 Pauline Ogrodzki1
Pauline Ogrodzki1 Stephen J. Forsythe2*
Stephen J. Forsythe2*- 1School of Science and Technology, Nottingham Trent University, Nottingham, United Kingdom
- 2foodmicrobe.com, Nottingham, United Kingdom
The Cronobacter genus is composed of seven species, within which a number of pathovars have been described. The most notable infections by Cronobacter spp. are of infants through the consumption of contaminated infant formula. The description of the genus has greatly improved in recent years through DNA sequencing techniques, and this has led to a robust means of identification. However some species are highly clonal and this limits the ability to discriminate between unrelated strains by some methods of genotyping. This article updates the application of three genotyping methods across the Cronobacter genus. The three genotyping methods were multilocus sequence typing (MLST), capsular profiling of the K-antigen and colanic acid (CA) biosynthesis regions, and CRISPR-cas array profiling. A total of 1654 MLST profiled and 286 whole genome sequenced strains, available by open access at the PubMLST Cronobacter database, were used this analysis. The predominance of C. sakazakii and C. malonaticus in clinical infections was confirmed. The majority of clinical strains being in the C. sakazakii clonal complexes (CC) 1 and 4, sequence types (ST) 8 and 12 and C. malonaticus ST7. The capsular profile K2:CA2, previously proposed as being strongly associated with C. sakazakii and C. malonaticus isolates from severe neonatal infections, was also found in C. turicensis, C. dublinensis and C. universalis. The majority of CRISPR-cas types across the genus was the I-E (Ecoli) type. Some strains of C. dublinensis and C. muytjensii encoded the I-F (Ypseudo) type, and others lacked the cas gene loci. The significance of the expanding profiling will be of benefit to researchers as well as governmental and industrial risk assessors.
Introduction
Cronobacter spp. is well known with respect to outbreaks of severe infant infections (necrotizing enterocolitis and meningitis) in neonatal intensive care units. However, the majority of Cronobacter infections are in the adult population with various symptoms including urinary tract infections (Holy and Forsythe, 2014; Patrick et al., 2014; Alsonosi et al., 2015). The organism is also a commensal member of the human body flora. Bacterial analysis of throat swabs from over 45,000 outpatients during the period 2005–2011 recovered the organism from every age group, with the highest frequency (8.7/1000 patients sampled) from infants less than 1 year of age (Holý et al., 2014). Cronobacter, then known as Enterobacter sakazakii, was the first foodborne pathogen which the FAO-WHO aimed to control through reducing neonatal and infant exposure to contaminated reconstituted infant formula (Food and Agriculture Organization of the United Nations [FAO], 2004). This resulted in both improved microbiological criteria at point of production and revised hygienic practices of preparation (Food and Agriculture Organization of the United Nations [FAO], 2004, 2006; Food and Agriculture Organization of the United Nations [FAO] and World Health Organization [WHO], 2008). In addition, the first expert committee meeting made various recommendations, including the need for an internationally validated detection and molecular typing methods for Cronobacter spp. and other relevant microorganisms (Food and Agriculture Organization of the United Nations [FAO], 2004).
This paper considers how, since 2004, these recommendations have been met through molecular typing methods based on the application of NGS over conventional methods. Reviews of wider aspects of Cronobacter, such as environmental fitness and virulence traits, have been recently published and therefore will not be considered in detail here (Forsythe et al., 2014; Almajed and Forsythe, 2016).
Powdered infant formula (PIF) has been the main vector associated with Cronobacter spp. and therefore has been the focus for the reduction in neonatal infections. Consequently, the Food and Agriculture Organization of the United Nations [FAO] (2004) encouraged the establishment of detection and molecular typing schemes which could be used to monitor sources of Cronobacter in PIF. An initial challenge was the differentiation of Cronobacter spp. (then known as Enterobacter sakazakii) from closely related organisms which could be co-recovered from infant formula and its ingredients, i.e., Franconibacter, Siccibacter and Enterobacter spp. (Stephan et al., 2014). Therefore an accurate taxonomic description of Cronobacter spp. was necessary for both reliable detection method development and for appropriate regulatory control. It should also be noted that other sources have been reported including the water used for reconstitution of PIF reconstitution, and also enteral feeding tubes (Hurrell et al., 2009b; Broge and Lee, 2013; Hariri et al., 2013; Ravisankar et al., 2014).
Unfortunately, some Cronobacter detection methods have been based on poorly characterized, even misidentified, strains (Jackson and Forsythe, 2016). The various Cronobacter species were initially defined according to the 16 Enterobacter sakazakii biotypes, however some of the biotype index strains were assigned the wrong Cronobacter species and this has limited further development of accurate phenotypic methods for Cronobacter identification (Iversen et al., 2008; Baldwin et al., 2009; Joseph et al., 2013a; Jackson and Forsythe, 2016). Additionally, the earlier reliance on phenotyping methods led to a number of mistaken identifications in the literature (Caubilla-Barron et al., 2007; Townsend S. et al., 2008; Blažková et al., 2015; Jackson et al., 2015a; Ogrodzki and Forsythe, 2015).
Such misidentifications can cause further confusion for risk management and the control of infection, as well as misinformation on current issues such as carriage of antibiotic resistance. Therefore reliable and robust means of identifying and typing Cronobacter isolates are required and should be internationally accessible.
Although 16S rDNA sequence analysis is generally applicable for bacterial species identification, it is not a reliable method for members of the Cronobacter genus as it is unable to reliably differentiate between the two species C. sakazakii and C. malonaticus (Iversen et al., 2008; Baldwin et al., 2009). In order to overcome this limitation, Joseph et al. (2012b) used representative strains across the genus which had been selected using multilocus sequence analysis (MLSA) of 7 housekeeping genes; ATP synthase b chain (atpD), elongation factor G (fusA), glutaminyl tRNA synthetase (glnS), glutamate synthase large subunit (gltB), DNA gyrase subunit B (gyrB), translation initiation factor IF-2 (infB) and phosphoenolpyruvate synthase A (ppsA). This DNA-sequence based approach overcame the preconceived grouping of strains based on phenotyping, and supported the recognition of two further Cronobacter species; C. universalis and C. condimenti (Joseph et al., 2012a).
There are a number of centralized MLST databases which are internationally available covering bacteria and fungi with standardized allele profile determination programs. The three major bacterial MLST databases are those at the Institute of Pasteur1, and the universities of Warwick2, and Oxford3. Initially MLST used laboratory-based sequencing of individual genes, but nowadays it is more reliant on in silico analysis of whole genomes. The analysis of uploaded Cronobacter genome sequences is through the ‘Bacterial Isolate Genomes Sequence Database’ (BIGSdb) facility4. Furthermore the inclusion of whole genomes has enabled the Cronobacter MLST database to be expanded to ribosomal-MLST (r-MLST; 53 genes) and core genome MLST (cg-MLST; 1836 genes) analysis to provide greater resolution between strains and deeper bacterial population studies (Maiden et al., 2013; Forsythe et al., 2014; Jolley and Maiden, 2014). Phylogenetic comparison of 7-loci MLST, rMLST, cg-MLST and whole genome analysis has confirmed the reliability and robustness of the 7-loci MLST scheme for speciation and subtyping within the Cronobacter genus as well as differentiation from related genera. Additionally, fusA allele phylogeny corresponds to whole genome phylogeny and can be used for initial speciation of Cronobacter isolates (Joseph et al., 2012c; Forsythe et al., 2014).
The use of MLST, based on NGS, has therefore supported defining the Cronobacter genus. The genus is within the family Enterobacteriaceae, with the nearest relatives being the Franconibacter and Siccibacter genera, as well as the more familiar genera of Enterobacter, Citrobacter and Pantoea (Stephan et al., 2014; Jackson et al., 2015b). According to the MLST phylogenetic analysis, the genus split from its nearest ancestor in the Enterobacteriaceae family ca. 45–68 million years ago, at the same time as the Salmonella genus diverged into its species and subspecies (McQuiston et al., 2008; Joseph and Forsythe, 2012).
Using in vitro virulence studies, it has been shown that Cronobacter spp. can invade intestinal and brain cells, as well as persist and even replicate in macrophages (Townsend et al., 2007; Townsend S.M. et al., 2008; Almajed and Forsythe, 2016). The most reported Cronobacter species in clinical cases are C. sakazakii and C. malonaticus in infant and adults, respectively (Forsythe et al., 2014; Alsonosi et al., 2015). A number of virulence traits have been proposed which may account for these clinical differences. Nevertheless, there have been insufficient large scale comparative whole genome studies to establish their contribution to severe clinical presentations. Of particular interest has been the predominance of C. sakazakii in neonatal infections, especially CC4 and ST12. However, studies have not identified traits unique to specific Cronobacter pathovars (Kucerova et al., 2010, 2011; Joseph and Forsythe, 2011; Joseph et al., 2012b; Masood et al., 2015). This could be due to unrecognized synergy between different virulence traits and environmental fitness traits leading to increased neonatal exposure and severity of infection (Hariri et al., 2013).
Cronobacter sakazakii strains are able to use sialic acid as a carbon source for growth (Joseph et al., 2013b). This could be a highly significant host-adaptation trait since sialic acid is found in breast milk, mucin and gangliosides (Almagro-Moreno and Boyd, 2009). Laboratory studies have shown that C. sakazakii is able to grow on both sialic acid and ganglioside (GM1) as a sole carbon source (Joseph et al., 2013b) and could enable the degradation of intestinal mucin, facilitating access to intestinal cells. Due to its association with brain development, sialic acid is an ingredient in PIF and therefore an additional C. sakazakii growth substrate following rehydration. In contrast, unlike other Cronobacter species, C. sakazakii strains are unable to grow on malonic acid. This organic acid is found in plants and the ability to utilize it is regarded as reflecting the initial plant-association of the Cronobacter genus (Schmid et al., 2009). The adaptation of C. sakazakii to a new ecosystem, with the subsequent loss of malonic acid utilization and gain in sialic acid utilization might therefore contribute to its pathogenicity toward neonates (Joseph et al., 2013b). The mechanisms and routes of exposure by which C. malonaticus infect adults more than infants are as yet unknown. In the Cronobacter genera, a total of 10 fimbrial families have been identified. C. sakazakii lack curli fimbriae, but are unique in encoding for β–fimbriae (Joseph et al., 2012b). The clinical significance of this has not been investigated, but might contribute to the greater predominance of C. sakazakii in infant infections through attachment to host cells. A range of other virulence traits have been reported. These include Cronobacter plasminogen activator (cpa) (Franco et al., 2011), iron utilization (Grim et al., 2012), outer membrane vesicle release causing host cell cytopathogenicity (Alzahrani et al., 2015; Kothary et al., 2017), as well as heavy metal resistance traits (copper, silver, zinc, tellurite) (Joseph et al., 2012b; Chaturvedi et al., 2015).
Cronobacter spp. produce capsular material composed of the O and K-antigens, colanic acid, Enterobacteriacae common antigen, and cellulose (Ogrodzki and Forsythe, 2015). The capsular material is composed of water-saturated, high molecular weight polysaccharides. It plays a role in virulence by enabling organisms to evade host response mechanisms (serum resistance and phagocytosis), as well as facilitating biofilm formation and desiccation survival (Willis and Whitfield, 2013). Cronobacter biofilms have been found in hospitals on equipment used to prepare infant formula, feeding bottles, and neonatal enteral feeding tubes (Iversen and Forsythe, 2003; Iversen et al., 2004; Kim et al., 2006; Hurrell et al., 2009a,b; Holy and Forsythe, 2014; Jackson et al., 2015a). The biofilms on enteral feeding tubes could act as loci for neonatal infection (Hurrell et al., 2009a,b). Under some growth conditions, the organism produces excessive capsule material which may protect the organism from disinfectants in food production environments, and enable long-term persistence under desiccation conditions, such as in PIF for over 2 years (Caubilla-Barron and Forsythe, 2007). It is therefore plausible that the strong association between C. sakazakii CC4 and neonatal meningitis is due to its environmental fitness of desiccation survival, resulting in its persistence in PIF and the environment of PIF manufacturing plants (Muller et al., 2013; Power et al., 2013; Sonbol et al., 2013; Fei et al., 2015, 2017). Therefore the greater incidence of C. sakazakii CC4 in PIF could lead to an increased risk of exposure and incidence of infection rather than being due to the organism encoding for specific pathogenicity traits.
A capsular profiling scheme for Cronobacter based on the K-antigen and colanic acid (CA) biosynthesis encoding genes has been proposed (Ogrodzki and Forsythe, 2015). This scheme was based on the analysis of 104 Cronobacter genomes and revealed that strains of C. sakazakii and C. malonaticus isolated from cases with the most severe neonatal clinical presentations of invasive meningitis and necrotizing enterocolitis (NEC), had a definable capsular profile which differed from strains associated with less severe clinical cases. They reported that all (n = 54) strains of C. sakazakii CC4 and ST12 strains known for being associated with severe neonatal infections of meningitis and NEC, had the capsular profile K2:CA2 (Joseph and Forsythe, 2011, 2012; Joseph et al., 2012c; Hariri et al., 2013; Forsythe et al., 2014). Of particular interest was that this particular capsule profile was also found in the isolates of the few C. sakazakii non-CC4 cerebral spinal fluid (CSF) cases, including a single C. malonaticus fatal case (Ogrodzki and Forsythe, 2015). Also of interest was that two C. turicensis strains encoding for sialic acid utilization also had the capsular profile K2:CA2. Since the earlier study by Ogrodzki and Forsythe (2015), a large number of Cronobacter genomes have become available and therefore a wider evaluation of capsular profiling across the genus is now feasible.
The establishment of internationally standardized molecular typing methods applicable across the Cronobacter genus is necessary given the severe outcomes of infections in neonates and the attributed link to contaminated PIF on the international market (Food and Agriculture Organization of the United Nations [FAO], 2004, 2006; Food and Agriculture Organization of the United Nations [FAO] and World Health Organization [WHO], 2008). Although it is generally possible to differentiate Cronobacter species by biochemical profiling, molecular methods are increasingly used as a more rapid and reliable tool to study bacterial genomic diversity and to track sources of infection. Since Cronobacter is ubiquitous, such typing schemes are applicable for both epidemiological and environmental investigations. For epidemiological analysis (i.e., tracing source and dissemination during an outbreak), PFGE with two restriction enzymes (Xba1 and Spe1) has in the past been the most common method (van Acker et al., 2001; Himelright et al., 2002). The method is limited, however, as not all Cronobacter strains can be typed due to intrinsic DNase activity, non-identical strains can give the same PFGE profile and the method does not give the relationship between strains (Centers for Disease Control and Prevention [CDC], 2010; Alsonosi et al., 2015). Due to these limitations of PFGE, the Centers for Disease Control and Prevention (CDC) is transitioning to using whole genome sequencing as the basis for PulseNet surveillance (Carleton and Gerner-Smidt, 2016; Nadon et al., 2017).
A number of typing methods for Cronobacter have been published, in particular PCR-probe O-antigen serotyping (Mullane et al., 2008; Jarvis et al., 2011; Sun et al., 2011). Initially 7 serotypes were defined for C. sakazakii and 2 in C. malonaticus. However, some strains of C. malonaticus were mis-identified as C. sakazakii by Sun et al. (2011) and were assigned C. sakazakii serotypes O:5 and O:6. Consequently, Blažková et al. (2015) proposed the O-antigen scheme should be revised with additional recognition of 7 new and 2 re-assigned serotypes. Chemical analysis of the O-polysaccharide units from many strains has confirmed the molecular Cronobacter serotypes. However three structures have been determined for C. sakazakii O:2 strains (Arbatsky et al., 2010; Czerwicka et al., 2010; Maclean et al., 2010). This is probably due to variants in the O-antigen genes which occur outside the target region of the PCR primers.
The PCR-probe O-serotyping has been superseded by allele profiling of gnd and galF (encoding 6-phosphogluconate dehydrogenase and UTP-glucose-1-phosphate uridylyltransferase subunits, respectively) (Ogrodzki and Forsythe, 2015). This DNA-sequence based approach is a more reliable and expansive method for O-antigen determination within Cronobacter. It has increased the definable number of serotypes in the Cronobacter genus from 18 to 34 (Ogrodzki and Forsythe, 2015).
MLST analysis has not only been able to support taxonomic revisions, but can differentiate strains to a greater degree than other methods; >500 defined STs compared with 34 for O-serotyping. It has also revealed a remarkably strong clonal nature in the Cronobacter genus, in particular within the clinically relevant C. sakazakii and C. malonaticus species (Baldwin et al., 2009; Joseph et al., 2012c). This has subsequently led to the recognition of particular pathovars (Joseph and Forsythe, 2011; Hariri et al., 2013; Sonbol et al., 2013; Forsythe et al., 2014). C. sakazakii CC4 is a DNA sequence defined evolutionary lineage which is especially associated with neonatal meningitis. C. sakazakii ST12 is associated with cases of NEC and not neonatal meningitis or septicaemia. C. sakazakii CC1 strains are primarily isolated from infant formula and clinical sources, whereas C. sakazakii ST8 are isolated from clinical and non-formula food sources. C. malonaticus adult infections are almost exclusively CC7.
While this clonal recognition is useful for the identification of Cronobacter pathovars, it is counter-productive for microbial source tracking as unrelated strains will occur in the same ST. This may explain the observation that the same PFGE pulsotype can be obtained for unrelated clinical C. sakazakii strains (Forsythe et al., 2014; Alsonosi et al., 2015). In order to address this issue, two further typing methods have been applied to discriminate strains within a given ST; capsule profiling and ‘CRISPRs’ and CRISPR-associated genes (cas) protein-coding genes (CRISPR-cas) array profiling. These were chosen as two independent typing tools as capsular genes do not follow phylogeny, and the CRISPR-cas array reflects the exposure of strains to phages and plasmids (Ogrodzki and Forsythe, 2015; Zeng et al., 2017).
There are a number of different CRISPR-cas systems, often named according to their first identification organism, i.e., E. coli (type I-E) and Yersinia pseudotuberculosis (type I-F) (Makarova et al., 2015). In general, CRISPR-cas systems may have up to three sections (a) cas genes (b) an AT-rich leader sequence upstream of the array and (c) a CRISPR array, composed of short (∼24–48 nucleotide) direct repeat sequences separated by similarly sized, unique spacers which are usually derived from mobile genetic elements such as bacteriophages and plasmids (Grissa et al., 2007b; Makarova et al., 2015). CRISPR-cas systems may have a number of roles such as adaptive immunity to phages and plasmids, as well as bacterial virulence and stress response (Barrangou et al., 2007; Shariat and Dudley, 2014).
Many applications have been identified for the CRISPR-cas system such as gene editing, evolutionary and phylogenetic studies, as well as genotyping for epidemiological investigations (Fricke et al., 2011; Makarova et al., 2015). CRISPR arrays may differ between closely related strains due to their different exposure histories to phages and plasmids, leading to differences in their spacer acquisitions. Therefore these loci can be used for molecular subtyping, offering greater discrimination between strains than MLST and PFGE, especially useful for highly clonal species, such as C. sakazakii.
This paper considers the diversity of Cronobacter across the genus according to 7-loci MLST, the association between capsule profile of the K-antigen and colanic acid biosynthesis genes with clinical presentation, and compares CRISPR-cas array profiling between the highly clonal Cronobacter species C. sakazakii with less clonal species C. dublinensis. Of particular relevance is the availability of over twice the number of genomes (280 compared with 104) since previous publications on capsule and CRISPR-cas array profiling (Forsythe et al., 2014; Ogrodzki and Forsythe, 2015, 2016).
Materials and Methods
Strains Used in This Study
A total of 1654 STd strains and 275 genomes were included in this study (Table 1). Strains from patients less than 1 year in age were defined as infant in origin, those from patients ages 1–15 were termed child, and those above 15 were regarded as adult isolates. Additional metadata can be obtained from the open access Cronobacter PubMLST database; http://pubmlst.org/cronobacter/.
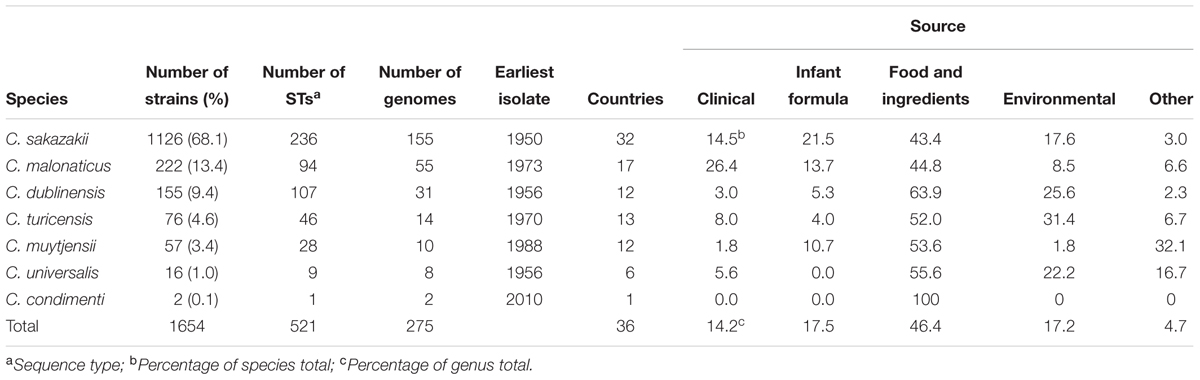
TABLE 1. Summary of Cronobacter isolates in the Cronobacter PubMLST database.
Seven Loci MLST Analysis
As per Forsythe et al. (2014), DNA sequences collated at http://pubmlst.org/cronobacter/ were investigated. Concatenated sequences of seven loci from 521 STs were downloaded in FASTA format using the Export/Sequences option. These sequences were aligned in MEGA version 6.05 using the ClustalW algorithm (Tamura et al., 2013). The final alignment spanned 3036 bp and was analyzed using the default pipeline in SplitsTree4 (UncorrectedP to calculate distances and NeighborNet to build the network) (Kloepper and Huson, 2008).
Goeburst Analysis
Phyloviz version 2.0 tool was used with goeBURST Full MST algorithm. Level 3 (TLV) was chosen which represents the Locus Variant level and removes of all the links greater than the number selected (Nascimento et al., 2017).
Bacterial Genome Analysis
As per Ogrodzki and Forsythe (2015, 2016). A total of 275 genomes were analyzed for this study, which were the total number of genomes available (March 2017). Of particular interest were 41 whole genome sequenced isolates were chosen as representatives of C. dublinensis and C. muytjensii. They were geographically dispersed over 12 countries and temporally spread over 50 years (Table 1). Additional metadata can be obtain from the open access Cronobacter PubMLST database; http://pubmlst.org/cronobacter/.
Defining CRISPR-cas Arrays
CrisprFinder tool was used to identify number of Crispr arrays, including the number and length of DR and spacers sequences; http://crispr.i2bc.paris-saclay.fr/ (Grissa et al., 2007a)
Phylogenetic Analysis
As per Ogrodzki and Forsythe (2015, 2016). DNA sequences were carefully curated prior to and after alignment and phylogenetic analyses in order to maximize the quality of the results using the satisfactory default parameters for the latter analyses. DNA sequences were aligned in MEGA version 6.05 using the ClustalW algorithm (Tamura et al., 2013) set to default parameters settings. The phylogenetic trees were generated using the Maximum Likelihood (ML) method based on the Tamura-Nei model with the additional parameters set to default settings. All phylogenetic trees are drawn to scale with branch lengths measured in the number of substitutions per site.
DNA Sequences
As per Ogrodzki and Forsythe (2016). Whole genome DNA sequences collated at http://pubmlst.org/cronobacter/ were investigated. In silico analyses were carried out using search options, such as BLAST, on the Cronobacter PubMLST portal accessible at: http://pubmlst.org/perl/bigsdb/bigsdb.pl?db=pubmlst_cronobacter_isolates.
DNA Annotation and Visualization Tools
As per Ogrodzki and Forsythe (2015, 2016). Nucleotide sequences were extracted from the corresponding genome assemblies in the Cronobacter PubMLST database.
Bacterial DNA sequences were investigated using the genome browser and annotation tool Artemis (Carver et al., 2012).
Results
Cronobacter Diversity and Source According to MLST
The MLST scheme for Cronobacter open access database5 was used as the source of strain sequences and metadata. At the time of writing this database contained over 1654 strain profiles, including >270 whole genomes, along with associated metadata such as source, date of isolation, and clinical presentations (Table 1). The strains had been collected from various sources (clinical, food and environmental) and countries over a 60 year period. Since the database also contains the metadata for over 1600 isolates, an informed understanding of the diversity and sources of the Cronobacter genus can be obtained. The database can also be used for the retrospective analysis of strains from earlier studies.
Investigating 1654 isolate entries in the Cronobacter MLST database revealed the temporal, geographic and source diversity of the organism (Table 1). Cronobacter strains have been isolated from 36 countries, and are from clinical (14.2%), infant formula (17.5%), food and food ingredients (46.4%), and environmental (17.2%). The earliest isolate (C. sakazakii NCIMB 8282) was isolated from dried milk powder in 1950.
Given the primary interest in Cronobacter is due to clinical infections, Goeburst analysis was used to visualize the diversity of clinical isolates according to their ST, patient age and site of isolation; Figure 1. The majority of strains were from C. sakazakii (68.1%) and C. malonaticus (13.4%). Out of a total of 236 defined STs for C. sakazakii, the major STs recovered from clinical sources were STs 1, 4, 8, and 12. ST4 was the most numerous (208 strains out of 1126) ST in the database. The ST4 isolates were primarily from infants (53%), and secondly adults (37%); Figure 1A. Other C. sakazakii STs of note were STs 1, 8, and 12 which were primarily composed of infant isolates; 79% (n = 14), 14% (n = 14) and 67% (n = 9), respectively. As shown in Figure 1B, the site of isolation for C. sakazakii ST4 was most frequently sputum (31%), CSF (16%) and feces (17%). Isolates of C. sakazakii ST8 were also from a range of sources (throat, fecal, sputum, CSF), whereas ST1 were from fewer sites; primarily CSF (36%) for ST1, and feces and throat (33% each) for ST12. C. malonaticus ST7 was the major (29% n = 199) C. malonaticus ST. Sixteen C. malonaticus ST7 strains had detailed clinical information. These had been recovered from throat, feces, and sputum samples; Figure 1B. The remaining 5 Cronobacter species together composed 18.5% of the database, and contained only 13 clinical isolates in total (Table 1).
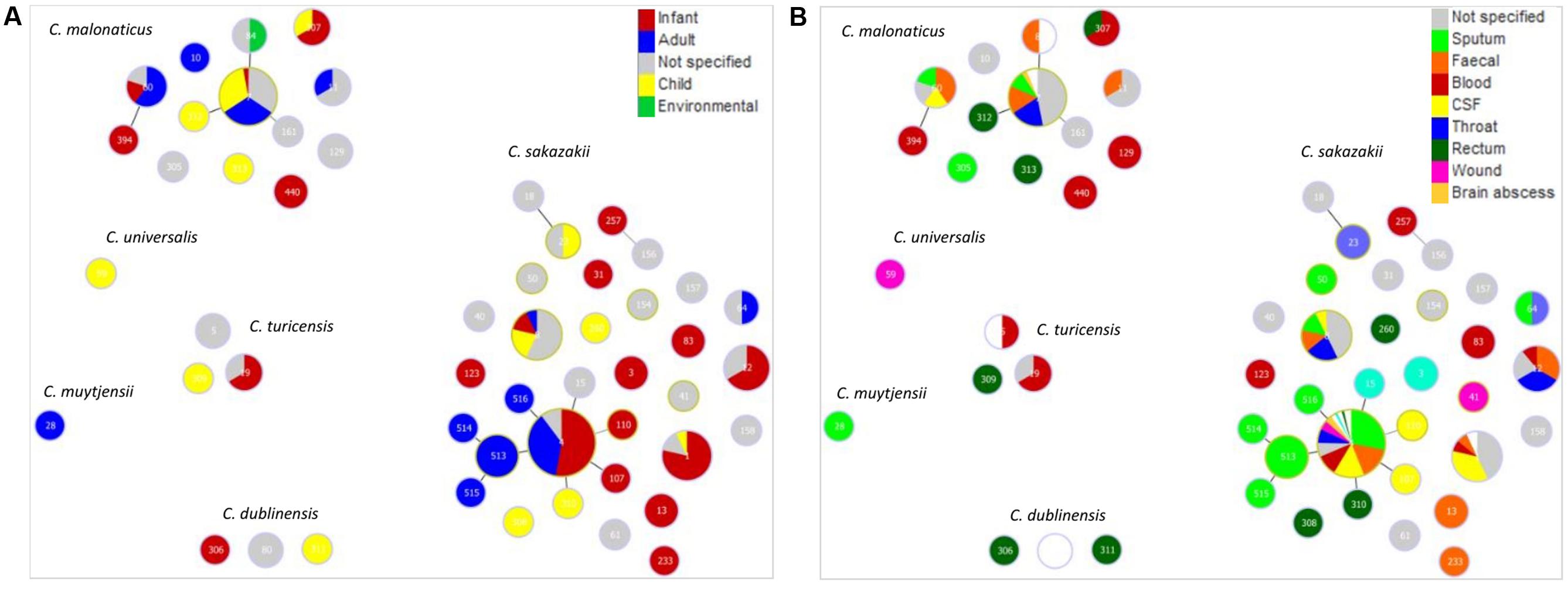
FIGURE 1. Genetic relationship of clinical Cronobacter isolates according to (A) patient age along with clinically relevant environment and (B) isolation site. The minimum spanning tree was constructed using goeBurst analysis of 53 STs from 234 strains. The size of the circle is proportional to the number of strains.
Capsule Profiling and Distribution of Sialic Acid Utilization Genes across the Cronobacter Genus
Table 2 summarizes the occurrence of K-antigen and colanic acid biosynthesis genes across 275 whole genome strains from the Cronobacter genus. The K2:CA2 profile, previously reported to be associated with neonatal meningitis cases, was primarily found in clinical isolates (n = 59/275). Most (n = 84/103) were C. sakazakii strains, in 16 STs. The K2:CA2 profile was found in 10 C. malonaticus strains of ST60 and ST307, of which 7 were clinical in origin. Previously only one C. malonaticus reported case of severe meningitis in an infant had been documented (Ogrodzki and Forsythe, 2015). This was a fatal meningitis case of an infant born with an EGA of 32 weeks in America (2011). The culture sequenced was a blood isolate, and was presumed to be the infectious organism of the brain. This single case was proposed by Ogrodzki and Forsythe (2015) as possible outlier evidence of K2:CA2 link to meningitis. In this paper a second C. malonaticus K2:CA2 isolate (ID 1494) is reported. This strain was isolated in China (2014) from the CSF of an infant born with an EGA of 36 weeks, who was fed fortified breast milk and developed clinical symptoms of meningitis on day 11. Of the remaining C. malonaticus strains with the K2:CA2 profile, three were from adults aged 26–82 years (no clinical symptoms available). There were no clinical details for the remaining 2 C. malonaticus strains. The K2:CA2 profile was also found in a total of 9 C. turicensis, C. dublinensis and C. universalis strains, none of which were clinical in origin. The second most frequent capsule profile was K1:CA1 (n = 89/275), of which 31 were clinical in origin but did not correspond to any specific clinical presentation (Table 2). This profile was not found in C. condimenti, C. universalis, or C. turicensis.
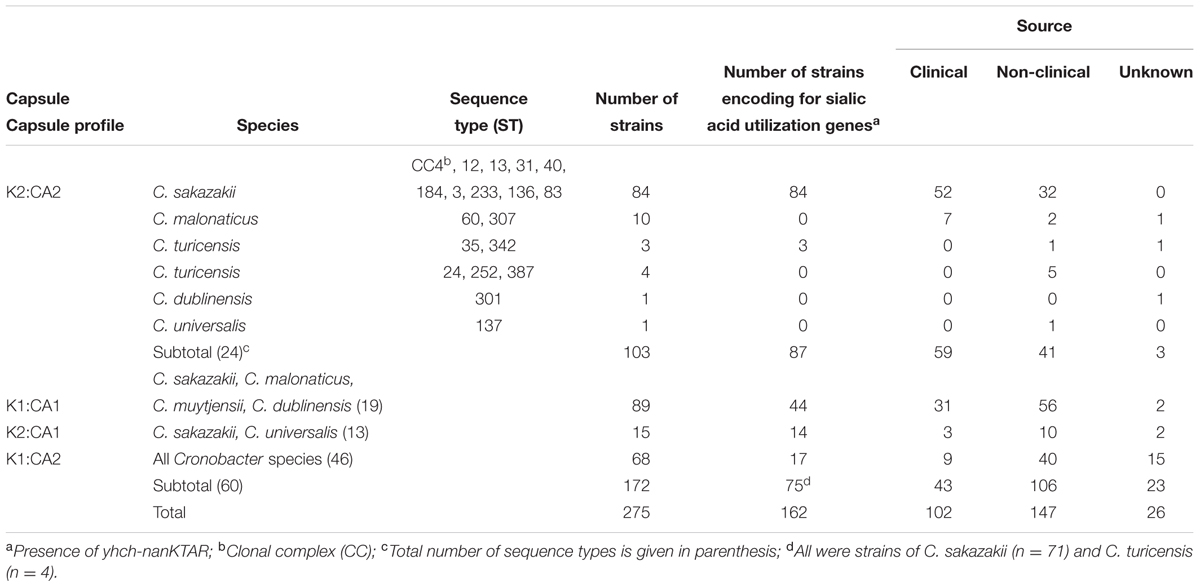
TABLE 2. Distribution of capsule profile (K-antigen and colanic acid genes) and presence of sialic acid utilization genes across the Cronobacter genus.
All C. sakazakii strains encoded for the sialic acid utilization genes (yhch-nanKTAR); Table 2. These genes were not found in any other species, except 7 (out of 14) C. turicensis strains, of which 3 had the K2:CA2 capsular profile and the remainder had the profile K1:CA2. The C. turicensis strains encoding for sialic acid utilization were in five different STs; 24, 35, 252, 342, and 387. The sialic acid utilization genes were not found in any other Cronobacter species.
CRISPR-cas Operon Architecture
Table 3 presents the detailed CRISPR-cas profiling of 100 isolates. The previously published data for the four clinically significant C. sakazakii pathovars (CC1, CC4, ST8 and ST12) are shown for comparative purposes with C. dublinensis and C. muytjensii. The latter two species were chosen for detailed analysis as they had not been the focus of earlier CRISPR-cas array studies (Ogrodzki and Forsythe, 2016; Zeng et al., 2017). C. muytjensii and C. dublinensis do not show such strong clonality compared with C. sakazakii and C. malonaticus resulting in the higher proportion of unique STs for the number of strains in the Cronobacter database (Table 1). It was therefore predicted that the diversity of the CRISPR-cas profiles might be greater in C. dublinensis and C. muytjensii. The genomes of C. dublinensis and C. muytjensii strains studied were widely temporally (58 years) and globally (11 countries) distributed in their origin and therefore representative of the diversity of the two species.
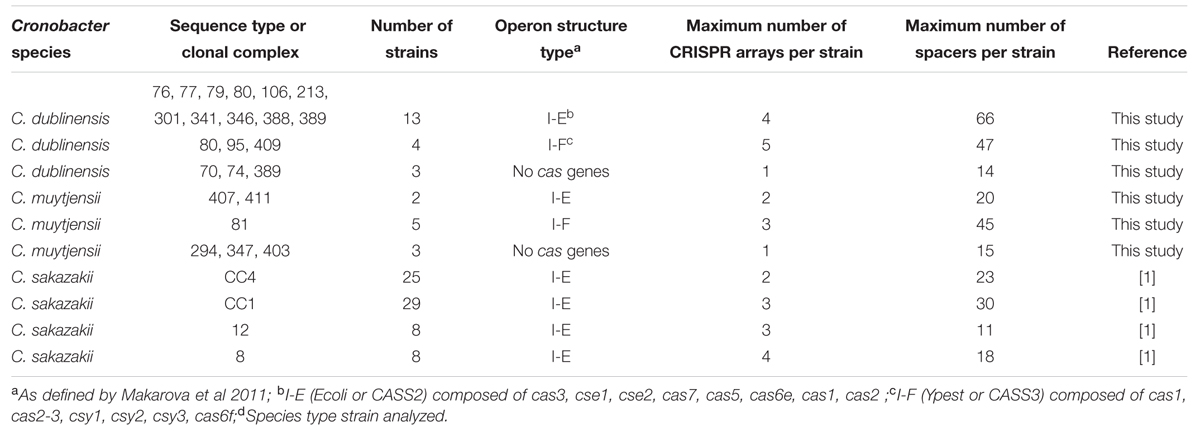
TABLE 3. CRISPR-cas operon structure and array profile variation in Cronobacter spp.
Twenty genomes of source distributed and ST diverse C. dublinensis strains were analyzed. Thirteen genomes revealed CRISPR-cas arrays with the same general I-E structure, also known as Ecoli or CASS2 type, due to the presence of cas3, cse1, cse2, cas7, cas5, cas6e, cas1, and cas2. There was a maximum of 4 CRISPR arrays per strain, with up to 66 spacers. Four genomes (ST 80, 95 and 409) contained I-F (Ypest or CASS3) type CRISPR-cas structure, encoding for cas1, cas2-3, csy1, csy2, csy3, and cas6f. The strains had up to 5 CRISPR arrays per strains, and up to 47 spacers. As shown in Table 3, the two strains from ST80 differed in their operon structure; one being I-E and the other I-F. Three genomes from STs 70, 74 and 389 did not encode for any cas genes, yet did encode for one CRISPR array composed of 14 spacers.
Ten C. muytjensii genomes which were diverse with respect to their temporal, source and ST were analyzed. Five ST81 strains contained the I-F (Ypest or CASS3) type CRISPR-cas structure, with a maximum of 5 CRISPR arrays, and 45 spacers per strain. Two genomes (ST 407 and ST411) revealed CRISPR-cas arrays with the general I-E (Ecoli or CASS2) type CRISPR-cas structure. There was a maximum of 2 CRISPR arrays per strain, with up to 20 spacers. Three genomes from STs 294, 347, and 403 did not encode for any cas genes, but did encode for one CRISPR array composed of 15 spacers.
For completeness of the relative comparative analysis, the CRISPR-cas profiles of the species types strains of the remaining three Cronobacter species, C turicensis, C. universalis, and C. condimenti, were determined and included in the analysis (Table 3). The type strains for C. universalis and C. condimenti did not contain any cas genes.
In order to investigate any phylogenetic relationship with the CRISPR-cas type across the C. dublinensis and C. muytjensii species, phylogenetic analysis of the Cronobacter genus based on 7-loci MLST is shown in Figure 2. The C. dublinensis species cluster is very diverse, compared with C. muytjensii, yet can be divided into two subgroups. The smaller subgroup contains the C. dublinensis subsp. dublinensis type strain, whereas C. dublinensis subsp. lactaridii and C. dublinensis subsp. lausanensis are in the larger cluster. There was no phylogenetic association between the distribution of the I-E and I-F type CRISPR-cas operon structures across C. dublinensis and C. muytjensis, or across the whole genus. In addition, the two C. dublinensis ST80 strains (isolated in 1979 and 2004 in United States and Switzerland respectively) differed in their CRISPR-cas type (Table 3).

FIGURE 2. MLST phylogenetic tree of C. dublinensis (16 STs) and C. muytjensii (6 STs) and species type strains of C. sakazakii, C. malonaticus, C. turicensis, C. universalis, and C. condimenti. The phylogenetic tree is drawn to scale with branch lengths measured in the number of substitutions per site. Scale bar represents nucleotide substitutions.
Discussion
Diversity of Cronobacter Species According to 7-Loci MLST
Due to initial concerns of the association between Cronobacter in PIF and infant infection, the organism became the focused attention for new identification and typing schemes. The primary goals being to reduce the risk of neonate exposure to the organism, and facilitate monitoring for the organism in PIF ingredients and associated manufacturing environments. The differentiation of the organism from closely related genera, such as Franconibacter and Siccibacter, has been achieved through a series of taxonomic revisions describing the Cronobacter genus (Stephan et al., 2014; Jackson et al., 2015b). While the considerable diversity within the Cronobacter genus has been recognized through DNA-sequence based typing schemes. These achievements have been through the application of NGS, and the centralization of the sequences in an open access database repository along with associated metadata. Therefore meeting the recommendations of the FAO-WHO expert committee as detailed in the Introduction (Food and Agriculture Organization of the United Nations [FAO], 2004).
The curated open access Cronobacter PubMLST database has been established at the University of Oxford for the genus along with associated metadata for each deposited strain; http://pubmlst.org/cronobacter/ (Forsythe et al., 2014). This database has enabled the recognition of identifiable Cronobacter clonal lineages within the genus as pathogenic variants, whereas others are primarily commensal organisms of the environment. The original 7-loci MLST scheme is congruent with both 53-loci rMLST and 1865-loci cg-MLST as well as whole genome phylogeny [Forsythe et al., 2014].
Although the Cronobacter PubMLST database cannot be used for direct enumeration purposes, the submission of new strains and sequences to the database will reflect the diversity of the genus with respect to global distribution and source variation. Additionally, the choice of strains genotyped and genome sequenced by different research groups will have been for different rationales, i.e., clinical significance, representative of species, or diversity study. Consequently, outbreak investigations may result in repeated isolation of indistinguishable strains from the same location being deposited in the database. Nevertheless this study of 1654 strains and 275 genomes is the most comprehensive to date and will serve as a reference for further specific studies and can be compared with the earlier analysis of 1000 strains (Forsythe et al., 2014).
All Cronobacter species have been isolated from food and food ingredients, and this source comprises 46.4% of the total profiled strains in the database (Table 1). These are primarily from starch-based ingredients, cake mixes, packet soup, salads, flavored teas as well as herbs and spices. Another source for Cronobacter spp. given in the database is the environment (17.2% entries) which included the manufacturing environment as well as the natural environment with isolates from water and soil. Despite reconstitution water being reported as the source of one reported serious C. sakazakii infection, water as a source of the bacterium has not received much attention (Hariri et al., 2013; Liu et al., 2013). The remaining strains in the database were clinical (14.2%) and PIF (17.5%) in origin. The major species were C. sakazakii and C. malonaticus (68.1 and 13.4%, respectively), which have been isolated from 32 countries, the earliest isolate being from dried milk powder in 1950 and the genome of which has been published (Masood et al., 2013).
The designation ‘clinical’ needs to be used with caution as it can mean a plethora of sources; symptomatic and asymptomatic patients, from normally sterile sites and non-sterile sites. For example, some strains of C. sakazakii isolated from neonatal feeding tubes are designated as ‘clinical’ as this was deemed more appropriate than ‘environmental,’ even though there were no associated infections reported. Consequently we have focused on isolates of clinical significance, such as those from CSF and blood with respect to proposing specific pathovars in the genus.
The four major pathovars in the Cronobacter genus are (i) C. sakazakii CC4, which is more predominantly associated with neonatal meningitis, (ii) C. sakazakii ST12 with neonatal necrotizing enterocolitis, (iii) C. sakazakii CC1 strains are primarily isolates from infant formula and clinical sources, and (iv) C. sakazakii ST8 were isolated from clinical and non-formula food sources (Forsythe et al., 2014). The reason for the predominance of these pathovars could be due to their persistence during the manufacture and storage of infant formula which would result in increased neonatal exposure, as well as encoding for virulence genes (Joseph and Forsythe, 2011).
The pathovar C. sakazakii CC4 comprises single (SLV), double (DLV) and triple (TLV) loci variants of C. sakazakii ST4. This clonal complex was the major one recovered from clinical sources (208/1126). They were primarily from infants (53%) as well as adults (37%) (Figure 1A). The reason for C. sakazakii CC4 association with neonatal meningitis is uncertain since no unique virulence traits had been determined in this ST (Masood et al., 2015). However, it could be linked to environmental fitness, in particular desiccation resistance accounting for the reported recovery of C. sakazakii CC4 strains from PIF and infant formula manufacturing plants in China, Ireland, Switzerland, Australia, and Germany (Craven et al., 2010; Reich et al., 2010; Jacobs et al., 2011; Sonbol et al., 2013; Yan et al., 2013). Fei et al. (2015) reported that, according to PFGE, environmental isolates of C. sakazakii CC4 from a PIF manufacturing plant in Switzerland were indistinguishable from those in the finished product. Therefore C. sakazakii CC4 may be an environmentally persistent clonal complex which due to increased neonatal exposure results in infant infections. Other C. sakazakii STs of isolated from infants include CC1 (79%), ST8 (14%), and ST12 (67%). The strains had been recovered from a number of sites; throat, sputum, feces, blood, and CSF.
Twenty-nine percent of C. malonaticus strains (n = 199) recorded in the database are in clonal complex 7 (Table 1). Strains in this complex have been isolated over the past 30 years. Strains of clinical origin had been recovered from throat, feces, and sputum samples; Figure 1B. It is of note that the only reported fatal neonatal meningitis cases which has been attributed to C. malonaticus were not from this ST. These will be discussed further below with respect to capsule profiling. The database only contained three C. malonaticus CC7 isolates from PIF, and there were no isolates from infant formula or milk powder manufacturing plants. This probably reflects a low incidence of this clonal complex in PIF and the manufacturing environment.
It is pertinent to emphasize that some of the predominant clinical STs were also found in the environment and this includes flies (Musca domestica and Sarcophaga haemorrhoidalis) which carry the pathovars C. sakazakii CC4 and ST8, as well as C. malonaticus ST7. Pava-Ripoll et al. (2012) reported that Cronobacter spp. were isolated from 14% of the flies they analyzed.
As given in Table 1, the remaining five species represented 18.5% of strains in the database. These 306 strains were composed of 191 STs. These species had primarily been isolated from environmental sites and are of less clinical relevance. The issue of whether only the clinically significance Cronobacter species need to be controlled in PIF has not been evaluated by regulatory authorities. However, the lack of epidemiological evidence of infection from all species may not be substantiated due to the frequent misidentification of Cronobacter strains following the routine use of phenotyping for identification (Jackson and Forsythe, 2016).
Diversity of Cronobacter Species According to Capsular Profiling
This new study enabled capsular profiling to be undertaken using genomes from across the whole Cronobacter genus, instead of being focussed on C. sakazakii and C. malonaticus as per the previous studies (Ogrodzki and Forsythe, 2015, 2016). Previously, the variation in the K-antigen and colanic acid biosynthesis encoding regions were profiled as a novel means of differentiating strains of C. sakazakii and C. malonaticus (Ogrodzki and Forsythe, 2015). The K-antigen region found was homologous to the well described K-antigen gene cluster from E. coli and was composed of three regions. However while E. coli has 60 K-antigen variants, the study of Cronobacer spp. by Ogrodzki and Forsythe (2015) found only two variants of the loci (designated K1 and K2). The K-antigen Region 1 (kpsEDCS) and Region 3 (kpsTM) were conserved across the two species, however there were only two variants of Region 2, designated as K1 and K2, which extended into the kpsS gene. The Region 2 variants differed in their length (2.28 and 3.78 kb) and CG% content (34.7 and 42.8%). They both encoded for glycosyltransferases genes for which there were no specific nearest matches (<50% similarity) in any BLAST search. Presumably the 2 variants reflect differences in the synthesized K-antigen which is exported to the cell surface, and also corresponds with the differences in the kpsS sequence.
This new study, using 275 genomes, shows the K-antigen region was present in all Cronobacter strains and species across the whole genus, with the exception of C. dublinensis ST389. Nevertheless there were no further variants have been found in this wider study compared to that of Ogrodzki and Forsythe (2015). The composition of the K-antigen specific capsular polysaccharide remains unknown, though it could be an important virulence or environmental fitness trait.
Previous analysis of the colanic acid biosynthesis region of C. sakazakii and C. malonaticus revealed there were two variants of the colanic acid biosynthesis gene cluster. CA2 differed from CA1 in the absence of the galE (encoding for UDP-N-acetyl glucosamine 4-epimerase). It is predictable that the chemical composition of the colanic acid produced by the two variants will differ, though the affect on virulence or environmental fitness trait is uncertain. The colanic acid biosynthesis region was present in all Cronobacter strains and species across the whole genus based on the analysis of 275 genomes. Nevertheless, as for the K-antigen region, there were no further variants found in this wider study. The variation is the K-antigen and colanic acid biosynthesis genes therefore do not follow the phylogeny of the genus.
Cronobacter sakazakii CC4 strains are associated with neonatal meningitis, and have the K2:CA2 capsule profile (Ogrodzki and Forsythe, 2015). Although this could be attributed to clonal inheritance, a limited number of non-C. sakazakii CC4 strains from meningitis cases have been collated from the PubMLST database. It is therefore notable that the K2:CA2 profile was also found in three other species C. malonaticus (n = 10), C. turicensis (n = 7), and C. universalis (n = 1) as well as the more distantly related C. dublinensis (n = 1); (Table 2). Therefore this capsule profile is neither phylogenetically nor clonaly related. In this new study, more strains of C. malonaticus ST60 and ST307 are shown to encode for the K2:CA2 profile. Previously only one C. malonaticus reported meningitis case had been documented (Ogrodzki and Forsythe, 2015). This strain (1569, ST307) was a blood isolate from a meningitis case in the United States, and was purported as being the infectious organism. This single case was taken as a possible outlier evidence for K2:CA2 profile link to meningitis. In this paper a second C. malonaticus K2:CA2 isolate of significance is reported. The strain Chcon-9 (ID 1494) was a CSF isolate, kindly deposited by Dr Jing-hua Cui (CDC Beijing). This second example was isolated in China (2014) from the CSF of an infant born with an EGA of 36 weeks, who had been fed fortified breast milk before developing clinical symptoms of meningitis on day 11. Strain Chcon-9 is ST60 not ST307 as per the previous C. malonaticus example. This latter observation further confirms the lack of congruence between the capsule profile and phylogeny. However, the remaining C. malonaticus ST60 strains of clinical origin also had the K2:CA2 profile and were from adults aged 26–82 years (no clinical symptoms available). The apparent correlation between capsule profile and severe infant meningitis gives a clear direction for further meningitis research with the bacterium.
None of the 55 C. malonaticus strains encoded for the sialic acid utilization genes (yhch-nanKTAR); (Table 2). This trait had previously been proposed as an important virulence trait in C. sakazakii given the occurrence of sialic acid in breast milk, mucin and gangliosides. Strains of C. sakazakii are able to grow on sialic acid and the ganglioside GM1 as a sole carbon source (Joseph et al., 2013b). This ability may explain the more frequent occurrence of C. sakazakii in severe meningitic infections compared with C. malonaticus.
Of the C. turicensis strains encoding for K2:CA2, none were clinical in origin (Table 2). The strains were from 5 STs which were not phylogenetically closely related (Joseph et al., 2012b). Three of these also encoded for the sialic acid utilization genes (yhch-nanKTAR), and a further 4 strains also encoded for these genes but had the capsule profile K1:CA2. These were the only strains outside the C. sakazakii species which encoded for the sialic acid utilization genes. Similar to the strains encoding for K2:CA2, the yhch-nanKTAR encoding strains which were identified in six different STs, clustered together according to phylogenetic analysis (Joseph et al., 2012c). The C. turicensis type strain lacked the sialic acid utilizing genes despite being closely related to these 6 STs. This would account for the earlier reported absence of sialic acid utilization in C. turicensis in laboratory studies by (Joseph et al., 2013b). Whether the acquisition of the sialic acid utilization trait by a phylogenetic cluster within C. turicensis is of ecological significance is uncertain given none of the strains were clinical in origin.
The K2:CA2 profile was also detected in one strain each of C. dublinensis (2030) and C. universalis (1883); (Table 2). Unfortunately the specific site of isolation and clinical presentation of C. dublinensis strain 2030 (synonym CDC 28–83) are unknown and was previously misidentified as C. sakazakii (Miled-Bennour et al., 2010). Currently this is the only ST301 strain in the Cronobacter PubMLST database, and hence the only one available for genome sequencing. The nearest related ST is ST70 for which there are 2 strains (1130 and 1462) in the database (Figure 2). Both strains were from Korea, one of which was from follow-up formula and the other was a food isolate (Chap et al., 2009). Neither of these strains have been sequenced to date. Whether other traits of significance occur in ST301 can be investigated in the future using comparative genomic analysis via the PubMLST Cronobacter database. This was considered as being outside the scope of this study. The single C. universalis strain (1883) encoding for K2:CA2 was isolated during a food survey in the Czech Republic (synonym DEM 3321) (Killer et al., 2015).
The occurrence of the K2:CA2 profile therefore does not follow phylogeny within the Cronobacter genus. Instead, there is only an association within C. sakazakii for severe cases of infection (NEC and meningitis) and C. malonaticus with severe cases of meningitis. Additionally, none of the C. turicensis strains encoding for K2:CA2 had been isolated from CSF. The co-occurrence of the sialic acid utilization genes did not occur in the two isolates of C. malonaticus isolated from meningitis cases. This observation does not discount the potential contribution of the sialic acid utilization genes in C. sakazakii meningitis, but does infer that it is not essential.
The Cronobacter genus is predicted to have evolved in the Palaeogene period of the Cenozoic era when early flowering plants evolved and coincides with the suspected natural plant habitat of the organism. The earliest branches of the genus lead to C. dublinensis and C. muytjensii, whereas C. sakazakii is predicted to have evolved more recently (Joseph and Forsythe, 2012; Grim et al., 2013). Possibly linked to this, Table 1 shows the considerable genetic diversity of C. dublinensis and C. muytjensii, as reflected by the relatively large number of STs for total number of strains (107/155 and 28/57) compared with the relative smaller number of C. sakazakii STs (236/1126) reflecting the considerable clonality within the latter species. Such clonality results in a low amount of genomic difference between unrelated strains and therefore limits the use of conventional genotyping methods such as PFGE.
Diversity of Cronobacter Species According to CRISPR-cas Array Profiling
CRISPR-cas array content has previously been strongly associated with sequence based phylogeny and hence could be used as a rapid lineage based detection method. This analysis has been applied to various Enterobacteriaceae (Yersinia, Salmonella and E. coli ) for phylogenetic, evolutionary and virulence related analysis (Makarova et al., 2015). It has also been considered as a discriminatory tool for epidemiological purposes since bacterial strains from the same geographical and temporal region should acquire the same spacers due to localized exposure to phages and plasmids, differentiating them from unrelated strains. In Cronobacter it was proposed that the strong clonal lineages of C. sakazakii resulted in a constraint in the CRISPR spacer array diversity (Ogrodzki and Forsythe, 2016). To consider if the CRISPR-cas array diversity varied across the genus, the arrays were determined for the less clonal species, C. dublinensis and C. muytjensii and compared with C. sakazakii.
Thirty C. muytjensii (n = 10) and C. dublinensis (n = 20) isolates were subject to detailed CRISPR-cas analysis. These two species were chosen since MLSA analysis had shown their greater diversity and more frequent novel sequence typing than the more clonal species C. sakazakii which has previously been analyzed. It was predicted that there would also be greater CRISPR-cas diversity in these two species given the probable reduction in clonal constraint of genomic recombination. The selected strains had been isolated from 11 countries over a 58 year period.
As shown in Table 3, both C. dublinensis and C. muytjensii included two CRISPR-cas operon architectures of I-E (Ecoli) and I-F (Ypseudo). The distribution of the operon types were not phylogenetically related as shown in Figure 2. There were a considerably high number of direct repeats and spacers in the two species, generating up to 5 CRISPR arrays and up to 66 spacers per strain compared to a maximum of 4 CRISPR arrays and 31 spacers per strain across 4 clonal C. sakazakii pathovars (Ogrodzki and Forsythe, 2016). Some strains in C. dublinensis and C. muytjensii lacked cas genes. The lack of cas genes was also found in the type strains of C. universalis and C. condimenti species.
Horizontal gene transfer of CRISPR and cas genes can occur between strains of the same species and even distant species and genera (Makarova et al., 2015). Subsequently not all strains within a species will necessarily possess the same sets of CRISPR-cas genes. As given in Table 3, within the Cronobacter genera there is a considerable variation in CRISPR-cas arrays, and even operon type and presence. Similarly, CRISPR-cas genes are present in enterohemorrhagic (EHEC) and Shiga toxin producing E. coli serotypes, but not the extra intestinal (ExPEC) E. coli phylogenetic group B2. Consequently, it has been proposed that the type I-E CRISPR-cas system could have alternative functions, such as gene expression and virulence (Touchon et al., 2011; Yin et al., 2013; García-Gutiérrez et al., 2015). Whether this relates to the variation in CRISPR-cas arrays in Cronobacter is currently unclear.
This is the first study to identify the wide-spread phylogenetic distribution of CRISPR-cas arrays across the Cronobacter genus, as opposed to just C. sakazakii (Ogrodzki and Forsythe, 2016; Zeng et al., 2017). This analysis has confirmed that differentiation within clonal lineages can be achieved using genotyping based on CRISPR-cas array variability.
Genotyping across the Cronobacter Genus
Due to the increasing number of genome and allele sequences (MLST alleles and whole genomes) deposited in the PubMLST Cronobacter database, this article not only updates researchers regarding the diversity of strains within the Cronobacter genus as revealed by 7-loci MLST, it has also investigated the occurrence of the capsule profiles and CRISPR-cas arrays beyond the initial studies of C. sakazakii and C. malonaticus.
Genotyping of Cronobacter spp. using MLST has considerably improved our understanding of the bacterial population diversity across the genus, and led to the recognition of clonal lineages and pathovars. However, like PFGE, MLST is unable to discriminate between unrelated strains within a clonal lineage (Alsonosi et al., 2015). Therefore more discriminatory DNA-sequence based methods need to be developed. Given the increasing availability and lowering costs of NGS tools, there is an increasing trend for genome-based genotyping methods, such as CRISPR-cas array profiling. Such analysis may also provide additional understanding of the diversity of the species and potential virulence mechanisms.
Given the expanding nature of the Cronobacter PubMLST database, the figures used were as of April 2017 and may differ in precise values when accessed later. Nevertheless the general consensus of Cronobacter diversity will be the same. The online Cronobacter PubMLST database has enabled open access to considerable information on Cronobacter isolates which can be interrogated by researchers, industry and regulatory authorities for taxonomic, and genotyping purposes. The curation of metadata of the isolates is standardized and therefore facilitates an international contribution to collating information.
Ethics Statement
All clinical data are taken from previous publications associated with the sequenced bacterial strains.
Author Contributions
PO: performed the genomic analysis and collated the data. SF: Initiated the study, wrote the manuscript, and managed the project. Both authors approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors gratefully thank Nottingham Trent University for their financial support (PO). This publication made use of the Cronobacter Multi Locus Sequence Typing website http://pubmlst.org/cronobacter/) developed by Keith Jolley and sited at the University of Oxford (Maiden et al., 2013). The development of this site has been funded by the Wellcome Trust. The authors thank the numerous collaborators who have submitted sequences to the open access Cronobacter PubMLST database.
Abbreviations
BIGSdb, Bacterial Isolate Genome Sequence database; Cas, CRISPR-associated protein-coding genes; CC, clonal complex; Cg-MLST, core genome multilocus sequence typing; CRISPR, Clustered regularly interspaced short palindromic repeats; EGA, estimated gestation age; MLST, multilocus sequence typing; NGS, next generation sequencing; PCR, polymerase chain reaction; PFGE, pulsed-field gel electrophoresis; PIF, powdered infant formula; r-MLST, ribosomal multilocus sequence typing; ST, sequence type.
Footnotes
- ^ http://bigsdb.pasteur.fr/
- ^ http://mlst.warwick.ac.uk/
- ^ https://pubmlst.org
- ^ https://pubmlst.org/cronobacter/
- ^ www.pubMLST.org/cronobacter/
References
Almagro-Moreno, S., and Boyd, E. F. (2009). Insights into the evolution of sialic acid catabolism among bacteria. BMC Evol. Biol. 9:118. doi: 10.1186/1471-2148-9-118
Almajed, F. S., and Forsythe, S. J. (2016). Cronobacter sakazakii clinical isolates overcome host barriers and evade the immune response. Microb. Pathog. 90, 55–63. doi: 10.1016/j.micpath.2015.11.014
Alsonosi, A., Hariri, S., Kajsik, M., Orieskova, M., Hanulik, V., Roderova, M., et al. (2015). The speciation and genotyping of Cronobacter isolates from hospitalised patients. Eur. J. Clin. Microbiol. Infect. Dis. 34, 1979–1988. doi: 10.1007/s10096-015-2440-8
Alzahrani, H., Winter, J., Boocock, D., De Girolamo, L., and Forsythe, S. J. (2015). Characterization of outer membrane vesicles from a neonatal meningitic strain of Cronobacter sakazakii. FEMS Microbiol. Lett. 362:fnv085. doi: 10.1093/femsle/fnv085
Arbatsky, N. P., Wang, M., Shashkov, A. S., Chizhov, A. O., Feng, L., Knirel, Y. A., et al. (2010). Structure of the O-polysaccharide of Cronobacter sakazakii O2 with a randomly O-acetylated l-rhamnose residue. Carbohydr. Res. 345, 2090–2094. doi: 10.1016/j.carres.2010.07.014
Baldwin, A., Loughlin, M., Caubilla-Barron, J., Kucerova, E., Manning, G., Dowson, C., et al. (2009). Multilocus sequence typing of Cronobacter sakazakii and Cronobacter malonaticus reveals stable clonal structures with clinical significance which do not correlate with biotypes. BMC Microbiol. 9:223. doi: 10.1186/1471-2180-9-223
Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., et al. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. doi: 10.1126/science.1138140
Blažková, M., Javùrková, B., Vlach, J. J., Göselová, S., Karamonová, L., Ogrodzki, P., et al. (2015). Diversity of O antigens within the genus Cronobacter: from disorder to order. Appl. Environ. Microbiol. 81, 5574–5582. doi: 10.1128/AEM.00277-15
Broge, T., and Lee, A. (2013). A case of Cronobacter (Enterobacter sakazakii) bacteremia in a breastfed infant. J. Pediatr. Infect. Dis. Soc. 2, e1–e2. doi: 10.1093/jpids/pit021
Carleton, H. A., and Gerner-Smidt, P. (2016). Whole-genome sequencing is taking over foodborne disease surveillance. Microbe Mag. 11, 311–317. doi: 10.1128/microbe.11.311.1
Carver, T., Harris, S. R., Berriman, M., Parkhill, J., and McQuillan, J. A. (2012). Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28, 464–469. doi: 10.1093/bioinformatics/btr703
Caubilla-Barron, J., and Forsythe, S. J. (2007). Dry stress and survival time of Enterobacter sakazakii and other Enterobacteriaceae in dehydrated powdered infant formula. J. Food Prot. 70, 2111–2117. doi: 10.4315/0362-028X-70.9.2111
Caubilla-Barron, J., Hurrell, E., Townsend, S., Cheetham, P., Loc-Carrillo, C., Fayet, O., et al. (2007). Genotypic and phenotypic analysis of Enterobacter sakazakii strains from an outbreak resulting in fatalities in a neonatal intensive care unit in France. J. Clin. Microbiol. 45, 3979–3985. doi: 10.1128/JCM.01075-07
Centers for Disease Control and Prevention [CDC] (2010). Multistate Outbreak of Human Salmonella Enteritidis Infections Associated with Shell Eggs. Available at: https://www.cdc.gov/salmonella/2010/shell-eggs-12-2-10.html [accessed May 2, 2017].
Chap, J., Jackson, P., Siqueira, R., Gaspar, N., Quintas, C., Park, J., et al. (2009). International survey of Cronobacter sakazakii and other Cronobacter spp. in follow up formulas and infant foods. Int. J. Food Microbiol. 136, 185–188. doi: 10.1016/j.ijfoodmicro.2009.08.005
Chaturvedi, N., Kajsik, M., Forsythe, S., and Pandey, P. N. (2015). Protein sequences insight into heavy metal tolerance in Cronobacter sakazakii BAA-894 encoded by plasmid pESA3. Arch. Microbiol. 197, 1141–1149. doi: 10.1007/s00203-015-1147-7
Craven, H. M., McAuley, C. M., Duffy, L. L., and Fegan, N. (2010). Distribution, prevalence and persistence of Cronobacter (Enterobacter sakazakii) in the nonprocessing and processing environments of five milk powder factories. J. Appl. Microbiol. 109, 1044–1052. doi: 10.1111/j.1365-2672.2010.04733.x
Czerwicka, M. M., Forsythe, S. J., Bychowska, A., Dziadziuszko, H., Kunikowska, D., Stepnowski, P., et al. (2010). Structure of the O-polysaccharide isolated from Cronobacter sakazakii 767. Carbohydr. Res. 345, 908–913. doi: 10.1016/j.carres.2010.01.020
Fei, P., Jiang, Y., Feng, J., Forsythe, S. J., Li, R., Zhou, Y., et al. (2017). Antibiotic and desiccation resistance of Cronobacter sakazakii and C. malonaticus isolates from powdered infant formula and processing environments. Front. Microbiol. 8:316. doi: 10.3389/fmicb.2017.00316
Fei, P., Man, C., Lou, B., Forsythe, S. J., Chai, Y., Li, R., et al. (2015). Genotyping and source tracking of Cronobacter sakazakii and C. malonaticus isolates from powdered infant formula and an infant formula production factory in China. Appl. Environ. Microbiol. 81, 5430–5439. doi: 10.1128/AEM.01390-15
Food and Agriculture Organization of the United Nations [FAO] (2004). Enterobacter sakazakii and other Microorganisms in Powdered Infant Formula, Microbiological Risk Assessment Series n. 6. Rome: Food and Agriculture Organization of the United Nations. doi: 10.1111/j.1740-8709.2004.00008.x
Food and Agriculture Organization of the United Nations [FAO] (2006). Expert Meeting on Enterobacter sakazakii and Salmonella in Powdered Infant Formula, Microbiological Risk Assessment Series n. 10. Geneva: World Health Organization.
Food and Agriculture Organization of the United Nations [FAO] and World Health Organization [WHO] (2008). Enterobacter sakazakii (Cronobacter spp.) in Powdered Follow-up Formulae, Microbiological Risk Assessment Series n. 15. Rome: FAO.
Forsythe, S. J., Dickins, B., and Jolley, K. A. (2014). Cronobacter, the emergent bacterial pathogen Enterobacter sakazakii comes of age; MLST and whole genome sequence analysis. BMC Genomics 15:1121. doi: 10.1186/1471-2164-15-1121
Franco, A. A., Kothary, M. H., Gopinath, G., Jarvis, K. G., Grim, C. J., Hu, L., et al. (2011). Cpa, the outer membrane protease of Cronobacter sakazakii, activates plasminogen and mediates resistance to serum bactericidal activity. Infect. Immun. 79, 1578–1587. doi: 10.1128/IAI.01165-10
Fricke, W. F., Mammel, M. K., McDermott, P. F., Tartera, C., White, D. G., Leclerc, J. E., et al. (2011). Comparative genomics of 28 Salmonella enterica isolates: evidence for CRISPR-mediated adaptive sublineage evolution. J. Bacteriol. 193, 3556–3568. doi: 10.1128/JB.00297-11
García-Gutiérrez, E., Almendros, C., Mojica, F. J. M., Guzmán, N. M., and García-Martínez, J. (2015). CRISPR content correlates with the pathogenic potential of Escherichia coli. PLOS ONE 10:e0131935. doi: 10.1371/journal.pone.0131935
Grim, C., Kothary, M., Gopinath, G., Jarvis, K., Beaubrun, J. J., McClelland, M., et al. (2012). Identification and characterization of Cronobacter iron acquisition systems. Appl. Environ. Microbiol. 78, 6035–6050. doi: 10.1128/AEM.01457-12
Grim, C. J., Kotewicz, M. L., Power, K. A., Gopinath, G., Franco, A. A., Jarvis, K. G., et al. (2013). Pan-genome analysis of the emerging foodborne pathogen Cronobacter spp. suggests a species-level bidirectional divergence driven by niche adaptation. BMC Genomics 14:366. doi: 10.1186/1471-2164-14-366
Grissa, I., Vergnaud, G., and Pourcel, C. (2007a). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Grissa, I., Vergnaud, G., and Pourcel, C. (2007b). The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 8:172. doi: 10.1186/1471-2105-8-172
Hariri, S., Joseph, S., and Forsythe, S. J. (2013). Cronobacter sakazakii ST4 strains and neonatal meningitis, United States. Emerg. Infect. Dis. 19, 175–177. doi: 10.3201/eid1901.120649
Himelright, I., Harris, E., and Lorch, V. A. M. (2002). Enterobacter sakazakii infections associated with the use of powdered infant formula–Tennessee, 2001. MMWR Morb. Mortal. Wkly. Rep. 51, 297–300.
Holy, O., and Forsythe, S. (2014). Cronobacter spp. as emerging causes of healthcare-associated infection. J. Hosp. Infect. 86, 169–177. doi: 10.1016/j.jhin.2013.09.011
Holý, O., Petrželová, J., Hanulík, V., Chromá, M., Matoušková, I., and Forsythe, S. J. (2014). Epidemiology of Cronobacter spp. isolates from patients admitted to the Olomouc University Hospital (Czech Republic). Epidemiol. Mikrobiol. Imunol. 63, 69–72.
Hurrell, E., Kucerova, E., Loughlin, M., Caubilla-Barron, J., and Forsythe, S. J. (2009a). Biofilm formation on enteral feeding tubes by Cronobacter sakazakii, Salmonella serovars and other Enterobacteriaceae. Int. J. Food Microbiol. 136, 227–231. doi: 10.1016/j.ijfoodmicro.2009.08.007
Hurrell, E., Kucerova, E., Loughlin, M., Caubilla-Barron, J., Hilton, A., Armstrong, R., et al. (2009b). Neonatal enteral feeding tubes as loci for colonisation by members of the Enterobacteriaceae. BMC Infect. Dis. 9:146. doi: 10.1186/1471-2334-9-146
Iversen, C., and Forsythe, S. (2003). Risk profile of Enterobacter sakazakii, an emergent pathogen associated with infant milk formula. Trends Food Sci. Technol. 14, 443–454. doi: 10.1016/S0924-2244(03)00155-9
Iversen, C., Lane, M., and Forsythe, S. J. (2004). The growth profile, thermotolerance and biofilm formation of Enterobacter sakazakii grown in infant formula milk. Lett. Appl. Microbiol. 38, 378–382. doi: 10.1111/j.1472-765X.2004.01507.x
Iversen, C., Mullane, N., McCardell, B., Tall, B. D., Lehner, A., Fanning, S., et al. (2008). Cronobacter gen. nov., a new genus to accommodate the biogroups of Enterobacter sakazakii, and proposal of Cronobacter sakazakii gen. nov., comb. nov., Cronobacter malonaticus sp. nov., Cronobacter turicensis sp. nov., Cronobacter muytjensii sp. nov., Cronobacter dublinensis sp. nov., Cronobacter genomospecies 1, and of three subspecies, Cronobacter dublinensis subsp. dublinensis subsp. nov., Cronobacter dublinensis subsp. lausannensis subsp. nov. and Cronobacter dublinensis subsp. lactaridi subsp. nov. Int. J. Syst. Evol. Microbiol. 58(Pt 6), 1442–1447. doi: 10.1099/ijs.0.65577-0
Jackson, E. E., Flores, J. P., Fernandez-Escartin, E., and Forsythe, S. J. (2015a). Reevaluation of a suspected Cronobacter sakazakii outbreak in Mexico. J. Food Prot. 78, 1191–1196. doi: 10.4315/0362-028X.JFP-14-563
Jackson, E. E., Masood, N., Ibrahim, K., Urvoy, N., Hariri, S., and Forsythe, S. J. (2015b). Description of Siccibacter colletis sp. nov., a novel species isolated from plant material, and emended description of Siccibacter turicensis. Int. J. Syst. Evol. Microbiol. 65(Pt 4), 1335–1341. doi: 10.1099/ijs.0.000108
Jackson, E. E., and Forsythe, S. J. (2016). Comparative study of Cronobacter identification according to phenotyping methods. BMC Microbiol. 16:146. doi: 10.1186/s12866-016-0768-6
Jacobs, C., Braun, P., and Hammer, P. (2011). Reservoir and routes of transmission of Enterobacter sakazakii (Cronobacter spp.) in a milk powder-producing plant. J. Dairy Sci. 94, 3801–3810. doi: 10.3168/jds.2011-4318
Jarvis, K. G., Grim, C. J., Franco, A. A., Gopinath, G., Sathyamoorthy, V., Hu, L., et al. (2011). Molecular characterization of Cronobacter lipopolysaccharide O-antigen gene clusters and development of serotype-specific PCR assays. Appl. Environ. Microbiol. 77, 4017–4026. doi: 10.1128/AEM.00162-11
Jolley, K. A., and Maiden, M. C. J. (2014). Using multilocus sequence typing to study bacterial variation: prospects in the genomic era. Future Microbiol. 9, 623–630. doi: 10.2217/fmb.14.24
Joseph, S., Cetinkaya, E., Drahovska, H., Levican, A., Figueras, M. J., and Forsythe, S. J. (2012a). Cronobacter condimenti sp. nov., isolated from spiced meat, and Cronobacter universalis sp. nov., a species designation for Cronobacter sp. genomospecies 1, recovered from a leg infection, water and food ingredients. Int. J. Syst. Evol. Microbiol. 62, 1277–1283. doi: 10.1099/ijs.0.032292-0
Joseph, S., Desai, P., Ji, Y., Cummings, C. A., Shih, R., Degoricija, L., et al. (2012b). Comparative analysis of genome sequences covering the seven Cronobacter species. PLOS ONE 7:e49455. doi: 10.1371/journal.pone.0049455
Joseph, S., Sonbol, H., Hariri, S., Desai, P., McClelland, M., and Forsythe, S. J. (2012c). Diversity of the Cronobacter genus as revealed by multilocus sequence typing. J. Clin. Microbiol. 50, 3031–3039. doi: 10.1128/JCM.00905-12
Joseph, S., and Forsythe, S. J. (2011). Predominance of Cronobacter sakazakii sequence type 4 in neonatal infections. Emerg. Infect. Dis. 17, 1713–1715. doi: 10.3201/eid1709.110260
Joseph, S., and Forsythe, S. J. (2012). Insights into the emergent bacterial pathogen Cronobacter spp., generated by multilocus sequence typing and analysis. Front. Microbiol. 3:397. doi: 10.3389/fmicb.2012.00397
Joseph, S., Hariri, S., and Forsythe, S. J. (2013a). Lack of continuity between Cronobacter biotypes and species as determined using multilocus sequence typing. Mol. Cell. Probes 27, 137–139. doi: 10.1016/j.mcp.2013.02.002
Joseph, S., Hariri, S., Masood, N., and Forsythe, S. (2013b). Sialic acid utilization by Cronobacter sakazakii. Microb. Inform. Exp. 3:3. doi: 10.1186/2042-5783-3-3
Killer, J., Skrivanova, E., Hochel, I., and Marounek, M. (2015). Multilocus sequence typing of Cronobacter strains isolated from retail foods and environmental samples. Foodborne Pathog. Dis. 12, 514–521. doi: 10.1089/fpd.2014.1884
Kim, H., Ryu, J.-H., and Beuchat, L. R. (2006). Attachment of and biofilm formation by Enterobacter sakazakii on stainless steel and enteral feeding tubes. Appl. Environ. Microbiol. 72, 5846–5856. doi: 10.1128/AEM.00654-06
Kloepper, T. H., and Huson, D. H. (2008). Drawing explicit phylogenetic networks and their integration into SplitsTree. BMC Evol. Biol. 8:22. doi: 10.1186/1471-2148-8-22
Kothary, M. H., Gopinath, G. R., Gangiredla, J., Rallabhandi, P. V., Harrison, L. M., Yan, Q. Q., et al. (2017). Analysis and characterization of proteins associated with outer membrane vesicles secreted by Cronobacter spp. Front. Microbiol. 8:134. doi: 10.3389/fmicb.2017.00134
Kucerova, E., Clifton, S. W., Xia, X.-Q., Long, F., Porwollik, S., Fulton, L., et al. (2010). Genome sequence of Cronobacter sakazakii BAA-894 and comparative genomic hybridization analysis with other Cronobacter species. PLOS ONE 5:e9556. doi: 10.1371/journal.pone.0009556
Kucerova, E., Joseph, S., and Forsythe, S. (2011). The Cronobacter genus: ubiquity and diversity. Qual. Assur. Saf. Crops Foods 3, 104–122. doi: 10.1111/j.1757-837X.2011.00104.x
Liu, H., Yang, Y., Cui, J., Liu, L., Liu, H., Hu, G., et al. (2013). Evaluation and implementation of a membrane filter method for Cronobacter detection in drinking water. FEMS Microbiol. Lett. 344, 60–68. doi: 10.1111/1574-6968.12155
Maclean, L. L., Vinogradov, E., Pagotto, F., Farber, J. M., and Perry, M. B. (2010). The structure of the O-antigen of Cronobacter sakazakii HPB 2855 isolate involved in a neonatal infection. Carbohydr. Res. 345, 1932–1937. doi: 10.1016/j.carres.2010.06.020
Maiden, M. C. J., Jansen van Rensburg, M. J., Bray, J. E., Earle, S. G., Ford, S. A., Jolley, K. A., et al. (2013). MLST revisited: the gene-by-gene approach to bacterial genomics. Nat. Rev. Microbiol. 11, 728–736. doi: 10.1038/nrmicro3093
Makarova, K. S., Wolf, Y. I., Alkhnbashi, O. S., Costa, F., Shah, S. A., Saunders, S. J., et al. (2015). An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 13, 722–736. doi: 10.1038/nrmicro3569
Masood, N., Moore, K., Farbos, A., Hariri, S., Paszkiewicz, K., Dickins, B., et al. (2013). Draft genome sequence of the earliest Cronobacter sakazakii sequence Type 4 strain, NCIMB 8272. Genome Announc. 1:e00782-13. doi: 10.1128/genomeA.00782-13
Masood, N., Moore, K., Farbos, A., Paszkiewicz, K., Dickins, B., McNally, A., et al. (2015). Genomic dissection of the 1994 Cronobacter sakazakii outbreak in a French neonatal intensive care unit. BMC Genomics 16:750. doi: 10.1186/s12864-015-1961-y
McQuiston, J. R., Herrera-Leon, S., Wertheim, B. C., Doyle, J., Fields, P. I., Tauxe, R. V., et al. (2008). Molecular phylogeny of the Salmonellae: relationships among Salmonella species and subspecies determined from four housekeeping genes and evidence of lateral gene transfer events. J. Bacteriol. 190, 7060–7067. doi: 10.1128/JB.01552-07
Miled-Bennour, R., Ells, T. C., Pagotto, F. J., Farber, J. M., Kérouanton, A., Meheut, T., et al. (2010). Genotypic and phenotypic characterisation of a collection of Cronobacter (Enterobacter sakazakii) isolates. Int. J. Food Microbiol. 139, 116–125. doi: 10.1016/j.ijfoodmicro.2010.01.045
Mullane, N., O’Gaora, P., Nally, J. E., Iversen, C., Whyte, P., Wall, P. G., et al. (2008). Molecular analysis of the Enterobacter sakazakii O-antigen gene locus. Appl. Environ. Microbiol. 74, 3783–3794. doi: 10.1128/AEM.02302-07
Muller, A., Stephan, R., Fricker-Feer, C., and Lehner, A. (2013). Genetic diversity of Cronobacter sakazakii isolates collected from a Swiss infant formula production facility. J. Food Prot. 76, 883–887. doi: 10.4315/0362-028X.JFP-12-521
Nadon, C., Van Walle, I., Gerner-smidt, P., Campos, J., Chinen, I., and Concepcion-acevedo, J. (2017). PulseNet International: vision for the implementation of whole genome sequencing (WGS) for global food- borne disease surveillance. Euro Surveill. 22:30544. doi: 10.2807/1560-7917.ES.2017.22.23.30544
Nascimento, M., Sousa, A., Ramirez, M., Francisco, A. P., Carriço, J. A., and Vaz, C. (2017). PHYLOViZ 2.0: providing scalable data integration and visualization for multiple phylogenetic inference methods. Bioinformatics 33, 128–129. doi: 10.1093/bioinformatics/btw582
Ogrodzki, P., and Forsythe, S. (2015). Capsular profiling of the Cronobacter genus and the association of specific Cronobacter sakazakii and C. malonaticus capsule types with neonatal meningitis and necrotizing enterocolitis. BMC Genomics 16:758. doi: 10.1186/s12864-015-1960-z
Ogrodzki, P., and Forsythe, S. J. (2016). CRISPR – cas loci profiling of Cronobacter sakazakii pathovars. Future Microbiol. 11, 1507–1519. doi: 10.2217/fmb-2016-0070
Patrick, M. E., Mahon, B. E., Greene, S. A., Rounds, J., Cronquist, A., Wymore, K., et al. (2014). Incidence of Cronobacter spp. infections, United States, 2003-2009. Emerg. Infect. Dis. 20, 1520–1523. doi: 10.3201/eid2009.140545
Pava-Ripoll, M., Pearson, R. E. G., Miller, A. K., and Ziobro, G. C. (2012). Prevalence and relative risk of Cronobacter spp., Salmonella spp., and Listeria monocytogenes associated with the body surfaces and guts of individual filth flies. Appl. Environ. Microbiol. 78, 7891–7902. doi: 10.1128/AEM.02195-12
Power, K. A., Yan, Q., Fox, E. M., Cooney, S., and Fanning, S. (2013). Genome sequence of Cronobacter sakazakii SP291, a persistent thermotolerant isolate derived from a factory producing powdered infant formula. Genome Announc. 1:e0008213. doi: 10.1128/genomeA.00082-13
Ravisankar, S., Syed, S. S., Garg, P., and Higginson, J. (2014). Is Cronobacter sakazakii infection possible in an exclusively breastfed premature neonate in the neonatal intensive care unit? J. Perinatol. 34, 408–409. doi: 10.1038/jp.2014.14
Reich, F., Konig, R., von Wiese, W., and Klein, G. (2010). Prevalence of Cronobacter spp. in a powdered infant formula processing environment. Int. J. Food Microbiol. 140, 214–217. doi: 10.1016/j.ijfoodmicro.2010.03.031
Schmid, M., Iversen, C., Gontia, I., Stephan, R., Hofmann, A., Hartmann, A., et al. (2009). Evidence for a plant-associated natural habitat for Cronobacter spp. Res. Microbiol. 160, 608–614. doi: 10.1016/j.resmic.2009.08.013
Shariat, N., and Dudley, E. G. (2014). CRISPRs: molecular signatures used for pathogen subtyping. Appl. Environ. Microbiol. 80, 430–439. doi: 10.1128/AEM.02790-13
Sonbol, H., Joseph, S., McAuley, C. M., Craven, H. M., and Forsythe, S. J. (2013). Multilocus sequence typing of Cronobacter spp. from powdered infant formula and milk powder production factories. Int. Dairy J. 30, 1–7. doi: 10.1016/j.idairyj.2012.11.004
Stephan, R., Grim, C. J., Gopinath, G. R., Mammel, M. K., Sathyamoorthy, V., Trach, L. H., et al. (2014). Re-examination of the taxonomic status of Enterobacter helveticus, Enterobacter pulveris and Enterobacter turicensis as members of the genus Cronobacter and their reclassification in the genera Franconibacter gen. nov. and Siccibacter gen. nov. as Francon. Int. J. Syst. Evol. Microbiol. 64(Pt 10), 3402–3410. doi: 10.1099/ijs.0.059832-0
Sun, Y., Wang, M., Liu, H., Wang, J., He, X., Zeng, J., et al. (2011). Development of an O-antigen serotyping scheme for Cronobacter sakazakii. Appl. Environ. Microbiol. 77, 2209–2214. doi: 10.1128/AEM.02229-10
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Touchon, M., Charpentier, S., Clermont, O., Rocha, E. P. C., Denamur, E., and Branger, C. (2011). CRISPR distribution within the Escherichia coli species is not suggestive of immunity-associated diversifying selection. J. Bacteriol. 193, 2460–2467. doi: 10.1128/JB.01307-10
Townsend, S., Hurrell, E., and Forsythe, S. (2008). Virulence studies of Enterobacter sakazakii isolates associated with a neonatal intensive care unit outbreak. BMC Microbiol. 8:64. doi: 10.1186/1471-2180-8-64
Townsend, S. M., Hurrell, E., Caubilla-Barron, J., Loc-Carrillo, C., and Forsythe, S. J. (2008). Characterization of an extended-spectrum beta-lactamase Enterobacter hormaechei nosocomial outbreak, and other Enterobacter hormaechei misidentified as Cronobacter (Enterobacter) sakazakii. Microbiology 154(Pt 12), 3659–3667. doi: 10.1099/mic.0.2008/021980-0
Townsend, S. M., Hurrell, E., Gonzalez-Gomez, I., Lowe, J., Frye, J. G., Forsythe, S., et al. (2007). Enterobacter sakazakii invades brain capillary endothelial cells, persists in human macrophages influencing cytokine secretion and induces severe brain pathology in the neonatal rat. Microbiology 153(Pt 10), 3538–3547. doi: 10.1099/mic.0.2007/009316-0
van Acker, J., de Smet, F., Muyldermans, G., Bougatef, A., Naessens, A., and Lauwers, S. (2001). Outbreak of necrotizing enterocolitis associated with Enterobacter sakazakii in powdered milk formula. J. Clin. Microbiol. 39, 293–297. doi: 10.1128/JCM.39.1.293-297.2001
Willis, L. M., and Whitfield, C. (2013). Structure, biosynthesis, and function of bacterial capsular polysaccharides synthesized by ABC transporter-dependent pathways. Carbohydr. Res. 378, 35–44. doi: 10.1016/j.carres.2013.05.007
Yan, Q., Power, K. A., Cooney, S., Fox, E., Gopinath, G. R., Grim, C. J., et al. (2013). Complete genome sequence and phenotype microarray analysis of Cronobacter sakazakii SP291: a persistent isolate cultured from a powdered infant formula production facility. Front. Microbiol. 4:256. doi: 10.3389/fmicb.2013.00256
Yin, S., Jensen, M. A., Bai, J., Debroy, C., Barrangou, R., and Dudley, E. G. (2013). The evolutionary divergence of Shiga toxin-producing Escherichia coli is reflected in clustered regularly interspaced short palindromic repeat (CRISPR) spacer composition. Appl. Environ. Microbiol. 79, 5710–5720. doi: 10.1128/AEM.00950-13
Keywords: Cronobacter, genotyping, genomes, MLST, capsule, CRISPR-cas profiles
Citation: Ogrodzki P and Forsythe SJ (2017) DNA-Sequence Based Typing of the Cronobacter Genus Using MLST, CRISPR-cas Array and Capsular Profiling. Front. Microbiol. 8:1875. doi: 10.3389/fmicb.2017.01875
Received: 29 June 2017; Accepted: 13 September 2017;
Published: 29 September 2017.
Edited by:
Giovanna Suzzi, Università degli Studi di Teramo, ItalyReviewed by:
Beatrix Stessl, Veterinärmedizinische Universität Wien, AustriaIrshad Sulaiman, United States Food and Drug Administration, United States
Songzhe Fu, University of New South Wales, Australia
Copyright © 2017 Ogrodzki and Forsythe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen J. Forsythe, c3RldmVmb3JzeXRoZUBmb29kbWljcm9iZS5jb20=