 Sadeeq Ur Rahman1,2,3
Sadeeq Ur Rahman1,2,3 Michael Stanton4
Michael Stanton4 Pat G. Casey1
Pat G. Casey1 Angela Spagnuolo5
Angela Spagnuolo5 Giuliano Bensi5
Giuliano Bensi5 Colin Hill1,2
Colin Hill1,2 Kevin P. Francis6
Kevin P. Francis6 Mark Tangney1,4,7
Mark Tangney1,4,7 Cormac G. M. Gahan1,2,7,8*
Cormac G. M. Gahan1,2,7,8*- 1APC Microbiome Institute, University College Cork, Cork, Ireland
- 2School of Microbiology, University College Cork, Cork, Ireland
- 3College of Veterinary Sciences and Animal Husbandry, Abdul Wali Khan University Mardan, Mardan, Pakistan
- 4Cork Cancer Research Centre, University College Cork, Cork, Ireland
- 5GSK Vaccines S.r.l., Siena, Italy
- 6PerkinElmer, Alameda, CA, United States
- 7SynBio Centre, University College Cork, Cork, Ireland
- 8School of Pharmacy, University College Cork, Cork, Ireland
Listeria monocytogenes is a Gram-positive facultative intracellular pathogen that is widely used as a model organism for the analysis of infection biology. In this context, there is a current need to develop improved reporters for enhanced bioluminescence imaging (BLI) of the pathogen in infection models. We have developed a click beetle red luciferase (CBR-luc) based vector (pPL2CBRopt) expressing codon optimized CBR-luc under the control of a highly expressed Listerial promoter (PHELP) for L. monocytogenes and have compared this to a lux-based system expressing bacterial luciferase for BLI of the pathogen using in vitro growth experiments and in vivo models. The CBR-luc plasmid stably integrates into the L. monocytogenes chromosome and can be used to label field isolates and laboratory strains of the pathogen. Growth experiments revealed that CBR-luc labeled L. monocytogenes emits a bright signal in exponential phase that is maintained during stationary phase. In contrast, lux-labeled bacteria produced a light signal that peaked during exponential phase and was significantly reduced during stationary phase. Light from CBR-luc labeled bacteria was more efficient than the signal from lux-labeled bacteria in penetrating an artificial tissue depth assay system. A cell invasion assay using C2Bbe1 cells and a systemic murine infection model revealed that CBR-luc is suited to BLI approaches and demonstrated enhanced sensitivity relative to lux in the context of Listeria infection models. Overall, we demonstrate that this novel CBR reporter system provides efficient, red-shifted light production relative to lux and may have significant applications in the analysis of L. monocytogenes pathogenesis.
Introduction
Listeria monocytogenes is an opportunistic facultative intracellular pathogen which is capable of withstanding harsh environmental conditions, including stresses encountered during the processing, packaging and storage of foods (Gahan and Hill, 2005). Following consumption of contaminated foods L. monocytogenes survives the conditions encountered in the gastrointestinal tract and can progress to systemic infection (listeriosis) (Gahan and Hill, 2014). In humans, the clinical manifestations of listeriosis include meningitis, encephalitis, and septicemia predominately in the immune-compromised host, or late-term spontaneous abortion in pregnant individuals. The mortality rate can reach 30% during common-source outbreaks in susceptible hosts (Mead et al., 2000). L. monocytogenes infection in mice has been used as an important model for the analysis of intracellular parasitism and subsequent immunity and this research has benefited from the development of molecular tools to analyze bacterial and host responses in vivo (Cossart and Toledo-Arana, 2008; Lebreton et al., 2016).
Bioluminescence imaging (BLI) is a non-invasive technology that permits the molecular analysis of cells through the expression of proteins that emit visible light (Byrne et al., 2013). The development of sensitive light detection systems has permitted the use of BLI for the visualization of cell activity or localization in small animal models and other model systems (Contag and Bachmann, 2002). The technology is particularly useful for the in vivo tracking of bacterial infectious agents in real time in small animal models (Cronin et al., 2012a,b). Engineering of bacteria to produce light has typically involved cloning and expression of a gene system (the bacterial lux system) that is naturally found in species of Vibrio and Photorhabdus (Gahan, 2012). However, eukaryotic luciferases from natural sources (e.g., firefly, click beetle, sea pansy) can also be expressed in bacteria and may offer alternatives to the use of bacterial Lux for bacterial imaging studies (Gahan, 2012; Tangney and Francis, 2012; Chang et al., 2014; Karimi et al., 2016).
The bacterial lux-based bioluminescent reporter systems are based on the expression of the bacterial lux operon (luxCDABE) to produce light (Belkin et al., 1996). This system does not require the addition of substrate as the LuxCDE proteins generate endogenous substrate in the bacterial cytoplasm (Gahan, 2012). In contrast, the use of eukaryotic bioluminescence systems (Luc enzymes) requires the addition of exogenous luciferin as substrate (Gahan, 2012; Vorobyeva et al., 2015). The emission spectra of these systems also varies, with the insect luciferase enzymes emitting light in the green-red range of the visible light spectrum, compared with the bacterial luciferase system that emits blue-green light (Contag and Bachmann, 2002). This has implications for whole body imaging experiments, as blue-green light is of a relatively short wavelength (400–490 nm) and therefore has a reduced ability to penetrate host tissues compared with light emitted in the red region of the spectrum (Rice et al., 2001; Zhao et al., 2005).
To date, BLI vectors utilized in L. monocytogenes have successfully exploited the bacterial lux operon for in vivo localization studies (Hardy et al., 2004, 2009; Disson et al., 2008; van Pijkeren et al., 2010; Poulsen et al., 2011; Cronin et al., 2012a; Bergmann et al., 2013) and for in situ analysis of microbial gene expression (Riedel et al., 2009; Sleator et al., 2009; Joyce and Gahan, 2010; Quereda et al., 2016). In the current study, we created a bioluminescence reporter system based upon a red-optimized click beetle luciferase enzyme (CBR-luc) for use in L. monocytogenes and compare this with a previously described lux system (Riedel et al., 2007). The CBR-luc system utilizes a Listeria-specific chromosomal integration vector (pPL2) (Lauer et al., 2002) and constitutive expression of the click beetle luciferase was achieved using the highly expressed listerial synthetic promoter (PHELP) described previously (Riedel et al., 2007). We envisage that this vector will have significant applications in the study of L. monocytogenes in the context of both in vivo and in vitro experimental systems.
Materials and Methods
Strains, Media, and Bacterial Growth
The strains and plasmids used in this study are listed in Table 1. E. coli TOP10 (Invitrogen, Paisley, United Kingdom) were used as a cloning host for the creation of pPL2CBRopt. E. coli was grown aerobically at 37°C in Luria-Bertani broth (LB) medium. L. monocytogenes EGDe (Glaser et al., 2001) and murinized L. monocytogenes EGDem (Monk et al., 2010) were grown aerobically at 30°C in brain heart infusion (BHI) medium (Oxoid, Basingstoke, United Kingdom). Clinical strains of L. monocytogenes strains 130029, 140030, and 150008 were obtained from clinical cases of listeriosis in Ireland (manuscript in preparation). The pH of the LB and BHI medium was buffered with 100 mM 3-(N-morpholino)propanesulfonic acid (pH 7.4) where outlined in the text. When required, antibiotics were added to the media as follows: for E. coli; chloramphenicol (30 μg/ml), ampicillin (100 μg/ml) and for L. monocytogenes; chloramphenicol (7.5 μg/ml). For measurement of growth curves, L. monocytogenes strains were grown overnight and diluted in fresh medium until the OD600 reached 0.05. Subsequently strains were grown shaking at 30°C and readings were taken using a spectrophotometer. To measure bioluminescence during growth over time, overnight cultures were diluted in fresh medium to OD600 0.05 and subsequently grown at 30°C. At selected time points, 50 μl of culture was mixed with 50 μl luciferin (PerkinElmer) with a final concentration of 150 μg/ml as described by the manufacturer.

TABLE 1. Strains and plasmids used in this study.
Synthesis, Cloning, and Integration of CBR
CBR was codon-optimized (CBRopt) for expression in Listeria and synthesized by GenScript (GenScript United States Inc., Piscataway, NJ, United States). The CBRopt gene (Gene Accession No. KY628997) was synthesized with an intact upstream constitutive Listerial promoter, highly expressed Listerial promoter (PHELP) (Riedel et al., 2007) and the fragment was sub-cloned into the pPL2 backbone (Lauer et al., 2002) in E. coli using Xho1 and Pst1 restriction enzymes resulting in pPL2CBRopt (see Figure 1). Plasmid DNA was isolated from E. coli using a QIAprep Spin Miniprep kit according to the manufacturer’s instructions (QIAGEN, Crawley, United Kingdom). The sequences were verified by sequencing through MWG Biotech AG (Ebersberg, Germany). Transformation of L. monocytogenes was performed as described (Park and Stewart, 1990). Successful integrants were screened for site-specific integration by PCR using template DNA of L. monocytogenes as described previously (Bron et al., 2006). Restriction endonucleases (Roche Diagnostic, Mannheim, Germany), T4 DNA ligase (Roche) and 2× PCR mixture (NEB, United Kingdom) were used as advised by the manufacturer. From each successful transformant one colony carrying the anticipated genotype was chosen for further bioluminescence analysis using an IVIS Lumina II system (PerkinElmer).
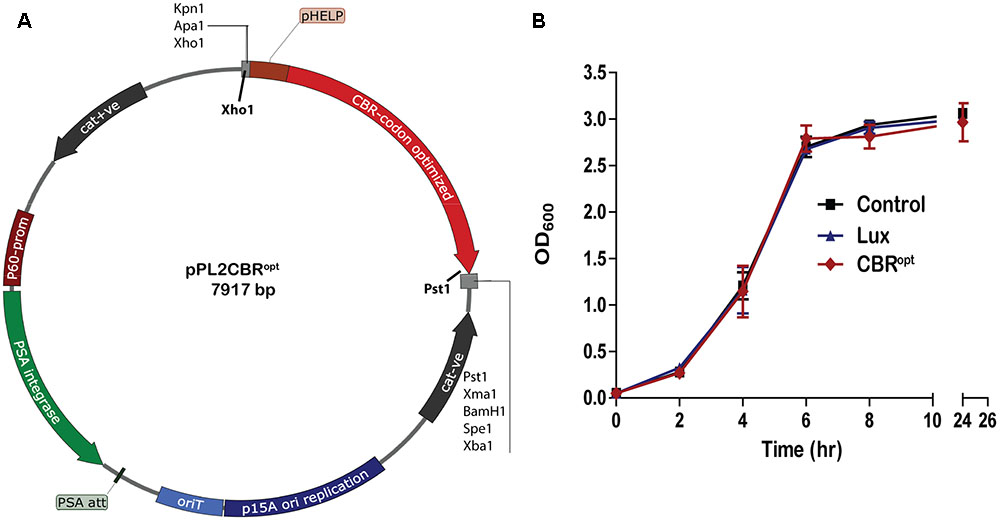
FIGURE 1. Cloning and expression of codon optimized click beetle luciferase. (A) Plasmid map of pPL2CBRopt expressing codon-optimized click beetle luciferase (CBRopt) from the Listerial highly expressed promoter (PHELP) (Riedel et al., 2007) in a pPL2 backbone (Lauer et al., 2002). (B) Growth rate of L. monocytogenes EGDe tagged with a bacterial lux (Riedel et al., 2007), CBRopt or an integrated vector control. Data and error bars represent mean and standard deviations, respectively, of triplicate samples. Each experiment was repeated at least three times in triplicate.
In Vitro Tissue Depth Model
Listeria monocytogenes EGDe::pPL2CBRopt, L. monocytogenes EGDe::pPL2lux (silent vector) or L. monocytogenes EGDe::pPL2 luxPHELP (expressing bacterial lux operon) (Table 1) were grown over night in BHI medium. Subsequently, cultures were grown in fresh BHI medium to a specified OD600. At specified OD600 (OD600 1 or 1.5) cells were transferred to 96-well round bottom plates for imaging. Luciferin (PerkinElmer) was added to L. monocytogenes EGDe::pPL2CBRopt (for in vitro 150 μg/ml, for in vivo 150 mg/kg body weight) according to the manufacturer’s instructions 15 min prior to imaging. Measurements of bioluminescence were repeated with intervening layers of uniformly cut meat slices (one to five layers, thickness varied from 2 to 10 mm depending on meat type) tested over the surface of the plate. Three different meat types (to reflect different physical and chemical parameters), chicken, serrano ham, and fresh sliced raw beef steak were tested. Images were acquired with up to 3 min exposure and processed by Living Image Software v4.2.
Cell Invasion
All the cell culture procedures were performed as described previously (Bron et al., 2006). C2Bbe1 cells (CRC-2012; American Type Culture Collection), a clone of the Caco2 human adeno carcinoma cell line, were used for the cell invasion assay. Briefly, C2Bbe1 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 4.5 g/l Glutamax (Gibco Laboratories, Grand Island, NY, United States), 10% fetal bovine serum (Gibco), 1% (vol/vol) non-essential amino acids (Gibco), 1% (vol/vol) penicillin–streptomycin (Gibco), and 0.01 mg/ml human transferrin (Calbiochem) at 37°C in a 5% CO2 environment. To perform the cell invasion assay, C2Bbe1 cells were trypsinized (Gibco), harvested by slow centrifugation at 500 × g for 5 min followed by resuspension in antibiotic free DMEM containing 10% fetal calf serum. Viable cells were counted using a standard trypan blue exclusion procedure. A known concentration of 1 × 105 viable cells per well were seeded onto a 24- or 12-well flat bottom tissue culture plate (Sarstedt, Leicester, United Kingdom). Subsequently, the plates were incubated for 48–72 h at 37°C in a 5% CO2 atmosphere until confluent. Listeria strains were grown overnight in BHI medium, re-inoculated into BHI medium and grown until the OD600 reached 1.0 (1 × 109 CFU/ml). Subsequently, bacterial cells were washed with antibiotic free DMEM medium twice and added to the C2Bbe1 cells at a multiplicity of infection of 100:1 followed by incubation at 37°C for 1 h in 5% CO2. The infection of C2Bbe1 cells was stopped after 1 h by adding gentamicin to a final concentration of 100 μg/ml, and cells were incubated further for 30 min to kill extracellular bacteria (Kim et al., 2004). Subsequently, the cells were washed three times with DMEM, and the levels of bioluminescence were measured using the IVIS Lumina II system (PerkinElmer). Immediately after imaging, the monolayers were lysed with 1 ml of ice-cold distilled water containing 0.01% triton (Sigma-Aldrich, St. Louis, MO, United States). The lysate was serially diluted in PBS and bacteria were plated onto BHI agar plates to determine the number of viable intracellular bacteria as CFU/ml.
Animals, Infection, and Image Acquisition
For in vivo experiment, 8- to 10-week-old female BALB/c mice (Envigo, United Kingdom) weighing approximately 18–21 g were housed in pathogen free conditions at 22°C with a 12/12 light cycle. Animals were randomly assigned to groups (n = 4) and provided with mouse chow and water ad libitum. Before experiments, mice were afforded an adaptation period of at least 7 days. For bacteria preparation and infection, overnight cultures of L. monocytogenes were pelleted by centrifugation (7000 × g for 5 min), washed twice with PBS, and used to infect mice (n = 24) intraperitoneally at 2 × 105 CFU in a total of 200 μl of PBS (Rea et al., 2004). The mice were followed for 72 h for imaging with 24 h intervals as described. After 24-, 48-, and 72-h post-infection, the infected mice were imaged following injection of luciferin (PerkinElmer, Inc., Boston, MA, United States) according to the manufacturer’s instructions at a dose of 150 mg/kg body weight administered via IP route as appropriate or imaged directly (mice infected with L. monocytogenes EGDe::pPL2luxPHELP). After whole body imaging, mice were sacrificed, and post-mortem resected organs were imaged using the IVIS system. Images were acquired with up to 5 min exposure and processed by Living Image Software v4.2 or v4.3.1.
Ethics Statement
All animal procedures were performed in accordance with the national ethical guidelines prescribed by the Health Products Regulatory Authority (HPRA). Protocols and all the murine experiments were approved by the animal ethics committee of University College Cork (AERR #2010/003 and #2012/015).
Statistical Analysis
All experiments were performed in triplicate at least two times. Data shown are for representative experiments. Statistical significance was based on either the Student’s t-test or ANOVA for comparison of groups (see Figures 2B, 6C). GraphPad Prism 5 or IBM-SPSS v22 was used for graphs and statistical analysis. Linear regressions were carried out using GraphPad Prism 5. In all analysis, p < 0.05 was considered as significant.
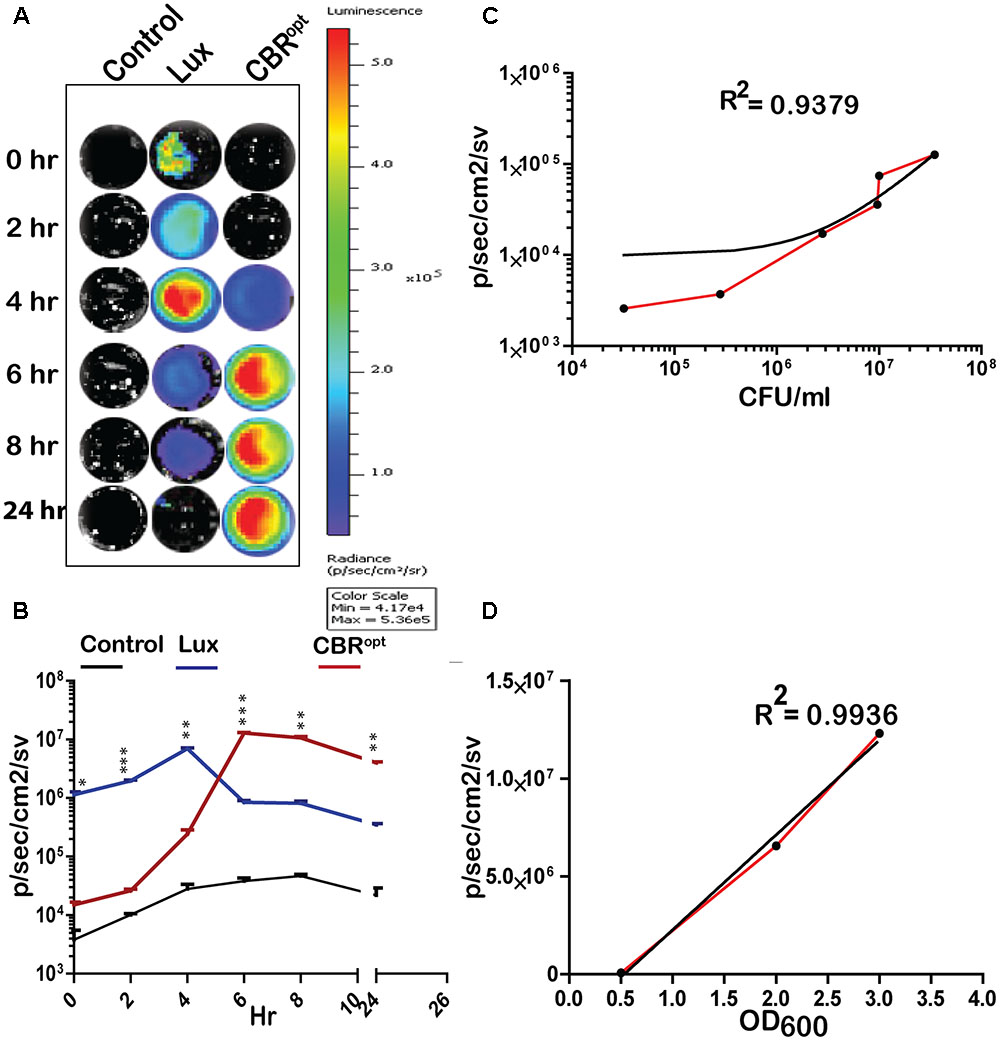
FIGURE 2. Luminescence of tagged L. monocytogenes during growth in media. (A) Luminescence imaging of luciferase-tagged L. monocytogenes strains. Luminescent images were taken using the PerkinElmer IVIS Lumina II system and processed using Living Image software as indicated in the text. The color bar represents bioluminescent signal intensity (in photons/s/cm2). (B) Luminescence (flux) of luciferase-tagged L. monocytogenes strains during in vitro growth in BHI broth. Data and error bars represent mean and standard deviation, respectively, of triplicate samples for each time point. A two-way ANOVA with Bonferroni post-test was used to compare groups. (C) Luciferase-tagged L. monocytogenes strains were grown to log phase and then serially diluted. Each dilution was imaged and also plated for CFU counts. The obtained data of total flux and CFU count was correlated. Statistical analysis showed a positive liner co-relation (with r2 = 0.94) between total flux emitted by CBRopt and the luminescent bacterial. (D) L. monocytogenes EGDe::pPL2CBRopt were grown in BHI medium and images were obtained by IVIS at given OD600 points. The data obtained was then analyzed that indicated a strong positive correlation between OD600 and total flux of the luminescent bacteria. Linear regression analysis was used to determine the relationship between flux and OD600 values in (C,D).
Results and Discussion
Construction and Functional Analysis of a Click Beetle Red Luciferase Reporter System
The main purpose of the work reported herein was the construction and functional analysis of the pPL2CBRopt bioluminescence vector (Figure 1A) using the backbone of the pPL2 integrating plasmid. pPL2 contains a PSA listeriophage integrase gene and attachment site that directs site-specific single copy integration into tRNAArg locus on the L. monocytogenes chromosome (Lauer et al., 2002). In our lab, we have previously successfully created pPL2-derivatives, pPL2lux and pPL2luxPHELP that allow BLI through expression of the bacterial lux operon (Bron et al., 2006; Riedel et al., 2007). Here we used a similar approach to create a vector expressing a CBR-luc with potential applications in deep tissue whole body imaging.
For this purpose, a codon optimized CBR-luc (CBRopt) gene for optimal expression in L. monocytogenes was synthesized under the influence of a constitutive synthetic promoter, the highly expressed Listeria promoter PHELP (Riedel et al., 2007). The synthesized construct was then sub-cloned into pPL2 using Xho1 and Pst1 restriction enzymes (Figure 1A). The sequence of the insert was verified and the novel vector was then transformed and integrated into the L. monocytogenes EGDe wild type strain and an engineered L. monocytogenes variant (EGDem) expressing a murine-specific InlA (Monk et al., 2010). The pPL2 vector contains a PSA listeriophage integrase gene and attachment site that directs site-specific single copy integration into the tRNAArg locus on the L. monocytogenes chromosome (Lauer et al., 2002). The successful integrants were then examined for integration using PCR and positive clones were screened phenotypically for light emission. A total of 10 candidate integrants were picked and screened for integration with results indicating that 9 (90%) were positive integrants. In parallel, we used the pPL2luxPHELP and pPL2lux (silent vector) (Riedel et al., 2007) as controls for integration and subsequent analysis of luminescence.
CBRopt Reporter Is Stably Integrated in L. monocytogenes and Does Not Impede Growth Rate
We tested the stability of the L. monocytogenes EGDe::pPL2 CBRopt and EGDem::pPL2CBRopt strains in the absence of chloramphenicol. L. monocytogenes EGDe::pPL2CBRopt and EGDem::pPL2CBRopt were sub-cultured for at least 50 generations in BHI medium without antibiotic selection. Dilutions of the last passage of the cultures were plated and the presence of the integrated plasmids in the resulting colonies was evaluated by scoring chloramphenicol resistant colonies. Our results indicated that all 50 colonies tested after 50 generations were found to retain chloramphenicol resistance indicating that the integrating plasmid was stably maintained without antibiotic selection pressure. Furthermore, at least 10 random chloramphenicol-resistant colonies were also tested phenotypically for light emission and the results indicated that all 10 tested colonies were bioluminescent.
Previous work has indicated that integration of lux-expressing pPL2 constructs does not influence bacterial growth rate relative to wild type bacteria (Riedel et al., 2007). In the current study we evaluated the growth behavior of L. monocytogenes EGDe::pPL2CBRopt relative to EGDe::pPL2luxPHELP (Riedel et al., 2007) and EGDe::pPL2 (Bron et al., 2006). Strains were grown in parallel under standard conditions (Figure 1B). Our data indicated that the growth of L. monocytogenes EGDe::pPL2CBRopt was normal relative to the other strains tested (Figure 1B). Furthermore, the growth rates of L. monocytogenes EGDe::pPL2CBRopt were also not affected in BHI (in which pH was adjusted to 7.4) or LB medium (data not shown).
CBRopt Can Be Used for Tagging Clinical L. monocytogenes Strains
Listeria monocytogenes EGDe is the most commonly used laboratory strain for the analysis of listerial pathogenesis (Glaser et al., 2001). However, there is increasing interest in the analysis of wild type clinical isolates in order to determine strain specific differences in pathogenesis (Maury et al., 2016). For this purpose, three clinical strains (Table 1), all originating from patients who suffered meningitis and isolated from cerebrospinal fluid (CSF) were transformed with pPL2CBRopt. Our results indicated that all three wild type clinical strains could be successfully tagged with our newly developed plasmid (Supplementary Figure S1). After 30 passages of these strains in vitro 50 colonies were tested and all retained chloramphenicol resistance indicating that the system is as stable as observed in the EGDe laboratory strain. We also determined that the in vitro growth rate of these wild type field isolates in BHI broth was not affected by the presence of the integrated plasmid (data not shown). We transformed the clinical isolates with pPL2luxPHELP in order to provide a comparison with the CBRopt system. Results indicated that bioluminescent profile followed the same light emission pattern as observed for the EGDe strain as described below (Supplementary Figure S1).
CBR Signals Are Brighter and Produce Greater Flux Than lux During In Vitro Growth
We determined the bioluminescence profile of L. monocytogenes EGDe::pPL2CBRopt over time. L. monocytogenes EGDe::pPL2 CBRopt was grown in BHI broth for 24 h to monitor bioluminescence (Figures 2A,B). No bioluminescence was detected at the initial stage of growth. The bioluminescence profile increased as bacteria grew and bioluminescence reached a peak following 6 h of bacterial growth and remained relatively high during stationary phase. We directly compared the bioluminescence of L. monocytogenes EGDe::pPL2CBRopt with the L. monocytogenes EGDe::pPL2luxPHELP strain as both bioluminescent vectors are constructed using the pPL2 backbone with expression of luciferase under the same PHELP promoter system (Riedel et al., 2007). Bioluminescence signals of bacterial luciferase (L. monocytogenes EGDe::pPL2luxPHELP) were higher during initial growth as compared to the isolate expressing CBRopt. However, the bioluminescence signal from L. monocytogenes EGDe::pPL2luxPHELP diminished quickly as bacteria entered the stationary phase and the overall bioluminescence expression pattern for this strain was similar to our previously reported data (Riedel et al., 2007). A decrease in the bioluminescence signal during stationary phase may be a feature of light expression from the luxABCDE system as the phenomenon has also been described for other Gram-positive bacteria including Streptococcus pneumonia (Francis et al., 2001) and Lactobacillus casei (Oozeer et al., 2005). It is possible that the diminished Lux signal during stationary phase may be related to decreased metabolic recycling of FMN to FMNH2 (an essential component for bacterial Lux activity), and ATP production (Francis et al., 2001; Andreu et al., 2010) This hypothesis is partially supported by a previous study which demonstrated a 100-fold increase in signal after providing a gene (frp) encoding a NADPH-FMN oxidoreductase in-trans with luxAB enzymes in a yeast model system (Szittner et al., 2003). However, this limitation would not influence the activity of CBR, which in our system exhibits relatively robust activity in late exponential phase and stationary phase.
In order to measure light intensity per cell, bioluminescence signals were measured in parallel with determination of the total number of viable bacterial CFU. Our results indicated that CBRopt signals were directly correlated with CFU (r2 = 0.9379; Figure 2C). A similar positive correlation was observed between the OD600 and total flux during growth in medium (Figure 2D). The strong positive correlation between CFU and total flux indicates that luminescence from CBR-luc tagged bacteria can successfully be used for accurate quantification of bacterial numbers during growth.
In order to directly compare signal intensity each luminescent strain was serially diluted until no luminescence signals were detectable (Figures 3A,B) and the CFU/ml of each representative dilution was calculated in order to determine relationship of total flux/CFU. The bioluminescence profile of both lux and CBR-luc luciferase reporters were indeed dependent on the number of the luminescent bacteria. Bioluminescence intensity was correlated with the number of bioluminescent bacteria both for EGDe::pPL2luxPHELP and EGDe::pPL2CBRopt during growth over time in BHI medium (Figure 3B) and the overall analysis indicated that CBRopt produces greater flux/CFU when compared with bacterial luciferase during growth in medium (p > 0.05) (Figure 3C).
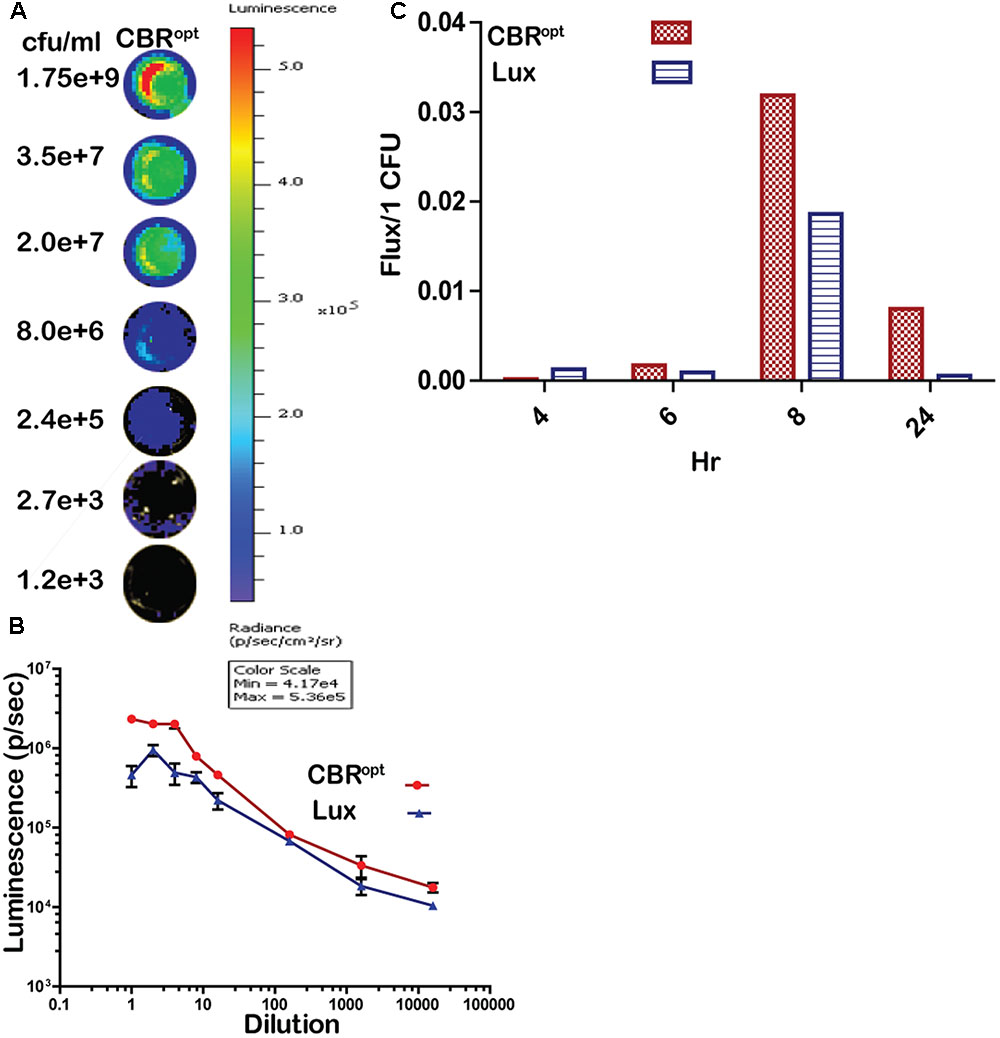
FIGURE 3. Luminescence per bacterium. (A) Luminescent images of CBRopt tagged L. monocytogenes following imaging of specified numbers bacteria (indicated as CFU/ml). L. monocytogenes EGDe::pPL2CBRopt were grown until mid-log phase. A 50-μl of culture in triplicate was then transferred to a 96-well plate and was serially diluted. Each dilution was imaged and also plated for CFU. A single representative well of the imaged bacterium is shown. (B) Correlation of the corresponding luminescence and CFU is shown. Data are expressed as total flux in photon/s. Data and error bars represent mean and standard deviation, respectively, of triplicate samples for each dilution. (C) CBRopt or lux-tagged L. monocytogenes were grown in BHI medium. At given time points (4, 6, 8, and 24 h), 50 μl of culture in triplicate was then transferred to 96-well plate and was serially diluted. Each dilution was imaged and also plated for CFU counts to determine the total flux per CFU.
Bioluminescence Profile of CBRopt in an In Vitro Model of Varied Tissue Depth
CBR-luc produces light of a wavelength (∼600 nm) that is within the red region of the visible light spectrum. As red light has greater penetrating power in animal tissues, we predicted that light generated by CBR-luc would penetrate mammalian tissues more effectively than that of lux (in the blue spectrum at around 480 nm). We compared the ability of the CBRopt and Lux luciferase signals to penetrate through varying depths of tissue using a previously described in vitro assay (Mason et al., 2016). Layers of cut meat slices were used to mimic varying levels of tissue depth. An aliquot of 50 μl of cells were deposited into a round bottom 96-well plate and light measurements were repeated following the addition of layers of meat slices to generate tissue depth as indicated in Figure 4. As expected a decrease in light detection was associated with an increase in tissue depth. Signal transmission and total flux of bacterial luciferase (L. monocytogenes EGDe::pPL2luxPHELP) quickly declined as the thickness of the tissue increased as compared to CBRopt (L. monocytogenes EGDe::pPL2CBRopt). In particular L. monocytogenes EGDe::pPL2CBRopt was clearly visible through multiple layers of raw beef steak whereas the penetrating power of light generated in L. monocytogenes EGDe::pPL2luxPHELP was poor and below detectable limits (Figures 4A,B).
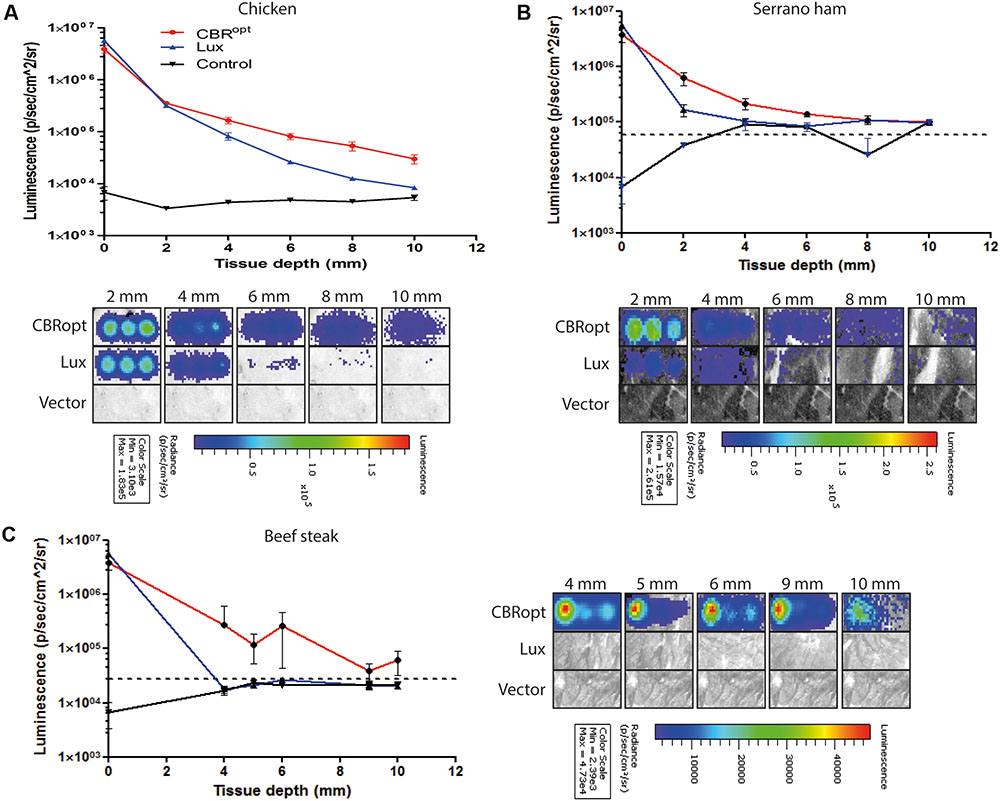
FIGURE 4. Ability of bioluminescence signals to penetrate an in vitro model tissue model. Luciferase-tagged and control (empty vector) L. monocytogenes strains were grown to log phase and were normalized to an identical OD600. A 50-μl of bacterial culture in triplicate was then transferred to a 96-well plate. Imaging was repeated with intervening layers of meat slices of various diameters. A representative experiment is shown. Attenuation of luminescence from bacterial luciferase is obvious in the red meat (beef steak) samples whereas CBRopt labeled bacteria produced a signal that was clearly visible through layers of red meat as indicated. The Figure indicates the identical experiment carried out using slices of (A) chicken, (B) serrano ham, and (C) fresh sliced raw beef steak. In all cases, error bars represent mean ± standard deviation. The dotted line represents luminescence from meat placed over a well containing media alone.
BLI of CBRopt during a Cell Invasion Assay
The pPL2luxPHELP vector has previously proved effective for the imaging of L. monocytogenes infection in cultured mammalian cells (Riedel et al., 2007). In the current study, we investigated the bioluminescence profile of L. monocytogenes EGDe::pPL2CBRopt during a cell invasion assay in order to determine the possibility of using this vector for monitoring and detection of EGDe in cell culture. For this purpose, L. monocytogenes EGDe::pPL2CBRopt, L. monocytogenes EGDe::pPL2luxPHELP and L. monocytogenes EGDe::pPL2lux were used to infect C2Bbe1 cells for 1 h and the levels of bioluminescence and number of the intracellular bacteria were determined (Figures 5A,B). The efficacy of invasion did not differ between the strains and bacteria were isolated at similar levels from each well. However, the bioluminescence intensity indicated that L. monocytogenes EGDe::pPL2CBRopt produced a consistently brighter signal and significantly higher flux than L. monocytogenes EGDe::pPL2luxPHELP in the context of in vitro infection (Figures 5A,B). This suggests that the pPL2CBRopt vector may have applications for BLI of Listeria during infection assays, perhaps with the potential for use in high throughput assay systems.
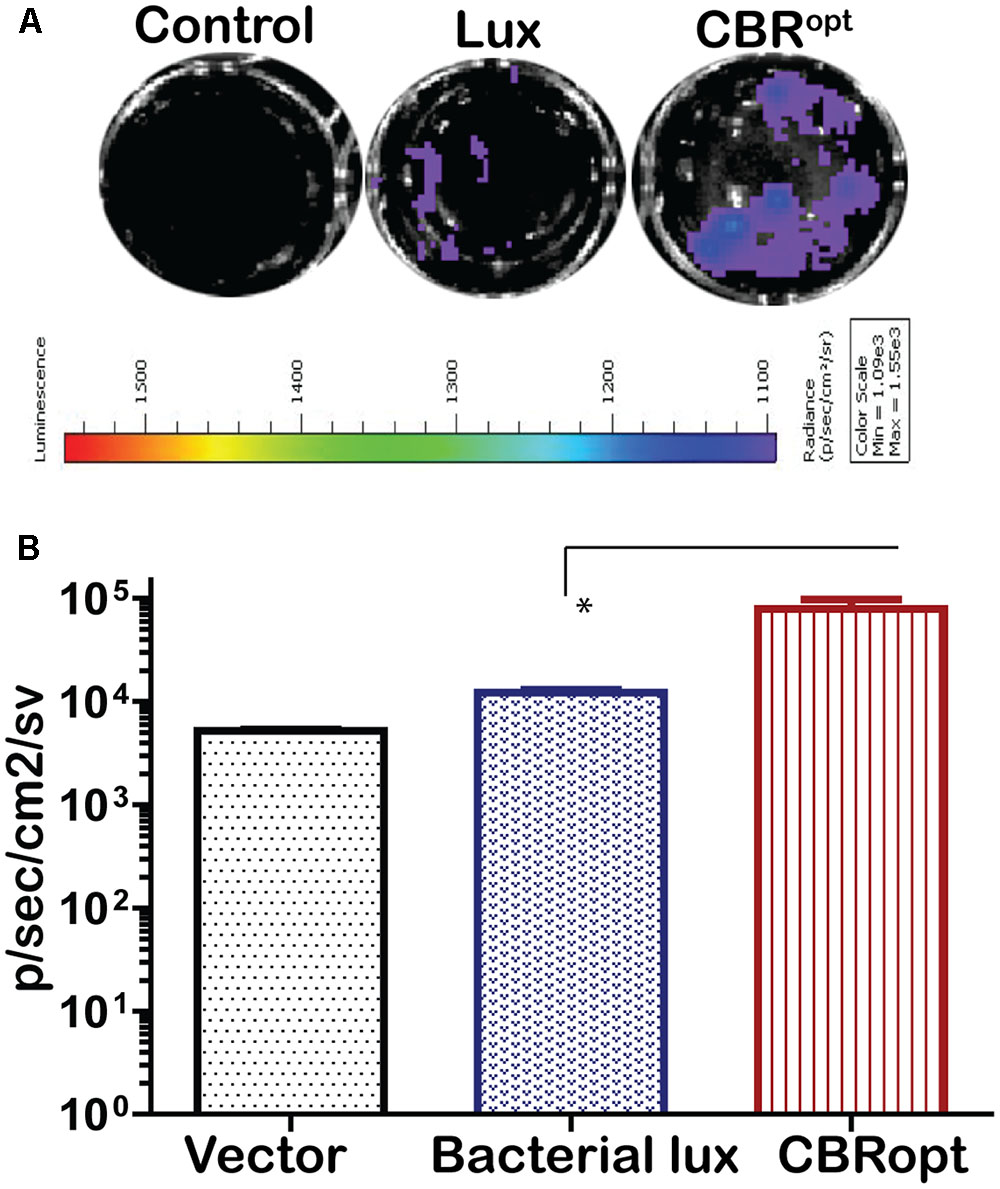
FIGURE 5. Comparison of luminescence during growth of L. monocytogenes in C2Bbe1 cell culture. (A) Luminescence images of luciferase-tagged L. monocytogenes or a control infecting C2Bbe1 cells after 1 h of infection. A 100 MOI infection rate was used. Cells were infected for about an hour, after that, extracellular bacteria were killed by gentamicin. The image was taken after washing superficial and non-invaded bacteria using. (B) Corresponding luminescence data of the luciferase-tagged L. monocytogenes or a control. CBRopt clearly emit higher flux than the bacterial lux. Data and error bar represent mean and standard deviation, respectively, of the triplicate samples. ∗p < 0.05 based upon ANOVA analysis.
CBRopt Facilitates Robust In Vivo Whole Body Imaging of Murine Infection
Murine infection is widely used for the analysis of factors contributing to the pathogenesis of L. monocytogenes (Lecuit, 2007; Cossart and Toledo-Arana, 2008). A number of pathogens such as Trypanosoma brucei (Van Reet et al., 2014), Mycobacterium tuberculosis (Chang et al., 2014), and Lactobacillus species (Daniel et al., 2015) have been previously tagged with CBR reporters that provided enhanced whole body images in murine infection model. Given the improved signal intensity mediated by CBR during in vitro growth, in particular during late exponential and stationary-phase, we examined the signal intensity from L. monocytogenes EGDe::pPL2CBRopt during murine infection. For this purpose, three groups of 12 mice were infected with murinized L. monocytogenes EGDem::pPL2CBRopt, murinized L. monocytogenes EGDem::pPL2luxPHELP, and murinized EGDem::pPL2lux with a standard inoculum (2 × 106 CFU/ml) via the intraperitoneal route. Four mice were left uninfected to serve as the control group. Four mice from each group were than imaged at 24 h intervals post-infection until 72 h. For the mice infected with L. monocytogenes EGDem::pPL2CBRopt luciferin was injected intraperitoneally 10 min prior to imaging. After imaging, mice were sacrificed and the resected organs (liver, spleen, and kidneys) were imaged for bioluminescence and further processed for counting the number of viable bacteria. Results of the whole body imaging of mice indicated that no bioluminescence was observed 24 h post-infection in any of the strains. No bioluminescence signals were detectable after dissecting the mice and imaging individual organs at this time point. This lack of bioluminescence correlated well with the lower number of viable bacteria recovered from the resected organs of the infected mice. Of these, the highest number of CFU was recovered from the spleen where CFU/ml were in the range of 1 × 103.
Notably, mice infected with L. monocytogenes EGDem::pPL2CBRopt produced detectable bioluminescence signals at 48 h post-infection, while no signals were detected from mice infected with L. monocytogenes EGDem::pPL2luxPHELP or the L. monocytogenes EGDem::pPL2lux control. We acknowledge that there was some variability in observable signals between individual mice (when imaged at the specified sensitivity) as noted in previous studies by other groups (Poulsen et al., 2011). The number of CFU in these animals reached approximately 1 × 106 CFU/ml in the spleen and liver. Importantly, there was no significant difference in CFU/ml among the infected strains recovered from the infected mice post-mortem in resected organs (results not shown). Interestingly, at 48 h infection bioluminescent signals were detected in the resected liver and spleen of mice infected with L. monocytogenes EGDem::pPL2luxPHELP, but no signals were detectable during whole body imaging, indicating that these signals were attenuated during whole body imaging, potentially as a consequence of the low penetrating power of the shorter wavelength light emitted by Lux. In contrast, bioluminescence signals were observed during whole body imaging of mice infected with L. monocytogenes EGDem::pPL2CBRopt at 48 h post-infection in addition to a clear signal from post-mortem resected organs (Figure 6).
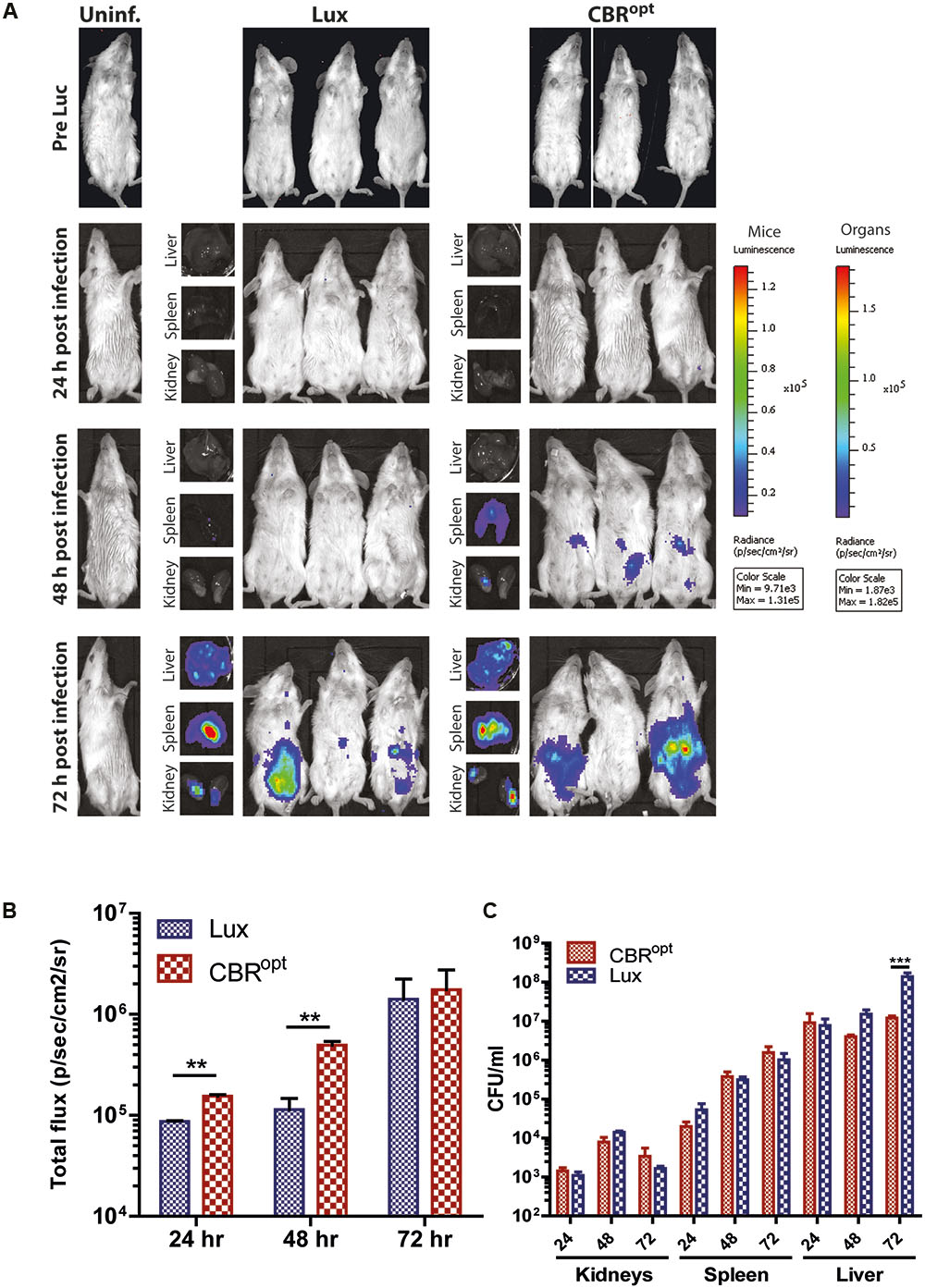
FIGURE 6. In vivo BLI. (A) In vivo BLI of sample mice infected with murinized L. monocytogenes EGDe tagged with either bacterial Lux or CBRopt. Mice (n = 4) were infected ip with L. monocytogenes at a dose of 2 × 106 and the infection was followed over time: 24, 48, and 72 h. Post-mortem-resected kidneys, spleen, and liver are shown along with the corresponding mouse. Light emission from CBRopt infected mice is clearly detectable at 48 h post-infection, unlike that of animal infected with the Lux-tagged strain. (B) Quantitative BLI readout from murine whole body imaging at given time points. Luminescence is shown as total flux (p/s/cm2/sv). (C) Bacterial count of post-mortem resected organs determined after plating homogenate. Data and error bars represent mean and standard deviation, respectively, of quadruplicate samples. Asterisks “∗∗” indicates p < 0.05 based upon Student’s t-test. ∗∗∗p < 0.01.
In a tandem study using infection with wild type (non-murinized) EGDe we also noted a clearly enhanced signal from mice infected with CBRopt labeled bacteria relative to mice infected with Lux-tagged EGDe (Supplementary Figure S2). In this experiment, bacterial burdens in the internal organs were comparable between the groups though the infectious load in the organs was noticeably lower than mice infected with EGDem (not shown). Again this suggests that the signal from CBRopt may provide greater sensitivity at low bacterial loads.
Overall, the ability to detect luminescence signals in mice infected with L. monocytogenes EGDem::pPL2CBRopt at 48 h suggested that pPL2CBRopt provides for increased sensitivity in the context of whole body imaging. At higher bacterial numbers on day 3 of infection the difference in signal intensity in whole body imaging between the Lux and CBR-labeled bacteria was less evident (Figure 6).
Conclusion
In conclusion, we have constructed a system for the stable chromosomal integration of constitutively expressed CBR luciferase in L. monocytogenes. In comparison to a L. monocytogenes strain expressing bacterial luciferase (LuxAB), CBR provides a greater luminescence signal during the stationary phase of growth and better penetrating power in an in vitro tissue depth model. We also noted enhanced sensitivity during murine infection and in an in vitro cell infection assay. A limitation of using CBR is the necessity to add luciferin substrate to the system. This adds an extra variable, as the availability of substrate in complex systems would certainly affect signal intensity. In contrast bacterial lux systems encode the capacity to generate endogenous substrate thereby potentially eliminating this extra variable. Given the different emission spectra of each system there is significant potential to utilize both in combination in order to measure dual bacterial populations in complex systems as has been described recently (Daniel et al., 2015). Overall, we suggest that the pPL2CBRopt vector may have significant applications for the study of L. monocytogenes in various model systems.
Author Contributions
SUR and CG wrote the paper. SUR, MS, PC, AS, GB, CH, KF, MT, and CG proofread the paper. SUR, MS, PC, AS, GB, CH, KF, MT, and CG designed experiments. SUR, MS, PC, and AS carried out experiments. SUR, MS, PC, AS, GB, CH, KF, MT, and CG analyzed experiments.
Funding
This work was funded by an Industry Academia Partnerships and Pathways grant (Vaccines and Imaging Partnership) as part of the European Commission Seventh Framework Programme (PIAP-GA-2013-612219-VIP). The authors acknowledge the financial support of the APC Microbiome Institute funded through Science Foundation Ireland (SFI) under Grant Number SFI/12/RC/2273.
Conflict of Interest Statement
AS and GB are permanent employees of the GSK group of companies. GB reports ownership of GSK restricted GSK shares.
The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01797/full#supplementary-material
FIGURE S1 | Bioluminescence profile of clinical listerial strains labeled with CBRopt reporter. EGDe-4b-130029:pPL2CBRopt, EGDe-4b-140030::pPL2CBRopt, and EGDe-1/2a-150008::pPL2CBRopt (see Table 1) were integrated with the newly developed codon optimized (CBRopt) reporter. The tagged strains were then grown in BHI medium and images were recorded at different time intervals. The overall bioluminescence profile is shown. Data and error bars represent mean and standard deviation, respectively, of triplicate samples for each time point. Asterisks “∗∗” indicates p < 0.05 based upon Student’s t-test.
FIGURE S2 |In vivo BLI of mice infected with wild type L. monocytogenes EGDe. (A) Whole body luminescence image of BALB/c mice infected with wild type L. monocytogenes EGDe tagged with either bacterial Lux or CBRopt. Mice were infected ip with a dose of 2 × 105 bacteria and were imaged for luminescence at 72 h post-infection. A clear and robust signal is detectable from mice infected with CBRopt tagged bacteria at this timepoint. A luminescent signal that exceeded background (uninfected animals) was detected in every L. monocytogenes EGDe CBRopt infected mouse. The number of CFU/ml recovered from infected mice were not significantly different between both groups (not shown). (B) Graph showing the signal intensity from the whole body images obtained at 72 h from infected mice. Luminescence is shown as total flux (p/s/cm2/sr). Data and error bars represent mean and standard error.
References
Andreu, N., Zelmer, A., Fletcher, T., Elkington, P. T., Ward, T. H., Ripoll, J., et al. (2010). Optimisation of bioluminescent reporters for use with mycobacteria. PLOS ONE 5:e10777. doi: 10.1371/journal.pone.0010777
Belkin, S., Smulski, D. R., Vollmer, A. C., Van Dyk, T. K., and LaRossa, R. A. (1996). Oxidative stress detection with Escherichia coli harboring a katG’::lux fusion. Appl. Environ. Microbiol. 62, 2252–2256.
Bergmann, S., Rohde, M., Schughart, K., and Lengeling, A. (2013). The bioluminescent Listeria monocytogenes strain Xen32 is defective in flagella expression and highly attenuated in orally infected BALB/cJ mice. Gut Pathog. 5:19. doi: 10.1186/1757-4749-5-19
Bron, P. A., Monk, I. R., Corr, S. C., Hill, C., and Gahan, C. G. (2006). Novel luciferase reporter system for in vitro and organ-specific monitoring of differential gene expression in Listeria monocytogenes. Appl. Environ. Microbiol. 72, 2876–2884. doi: 10.1128/AEM.72.4.2876-2884.2006
Byrne, W. L., DeLille, A., Kuo, C., de Jong, J. S., van Dam, G. M., Francis, K. P., et al. (2013). Use of optical imaging to progress novel therapeutics to the clinic. J. Control. Release 172, 523–534. doi: 10.1016/j.jconrel.2013.05.004
Chang, M., Anttonen, K. P., Cirillo, S. L., Francis, K. P., and Cirillo, J. D. (2014). Real-time bioluminescence imaging of mixed mycobacterial infections. PLOS ONE 9:e108341. doi: 10.1371/journal.pone.0108341
Contag, C. H., and Bachmann, M. H. (2002). Advances in in vivo bioluminescence imaging of gene expression. Annu. Rev. Biomed. Eng. 4, 235–260. doi: 10.1146/annurev.bioeng.4.111901.093336
Cossart, P., and Toledo-Arana, A. (2008). Listeria monocytogenes, a unique model in infection biology: an overview. Microbes Infect. 10, 1041–1050. doi: 10.1016/j.micinf.2008.07.043
Cronin, M., Akin, A. R., Collins, S. A., Meganck, J., Kim, J. B., Baban, C. K., et al. (2012a). High resolution in vivo bioluminescent imaging for the study of bacterial tumour targeting. PLOS ONE 7:e30940. doi: 10.1371/journal.pone.0030940
Cronin, M., Stanton, R. M., Francis, K. P., and Tangney, M. (2012b). Bacterial vectors for imaging and cancer gene therapy: a review. Cancer Gene Ther. 19, 731–740. doi: 10.1038/cgt.2012.59
Daniel, C., Poiret, S., Dennin, V., Boutillier, D., Lacorre, D. A., Foligne, B., et al. (2015). Dual-color bioluminescence imaging for simultaneous monitoring of the intestinal persistence of Lactobacillus plantarum and Lactococcus lactis in living mice. Appl. Environ. Microbiol. 81, 5344–5349. doi: 10.1128/AEM.01042-15
Disson, O., Grayo, S., Huillet, E., Nikitas, G., Langa-Vives, F., Dussurget, O., et al. (2008). Conjugated action of two species-specific invasion proteins for fetoplacental listeriosis. Nature 455, 1114–1118. doi: 10.1038/nature07303
Francis, K. P., Yu, J., Bellinger-Kawahara, C., Joh, D., Hawkinson, M. J., Xiao, G., et al. (2001). Visualizing pneumococcal infections in the lungs of live mice using bioluminescent Streptococcus pneumoniae transformed with a novel gram-positive lux transposon. Infect. Immun. 69, 3350–3358. doi: 10.1128/iai.69.5.3350-3358.2001
Gahan, C., and Hill, C. (2005). Gastrointestinal phase of Listeria monocytogenes infection. J. Appl. Microbiol. 98, 1345–1353. doi: 10.1111/j.1365-2672.2005.02559.x
Gahan, C. G. (2012). The bacterial lux reporter system: applications in bacterial localisation studies. Curr. Gene Ther. 12, 12–19. doi: 10.2174/156652312799789244
Gahan, C. G., and Hill, C. (2014). Listeria monocytogenes: survival and adaptation in the gastrointestinal tract. Front. Cell. Infect. Microbiol. 4:9. doi: 10.3389/fcimb.2014.00009
Glaser, P., Frangeul, L., Buchrieser, C., Rusniok, C., Amend, A., Baquero, F., et al. (2001). Comparative genomics of Listeria species. Science 294, 849–852.
Hardy, J., Chu, P., and Contag, C. H. (2009). Foci of Listeria monocytogenes persist in the bone marrow. Dis. Model Mech. 2, 39–46. doi: 10.1242/dmm.000836
Hardy, J., Francis, K. P., DeBoer, M., Chu, P., Gibbs, K., and Contag, C. H. (2004). Extracellular replication of Listeria monocytogenes in the murine gall bladder. Science 303, 851–853. doi: 10.1126/science.1092712
Joyce, S. A., and Gahan, C. G. (2010). Molecular pathogenesis of Listeria monocytogenes in the alternative model host Galleria mellonella. Microbiology 156(Pt 11), 3456–3468. doi: 10.1099/mic.0.040782-0
Karimi, S., Ahl, D., Vagesjo, E., Holm, L., Phillipson, M., Jonsson, H., et al. (2016). In vivo and in vitro detection of luminescent and fluorescent Lactobacillus reuteri and application of red fluorescent mCherry for assessing plasmid persistence. PLOS ONE 11:e0151969. doi: 10.1371/journal.pone.0151969
Kim, H., Boor, K. J., and Marquis, H. (2004). Listeria monocytogenes σB contributes to invasion of human intestinal epithelial cells. Infect. Immun. 72, 7374–7378. doi: 10.1128/IAI.72.12.7374-7378.2004
Lauer, P., Chow, M. Y., Loessner, M. J., Portnoy, D. A., and Calendar, R. (2002). Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J. Bacteriol. 184, 4177–4186. doi: 10.1128/JB.184.15.4177-4186.2002
Lebreton, A., Stavru, F., Brisse, S., and Cossart, P. (2016). 1926-2016: 90 Years of listeriology. Microbes Infect. 18, 711–723. doi: 10.1016/j.micinf.2016.10.009
Lecuit, M. (2007). Human listeriosis and animal models. Microbes Infect. 9, 1216–1225. doi: 10.1016/j.micinf.2007.05.009
Mason, E. A., Lopez, R., and Mason, R. P. (2016). Wavelength shifting of chemiluminescence using quantum dots to enhance tissue light penetration. Opt. Mater. Express 6, 1384–1392. doi: 10.1364/OME.6.001384
Maury, M. M., Tsai, Y. H., Charlier, C., Touchon, M., Chenal-Francisque, V., Leclercq, A., et al. (2016). Uncovering Listeria monocytogenes hypervirulence by harnessing its biodiversity. Nat. Genet. 48, 308–313. doi: 10.1038/ng.3501
Mead, P. S., Slutsker, L., Dietz, V., and McCaig, L. F. (2000). Food-related illness and death in the United States. Emerg. Infect. Dis. 5, 607–625. doi: 10.3201/eid0505.990502
Monk, I. R., Casey, P. G., Hill, C., and Gahan, C. G. (2010). Directed evolution and targeted mutagenesis to murinize Listeria monocytogenes internalin A for enhanced infectivity in the murine oral infection model. BMC Microbiol. 10:318. doi: 10.1186/1471-2180-10-318
Oozeer, R., Furet, J., Goupil-Feuillerat, N., Anba, J., Mengaud, J., and Corthier, G. (2005). Differential activities of four Lactobacillus casei promoters during bacterial transit through the gastrointestinal tracts of human-microbiota-associated mice. Appl. Environ. Microbiol. 71, 1356–1363. doi: 10.1128/AEM.71.3.1356-1363.2005
Park, S. F., and Stewart, G. S. (1990). High-efficiency transformation of Listeria monocytogenes by electroporation of penicillin-treated cells. Gene 94, 129–132. doi: 10.1016/0378-1119(90)90479-B
Poulsen, K. P., Faith, N. G., Steinberg, H., and Czuprynski, C. J. (2011). Pregnancy reduces the genetic resistance of C57BL/6 mice to Listeria monocytogenes infection by intragastric inoculation. Microb. Pathog. 50, 360–366. doi: 10.1016/j.micpath.2011.02.003
Quereda, J. J., Dussurget, O., Nahori, M. A., Ghozlane, A., Volant, S., Dillies, M. A., et al. (2016). Bacteriocin from epidemic Listeria strains alters the host intestinal microbiota to favor infection. Proc. Natl. Acad. Sci. U.S.A. 113, 5706–5711. doi: 10.1073/pnas.1523899113
Rea, R. B., Gahan, C. G., and Hill, C. (2004). Disruption of putative regulatory loci in Listeria monocytogenes demonstrates a significant role for Fur and PerR in virulence. Infect. Immun. 72, 717–727. doi: 10.1128/IAI.72.2.717-727.2004
Rice, B. W., Cable, M. D., and Nelson, M. B. (2001). In vivo imaging of light-emitting probes. J. Biomed. Opt. 6, 432–440. doi: 10.1117/1.1413210
Riedel, C. U., Monk, I. R., Casey, P. G., Morrissey, D., O’Sullivan, G. C., Tangney, M., et al. (2007). Improved luciferase tagging system for Listeria monocytogenes allows real-time monitoring in vivo and in vitro. Appl. Environ. Microbiol. 73, 3091–3094. doi: 10.1128/aem.02940-06
Riedel, C. U., Monk, I. R., Casey, P. G., Waidmann, M. S., Gahan, C. G., and Hill, C. (2009). AgrD-dependent quorum sensing affects biofilm formation, invasion, virulence and global gene expression profiles in Listeria monocytogenes. Mol. Microbiol. 71, 1177–1189. doi: 10.1111/j.1365-2958.2008.06589.x
Sleator, R. D., Watson, D., Hill, C., and Gahan, C. G. (2009). The interaction between Listeria monocytogenes and the host gastrointestinal tract. Microbiology 155(Pt 8), 2463–2475. doi: 10.1099/mic.0.030205-0
Szittner, R., Jansen, G., Thomas, D. Y., and Meighen, E. (2003). Bright stable luminescent yeast using bacterial luciferase as a sensor. Biochem. Biophys. Res. Commun. 309, 66–70. doi: 10.1016/S0006-291X(03)01530-4
Tangney, M., and Francis, K. P. (2012). In vivo optical imaging in gene & cell therapy. Curr. Gene Ther. 12, 2–11. doi: 10.2174/156652312799789299
van Pijkeren, J. P., Morrissey, D., Monk, I. R., Cronin, M., Rajendran, S., O’Sullivan, G. C., et al. (2010). A novel Listeria monocytogenes-based DNA delivery system for cancer gene therapy. Hum. Gene Ther. 21, 405–416. doi: 10.1089/hum.2009.022
Van Reet, N., Van de Vyver, H., Pyana, P. P., Van der Linden, A. M., and Buscher, P. (2014). A panel of Trypanosoma brucei strains tagged with blue and red-shifted luciferases for bioluminescent imaging in murine infection models. PLOS Negl. Trop. Dis. 8:e3054. doi: 10.1371/journal.pntd.0003054
Vorobyeva, A. G., Stanton, M., Godinat, A., Lund, K. B., Karateev, G. G., Francis, K. P., et al. (2015). Development of a bioluminescent nitroreductase probe for preclinical imaging. PLOS ONE 10:e0131037. doi: 10.1371/journal.pone.0131037
Keywords: Listeria monocytogenes, click beetle luciferase, bioluminescence, in vivo imaging, pathogen, virulence, clinical
Citation: Ur Rahman S, Stanton M, Casey PG, Spagnuolo A, Bensi G, Hill C, Francis KP, Tangney M and Gahan CGM (2017) Development of a Click Beetle Luciferase Reporter System for Enhanced Bioluminescence Imaging of Listeria monocytogenes: Analysis in Cell Culture and Murine Infection Models. Front. Microbiol. 8:1797. doi: 10.3389/fmicb.2017.01797
Received: 15 May 2017; Accepted: 05 September 2017;
Published: 26 September 2017.
Edited by:
Thomas Dandekar, University of Würzburg, GermanyReviewed by:
Olivier Dussurget, University Paris Diderot, Institut Pasteur, INSERM, INRA, FranceMartijn van Griensven, Technische Universität München, Germany
Copyright © 2017 Ur Rahman, Stanton, Casey, Spagnuolo, Bensi, Hill, Francis, Tangney and Gahan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cormac G. M. Gahan, Yy5nYWhhbkB1Y2MuaWU=