 Mareen Morawe1*
Mareen Morawe1* Henrike Hoeke2,3Dirk K. Wissenbach4
Henrike Hoeke2,3Dirk K. Wissenbach4 Guillaume Lentendu5
Guillaume Lentendu5 Tesfaye Wubet6
Tesfaye Wubet6 Eileen Kröber7
Eileen Kröber7 Steffen Kolb1,7*
Steffen Kolb1,7*- 1Department of Ecological Microbiology, University of Bayreuth, Bayreuth, Germany
- 2Department of Molecular Systems Biology, Helmholtz Centre for Environmental Research, Leipzig, Germany
- 3Department of Pharmaceutical and Medicinal Chemistry, Institute of Pharmacy, University of Leipzig, Leipzig, Germany
- 4Institute of Forensic Medicine, University Hospital Jena, Jena, Germany
- 5Department of Ecology, University of Kaiserslautern, Kaiserslautern, Germany
- 6Department of Soil Ecology, Helmholtz Centre for Environmental Research, Leipzig, Germany
- 7Institute of Landscape Biogeochemistry, Leibniz Centre for Landscape Research, Müncheberg, Germany
Methanol is an abundant atmospheric volatile organic compound that is released from both living and decaying plant material. In forest and other aerated soils, methanol can be consumed by methanol-utilizing microorganisms that constitute a known terrestrial sink. However, the environmental factors that drive the biodiversity of such methanol-utilizers have been hardly resolved. Soil-derived isolates of methanol-utilizers can also often assimilate multicarbon compounds as alternative substrates. Here, we conducted a comparative DNA stable isotope probing experiment under methylotrophic (only [13C1]-methanol was supplemented) and combined substrate conditions ([12C1]-methanol and alternative multi-carbon [13Cu]-substrates were simultaneously supplemented) to (i) identify methanol-utilizing microorganisms of a deciduous forest soil (European beech dominated temperate forest in Germany), (ii) assess their substrate range in the soil environment, and (iii) evaluate their trophic links to other soil microorganisms. The applied multi-carbon substrates represented typical intermediates of organic matter degradation, such as acetate, plant-derived sugars (xylose and glucose), and a lignin-derived aromatic compound (vanillic acid). An experimentally induced pH shift was associated with substantial changes of the diversity of active methanol-utilizers suggesting that soil pH was a niche-defining factor of these microorganisms. The main bacterial methanol-utilizers were members of the Beijerinckiaceae (Bacteria) that played a central role in a detected methanol-based food web. A clear preference for methanol or multi-carbon substrates as carbon source of different Beijerinckiaceae-affiliated phylotypes was observed suggesting a restricted substrate range of the methylotrophic representatives. Apart from Bacteria, we also identified the yeasts Cryptococcus and Trichosporon as methanol-derived carbon-utilizing fungi suggesting that further research is needed to exclude or prove methylotrophy of these fungi.
Introduction
Methanol is an abundant volatile organic compound (VOC) in the troposphere, i.e., reaches mixing ratios of up to 10 ppb. Due to its high reactivity, methanol impacts on the oxidative capacity, the formation of ozone, and pools of important organic reactants in the atmosphere (Galbally and Kirstine, 2002; Wohlfahrt et al., 2015). Atmospheric methanol mainly originates from growing plants and decaying plant material but originates to a smaller extent from reactions of methyl peroxy radicals in the troposphere (Fall and Benson, 1996; Warneke et al., 1999; Galbally and Kirstine, 2002; Millet et al., 2008; Wohlfahrt et al., 2015). Large sinks for atmospheric methanol are reactions with hydroxyl radicals and oceanic uptake, including contributions of microorganisms (Millet et al., 2008). The role of methylotrophic microorganisms in terrestrial ecosystems as global sinks of methanol is nonetheless undisputed (Kolb, 2009; Stacheter et al., 2013; Wohlfahrt et al., 2015). However, their environmental controls, their distribution in the phyllo- and rhizosphere, and their diversity in different climate zones are largely unresolved (Kolb, 2009; Kolb and Stacheter, 2013; Stacheter et al., 2013; Wohlfahrt et al., 2015).
Microbial methanol-utilizers were discovered in the late 19th century and can be aerobic or anaerobic (Loew, 1892; Chistoserdova et al., 2009; Kolb, 2009; Chistoserdova and Lidstrom, 2013; Hedderich and Withman, 2013). Aerobic methanol-utilizers are phylogenetically diverse and affiliate with gram-negative Alpha-, Beta-, and Gammaproteobacteria and Verrucomicrobia and with gram-positive Actinobacteria and Firmicutes, as well as with some fungi, i.e., ascomycetous yeasts and molds (Kolb, 2009; Gvozdev et al., 2012; Chistoserdova and Lidstrom, 2013; Kolb and Stacheter, 2013; Sharp et al., 2014).
Although methanol-utilizers were among the first microorganisms that were targeted in the environment using molecular tools two decades ago, an understanidng of their global biogeography has only just started to grow (Holmes et al., 1995; Kolb and Stacheter, 2013). The sensitivity of the environmental detection of low-abundant, one-carbon (C1) compound converting microorganisms has been improved by the detection of key genes of methylotrophy (Holmes et al., 1995; McDonald and Murrell, 1997; Kolb and Stacheter, 2013). The initial enzymatic step of microbial methanol utilization is the oxidation of methanol to formaldehyde. For this reaction, at least three different enzymes occur in Bacteria (Kolb and Stacheter, 2013). In methylotrophic fungi, methanol is oxidized by alternative oxidoreductases (Gvozdev et al., 2012). The most prominent methanol oxidoreductase is the pyrroloquinoline quinone (PQQ)-dependent methanol dehydrogenase (MDH) encoded by the genes mxaFI. Burkholderiales possess an isoenzyme encoded by mdh2 (Kalyuzhnaya et al., 2008). In many Bacteria, xoxF (synonymous to mxaF’) is present, and a subtype (belonging to clade xoxF4) encodes a functional MDH that has been recently used to detect xoxF-possessing Bacteria in coastal environments (Chistoserdova, 2011; Taubert et al., 2015). Based on this broad spectrum of known enzymes, several gene markers have been developed. However, only mxaF and xoxF are currently well-established functional gene markers that have been used in environmental surveys of methanol-utilizers, whereby no universal primers covering all five xoxF clades exist (McDonald and Murrell, 1997; Moosvi et al., 2005; Neufeld et al., 2007; Stacheter et al., 2013; Taubert et al., 2015). Knowing the limitations of available functional markers, we complemented the detection of mxaF/xoxF and bacterial 16S rRNA genes in combination with a stable isotope labeling apporach to identify methanol-utilizers in a forest soil.
The taxonomic diversity of methanol-utilizers in temperate forest soils is affected by soil pH and the presence of trees, but a detailed understanding of driving factors in these ecosystems is lacking (Kolb, 2009; Degelmann et al., 2010; Stacheter et al., 2013). Soil-derived methanol-utilizers often grow on multi-carbon compounds (Kolb, 2009). Thus, one may speculate that such facultatively methylotrophic microorganisms might occupy different ecological niches with regard to their alternative substrates in the organic compound-rich top layer of forest soils, which would enable them to permanently establish in a complex soil community along with other methanol-utilizers and non-methylotrophic heterotrophs.
Few studies have begun to gain a more detailed understanding of methanol-utilizers in soils (Radajewski et al., 2000, 2002; Lueders et al., 2004; Stacheter et al., 2013). The limited molecular detection based on gene markers in most of the previous studies on soil methylotrophs might have led to an underestimation of the taxonomic biodiversity of these methylotrophs. Thus, the current scientific view on terrestrial methanol-utilizers is still largely based on pure cultures and few stable isotope probing (SIP) experiments (Radajewski et al., 2002; Lueders et al., 2004; Kolb, 2009).
In our study, comparative DNA SIP experiments with several 13C-isotopologs of potential alternative multi-carbon substrates and with [13C1]-methanol in separate treatments were conducted in an acidic aerated forest soil. Thereby, we focussed on the substrate utilization of methanol-utilizing methylotrophs under mixed substrate conditions (i.e., the presence of methanol and an alternative [13Cu]-substrate). To better understand the role of soil pH for niche partitioning of detectable soil methanol-utilizers, we conducted a methanol-supplemented SIP experiment under the acidic in situ and artificially induced pH-neutral conditions, at which most soil-derived isolates have their growth optimum. The specific objectives of this study were (i) to identify aerobic bacteria and fungi that assimilated methanol-derived carbon, (ii) to evaluate the significance of soil pH on indigenous methanol-utilizers, and (iii) to resolve alternative substrate spectra of active methanol-utilizers using common soil organic compounds.
Materials and Methods
Study Site and Soil Sampling
The study site was located in a temperate German forest (Steigerwald, 49°52′ N, 10°28′ E) dominated by European beech (Fagus sylvatica). The characteristics of the soil are described elsewhere (Degelmann et al., 2009). Soil samples were taken from the upper part of the soil (10 cm, O plus A horizons) without the litter layer. Five sampling sites were chosen that reflected the general characteristics of the sampling area (saplings, dead wood, clearing, shady, and old beeches). Samples were taken in August 2013 (for the substrate SIP experiment) and in September 2014 (for the pH shift SIP experiment). Fresh forest soil samples were sieved (2 mm) and equally pooled to further prepare the soil slurries.
[13Cu]-Substrates for SIP Experiments
Filter-sterilized 40 mM stock solutions of methanol and multi-carbon substrates (i.e., acetate, glucose, xylose, and vanillic acid) were prepared with either the [13C]-isotopolog (‘labeled,’ 99 atom% C) or the [12C]-isotopolog (i.e., ‘unlabeled,’ natural abundance of 13C). All multi-carbon substrate stock solutions also included 40 mM [12C]-methanol. The isotopologs were fully labeled (i.e., [13Cu]) except for vanillic acid, in which only the aromatic ring carbon atoms were [13C]-labeled (i.e., [13C1-6]). CO2 treatments were set up with either [13C]-CO2 (‘labeled,’ 99 atom% C; <3 atom% 18O) or [12C]-CO2. For more detailed information, refer to the Supplementary Information on Materials and Methods.
Substrate SIP Experiment under Mixed Substrate Conditions
Soil slurries were prepared by mixing 50 g of freshly sieved soil with 40 ml of trace element solution (in 1 L of sterile water: HCl, 50 μM; FeCl2, 5 μM; ZnCl2, MnCl2, CoCl2, 50 μM; Na2MoO4, 0.15 μM; H3BO3, NiCl2, 0.10 μM; CuCl2, 0.01 μM; after Whittenbury et al., 1970) and initially homogenized by hand shaking. Incubations were conducted as soil slurries to achieve homogenous physicochemical conditions (e.g., substrate concentrations, redox conditions, and pH) and to provide a sufficent distribution of supplemented substrates as well as a balanced distribution of microorganisms to minimize heterogeneity in collected sub-samples.
Incubations were performed in duplicates for each approach (control, 12C, and 13C) on an end-over-end shaker at 20°C in the dark. Oxic conditions were maintained by placing a large gas phase inside the flasks (the ratio of gas phase to slurry volume was 12:1) and by daily opening for few minutes before re-sealing, allowing gas phase exchange. The O2 concentrations were monitored to ensure oxic conditions.
Substrates (i.e., methanol, acetate, and sugars; 1 ml) and methane were daily supplemented to a final concentration of 1 mM and 200 ppm, respectively. Vanillic acid (1 ml, 1 mM final concentration) was supplemented if it was no longer detected. In order to obtain mixed substrate conditions methanol (12C) was supplemented in combination with alternative multi-carbon substrates (12C or 13C) at the same concentrations (1 mM, final concentration). Thus, treatments solely supplemented with methanol served as multi-carbon substrate controls. Unsupplemented control treatments (i.e., solely 1 ml trace element solution was supplemented) served as methanol control treatments and lacked any other substrate supplementation besides methane. Methane was supplemented to support also methanotrophic organisms in the soil, which might also be important methanol-utilizers (such as the methanotrophic USCα group, Degelmann et al., 2010). CO2 incubations were supplemented with 10% CO2 in the headspace (approximately 7 mM total concentration) and opened if the O2 concentration was below 10%. The purpose of the CO2 treatments was to evaluate the cross-feeding effects through the assimilation of [13C]-CO2.
For more detailed information and an overview over the experimental set-up (Supplementary Figure S1), refer to the Supplementary Information on Materials and Methods.
pH Shift SIP Experiment under Methylotrophic Conditions
Two treatments were conducted, mimicking acidic in situ and elevated pH-neutral conditions. Soil slurries were prepared according to the substrate SIP experiment. The treatments for in situ pH 4 were prepared by mixing 50 g freshly sieved soil and 40 ml of trace element solution in one incubation flask. The treatments for pH 7 were prepared by mixing 300 g freshly sieved soil with 240 ml of trace element solution. Then, the pH was adjusted to 7 with sterile NaOH, and the solution was mixed until the pH remained constant. A total of 90 ml of the pH adjusted slurry (corresponding with the volume of a slurry consisting of 50 g soil and 40 ml trace element solution) was placed into each incubation flask. Methanol was supplemented daily to a final concentration of 1 mM per pulse. Control treatments were only supplemented with the same volume of trace element solution. Daily aliquots were taken, and the pH was monitored to avoid changes and re-adjusted when necessary. The pH shift SIP experiment treatments lacked methane supplementation. Thus, a putatively supporting effect of methane on methylotrophs under in situ conditions could be evaluated by the comparison of all methanol treatments of both SIP experiments conducted, i.e., substrate SIP and pH shift SIP experiment. For more detailed information, refer to the Supplementary Information on Materials and Methods.
Chemical Analytics
The pH value was determined in soil slurry aliquots. Gases were measured by gas chromatography using thermal conductivity (O2, CO2) and a flame ionization detector (methane). The amount of [13C]-CO2 was determined using GC mass spectrometry. The conversion of supplemented multi-carbon compounds was monitored by high-performance liquid chromatography (HPLC) using the refractive index and a diode array detector. Details can be found in the Supplementary Information on Materials and Methods.
Nucleic Acid Extraction and Separation of ‘Heavy’ (H), ‘Middle’ (M), and ‘Light’ (L) DNA by Density Gradient Centrifugation
Nucleic acids were extracted from two 0.5 g soil slurry samples of each replicate according to Griffiths et al. (2000). DNA was precipitated, purified from co-extracted RNA by RNase treatment and quantified with Quant-iT-Pico Green (Invitrogen, Carlsbad, CA, United States).
DNA SIP was performed according to the protocol of Neufeld et al. (2007). Equally pooled DNA from the t0, 12C and 13C treatments (5–10 μg) was added to CsCl-containing gradients [buoyant density (BD) 1.732 ± 0.0006 g ml-1]. Isopycnic centrifugation was performed (44 100 rpm, i.e., 177 000 gav, at 20°C for 40 h; rotor VTi65.2; Beckmann, Fullerton, CA, United States) to separate DNA by its BDs. Gradients were separated into 10 fractions (450 μl each), and the BD of each fraction was determined by repeated weighing at 20°C. DNA was precipitated with glycogen (10 mg ml-1) and polyethylene glycol and quantified.
According to the reported BD for non-labeled and fully labeled DNA (Lueders et al., 2004), fractions 1 to 10 were separately pooled into ‘heavy’ (H) fractions (BD ≥ 1.730 g ml-1), ‘middle’ (M) fractions (BD between 1.730 and 1.715 g ml-1), and ‘light’ (L) fractions (BD ≤ 1.715 g ml-1).
Barcoded Amplicon Pyrosequencing of 16S rRNA Genes, mxaF/xoxF, and ITS
DNA from the pooled fractions was used for amplicon pyrosequencing. For bacterial genes (i.e., 16S rRNA and mxaF/xoxF), a two-step PCR approach was performed to decrease bias (Berry et al., 2011). In brief, amplicons derived from a first PCR with ‘conventional’ primers (i.e., untagged primers) were subsequently amplified with ‘barcoded’ primers (i.e., primers with an additional barcode sequences at the 5′ terminus) to obtain amplicons with distinguishable nucleotide tags (details in Supplementary Information on Materials and Methods).
Bacterial 16S rRNA gene fragments were amplified using the primers 341f and 785-805r, which had the best overall bacterial sequence and phylum coverage (Muyzer et al., 1998; Herlemann et al., 2011; Klindworth et al., 2013). In order to amplify mxaF/xoxF gene sequences the primer pairs ‘mxaF1’ (1003f/1555r; McDonald and Murrell, 1997; Neufeld et al., 2007) and ‘mxaF2’ (mxaF_for/mxaF_rev; Moosvi et al., 2005) were used. Simultaneous amplification of mxaF and xoxF is assumed since in a previous study xoxF gene sequences were also amplified using the primer pair ‘mxaF1’ (Stacheter et al., 2013). Trials to amplify pmoA (encodes the beta-subunit of particulate methane monooxygenase) genes were not sufficiently successful, i.e., only a sparse smear or weak bands were visible after PCR and purification led to loss of amplicons. Thus, this gene was no longer analyzed. Barcoded amplicon pools were pyrosequenced at the Göttingen Genomics Laboratory using a Roche GS-FLX 454 Sequencer and GSL FLX Titanium series reagents (Roche Diagnostics GmbH, Mannheim, Germany) as previously described (Stacheter et al., 2013). Fungal ITS fragments (internal transcibed spacer) were amplified using the primers ITS1F and ITS4 (White et al., 1990; Gardes and Bruns, 1993). The amplicons obtained were equimolarly pooled and pyrosequenced at the Department of Soil Ecology (UFZ, Halle, Germany) as previously described (Wubet et al., 2012). For detailed information on the primer sequences, PCR conditions, strategies and performance, please refer to the Supplementary Information on Materials and Methods.
Read Filtering and Clustering
The reads of bacterial genes were trimmed to nearly equal sequence lengths (446 bp for 16S rRNA, 440 bp for mxaF), amplicon pyrosequencing errors were corrected using ACACIA, and potential 16S rRNA chimeric sequences were sorted out using the UCHIME algorithm implemented in USEARCH and the latest RDP Gold database for high-quality 16S rRNA gene reference sequences (Edgar et al., 2011; Bragg et al., 2012). Using JAguc v2.1, the sequences were clustered into operational taxonomic units (OTUs) applying the UPGMA model (Nebel et al., 2011). The OTUs of 16S rRNA were clustered at the family level using 90.1% as the pairwise similarity cut-off value (to ensure sufficient sampling depth), and mxaF OTUs were clustered with a cut-off value of 90%, which was higher than that previously reported, to obtain a higher diversity (Yarza et al., 2010; Stacheter et al., 2013). Representative sequences (i.e., longest sequence, in the case of identical lengths the sequence was randomly chosen by the program) of each OTU were used for further taxonomic affiliation. 16S rRNA phylotypes were primarily affiliated using a local nucleotide BLAST and affiliation was checked by a online megaBLAST that uses a nucelotide database updated daily and a phylogenetic tree generated using MEGA Version 6.06 (Tamura et al., 2013). The phylogenetic affiliation of mxaF phylotypes was determined using manual BLAST (megaBLAST) and phylogenetic treeing.
For the detailed resolution of OTU16S438 the filtered 16S rRNA gene dataset (also used in the JAguc analysis) and all sequences of OTU16S438 were combined and clustered using QIIME at a species-level cut-off. For more detailed information refer to the Supplementary Information on Materials and Methods.
Reads of fungal ITS genes were demultiplexed and quality trimmed using MOTHUR, normalized (1503 counts per sample), and checked for chimeric sequences using UCHIME (Schloss et al., 2009; Edgar et al., 2011). The sequences were clustered into OTUs using CD-HIT-EST at a 97% pairwise similarity cut-off value (Fu et al., 2012). Representative sequences were classified against the dynamic UNITE database (v7 release 01.08.2015; Kõljalg et al., 2013) using a MOTHUR-implemented classifier of Wang et al. (2007). For detailed information, please refer to the Supplementary Information on Materials and Methods.
Identification of ‘13C-Labeled’ Phylotypes
The ‘13C-label’ of phylotypes was determined by analyzing the relative abundances of phylotypes in the amplicon read libraries of the H, M, and L fractions of the [12C] and [13C] treatments of each gene dataset (i.e., 16S rRNA gene, mxaF, and ITS). The phylotypes that occurred only once within the complete dataset of all amplicon libraries were considered erroneous and removed, whereas singletons in each individual amplicon library were preserved (not applied for the ITS dataset, in which phylotypes with fewer than three reads were removed in the previous read-filtering step). A comparison of the relative abundances in the H fractions of [13C] treatments with those in the H fractions of [12C] treatments and a comparison of the H and L fractions of the [13C] treatment were conducted. This procedure minimizes the identification of false-positive phylotypes due to the migration of light DNA into the H fractions (Lueders et al., 2004; Dallinger and Horn, 2014). The following criteria had to be met to classify a phylotype as ‘labeled’: (1) The abundance in the appropriate fraction (i.e., H or M fraction) of the [13C] treatment was higher than that in the corresponding fraction of the [12C] treatment; (2) the abundance in the L fraction was lower than that in the H or M fraction of the [13C] treatment; (3) the abundance in the H or M fraction of the [13C] treatment was ≥0.5%; and (4) the difference in the abundance in the compared fractions of the [13C] treatment was ≥0.1% compared to that of the [12C] treatment. Phylotypes that met all these criteria were considered ‘potentially labeled’ and were the basis for the calculation of the ‘labeling proportion’ (LP). The LP serves as an indicator for the relative importance of different bacterial taxa assimilating the supplemented [13Cu]-substrate (directly or indirectly) and is not a proxy for the amount of incorporated 13C into the DNA. The LP of a certain potentially labeled phylotype x was
with RA is the relative abundance, n is the number of all ‘potentially labeled’ phylotypes, is the sum of all relative abundances of ‘potentially labeled’ taxa in the M or H gradient fraction of the [13C] treatment, and is the relative abundance of a certain phylotype x in the M or H gradient fraction of the [13C] treatment. A threshold value of 5% was used to distinguish between labeled taxa of greater (i.e., LPx ≥ 5%) or minor (i.e., LPx < 5%) importance (Dallinger and Horn, 2014). The phylotypes that were identified as labeled in the M fraction were considered as ‘weakly labeled’ for two possible reasons: (i) not fully labeled DNA and (ii) fully labeled DNA of organisms with very low GC content (<40%). Nonetheless, we expected that the general genome GC content is higher than 40% and is similar for the majority of microorganisms of this environment as previous genome studies suggest (Foerstner et al., 2005).
Quantification of 16S rRNA Genes, mxaF, and mmoX in the pH Shift SIP Experiment
The gene fragments were quantified in duplicates on an iQ5 Real-Time qPCR cycler (BioRad, Munich, Germany) with primer sets specific for Bacteria and mxaF (Moosvi et al., 2005) and mmoX (Kolb et al., 2005) using internal standards. According to published protocols, all qPCR measurements were inhibitor corrected because co-extracted humic acids were obvious and inhibition was well-recorded (Degelmann et al., 2010; Zaprasis et al., 2010) (detailed information in Supplementary Information on Materials and Methods).
Nucleotide Sequence Accession Numbers
Representative sequences of labeled phylotypes derived from barcoded amplicon pyrosequencing were deposited in EMBL under accession numbers LT607885 to LT607955 (16S rRNA gene), LT607956 to LT608017 (mxaF), and LT608018 to LT608119 (ITS). All raw pyrosequencing datasets were deposited in the ENA Short Read Archive under the study accession number ERP016444, including the 16S rRNA gene, mxaF and ITS datasets.
Results
Identification of Microorganisms Utilizing Methanol-Derived Carbon in an Acidic Forest Soil
A successful labeling of Bacteria and fungi was proven by the dissimilar composition of phylotypes of [12C] and [13C] H and M fractions (Supplementary Figure S1). In addition, the carbon recovery rates for [13C]-CO2 revealed that the supplemented substrates were not only dissimilated but were also assimilated (Supplementary Figures S3, S4).
The [13C1]-methanol treatment of the substrate SIP experiment revealed one labeled phylotype (OTU16S438) dominating the H fraction. This phylotype was also highly labeled in the M fraction and was affiliated to Beijerinckiaceae, i.e., it was closely related to methylotrophic species such as Methylovirgula ligni (Figure 1A and Supplementary Tables S1, S2). Further minor and weakly labeled phylotypes were found among a wide range of bacterial phyla (i.e., minor labeled, LP < 2%: Acidobacteria, Proteobacteria, and Verrucomicrobia; weakly labeled: Armatimonadetes, Planctomycetes, Alphaproteobacteria, and Verrucomicrobia) (Figure 1A and Supplementary Table S1). The sequence identities of these labeled phylotypes to their closest related cultured species ranged from 83 to 99% (Supplementary Table S2), suggesting that hitherto unrecognized methanol-utilizers or methanol-derived carbon-utilizers occurred.
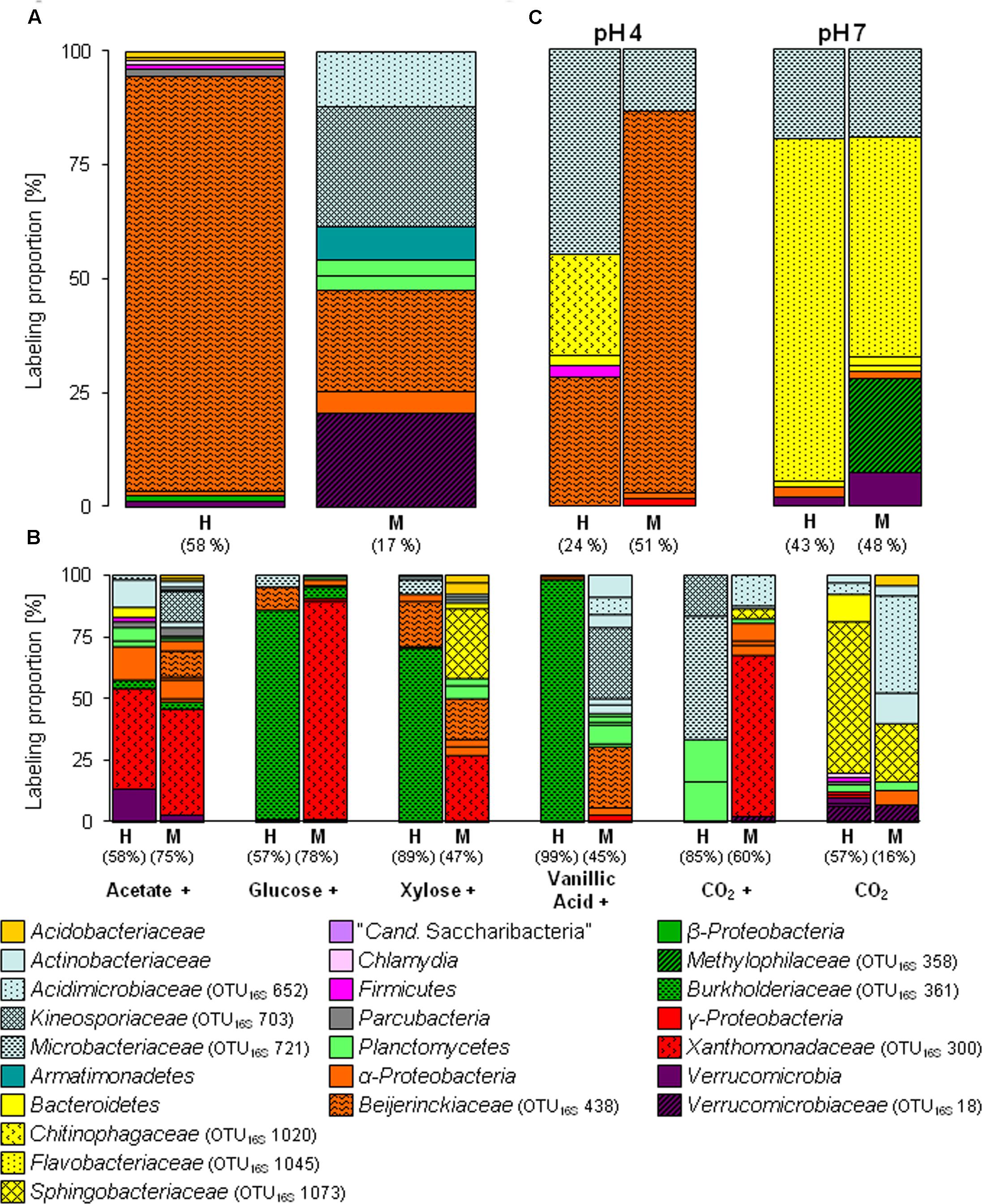
FIGURE 1. Labeling proportions (LPs) and identities of bacterial 16S rRNA phylotypes in the ‘heavy’ and ‘middle’ fractions of [13C1]-methanol (A) and [13Cu]-substrates (B) treatments of the substrate SIP experiment, and treatments of the pH SIP experiment (C). Cross, additional [12C]-methanol supplementation in substrate treatments. Equal colors, the same phylum affiliation. ‘H’ and ’M,’ ‘heavy’ and ‘middle’ fractions, respectively. Values in brackets, contribution of labeled phylotypes to the total number of sequences. The ‘labeling proportions’ are indicators for the relative importance of different taxa assimilating the supplemented 13C (directly or indirectly). More detailed taxonomic information can be found in Supplementary Tables S16, S17.
The [13C1]-methanol treatment of the pH shift SIP experiment revealed a slightly different distribution of labeled phylotypes compared to the substrate SIP experiment suggesting an influence of methane on the methanol-utilizer community in the forest soil. In addition to the Beijerinckiaceae-phylotype (OTU16S438), also phylotypes affiliated to Microbacteriaceae (OTU16S721, 99% sequence identity to Leifsonia xyli) and Chitinophagaceae (OTU16S1020, 96% sequence identity to Chitinophaga sp.) were labeled (Figure 1C and Supplementary Table S6). Further minor or weakly labeled phylotypes belonged to Sphingobacteriales, Paenibacillaceae, Sphingomonadaceae, and Xanthomonadaceae (Rhodanobacter) (Figure 1C and Supplementary Table S6).
Labeled Phylotypes Based on MDH Gene Markers
The specific primers used in our study were assumed to target mxaF as well as xoxF gene sequences as a previous study on temperate soils indicated (Stacheter et al., 2013). However, no xoxF-affiliated phylotype was labeled in any treatment (including also [13Cu]-substrate treatments of the substrate SIP experiment), and potentially further present xoxF-containing methylotrophs might have been overlooked. The labeled phylotypes of the [13C1]-methanol treatment of the substrate SIP experiment were dominated by three phylotypes belonging to Methylobacterium (OTUmxaF40) and Hyphomicrobium (OTUsmxaF185 and 210), whereas in the [13C1]-methanol treatment of the pH shift SIP experiment labeled mxaF phyloytpes belonged mainly to Hyphomicrobium.
Although Beijerinckiaceae appeared to be the dominantly labeled bacterial phylotype based on the 16S rRNA genes, only one Beijerinckiaceae-related mxaF phylotype (OTUmxaF144) was labeled in the substrate SIP experiment (Supplementary Table S3 and Figure 2A). Methane might have stimulated this mxaF-phylotype since its LP was lower in the methane-free treatment of the pH shift SIP experiment. In this treatment another Beijerinckiaceae-phylotype (OTUmxaF338) and a Methylorhabdus-affiliated phylotype (OTUmxaF18) were weakly labeled (Figure 2A and Supplementary Table S8, Figures S5–S7, S9).
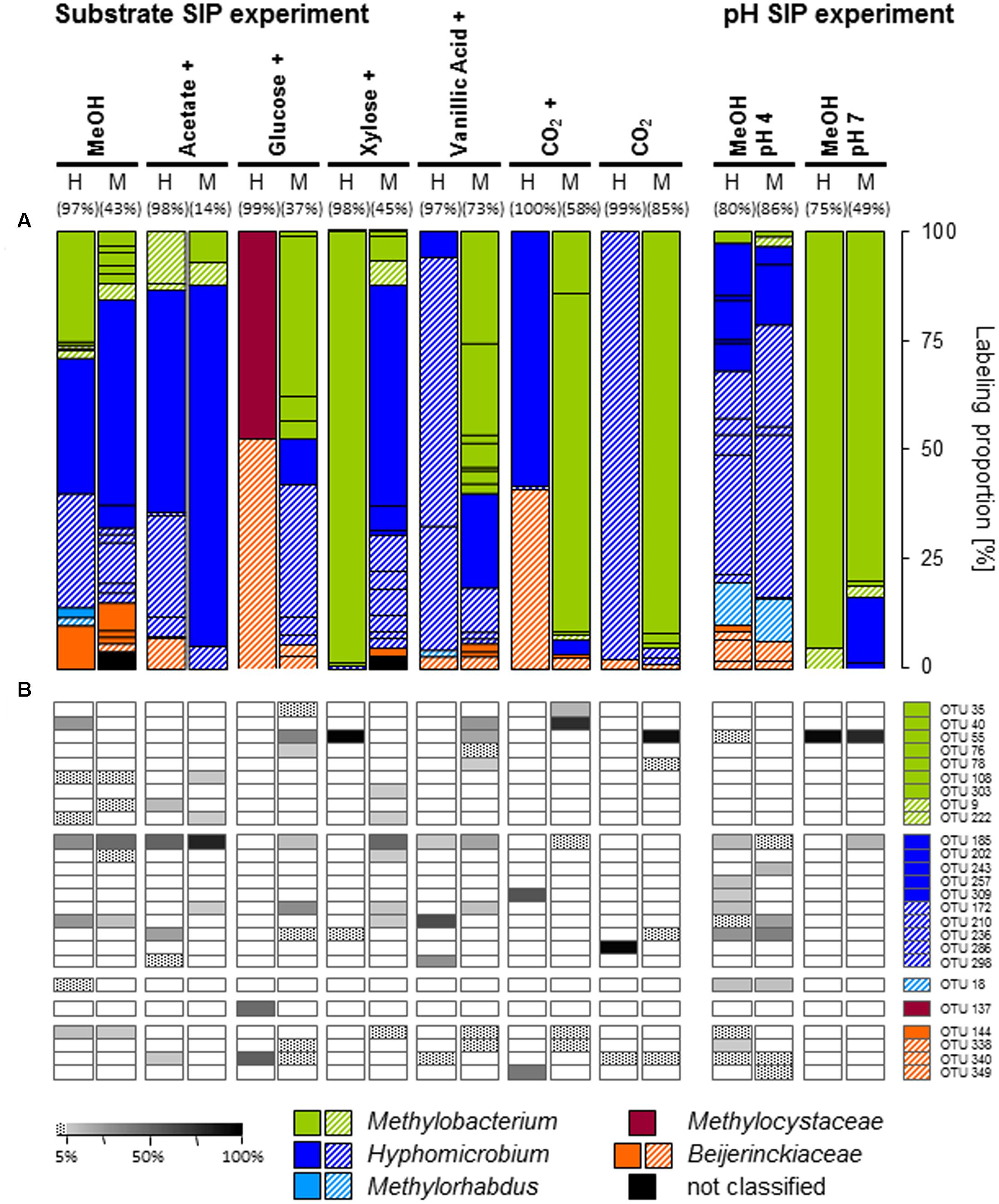
FIGURE 2. Labeling proportions and identities of mxaF phylotypes in the ‘heavy’ and ‘middle’ fractions of all labeled mxaF phylotypes in substrate or pH treatments (A) and the relative frequencies of labeled phylotypes (i.e., gray shades, ‘labeling proportion’ >5%; dotted, ‘labeling proportion’ <5%) (B). Cross, additional [12C]-methanol supplementation in the substrate SIP experiment. Equal colors, the same family affiliation. Shading, ambiguous affiliation (i.e., sequence identity with BLASTn <90% and an ambiguous position in the phylogenetic tree). ‘H’ and ’M,’ ‘heavy’ and ‘middle’ fractions, respectively. Values in brackets, contribution of labeled OTUs to the total number of sequences. The ‘labeling proportions’ are indicators for the relative importance of different taxa assimilating the supplemented 13C (directly or indirectly). More detailed taxonomic information can be found in Supplementary Table S15.
Fungi Assimilating Methanol-Derived Carbon
Apart from Bacteria also fungi assimilated methanol-derived carbon (Figure 3A and Supplementary Table S4). In the [13C1]-methanol treatment of the substrate SIP experiment, one abundantly labeled phylotype was affiliated to the basidiomycetous yeast Cryptococcus (OTUITS46). Further labeled phylotypes (LP ≥ 5%) were affiliated to Ascomycota (OTUsITS10, 32, and 2) and Zygomycota (OTUITS5; Mortierella sp., 98% sequence identity) (Figure 3A and Supplementary Tables S4, S5). A weak label was observed for phylotypes affiliated to Zygomycota (OTUITS112), Basidiomycota (OTUsITS22 and 27), and Ascomycota (OTUITS135) (Figure 3A and Supplementary Tables S4, S5). Consistently with the [13C1]-methanol treatment of the substrate SIP experiment, the zygomycetous phylotypes of the [13C1]-methanol treatment of the pH shift SIP experiment were affiliated to the genus Mortierella (Figure 3C and Supplementary Tables S5, S10). In contrast, further labeled fungal phylotypes were affiliated to Basidiomycota (OTUsITS30, 15, 12, and 7) (Figure 3C and Supplementary Table S10), and a remarkable number of weakly labeled phylotypes were affiliated to Ascomycota, Basidiomycota, and Zygomycota (Figure 3C and Supplementary Table S10).
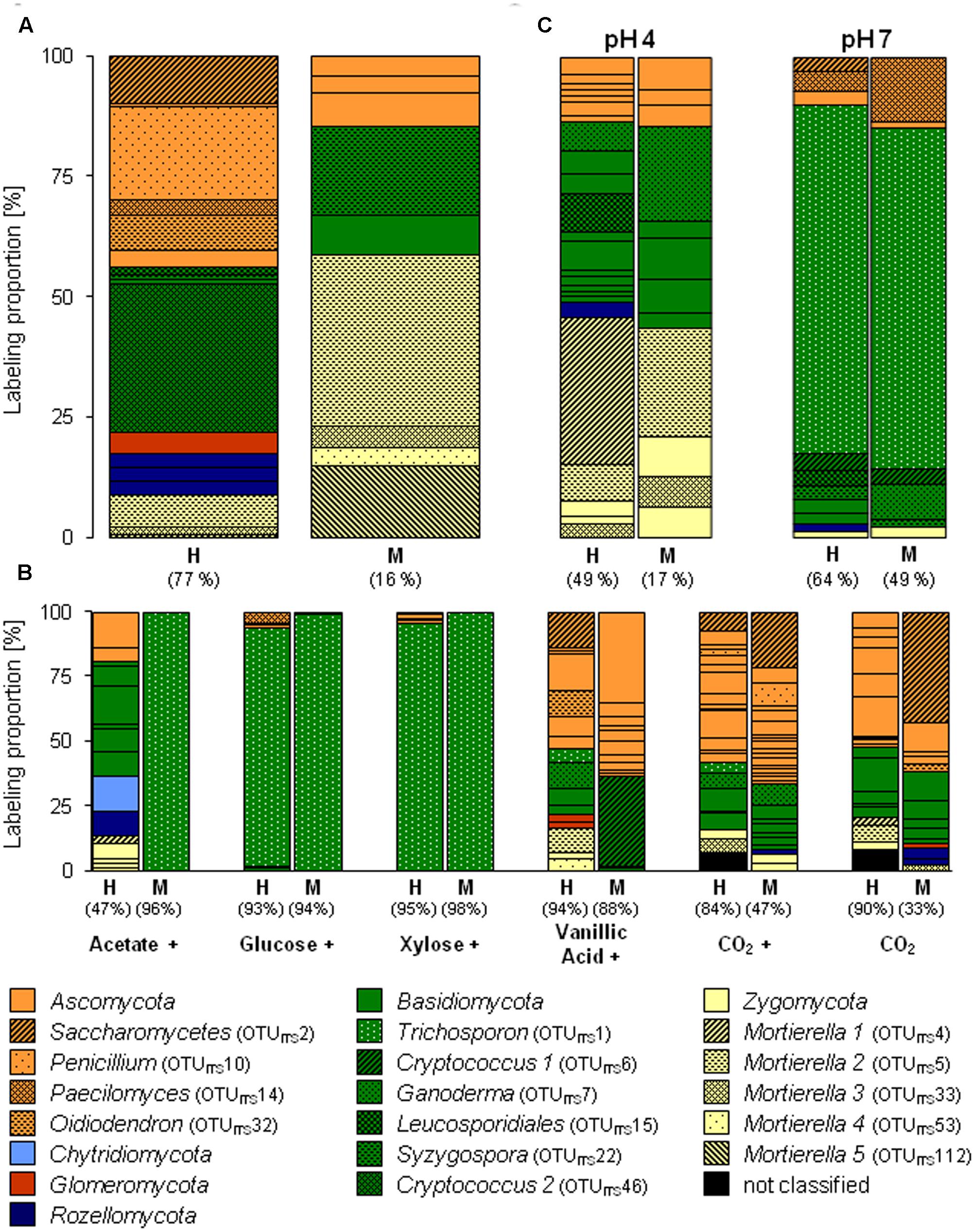
FIGURE 3. Labeling proportions and identities of fungal ITS phylotypes in the ‘heavy’ and ‘middle’ fractions of [13C1]-methanol (A) and different [13Cu]-substrates (B) treatments of the substrate SIP experiment, and of the treatments of the pH SIP experiment (C). Cross, additional [12C]-methanol supplementation in substrate treatments. Equal colors, the same phylum affiliation. ‘H’ and ‘M,’ ‘heavy’ and ‘middle’ fractions, respectively. Values in brackets, contribution of labeled OTUs to the total number of sequences. The ‘labeling proportions’ are indicators for the relative importance of different taxa assimilating the supplemented 13C (directly or indirectly).
Effect of pH on the Abundance of Bacteria and Methylotrophs
The quantification of the bacterial abundance (16S rRNA gene numbers) and mxaF gene numbers revealed an increase in both pH 7 treatments (i.e., the unsupplemented controls and methanol treatments), demonstrating the general growth restrictions at the in situ pH 4 for Bacteria and methylotrophs (Figure 4). However, a decrease of mmoX gene numbers in the pH 7 treatment occurred, whereas the values remained constant in the pH 4 treatments (Supplementary Figure S2) suggesting that the acidic in situ pH conditions were advantageous for methane-utilizing Beijerinckiaceae. Since the amplicons were not checked for their identity, a detection of non-target sequences cannot be excluded and thus this result should be regarded with caution.
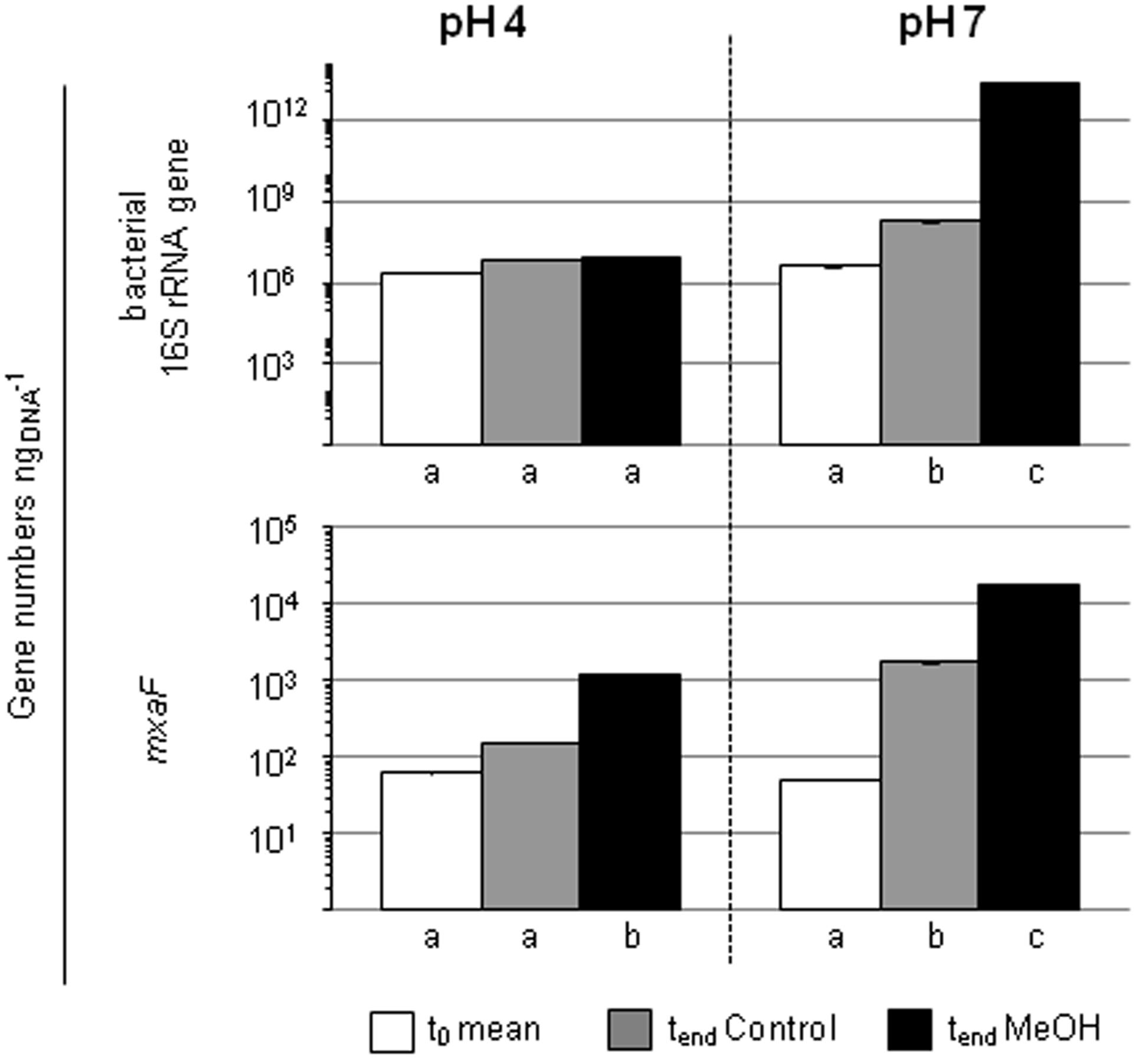
FIGURE 4. Gene numbers of 16S rRNA genes and mxaF of treatments with different pH in the pH shift SIP experiment. Columns, mean values of the experimental replicates. Error bars, standard deviation; if not visible, the variability between replicates was below 0.5%. Different letters, significant differences between samples (t-test; normal distribution was assumed based on the Shapiro-Wilk-test; n = 3).
Identification of Autochthonous Microorganisms that Utilized Methanol-Derived Carbon under Elevated pH Conditions
As expected several different phylotypes were labeled under the elevated pH condition indicating again the growth restricting conditions of the in situ acidic soil. In accordance with a generally increasing proportion of Bacteroidetes (Supplementary Figure S3A) in the pH 7 treatment, the most abundantly labeled phylotypes affiliated with Flavobacteriaceae (OTU16S1045, 99% sequence identity to Chryseobacterium sp.) and Microbacteriaceae (OTU16S721, 99% sequence identity to Leifsonia xyli) (Figure 1C and Supplementary Tables S2, S7). Weakly labeled phylotypes (i.e., LP < 3%) were affiliated to Sphingobacteriaceae, Caulobacteraceae, and Verrucomicrobia with partially low sequence identities to the next related sequence of a cultured isolate (Supplementary Table S2). A Methylophilaceae-related phylotype (OTU16S358) was only weakly labeled but with a high LP of approximately 20% (Figure 1C and Supplementary Tables S2, S7). This phylotype was not labeled in the pH 4 treatment (Figures 1A,C), which suggests a decreased competitiveness and initial low abundance under in situ conditions. Further weakly labeled phylotypes were affiliated with Bacteroidetes, Alphaproteobacteria, and Verrucomicrobia (Figure 1C and Supplementary Tables S2, S7, Figure S10).
In contrast to the aforementioned findings on labeled mxaF-phylotypes, a Methylobacterium-related phylotype (OTUmxaF55) was highly abundant indicating the preference of neutral pH conditions (Figure 2A and Supplementary Table S9).
A preference of a neutral pH was also observed for one fungal phylotype affiliated to the yeast Trichosporon (OTUITS1), that was abundantly labeled under an elevated pH. Further weakly labeled fungal phylotypes were affiliated to Ascomycota and Basidiomycota (Figure 3C and Supplementary Table S11), which suggests that these taxa did not play a dominant role in methanol assimilation.
The Substrate Range of Methanol-Derived Carbon-Utilizing Microorganisms
As expected, several taxa not known to include methylotrophs such as Xanthomonadaceae (OTU16S300, Rhodanobacter), Burkholderiaceae (OTU16S361, Burkholderia), and Sphingobacteriaceae (OTU16S1073, Mucilaginibacter) were labeled in the multi-carbon substrate treatments. If these microorganisms dissimilated methanol without assimilating carbon remains speculative. ‘True’ methanol and/or methanol-derived carbon utilizers (i.e., assimilation of carbon from methanol) were identified in the [13C1]-methanol treatment. The congruent detection of a phylotype in both, the methanol and an alternative substrate treatment, was considered to estimate the putative substrate range of methanol-derived carbon utilizers.
The Beijerinckiaceae-phylotype (OTU16S438) was labeled in all treatments with multi-carbon compounds, which suggests that this phylotype assimilates carbon derived from acetate, sugars and even aromatic compounds in the presence of methanol (Figure 5). A more detailed analysis on species-level revealed that several phylotypes were grouped together by OTU16S438 and that clearly different trophic types belonged to this OTU (Figure 6). Species-level phylotypes that were identified as obligately methylotrophic (A, B, and D) were closely affiliated with known methylotrophs of Beijerinckiaceae (i.e., Methylorosula, Methyloferula, and Methylovirgula) and Hyphomicrobiaceae (i.e., Hyphomicrobium). Phylotype C was identified as restricted facultatively methylotrophic and was closely affiliated with Methylocella. Further phylotypes (E, F, G, H, and I) were affiliated with several members of Rhizobiales but were apparently non-methylotrophic. Low LPs and sometimes only a weak labeling of the Beijerinckiaceae-affiliated phylotype (family-level) suggested a higher competition or slower growth rates with multi-carbon substrates. A potentially occuring cross-feeding via the assimilation of 13CO2 can be considered as negligible because OTU16S438 was not labeled in the 13CO2 treatments (Figures 1B, 5). However, the utilization of methanol cannot fully be excluded for the apparent non-methylotrophic phylotypes since SIP analysis cannot resolve the sole dissimilation of methanol or the indirect carbon assimilation via CO2.

FIGURE 5. Bacterial (A) and fungal (B) phylotypes labeled in the methanol treatment of the substrate SIP experiment and their congruent labeling in other treatments of both SIP experiments. Black, LP > 5%; gray, LP < 5%; white, not labeled. The phylotypes that were only labeled in treatments with multi-carbon substrates or in the pH shift SIP experiment are not presented. Cross, additional [12C]-methanol supplementation in substrate treatments. Equal colors, the same phylum affiliation. ‘H’ and ‘M,’ ‘heavy’ and ‘middle’ fractions, respectively.
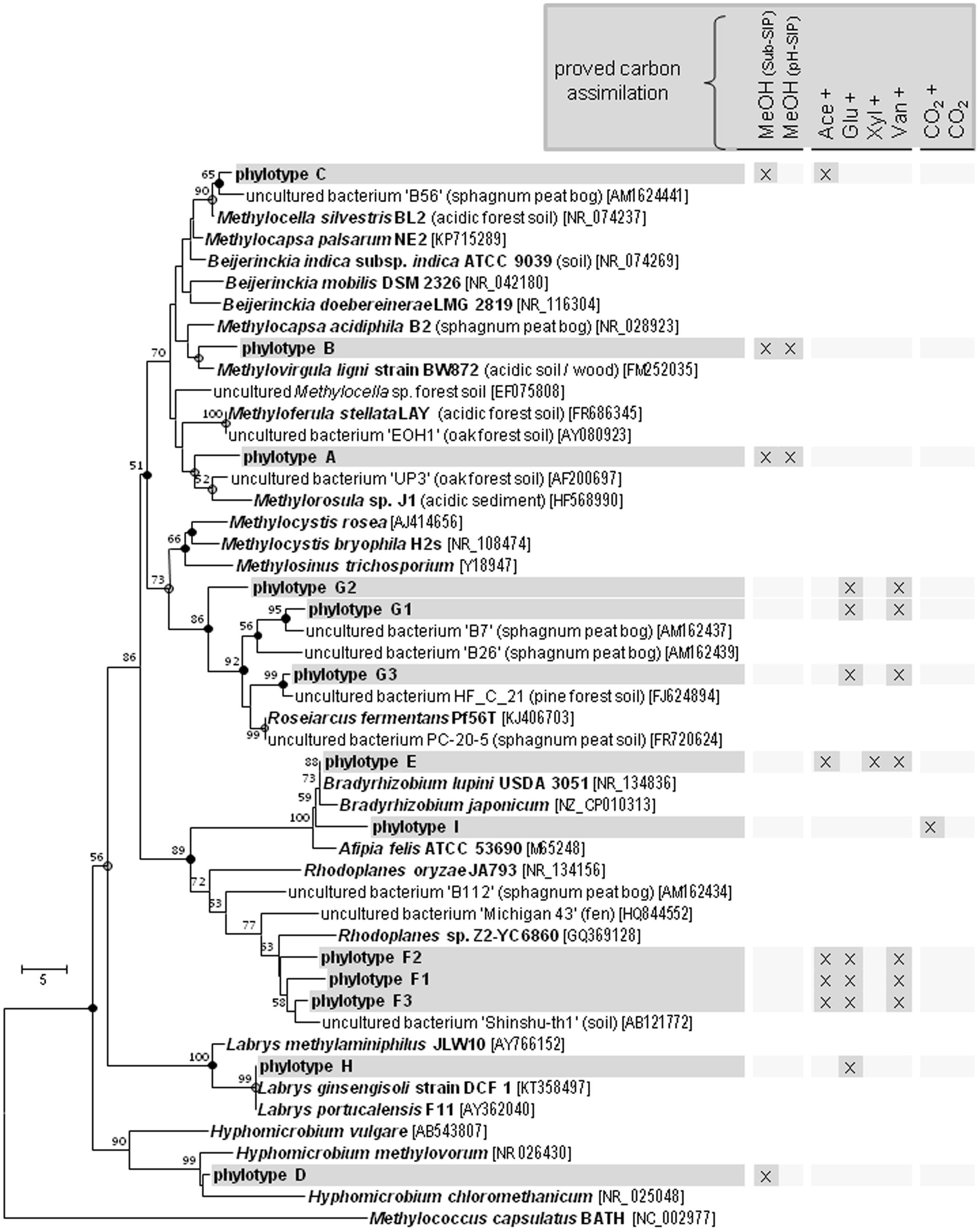
FIGURE 6. Species-Level resolution of the Beijerinckiaceae-affiliated phylotype OTU16S438. Species-level phylotypes of the Beijerinckiaceae-affiliated OTU16S438 ( , phylotype A to I based on species level similarity cut-off) and their putative substrate utilization. Outgroup, 16S rRNA sequence of Methylococcus capsulatus BATH. Bootstrap were based on 1000 replicated calculations. The trees was calculated with the neighborjoining method. Dots at the nodes, congruent nodes with trees caculated with the maximum likelihood and maximum parsimony method (●, true for three methods;
, phylotype A to I based on species level similarity cut-off) and their putative substrate utilization. Outgroup, 16S rRNA sequence of Methylococcus capsulatus BATH. Bootstrap were based on 1000 replicated calculations. The trees was calculated with the neighborjoining method. Dots at the nodes, congruent nodes with trees caculated with the maximum likelihood and maximum parsimony method (●, true for three methods;  , true for two methods). If known the isolation origin of each sequence is given in brackets. Accession numbers are given in squared brackets. The bar indicates 5 changes per nucleotide. ‘Sub-SIP,’ substrate SIP experiment; ‘pH-SIP,’ pH shift SIP experiment; cross, additional [12C]-methanol supplementation in substrate treatments of the substrate SIP experiment.
, true for two methods). If known the isolation origin of each sequence is given in brackets. Accession numbers are given in squared brackets. The bar indicates 5 changes per nucleotide. ‘Sub-SIP,’ substrate SIP experiment; ‘pH-SIP,’ pH shift SIP experiment; cross, additional [12C]-methanol supplementation in substrate treatments of the substrate SIP experiment.
Further alphaproteobacterial methanol-derived carbon-assimilating phylotypes exhibited both a broad substrate range (OTU16S449; Sphingomonadaceae) and a narrow substrate range (OTU16S467; Acetobacteraceae) (Figure 5). Interestingly, most of the 16S rRNA phylotypes that were only labeled in the pH 7 treatment were not labeled at the acidic in situ pH (Figure 5) suggesting an inhibition or even growth-restriction and thus no utilization of any multi-carbon substrate. Only the Microbacteriaceae-phylotype (OTU16S721, Leifsonia spp.) exhibited a broad substrate and pH range (Figure 5).
All mxaF phylotypes that were labeled in the methanol treatment of the substrate SIP experiment were also labeled in the multi-carbon substrate treatments (Figure 2B and Supplementary Table S3). A Hyphomicrobium-phylotype (OTUmxaF185) exhibited the broadest substrate range and was detectable in all treatments supplemented with methanol (12C and 13C). A labeling in both the acetate and vanillic acid treatments was observed with high LPs for acetate indicating a preference for this substrate (Figure 2B). The weak labeling in both sugar treatments suggested a low carbon assimilation rate and/or a general higher competitive pressure through non-methylotrophic heterotrophs (Figure 2B). OTUmxaF185 was also labeled in the pH shift SIP experiment, but a weaker labeling at pH 7 indicated a preference for acidic conditions (Figure 2B and Supplementary Tables S8, S9). The weak labeling of a Beijerinckiaceae-phylotype (OTUmxaF144) in treatments containing xylose, vanillic acid, or CO2 plus methanol (Figure 2B) suggested facultative methylotrophy with a preference for methanol and/or a slower growth rate on multi-carbon substrates. Apart from the already mentioned methylotrophic families a Methylocystaceae-affiliated phylotype (OTUmxaF137) was labeled in one multi-carbon substrate treatment (i.e., the treatments with [13C6]-glucose, Figure 2A). This phylotype might be only little competitve under methylotrophic conditions. Only a few mxaF phylotypes of Methylobacterium and Hyphomicrobium were solely labeled in multi-carbon substrate treatments. This observation suggests a preference for multi-carbon substrates over methanol by these phylotypes or a weak competiveness for methanol (Figure 2B). Under experimentally elevated pH another Methylobacterium-phylotype (OTUmxaF55) was abundantly labeled. Its facultatively methylotrophic lifestyle can be concluded from the fact that it was labeled in both sugar and vanillic acid treatments (Figure 2B and Supplementary Table S9). Obviously, OTUmxaF55 had a preference for more neutral pH conditions, because it was only weakly labeled in the pH 4 treatment (Figure 2B and Supplementary Tables S8, S9).
The fungus Cryptococcus (OTUITS46; Basidiomycota) was abundant in the H fraction of labeled fungal phylotypes in the [13C1]-methanol treatment of the substrate SIP experiment (Figures 3, 5). Although a general increase in the relative abundance of Cryptococcus-related phylotypes was observed in the vanillic acid treatment (Supplementary Table S12), another Cryptococcus phylotype (OTUITS6) was weakly labeled in the vanillic acid treatment (Figure 5). An assimilation of the methyl group of aromatic compounds is conceivable, as well as the possibility of utilizing carbon derived from the breakdown products of vanillic acid.
Three ascomycetous phylotypes (OTUITS2, Saccharomycetes; OTUITS32, Oidiodendron; and OTUITS5, Mortierella) were labeled in the methanol and vanillic acid treatments and Mortierella-related phylotypes were detectable with low LPs in both methanol treatments at the in situ pH and the vanillic acid treatment (Figures 3, 5). The broadest substrate range was revealed by a Trichosporon-phylotype (OTUITS1) labeled in acetate, sugar and vanillic acid treatments. Moreover, methanol utilization cannot be excluded for this phylotype because it was also labeled in the pH 7 treatment of the pH shift SIP experiment, which suggests an assimilation of methanol or at least the utilization of methanol-derived carbon (Figures 3, 5).
Discussion
Stable isotope probing combined with high-throughput sequencing have expanded our knowledge on the phylogenetic diversity and global distribution of methylotrophs in terrestrial ecosystems (e.g., Kolb and Stacheter, 2013; Stacheter et al., 2013; Chistoserdova, 2015). However, their significant role in ecosystem-level methanol cycling has been largely neglected, although terrestrial ecosystems are strong sources and sinks. Thus, methylotrophs have a direct impact on global methanol fluxes and consequently on the global atmospheric chemistry (Galbally and Kirstine, 2002; Kolb, 2009; Stacheter et al., 2013). Furthermore, the role of methylotrophic yeasts and fungi, has hardly been investigated in soils as well as the impact of methanol-derived carbon on the soil microbial food web (Lueders et al., 2004; Kolb, 2009).
The Dominant Methanol-Utilizing Bacteria Possess a Restricted Substrate Range
The main bacterial methanol-utilizers were affiliated with Beijerinckiaceae. This finding is consistent with studies on other acidic soils and species descriptions of acidophilic methylotrophic Beijerinckiaceae (Radajewski et al., 2000, 2002; Dedysh et al., 2001, 2006; Morris et al., 2002; Marín and Arahal, 2013 and references therein). The genera Chelatococcus and Camelimonas were excluded from all considerations on Beijerinckiaceae, since they are not capable of C1 compound utilization (Dedysh et al., 2016). Beijerinckiaceae comprises strains with remarkably different metabolic capacities including chemoheterotrophy (Beijerinckia), facultative methylotrophy (Beijerinckia, Methylorosula, Methylovirgula), ‘restricted’ facultative methanotrophy (Methylocella, Methylocapsa), and obligate methanotrophy (Methylocapsa, Methyloferula) (Marín and Arahal, 2013 and references therein; Tamas et al., 2014; Dedysh et al., 2016).
The metabolic behavior of facultative methylotrophs under in situ conditions has not been resolved, since multi-carbon utilization studies are often conducted as a comparison between methylotrophic (only C1 compounds being supplemented) and multi-carbotrophic (only multi-carbon compounds being supplemented) conditions. Such studies on pure cultures of Methylobacterium extorquens AM1 revealed a high methanol oxidation capacity in the presence of alternative multi-carbon substrates (i.e., multi-carbotrophic followed by methylotrophic conditions) or mixed substrate conditions suggesting that methanol is primarily used for energy conservation (Bosch et al., 2008; Skovran et al., 2010; Peyraud et al., 2011, 2012). Regrettably, such carbon flux studies are not available for members of the Beijerinckiaceae but we assume a similar regulation phenomenon in our experiment. We detected a Beijerinckiaceae-affiliated phylotype (OTU16S438), that comprises different trophic types on species-level resolution. However, due to the limited phylogenetic resolution of the analyzed 16S rRNA gene fragment (444 bp) and the close relation of Rhizobiales members, an unequivocal determination of the genus or species would still be rather vague. The species-level phylotypes A, B, C, and D were closely affiliated with known methylotrophs. The phylotypes A, B, and D were apparently limited in their substrate range to methanol only. The phylotype C revealed a restricted substrate range including methanol and acetate and was closely related with Methylocella, which was the first genus reported as facultatively methanotrophic (Dedysh et al., 2005a). Among the facultatively methylotrophic Beijerinckiaceae the documented multi-carbon substrate range is also usually limited to only a few carboxylic acids (Dedysh et al., 2005a; Vorobev et al., 2009; Marín and Arahal, 2013). There exist only two exceptions – (i) Beijerinckia mobilis, that is to date the only known methanol-utilizer of the metabolically versatile genus Beijerinckia, and (ii) Methylorosula polaris, which exhibits a broad substrate range including sugars and polysaccharides (Dedysh et al., 2005b; Berestovskaya et al., 2012; Marín and Arahal, 2013). However, in our substrate SIP experiment none of these facultative methylotrophs assimilated carbon from multi-carbon substrates.
Apart from a direct substrate utilization several methylotrophic Beijerinckiaceae assimilate carbon at the level of CO2 such as Methyloferula, Methylorosula, and Methylovirgula (Marín and Arahal, 2013 and references therein; Vorobev et al., 2011). Thus, the oxidation of endogenous or supplemented substrates to CO2 by chemoheterotrophic microorganisms might have supported methylotrophic Beijerinckiaceae and could have led to a dilution of a conceivable 13C-signal, although we tried to minimize such an effect by several experimental measures.
A potentional to utilize methane was somewhat likely for some of the labeled Beijerinckiaceae-associated phylotypes since mmoX genes (encoding for Beijerinckiaceae-associated sMMO) were detectable by qPCR (Dedysh et al., 2005a; Vorobev et al., 2011; Marín and Arahal, 2013). However, a final proof of their identities by sequencing of the amplicon is missing. Additionally, a growth stimulating effect of methane was also observed when comparing both methanol treatments of both SIP experiments (i.e., the substrate SIP and the pH shift SIP experiment), which revealed a higher LP when methane was available. Taken together, our findings suggest that (i) Beijerinckiaceae-affiliated taxa were important methanol-utilizers in the forest soil, (ii) that their substrate range might be strongly limited to methanol under in situ conditions, and (iii) that the capability of methanol utilization defines their ecological niche in a complex forest soil microbiome.
All apparently non-methylotrophic species-level members of the OTU16S438 (phylotypes E to I) affiliated with different members of Rhizobiales. For these phylotypes an assimilation of carbon derived from methanol could not be confirmed.
The phylotypes E and I were affiliated to Bradyrhizobiaceae that are trophically versatile. Methylotrophy cannot be fully excluded since Bradyrhizobium species posses xoxF genes and MDH activity but exhibit only weak growth (Kaneko et al., 2002; Sudtachat et al., 2009; Fitriyanto et al., 2011; de Souza et al., 2013). Thus, the low concentrations of methanol (i.e., 1 mM per pulse) led likely to a low 13C incorporation and might have prevented the verification of methanol utilization. The substrate range of the phylotype E included acetate, xylose, and vanillic acid. Utilization of methylated aromatic compounds causes an upregulated expression of C1 metabolism genes such as xoxF (Ito et al., 2006). The observed ability of Bradyrhizobium species to fix CO2 is in accordance with the detection of phylotype I in the CO2 treatments (Masuda et al., 2010). Thus, CO2 fixation might have caused a dilution of the 13C signal and thus prevented the proof of methanol utilization.
Phylotype H was affiliated to Labriaceae (Labrys). Isolates of this family possess xoxF-like genes (Beck et al., 2015). The only methylotrophic species reported to date is Labrys methylaminiphilus that utilizes methylamine and several monosaccharides, but not methanol (Miller et al., 2005). However, also L. monachus can utilize methanol to a certain extent (Miller et al., 2005). Thus, the genus Labrys might comprise hitherto unknown methanol utilizers.
The phylotypes F and G were closely affiliated to environmental sequences. Classification revealed an affiliation with Rhodoplanes (Hyphomicrobiaceae). Nonetheless, several reads of phylotype F were closely related with Methylocystaceae (Methylosinus) and Xanthobacteraceae (Variibacter, Pseudolabrys) rendering an unambiguous affiliation hardly possible. Interestingly, studies in peat bogs addressing methanotrophs identified also sequences somehow affiliated to Rhodoplanes by using methanotrophic specific probes (i.e., Methylosinus specific) but were also not able to verify methylotrophy of these taxa (McDonald et al., 1996, 1999). Thus, this cluster of Rhodoplanes-affiliated sequences remains enigmatic regarding methylotrophy. Another still enigmatic taxon is phylotype G that was affiliated with several environmental sequences that were in turn affiliated with Methylocystaceae and Beijerinckiaceae. Within this cluster, only one species (Roseiarcus fermentans) has been taxonomically described. However, this species cannot utilize C1 compounds (Kulichevskaya et al., 2014).
Discrepancy of Identified Methanol-Utilizers Based on 16S rRNA Gene and mxaF
Most mxaF phylotypes were affiliated with Methylobacteriaceae and Hyphomicrobiaceae instead of Beijerinckiaceae, and no significant similarity to mxaF from a previous study on a forest soil or mxaF genes of Methylovirgula ligni was evident (Radajewski et al., 2002; Vorobev et al., 2009). Beijerinckiaceae can harbor mxaF sequences, which are similar to that of Methylobacterium or Hyphomicrobium due to horizontal gene transfer (HGT) events that occurred during the evolution of this family (Lau et al., 2013; Tamas et al., 2014).
Interestingly, one mxaF phylotype affiliated with Methylocystaceae (OTUmxaF137) was labeled under mixed substrate conditions with glucose although Methylocystaceae were not abundant in the bacterial community based on both gene markers (16S rRNA and mxaF, Supplementary Figures S2, S8). Methylocystaceae comprise restricted facultative methanotrophs, i.e., strains of the Methylocystis slowly grow on acetate or ethanol (Belova et al., 2011). However, the utilization of glucose has never been reported. Thus, it is possible that OTUmxaF137 is a hitherto unknown glucose-utilizing member of Methylocystaceae or the specific mxaF genotype has been horizontally transferred to the methanotrophic sister genera.
Another aspect is the lack of detection of xoxF genes. Both genes possess different functions and phylogenetic distributions. At the time conducting our experiments an unequivocal grouping between mxaF genes and five distinct clades of xoxF genes (xoxF1 to xoxF5) was known (Chistoserdova, 2011; Keltjens et al., 2014). Only recently, xoxF gene sequences were analyzed in detail, which enabled the development of divergent xoxF primers for the different clades (Taubert et al., 2015). Thus, targeting both mxaF and xoxF genes with only one primer pair might result in biased amplification results toward mxaF.
Putative Fungal Methanol-Utilizers
Until now, only a limited number of methylotrophic fungi belonging to yeasts or molds (mainly Ascomycota), have been reported (Kolb, 2009; Kolb and Stacheter, 2013). In our study, fungal phylotypes that assimilated methanol-derived carbon were basidiomycetous yeasts (Tremellomycetes), in particular Cryptococcus and Trichosporon, and the zygomycetous genus Mortierella. These genera are globally abundant in soil and are saprotrophs that utilize degradation products of plants (Botha, 2011; Voříšková and Baldrian, 2013). Cryptococcus species might be involved in methane cycling (Takishita et al., 2006) and can be associated with bacterial methylotrophs (Methylorosula sp.) (Delavat et al., 2013). For Trichosporon methylotrophy has been documented (Kaszycki et al., 2006). The zygomycetous Mortierella are widespread and are generalistic saprotrophic fungi with the ability to degrade complex plant material (Dix and Webster, 1995; Kjøller and Struwe, 2002; Hanson et al., 2008; Buée et al., 2009). Since Cryptococcus and Mortierella are saprotrophic and have a broad range of substrates, a labeling by cross feeding on labeled bacteria might have been an alternative route of carbon assimilation. Nevertheless, an assimilation of methanol cannot be excluded rendering these fungi new candidate methylotrophs that need further experimental attention.
Influence of an Elevated pH on the Indigenous Methanol-Utilizing Microbiome
Soil is not homogeneous and microscale habitats exist. For example, within few millimetres of soil, pH values can differ up to one pH unit (Or et al., 2007). Thus, we wanted to address if and how the indigenous methanol-utilizers might have been affected to elevated pH values. The total bacterial community was significantly influenced by the increased pH as expected (Supplementary Table S13), and the active methanol-utilizing taxa shifted compared to those under in situ pH.
We identified phylotypes of the phyla Bacteroidetes, Actinobacteria, and Betaproteobacteria as methanol-utilizers at a neutral pH. Among the Bacteroidetes, a small number of methylotrophs belonging to Flavobacteriia and Sphingobacteriia have been reported but none among Flavobacteriaceae (Boden et al., 2008; Madhaiyan et al., 2010; Kolb and Stacheter, 2013). Among the Actinobacteria, strains of Leifsonia are methanol utilizers (Hung et al., 2011). The detected Betaproteobacteria-affiliated phylotype was weakly labeled at neutral pH, but was affiliated to the well-known methylotrophic Methylophilaceae, of which isolates are neutro- to alcaliphilic (Doronina et al., 2013). Very likely, this phylotype had a lower competitiveness compared with the main bacterial methanol utilizers – i.e., Beijerinckiaceae. Although Methylophilaceae have been reported to be trophically versatile (Doronina et al., 2013), we did not detect this phylotype in any multi-carbon substrate treatment, which supports the hypothesis that this phylotype thrived in situ under unfavorable conditions.
Although the pH shift did not significantly affect the total fungal community (Supplementary Table S14), a reducing effect of the elevated pH on the alpha diversity of active methanol-derived carbon-utilizing fungi was likely based on the observation of a lower number of labeled phylotypes at neutral pH. Cryptococcus and Mortierella comprise several acid-tolerant species, which likely can also grow under neutral pH conditions (Gross and Robbins, 2000). However, their pH optima seem to be restricted to values < 7, which may explain why Trichosporon outcompeted Cryptococcus and Mortierella species at pH 7 (Gross and Robbins, 2000). The only known methylotrophic Trichosporon strain has a growth optimum of pH 8 (Kaszycki et al., 2006). Methylotrophy was not tested at an acidic pH. Since the Trichosporon phylotype also incorporated carbon from [13Cu]-glucose and [13Cu]-xylose at pH 4, a broader pH optimum of this taxon of potentially methylotrophic soil yeasts is likely.
A Methanol-Driven Microbial Food Web in the Investigated Acidic Forest Soil
The detection of several other bacterial and fungal organisms assimilating methanol or methanol-derived carbon suggested a tight trophic link between Beijerinckiaceae and other microorganims in the soil. On the one hand Beijerinckiacea might have provided carbon sources for several bacterial taxa (i.e., Acidobacteria, Planctomycetes, non-methanotrophic Verrucomicrobia and Actinobacteria), resulting in a weak label in methanol treatments. Planctomycetes and Verrucomicrobia can degrade extracellular polysaccharides (EPSs) produced by Beijerinckiaceae (Wang et al., 2015). Peat-derived Acidobacteria were enriched with methanol, glucose, or xylan; but isolation on solely methanol was unsuccessful (Pankratov et al., 2008) suggesting an indirect stimulation. Edaphobacter aggregans that grows only in co-culture with Methylocella silvestris suggesting methylotrophic Beijerinckiaceae as effective suppliers of carbon through EPS formation (Koch et al., 2008). Actinobacteria might also utilize other compounds of Beijerinckiaceae. Kineosporia spp. (OTU16S703, Actinomycetales) can grow on DNA (Kudo et al., 1998) and therefore both DNA and EPS from Beijerinckiaceae might have served as carbon sources. Furthermore, autotrophic growth has been reported for some Acidimicrobiales (Johnson et al., 2009) and methanotrophic Verrucomicrobia (Khadem et al., 2011; Sharp et al., 2014). Since we detected Verrucomicrobia by SIP in treatments that were supplemented with 13CO2, cross labeling through 13CO2 was somewhat likely, although we regularly exchanged the headspace atmosphere to minimize this experimental artifact.
On the other hand, soil Beijerinckiaceae might feed on methanol released by fungi during lignin decomposition (Messner et al., 2003). An example, for which such a trophic link has been suggested is Methylovirgula ligni isolated from a decaying wood that was substantially colonized by a white-rot fungus (Folman et al., 2008; Vorobev et al., 2009). The same might be true for members of the Actinobacteria (i.e., Actinomycetes) and Planctomycetes (i.e., Phycisphaerae) of which both members of both phyla might be involved in biopolymer degradation (McCarthy, 1978; Suneetha and Khan, 2010; Bienhold et al., 2013). Thus, it is conceivable that the fungal and bacterial activity increased the local concentration of methanol when degrading plant residues and thus support methylotrophic Beijerinckiaceae.
Conclusion
Our study revealed acidotolerant Beijerinckiaceae as the main bacterial methanol sink in a decidous forest soil and highlights their importance for the conversion of methanol in forest soils. These methanol-utilizing Bacteria revealed a clear preference for C1 compounds that likely enabled them to establish in a complex soil microbiome. The utilization of methanol as sole energy source of various taxa of this family cannot be excluded. We also detected soil yeasts, such as Cryptococcus and Trichosporon, and saprotrophic Mortierella, which suggests that these fungi need to be carefully checked if they are indeed not able to grow on methanol. A putative carbon cross-feeding due to secondarily 13C-assimilation especially for fungal species (saprotrophic fungi) cannot be excluded. Nonetheless, the headspace of experimental flasks were reguarly flushed with fresh air and 12CO2 was added to dilute formed to prevent 13CO2 labeling through this compound. The observed and discussed aspects of the interaction of Beijerinckiaceae, yeasts, fungi, and non-methylotrophic heterotrophic Bacteria suggests that these microorganisms are tightly trophically linked through methanol release from plant organs and residues in the surface soil horizons of deciduous temperate forests. Eventually, we provided evidence that soil pH and the substrate spectrum are crucial factors that define the ecological niches of soil methanol utilizers.
Author Contributions
MM conducted the experiments and wrote the first draft of the manuscript. HH, DKW, GL, and TW analyzed ITS data and contributed to interretation of fungi-associated results. EK supported bioinformatic amplicon analysis and was involved in the interpretation of the data. SK conceptualized the study and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was funded by grants of the Deutsche Forschungsgemeinschaft (DFG Ko 2912/5-1) and funds of the Institute of Landscape Biogeochemistry at the Leibniz Centre for Agricultural Research e.V. (ZALF) (Müncheberg, Germany). We thank Beatrix Schnabel for technical assistance and members of the Göttingen Genomics Lab (University of Göttingen; Germany) for technical assistance with pyrosequencing. The authors are very grateful for Harold L. Drake, who hosted and supported the project at the Department of Ecological Microbiology of the University of Bayreuth (Germany).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01361/full#supplementary-material
References
Beck, D. A., McTaggart, T. L., Setboonsarng, U., Vorobev, A., Goodwin, L., Shapiro, N., et al. (2015). Multiphyletic origins of methylotrophy in Alphaproteobacteria, exemplified by comparative genomics of Lake Washington isolates. Environ. Microbiol. 3, 547–554. doi: 10.1111/1462-2920.12736
Belova, S. E., Baani, M., Suzina, N. E., Bodelier, P. L. E., Liesack, W., and Dedysh, S. N. (2011). Acetate utilization as a survival strategy of peat-inhabiting Methylocystis spp. Environ. Microbiol. Rep. 3, 36–46. doi: 10.1111/j.1758-2229.2010.00180.x
Berestovskaya, J. J., Kotsyurbenko, O. R., Tourova, T. P., Kolganova, T. V., Doronina, N. V., Golyshin, P. N., et al. (2012). Methylorosula polaris gen. nov., sp. nov., an aerobic, facultatively methylotrophic psychrotolerant bacterium from tundra wetland soil. Int. J. Syst. Evol. Microbiol. 62, 638–646. doi: 10.1099/ijs.0.007005-0
Berry, D., Ben Mahfoudh, K., Wagner, M., and Loy, A. (2011). Barcoded primers used in multiplex amplicon pyrosequencing bias amplification. Appl. Environ. Microbiol. 77, 7846–7849. doi: 10.1128/AEM.05220-11
Bienhold, C., Pop Ristova, P., Wenzhöfer, F., Dittmar, T., and Boetius, A. (2013). How deep-sea wood falls sustain chemosynthetic life. PLoS ONE 8:e53590. doi: 10.1371/journal.pone.0053590
Boden, R., Thomas, E., Savani, P., Kelly, D. P., and Wood, A. P. (2008).Novel methylotrophic bacteria isolated from the River Thames (London, UK). Environ. Microbiol. 10, 3225–3236. doi: 10.1111/j.1462-2920.2008.01711.x
Bosch, G., Skovran, E., Xia, Q., Wang, T., Taub, F., Miller, J. A., et al. (2008). Comprehensive proteomics of Methylobacterium extorquens AM1 metabolism under single carbon and non methylotrophic conditions. Proteomics 8, 3494–3505. doi: 10.1002/pmic.200800152
Botha, A. (2011). The importance and ecology of yeasts in soil. Soil Biol. Biochem. 43, 1–8. doi: 10.1016/j.soilbio.2010.10.001
Bragg, L., Stone, G., Imelfort, M., Hugenholtz, P., and Tyson, G. W. (2012). Fast, accurate error-correction of amplicon pyrosequences using Acacia. Nat. Methods 9, 425–426. doi: 10.1038/nmeth.1990
Buée, M., Reich, M., Murat, C., Morin, E., Nilsson, R. H., Uroz, S., et al. (2009). 454 pyrosequencing analyzes of forest soils reveal an unexpectedly high fungal diversity. New Phytol. 184, 449–456. doi: 10.1111/j.1469-8137.2009.03003.x
Chistoserdova, L. (2011). Modularity of methylotrophy, revisited. Environ. Microbiol. 13, 2603–2622. doi: 10.1111/j.1462-2920.2011.02464.x
Chistoserdova, L. (2015). Methylotrophs in natural habitats: current insights through metagenomics. Appl. Microbiol. Biotechnol. 99, 5763–5779. doi: 10.1007/s00253-015-6713-z
Chistoserdova, L., Kalyuzhnaya, M. G., and Lidstrom, M. E. (2009). The expanding world of methylotrophic metabolism. Annu. Rev. Microbiol. 63, 477–499. doi: 10.1146/annurev.micro.091208.073600
Chistoserdova, L., and Lidstrom, M. E. (2013). “Aerobic methylotrophic prokaryotes,” in The Prokaryotes – Prokaryotic Physiology and Biochemistry, eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (New York, NY: Springer), 267–285.
Dallinger, A., and Horn, M. A. (2014). Agricultural soil and drilosphere as reservoirs of new and unusual assimilators of 2,4-dichlorophenol carbon. Environ. Microbiol. 16, 84–100. doi: 10.1111/1462-2920.12209
de Souza, J. A. M., Carrareto Alves, L. M., de Mello Varani, A., and de Macedo Lemos, E. G. (2013). “The family Bradyrhizobiaceae,” in The Prokaryotes – Alphaproteobacteria and Betaproteobacteria, eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (New York, NY: Springer), 135–154.
Dedysh, S. N., Derakshani, M., and Liesack, W. (2001). Detection and enumeration of methanotrophs in acidic Sphagnum peat by 16S rRNA fluorescence in situ hybridization, including the use of newly developed oligonucleotide probes for Methylocella palustris. Appl. Environ. Microbiol. 67, 4850–4857. doi: 10.1128/AEM.67.10.4850-4857.200
Dedysh, S. N., Haupt, E. S., and Dunfield, P. F. (2016). Emended description of the family Beijerinckiaceae and transfer of the genera Chelatococcus and Camelimonas to the family Chelatococcaceae fam. nov. Int. J. Syst. Evol. Microbiol. 66, 3177–3182. doi: 10.1099/ijsem.0.001167
Dedysh, S. N., Knief, C., and Dunfield, P. F. (2005a). Methylocella species are facultatively methanotrophic. J. Bacteriol. 187, 4665–4670. doi: 10.1128/JB.187.13.4665-4670.2005
Dedysh, S. N., Pankratov, T. A., Belova, S. E., Kulichevskaya, I. S., and Liesack, W. (2006). Phylogenetic analysis and in situ identification of bacteria community composition in an acidic Sphagnum peat bog. Appl. Environ. Microbiol. 72, 2110–2117. doi: 10.1128/AEM.72.3.2110-2117.2006
Dedysh, S. N., Smirnova, K. V., Khmelenina, V. N., Suzina, N. E., Liesack, W., Yuri, A., et al. (2005b). Methylotrophic autotrophy in Beijerinckia mobilis. J. Bacteriol. 187, 3884–3888.
Degelmann, D. M., Borken, W., Drake, H. L., and Kolb, S. (2010). Different atmospheric methane-oxidizing communities in European beech and Norway spruce soils. Appl. Environ. Microbiol. 76, 3228–3235. doi: 10.1128/AEM.02730-09
Degelmann, D. M., Borken, W., and Kolb, S. (2009). Methane oxidation kinetics differ in European beech and Norway spruce soils. Eur. J. Soil Sci. 60, 499–506. doi: 10.1111/j.1365-2389.2009.01138.x
Delavat, F., Lett, M. C., and Lièvremont, D. (2013). Yeast and bacterial diversity along a transect in an acidic, As–Fe rich environment revealed by cultural approaches. Sci. Total Environ. 46, 823–828. doi: 10.1016/j.scitotenv.2013.06.023
Doronina, N., Kaparullina, E., and Trotsenko, Y. (2013). “The family Methylophilaceae,” in The Prokaryotes – Alphaproteobacteria and Betaproteobacteria, eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (New York, NY: Springer), 869–880.
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Fall, R., and Benson, A. A. (1996). Leaf methanol – the simplest natural product from plants. Trends Plant Sci. 1, 296–301. doi: 10.1016/S1360-1385(96)88175-0
Fitriyanto, N. A., Fushimi, M., Matsunaga, M., Pertiwiningrum, A., Iwama, T., and Kawai, K. (2011). Molecular structure and gene analysis of Ce3+ induced methanol dehydrogenase of Bradyrhizobium sp. MAFF211645. J. Biosci. Bioeng. 6, 613–617. doi: 10.1016/j.jbiosc.2011.01.015
Foerstner, K. U., von Mering, C., Hooper, S. D., and Bork, P. (2005). Environments shape the nucleotide composition of genomes. EMBO Rep. 6, 1208–1213. doi: 10.1038/sj.embor.7400538
Folman, L. B., Klein Gunnewiek, P. J., Boddy, L., and de Boer, W. (2008). Impact of white-rot fungi on numbers and community composition of bacteria colonizing beech wood from forest soil. FEMS Microbiol. Ecol. 63, 181–191. doi: 10.1111/j.1574-6941.2007.00425.x
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Galbally, I. E., and Kirstine, W. (2002). The production of methanol by flowering plants and the global cycle of methanol. J. Atmos. Chem. 43, 195–229. doi: 10.1023/A:1020684815474
Gardes, M., and Bruns, T. D. (1993). ITS primers with enhanced specificity for basidiomycetes - application to the identification of mycorrhizae and rusts. Mol. Ecol. 2, 113–118. doi: 10.1111/j.1365-294X.1993.tb00005.x
Griffiths, R. I., Whiteley, A. S., O’Donnell, A. G., and Bailey, M. J. (2000). Rapid method for coextraction of DNA and RNA from natural environments for analysis of ribosomal DNA- and rRNA-based microbial community composition. Appl. Environ. Microbiol. 66, 5488–5491. doi: 10.1128/AEM.66.12.5488-5491.2000
Gross, S., and Robbins, E. I. (2000). Acidophilic and acid-tolerant fungi and yeasts. Hydrobiologia 433, 91–109. doi: 10.1023/A:1004014603333
Gvozdev, A. R., Tukhvatullin, I. A., and Gvozdev, R. I. (2012). Quinone-dependent alcohol dehydrogenases and FAD-dependent alcohol oxidases. Biochemistry 77, 843–856. doi: 10.1134/S0006297912080056
Hanson, C. A., Allison, S. D., Bradford, M. A., Wallenstein, M. D., and Treseder, K. K. (2008). Fungal taxa target different carbon sources in forest soil. Ecosystems 11, 1157–1167. doi: 10.1007/s10021-008-9186-4
Hedderich, R., and Withman, W. B. (2013). “Physiology and biochemistry of the methane-producing Archaea,” in The Prokaryotes – Prokaryotic Physiology and Biochemistry, eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (New York, NY: Springer), 635–662.
Herlemann, D. P., Labrenz, M., Jürgens, K., Bertilsson, S., Waniek, J. J., and Andersson, A. F. (2011). Transitions in bacterial communities along the 2000km salinity gradient of the Baltic Sea. ISME J. 5, 1571–1579. doi: 10.1038/ismej.2011.41
Holmes, A. J., Costello, A., Lidstrom, M. E., and Murrell, J. C. (1995). Evidence that particulate methane monooxygenase and ammonia monooxygenase may be evolutionarily related. FEMS Microbiol. Lett. 132, 203–208. doi: 10.1111/j.1574-968.1995.tb07834.x
Hung, W.-L., Wade, W. G., Boden, R., Kelly, D. P., and Wood, A. P. (2011). Facultative methylotrophs from the human oral cavity and methylotrophy in strains of Gordonia, Leifsonia, and Microbacterium. Arch. Microbiol. 193, 407–417. doi: 10.1007/s00203-011-0689-6
Ito, N., Itakura, M., Eda, S., Saeki, K., Oomori, H., Yokoyama, T., et al. (2006). Global gene expression in Bradyrhizobium japonicum cultured with vanillin, vanillate, 4-hydroxybenzoate and protocatechuate. Microbes Environ. 21, 240–250. doi: 10.1264/jsme2.21.240
Johnson, D. B., Bacelar-Nicolau, P., Okibe, N., Thomas, A., and Hallberg, K. B. (2009). Ferrimicrobium acidiphilum gen. nov., sp. nov. and Ferrithrix thermotolerans gen. nov., sp. nov.: heterotrophic, iron-oxidizing, extremely acidophilic Actinobacteria. Int. J. Syst. Evol. Microbiol. 59, 1082–1089. doi: 10.1099/ijs.0.65409-0
Kalyuzhnaya, M. G., Hristova, K. R., Lidstrom, M. E., and Chistoserdova, L. (2008). Characterization of a novel methanol dehydrogenase in representatives of Burkholderiales: implications for environmental detection of methylotrophy and evidence for convergent evolution. J. Bacteriol. 190, 3817–3823. doi: 10.1128/JB.00180-08
Kaneko, T., Nakamura, Y., Sato, S., Minamisawa, K., Uchiumi, T., Sasamoto, S., et al. (2002). Complete genomic sequence of nitrogen-fixing symbiotic bacterium Bradyrhizobium japonicum USDA110. DNA Res. 9, 189–197. doi: 10.1093/dnares/9.6.189
Kaszycki, P., Czechowska, K., Petryszak, P., Miêdzobrodzki, J., Pawlik, B., and Kołoczek, H. (2006). Methylotrophic extremophilic yeast Trichosporon sp.: a soil-derived isolate with potential applications in environmental biotechnology. Acta Biochim. Pol. 53, 463–473.
Keltjens, J. T., Pol, A., Reimann, J., and Op den Camp, H. J. (2014). PQQ-dependent methanol dehydrogenases: rare-earth elements make a difference. Appl. Microbiol. Biotechnol. 98, 6163–6183. doi: 10.1007/s00253-014-5766-8
Khadem, A. F., Pol, A., Wieczorek, A., Mohammadi, S. S., Francoijs, K. J., Stunnenberg, H. G., et al. (2011). Autotrophic methanotrophy in Verrucomicrobia: Methylacidiphilum fumariolicum SolV uses the Calvin-Benson-Bassham cycle for carbon dioxide fixation. J. Bacteriol. 193, 4438–4446. doi: 10.1128/JB.00407-11
Kjøller, A. H., and Struwe, S. (2002). “Fungal communities, succession, enzymes, and decomposition,” in Enzymes in the Environment: Activity, Ecology and Applications, eds R. G. Burns and R. P. Dick (New York, NY: Marcel Dekker, Inc), 267–284.
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1. doi: 10.1093/nar/gks808
Koch, I. H., Gich, F., Dunfield, P. F., and Overmann, J. (2008). Edaphobacter modestus gen. nov., sp. nov., and Edaphobacter aggregans sp. nov., Acidobacteria isolated from alpine and forest soils. Int. J. Syst. Evol. Microbiol. 58, 1114–1122. doi: 10.1099/ijs.0.65303-0
Kolb, S. (2009). Aerobic methanol-oxidizing Bacteria in soil. FEMS Microbiol. Lett. 300, 1–10. doi: 10.1111/j.1574-6968.2009.01681.x
Kolb, S., Knief, C., Dunfield, P. F., and Conrad, R. (2005). Abundance and activity of uncultured methanotrophic bacteria involved in the consumption of atmospheric methane in two forest soils. Environ. Microbiol. 7, 1150–1161. doi: 10.1111/j.1462-2920.2005.00791.x
Kolb, S., and Stacheter, A. (2013). Prerequisites for amplicon pyrosequencing of microbial methanol-utilizers in the environment. Front. Microbiol. 4:268. doi: 10.3389/fmicb.2013.00268
Kõljalg, U., Nilsson, R. H., Abarenkov, K., Tedersoo, L., Taylor, A. F., Bahram, M., et al. (2013). Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 22, 5271–5277. doi: 10.1111/mec.12481
Kudo, T., Matsushima, K., Itoh, T., Sasaki, J., and Suzuki, K. (1998). Description of four new species of the genus Kineosporia: Kineosporia succinea sp. nov., Kineosporia rhizophila sp. nov., Kineosporia mikuniensis sp. nov. and Kineosporia rhamnosa sp. nov., isolated from plant samples, and amended description of the genus Kineosporia. Int. J. Syst. Evol. Microbiol. 48, 1245–1255. doi: 10.1099/00207713-48-4-1245
Kulichevskaya, I. S., Danilova, O. V., Tereshina, V. M., Kevbrin, V. V., and Dedysh, S. N. (2014). Descriptions of Roseiarcus fermentans gen. nov., sp. nov., a bacteriochlorophyll a-containing fermentative bacterium related phylogenetically to alphaproteobacterial methanotrophs, and of the family Roseiarcaceae fam. nov. Int. J. Syst. Evol. Microbiol. 64, 2558–2565. doi: 10.1099/ijs.0.064576-0
Lau, E., Fisher, M. C., Steudler, P. A., and Cavanaugh, C. M. (2013). The methanol dehydrogenase gene, mxaF, as a functional and phylogenetic marker for proteobacterial methanotrophs in natural environments. PLoS ONE 8:e56993. doi: 10.1371/journal.pone.0056993
Loew, O. (1892). Ueber einen Bacillus, welcher ameisensäure und formaldehyd assimiliren kann. Centralbl. Bakteriol. 12, 462–465.
Lueders, T., Manefield, M., and Friedrich, M. W. (2004). Enhanced sensitivity of DNA- and rRNA-based stable isotope probing by fractionation and quantitative analysis of isopycnic centrifugation gradients. Environ. Microbiol. 6, 73–78. doi: 10.1046/j.1462-2920.2003.00536.x
Madhaiyan, M., Poonguzhali, S., Lee, J.-S., Senthilkumar, M., Lee, K. C., and Sundaram, S. (2010). Mucilaginibacter gossypii sp. nov. and Mucilaginibacter gossypiicola sp. nov., plant-growth-promoting bacteria isolated from cotton rhizosphere soils. J. Syst. Evol. Microbiol. 60, 2451–2457. doi: 10.1099/ijs.0.018713-0
Marín, I., and Arahal, D. R. (2013). “The family Beijerinckiaceae,” in The Prokaryotes – Alphaproteobacteria and Betaproteobacteria, eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (New York, NY: Springer), 115–133.
Masuda, S., Eda, S., Sugawara, C., Mitsui, H., and Minamisawa, K. (2010). The cbbL gene is required for thiosulfate-dependent autotrophic growth of Bradyrhizobium japonicum. Microbes Environ. 25, 220–223.
McCarthy, A. J. (1978). Lignocellulose-degrading Actinomycetes. FEMS Microbiol. Rev. 46, 145–163. doi: 10.1111/j.1574-6968.1987.tb02456.x
McDonald, I. R., Hall, G. H., Pickup, R. W., and Murrell, J. C. (1996). Methane oxidation potential and preliminary analysis of methanotrophs in blanket bog peat using molecular ecology techniques. FEMS Microbiol. Ecol. 21, 197–211. doi: 10.1111/j.1574-6941.1996.tb00347.x
McDonald, I. R., and Murrell, J. C. (1997). The methanol dehydrogenase structural gene mxaF and its use as a functional gene probe for methanotrophs and methylotrophs. Appl. Environ. Microbiol. 63, 3218–3224.
McDonald, I. R., Upton, M., Hall, G., Pickup, R. W., Edwards, C., Saunders, J. R., et al. (1999). Molecular ecological analysis of methanogens and methanotrophs in blanket bog peat. Microb. Ecol. 38, 225–233. doi: 10.1007/s002489900172
Messner, K., Fackler, K., Lamaipis, P., Gindl, W., Srebotnik, E., and Watanabe, T. (2003). “Overview of white-rot research: where we are today,” in Wood Deterioration and Preservation: Advances in Our Changing World (ACS Symposium Series 845), eds B. Goodell, D. D. Nicholas, and T. R. Schultz (Washington, DC: American Chemical Society), 73–96.
Miller, J. A., Kalyuzhnaya, M. G., Noyes, E., Lara, J. C., Lidstrom, M. E., and Chistoserdova, L. (2005). Labrys methylaminiphilus sp. nov., a novel facultatively methylotrophic bacterium from a freshwater lake sediment. Int. J. Syst. Evol. Microbiol. 55, 1247–1253. doi: 10.1099/ijs.0.63409-0
Millet, D. B., Jacob, D. J., Custer, T. G., de Gouw, J. A., Goldstein, A. H., Karl, T., et al. (2008). New constraints on terrestrial and oceanic sources of atmospheric methanol. Atmos. Chem. Phys. 8, 6887–6905. doi: 10.5194/acp-8-6887-2008
Moosvi, S. A., McDonald, I. R., Pearce, D. A., Kelly, D. P., and Wood, A. P. (2005). Molecular detection and isolation from Antarctica of methylotrophic bacteria able to grow with methylated sulfur compounds. Syst. Appl. Microbiol. 28, 541–554. doi: 10.1016/j.syapm.2005.03.002
Morris, S. A., Radajewski, S., Willison, T. W., and Murrell, J. C. (2002). Identification of the functionally active methanotroph population in a peat soil microcosm by stable-isotope probing. Appl. Environ. Microbiol. 68, 1446–1453. doi: 10.1128/AEM.68.3.1446-1453.2002
Muyzer, G., Brinkhoff, T., Nuebel, U., Santegoeds, C., Schäfer, H., and Waver, C. (1998). “Denaturing gradient gel electrophoresis (DGGE) in microbial ecology,” in Molecular Microbial Ecology Manual, eds A. D. L. Akkermans, J. D. van Elsas, and F. J. de Bruijn (Dordrecht: Kluwer Academic Publishers), 1–27.
Nebel, M. E., Wild, S., Holzhauser, M., Hüttenberger, L., Reitzig, R., Sperber, M., et al. (2011). Jaguc – a software package for environmental diversity analyzes. J. Bioinform. Comput. Biol. 9, 749–773. doi: 10.1142/S0219720011005781
Neufeld, J. D., Vohra, J., Dumont, M. G., Lueders, T., Manefield, M., Friedrich, M. W., et al. (2007). DNA stable-isotope probing. Nat. Protoc. 2, 860–866. doi: 10.1038/nprot.2007.109
Or, D., Smets, B. F., Wraith, J. M., Dechesne, A., and Friedman, S. P. (2007). Physical constraints affecting bacterial habitats and activity in unsaturated porous media – a review. Adv. Water Resour. 30, 1505–1527. doi: 10.1016/j.advwatres.2006.05.025
Pankratov, T. A., Serkebaeva, Y. M., Kulichevskaya, I. S., Liesack, W., and Dedysh, S. N. (2008). Substrate-induced growth and isolation of Acidobacteria from acidic Sphagnum peat. ISME J. 2, 551–560. doi: 10.1038/ismej.2008.7
Peyraud, R., Kiefer, P., Christen, P., Portais, J.-C., and Vorholt, J. A. (2012). Co-Consumption of methanol and succinate by Methylobacterium extorquens AM1. PLoS ONE 7:e48271. doi: 10.1371/journal.pone.0048271
Peyraud, R., Schneider, K., Kiefer, P., Massou, S., Vorholt, J. A., and Portais, J.-C. (2011). Genome-scale reconstruction and system level investigation of the metabolic network of Methylobacterium extorquens AM1. BMC Syst. Biol. 5:189. doi: 10.1186/1752-0509-5-189
Radajewski, S., Ineson, P., Parekh, N. R., and Murrell, J. C. (2000). Stable-isotope probing as a tool in microbial ecology. Nature 403, 646–649. doi: 10.1038/35001054
Radajewski, S., Webster, G., Reay, D. S., Morris, S. A., Ineson, P., Nedwell, D. B., et al. (2002). Identification of active methylotroph populations in an acidic forest soil by stable-isotope probing. Microbiology 148, 2331–2342. doi: 10.1099/00221287-148-8-2331
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Sharp, C. E., Smirnova, A. V., Graham, J. M., Stott, M. B., Khadka, R., Moore, T. R., et al. (2014). Distribution and diversity of Verrucomicrobia methanotrophs in geothermal and acidic environments. Environ. Microbiol. 16, 1867–1878. doi: 10.1111/1462-2920.12454
Skovran, E., Crowther, G. J., Guo, X., Yang, S., and Lidstrom, M. E. (2010). A systems biology approach uncovers cellular strategies used by Methylobacterium extorquens AM1 during the switch from multi- to single-carbon growth. PLoS ONE 5:e14091. doi: 10.1371/journal.pone.0014091
Stacheter, A., Noll, M., Lee, C. K., Selzer, M., Glowik, B., Ebertsch, L., et al. (2013). Methanol oxidation by temperate soils and environmental determinants of associated methylotrophs. ISME J. 7, 1051–1064. doi: 10.1038/ismej.2012.167
Sudtachat, N., Ito, N., Itakura, M., Masuda, S., Eda, S., Mitsui, H., et al. (2009). Aerobic vanillate degradation and C1 compound metabolism in Bradyrhizobium japonicum. Appl. Environ. Microbiol. 75, 5012–5017. doi: 10.1128/AEM.00755-09
Suneetha, V., and Khan, Z. A. (2010). “Actinomycetes: sources for soil enzymes”, in Soil Enzymology 22, eds G. Shukla and A. Varma (Berlin: Springer), 259–269.
Takishita, K., Tsuchiya, M., Reimer, J. D., and Maruyama, T. (2006). Molecular evidence demonstrating the basidiomycetous fungus Cryptococcus curvatus is the dominant microbial eukaryote in sediment at the Kuroshima knoll methane seep. Extremophiles 10, 165–169. doi: 10.1007/s00792-005-0495-7
Tamas, I., Smirnova, A. V., He, Z., and Dunfield, P. F. (2014). The (d)evolution of methanotrophy in the Beijerinckiaceae – a comparative genomics analysis. ISME J. 8, 369–382. doi: 10.1038/ismej.2013.145
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Taubert, M., Grob, C., Howat, A. M., Burns, O. J., Dixon, J. L., Chen, Y., et al. (2015). XoxF encoding an alternative methanol dehydrogenase is widespread in coastal marine environments. Environ. Microbiol. 17, 3937–3948. doi: 10.1111/1462-2920.12896
Voříšková, J., and Baldrian, P. (2013). Fungal community on decomposing leaf litter undergoes rapid successional changes. ISME J. 7, 477–486. doi: 10.1038/ismej.2012.116
Vorobev, A. V., Baani, M., Doronina, N. V., Brady, A. L., Liesack, W., Dunfield, P. F., et al. (2011). Methyloferula stellata gen. nov., sp. nov., an acidophilic, obligately methanotrophic bacterium that possesses only a soluble methane monooxygenase. Int. J. Syst. Evol. Microbiol. 61, 2456–2463. doi: 10.1099/ijs.0.028118-0
Vorobev, A. V., de Boer, W., Folman, L. B., Bodelier, P. L. E., Doronina, N. V., Suzina, N. E., et al. (2009). Methylovirgula ligni gen. nov., sp. nov., an obligately acidophilic, facultatively methylotrophic bacterium with a highly divergent mxaF gene. Int. J. Syst. Evol. Microbiol. 59, 2538–2545. doi: 10.1099/ijs.0.010074-0
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Wang, X., Sharp, C. E., Jones, G. M., Grasby, S. E., Brady, A. L., and Dunfield, P. F. (2015). Stable-isotope probing identifies uncultured Planctomycetes as primary degraders of a complex heteropolysaccharide in soil. Appl. Environ. Microbiol. 81, 4607–4615. doi: 10.1128/AEM.00055-15
Warneke, C., Karl, T., Judmaier, H., Hansel, A., Jordan, A., and Lindinger, W. (1999). Acetone, methanol, and other partially oxidized volatile organic emissions from dead plant matter by abiological processes: significance for atmospheric HOx chemistry. Glob. Biogeochem. Cycles 13, 9–17. doi: 10.1029/98GB02428
White, T. J., Bruns, T. D., Lee, S. B., and Taylor, J. W. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in PCR Protocols – a Guide to Methods and Applications, eds M. A. Innis, D. H. Gelfand, J. J. Sninsky, and T. J. White (Zürich: Academic Press), 315–322.
Whittenbury, R., Philips, K. C., and Wilkinson, J. F. (1970). Enrichment, isolation, and some properties of methane-utilizing bacteria. J. Gen. Microbiol. 61, 205–218. doi: 10.1099/00221287-61-2-205
Wohlfahrt, G., Amelynck, C., Ammann, C., Arneth, A., Bamberger, I., Goldstein, A. H., et al. (2015). An ecosystem-scale perspective of the net land methanol flux: synthesis of micrometeorological flux measurements. Atmos. Chem. Phys. 15, 7413–7427. doi: 10.5194/acp-15-7413-2015
Wubet, T., Christ, S., Schöning, I., Boch, S., Gawlich, M., Schnabel, B., et al. (2012). Differences in soil fungal communities between European beech (Fagus sylvatica L.) dominated forests are related to soil and understory vegetation. PLOS ONE 7:e47500. doi: 10.1371/journal.pone.0047500
Yarza, P., Ludwig, W., Euzéby, J., Amann, R., Schleifer, K.-H., Glöckner, F. O., et al. (2010). Update of the all-species living tree project based on 16S and 23S rRNA sequence analyzes. Syst. Appl. Microbiol. 33, 291–299. doi: 10.1016/j.syapm.2010.08.001
Keywords: DNA stable isotope probing, mxaF, bacterial 16S rRNA gene, fungal ITS, pH, high-throughput sequencing
Citation: Morawe M, Hoeke H, Wissenbach DK, Lentendu G, Wubet T, Kröber E and Kolb S (2017) Acidotolerant Bacteria and Fungi as a Sink of Methanol-Derived Carbon in a Deciduous Forest Soil. Front. Microbiol. 8:1361. doi: 10.3389/fmicb.2017.01361
Received: 18 March 2017; Accepted: 05 July 2017;
Published: 24 July 2017.
Edited by:
Svetlana N. Dedysh, Winogradsky Institute of Microbiology (RAS), RussiaReviewed by:
Marc Gregory Dumont, University of Southampton, United KingdomMarina G. Kalyuzhanaya, San Diego State University, United States
Copyright © 2017 Morawe, Hoeke, Wissenbach, Lentendu, Wubet, Kröber and Kolb. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mareen Morawe, bWFyZWVuLm1vcmF3ZUB1bmktYmF5cmV1dGguZGU= Steffen Kolb, a29sYkB6YWxmLmRl