
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 14 September 2017
Sec. Fungi and Their Interactions
Volume 8 - 2017 | https://doi.org/10.3389/fmicb.2017.01772
 Sergio Triana1,2,3
Sergio Triana1,2,3 Hans de Cock4
Hans de Cock4 Robin A. Ohm4Giovanna Danies1Han A. B. Wösten4
Robin A. Ohm4Giovanna Danies1Han A. B. Wösten4 Silvia Restrepo1
Silvia Restrepo1 Andrés F. González Barrios2*
Andrés F. González Barrios2* Adriana Celis1,4*
Adriana Celis1,4*Malassezia species are lipophilic and lipid-dependent yeasts belonging to the human and animal microbiota. Typically, they are isolated from regions rich in sebaceous glands. They have been associated with dermatological diseases such as seborrheic dermatitis, pityriasis versicolor, atopic dermatitis, and folliculitis. The genomes of Malassezia globosa, Malassezia sympodialis, and Malassezia pachydermatis lack the genes related to fatty acid synthesis. Here, the lipid-synthesis pathways of these species, as well as of Malassezia furfur, and of an atypical M. furfur variant were reconstructed using genome data and Constraints Based Reconstruction and Analysis. To this end, the genomes of M. furfur CBS 1878 and the atypical M. furfur 4DS were sequenced and annotated. The resulting Enzyme Commission numbers and predicted reactions were similar to the other Malassezia strains despite the differences in their genome size. Proteomic profiling was utilized to validate flux distributions. Flux differences were observed in the production of steroids in M. furfur and in the metabolism of butanoate in M. pachydermatis. The predictions obtained via these metabolic reconstructions also suggested defects in the assimilation of palmitic acid in M. globosa, M. sympodialis, M. pachydermatis, and the atypical variant of M. furfur, but not in M. furfur. These predictions were validated via physiological characterization, showing the predictive power of metabolic network reconstructions to provide new clues about the metabolic versatility of Malassezia.
Malassezia species are lipophilic and lipid-dependent yeasts that are frequently encountered in the human and animal microbiota. Usually, they are isolated from regions rich in sebaceous glands. They have been associated with dermatological diseases such as seborrheic dermatitis (SD), pityriasis versicolor, atopic dermatitis, and folliculitis (Boekhout, 2010). The increasing number of Malassezia isolations from systemic infections shows that members of this genus are emerging opportunistic agents (Gaitanis et al., 2012; Arendrup et al., 2014).
The lipid dependency of the Malassezia species has been confirmed through the genome sequences of 14 species within the genus (Xu et al., 2007; Gioti et al., 2013; Triana et al., 2015; Wu et al., 2015). The genomes lack the cytosolic fatty acid synthase (FAS) gene, thus explaining why they cannot synthesize the fatty acid palmitate de novo. On the other hand, lipase, phospholipase, and sphingomyelinase genes that are involved in the release of fatty acids from the host are present, enabling lipid synthesis in Malassezia species (Xu et al., 2007). Genes homologous to FAS genes have also been identified, but they have been predicted to have different functions (Xu et al., 2007; Triana et al., 2015).
The Malassezia genus shows different lipid-assimilation phenotypes as well as differences in the number of lipids that are required in the growth medium. Malassezia furfur usually assimilates different kinds of Tween including Tween 20, 40, 60, and 80. However, atypical strains have been identified, including strains that can only assimilate Tween 80 (Gonzalez et al., 2009), and strains that are reported to be lipid-independent (Zinkeviciene et al., 2012). In the latter case, lipid-independent growth was only tested in Sabouraud glucose agar medium, which still contains lipids (Wu et al., 2015). Malassezia pachydermatis was also believed to be lipid-independent (Juntachai et al., 2009). However, recent findings show that this species is actually a more versatile lipid-dependent yeast also lacking a FAS gene (Triana et al., 2015; Wu et al., 2015). The Malassezia assimilation assay is widely used to determine the lipid requirement of strains (Boekhout, 2010). For instance, growth on Tween is indicative of a role being played by external lipase(s) that release the fatty acid tail from the non-ionic detergent. Analysis of the genomes of Malassezia spp. revealed a collection of genes encoding lipases, phospholipases, and sphingomyelinases (Xu et al., 2007; Gioti et al., 2013; Triana et al., 2015; Wu et al., 2015) that are most likely involved in the release of fatty acids from a variety of lipid compounds, for example, such as those produced by the sebaceous glands in the skin of the host. Uptake of these fatty acids and their subsequent use in various lipid-biosynthesis routes is required to sustain the growth of Malassezia species.
Genomic data and Constraints Based Reconstruction and Analysis (COBRA)-based models (Murabito et al., 2009) can be used to reconstruct the cellular metabolism of an organism in a mathematical model. Such networks can be used to predict a cell’s behavior under different conditions or disorders (Orth et al., 2014). Flux balance analysis (FBA) models are constraint-based approaches suitable for studying the range of possible phenotypes of a metabolic system (Murabito et al., 2009).
The aim of this work was to investigate the genomic and metabolic differences between reference strains of four Malassezia species and an atypical M. furfur strain. To this end, we used metabolic modeling, using genomics and proteomics data. The results show flux differences in the production of steroid in M. furfur and the metabolism of butanoate in M. pachydermatis. In addition, defects in the assimilation of palmitic acid in Malassezia globosa, Malassezia sympodialis, M. pachydermatis, and the atypical variant of M. furfur (4DS), were suggested, but not for M. furfur CBS 1878. These predictions were validated via culturing on defined media.
The reference Malassezia strains M. globosa CBS 7986, M. sympodialis CBS 7222, M. pachydermatis CBS 1879, and M. furfur CBS 1878 purchased from Fungal Biodiversity Center (Westerdijk Institute, Utrecht, Netherlands), as well as a previously reported isolate of M. furfur with atypical assimilation of Tween 80 (4DS) (from now on referred to as atypical M. furfur) (Gonzalez et al., 2009) were used in this study and recovered in modified Dixon agar (mDixon): 36 g L-1 mycosel agar (BD, United States), 20 g L-1 Ox-bile (Sigma–Aldrich, United States), 36 g L-1 malt extract (Oxoid, UK), 2 mL L-1 glycerol (Sigma–Aldrich, United States), 2 mL L-1 oleic acid (Sigma–Aldrich, United States), and 10 mL L-1 Tween 40 (Sigma–Aldrich, United States) for 4–5 days at 33°C (Boekhout, 2010). The genomes of M. globosa (Xu et al., 2007), M. pachydermatis (Triana et al., 2015), and M. sympodialis (Gioti et al., 2013) have been sequenced and are available in GenBank under the accession numbers GCA_000181695.1, GCA_001278385.1, and GCA_000349305.2. For the phylogenetic analysis the Malassezia genomes reported by Wu et al. (2015) and the Ustilago maydis genome (Kämper et al., 2006) were used, these are available in GenBank under BioProject ID: PRJNA286710 and accession number GCA_000328475.2, respectively.
The M. furfur strains that were sequenced in this study were recovered in modified mDixon for 4–5 days at 33°C (Boekhout, 2010). Genomic DNA was extracted as described (Gonzalez et al., 2009).
DNA from the M. furfur strains was sequenced at the Beijing Genomics Institute (Shenzhen) using the Illumina HiSeq 2000 platform. Two runs of 100-bp paired-end reads and 200-bp insert-size libraries were undertaken following standard Illumina protocols. The quality of the reads was analyzed using FastQC software (Andrews, 2010) and trimmed and filtered based on quality using Flexbar (Dodt et al., 2012). De novo assembly was performed using the CLC Assembly Cell software (CLC bio, 2014) using default parameters. The resulting contigs were scaffolded using SSPACE_Basic script (Boetzer et al., 2011), discarding scaffolds <1,000 bp. Gaps in the scaffolds were filled with the GapFiller script (Boetzer and Pirovano, 2012). Assembly statistics such as N50, N75, L50, L75, and GC content, were computed with the QUAST software (Gurevich et al., 2013). The average genome coverage was calculate by mapping the reads to the assembly with Bowtie (Langmead et al., 2009) using default parameters and calculating the coverage per base using BEDTools (Quinlan and Hall, 2010). This whole-genome shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession numbers MATP00000000 and LMYL00000000. The version described herein are LMYL01000000 and MATP01000000.
To determine the genome similarity among the species and to identify the presence of repetitions in the genomic and protein level, we did the following. The assemblies of the M. furfur strains were aligned with each other and with the genomes of M. globosa (Xu et al., 2007), M. sympodialis (Gioti et al., 2013), and M. pachydermatis (Triana et al., 2015) using Nucmer (a maximum gap between two adjacent matches in a cluster of 90 bp and a minimum length of a maximal exact match of 20 bp) and Promer (a maximum gap between two adjacent matches in a cluster of 30 amino acids and a minimum length of a maximal exact match of six amino acids), which evaluates the six-frame translation of the nucleotide sequence. Mummer (Delcher et al., 2002) alignments were plotted and the genome coverage per nucleotide was calculated with the BEDTools suite Coverage tool (Quinlan and Hall, 2010). The percentage of matches in each pairwise comparison was computed using custom python scripts.
The assemblies of the M. furfur strains were annotated using the MAKER 2 pipeline (Cantarel et al., 2007). To this end, a set of 109,264 previously reported Ustilaginomycotina proteins from NCBI protein database was used. In addition, genes predicted by CEGMA (Parra et al., 2007) and GeneMark (Borodovsky and Lomsadze, 2011), 1,413 expressed sequence tags (ESTs) from Malassezia spp., and results from the ab initio gene predictors SNAP (Korf, 2004) and Augustus (Stanke et al., 2004) were used as genetic evidence for the annotation. MAKER was run two consecutive times. The first run included proteins, ESTs, and predicted genes to identify genes within the scaffolds. The first output obtained from MAKER was converted into a model for SNAP and a training set for Augustus. Subsequently, the ab initio results were provided as an input model for the second MAKER run. The statistics of the resulting annotation were calculated with genome tools (Gremme et al., 2013).
Functional annotation of the predicted genes was performed using Blast2GO (Conesa and Götz, 2008), which included BlastX (Altschul et al., 1990) and an InterProScan annotation (Quevillon et al., 2005). To determine the number of duplicated proteins in the M. furfur genomes, CD-Hit (Li and Godzik, 2006) was run with an identity threshold of 90%. Non-coding repeated sequences within the genomes were analyzed using RepeatMasker1 by running them against the RepBase library.
To assess whether genes involved in the formation of free fatty acid precursors are absent in the M. furfur genomes, as is the case for M. globosa, M. sympodialis, and M. pachydermatis, 2,382 fungal and bacterial genes encoding FAS and 954 polyketide synthase (PKS) genes were compared with the predicted genes and proteins of the genomes using blastp, blastn, tblastn from the blast suite version 2.2.29 (Altschul et al., 1990) (parameters by default) and phmmer and hmmsearch from the HMMER software, version 3.1b22. Further validation of missing InterProScan domain was manually done by comparing the missing domain sequence from the Malassezia species presenting the domain with the predicted protein of the other species.
To estimate the phylogeny of the available Malassezia species (Xu et al., 2007; Gioti et al., 2013; Triana et al., 2015; Wu et al., 2015), genes were predicted on each genome assembly using Augustus 3.0.2 (Stanke et al., 2004) with the parameters that were previously optimized for M. furfur CBS 1878. U. maydis was used as an outgroup. Highly conserved genes were identified using BUSCO 2.0 (Simão et al., 2015) using the gene set “fungi odb9.” These sequences of each species were concatenated, aligned using MAFFT version 7.309 (Katoh and Standley, 2013), and the well-aligned regions were extracted using Gblocks 0.91b (Castresana, 2000). This resulted in 62,988 amino acid positions. FastTree 2.1.9 (Price et al., 2010) was used to reconstruct the species tree.
The predicted proteins were compared to the KEGG database (Kanehisa and Goto, 2000) using the KAAS server (Moriya et al., 2007) and Blast2GO (Conesa and Götz, 2008) to obtain corresponding Enzyme Commission (EC) numbers. These numbers were used to retrieve the associated reactions from KEGG and to map the corresponding metabolic pathways. The directionality of each reaction was determined using the literature, the MetaCyc database (Caspi et al., 2014), and the Gibbs free energy obtained with the group contribution method (Jankowski et al., 2008). The reaction nomenclature was converted to the Metanetx identifiers in order to have a more cohesive nomenclature in the network (Ganter et al., 2013). Furthermore, the metabolic core was determined as the reactions shared among the five strains.
The predicted enzymes were analyzed with the subCELlular LOcalization predictor (CELLO; Yu et al., 2004), a peptide localization predictor that uses support vector machines based on n-peptide compositions. The significant compartment was selected and added to the reactions of the enzyme. Transport reactions were added to the network according to the genome annotation and the literature review. The metabolites with production and consumption problems were identified and missing data were imputed using an iterative approach with the GapFind and GapFill algorithms (Boetzer and Pirovano, 2012) implemented in the General Algebraic Modeling System (GAMS) (Bruce, 2013) using the minimal media defined for iMM904 Saccharomyces cerevisiae (Zomorrodi and Maranas, 2010) supplemented with oleic acid and glucose as the sole carbon source.
In addition to a manual curation based on the literature and visualization in Cytoscape (Shannon et al., 2003), an in-house exchange-reaction database and the complete metabolic reactions’ repository for Metanetx (Ganter et al., 2013) were used to detect unconnected metabolites, and to calculate the topological statistics of the network.
A stoichiometric matrix (S) was obtained from the metabolic network using in-house Perl scripts to obtain a system of linear equations, S × v = 0, where v is the flux vector. System constraints included the lower and upper bounds of reaction fluxes. The system allowed 0.000–1,000 mmol gDW-1 h-1 for irreversible reactions and -1,000 to 1,000 mmol gDW-1 h-1 for reversible reactions. The system was solved to identify the theoretical limits for different fluxes in the metabolic system using GAMS (GAMS Development Corp., Washington, DC, United States) software. The linear programming presented here was developed with solver CPLEX 12.6.0.0 with an optimization tolerance of 10-6. A modified biomass production reaction (Supplementary Table S1) of iMM904 S. cerevisiae as an objective function (Zomorrodi and Maranas, 2010) were fixed as parameters for the optimization process. Furthermore the same media used in the GapFill process, the minimal media defined for iMM904 S. cerevisiae (Zomorrodi and Maranas, 2010) supplemented with oleic acid and glucose (uptake rate of 10 mmol gDW-1 h-1) in anaerobic conditions (oxygen uptake rate of 2 mmol gDW-1h-1) was used for the simulation. This optimization problem aimed to solve the maximization of the flux through the reaction of biomass used as objective functions. The resulting flux distributions were filtered with a cutoff of ±0.05 mmol gDW-1 h-1 to allow a better visualization of the reactions that can carry fluxes and plotted as a heatmap in R (R Core Team, 2013).
Protein extraction was carried out as described (Kim et al., 2010). Strains were grown at 180 rpm and 33°C on DB (20 g L-1 Ox-bile, Sigma–Aldrich, United States), 36 g L-1 malt extract (Oxoid, UK), 6 g L-1 peptone (Oxoid, UK), 2 mL L-1 glycerol (Sigma–Aldrich, United States), 2 mL L-1 oleic acid (Sigma–Aldrich, United States), 10 mL L-1 Tween 40 (Sigma–Aldrich, United States), and 500 mg L-1 chloramphenicol (Sigma–Aldrich, United States). Aliquots of 5 mL were taken in the early exponential and early stationary phase and centrifuged at 26,000 g for 10 min. The resulting pellet was washed three times with PBS, after which it was resuspended in extraction buffer [1:10 cell to extraction buffer v/v ratio; 125 mM ammonium bicarbonate (Sigma–Aldrich, United States), 20 mM 𝜀-aminocaproic acid (Sigma–Aldrich, United States), 5 mM ethylenediaminetetraacetic acid (Sigma–Aldrich, United States), and 1 mM phenylmethylsulfonyl fluoride (Thermo Fisher Scientific, United States)]. Cells were disrupted by vortexing for 10 min with 4-mm silica beads and centrifuged at 26,000 g and 4°C for 10 min. Proteins in the supernatant were precipitated with 20% TCA (Sigma–Aldrich, United States) (Wood, 1991). The amount of protein was quantified using a Nanodrop (Thermo Fisher Scientific, United States) and visualized by SDS-PAGE electrophoresis. The protein extraction was carried out five times per sample.
The extracted proteins were sent to the proteomic center of the University of California at Davis3. Protein profiling for each sample was carried out using the mass spectrometer Michrom HPLC Paradigm type, the Q-mass spectrometer ionization Proxeon Exactive nano-spray, and the Easy-LC II HPLC.
The identification and annotation of the proteins was performed using Scaffold (Proteome Software Inc., Portland, OR, United States) and proteins were compared against the Uniprot Ustilaginomycotina proteins and the Malassezia spp. predicted proteins. The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE (Vizcaíno et al., 2016) partner repository with the dataset identifier PXD004523. The resulting proteins were clustered with CD-Hit to compare the proteins among the samples. These clusters were analyzed statistically with R using the normalized total spectra count (R Core Team, 2013). To validate the network, proteins related to the reactions in the genomic model were selected (excluding those metabolic and transport reactions added with GapFill that did not have an associated enzyme) and compared to the abundance of the proteins in each extract using at least two replicates with 95% identity.
To determine the growth on different Tween varieties and with fatty acids, the strains were first grown on mDixon at 33°C for 7 days. The fungal cells were suspended in 3 ml of water with 0.1% Tween 80 used to inoculate 27 mL of minimal medium (MM) [containing per liter: 10 mL K-buffer pH 7.0 (200 g L-1 K2HPO4; Sigma–Aldrich, United States), 145 g L-1 KH2PO4 (Sigma–Aldrich, United States), 20 mL M-N (30 g L-1 MgSO4.7H2O; Sigma–Aldrich, United States), 15 g L-1 NaCl (Sigma–Aldrich, United States), 1 mL 1% CaCl2.2H2O (Sigma–Aldrich, United States) (w/v), 10 mL 20% glucose (Sigma–Aldrich, United States) (w/v), 10 mL 0.01% FeSO4 (Sigma–Aldrich, United States) (w/v), 5 mL spore elements (100 mg L-1 ZnSO4.7H2O; Sigma–Aldrich, United States), 100 mg L-1 CuSO4.5H2O (Sigma–Aldrich, United States), 100 mg L-1 H3BO3 (Sigma–Aldrich, United States), 100 mg L-1 MnSO4.H2O (Sigma–Aldrich, United States), l00 mg L-1 Na2MoO4.2H2O (Sigma–Aldrich, United States), and 2.5 mL 20% NH4NO3 (Sigma–Aldrich, United States) (w/v)] containing 4 mM Tween [20, 40, 60 or 80 (Sigma–Aldrich, United States)], or 4 mM oleic acid (Carlo Erba), or palmitic acid (Merck) supplemented with 1% Brij-58 (Sigma–Aldrich, United States), an emulsifier that is not metabolized. It did not support the growth of the FAS mutant of the yeast S. cerevisiae (Schweizer and Bolling, 1970) that was grown for 3 days at 33°C. Subsequently, 0.3 mL was used to inoculate 29.7 mL of fresh MM containing either 4 mM Tween 20, 40, 60, 80, oleic acid, and/or palmitic acid in 1% Brij-58, with mDixon broth as the positive control. Growth was followed during 8 days in the medium containing Tween 40, palmitic acid, oleic acid, or mixtures of palmitic and oleic acid by determining the colony-forming unit (CFU) by plating on mDixon plates with subsequent incubation at 33°C. In the medium containing Tweens and oleic acid, the optical density (OD) at 600 nm was measured and plating aliquots of the liquid cultures on mDixon plates at 33°C was used to determine the viability of the cells after 8 days of growth. The fatty acids used for culturing were analyzed for composition via a gas chromatography–flame ionization detector; separation was reached using an RTX-Wax column (30 m × 0.25 mm × 0.5 μm) of RESTEK®. FAMEs were identified by comparing their retention times with those identified with a Supelco® 37 Component FAME Mix standard. Quantification was intended as a relative concentration. Palmitic acid (Merck) contained 98% palmitic acid and 2% elaidic acid, an unsaturated acid. The oleic acid (Carlo Erba) contained 78% oleic acid and, in addition, we detected polyunsaturated fatty acids (10% of linoleic acid), unsaturated fatty acids (3% palmitoleic acid and 2% elaidic acid), and saturated fatty acids (6% of palmitic acid and 1% heptadecanoic acid).
The draft genomes of M. furfur CBS 1878 and the atypical M. furfur 4DS were assembled from a shotgun Illumina HiSeq 2000-Paired data set using the CLC-assembler (CLC bio, 2014). The assemblies yielded 2,084 scaffolds (N50 = 23 kb) and 3,577 scaffolds (N50 = 42 kb), respectively, corresponding to nuclear genomes of 14.19 and 10.38 Mb. The summary of the genome assembly statistics calculated by QUAST (Gurevich et al., 2013) is shown in Table 1.
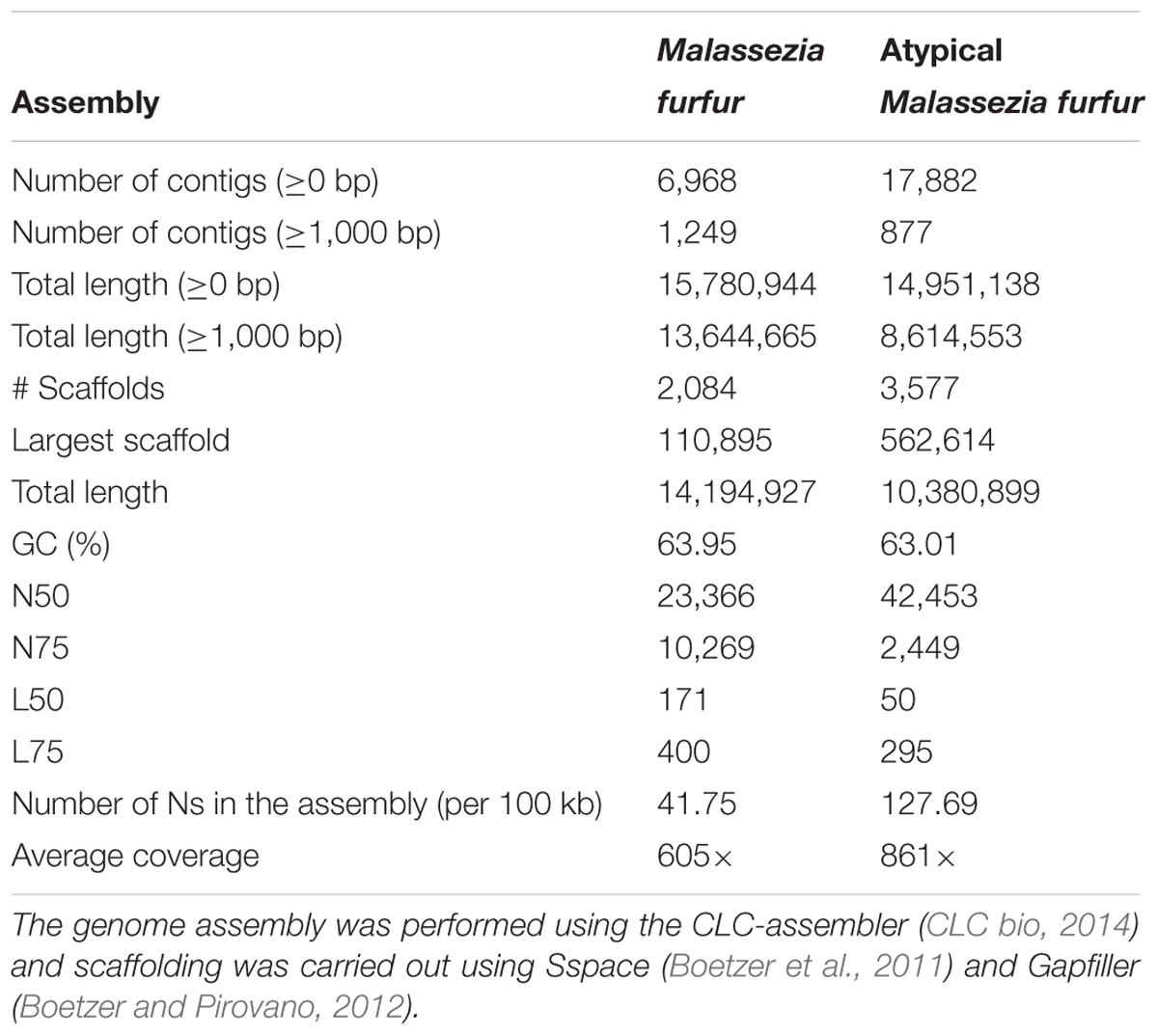
TABLE 1. Assembly statistics calculated by QUAST (Gurevich et al., 2013) and BEDtools (Quinlan and Hall, 2010).
A pairwise Nucmer comparison at the nucleotide level showed that M. furfur and the atypical M. furfur had more repetitions and/or duplications in their genomes than M. globosa, M. pachydermatis, and M. sympodialis did (Supplementary Figure S1 and Table 2), with 30.8 and 7% of the genomes of M. furfur and of the atypical M. furfur strains, respectively, representing multiple matches or repetitions. In contrast, less than 1% of the genomes of M. globosa, M. pachydermatis, and M. sympodialis corresponded to multiple matches.
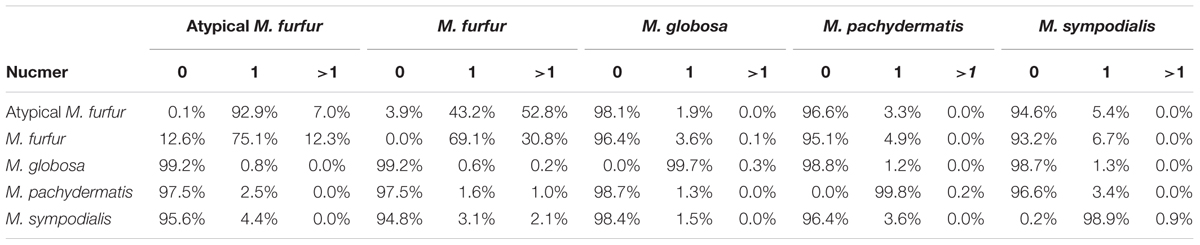
TABLE 2. Percentage of the genome with zero (0), one (1), or multiple (>1) matches in the Nucmer genome alignment (Boetzer and Pirovano, 2012) with a maximum gap between two adjacent matches in a cluster of 90 bp and a minimum length of a maximal exact match of 20 bp.
The Nucmer alignment showed significant sequence divergence between the four species. The biggest difference was observed for M. globosa, which only showed 0.8% identity using exact matches of at least 20 bp when compared to M. furfur or the atypical M. furfur genomes. This was <1.2% when the M. globosa genome was compared to M. pachydermatis and M. sympodialis. As expected, the most similar strains were M. furfur and the atypical M. furfur strains with a similarity >80% (Table 2). A higher degree of similarity was observed when evaluating the six translation frames using Promer (Supplementary Figure S2 and Table 3). The biggest differences were found when M. furfur and M. sympodialis were compared, with 70.7% zero matches. The second highest difference was between M. globosa and M. furfur, with approximately 48% of zero matches. As expected, the highest similarity was observed in the case of the M. furfur strains with only 3.4% of zero matches.
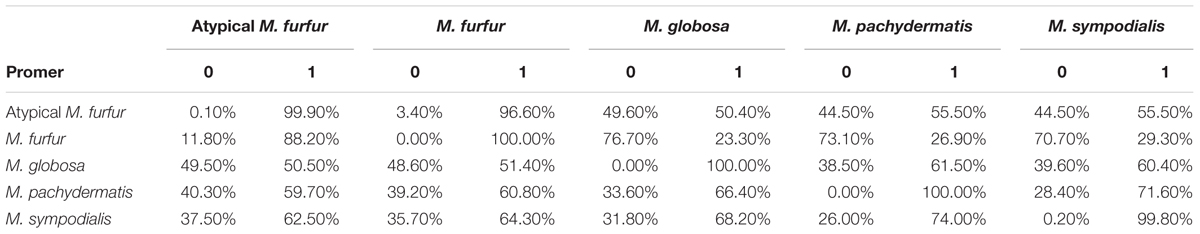
TABLE 3. Percentage of the genome with zero (0), or one or more (1) matches in the Promer genome alignment (Delcher et al., 2002) with a maximum gap between two adjacent matches in a cluster of 30 amino acids and a minimum length of a maximal exact match of six amino acids.
To establish the taxonomic position of the two reported M. furfur sequences in this study with the already published Malassezia genomes, we built a phylogenetic tree using highly conserved genes (Figure 1). This phylogeny showed that atypical M. furfur clustered with M. furfur CBS 7982 and our sequence of M. furfur CBS 1878 clustered with the previously reported sequence of this strain.

FIGURE 1. Phylogenetic analysis of the Malassezia genus reconstructed from highly conserved genes identified with BUSCO 2.0 (Simão et al., 2015). (1) Xu et al., 2007, (2) Gioti et al., 2013, (3) Triana et al., 2015, (4) Wu et al., 2015, (5) Kämper et al., 2006.
A total of 10,203 and 12,131 protein-encoding genes were predicted for M. furfur and the atypical M. furfur strain using multiple lines of evidence (ab initio predictors, ESTs from M. globosa, and protein alignments). Functional annotation showed that both M. furfur strains contained twice the amount of proteins when compared to the other Malassezia species. Yet, the number of EC numbers and reactions were similar, as can be seen in Figure 2. The average genome length, exon length, and the number of exons per gene for the five genomes analyzed are presented in Figure 2.
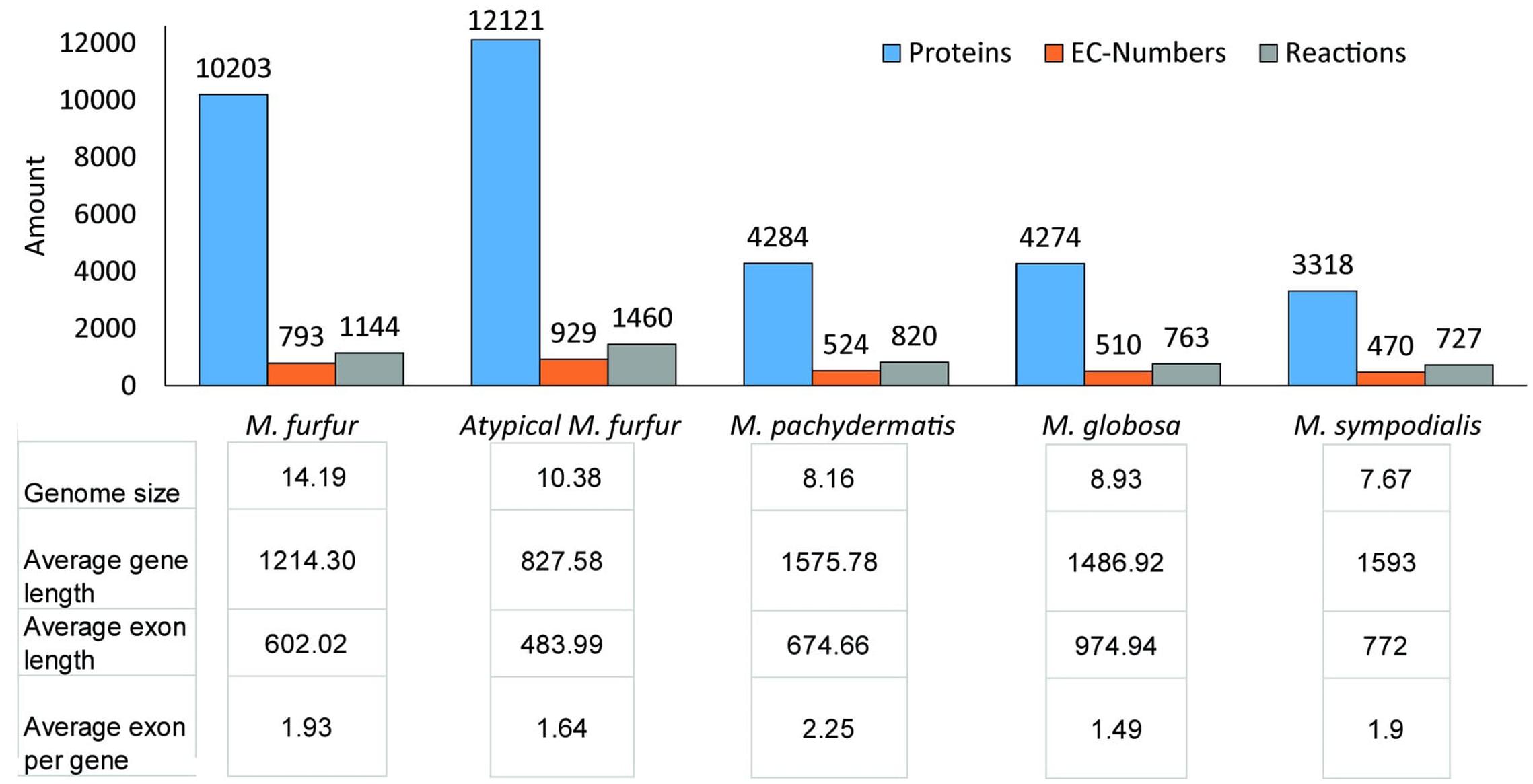
FIGURE 2. Annotation data for each species: for the M. furfur strains, the predicted proteins were obtained from MAKER (Cantarel et al., 2007). For all the strains, the ECs were obtained from KAAS (KEGG Automatic Annotation Server) (Moriya et al., 2007) and Blast2GO (Conesa and Götz, 2008), the reactions were retrieved from the KEGG database, and the annotation statistics were calculated with genome tools (Gremme et al., 2013).
The genomes of M. furfur and the atypical M. furfur contained 7,570 and 10,434 protein clusters, respectively, as predicted with CD-Hit (Li and Godzik, 2006). The total number of non-coding repetitions found using RepeatMasker was approximately 1.61 and 1.32% for M. furfur and the atypical M. furfur strain, respectively. Most repetitions were found to be low-complexity repeats and long terminal repeat elements.
To search for genes related to lipid biosynthesis, a total of 2,382 FASs (i.e., 179 fungal and 2,203 bacterial) were compared with the predicted proteins of the M. furfur strains using blastp, blastn, and tblastn (Altschul et al., 1990) and phmmer and hmmsearch (see text footnote 2). No FAS genes were identified in the two genomes, but several PKS genes (three in M. furfur and one in atypical M. furfur) were found (Figure 3). The PKSs from the five species shared most of the domains, with the exception of M. globosa, which lacked the S-adenosyl-L-methionine-dependent methyltransferase domain, and M. furfur, which lacked the ketoreductase domain, acyl carrier protein-like domain, NAD-binding domain, and the thioester reductase-like domain in its three predicted PKSs. Out of 954 fungal homologs, the PKS of the basidiomycete Paxillus involutus was the most similar to that of the atypical M. furfur, with an identity of 31%. The most similar PKSs to those of Malassezia were found in basidiomycetes. The closest homolog of the PKS of M. furfur was from Hydnomerulius pinastri, with an identity of 29.8%, while an Auricularia delicata homolog was most similar to the PKS of M. pachydermatis and M. sympodialis, showing 29.9 and 31% similarity, respectively. In the case of M. globosa, the most similar PKS was that of Coniophora puteana, with an identity of 26.8%.
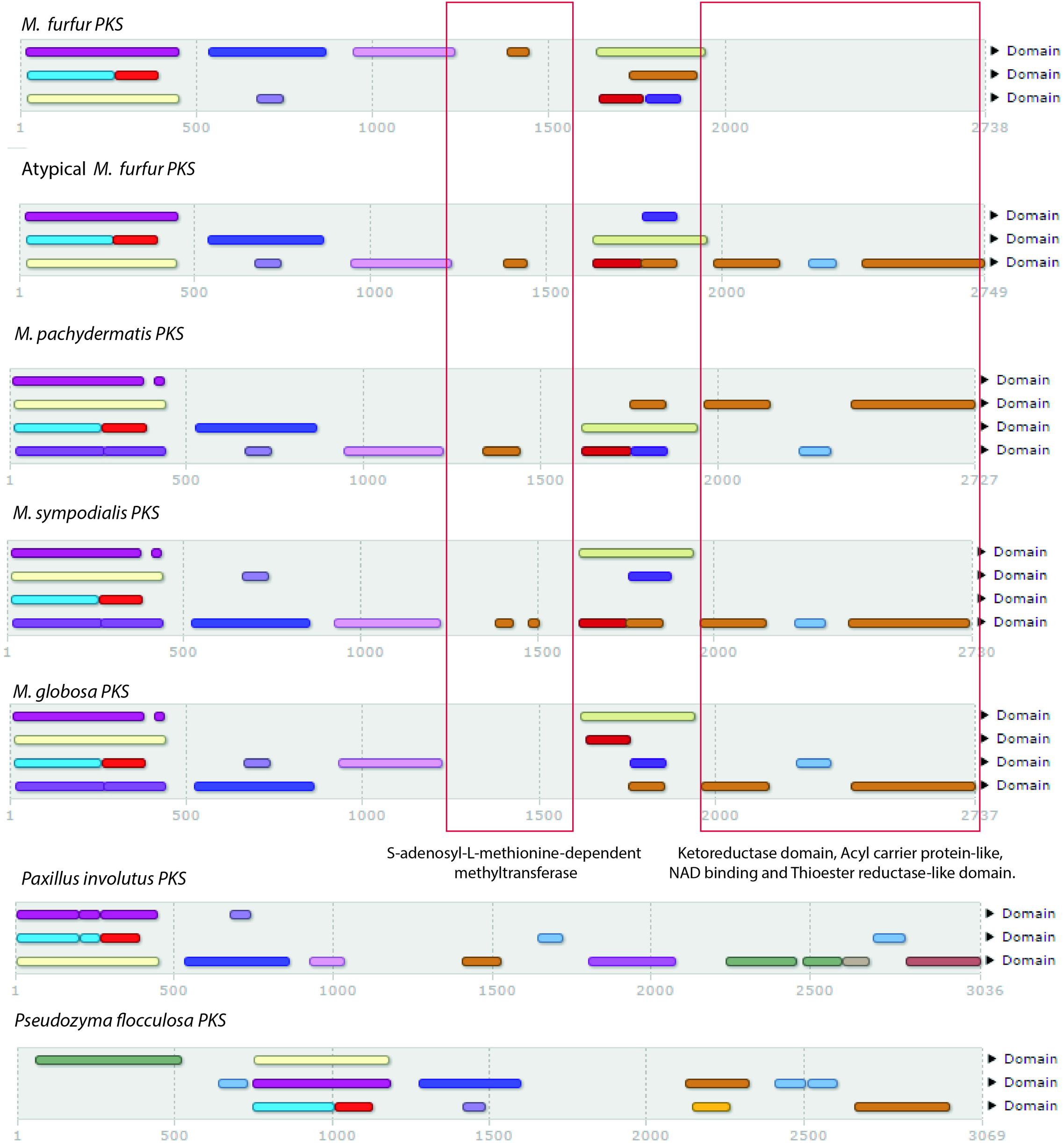
FIGURE 3. Domains obtained with InterProScan (Quevillon et al., 2005) of the five polyketide synthases (PKSs) of Malassezia species compared with Paxillus involutus and Pseudozyma flocculosa.
The distribution of the genes in the genomes of the five Malassezia strains was similar when analyzing 13 categories of metabolic genes (Figure 4). The metabolic core (Supplementary Figure S3) represented 628 reactions that were shared between the strains. These were mainly distributed in the carbohydrate and amino acid metabolism, and lipid metabolism. When examining the lipid metabolism in more detail, arachidonic acid biosynthesis was only present in the atypical M. furfur. Differences in the fungal steroid biosynthesis were also found. Around 10 reactions involved in this pathway were present in the M. furfur strains, which was higher than those in the other species with less than six reactions. Similarly, 36 reactions associated with fatty acid degradation were detected in atypical M. furfur, a higher number than that found in the other species studied (23–31 reactions) (Figure 4).
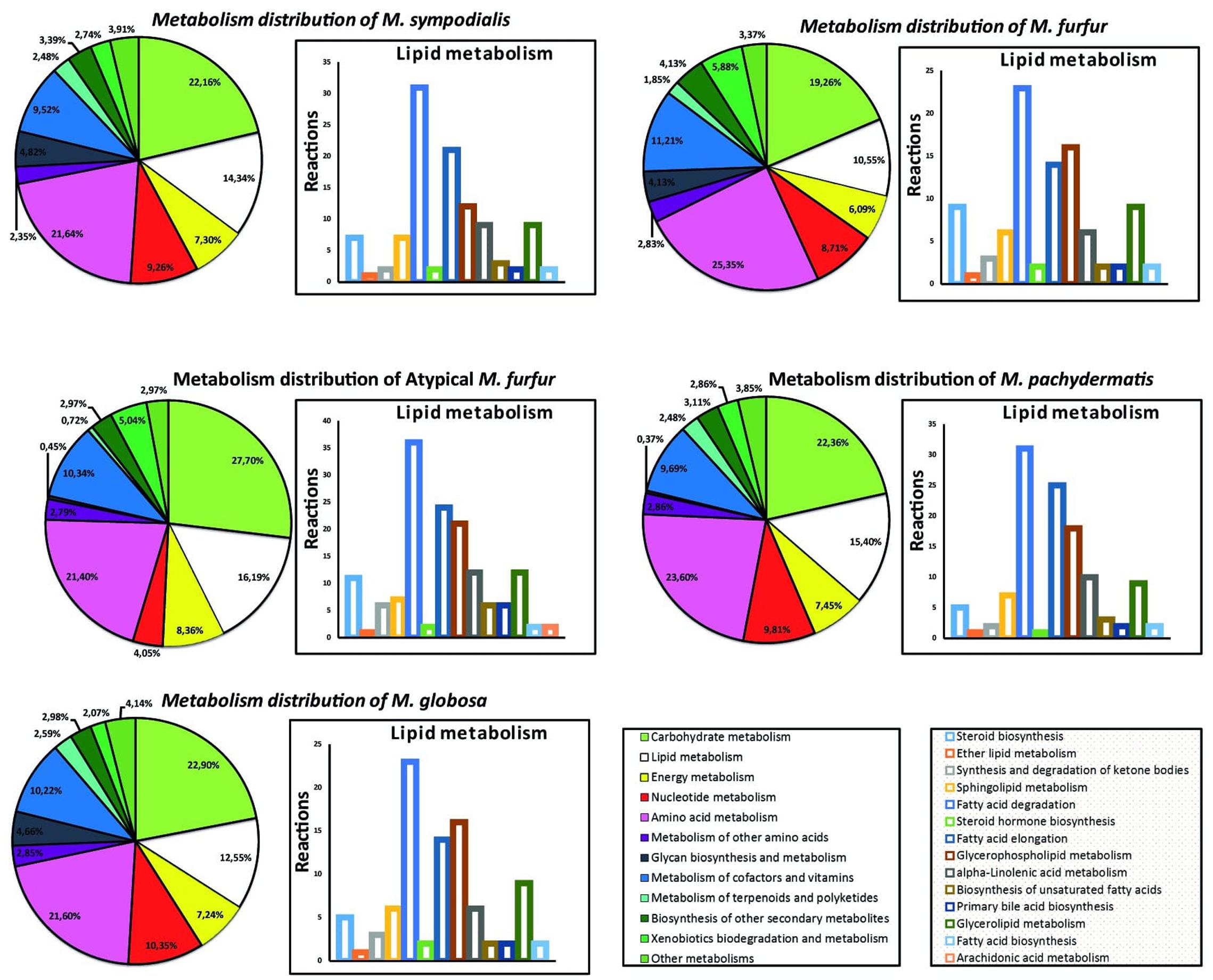
FIGURE 4. Metabolism distribution predicted from each species and an expanded distribution for the lipid metabolism.
The database search and the group contribution method used to calculate the Gibbs free energy predicted that 15–26% of the reactions of the five Malassezia strains were irreversible. Compartmentalization analyses showed a similar distribution of reactions among the species. However, differences were observed in the number of reactions when the M. furfur strains were compared to the other species. This corresponded to cytoplasmic and mitochondrial reactions (Figure 5). After the gap-filling approach to reduce the network pathologies, around 250 reactions were added to M. globosa and M. sympodialis, 300 to M. pachydermatis, 600 to M. furfur, and 660 to the atypical M. furfur strain. The final networks ended up with 2,162 metabolites in M. globosa, 2,303 in M. sympodialis, 1,838 in M. pachydermatis, 3,103 in M. furfur, and 3,642 in the atypical M. furfur strain. The final metabolic networks (Supplementary Data Sheet 2) were visualized in Cytoscape using the compartment as a discrete mapping category to color the nodes. A topological analysis was performed (Figure 6). The clustering coefficients were around 0.14 and 0.18 and the diameter of the networks ranged between 10 and 15 nodes.
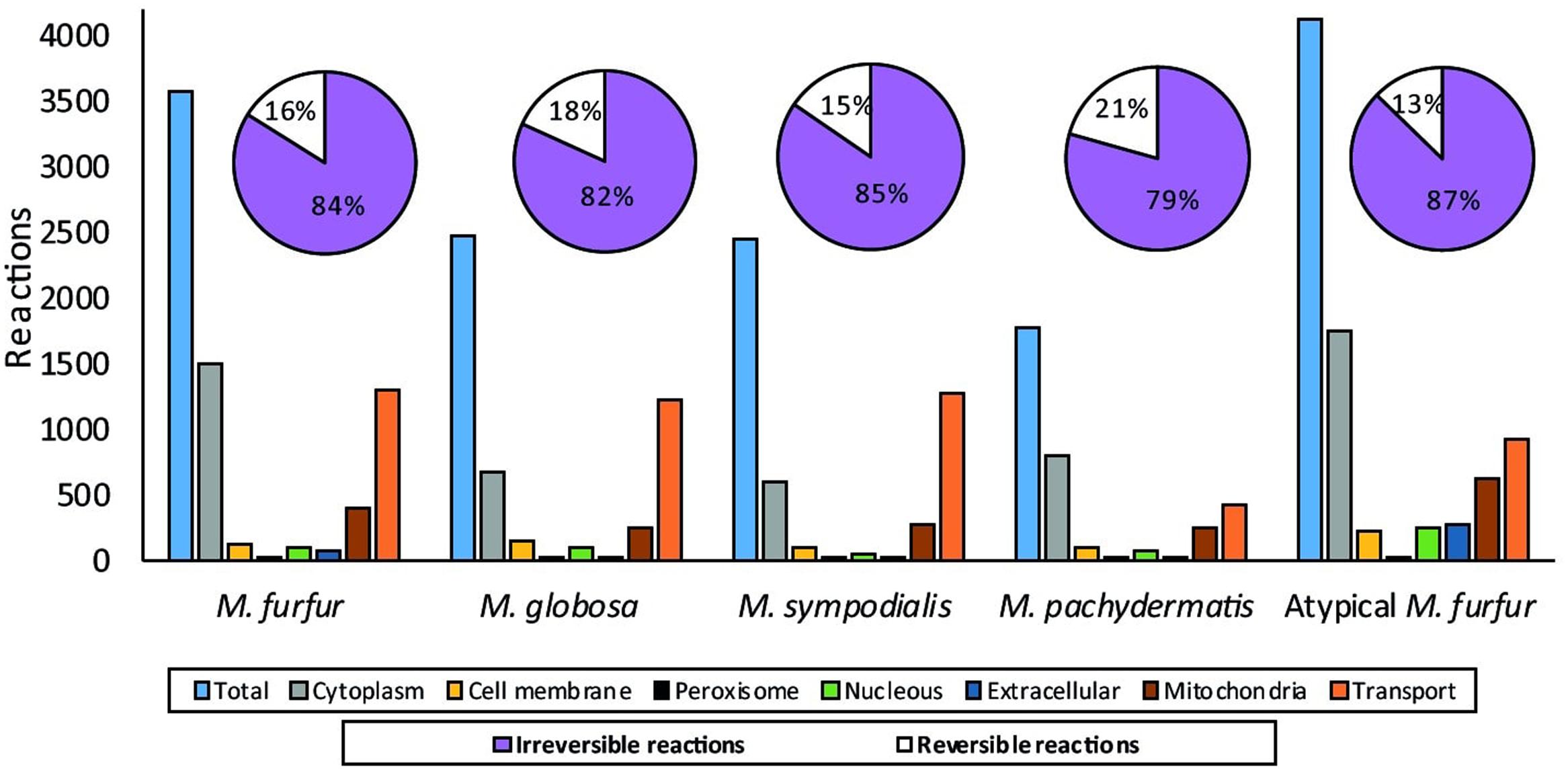
FIGURE 5. Distribution of the reactions after compartmentalization with the subCELlular LOcalization predictor (CELLO) (Yu et al., 2004) and the percentage of reversible and irreversible reactions determined from the Gibbs free energy of the reactions calculated with the group contribution approach and from the literature.
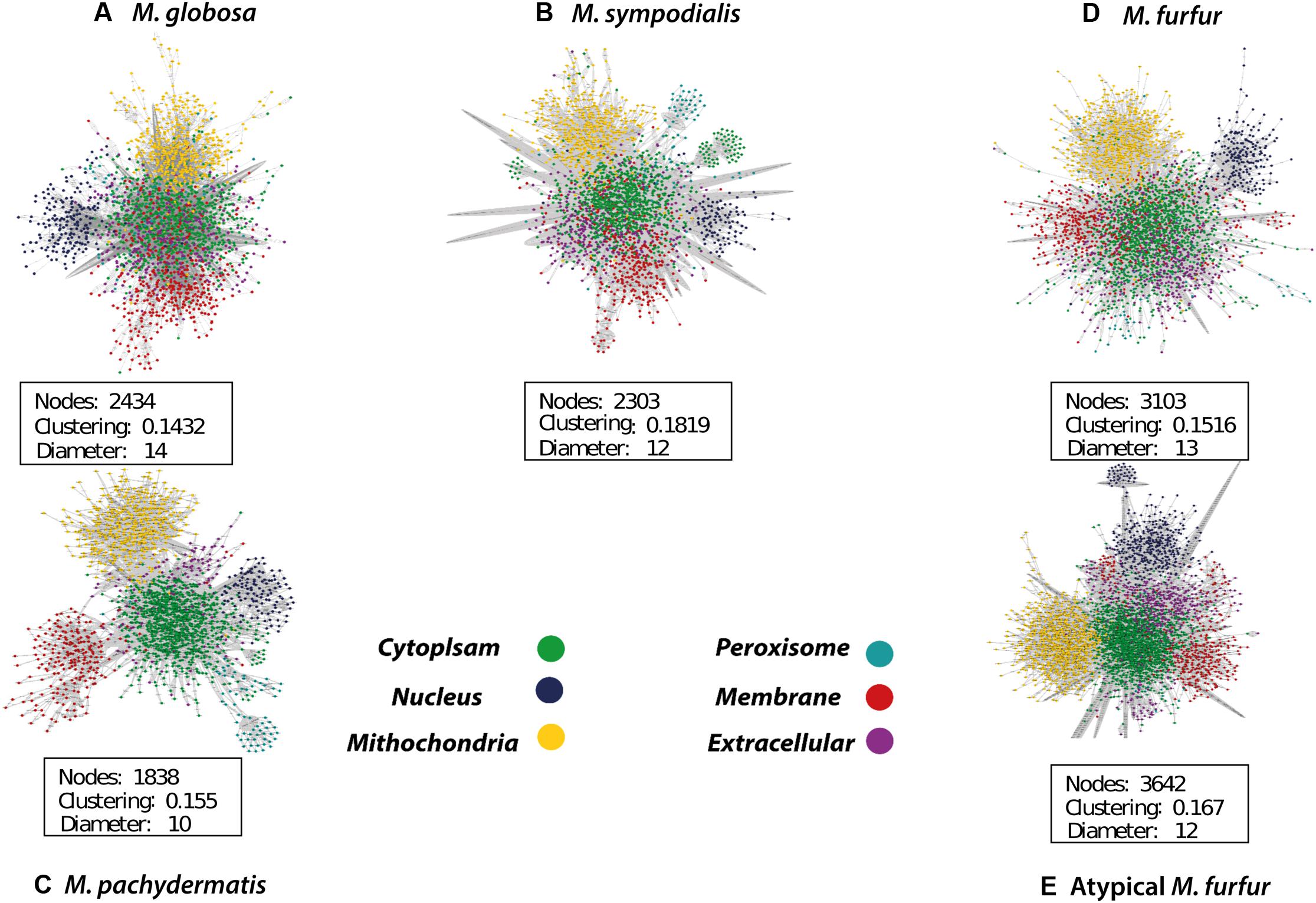
FIGURE 6. Compartmentalized metabolic network of Malassezia globosa (A), Malassezia sympodialis (B), Malassezia pachydermatis (C), Malassezia furfur (D), and atypical Malassezia furfur (E) visualized in Cytoscape (Shannon et al., 2003).
FBA was carried out and the resulting predicted biomass fluxes that were used as objective functions are shown in Table 4. The highest biomass production was found in the atypical M. furfur strain at 2.84 mmol gDW-1 h-1 and the lowest was in M. sympodialis at 0.089 mmol gDW-1 h-1. The number of reactions that that carried flux varied among the species, ranging from 36% in the atypical M. furfur strain to 42% in the M. sympodialis strain.

TABLE 4. Predicted biomass flux with the flux balance analysis.
The flux distribution of each network is represented in Figure 7 for visualization as a heatmap, the resulting clusters were found based in the flux carried by each reaction, with M. globosa and M. sympodialis having the most similar flux distribution. The main differences between M. furfur and the other strains were reactions involved in valine, leucine, and isoleucine degradation, and pyrimidine and purine metabolism. Differences in the lipid-metabolism reactions were also found. M. furfur displayed high fluxes in reactions related to fatty acid degradation and elongation, which involved the conversion of hydroxyacyl-CoA to trans-2-enoyl-CoA. These were higher in comparison to those observed in the atypical M. furfur strain. Several reactions associated with fungal steroid biosynthesis were found in the atypical M. furfur and M. furfur isolates. Despite the atypical M. furfur isolate having a larger number of these reactions, they had higher fluxes in the M. furfur strain. In the atypical M. furfur strain, higher fluxes were found in reactions involved in the degradation of maltose, fructose, and starch. M. globosa and M. sympodialis showed similar metabolic behavior. However, some differences in M. sympodialis corresponding to changes in the pyruvate and gluconeogenesis pathways, as well as the pathways involved in the degradation of long-chain fatty acids into acyl-CoA, and in the conversion of hydroxyacyl-CoA to trans-2-enoyl-CoA were found. Finally, in M. pachydermatis, there were differences in the core metabolism of pyruvate and butanoate, and in the biosynthesis of phenylalanine, tyrosine, and tryptophan.

FIGURE 7. Flux distribution resulting from the flux balance analysis (FBA) of each metabolic network reaction filtered for visualization with a cutoff of ±0.05 mmol gDW-1 h-1.
As lipid assimilation differs among Malassezia species, and lipid metabolism may be related to the pathogenesis of these yeasts, differences in lipid assimilation among the species was further evaluated in the model. In reactions involve in fatty acid biosynthesis such as MNXR2003 (ATP + hexadecanoic acid + CoA ⇔ AMP + palmitoyl-CoA + diphosphate), involved in the conversion of hexadecanoic acid to palmitoyl-CoA, higher fluxes were found in M. furfur, even though all the strains’ genomes contain the genes that codify the enzyme that catalyzes these reactions.
An average number of 8,864 ± 2,400 M. furfur peptides were found during the exponential phase (T1), corresponding to an average of 1,865 ± 154 proteins. The lowest number of peptides (828 ± 456) was obtained with M. globosa in the stationary phase (T2), corresponding to an average of 116 ± 60 proteins. Consensus proteins were defined as proteins present in at least two replicates with a quantitative identity of ≥95%. The number of these proteins differed among the strains. M. furfur in the stationary phase had the highest number (1,539 consensus proteins), while M. globosa presented the lowest number in the exponential phase (201 consensus proteins). The number of reactions was higher in atypical M. furfur in the exponential phase, with 841 reactions, as compared to the other species (Table 5). The principal component analysis (PCA) showed that the proteome profiles behaved differently among the species, with the samples from atypical M. furfur and M. globosa having the most similar profiles. In addition, the clustering and PCA analyses also showed that the replicates behaved similarly within each species in both the stationary and exponential phases (Figure 8).
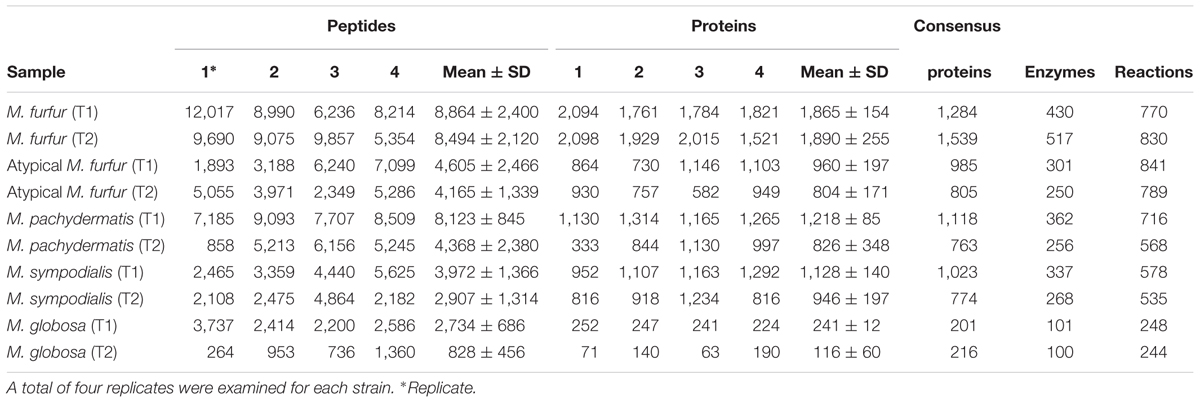
TABLE 5. Proteomic profiling results of five strains of Malassezia during the exponential (T1) and stationary (T2) phase.
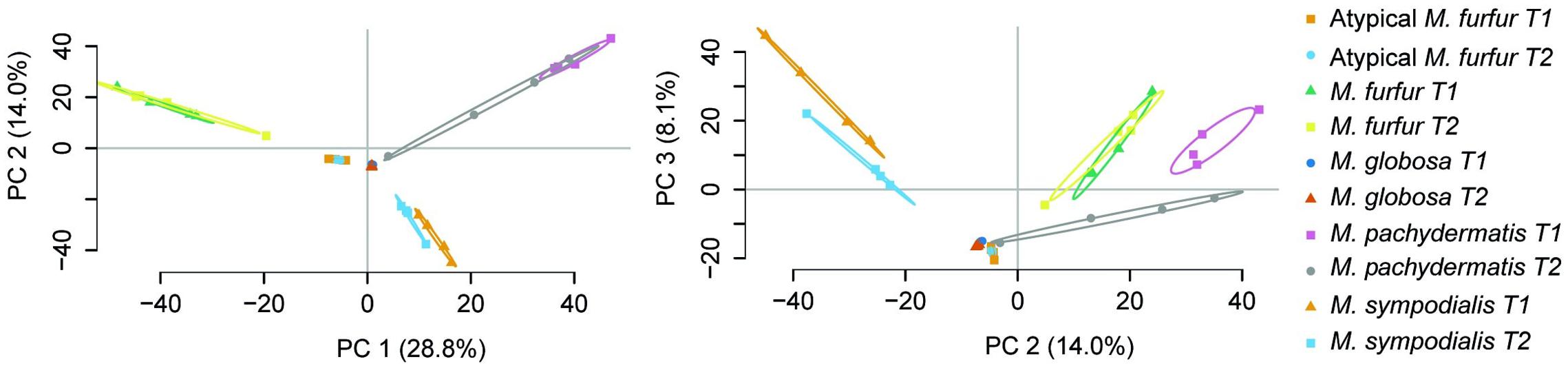
FIGURE 8. Principal component analysis (PCA) of the expressed protein cluster for the five strains at T1 (exponential phase) and T2 (stationary phase) with four replicates.
Protein validation of the models was conducted and expressed as the percentage of proteins predicted by the model that were detected by proteomic profiling (expressed proteins) as well as the percentage of expressed enzymes that were found to be catalyzing an reaction carrying flux in the model (Figure 9). Based on the validation and taking into account the percentage of the total reactions that can be experimentally validated (83% in M. furfur, 80% in M. globosa, 73% in M. sympodialis, 79% in M. pachydermatis, and 78% in Atypical M. furfur). The most successful model was that of M. pachydermatis, where ∼40 and 30% of the predicted proteins were expressed in the exponential and stationary phase, respectively. The less accurate models were those of M. globosa and of the atypical M. furfur, with ∼10% of the predicted proteins being found. Similarly, when evaluating the number of predicted enzymes that were expressed, M. pachydermatis showed the best prediction model with ∼90% of its expressed enzymes predicted by the flux distribution in both the exponential and stationary phases. M. globosa, on the contrary, only showed ∼50% of its expressed enzymes, as predicted by the model under both growth phases.
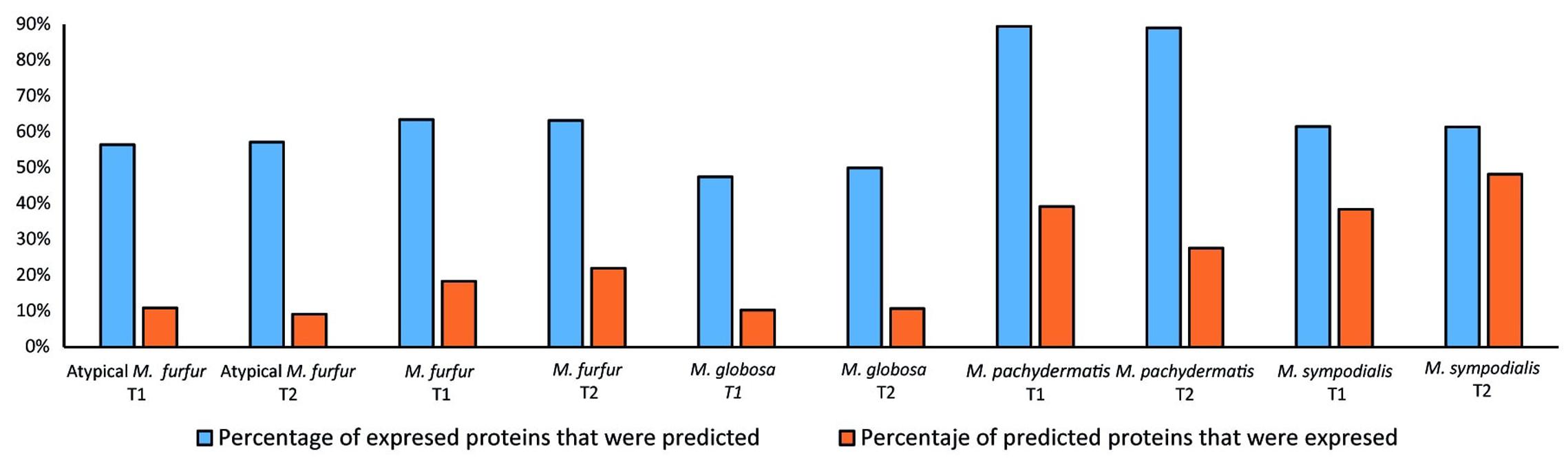
FIGURE 9. Protein validation as the percentage of enzymes predicted by the model that were expressed and the percentage of enzymes expressed that were predicted.
We performed a physiological characterization of these species. We used MM instead of rich media such as Sabouraud or potato dextrose agar, since these media contain small amounts of lipids that can sustain the growth of M. pachydermatis (Wu et al., 2015). Culturing was performed in liquid MM in the presence of oleic acid, palmitic acid, or Tween. Strains were first grown in mDixon containing a variety of lipid sources and Tween 80. To prevent subsequent growth in MM due to the presence of residual lipids either from mDixon or associated with cells, we performed a two-phase growth in MM. First, cells were diluted into MM containing oleic acid, palmitic acid, or one of the Tween sources. After 3 days, these cells were diluted again in fresh MM with oleic acid, palmitic acid, or Tween. The results are described in Table 6 and in Supplementary Figures S4 and S5. M. furfur indeed can assimilate palmitic acid or oleic acid as well as all Tween variants (Table 6 and Supplementary Figures S4, S5), including Tween 40, which represents the C16:0 fatty acid donor in this respect.
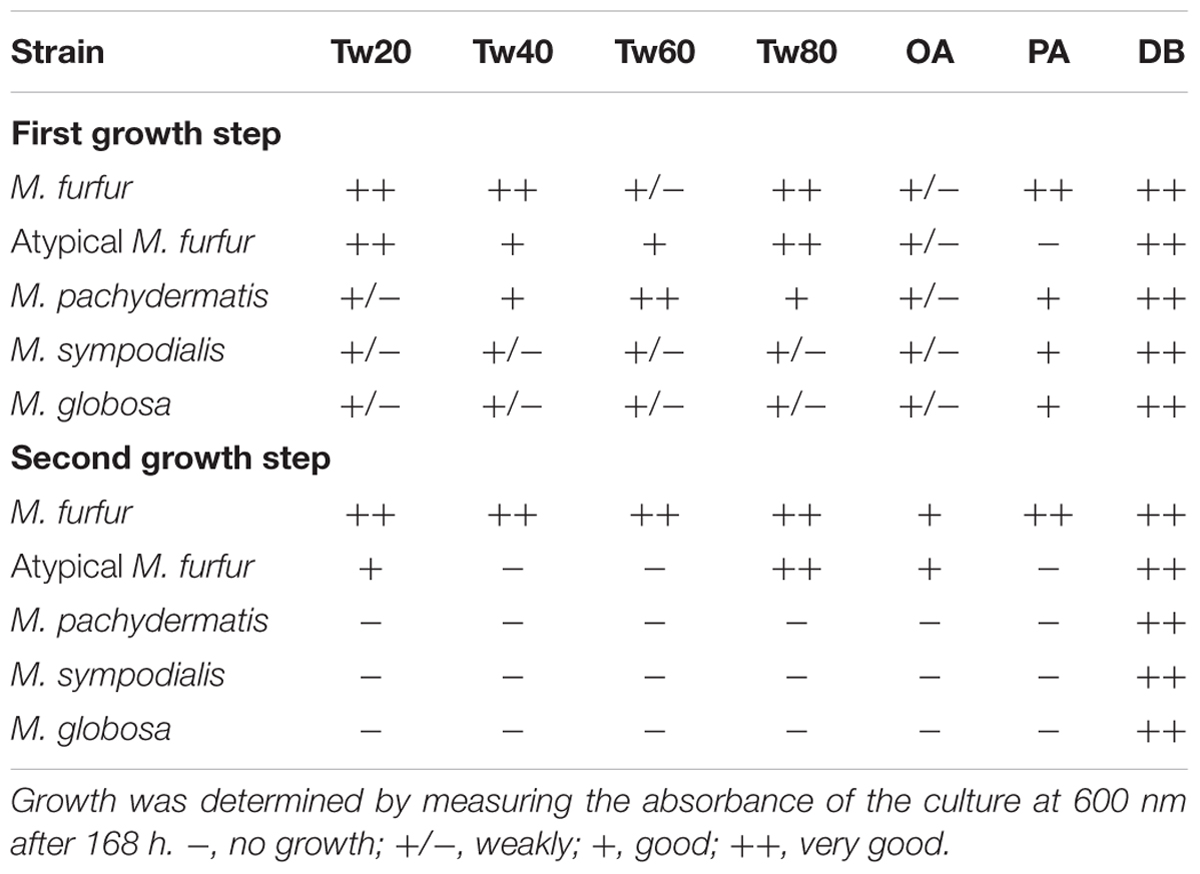
TABLE 6. Physiological characterization of Malassezia spp. by culturing in liquid minimal medium (MM) containing either Tw20 (Tween 20), Tw40 (Tween 40), Tw60 (Tween 60), Tw80 (Tween 80), OA (oleic acid), PA (palmitic acid), or DB (Dixon broth) during the first and second growth step.
The atypical M. furfur strain, however, was only able to assimilate Tween 80, Tween 20, and oleic acid. We observed that M. pachydermatis, M. globosa, and M. sympodialis were able to grow in the first step in MM, however, these strains were not able to grow in the second step in this defined medium supplemented with oleic acid, palmitic acid, or any of the Tween sources (Table 6 and Supplementary Figures S4, S5). However, growth of these latter strains was sustained in the mDixon medium. Interestingly, whereas M. furfur was able to maintain growth in MM with palmitic acid, the atypical M. furfur, M. pachydermatis, M. globosa, and M. sympodialis growth (as determined by the CFU) declined rapidly in the second growth step (Supplementary Figure S5).
We determined whether M. pachydermatis and atypical M. furfur could sustain growth in MM with a mixture of palmitic acid and oleic acid (Supplementary Figure S6). Interestingly, we observed that both strains were then indeed able to maintain growth or survive in this mixture of saturated and unsaturated fatty acids.
The lipid-dependent and lipophilic life style of Malassezia spp. seems to involve selection pressure to adapt to skin environments rich in lipid sources in human and animal hosts. Many factors can disturb the normal status of these yeast species and lead to disease. Approaches to understanding how this disturbance occurs provide clues to managing the negative impacts on the host. Omics studies are necessary to fully comprehend these mechanisms (Jo et al., 2016). In this study, the genomes of the previously reported M. furfur CBS 1878 (Wu et al., 2015) and an isolate of M. furfur with atypical assimilation of Tween 80 were sequenced. Furthermore, the metabolic networks of five Malassezia strains were reconstructed via genome annotation, reaction directionality, compartmentalization, and manual curation. This allowed us to elucidate differences in the genomes, in the metabolism of fungal steroids, and other pathways among the four Malassezia species that were studied. Moreover, we carried out an FBA on the metabolic reconstructions to measure the potential of each strain to produce biomass. We observed differences in the flux distribution among the species, with a variation in the fluxes of reactions related to lipid metabolism. Furthermore, proteomic profiling was used to validate the results. The validation showed that most of the proteins expressed in the proteomic profiling were predicted. However, not all the model’s predictions were corroborated by this approach. Future high-throughput proteomic, metabolomic, and modeling approaches are needed to validate the models.
The genome assemblies of M. furfur and the atypical M. furfur resulted in a genome size of 14.19 and 10.3 Mb, respectively, approximately double the size of the genomes of previously sequenced species [M. globosa (Xu et al., 2007), M. sympodialis (Gioti et al., 2013), and M. pachydermatis (Triana et al., 2015)]. Differences in the genome size of M. furfur CBS 1878 (14.19 Mb) in comparison to that of the same strain reported by Wu et al. (2015) (13.5 Mb) could be due to the assembly and scaffolding methods used in each study. The total length of the resulting contigs longer than 1,000 bp in our assembly is around 13.6 Mb (Table 1); a similar size to the previously reported size of 13.5 Mb. Phylogenetic analysis conducted in this study established that our M. furfur strains are part of the Malassezia cluster A that has been previously described. This cluster consists of several strains of this species that were suggested to be a species complex (Wu et al., 2015). Particularly, atypical M. furfur (10.3 Mb) clustered with M. furfur 7982 (7.7 Mb), thus showing them to be distantly related to other strains, and suggesting that they are implicated in divergence events (Wu et al., 2015). More analysis is required to define the relation of the atypical strain and how it may be implicated in those events.
Additionally, the assembled genomes showed a greater number of scaffolds, indicating a possible fragmentation of the genome (de Wit et al., 2012). The fragmentation was also evidenced by the reduction in the average gene size. The fragmentation present in the genome sequence assembly may cause a bias when comparing it to the other genomes and may further generate false-positive protein annotations (Klassen and Currie, 2012; Faino et al., 2015). However, the CD-Hit and EC results provided support for our protein dataset. The CD-Hit analysis showed that the number of protein clusters was close to the number of proteins that, although higher when compared to those of the other species, did not collapse after clustering. Also, the finding of new EC numbers in the genomes, as well as EC numbers previously reported in other Malassezia genomes, provided support for our annotation. Additionally, the metabolic reconstruction approach allowed us to reduce the fragmentation bias by determining the possible reactions of each protein according to the domains of each gene. The genome assembly could further be improved by resequencing these two genomes with a technique that generates longer reads such as PacBio (English et al., 2012). Different mechanisms might explain the observed genome size differences. The options might include the presence of non-coding repetitions, possible protein duplications in the genomes, and/or the presence of new proteins. The first explanation could refer to mobile genetic elements such as transposons and retrotransposons as well as low-complex repetitions, as has been reported in other haploid fungi (Galagan, 2005). However, the analyses showed that less than 2% of the genomes presented these kinds of repetitions. To assess whether protein duplications or an increase in the number of unique proteins may explain the increase in the genome size of both M. furfur strains studied, we annotated the assemblies. The number of predicted proteins was higher in the genomes of M. furfur and the atypical M. furfur than in the genomes of M. globosa, M. sympodialis, and M. pachydermatis. Differences in the number of predicted proteins could not solely be explained by protein duplications, as mentioned above. The number of protein clusters detected by CD-Hit correlates with the number of total proteins. Among the number of predicted proteins found in M. furfur and in the atypical M. furfur strain (10,203 and 12,121, respectively) 7,570 and 10,434 protein clusters, respectively, were detected. Thus, although there are indeed protein duplications, most of the predicted proteins represent unique proteins in the genomes of M. furfur and the atypical M. furfur strains. This is further supported in the case of enzymes by the fact that the unique EC numbers and reactions in both M. furfur strains were higher than those in the other species studied. This diversification in the number of predicted proteins may provide evidence for the metabolic versatility found in M. furfur. The increase in unique proteins and the genetic diversification present in the M. furfur strains may be due to mating of the yeast, since bipolar mating systems have been previously proposed in Malassezia spp. (Wu et al., 2015), and sexual reproduction has been proposed to promote genetic variation in other pathogenic fungi (Ene and Bennett, 2014; Heitman et al., 2014). These special characteristics can be an advantage in terms of being able to easily adapt to different body sites, even under adverse conditions, such as blood in the case of a fungemia, which this species has been associated with as well (Barber et al., 1993). However, the analysis of metagenomic datasets from different sites on healthy human skin showed that M. furfur is less frequently detected than M. globosa, M. restricta, and M. sympodialis (Wu et al., 2015). This pattern is suggestive of the metabolic profile of this species leading to a strong and intimate relation with the host due to its complex lipid requirements.
As expected, the FAS was not found in the genomes of either M. furfur or the atypical M. furfur strains. The PKSs were found to be conserved among the Malassezia species, with the exception of M. globosa, which lacked one domain (Xu et al., 2007), and the M. furfur isolate, which lacked several domains in its three predicted PKSs. Mycobacterium tuberculosis PKSs have been associated with the biosynthesis of unique lipids or glycolipid conjugates (Quadri, 2014), and in Streptomyces griseus, with the synthesis of phenolic lipids (Funabashi et al., 2008). Further studies are needed to determine the importance of these unique PKSs in Malassezia and their role in the lipid dependency of this genus. They are clearly different from other fungi with an identity of less than 32% with the phylogenetically related basidiomycete P. involutus (even with this was the most similar enzyme).
Even though there were differences in the number of predicted reactions among the species, the proportion in each pathway was similar. Carbohydrate and amino acid metabolism, which are part of the core metabolism of an organism, were the most abundant, as is the case for other fungi such as Aspergillus oryzae (Vongsangnak et al., 2008) and Mortierella alpine (Ye et al., 2015). With respect to fungal steroid biosynthesis, we found that the atypical M. furfur strain had the highest number of reactions, followed by M. furfur. These reactions were almost negligible in the other three species studied. Differences in the fungal steroid biosynthesis between M. furfur and the other three Malassezia species studied may be explained by (i) the production of steroid-like fungal hormones in M. furfur for sexual reproduction, as happens in ascomycetes (Gooday, 1974); (ii) fungal steroids perhaps being a constitutive component of M. furfur, as is the case of the fruiting body of the basidiomycete Sarcodon joedes (Liu et al., 2013); and most likely, (iii) steroids in M. furfur perhaps acting as secondary metabolites, as previously reported in the basidiomycete Polyporus ellisii (Wang et al., 2012). Finally, the differences in arachidonic acid lipid metabolism found in the atypical M. furfur strain may be related to the presence of precursors of eicosanoids that may act as an alternative lipid compound in this strain (Bajpai et al., 1991). In addition, arachidonic acid may act as a mediator of skin inflammation—a previously reported role of M. furfur (Plotkin et al., 1998).
The topological features of the networks were used to assess their robustness. The node degree distribution (Supplementary Figures S7, S8) fits a power law regression and we showed that the five networks had free-scale topologies and non-random behavior (Barabási and Oltvai, 2004). The higher clustering and diameter of M. sympodialis and of the M. furfur strain (M. furfur CBS 1878) networks imply a more complex network. Yet, the clustering and diameter of these two networks were lower than for other yeast metabolic networks such as those of S. cerevisiae (Zomorrodi and Maranas, 2010). This may be due to the reduction in the number of connections among metabolites in each compartment when these are compartmentalized and subdivided as a representation of organelles.
The biomass fluxes found in our study were similar to those reported in other yeasts such as S. cerevisiae (0.7388 mmol gDW-1 h-1) (Pitkänen et al., 2014), but because of the lack of biomass data for Malassezia species further experimental validation of the growth rate in each species is necessary. The FBA allowed us to observe that even though the atypical M. furfur had a higher number of reactions for the steroid biosynthesis pathways, these had higher fluxes in the given condition in M. furfur strain CBS 1878. In addition, we found that M. furfur strain CBS 1878 displayed high fluxes in reactions related to the degradation and elongation of fatty acids such as the conversion of hydroxyacyl-CoA to trans-2-enoyl-CoA (Autio et al., 2008) than the atypical M. furfur strain did, and this may explain the differences in lipid assimilation observed between these two strains. Strikingly, the FBA analysis suggested an important difference in the use of palmitate. M. furfur had a high flux in the CoA activation of palmitate, whereas the other species lacked this activity. FASs are required in most organisms to synthesize fatty acids such as the end product palmitate (C16:0) in the cytosol and all Malassezia spp. lack a FAS complex and are not able to produce palmitate (Triana et al., 2015; Wu et al., 2015). This phenotype can be complemented with external lipids and Tween, which can act as donors for fatty acids, and after uptake, are directly used and/or elongated into long-chain- or very long-chain fatty acids, desaturated, or degraded via β-oxidation in the peroxisome. We observed that M. furfur is able to grow in MM supplemented with Tween 40 or with palmitate, indicating that palmitate is indeed used, and this is in accordance with the FBA predictions. The physiological analysis of the atypical M. furfur strain indicates that it cannot use palmitate since the second growth step in liquid medium supplemented with Tween 40 or with palmitate was not sustained. Similar results were obtained for M. pachydermatis, M. globosa, and M. sympodialis (Supplementary Figures S4, S5), which is in agreement with the FBA predictions. These findings could suggest that the transport and activation of palmitic acid in palmitic acid CoA is happening in M. furfur, however, could be hampered in other strains. More studies such flux variability analysis are required to clarify this. However, previous in silico analysis of the fatty acid metabolism of M. globosa using integrated microbial genomes confirmed the presence of the complete β-oxidation pathway for the degradation of saturated fatty acids (James et al., 2013). These differences could be related to differences in the fluxes in reactions related to this pathway in M. furfur. The observation that these latter four strains did grow in the first step in MM indicates that residual lipids from mDixon and/or associated with cells are sufficient to support the first step. These lipids are depleted in the second growth step in MM, which allows for a real analysis of lipid dependency. The observation that the atypical M. furfur cannot grow in MM with Tween 20, 40, and 60, the donors of C12:0, C16:0, and C18:0 fatty acids, respectively, suggests that these saturated fatty acids cannot be further elongated and/or desaturated. All strains except M. furfur and the atypical variant were not able to grow in MM supplemented with a single Tween or with fatty acid species, whereas they did grow in mDixon. This observation might be explained if we assume that the strains require a mixture of saturated and unsaturated fatty acids, as was similarly observed with fasl, olel (FAS and fatty acid desaturase minus) mutants of S. cerevisiae (Henry, 1973). Further analysis showed that the atypical M. furfur and M. pachydermatis indeed was capable to grow or survive in MM with a mixture of palmitic acid and oleic acid.
Other studies suggested that M. globosa and M. restricta are not capable of synthesizing unsaturated fatty acids due to the lack of a Δ 9-desaturase (EC 1.14.19.2) gene (Xu et al., 2007; Boekhout, 2010), which is involved in the desaturation of palmitic acid and stearic acid to palmitoleic acid and oleic acid, respectively. However, Gioti et al. (2013) found this desaturase gene in the genome of M. sympodialis. We also found the same gene in M. furfur, atypical M. furfur, and M. pachydermatis, suggesting the ability of these species to produce unsaturated fatty acids such as oleic acid, thus providing these species with an advantage in terms of their metabolic versatility to rapidly adapt to regions in which the availability of such fatty acid sources is limited. This ability is missing in M. globosa and M. restricta, which do require these unsaturated fatty acids as additional sources from their host to support their growth. These particular differences highlight the differences in the pathogenic role of these species in the development of certain dermatological diseases in which different species have epidemiological relevance, as is the case for M. globosa and dandruff/SD (Boekhout, 2010). In this regard, more research is required to determine how these different Malassezia spp. use external fatty acids.
The lipid-metabolism reactions among the other species were also divergent. Higher activity in the reactions involved in the degradation of long-chain fatty acids was present in M. sympodialis but not in M. globosa or M. pachydermatis. These differences may be associated with the phenotypic differences of each species, reflected by the differential lipid assimilation or the differential use of Tweens. We should, however, explore more deeply the physiology and regulation of β-oxidation in this yeast (Yarrow, 1998; Boekhout, 2010).
The higher number of reactions related to the butanoate metabolism found in M. pachydermatis may be related to the lipid-assimilation versatility of this species. This metabolism is closely related to the synthesis of type II and type III polyketides (which may be precursors of unique lipids) as well as to the fatty acid metabolism (Rawlings, 1998; Charrier et al., 2006). Thus, the higher activity of reactions involved in the metabolism of butanoate in M. pachydermatis may be reflected in the production of lipids that may not be produced by the other Malassezia species and that may be corroborated by lipid profiling of the yeasts.
The proteomic profiling allowed us to validate, on average, 30% of the predicted proteins of the model, implying that the other enzymes that are supposed to be expressed according to the FBA are not present or that their concentrations are substantially low. An average of 70% of the proteins expressed in all the samples were in fact predicted. Given that this technique is still not widely used to validate these kinds of models, the cutoff points and expected percentages are not well known. Furthermore, issues such as the identification of large-scale protein datasets, small protein concentration detection, and the extraction of low soluble proteins such as membrane proteins may affect the profiling (Marcotte, 2007; Yates, 2013).
Together, the metabolic reconstruction and modeling showed versatility within the genus of Malassezia. Flux differences were suggested in the production of steroids in M. furfur and in butanoate metabolism in M. pachydermatis. The assimilation defects of palmitic acid were suggested in M. globosa, M. sympodialis, M. pachydermatis, and in the atypical variant of M. furfur. The capability of M. furfur to assimilate palmitic acid was confirmed via culturing on defined media.
ST, AC, AG, HdC, and SR contributed to the design of the work. ST, AC, and RO performed the experiments. All authors were involved in the analysis and interpretation of data. ST and AC wrote the manuscript. HdC, HW, SR, and AG made revisions. All authors approved the version to be published and agreed to be accountable for all aspects of the work.
This work was supported by Colciencias grant No. 120465741393 and the Netherlands fellowship program NFP-phd.14/99.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer LK and handling Editor declared their shared affiliation.
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01772/full#supplementary-material
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Andrews, S. (2010). FastQC: A Quality Control Tool for High throughput Sequence Data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Arendrup, M. C., Boekhout, T., Akova, M., Meis, J. F., Cornely, O. A., and Lortholary, O. (2014). ESCMID† and ECMM‡ joint clinical guidelines for the diagnosis and management of rare invasive yeast infections. Clin. Microbiol. Infect. 20, 76–98. doi: 10.1111/1469-0691.12360
Autio, K. J., Guler, J. L., Kastaniotis, A. J., Englund, P. T., and Hiltunen, J. K. (2008). The 3-hydroxyacyl-ACP dehydratase of mitochondrial fatty acid synthesis in Trypanosoma brucei. FEBS Lett. 582, 729–733. doi: 10.1016/j.febslet.2008.01.051
Bajpai, P. K., Bajpai, P., and Ward, O. P. (1991). Arachidonic acid production by fungi. Appl. Environ. Microbiol. 57, 1255–1258.
Barabási, A.-L., and Oltvai, Z. N. (2004). Network biology: understanding the cell’s functional organization. Nat. Rev. Genet. 5, 101–113. doi: 10.1038/nrg1272
Barber, G. R., Brown, A. E., Kiehn, T. E., Edwards, F. F., and Armstrong, D. (1993). Catheter-related Malassezia furfur fungemia in immunocompromised patients. Am. J. Med. 95, 365–370. doi: 10.1016/0002-9343(93)90304-8
Boekhout, T. (ed.). (2010). Malassezia and the Skin: Science and Clinical Practice. Berlin: Springer.
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D., and Pirovano, W. (2011). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. doi: 10.1093/bioinformatics/btq683
Boetzer, M., and Pirovano, W. (2012). Toward almost closed genomes with GapFiller. Genome Biol. 13:R56. doi: 10.1186/gb-2012-13-6-r56
Borodovsky, M., and Lomsadze, A. (2011). “Eukaryotic gene prediction using GeneMark.hmm-E and GeneMark-ES,” in Current Protocols in Bioinformatics, eds A. D. Baxevanis, G. A. Petsko, L. D. Stein, and G. D. Stormo (Hoboken, NJ: John Wiley & Sons, Inc.).
Bruce, A. M. (2013). McCarl Expanded GAMS User Guide, GAMS Release 24.2.1. Washington, DC: GAMS Development Corporation.
Cantarel, B. L., Korf, I., Robb, S. M. C., Parra, G., Ross, E., Moore, B., et al. (2007). MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196. doi: 10.1101/gr.6743907
Caspi, R., Altman, T., Billington, R., Dreher, K., Foerster, H., Fulcher, C. A., et al. (2014). The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 42, D459–D471. doi: 10.1093/nar/gkt1103
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Charrier, C., Duncan, G. J., Reid, M. D., Rucklidge, G. J., Henderson, D., Young, P., et al. (2006). A novel class of CoA-transferase involved in short-chain fatty acid metabolism in butyrate-producing human colonic bacteria. Microbiology 152, 179–185. doi: 10.1099/mic.0.28412-0
Conesa, A., and Götz, S. (2008). Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics 2008:619832. doi: 10.1155/2008/619832
de Wit, P. J. G. M., van der Burgt, A., Ökmen, B., Stergiopoulos, I., Abd-Elsalam, K. A., Aerts, A. L., et al. (2012). The genomes of the fungal plant pathogens Cladosporium fulvum and Dothistroma septosporum reveal adaptation to different hosts and lifestyles but also signatures of common ancestry. PLOS Genet. 8:e1003088. doi: 10.1371/journal.pgen.1003088
Delcher, A. L., Phillippy, A., Carlton, J., and Salzberg, S. L. (2002). Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 30, 2478–2483. doi: 10.1093/nar/30.11.2478
Dodt, M., Roehr, J., Ahmed, R., and Dieterich, C. (2012). FLEXBAR—flexible barcode and adapter processing for next-generation sequencing platforms. Biology 1, 895–905. doi: 10.3390/biology1030895
Ene, I. V., and Bennett, R. J. (2014). The cryptic sexual strategies of human fungal pathogens. Nat. Rev. Microbiol. 12, 239–251. doi: 10.1038/nrmicro3236
English, A. C., Richards, S., Han, Y., Wang, M., Vee, V., Qu, J., et al. (2012). Mind the gap: upgrading genomes with pacific biosciences RS long-read sequencing technology. PLOS ONE 7:e47768. doi: 10.1371/journal.pone.0047768
Faino, L., Seidl, M. F., Datema, E., van den Berg, G. C. M., Janssen, A., Wittenberg, A. H. J., et al. (2015). Single-molecule real-time sequencing combined with optical mapping yields completely finished fungal genome. mBio 6:e00936-15. doi: 10.1128/mBio.00936-15
Funabashi, M., Funa, N., and Horinouchi, S. (2008). Phenolic lipids synthesized by Type III polyketide synthase confer penicillin resistance on Streptomyces griseus. J. Biol. Chem. 283, 13983–13991. doi: 10.1074/jbc.M710461200
Gaitanis, G., Magiatis, P., Hantschke, M., Bassukas, I. D., and Velegraki, A. (2012). The Malassezia genus in skin and systemic diseases. Clin. Microbiol. Rev. 25, 106–141. doi: 10.1128/CMR.00021-11
Galagan, J. E. (2005). Genomics of the fungal kingdom: insights into eukaryotic biology. Genome Res. 15, 1620–1631. doi: 10.1101/gr.3767105
Ganter, M., Bernard, T., Moretti, S., Stelling, J., and Pagni, M. (2013). MetaNetX.org: a website and repository for accessing, analysing and manipulating metabolic networks. Bioinformatics 29, 815–816. doi: 10.1093/bioinformatics/btt036
Gioti, A., Nystedt, B., Li, W., Xu, J., Andersson, A., Averette, A. F., et al. (2013). Genomic insights into the atopic eczema-associated skin commensal yeast Malassezia sympodialis. mBio 4:e00572-12. doi: 10.1128/mBio.00572-12
Gonzalez, A., Sierra, R., Cardenas, M. E., Grajales, A., Restrepo, S., Cepero de Garcia, M. C., et al. (2009). Physiological and molecular characterization of atypical isolates of Malassezia furfur. J. Clin. Microbiol. 47, 48–53. doi: 10.1128/JCM.01422-08
Gooday, G. W. (1974). Fungal sex hormones. Annu. Rev. Biochem. 43, 35–49. doi: 10.1146/annurev.bi.43.070174.000343
Gremme, G., Steinbiss, S., and Kurtz, S. (2013). GenomeTools: a comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans. Comput. Biol. Bioinform. 10, 645–656. doi: 10.1109/TCBB.2013.68
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Heitman, J., Carter, D. A., Dyer, P. S., and Soll, D. R. (2014). Sexual reproduction of human fungal pathogens. Cold Spring Harb. Perspect. Med. 4:a019281. doi: 10.1101/cshperspect.a019281
Henry, S. A. (1973). Death resulting from fatty acid starvation in yeast. J. Bacteriol. 116, 1293–1303.
James, A. G., Abraham, K. H., Cox, D. S., Moore, A. E., and Pople, J. E. (2013). Metabolic analysis of the cutaneous fungi Malassezia globosa and M. restricta for insights on scalp condition and dandruff. Int. J. Cosmet. Sci. 35, 169–175. doi: 10.1111/ics.12022
Jankowski, M. D., Henry, C. S., Broadbelt, L. J., and Hatzimanikatis, V. (2008). Group contribution method for thermodynamic analysis of complex metabolic networks. Biophys. J. 95, 1487–1499. doi: 10.1529/biophysj.107.124784
Jo, J.-H., Kennedy, E. A., and Kong, H. H. (2016). Topographical and physiological differences of the skin mycobiome in health and disease. Virulence 8, 324–333. doi: 10.1080/21505594.2016.1249093
Juntachai, W., Oura, T., Murayama, S. Y., and Kajiwara, S. (2009). The lipolytic enzymes activities of Malassezia species. Med. Mycol. 47, 477–484. doi: 10.1080/13693780802314825
Kämper, J., Kahmann, R., Bölker, M., Ma, L.-J., Brefort, T., Saville, B. J., et al. (2006). Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 444, 97–101. doi: 10.1038/nature05248
Kanehisa, M., and Goto, S. (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kim, H. J., Kim, E. T., Lim, C. Y., Park, C., Kang, B. T., Kim, J. W., et al. (2010). The immunoglobulin G response to Malassezia pachydermatis extracts in atopic and non-atopic dogs. Can. Vet. J. 51, 869–872.
Klassen, J. L., and Currie, C. R. (2012). Gene fragmentation in bacterial draft genomes: extent, consequences and mitigation. BMC Genomics 13:14. doi: 10.1186/1471-2164-13-14
Korf, I. (2004). Gene finding in novel genomes. BMC Bioinformatics 5:59. doi: 10.1186/1471-2105-5-59
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. doi: 10.1186/gb-2009-10-3-r25
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Liu, H.-W., Hu, L., Zhang, A.-L., and Gao, J.-M. (2013). Steroids and phenolic constituents from the fruiting bodies of the basidiomycete Sarcodon joedes. Nat. Prod. Res. 27, 80–84. doi: 10.1080/14786419.2012.656112
Marcotte, E. M. (2007). How do shotgun proteomics algorithms identify proteins? Nat. Biotechnol. 25, 755–757. doi: 10.1038/nbt0707-755
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185. doi: 10.1093/nar/gkm321
Murabito, E., Simeonidis, E., Smallbone, K., and Swinton, J. (2009). Capturing the essence of a metabolic network: a flux balance analysis approach. J. Theor. Biol. 260, 445–452. doi: 10.1016/j.jtbi.2009.06.013
Orth, J. D., Conrad, T. M., Na, J., Lerman, J. A., Nam, H., Feist, A. M., et al. (2014). A comprehensive genome-scale reconstruction of Escherichia coli metabolism–2011. Mol. Syst. Biol. 7, 535–535. doi: 10.1038/msb.2011.65
Parra, G., Bradnam, K., and Korf, I. (2007). CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067. doi: 10.1093/bioinformatics/btm071
Pitkänen, E., Jouhten, P., Hou, J., Syed, M. F., Blomberg, P., Kludas, J., et al. (2014). Comparative genome-scale reconstruction of gapless metabolic networks for present and ancestral species. PLOS Comput. Biol. 10:e1003465. doi: 10.1371/journal.pcbi.1003465
Plotkin, L. I., Mathov, I., Squiquera, L., and Leoni, J. (1998). Arachidonic acid released from epithelial cells by Malassezia furfur phospholipase A2: a potential pathophysiologic mechanism. Mycologia 90, 163–169. doi: 10.2307/3761291
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 – approximately maximum-likelihood trees for large alignments. PLOS ONE 5:e9490. doi: 10.1371/journal.pone.0009490
Quadri, L. E. N. (2014). Biosynthesis of mycobacterial lipids by polyketide synthases and beyond. Crit. Rev. Biochem. Mol. Biol. 49, 179–211. doi: 10.3109/10409238.2014.896859
Quevillon, E., Silventoinen, V., Pillai, S., Harte, N., Mulder, N., Apweiler, R., et al. (2005). InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–W120. doi: 10.1093/nar/gki442
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
R Core Team (2013). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rawlings, B. J. (1998). Biosynthesis of fatty acids and related metabolites. Nat. Prod. Rep. 15, 275–308. doi: 10.1039/a815275y
Schweizer, E., and Bolling, H. (1970). A Saccharomyces cerevisiae mutant defective in saturated fatty acid biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 67, 660–666. doi: 10.1073/pnas.67.2.660
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Stanke, M., Steinkamp, R., Waack, S., and Morgenstern, B. (2004). AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–W312. doi: 10.1093/nar/gkh379
Triana, S., González, A., Ohm, R. A., Wösten, H. A., de Cock, H., Restrepo, S., et al. (2015). Draft genome sequence of the animal and human pathogen Malassezia pachydermatis strain CBS 1879. Genome Announc. 3:e01197-15. doi: 10.1128/genomeA.01197-15.Copyright
Vizcaíno, J. A., Csordas, A., del-Toro, N., Dianes, J. A., Griss, J., Lavidas, I., et al. (2016). 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456. doi: 10.1093/nar/gkv1145
Vongsangnak, W., Olsen, P., Hansen, K., Krogsgaard, S., and Nielsen, J. (2008). Improved annotation through genome-scale metabolic modeling of Aspergillus oryzae. BMC Genomics 9:245. doi: 10.1186/1471-2164-9-245
Wang, S., Zhang, L., Liu, L.-Y., Dong, Z.-J., Li, Z.-H., and Liu, J.-K. (2012). Six novel steroids from culture of basidiomycete Polyporus ellisii. Nat. Prod. Bioprospect. 2, 240–244. doi: 10.1007/s13659-012-0058-4
Wood, E. (1991). Protein purification methods: a practical approach. Biochem. Educ. 19, 39–40. doi: 10.1016/0307-4412(91)90149-3
Wu, G., Zhao, H., Li, C., Rajapakse, M. P., Wong, W. C., Xu, J., et al. (2015). Genus-wide comparative genomics of Malassezia delineates its phylogeny, physiology, and niche adaptation on human skin. PLOS Genet. 11:e1005614. doi: 10.1371/journal.pgen.1005614
Xu, J., Saunders, C. W., Hu, P., Grant, R. A., Boekhout, T., Kuramae, E. E., et al. (2007). Dandruff-associated Malassezia genomes reveal convergent and divergent virulence traits shared with plant and human fungal pathogens. Proc. Natl. Acad. Sci. U.S.A. 104, 18730–18735. doi: 10.1073/pnas.0706756104
Yarrow, D. (1998). “Methods for the isolation, maintenance and identification of yeasts,” in The Yeasts, 4th Edn, ed. C. P. Kurtzman and J. W. Fell (Amsterdam: Elsevier), 77–100.
Yates, J. R. (2013). The revolution and evolution of shotgun proteomics for large-scale proteome analysis. J. Am. Chem. Soc. 135, 1629–1640. doi: 10.1021/ja3094313
Ye, C., Xu, N., Chen, H., Chen, Y. Q., Chen, W., and Liu, L. (2015). Reconstruction and analysis of a genome-scale metabolic model of the oleaginous fungus Mortierella alpina. BMC Syst. Biol. 9:1. doi: 10.1186/s12918-014-0137-8
Yu, C.-S., Lin, C.-J., and Hwang, J.-K. (2004). Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. 13, 1402–1406. doi: 10.1110/ps.03479604
Zinkeviciene, A., Norkunas, V., and Citavicius, D. (2012). Atypical non-lipid-dependent strains of Malassezia furfur. Cent. Eur. J. Biol. 7, 241–249. doi: 10.2478/s11535-012-0018-3
Zomorrodi, A. R., and Maranas, C. D. (2010). Improving the iMM904 S. cerevisiae metabolic model using essentiality and synthetic lethality data. BMC Syst. Biol. 4:178. doi: 10.1186/1752-0509-4-178
Keywords: Malassezia, genome, metabolic reconstruction, FBA, lipid metabolism
Citation: Triana S, de Cock H, Ohm RA, Danies G, Wösten HAB, Restrepo S, González Barrios AF and Celis A (2017) Lipid Metabolic Versatility in Malassezia spp. Yeasts Studied through Metabolic Modeling. Front. Microbiol. 8:1772. doi: 10.3389/fmicb.2017.01772
Received: 14 July 2017; Accepted: 31 August 2017;
Published: 14 September 2017.
Edited by:
Charley Christian Staats, Federal University of Rio Grande do Sul (UFRGS), BrazilReviewed by:
Macit Ilkit, Çukurova University, TurkeyCopyright © 2017 Triana, de Cock, Ohm, Danies, Wösten, Restrepo, González Barrios and Celis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrés F. González Barrios, YW5kZ29uemFAdW5pYW5kZXMuZWR1LmNv Adriana Celis, YWNlbGlzQHVuaWFuZGVzLmVkdS5jbw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.