2016; Bansal et al., 2017; Botella et al., 2017). In a comparative study, M. tuberculosis and M. smegmatis responses to acid nitrosative stress, mimicking the macrophage environment, were determined by transcriptional profiling (Cossu et al., 2013). Rv1685c in M. tuberculosis and its ortholog MSMEG_3765 in M. smegmatis, both annotated as TetR transcriptional regulators, were found to be up-regulated. Members of the TetR family of transcriptional regulators are very common in bacteria and are involved in the control of efflux pumps, along with other cellular activities (Cuthbertson and Nodwell, 2013; Deng et al., 2013; Xu, 2016).
In this study, we show that MSMEG_3765 is co-transcribed with MSMEG_3762 and MSMEG_3763 (encoding an ABC transporter system), and that the MSMEG_3762/63/65 operon is regulated by MSMEG_3765. Given that this region and regulatory motif is conserved in M. tuberculosis, it is likely that the equivalent regulator in M. tuberculosis (Rv1685c) also controls the orthologous region in M. tuberculosis (Rv1687c/86c/85c). This is supported by the observation that the Rv1687c promoter is de-repressed in ΔMSMEG_3765 genetic background (Figure 4C). Understanding the function of MSMEG_3762/63/65 and Rv1687c/86c/85c is crucial to identifying the physiological role of this tightly regulated system.
In addition to up-regulation by acid-nitrosative stress, Rv1687c/86c/85c have been shown to be induced upon exposure to triclosan and lupulone, compounds that show potential as anti-mycobacterial agents, therefore it is feasible to speculate that the TetR-regulated ABC transporter is involved in drug efflux (Betts et al., 2003; Wei et al., 2014). A recent study, however, has shown that over-expression of Rv1686c-Rv1687c did not result in an increase in MIC to these compounds and further work is needed to investigate the role of this cluster in drug efflux (Gomez et al., 2016). The observation that other mycobacterial ABC transporters are involved in drug resistance in clinical isolates highlights the importance of extending our knowledge of the function and regulation of this cluster (Hao et al., 2011; Wang et al., 2013). Given the observation that the ABC transporter is up-regulated in acid-nitrosative stress, and in M. tuberculosis after macrophage infection (Schnappinger et al., 2003; Rohde et al., 2007) it is tempting to speculate that over-expression of the transporter may be functionally significant in the export of toxic compounds generated under these conditions, although ABC transporters may have multiple other roles in virulence including secretion of enzymes and siderophores, and the import of nutrients, ions and osmoprotective moieties (Lewis et al., 2012).
Of the 52 TetR regulators in the M. tuberculosis genome only a small percentage have been characterized (Balhana et al., 2015). In this study we have predicted and verified a novel regulatory motif, this is a 36 bp imperfect palindrome at the 5′ intergenic region extending into the coding sequence of M. tuberculosis Rv1687c, M. marinum MMAR_2486, M. avium subsp. paratuberculosis MAP_1393c, M. avium MAV_3085, and M. smegmatis MSMEG_3762 genes (Figure 3). Short (16 bp) palindromic motifs are frequently described for this family of regulators, although there are exceptions. EthR, a TetR regulator involved in ethionamide bio-activation binds to a 55 bp operator containing imperfect direct repeats (Engohang-Ndong et al., 2004). We have unequivocally demonstrated binding of purified recombinant MSMEG_3765 to a short 36 bp fragment containing this motif and shown that this binding is abrogated when the motif is truncated at the 3′ end. Further work is necessary in order to determine the affinity of the regulator for the motif and the ligand of this tightly regulated system involved in the mycobacterial response to antimicrobial drugs and the macrophage intracellular environment.
Author Contributions
FP, BDS, and LM performed the experimental work under the supervision of MS, and FP performed the microarrays and GFP assays under the supervision of SW, at the University of Sussex. SK and FP performed the bioinformatics work. MS and SK wrote the manuscript with a consistent contribution of FP, LM, and SW.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
This work was partially supported by PON03PE_00060_2 – Salute, Biotecnologie e Agroalimentare.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.02039/full#supplementary-material
Footnotes
References
Agrawal, P., Miryala, S., and Varshney, U. (2015). Use of Mycobacterium smegmatis deficient in ADP-ribosyltransferase as surrogate for Mycobacterium tuberculosis in drug testing and mutation analysis. PLOS ONE 10:e0122076. doi: 10.1371/journal.pone.0122076
Andreu, N., Zelmer, A., Fletcher, T., Elkington, P. T., Ward, T. H., Ripoll, J., et al. (2010). Optimisation of bioluminescent reporters for use with mycobacteria. PLOS ONE 5:e10777. doi: 10.1371/journal.pone.0010777
Awuh, J. A., and Flo, T. H. (2017). Molecular basis of mycobacterial survival in macrophages. Cell Mol. Life Sci. 74, 1625–1648. doi: 10.1007/s00018-016-2422-8
Ayabina, D., Hendon-Dunn, C., Bacon, J., and Colijn, C. (2016). Diverse drug-resistant subpopulations of Mycobacterium tuberculosis are sustained in continuous cultures. J. R. Soc. Interface 13:20160745. doi: 10.1098/rsif.2016.0745
Bacon, J., and Marsh, P. D. (2007). Transcriptional responses of Mycobacterium tuberculosis exposed to adverse conditions in vitro. Curr. Mol. Med. 7, 277–286.
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. doi: 10.1093/nar/gkp335
Balhana, R. J. C., Singla, A., Sikder, M. H., Withers, M., and Kendall, S. L. (2015). Global analyses of TetR family transcriptional regulators in mycobacteria indicates conservation across species and diversity in regulated functions. BMC Genomics 16:479. doi: 10.1186/s12864-015-1696-9
Bansal, R., Anil Kumar, V., Sevalkar, R. R., Singh, P. R., and Sarkar, D. (2017). Mycobacterium tuberculosis virulence-regulator PhoP interacts with alternative sigma factor SigE during acid-stress response. Mol. Microbiol. 104, 400–411. doi: 10.1111/mmi.13635
Betts, J. C., McLaren, A., Lennon, M. G., Kelly, F. M., Lukey, P. T., Blakemore, S. J., et al. (2003). Signature gene expression profiles discriminate between isoniazid-, thiolactomycin-, and triclosan-treated Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 47, 2903–2913. doi: 10.1128/AAC.47.9.2903-2913.2003
Botella, H., Vaubourgeix, J., Lee, M. H., Song, N., Xu, W., Makinoshima, H., et al. (2017). Mycobacterium tuberculosis protease MarP activates a peptidoglycan hydrolase during acid stress. EMBO J. 36, 536–548. doi: 10.15252/embj.201695028
Butler, R. E., Cihlarova, V., and Stewart, G. R. (2010). Effective generation of reactive oxygen species in the mycobacterial phagosome requires K+ efflux from the bacterium. Cell Microbiol. 12, 1186–1193. doi: 10.1111/j.1462-5822.2010.01463.x
Carroll, P., and James, J. (2009). Assaying promoter activity using LacZ and GFP as reporters. Methods Mol. Biol. 465, 265–277. doi: 10.1007/978-1-59745-207-6_18
Colangeli, R., Haq, A., Arcus, V. L., Summers, E., Magliozzo, R. S., McBride, A., et al. (2009). The multifunctional histone-like protein Lsr2 protecs mycobacteria against reactive oxygen intermediates. Proc. Natl. Acad. Sci. U.S.A. 106, 4414–4418. doi: 10.1073/pnas.0810126106
Cossu, A., Sechi, L. A., Bandino, E., Zanetti, S., and Rosu, V. (2013). Expression profiling of Mycobacterium tuberculosis H37Rv and Mycobacterium smegmatis in acid-nitrosative multi-stress displays defined regulatory networks. Microb. Pathog. 65, 89–96. doi: 10.1016/j.micpath.2013.10.004
Cuthbertson, L., and Nodwell, J. R. (2013). The TetR family of regulators. Microbiol. Mol. Biol. Rev. 77, 440–475. doi: 10.1128/MMBR.00018-13
Deng, W., Li, C., and Xie, J. (2013). The underling mechanism of bacterial TetR/AcrR family transcriptional repressors. Cell. Signal. 25, 1608–1613. doi: 10.1016/j.cellsig.2013.04.003
Dhandayuthapani, S., Via, L. E., Thomas, C. A., Horowitz, P. M., Deretic, D., and Deretic, V. (1995). Green fluorescent protein as a marker for gene expression and cell biology of mycobacterial interactions with macrophages. Mol. Microbiol. 17, 901–912. doi: 10.1111/j.1365-2958.1995.mmi_17050901.x
Dheda, K., Gumbo, T., Maartens, G., Dooley, K. E., McNerney, R., Murray, M., et al. (2017). The epidemiology, pathogenesis, transmission, diagnosis, and management of multidrug-resistant, and incurable tuberculosis. Lancet Respir. Med. doi: 10.1016/S2213-2600(17)30079-6 [Epub ahead of print].
Ehrt, S., and Schnappinger, D. (2009). Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell Microbiol. 11, 1170–1178. doi: 10.1111/j.1462-5822.2009.01335.x
Engohang-Ndong, J., Baillat, D., Aumercier, M., Bellefontaine, F., Besra, G. S., Locht, C., et al. (2004). EthR, a repressor of the TetR/CamR family implicated in ethionamide resistance in mycobacteria, octamerizes cooperatively on its operator. Mol. Microbiol. 51, 175–188. doi: 10.1046/j.1365-2958.2003.03809.x
Evangelopoulos, D., da Fonseca, J. D., and Waddell, S. J. (2015). Understanding anti-tuberculosis drug efficacy: rethinking bacterial populations and how we model them. Int. J. Infect. Dis. 32, 76–80. doi: 10.1016/j.ijid.2014.11.028
Gan, H., Lee, J., Ren, F., Chen, M., Kornfeld, H., and Remold, H. G. (2008). Mycobacterium tuberculosis blocks crosslinking of annexin-1 and apoptotic envelope formation on infected macrophages to maintain virulence. Nat. Immunol. 9, 1189–1197. doi: 10.1038/ni.1654
Garton, N. J., and O’Hare, H. M. (2013). Tuberculosis: feeding the enemy. Chem. Biol. 20, 971–972. doi: 10.1016/j.chembiol.2013.08.001
Gomez, A., Andreu, N., Ferrer-Navarro, M., Yero, D., and Gilbert, I. (2016). Triclosan-induced genes Rv1686c-Rv1687c and Rv3161c are not involved in triclosan resistance in Mycobacterium tuberculosis. Sci. Rep. 6:26221. doi: 10.1038/srep26221
Green, J., Rolfe, M. D., and Smith, L. J. (2014). Transcriptional regulation of bacterial virulence gene expression by molecular oxygen and nitric oxide. Virulence 5, 794–809. doi: 10.4161/viru.27794
Hao, P., Shi-Liang, Z., Ju, L., Ya-Xin, D., Biao, H., Xu, W., et al. (2011). The role of ABC efflux pump, Rv1456c-Rv1457c-Rv1458c, from Mycobacterium tuberculosis clinical isolates in China. Folia Microbiol. 56, 549–553. doi: 10.1007/s12223-011-0080-7
Kendall, S. L., Movahedzadeh, F., Rison, S. C. G., Wernisch, L., Parish, T., Duncan, K., et al. (2004). The Mycobacterium tuberculosis dosRS two-component system is induced by multiple stresses. Tuberculosis (Edinb). 84, 247–255. doi: 10.1016/j.tube.2003.12.007
Lewis, V. G., Ween, M. P., and McDevitt, C. A. (2012). The role of ATP-binding cassette transporters in bacterial pathogenicity. Protoplasma 249, 919–942. doi: 10.1007/s00709-011-0360-8
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Malone, K. M., and Gordon, S. V. (2016). Antibiotic methylation: a new mechanism of antimicrobial resistance. Trends Microbiol. 24, 771–772. doi: 10.1016/j.tim.2016.08.003
Namouchi, A., Gomez-Munoz, M., Frye, S. A., Moen, L. V., Rognes, T., Tønjum, T., et al. (2016). The Mycobacterium tuberculosis transcriptional landscape under genotoxic stress. BMC Genomics 17:791. doi: 10.1186/s12864-016-3132-1
Parish, T., Gordhan, B. G., McAdam, R. A., Duncan, K., Mizrahi, V., and Stoker, N. G. (1999). Production of mutants in amino acid biosynthesis genes of Mycobacterium tuberculosis by homologous recombination. Microbiology 145, 3497–3503. doi: 10.1099/00221287-145-12-3497
Rohde, K. H., Abramovitch, R. B., and Russell, D. G. (2007). Mycobacterium tuberculosis invasion of macrophages: linking bacterial gene expression to environmental cues. Cell Host Microbe 2, 352–364. doi: 10.1016/j.chom.2007.09.006
Salina, E. G., Waddell, S. J., Hoffman, N., Rosenkrands, I., Butcher, P. D., and Kaprelyants, A. S. (2014). Potassium availability triggers Mycobacterium tuberculosis transition to, and resuscitation from, non-culturable (dormant) states. Open Biol. 4:140106. doi: 10.1098/rsob.140106
Schnappinger, D., Ehrt, S., Voskuil, M. I., Liu, Y., Mangan, J. A., Monahan, I. M., et al. (2003). Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J. Exp. Med. 198, 693–704. doi: 10.1084/jem.20030846
Tailleux, L., Waddell, S. J., Pelizzola, M., Mortellaro, A., Withers, M., Tanne, A., et al. (2008). Probing host pathogen cross-talk by transcriptional profiling of both Mycobacterium tuberculosis and infected human dendritic cells and macrophages. PLOS ONE 3:e1403. doi: 10.1371/journal.pone.0001403
Trauner, A., Lougheed, K. E., Bennet, M. H., Hingley-Wilson, S. M., and Williams, H. D. (2012). The dormancy regulator DosR controls ribosome stability in hypoxic mycobacteria. J. Biol. Chem. 287, 24053–24063. doi: 10.1074/jbc.M112.364851
Valdivia, R. H., Hromockyj, A. E., Monack, D., Ramakrishnan, L., and Falkow, S. (1996). Applications for green fluorescent protein (GFP) in the study of host-pathogen interactions. Gene 173, 47–52. doi: 10.1016/0378-1119(95)00706-7
Voskuil, M. L., Bartek, I. L., Visconti, K., and Schoolnik, G. K. (2011). The response of Mycobacterium tuberculosis to reactive oxygen and nitrogen species. Front. Microbiol. 2:105. doi: 10.3389/fmicb.2011.00105
Waddell, S. J., and Butcher, P. D. (2010). Use of DNA arrays to study transcriptional responses to antimycobacterial compounds. Methods Mol. Biol. 642, 75–91. doi: 10.1007/978-1-60327-279-7_6
Wang, K., Pei, H., Huang, B., Zhu, X., Zhang, J., Zhou, B., et al. (2013). The expression of ABC efflux pump, Rv1217c-Rv1218c, and its association with multidrug resistance of Mycobacterium tuberculosis in China. Curr. Microbiol. 66, 222–226. doi: 10.1007/s00284-012-0215-3
Wei, J., Liang, J., Shi, Q., Yuan, P., Meng, R., Tang, X., et al. (2014). Genome-wide transcription analyses in Mycobacterium tuberculosis treated with lupulone. Braz. J. Microbiol. 45, 333–341. doi: 10.1590/S1517-83822014005000032
Keywords: mycobacteria, TetR-family, transcription factors, binding motif, efflux pump, acid-nitrosative response, microarray analysis
Citation: Perrone F, De Siena B, Muscariello L, Kendall SL, Waddell SJ and Sacco M (2017) A Novel TetR-Like Transcriptional Regulator Is Induced in Acid-Nitrosative Stress and Controls Expression of an Efflux Pump in Mycobacteria. Front. Microbiol. 8:2039. doi: 10.3389/fmicb.2017.02039
Received: 24 July 2017; Accepted: 05 October 2017;
Published: 23 October 2017.
Edited by:
Biswarup Mukhopadhyay, Virginia Tech, United StatesReviewed by:
Martin I. Voskuil, University of Colorado Denver, United StatesTanya Parish, Infectious Disease Research Institute, United States
Copyright © 2017 Perrone, De Siena, Muscariello, Kendall, Waddell and Sacco. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margherita Sacco, bWFyZ2hlcml0YS5zYWNjb0B1bmljYW1wYW5pYS5pdA==