 Yue-Hui Hong1
Yue-Hui Hong1 Cong-Cong Ye1Qian-Zhi Zhou1
Cong-Cong Ye1Qian-Zhi Zhou1 Xiao-Ying Wu2Jian-Ping Yuan1Juan Peng1Hailin Deng1*
Xiao-Ying Wu2Jian-Ping Yuan1Juan Peng1Hailin Deng1* Jiang-Hai Wang1*
Jiang-Hai Wang1*- 1Guangdong Provincial Key Laboratory of Marine Resources and Coastal Engineering/South China Sea Bioresource Exploitation and Utilization Collaborative Innovation Center, School of Marine Sciences, Sun Yat-sen University, Guangzhou, China
- 2State Key Laboratory of Conservation and Utilization of Subtropical Agro-Bioresources, College of Natural Resources and Environment, South China Agricultural University, Guangzhou, China
Petroleum pollution is a severe environmental issue. Comprehensively revealing the genetic backgrounds of hydrocarbon-degrading microorganisms contributes to developing effective methods for bioremediation of crude oil-polluted environments. Marine bacterium Achromobacter sp. HZ01 is capable of degrading hydrocarbons and producing biosurfactants. In this study, the draft genome (5.5 Mbp) of strain HZ01 has been obtained by Illumina sequencing, containing 5,162 predicted genes. Genome annotation shows that “amino acid metabolism” is the most abundant metabolic pathway. Strain HZ01 is not capable of using some common carbohydrates as the sole carbon sources, which is due to that it contains few genes associated with carbohydrate transport and lacks some important enzymes related to glycometabolism. It contains abundant proteins directly related to petroleum hydrocarbon degradation. AlkB hydroxylase and its homologs were not identified. It harbors a complete enzyme system of terminal oxidation pathway for n-alkane degradation, which may be initiated by cytochrome P450. The enzymes involved in the catechol pathway are relatively complete for the degradation of aromatic compounds. This bacterium lacks several essential enzymes for methane oxidation, and Baeyer-Villiger monooxygenase involved in the subterminal oxidation pathway and cycloalkane degradation was not identified. These results suggest that strain HZ01 degrades n-alkanes via the terminal oxidation pathway, degrades aromatic compounds primarily via the catechol pathway and cannot perform methane oxidation or cycloalkane degradation. Additionally, strain HZ01 possesses abundant genes related to the metabolism of secondary metabolites, including some genes involved in biosurfactant (such as glycolipids and lipopeptides) synthesis. The genome analysis also reveals its genetic basis for nitrogen metabolism, antibiotic resistance, regulatory responses to environmental changes, cell motility, and material transport. The obtained genome data provide us with a better understanding of hydrocarbon-degrading bacteria, which may contribute to the future design of rational strategies for bioremediation of petroleum-polluted marine environments.
Introduction
Petroleum pollution in marine environments mainly caused by anthropogenic activities is a serious environmental issue due to its negative impacts on human health and ecosystems. For instance, the “Deep Water Horizon” oil spill accident in the Gulf of Mexico was one of the most dramatic pollution events, which had resulted in a serious damage to the marine sites (Xue et al., 2015). The maintenance of ecological balance imperatively requires the development of effective ways to remediate crude oil-polluted environments. Among the proposed remediation techniques for the treatment of marine oil pollution, microbial remediation has been regarded as one of the most reliable strategies for the thorough elimination of petroleum contaminants (Hassanshahian et al., 2012). Revealing the genomic backgrounds of hydrocarbon-degrading bacteria contributes to developing effective methods to reduce oil contamination and mitigate its environmental damage. High-throughput sequencing is providing us with novel knowledge on the underlying mechanisms in microorganisms conducting oil degradation. So far, the genomes of a few hydrocarbon-degrading bacteria have been analyzed in depth. These genomes exhibit various characteristic differences. This is in accord with the inference that the degradation properties of hydrocarbon-degrading bacteria are generally different (Rojo, 2009). For instance, Alcanivorax borkumensis SK2 contains a streamlined genome with few energy production-related genes and mobile genetic elements, but with abundant genes related to oil degradation (Schneiker et al., 2006). Compared with strain SK2, Oleispira antarctica RB-8 has a larger genome with massive gene-transfer events (Kube et al., 2013). In view of the characteristic differences among diverse oil-degrading bacteria and the complexity of petroleum degradation, genome-wide elucidation of the entire degradation mechanisms of petroleum hydrocarbons is still in a relatively early stage.
Some strains belonging to the genus Achromobacter are regarded as representative bacteria with bioremediation potential, as diverse bioremediation properties of this bacterial population have been reported, e.g., biphenyl catabolism (Furukawa et al., 1989), arsenite oxidation (Cai et al., 2009), haloaromatic acid degradation (Jencova et al., 2008), detoxification of chromium-containing slag (Chai et al., 2010), and hydrocarbon degradation (Deng et al., 2014). Many genome sequences of Achromobacter spp. are already known, and some characteristics of Achromobacter strains have been deeply analyzed at the genomic level. For instance, the pathogenic mechanisms of opportunistic pathogen Achromobacter xylosoxidans NH44784-1996 has been revealed using complete genome sequencing (Jakobsen et al., 2013). However, genome-wide researches on elucidating (i) the pathways of hydrocarbon degradation, (ii) the biosynthesis of secondary metabolites, and (iii) the genetic basis for environment adaptation in hydrocarbon-degrading Achromobacter spp. are currently lacking.
Achromobacter sp. HZ01, isolated from the crude oil-polluted seawater in the South China Sea, is capable of degrading petroleum hydrocarbons and producing biosurfactants (Deng et al., 2014, 2016), exhibiting a good potential for various applications. A de novo transcriptome of strain HZ01 regarding hydrocarbon degradation was previously reported, and some functional genes and pathways were analyzed (Hong et al., 2016). However, RNA-seq-based transcriptomics cannot cover all genes and pathways because it focuses on the expressed genes under specific conditions. To reveal the genetic background of strain HZ01 more comprehensively, it is necessary to carry out whole genome sequencing.
In this study, we report the draft genome of strain HZ01. The genome analysis contributed to better understanding its genetic basis for petroleum degradation, production of secondary metabolites, antibiotic resistance, and some other important physiological functions. The present work may also provide a basis for developing a cost-effective and eco-friendly method to remediate crude oil-contaminated marine environments.
Materials and Methods
Strain and Carbon Source Utilization
Strain HZ01 was isolated from crude oil-polluted seawater at the Daya Bay, South China Sea, as previously described (Deng et al., 2014). It was deposited in the China Center for Type Culture Collection with the preservation number of “CCTCC AB 2013198.”
Medium A (pH 7.5) contained (g/L): NH4NO3, 2.5; Na2HPO4⋅12H2O, 2; KH2PO4, 1; MgSO4⋅7H2O, 0.2; NaCl, 10; and trace element solution (1 mL/L). Medium A was supplemented with single carbon source (glucose, D-fructose, D-galactose, lactose, sucrose, D-mannose, D-maltose, mannitol, pyruvic acid, glycerol, and citric acid, respectively). The trace element solution was composed of (mg/L): CaCl2, 20; CuSO4, 0.5; MnSO4⋅H2O, 0.5; FeCl3, 30; and ZnSO4⋅7H2O, 10. Strain HZ01 was incubated in the Luria–Bertani (LB) medium at 28°C and 150 rpm for 16 h. Bacterial cells in the logarithmic phase were collected by centrifugation (2,600 g) at room temperature for 2 min. Then, the cells were resuspended and inoculated into medium A with an inoculum dose of 10% (v/v), followed by incubation at 28°C and 150 rpm. At indicated time points, the optical densities (OD600) of the culture broths were measured using a spectrophotometer. The medium without bacteria inoculation at each time point served as a negative control. The obtained OD600 values were used for plotting growth curves, to determine whether strain HZ01 was capable of utilizing an indicated compound as the sole carbon and energy source. The experiments were performed in triplicate with two repetitions.
Emulsification Activity of the Culture Broth
Strain HZ01 was incubated in the LB medium at 28°C and 150 rpm for 16 h, followed by centrifugation (2,600 g) at room temperature for 2 min. The cell pellets were resuspended and inoculated into medium A with an inoculum dose of 10% (v/v). Citric acid (40 g/L) was used as the sole source of carbon and energy. The medium without bacteria inoculation served as a negative control. After incubation at 28°C and 150 rpm for 3 days, the culture broth was subjected to centrifugation (12,000 g) at room temperature for 2 min. Three milliliters of the resulting supernatants were mixed with an equal volume of soybean oil, coconut oil, olive oil, diesel oil, kerosene, and hexane, respectively. Three milliliters of sodium dodecyl sulfate (SDS; 0.5 g/L) were also mixed with those compounds, respectively, serving as positive controls. After being vortexed for 2 min, the mixtures were kept to settle at room temperature for 24 h, followed by the observation of emulsification layers. The experiments were performed in duplicate with two repetitions.
Genome Sequencing
Strain HZ01 was incubated in the LB medium at 28°C and 150 rpm for 16 h before DNA extraction. The genomic DNA was extracted using the phenol-chloroform-isoamyl alcohol method (Sambrook and Russell, 2001). After quality verification of the isolated DNA, genome sequencing libraries with insert sizes of 500 and 800 bp were constructed, respectively. The Illumina HiSeq 2500 sequencing platform was employed to perform the genome sequencing using the paired-end (PE250) sequencing strategy.
Sequence Assembly and Annotation
High-quality reads were obtained after quality control and elimination of adaptor sequences and low-quality reads, followed by sequence assembly into scaffolds using the Newbler v2.9 assembly tool (Roche Diagnostics). The CheckM (Parks et al., 2015) was employed to estimate the completeness of the draft genome. Protein-coding genes were predicted by the Genemark (Besemer et al., 2001). Gene annotation was performed by similarity searches (E-value ≤ 10-5) against the non-redundant database (NCBI-nr1), the Kyoto Encyclopedia of Genes and Genomes (KEGG2), and the evolutionary genealogy of genes: Non-supervised Orthologous Groups (eggNOG) (Jensen et al., 2008). Additionally, pathway-based functional annotation of the predicted genes was conducted using the KEGG Automatic Annotation Server (KAAS) (Moriya et al., 2007).
Genome Comparisons and Gene Cluster Prediction
The general genome features of strain HZ01 and some other strains were compared. The genome synteny was analyzed by using the MUMmer (Kurtz et al., 2004) with the genomes of Achromobacter xylosoxidans A8 and Achromobacter xylosoxidans NH44784-1996 as a reference, respectively. Two-tailed Fisher exact test was employed to evaluate the differences in gene abundance of COG categories between the genome of strain HZ01 and other genomes in the Integrated Microbial Genomes (IMG) database (Markowitz et al., 2014).
The analysis of core and pan genome was carried out as previously described (Xiao et al., 2016; Ibrahim et al., 2017). The genome data of eight strains (including all Achromobacter species) were from the NCBI database. The core genome contains genes (core genes) present in the genomes of all indicated organisms. The pan genome is composed of core genes and a dispensable genome including genes unique to a genome and genes contained in two or more species (Medini et al., 2005). Single-copy genes were screened out according to the core and pan genome analysis. Protein sequence alignment was performed using the MUSCLE v3.8.31 (Edgar, 2004). The Treebest v1.9.2 (Vilella et al., 2009) was employed to construct a phylogenetic tree based on the core genes using the neighbor-joining method. Additionally, a phylogenetic tree based on the 16S rRNA genes was constructed using the neighbor-joining method. The sequences of 16S rRNA genes were from the NCBI database.
The antibiotics and secondary metabolite analysis shell (antiSMASH) (Weber et al., 2015) was employed to predict the gene clusters related to secondary metabolite biosynthesis.
Analysis of Drug Resistance
To identify the antibiotic resistance genes in strain HZ01, homology alignment was performed using blastp (E-value < 10-5; percent identity ≥ 40%) against the Antibiotic Resistance Genes Database (ARDB) (Liu and Pop, 2009). The best hit was selected as the annotation of a query sequence.
The antibiotic resistance profile of strain HZ01 was determined by the Kirby-Bauer disk diffusion method (Cockerill et al., 2013) using Escherichia coli ATCC 25922 as a quality control. Briefly, bacterial cells were incubated in the LB medium for 16 h, followed by centrifugation (2,600 g) at room temperature for 2 min. The cell pellets were resuspended using sterile normal saline and adjusted to 0.5 McFarland standards. Then, the cells were spread onto the Mueller Hinton Agar (Guangdong Huankai Microbial Sci. & Tech. Co., Ltd., Guangzhou, China) plates using sterile cotton swabs. After being dried at room temperature for 5 min, the plates were placed with antibiotic disks (Hangzhou Microbial Reagent Co., Ltd., Hangzhou, China), followed by incubation at 35°C for 18 h. The inhibition zones were then measured, and the results were interpreted according to the standards of Clinical and Laboratory Standards Institute (CLSI) (Cockerill et al., 2013). The experiments were performed in triplicate with two repetitions.
Data Deposition
The raw reads generated from genome sequencing were deposited in the Sequence Read Archive (SRA) under accession number SRP073408. The Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under accession number LWKV00000000. The version described in this paper is version LWKV01000000.
Results
General Features of the Draft Genome
Genome sequencing generated 1.87 and 1.34 M pairs of clean reads for libraries A and B, respectively, providing a 140 × coverage of the genome. The quality control information of the Illumina sequencing was shown in Supplementary Table S1 and Figure S1. After sequence assembly, the draft genome (5,532,918 bp; with a GC content of 68.1%) of strain HZ01 was obtained, containing 12 scaffolds (Table 1 and Figure 1). The genome contained 5,162 predicted genes with an average length of 990 bp, including 4 rRNA and 54 tRNA genes.
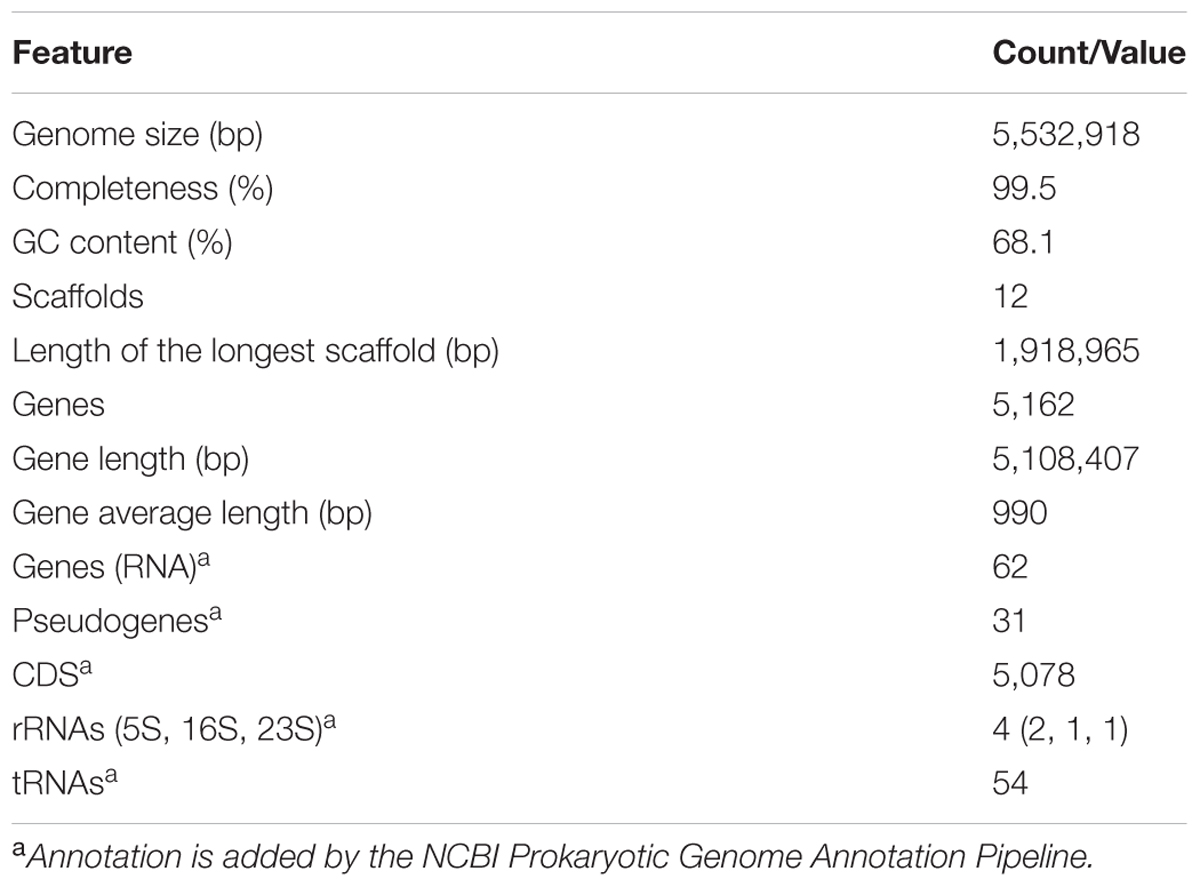
TABLE 1. General features of the Achromobacter sp. HZ01 genome.
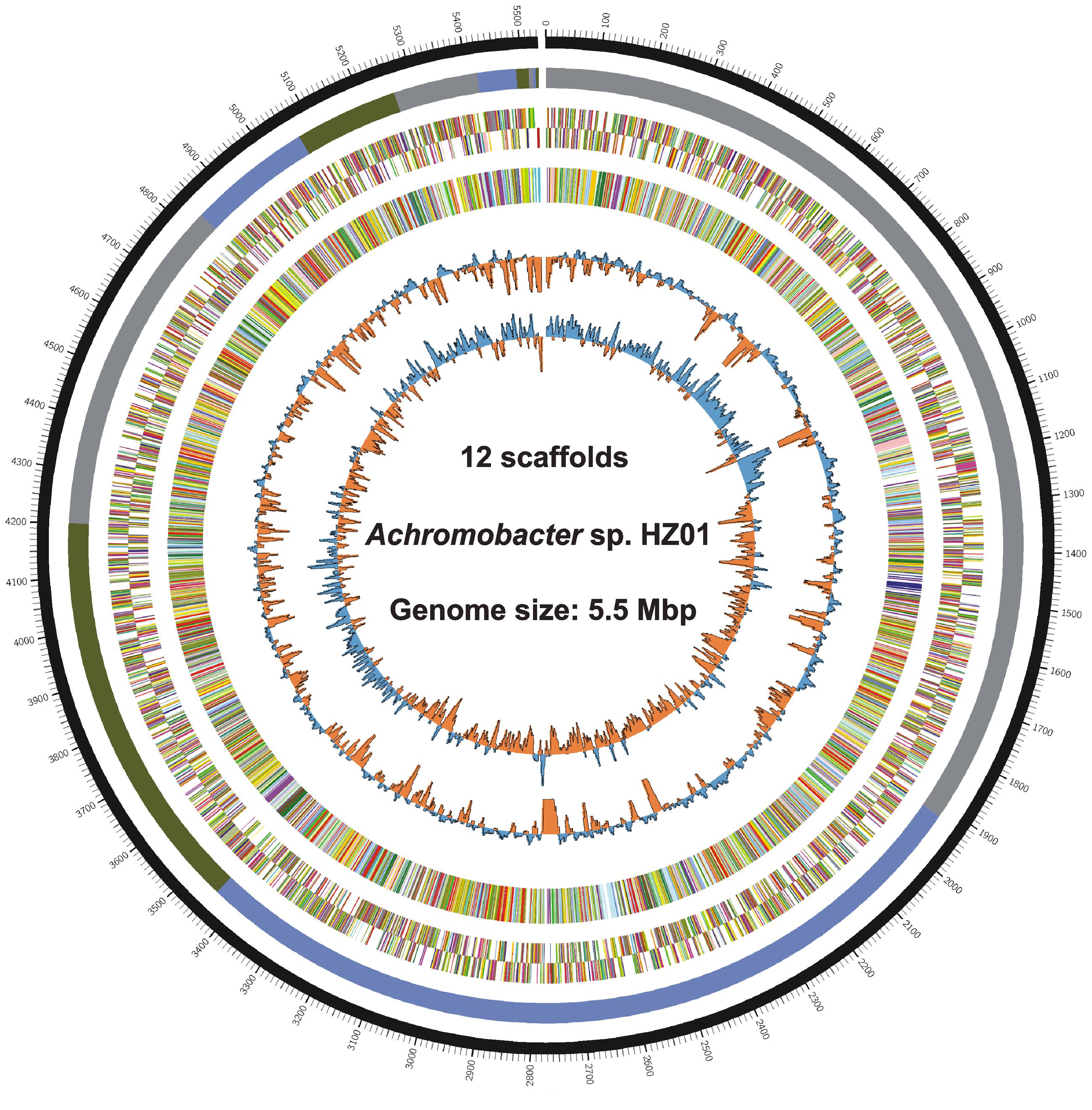
FIGURE 1. Circular chromosome of Achromobacter sp. HZ01. The circular genome map from the outside inward: ring 1, genomic position; ring 2, assembled scaffolds in order of size; rings 3 and 4, predicted genes on the forward (ring 3) and the reverse (ring 4) strands; ring 5, genes assigned to COG categories; ring 6, GC content; ring 7, GC skew. The circular map was generated by Circos v0.65 (Krzywinski et al., 2009).
A total of 5,081 genes were annotated in the NCBI-nr database (Supplementary Table S2). Annotation using the KAAS revealed that more genes were enriched in “amino acid metabolism” (Supplementary Figure S2). More specifically, “ABC transporters” was the most abundant subcategories among the annotated metabolic pathways, followed by “biosynthesis of amino acids,” “two-component system,” and “carbon metabolism” (Supplementary Figure S3). Annotation in the eggNOG database showed that most genes were assigned to “amino acid transport and metabolism,” “general function prediction only,” “transcription,” “function unknown,” and “inorganic ion transport and metabolism” (Supplementary Table S3).
Carbon Source Utilization
It has been demonstrated that strain HZ01 is capable of utilizing NH4NO3 as a nitrogen source and using hydrophobic n-alkanes, anthracene, phenanthrene, and pyrene as the carbon source, respectively (Deng et al., 2014). In this study, NH4NO3 was employed as the sole nitrogen source to investigate the utilization of water-soluble carbon sources. The results showed that active growth was obtained using citric acid or pyruvic acid as the sole carbon source, respectively (Figures 2A,C). When the concentration of carbon sources was set at 40 g/L, the OD600 values (after the time point of 4 h) of the glucose- or lactose-treated group were a little higher than those of other groups, except the citric acid- or pyruvic acid-treated group (Figure 2B). However, when the concentration was set at 20 g/L, none of the carbon sources, other than citric acid and pyruvic acid, could be efficiently utilized (Figures 2C,D). The inefficient utilization of lactose in strain HZ01 is consistent with the property of non-lactose fermenting strains of Achromobacter xylosoxidans (De Baets et al., 2007). The limited carbon source range was similar to the inability of hydrocarbon-degrading Alcanivorax borkumensis SK2 to use common carbohydrates for growth (Schneiker et al., 2006), while Celeribacter indicus P73T is capable of utilizing many sugars (Cao et al., 2015).
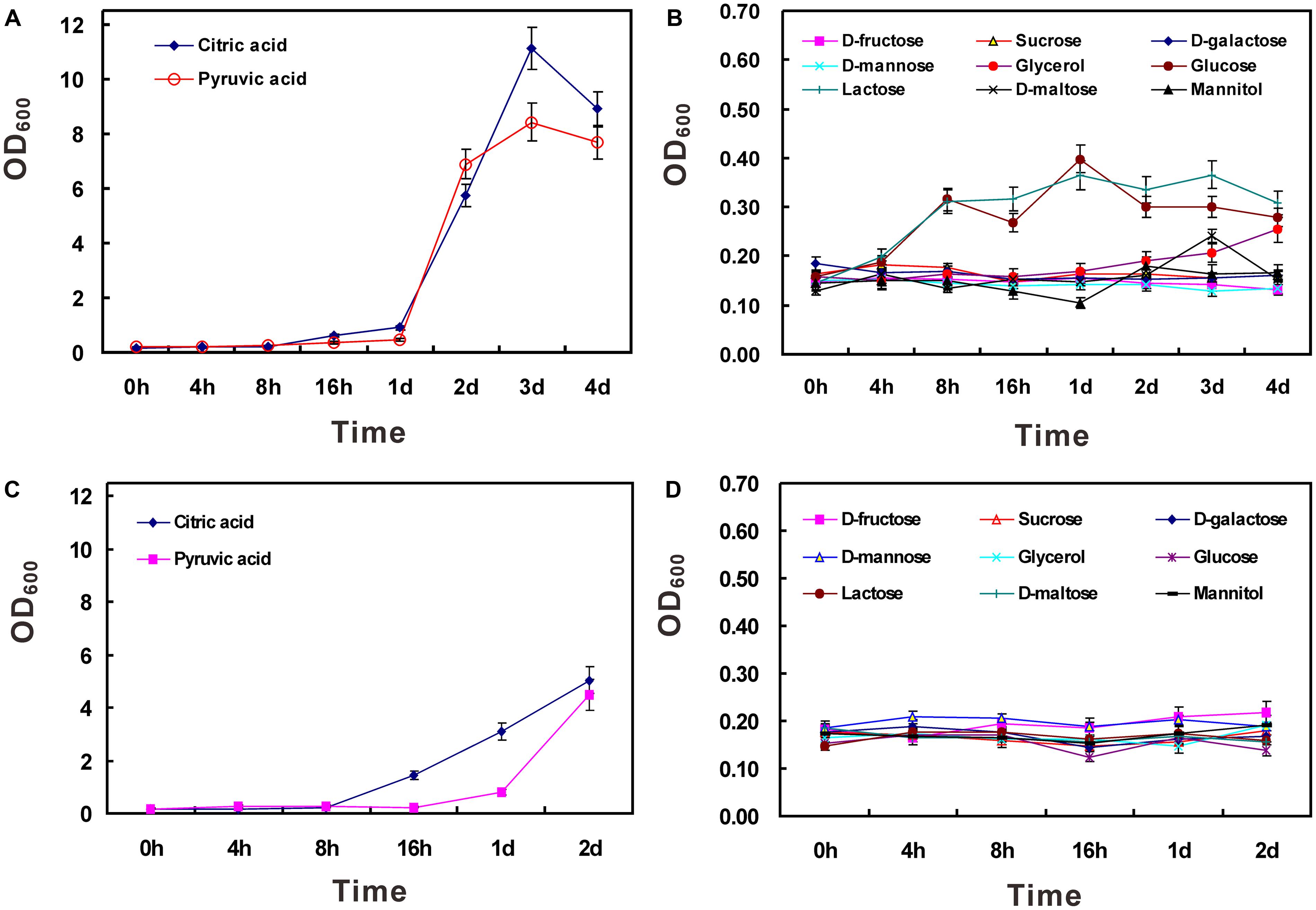
FIGURE 2. Utilization of carbon sources in strain HZ01. The incubation was performed using medium A supplemented with single carbon source at the concentration of 40 (A,B) and 20 g/L (C,D), respectively. Data are presented as mean ± standard deviation (SD) involving triplicate assays. OD600, optical density at 600 nm; h, hour; d, day.
A total of 430 genes in strain HZ01 were assigned to the “carbohydrate metabolism” pathway by KAAS annotation (Supplementary Table S4). Specifically, abundant genes were enriched in “pyruvate metabolism,” which might explain the bacterial efficient utilization of pyruvic acid (Figures 2A,C). An intact citrate cycle (TCA cycle; Supplementary Table S4 and Figure S4) pathway was identified in strain HZ01, which was consistent with its capability to efficiently utilize citric acid (Figures 2A,C). Although most enzymes involved in the “glycolysis/gluconeogenesis” pathway were identified in strain HZ01, genes coding for hexokinase and its homologs were not included in the genome. The hexokinase is a key enzyme of glycolysis and catalyzes the irreversible phosphorylation of glucose, mannose, and fructose. In addition to the lack of hexokinase, fructokinase was also not identified. Thus, fructose cannot be converted to fructose-6-phosphate in strain HZ01. Besides, the gene coding for 6-phosphofructokinase-1, another key enzyme that converted fructose-6-phosphate to 1,6-fructose-biphosphate, was not identified in the genome (Supplementary Figure S5). This feature was similar to that of hydrocarbon-degrading Polymorphum gilvum SL003B-26A1T (Nie et al., 2012). In strain SL003B-26A1T, the lack of 6-phosphofructokinase-1 was complemented by the pentose phosphate (PP) pathway to generate glyceraldehyde-3-phosphate that subsequently involved in the downstream steps of glycolysis. A total of 26 genes in strain HZ01 were annotated as the enzymes involved in the PP pathway (Supplementary Table S4), however, several important enzymes, such as glucose-6-phosphate dehydrogenase and 6-phosphogluconolactonase, were not present in this bacterium (Supplementary Figure S6). Thus, strain HZ01 cannot convert glucose to glyceraldehyde-3-phosphate through the intermediate product 1,6-fructose-biphosphate or via the PP pathway. Several oil-degrading microorganisms, such as Geobacillus thermodenitrificans NG80-2 and Desulfatibacillum alkenivorans AK-01, harbor a comparatively complete glycolysis pathway through the 1,6-fructose-biphosphate (Feng et al., 2007; Callaghan et al., 2012). Hydrocarbon-degrading Celeribacter indicus P73T contains all the enzymes (except glucose-6-phosphatase) needed in glycolysis/gluconeogenesis, as well as all the enzymes required for the PP pathway and the TCA cycle (Cao et al., 2015). These evidences indicate the differences of carbohydrate metabolism among oil-degrading bacteria. Additionally, strain HZ01 lacks lactose permease and β-galactosidase for the metabolism of lactose. Galactose cannot be processed by glycolysis in this strain partially due to its lack of two essential enzymes (galactokinase and galactose-1-phosphate uridylyltransferase), which convert the galactose to glucose-1-phosphate. The absence of beta-fructofuranosidase and mannitol dehydrogenase might explain the inefficient utilization of sucrose and mannitol, respectively.
Genome annotation suggests that carbohydrate transporters in strain HZ01 are relatively rare (Supplementary Table S5), which may be another reason explaining its inability to utilize several common carbohydrates as the sole carbon source for growth (Figure 2). For instance, strain HZ01 lacks FrcB, FrcC, and FrcA for fructose transport. The glucose/arabinose transport system, consisting of GlcS, GlcU, GlcT, and GlcV, are not present in the genome. Among the proteins SmoE, SmoF, SmoG, and SmoK for sorbitol/mannitol transport, SmoG is not contained in strain HZ01. The sucrose-specific IIA component for sucrose uptake is absent. The LacE, LacF, LacG, and LacK for lactose/L-arabinose transport were also not identified. Among the proteins MalE, MalF, MalG, and MalK for maltose/maltodextrin transport, only MalK was identified (Supplementary Table S5).
In summary, lacking some important enzymes and carbohydrate transporters in strain HZ01 is in accord with its inability to efficiently utilize several common carbohydrates for growth, such as glucose, fructose, mannose, galactose, lactose, sucrose, and mannitol (Figure 2). The inefficient utilization of these compounds is consistent with the low carbohydrate availability in the marine environment where strain HZ01 was isolated (Nie et al., 2012; Deng et al., 2014). This genome feature may result from the long-term evolution for adaptation to its environment. Although strain HZ01 is not capable of utilizing some common carbohydrates, it can use hydrocarbons as carbon sources (Deng et al., 2014).
Genetic Basis for Petroleum Degradation
Emulsification of Biosurfactants
Biosurfactants, which are mainly derived from the secondary metabolites of microorganisms (Nie et al., 2012), contain hydrophilic and hydrophobic groups in their structure. They harbor surface activities and emulsification activities that increase the bioavailability and degradation rates of hydrophobic substrates, including hydrocarbons (Desai and Banat, 1997). They are a structurally diverse class of compounds, with glycolipids and lipopeptides reported as two primary isolated families. So far, researches regarding the effects of biosurfactants on the bioremediation properties of Achromobacter strains are scarce. It has been demonstrated that surfactants SDS, Tween 60 and Tween 80 (derived from chemical processes) can enhance the detoxification of chromium-containing slag by Achromobacter sp. CH-1 (Chai et al., 2009).
Strain HZ01 is capable of producing biosurfactants that efficiently emulsify diverse hydrophobic compounds, exhibiting the potential for bioremediation (Deng et al., 2016). It has been demonstrated that citric acid is related to biosurfactant production (Pirog et al., 2008). In this work, the emulsification activities of the culture broth using citric acid as the sole carbon source were investigated. Although the fermentation broth was not concentrated, stable emulsions were formed using soybean oil, coconut oil, diesel oil, kerosene, and hexane as emulsion substrates, respectively (Figure 3), indicating that biosurfactants might exist in the fermentation broth. Further studies are needed to confirm the biosurfactant production in strain HZ01 using citric acid as the sole carbon source. Diesel oil and kerosene, both derived from petroleum, contain diverse hydrocarbons. The emulsification of diesel oil, kerosene, and hexane suggests that the fermentation products produced by strain HZ01 may promote its utilization of hydrocarbons.

FIGURE 3. Emulsification activity of the fermentation broth using citric acid as the sole source of carbon and energy. The cell-free supernatant of the fermentation broth was used in the experiments. SDS at the concentration of 0.5 g/L was used as a positive control. -, fermentation medium without bacteria inoculation incubated for 3 days; +, incubation for 3 days with bacteria inoculation.
Genome annotation revealed that a total of 197 genes were assigned to “secondary metabolites biosynthesis, transport, and catabolism” (Supplementary Table S6). The genome harbors candidate genes associated with biosurfactant production, such as 3-oxoacyl-ACP reductase, acyltransferase, phosphomannomutase, and glycosyltransferase (Supplementary Table S7), which are essential for glycolipid synthesis (Maier and Soberon-Chavez, 2000). Eleven genes were annotated as LuxR family transcriptional regulators that play an important role in the regulation of glycolipid synthesis (Satpute et al., 2010). Genes coding for cell envelope biogenesis protein OmpA (gene_2469) and outer membrane protein OmpAb (gene_3528) were identified. OmpA is an active constituent of alasan (a bioemulsifier) and has been demonstrated to be involved in the degradation of hydrophobic compounds, including hydrocarbons (Walzer et al., 2006). OmpH (gene_1336), which is possibly related to bioemulsifier production (Schneiker et al., 2006), is also present in strain HZ01. Generally, lipopeptides are synthesized through the ribosome-independent pathway using non-ribosomal peptide synthetases (NRPSs) (Roongsawang et al., 2010). One gene (gene_1007) was assigned to “non-ribosomal peptide structures.” Additionally, thioesterases and peptide synthetases, both related to lipopeptide biosynthesis (Yao et al., 2003; Moyne et al., 2004), were also identified (Supplementary Table S7). Strain HZ01 was previously reported to produce a cyclic lipopeptide containing the peptide chain of Gly-Gly-Leu-Met-Leu-Leu linked to a C21 fatty acid chain (Deng et al., 2016). We infer that the biosynthetic pathway of this biosurfactant includes three major processes (Supplementary Figure S7), i.e., (a) a 3-hydroxy-heneicosanoic acid is derived from specific carbon sources via fatty acid biosynthesis; (b) the hexapeptide is obtained by a series of enzymatic condensations from the N-terminal of Leu to the C-terminal of Gly; and (c) the 3-hydroxy-heneicosanoic acid may undergo an enzymatic condensation process, being incorporated at the C- and N-terminals of the hexapeptide to produce a cyclic lipopeptide. Further studies are needed to clarify this biosynthetic pathway.
In summary, the identified genes provide a genetic basis for biosurfactant production in strain HZ01, which may facilitate its uptake and degradation of petroleum hydrocarbons.
Degradation Pathways of n-Alkanes
Petroleum is a complex mixture primarily containing saturated hydrocarbons, PAHs and asphaltenes, of which alkanes are its major constituents (Rojo, 2009). Strain HZ01 is capable of degrading C12–C27 n-alkanes and PAHs (Deng et al., 2014). Its genome contains numerous genes for hydrocarbon degradation. Methyl-accepting chemotaxis proteins (MCP; gene_441, gene_1384, gene_1386, gene_1387, and gene_2249), related to alkane chemotaxis (Smits et al., 2003), may be beneficial for strain HZ01 to seek hydrocarbons as substrates. Cytochrome o ubiquinol oxidase (Cyo; gene_2136, gene_2137, gene_2138, gene_2181, gene_2182, gene_2183, and gene_2184) exhibits a negative regulation over the expression of alkane degradation genes, and there is a marked correlation between the Cyo levels and the repression extents of alkane degradation pathway (Dinamarca et al., 2003). Transcriptome sequencing showed that the transcriptional level of Cyo in strain HZ01 was not affected during petroleum degradation (Hong et al., 2016). The Cyo is not reported in hydrocarbon-degrading Alcanivorax borkumensis SK2 (Schneiker et al., 2006), Oleispira antarctica RB-8 (Kube et al., 2013), Geobacillus thermodenitrificans NG80-2 (Feng et al., 2007), Polymorphum gilvum SL003B-26A1T (Nie et al., 2012), or Desulfatibacillum alkenivorans AK-01 (Callaghan et al., 2012).
n-Alkanes are usually subjected to degradation via the terminal or subterminal oxidation pathway (Rojo, 2009). In the terminal oxidation pathway, alkane degradation is initiated by oxygenation of a terminal methyl group to generate a primary alcohol, which is converted into its corresponding aldehyde and is further oxidized to a fatty acid. Subsequently, the fatty acid combines with CoA and is subjected to β-oxidation to produce acetyl-CoA. The acetyl-CoA is further processed by the TCA cycle to finally generate H2O, CO2, and energy. Diverse enzymes involved in this process were identified in strain HZ01, such as oxygenases, hydroxylases, alcohol dehydrogenases, aldehyde dehydrogenases, and acyl-CoA synthetases (Supplementary Table S8). Representative alkane hydroxylase AlkB and its homologs that initiated the oxidation of medium-chain alkanes were not identified in the genome, which was consistent with the genome features of oil-degrading Geobacillus thermodenitrificans NG80-2 (Feng et al., 2007) and Polymorphum gilvum SL003B-26A1T (Nie et al., 2012). The lack of AlkB and its homologs in hydrocarbon-degrading marine bacteria is not uncommon (Venter et al., 2004). Some bacteria use the enzymes of cytochrome P450 family to perform similar functions as that of AlkB and its homologs (Rojo, 2009). One gene coding for cytochrome P450 (gene_1029) in strain HZ01 was identified. Its sequence showed an identity of 60% to the sequence of cytochrome P450 monooxygenase in Pseudomonas sp. Lz4W (GenBank accession no. EMI05095.1), suggesting that the cytochrome P450 of strain HZ01 may have similar functions as that of strain Lz4W. RNA-seq demonstrated that two transcripts of cytochrome P450 were up-regulated after petroleum treatment (Hong et al., 2016). Additionally, ferredoxin and ferredoxin reductase, required for the electron transfer from NAD(P)H to cytochrome P450, were also present in the genome (Supplementary Table S8), contributing to the alkane oxidation. Thus, the cytochrome P450 may play an important role in the initial oxidation of n-alkanes in strain HZ01. Comparative analysis revealed that, although strains NG80-2 and SL003B-26A1T lacked AlkB and its homologs, long-chain alkane monooxygenase LadA was included in their genomes for alkane degradation (Feng et al., 2007; Nie et al., 2012). LadA belongs to the bacterial luciferase family and plays an important role in the initial oxidation of C15–C36 n-alkanes (Feng et al., 2007). On the contrary, Alcanivorax borkumensis SK2 and Oleispira antarctica RB-8 contained AlkB and its homologs, as well as cytochrome P450, whereas LadA was not identified (Schneiker et al., 2006; Kube et al., 2013). LadA was also not identified in strain HZ01 by genome sequencing. Nevertheless, RNA-seq showed that strain HZ01 contained one transcript coding for luciferase-like monooxygenase family protein 1 (Hong et al., 2016), which might have the functions similar to those of LadA.
Fatty acids are the major intermediate products of alkane degradation and are processed by β-oxidation to generate acetyl-CoA (Rojo, 2009). Besides acyl-CoA synthetase, which catalyzes the conjugation of fatty acid to CoA, fatty acid hydroxylase (gene_696, gene_1843, and gene_2856) is also contained in strain HZ01. The fatty acid hydroxylase catalyzes the hydroxylation of fatty acids, generating hydroxy fatty acids. Subsequently, the hydroxy fatty acids are converted into dicarboxylic acids, followed by β-oxidation (Coon, 2005). A total of 40 genes in strain HZ01 were assigned to the “fatty acid degradation” pathway, including the genes related to β-oxidation (Supplementary Table S9 and Figure S8). RNA-seq demonstrated that the genes involved in β-oxidation were activated after petroleum treatment (Hong et al., 2016). In addition, an intact TCA cycle pathway for processing the resulting acetyl-CoA is present in strain HZ01 (Supplementary Figure S4).
A total of 35 genes were assigned to the “methane metabolism” pathway (Supplementary Table S10). For instance, strain HZ01 contained fdoG, fdfH, fdoH, and fdoI coding for formate dehydrogenase subunits. However, some enzymes essential for methane oxidation were not identified, including methane monooxygenase, methanol dehydrogenase, and formaldehyde dehydrogenase. Strain HZ01 contains esterases, but the Baeyer-Villiger monooxygenase, an essential enzyme involved in the cycloalkane degradation and the subterminal oxidation of n-alkanes, was not included in the genome. The identified genes suggest that (i) subterminal oxidation pathway is not present in strain HZ01; (ii) this bacterium cannot perform methane oxidation or cycloalkane degradation; and that (iii) the degradation of n-alkanes in strain HZ01 is performed via the terminal oxidation pathway (Figure 4). The results of genome sequencing further verify the pathway for n-alkane degradation predicted by RNA-seq (Hong et al., 2016). The metabolic reaction initiated by fatty acid hydroxylase (Figure 4) was not included in the previous predicted pathway. Besides, RNA-seq showed that the following steps of terminal oxidation pathway were activated after strain HZ01 was treated with petroleum for 16 h: (i) the initial oxidation of n-alkanes; (ii) reactions catalyzed by dehydrogenases and acyl-CoA synthases; and (iii) fatty acid β-oxidation (Hong et al., 2016).
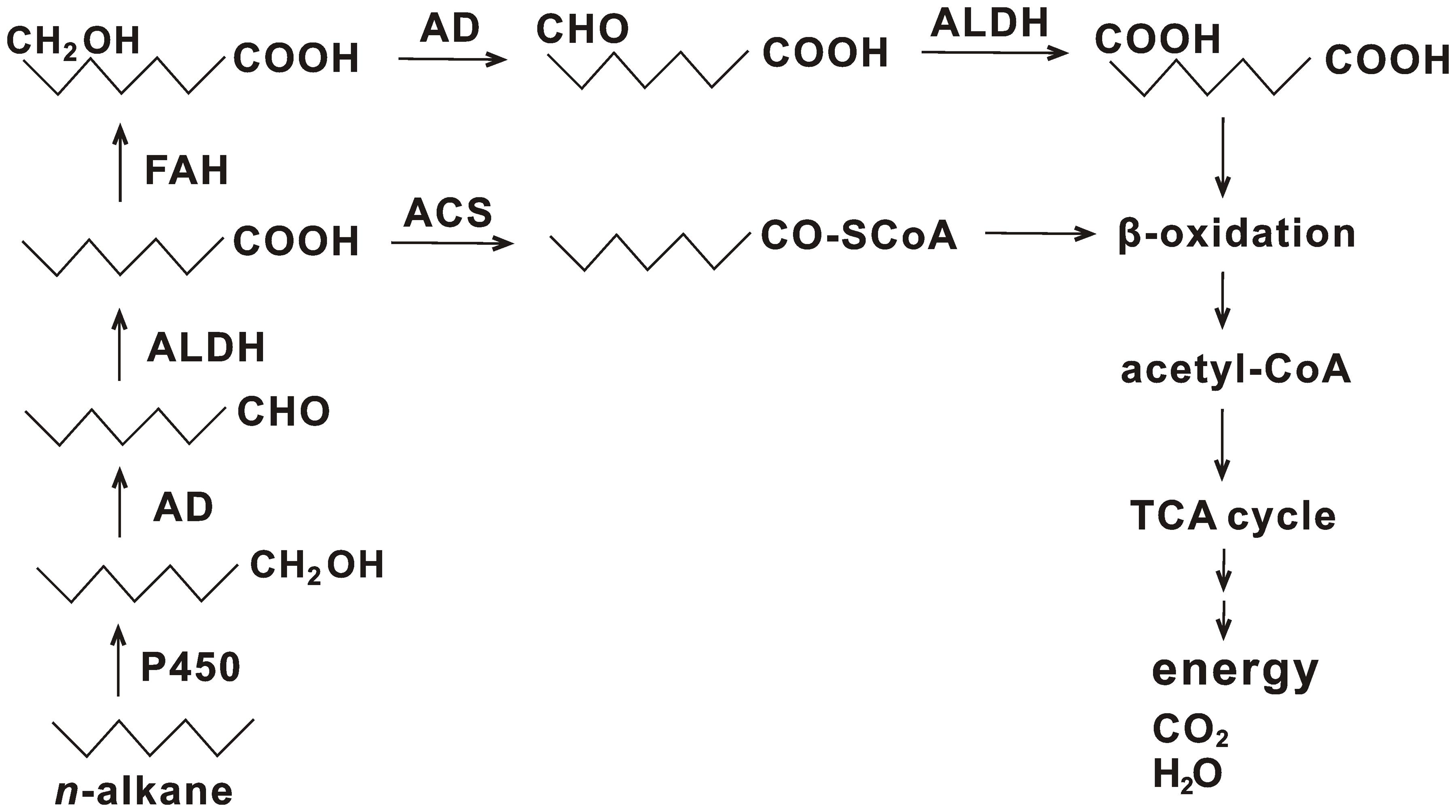
FIGURE 4. Terminal oxidation pathway for n-alkane degradation in Achromobacter sp. HZ01. P450, cytochrome P450; AD, alcohol dehydrogenase; ALDH, aldehyde dehydrogenase; ACS, acyl-CoA synthetase; FAH, fatty acid hydroxylase; TCA cycle, citrate cycle.
Degradation Pathways of Aromatic Compounds
In the previous RNA-seq, some transcripts related to the degradation of aromatic compounds were identified in strain HZ01 (Hong et al., 2016). Nevertheless, the potential pathways for degradation of aromatic compounds in this bacterium have not been revealed so far.
Genome sequencing of this study showed that abundant genes related to the degradation of aromatic compounds were contained in strain HZ01. For instance, a total of 28 genes were assigned to “degradation of aromatic compounds” (Supplementary Table S11). During the degradation of aromatic compounds, reactive dihydroxylated intermediates, such as catechol and protocatechuate, will be produced. These intermediates are further degraded via the intradiol (ortho) or extradiol (meta) ring cleavage (Strachan et al., 1998). The resulting products of the ortho-pathway (acetyl-CoA and succinyl-CoA) and the meta-pathway (pyruvate and acetaldehyde) are further processed by the TCA cycle (Pessione et al., 1999).
The homogentisate pathway, the protocatechuate and catechol branches of the β-ketoadipate pathway, and the phenylacetate pathway are four common pathways for the degradation of aromatic compounds (Jimenez et al., 2002). The genome sequencing of this work showed that only PhaA and PhaB, both belonging to the enoyl-CoA hydratase/isomerase family proteins, were identified in the phenylacetate pathway (Figure 5).
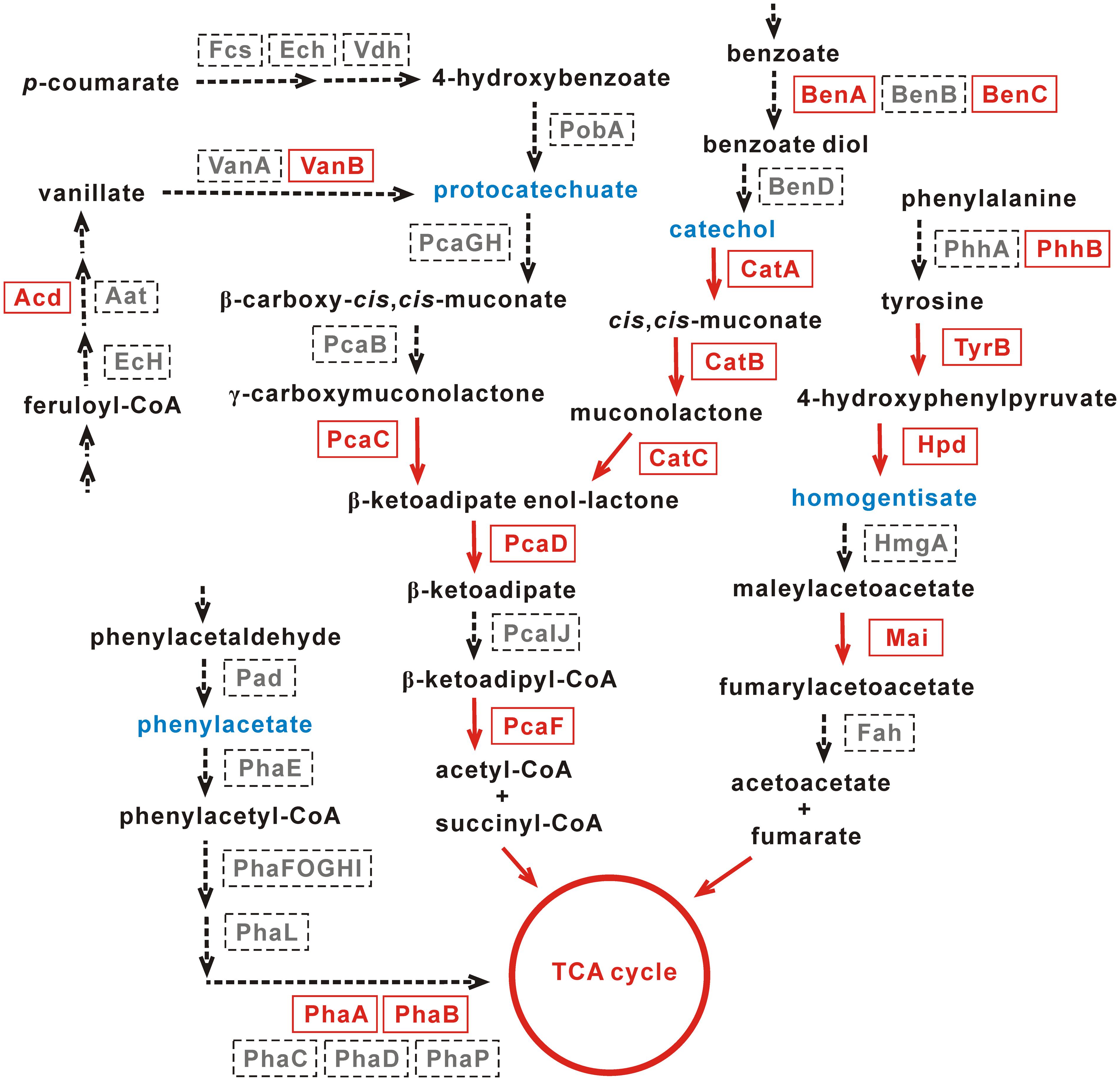
FIGURE 5. Pathways for the degradation of aromatic compounds in Achromobacter sp. HZ01. The enzymes present and absent in strain HZ01 are shown in red fonts and gray fonts, respectively. The red arrows indicate that strain HZ01 contains corresponding metabolic steps. The dotted arrows show the lacking steps in strain HZ01. Blue fonts represent the key intermediate products. Not all the upper-stream steps are shown in the figure.
Protocatechuate and catechol are representative intermediates of the protocatechuate and catechol branches of the β-ketoadipate pathway, respectively, and these two intermediates are generally produced during the degradation of aromatic compounds. For instance, the catechol intermediate may be generated from benzoate, benzamide, (chloro)-biphenyl, benzonitrile, benzaldehyde, salicylate, and mandelate (Chain et al., 2006). Both the protocatechuate and catechol branches will produce β-ketoadipate enol-lactone, which is sequentially processed by PcaD (3-oxoadipate enol-lactonase; gene_4113), PcaIJ (not identified) and PcaF (beta-ketoadipyl CoA thiolase; gene_443). Then, acetyl-CoA and succinyl-CoA are generated and further metabolized by TCA cycle (Jimenez et al., 2002). PcaC (4-carboxymuconolactone decarboxylase; gene_4173) catalyzed the production of β-ketoadipate enol-lactone from γ-carboxymuconolactone and was identified in the protocatechuate branch (Figure 5). Nevertheless, PcaGH and PcaB were not identified. In the upper-stream of the protocatechuate branch, Acd (acyl-CoA dehydrogenase) and VanB (vanillate monooxygenase ferredoxin subunit) are present in strain HZ01. Acd and VanB play important roles in the production of vanillate from feruloyl-CoA and protocatechuate from vanillate, respectively.
So far, some Achromobacter strains capable of degrading aromatic compounds have been reported. Catechol dioxygenases play an important role in the degradation of aromatic compounds (Moon et al., 1997). Similar to strain HZ01, Achromobacter sp. DMS1 and Achromobacter xylosoxidans DN002 contain catechol 1,2-dioxygenase (Supplementary Table S12). Achromobacter xylosoxidans KF701, strain DN002 and Achromobacter xylosoxidans T7 contain catechol 2,3-dioxygenase, which is not found in strain HZ01. The catechol branch of the β-ketoadipate pathway is comparatively intact than the protocatechuate branch in strain HZ01. CatA (catechol 1,2-dioxygenase; gene_376) performs the intradiol cleavage of catechol and some of its derivatives. This enzyme also catalyzes the extradiol cleavage of meta-substituted substrates, such as 3-methoxycatechol and 3-methylcatechol (Strachan et al., 1998). Within the catechol branch, CatA transforms catechol into cis,cis-muconate, which is processed by CatB (muconate cycloisomerase; gene_772) to generate muconolactone (Figure 5). Then, CatC (muconolactone D-isomerase; gene_4111) catalyzes muconolactone to produce β-ketoadipate enol-lactone. In the upper-stream of catechol, BenA (benzoate/toluate 1,2-dioxygenase subunit alpha; gene_3355 and gene_3376) and BenC (benzoate/toluate 1,2-dioxygenase reductase component; gene_2862) were identified, but BenB and BenD were not present. BenABC can transform benzoate into benzoate diol, which is further metabolized by BenD to generate catechol (Figure 5).
Regarding the homogentisate pathway, only Mai (maleylacetoacetate isomerase; gene_2657 and gene_5130) was identified in the downstream of homogentisate. This enzyme catalyzes maleylacetoacetate to generate fumarylacetoacetate (Figure 5). In the upper-stream of homogentisate, PhhB (pterin-4-alpha-carbinolamine dehydratase; gene_3287), TyrB (aromatic amino acid aminotransferase; gene_533) and Hpd (4-hydroxyphenylpyruvate dioxygenase; gene_3903) were identified, but PhhA was not found. PhhA and PhhB play an important role in the transformation of phenylalanine into tyrosine (Jimenez et al., 2002). Then, the tyrosine is catalyzed by TyrB to generate 4-hydroxyphenylpyruvate, which is transformed into homogentisate by Hpd (Figure 5).
Additionally, the genes coding for benzoate/toluate 1,2-dioxygenase subunit alpha (gene_3355 and gene_3376) and benzoate/toluate 1,2-dioxygenase reductase component (gene_2862) were identified in strain HZ01. RNA-seq showed that the expression levels of benzoate 1,2-dioxygenase subunit alpha and benzoate 1,2-dioxygenase large subunit were not affected by petroleum treatment (Hong et al., 2016). The benzoate 1,2-dioxygenase system performs its functions via catalyzing the double hydroxylation of benzoates with the incorporation of two oxygen atoms (Yamaguchi and Fujisawa, 1980). Salicylate hydroxylase (gene_2661), a key enzyme in naphthalene catabolism, catalyzes the decarboxylation and hydroxylation of salicylate, generating catechol, H2O, and CO2 (You et al., 1991). Glutathione S-transferase (GST), related to PAHs degradation (Xia and Min, 2003), were also identified in strain HZ01 (Supplementary Table S8). Laccases (gene_3832), a family of blue multicopper oxidases, play an important role in the oxidation of numerous aromatic compounds (Fang et al., 2011). In addition, 150 genes in strain HZ01 were assigned to “xenobiotics biodegradation and metabolism” using KAAS annotation (Supplementary Table S13), such as genes related to the degradation of fluorobenzoate, toluene, xylene, nitrotoluene, and dioxin. Several genes encoding GntR family transcriptional regulators and MarR family transcriptional regulators related to aromatic compound degradation were also identified.
Strain HZ01 has been reported to degrade anthracene, phenanthrene, and pyrene (Deng et al., 2014). The genome sequencing in this study further suggests that strain HZ01 has the potential for bioremediation of aromatic-compounds-contaminated environments. Since the enzymes involved in the catechol pathway are comparatively complete, this pathway may play a major role in the degradation of aromatic compounds in strain HZ01. Further studies are needed to elucidate the aromatic compounds degradation in this bacterium.
Unsaturated fatty acids are related to the fluidity of cell membrane and contribute to the adaptation of bacteria to low-temperature environments and the low solubility of substrates (including hydrocarbons). A total of 12 genes in the genome of strain HZ01 were assigned to “biosynthesis of unsaturated fatty acids.” For instance, gene_4447 encodes fatty acid desaturase, which is existent in almost all organisms and is a key enzyme in the biosynthesis of fatty acids (Alonso et al., 2003). These genes play important roles in the fatty acid synthesis in strain HZ01 and may enhance its capability for hydrocarbon degradation. Other candidate genes associated with the degradation of petroleum components were shown in Supplementary Table S8, mainly including the genes coding for dehydrogenases, rubredoxins, oxidoreductases, monooxygenases, and dioxygenases. In summary, the identified genes and pathways provide a genetic basis for petroleum degradation in strain HZ01.
Besides, the analysis contents of this study also include (i) genomic comparison, (ii) amino acid metabolism, (iii) nitrogen metabolism, (iv) biosynthesis of other secondary metabolites, (v) antibiotic resistance, (vi) two-component systems, (vii) cell motility, and (viii) membrane transport (Supplementary Results and Discussion). According to the genes identified by genome sequencing, the material transport and metabolic pathways in Achromobacter sp. HZ01 were depicted in Figure 6.
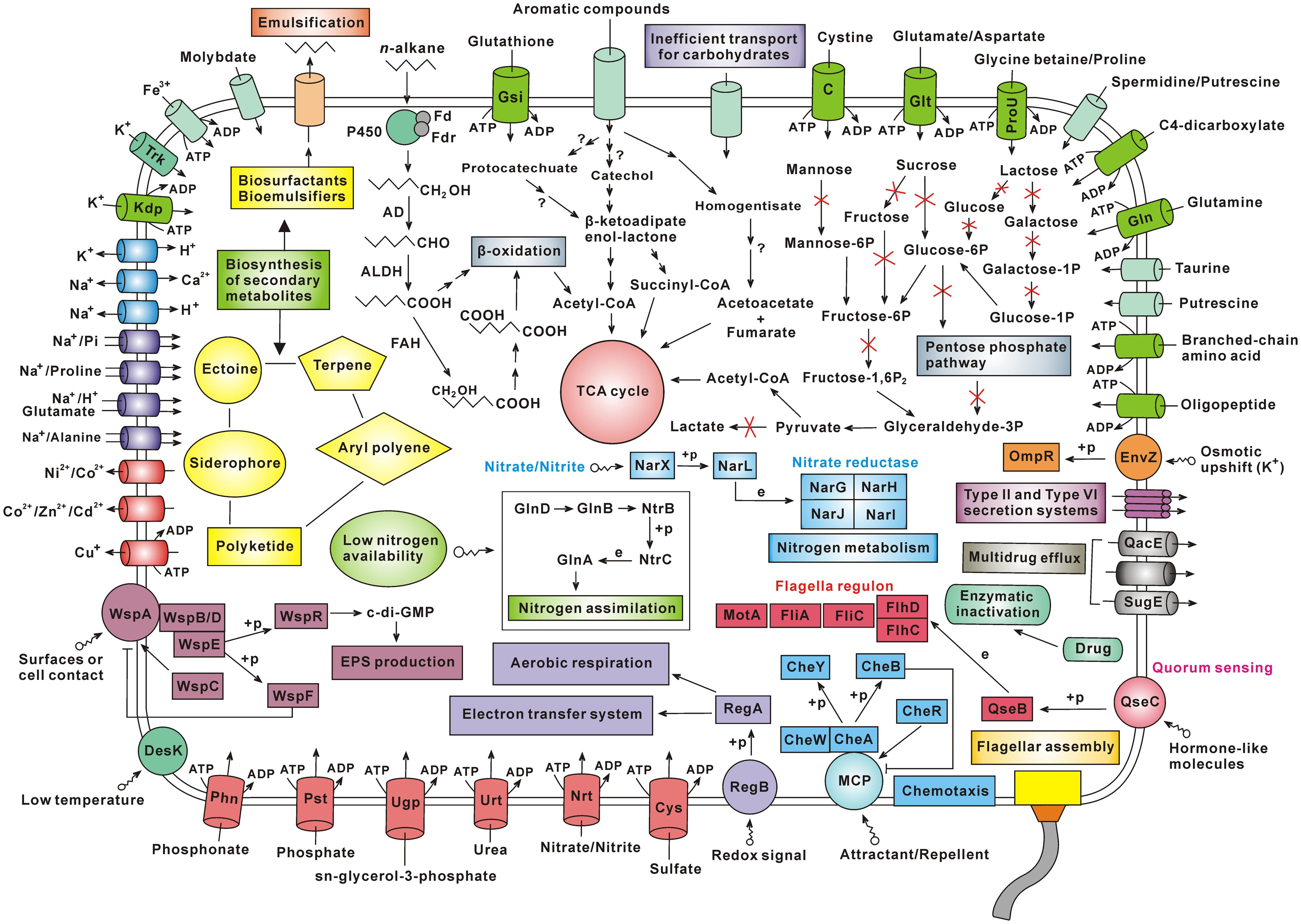
FIGURE 6. Schematic overview of the metabolism and material transport in Achromobacter sp. HZ01. Primary information: (i) the n-alkane degradation in strain HZ01 is performed via the terminal oxidation pathway, and the catechol pathway may play a major role in the degradation of aromatic compounds; (ii) the inefficient carbohydrate transport and the lack of some key enzymes account for the bacterial inability to utilize several common carbohydrates for growth; (iii) strain HZ01 harbors the genes related to biosynthesis of secondary metabolites; (iv) the membrane transporters are essential for nutrient uptake and substance export; (v) strain HZ01 contains important two-component systems for the responses to environmental changes; (vi) the chemotaxis and flagellar assembly are beneficial for pursuing nutrients and avoiding environmental damages; (vii) besides through the enzymatic inactivation of antibiotics, strain HZ01 contains efflux pumps for elimination of antibiotics. Not all the metabolic pathways and transporters are shown in the figure. Import or export of solutes is indicated by the direction of the arrow through the transporter. The arrow with a red X shape indicates that the metabolic step is absent. The arrow with a question mark indicates that it is unsure whether the corresponding step can be completed. Abbreviations: Fd, ferredoxin; Fdr, ferredoxin reductase; P450, cytochrome P450; AD, alcohol dehydrogenase; ALDH, aldehyde dehydrogenase; FAH, fatty acid hydroxylase; +p, phosphorylation; e, expression; Trk, potassium transporter Trk; Kdp, Kdp system for potassium transport; EPS, extracellular polysaccharide; Phn, phosphonate transport system; Pst, phosphate transport system; Ugp, sn-glycerol-3-phosphate transport system; Urt, urea transport system; Nrt, nitrate/nitrite transport system; Cys, sulfate transport system; MCP, methyl-accepting chemotaxis protein; Gln, glutamine transport system; ProU, glycine betaine/proline transport system; Glt, glutamate/aspartate transport system; C, cystine transport system; Gsi, glutathione transport system.
Discussion
It has been demonstrated that Achromobacter sp. HZ01 is capable of degrading petroleum hydrocarbons and adapting to a wide range of salinity (Deng et al., 2014). The genome analysis of this study verifies its genetic basis for these properties (see the results above and Supplementary Results), which further indicates that strain HZ01 has a good potential for bioremediation of crude oil-polluted marine environments. Achromobacter strains are representative bacteria with bioremediation potential (Supplementary Table S12), and some of their degradation properties have been analyzed (Furukawa et al., 1989; Cai et al., 2009). Among the reported Achromobacter strains, Achromobacter xylosoxidans XL and Achromobacter sp. 4(2010) have some similarities with strain HZ01 in alkane degradation. Strain XL is capable of degrading C12–C23 and C27–C43 alkanes (Si et al., 2011), and strain 4(2010) can degrade diesel oil (Kaczorek et al., 2013), an oil mixture containing diverse hydrocarbons. However, their pathways for alkane degradation have not been determined. To our knowledge, genome-wide researches regarding the bioremediation properties of Achromobacter strains are very rare (Supplementary Table S12). Our work is the first genome analysis systematically elucidating the pathways of hydrocarbon degradation in bacterial strains belonging to the genus Achromobacter. The obtained data expand our insights into the bioremediation properties of Achromobacter strains. It also provides valuable genome information for developing a cost-effective and eco-friendly method for bioremediation. For instance, the genome sequencing indicates that strain HZ01 lacks long-chain alkane monooxygenase LadA, which is related to the degradation of C15–C36 alkanes (Feng et al., 2007). Although strain HZ01 is capable of degrading C12–C27 n-alkanes (Deng et al., 2014), cloning the LadA into this bacterium may enhance its capability for n-alkane degradation. In other words, based on comprehensively understanding the genetic background of strain HZ01, gene engineering technique may be employed to improve its hereditary features, which will undoubtedly enhance its possibility for practical applications in bioremediation.
In summary, the genome sequencing and genome-based functional analysis of Achromobacter sp. HZ01 provide us with deep insights into its genetic basis for (i) major metabolisms, (ii) petroleum degradation, (iii) biosynthesis of secondary metabolites, (iv) antibiotic resistance, (v) bacterial responses to environmental changes, (vi) cell motility, and (vii) material transport and secretion. The obtained genome data contribute to developing rational strategies for bioremediation of petroleum-polluted marine environments. The results also provide a valuable reference for the genomics, transcriptomics and proteomics of other microorganisms.
Author Contributions
Y-HH, J-HW, and HD designed the experiments. Y-HH, C-CY, and Q-ZZ performed the experiments. Y-HH, J-HW, HD, X-YW, J-PY, and JP interpreted the experimental data. Y-HH and J-HW wrote the manuscript. Y-HH, J-HW, and HD revised the manuscript.
Funding
This work was jointly supported by the funds of Guangdong Research and Construction of Public Service Abilities (No. 2014B020204004 and 2017B020218004), the National Basic Research Program of China (973 program) (No. 2012CB956004), and the Research Fund Program of Guangdong Provincial Key Laboratory of Marine Resources and Coastal Engineering.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01507/full#supplementary-material
Footnotes
References
Alonso, D. L., Garcia-Maroto, F., Rodriguez-Ruiz, J., Garrido, J. A., and Vilches, M. A. (2003). Evolution of the membrane-bound fatty acid desaturases. Biochem. Syst. Ecol. 31, 1111–1124. doi: 10.1016/s0305-1978(03)00041-3
Besemer, J., Lomsadze, A., and Borodovsky, M. (2001). GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29, 2607–2618. doi: 10.1093/nar/29.12.2607
Cai, L., Rensing, C., Li, X., and Wang, G. (2009). Novel gene clusters involved in arsenite oxidation and resistance in two arsenite oxidizers: Achromobacter sp. SY8 and Pseudomonas sp. TS44. Appl. Microbiol. Biotechnol. 83, 715–725. doi: 10.1007/s00253-009-1929-4
Callaghan, A. V., Morris, B. E., Pereira, I. A., McInerney, M. J., Austin, R. N., Groves, J. T., et al. (2012). The genome sequence of Desulfatibacillum alkenivorans AK-01: a blueprint for anaerobic alkane oxidation. Environ. Microbiol. 14, 101–113. doi: 10.1111/j.1462-2920.2011.02516.x
Cao, J., Lai, Q., Yuan, J., and Shao, Z. (2015). Genomic and metabolic analysis of fluoranthene degradation pathway in Celeribacter indicus P73T. Sci. Rep. 5:7741. doi: 10.1038/srep07741
Chai, L., Chen, L., Huang, Y., Shu, Y., and Su, Y. (2009). Influence of surfactants on detoxification of chromium-containing slag by Achromobacter sp. CH-1. J. Cent. South Univ. Sci. Technol. 40, 41–47.
Chai, L., Wang, Y., Yang, Z., Wang, Q., and Wang, H. (2010). Detoxification of chromium-containing slag by Achromobacter sp. CH-1 and selective recovery of chromium. Trans. Nonferr. Metal Soc. China 20, 1500–1504. doi: 10.1016/s1003-6326(09)60328-9
Chain, P. S., Denef, V. J., Konstantinidis, K. T., Vergez, L. M., Agullo, L., Reyes, V. L., et al. (2006). Burkholderia xenovorans LB400 harbors a multi-replicon, 9.73-Mbp genome shaped for versatility. Proc. Natl. Acad. Sci. U.S.A. 103, 15280–15287. doi: 10.1073/pnas.0606924103
Cockerill, F., Patel, J., Alder, J., Bradford, P., Dudley, M., and Eliopoulos, G. (2013). Performance Standards for Antimicrobial Susceptibility Testing: Twenty-Third Informational Supplement; M100-S23. Wayne, PA: CLSI.
Coon, M. J. (2005). Omega oxygenases: nonheme-iron enzymes and P450 cytochromes. Biochem. Biophys. Res. Commun. 338, 378–385. doi: 10.1016/j.bbrc.2005.08.169
De Baets, F., Schelstraete, P., Van Daele, S., Haerynck, F., and Vaneechoutte, M. (2007). Achromobacter xylosoxidans in cystic fibrosis: prevalence and clinical relevance. J. Cyst. Fibros. 6, 75–78. doi: 10.1016/j.jcf.2006.05.011
Deng, M. C., Li, J., Hong, Y. H., Xu, X. M., Chen, W. X., Yuan, J. P., et al. (2016). Characterization of a novel biosurfactant produced by marine hydrocarbon-degrading bacterium Achromobacter sp. HZ01. J. Appl. Microbiol. 120, 889–899. doi: 10.1111/jam.13065
Deng, M. C., Li, J., Liang, F. R., Yi, M., Xu, X. M., Yuan, J. P., et al. (2014). Isolation and characterization of a novel hydrocarbon-degrading bacterium Achromobacter sp. HZ01 from the crude oil-contaminated seawater at the Daya Bay, southern China. Mar. Pollut. Bull. 83, 79–86. doi: 10.1016/j.marpolbul.2014.04.018
Desai, J. D., and Banat, I. M. (1997). Microbial production of surfactants and their commercial potential. Microbiol. Mol. Biol. Rev. 61, 47–64.
Dinamarca, M. A., Aranda-Olmedo, I., Puyet, A., and Rojo, F. (2003). Expression of the Pseudomonas putida OCT plasmid alkane degradation pathway is modulated by two different global control signals: evidence from continuous cultures. J. Bacteriol. 185, 4772–4778. doi: 10.1128/JB.185.16.4772-4778.2003
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Fang, Z., Li, T., Wang, Q., Zhang, X., Peng, H., Fang, W., et al. (2011). A bacterial laccase from marine microbial metagenome exhibiting chloride tolerance and dye decolorization ability. Appl. Microbiol. Biotechnol. 89, 1103–1110. doi: 10.1007/s00253-010-2934-3
Feng, L., Wang, W., Cheng, J., Ren, Y., Zhao, G., Gao, C., et al. (2007). Genome and proteome of long-chain alkane degrading Geobacillus thermodenitrificans NG80-2 isolated from a deep-subsurface oil reservoir. Proc. Natl. Acad. Sci. U.S.A. 104, 5602–5607. doi: 10.1073/pnas.0609650104
Furukawa, K., Hayase, N., Taira, K., and Tomizuka, N. (1989). Molecular relationship of chromosomal genes encoding biphenyl/polychlorinated biphenyl catabolism: some soil bacteria possess a highly conserved bph operon. J. Bacteriol. 171, 5467–5472. doi: 10.1128/jb.171.10.5467-5472.1989
Hassanshahian, M., Emtiazi, G., and Cappello, S. (2012). Isolation and characterization of crude-oil-degrading bacteria from the Persian Gulf and the Caspian Sea. Mar. Pollut. Bull. 64, 7–12. doi: 10.1016/j.marpolbul.2011.11.006
Hong, Y. H., Deng, M. C., Xu, X. M., Wu, C. F., Xiao, X., Zhu, Q., et al. (2016). Characterization of the transcriptome of Achromobacter sp. HZ01 with the outstanding hydrocarbon-degrading ability. Gene 584, 185–194. doi: 10.1016/j.gene.2016.02.032
Ibrahim, M., Subramanian, A., and Anishetty, S. (2017). Comparative pan genome analysis of oral Prevotella species implicated in periodontitis. Funct. Integr. Genomics doi: 10.1007/s10142-017-0550-3 [Epub ahead of print].
Jakobsen, T. H., Hansen, M. A., Jensen, P. O., Hansen, L., Riber, L., Cockburn, A., et al. (2013). Complete genome sequence of the cystic fibrosis pathogen Achromobacter xylosoxidans NH44784-1996 complies with important pathogenic phenotypes. PLoS ONE 8:e68484. doi: 10.1371/journal.pone.0068484
Jencova, V., Strnad, H., Chodora, Z., Ulbrich, P., Vlcek, C., Hickey, W. J., et al. (2008). Nucleotide sequence, organization and characterization of the (halo)aromatic acid catabolic plasmid pA81 from Achromobacter xylosoxidans A8. Res. Microbiol. 159, 118–127. doi: 10.1016/j.resmic.2007.11.018
Jensen, L. J., Julien, P., Kuhn, M., von Mering, C., Muller, J., Doerks, T., et al. (2008). eggNOG: automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 36, D250–D254. doi: 10.1093/nar/gkm796
Jimenez, J. I., Minambres, B., Garcia, J. L., and Diaz, E. (2002). Genomic analysis of the aromatic catabolic pathways from Pseudomonas putida KT2440. Environ. Microbiol. 4, 824–841. doi: 10.1046/j.1462-2920.2002.00370.x
Kaczorek, E., Salek, K., Guzik, U., Dudzinska-Bajorek, B., and Olszanowski, A. (2013). The impact of long-term contact of Achromobacter sp. 4 (2010) with diesel oil – Changes in biodegradation, surface properties and hexadecane monooxygenase activity. Int. Biodeterior. Biodegradation 78, 7–16. doi: 10.1016/j.ibiod.2012.12.003
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kube, M., Chernikova, T. N., Al-Ramahi, Y., Beloqui, A., Lopez-Cortez, N., Guazzaroni, M. E., et al. (2013). Genome sequence and functional genomic analysis of the oil-degrading bacterium Oleispira antarctica. Nat. Commun. 4:2156. doi: 10.1038/ncomms3156
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Liu, B., and Pop, M. (2009). ARDB—antibiotic resistance genes database. Nucleic Acids Res. 37, D443–D447. doi: 10.1093/nar/gkn656
Maier, R. M., and Soberon-Chavez, G. (2000). Pseudomonas aeruginosa rhamnolipids: biosynthesis and potential applications. Appl. Microbiol. Biotechnol. 54, 625–633. doi: 10.1007/s002530000443
Markowitz, V. M., Chen, I. M., Palaniappan, K., Chu, K., Szeto, E., Pillay, M., et al. (2014). IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 42, D560–D567. doi: 10.1093/nar/gkt963
Medini, D., Donati, C., Tettelin, H., Masignani, V., and Rappuoli, R. (2005). The microbial pan-genome. Curr. Opin. Genet. Dev. 15, 589–594. doi: 10.1016/j.gde.2005.09.006
Moon, J., Kang, E., Min, K. R., Kim, C. K., Min, K. H., Lee, K. S., et al. (1997). Characterization of the gene encoding catechol 2,3-dioxygenase from Achromobacter xylosoxidans KF701. Biochem. Biophys. Res. Commun. 238, 430–435. doi: 10.1006/bbrc.1997.7312
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185. doi: 10.1093/nar/gkm321
Moyne, A. L., Cleveland, T. E., and Tuzun, S. (2004). Molecular characterization and analysis of the operon encoding the antifungal lipopeptide bacillomycin D. FEMS Microbiol. Lett. 234, 43–49. doi: 10.1016/j.femsle.2004.03.011
Nie, Y., Tang, Y. Q., Li, Y., Chi, C. Q., Cai, M., and Wu, X. L. (2012). The genome sequence of Polymorphum gilvum SL003B-26A1(T) reveals its genetic basis for crude oil degradation and adaptation to the saline soil. PLoS ONE 7:e31261. doi: 10.1371/journal.pone.0031261
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Pessione, E., Divari, S., Griva, E., Cavaletto, M., Rossi, G. L., Gilardi, G., et al. (1999). Phenol hydroxylase from Acinetobacter radioresistens is a multicomponent enzyme. Purification and characterization of the reductase moiety. Eur. J. Biochem. 265, 549–555. doi: 10.1046/j.1432-1327.1999.00720.x
Pirog, T. P., Korzh, Y. V., Shevchuk, T. A., and Tarasenko, D. A. (2008). Peculiarities of C2 metabolism and intensification of the synthesis of surface-active substances in Rhodococcus erythropolis EK-1 grown in ethanol. Microbiology 77, 665–673. doi: 10.1134/s0026261708060039
Rojo, F. (2009). Degradation of alkanes by bacteria. Environ. Microbiol. 11, 2477–2490. doi: 10.1111/j.1462-2920.2009.01948.x
Roongsawang, N., Washio, K., and Morikawa, M. (2010). Diversity of nonribosomal peptide synthetases involved in the biosynthesis of lipopeptide biosurfactants. Int. J. Mol. Sci. 12, 141–172. doi: 10.3390/ijms12010141
Sambrook, J., and Russell, D. W. (2001). Molecular Cloning: a Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Satpute, S. K., Bhuyan, S. S., Pardesi, K. R., Mujumdar, S. S., Dhakephalkar, P. K., Shete, A. M., et al. (2010). Molecular genetics of biosurfactant synthesis in microorganisms. Adv. Exp. Med. Biol. 672, 14–41. doi: 10.1007/978-1-4419-5979-9_2
Schneiker, S., Martins dos Santos, V. A., Bartels, D., Bekel, T., Brecht, M., Buhrmester, J., et al. (2006). Genome sequence of the ubiquitous hydrocarbon-degrading marine bacterium Alcanivorax borkumensis. Nat. Biotechnol. 24, 997–1004. doi: 10.1038/nbt1232
Si, M., Zhao, Y., and Su, T. (2011). Isolation and identification of a high-efficiency alkane-degrading Achromobacter insolitus XL strain and its degradation characteristics. J. Soil Sci. 42, 562–567.
Smits, T. H., Witholt, B., and van Beilen, J. B. (2003). Functional characterization of genes involved in alkane oxidation by Pseudomonas aeruginosa. Antonie Van Leeuwenhoek 84, 193–200. doi: 10.1023/A:1026000622765
Strachan, P. D., Freer, A. A., and Fewson, C. A. (1998). Purification and characterization of catechol 1,2-dioxygenase from Rhodococcus rhodochrous NCIMB 13259 and cloning and sequencing of its catA gene. Biochem. J. 333(Pt 3), 741–747. doi: 10.1042/bj3330741
Venter, J. C., Remington, K., Heidelberg, J. F., Halpern, A. L., Rusch, D., Eisen, J. A., et al. (2004). Environmental genome shotgun sequencing of the Sargasso Sea. Science 304, 66–74. doi: 10.1126/science.1093857
Vilella, A. J., Severin, J., Ureta-Vidal, A., Heng, L., Durbin, R., and Birney, E. (2009). EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 19, 327–335. doi: 10.1101/gr.073585.107
Walzer, G., Rosenberg, E., and Ron, E. Z. (2006). The Acinetobacter outer membrane protein A (OmpA) is a secreted emulsifier. Environ. Microbiol. 8, 1026–1032. doi: 10.1111/j.1462-2920.2006.00994.x
Weber, T., Blin, K., Duddela, S., Krug, D., Kim, H. U., Bruccoleri, R., et al. (2015). antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 43, W237–W243. doi: 10.1093/nar/gkv437
Xia, Y., and Min, H. (2003). Identification, cloning and sequencing of GST gene of bacterium degrading poly-aromatic hydrocarbons. Wei Sheng Wu Xue Bao 43, 691–697.
Xiao, B., Sun, Y.-F., Lian, B., and Chen, T. M. (2016). Complete genome sequence and comparative genome analysis of the Paenibacillus mucilaginosus K02. Microb. Pathog. 93, 194–203. doi: 10.1016/j.micpath.2016.01.016
Xue, J., Yu, Y., Bai, Y., Wang, L., and Wu, Y. (2015). Marine oil-degrading microorganisms and biodegradation process of petroleum hydrocarbon in marine environments: a review. Curr. Microbiol. 71, 220–228. doi: 10.1007/s00284-015-0825-7
Yamaguchi, M., and Fujisawa, H. (1980). Purification and characterization of an oxygenase component in benzoate 1,2-dioxygenase system from Pseudomonas arvilla C-1. J. Biol. Chem. 255, 5058–5063.
Yao, S., Gao, X., Fuchsbauer, N., Hillen, W., Vater, J., and Wang, J. (2003). Cloning, sequencing, and characterization of the genetic region relevant to biosynthesis of the lipopeptides iturin A and surfactin in Bacillus subtilis. Curr. Microbiol. 47, 272–277. doi: 10.1007/s00284-002-4008-y
Keywords: Achromobacter, bioremediation, genome, hydrocarbon, petroleum pollution, strain HZ01
Citation: Hong Y - H, Ye C-C, Zhou Q-Z, Wu X-Y, Yuan J-P, Peng J, Deng H and Wang J-H (2017) Genome Sequencing Reveals the Potential of Achromobacter sp. HZ01 for Bioremediation. Front. Microbiol. 8:1507. doi: 10.3389/fmicb.2017.01507
Received: 02 May 2017; Accepted: 27 July 2017;
Published: 09 August 2017.
Edited by:
Chaomin Sun, Institute of Oceanology (CAS), ChinaReviewed by:
Wenli Chen, Huazhong Agricultural University, ChinaNaresh Singhal, University of Auckland, New Zealand
Copyright © 2017 Hong, Ye, Zhou, Wu, Yuan, Peng, Deng and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiang-Hai Wang, d2FuZ2poYWlAbWFpbC5zeXN1LmVkdS5jbg== Hailin Deng, ZGVuZ2hsaW4zQG1haWwuc3lzdS5lZHUuY24=